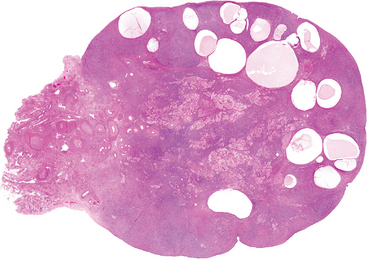
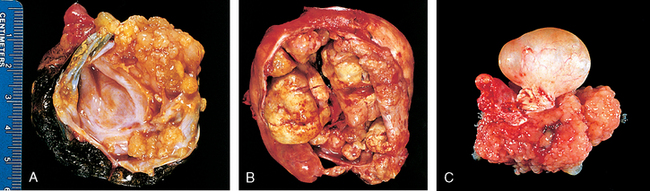
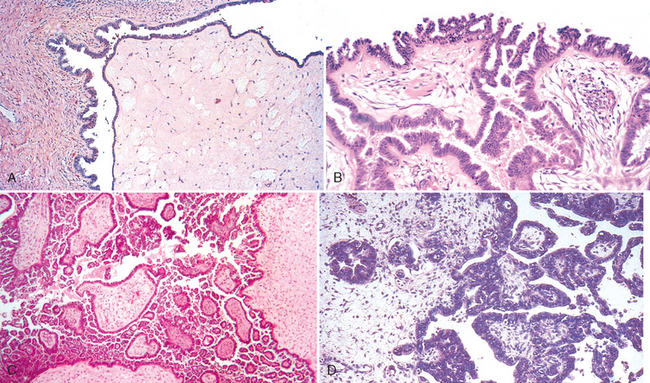
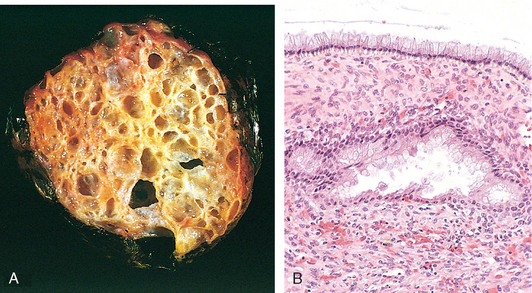
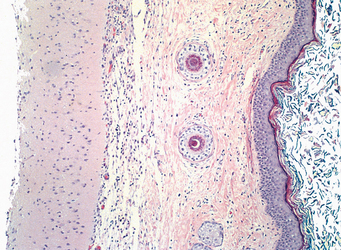
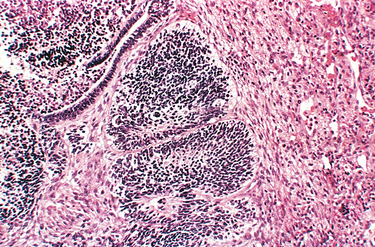
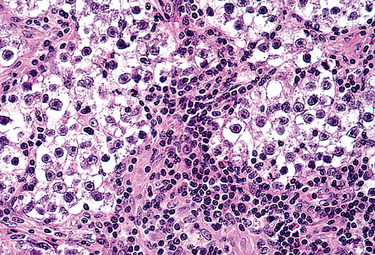
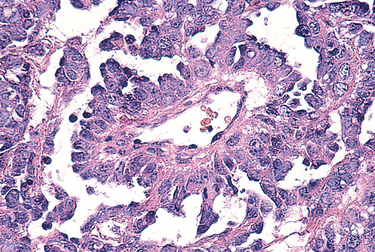
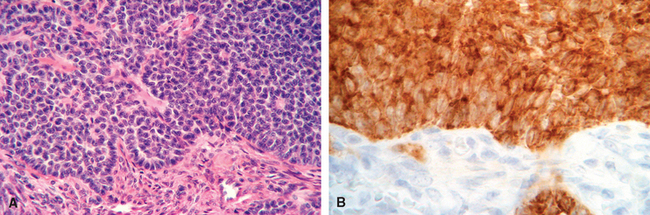
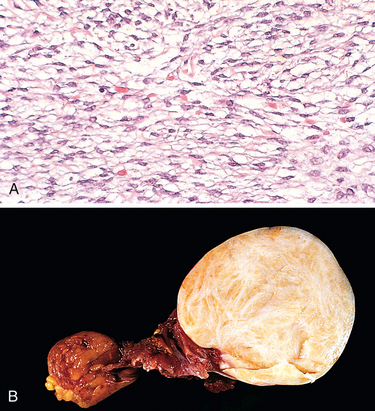
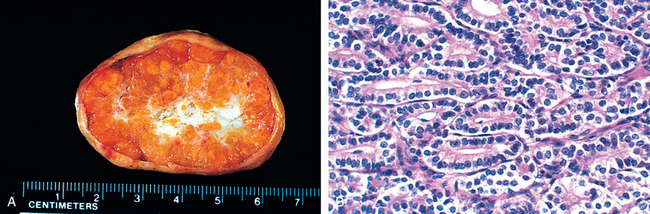
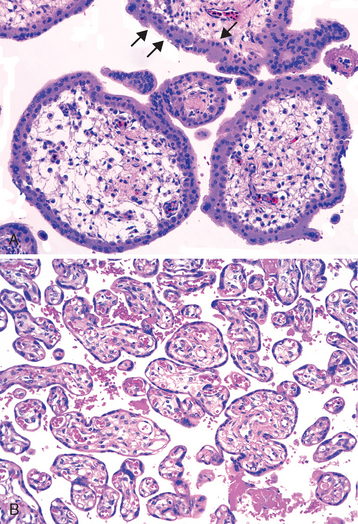
1 Robboy S, et al, editors. Embryology of the Female Genital Tract, 5th ed., New York: Springer-Verlag, 2002.
2 Kurita T, et al. Epithelial-stromal tissue interaction in paramesonephric (müllerian) epithelial differentiation. Dev Biol. 2001;240:194.
3 Quade BJ, et al. Expression of the p53 homologue p63 in early cervical neoplasia. Gynecol Oncol. 2001;80:24.
4 Malasanos TH. Sexual development of the fetus and pubertal child. Clin Obstet Gynecol. 1997;40:153.
5 Ince TA, et al. p63 coordinates anogenital modeling and epithelial cell differentiation in the developing female urogenital tract. Am J Pathol. 2002;161:1111.
6 Richart RM. Cervical intraepithelial neoplasia. Pathol Annu. 1973;8:301.
7 Kurman R, editor. Blaustein’s Pathology of the Female Genital Tract, 5th ed., New York: Springer-Verlag, 2002.
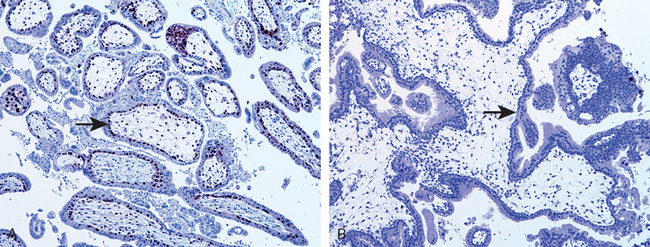
8 Benirschke KP, Baergen RN. Pathology of the Human Placenta, 5th ed. New York: Springer, 2006.
9 McMillan A, et al, editors. Clinical Practice in Sexually Transmissible Infections. London: WB Saunders, 2002.
10 Xu F, et al. Seroprevalence and coinfection with herpes simplex virus type 1 and type 2 in the United States, 1988–1994. J Infect Dis. 2002;185:1019.
11 Stanberry LR, et al. Prospects for control of herpes simplex virus disease through immunization. Clin Infect Dis. 2000;30:549.
12 Pararas MV, et al. Preterm birth due to maternal infection: causative pathogens and modes of prevention. Eur J Clin Microbiol Infect Dis. 2006;25:562.
13 Pinto AP, et al. Allelic imbalance in lichen sclerosus, hyperplasia, and intraepithelial neoplasia of the vulva. Gynecol Oncol. 2000;77:171.
14 de Koning MN, et al. Prevalence of mucosal and cutaneous human papillomaviruses in different histologic subtypes of vulvar carcinoma. Mod Pathol. 2008;21:334.
15 Vanin K, et al. Overexpression of wild-type p53 in lichen sclerosus adjacent to human papillomavirus-negative vulvar cancer. J Invest Dermatol. 2002;119:1027.
16 Jones RW, et al. Vulvar intraepithelial neoplasia: aspects of the natural history and outcome in 405 women. Obstet Gynecol. 2005;106:1319.
17 Belousova IE, et al. Vulvar Toker cells: the long-awaited missing link: a proposal for an origin-based histogenetic classification of extramammary Paget disease. Am J Dermatopathol. 2006;28:84.
18 Willman JH, et al. Vulvar clear cells of Toker: precursors of extramammary Paget’s disease. Am J Dermatopathol. 2005;27:185.
19 Schrager S, Potter BE. Diethylstilbestrol exposure. Am Fam Physician. 2004;69:2395.
20 Hilgers RD, et al. Embryonal rhabdomyosarcoma (botryoid type) of the vagina. A clinicopathologic review. Am J Obstet Gynecol. 1970;107:484.
21 Kaewsrichan J, et al. Selection and identification of anaerobic Lactobacilli producing inhibitory compounds against vaginal pathogens. FEMS Immunol Med Microbiol. 2006;48:75.
22 Munoz N, et al. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N Engl J Med. 2003;348:518.
23 Schiffman M, et al. Human papillomavirus and cervical cancer. Lancet. 2007;370:890.
24 Franco EL, et al. Epidemiology of acquisition and clearance of cervical human papillomavirus infection in women from a high-risk area for cervical cancer. J Infect Dis. 1999;180:1415.
25 Ostor AG. Natural history of cervical intraepithelial neoplasia: a critical review. Int J Gynecol Pathol. 1993;12:186.
26 Moscicki AB, et al. Risk of high-grade squamous intraepithelial lesion in HIV-infected adolescents. J Infect Dis. 2004;190:1413.
27 Saslow D, et al. American Cancer Society guideline for the early detection of cervical neoplasia and cancer. J Low Genit Tract Dis. 2003;7:67.
28 Wright TCJr., et al. Interim guidance for the use of human papillomavirus DNA testing as an adjunct to cervical cytology for screening. Obstet Gynecol. 2004;103:304.
29 Wright TCJr., et al. 2006 consensus guidelines for the management of women with abnormal cervical cancer screening tests. Am J Obstet Gynecol. 2007;197:346.
30 Cutts FT, et al. Human papillomavirus and HPV vaccines: a review. Bull World Health Organ. 2007;85:719.
31 Jabbour HN, et al. Endocrine regulation of menstruation. Endocr Rev. 2006;27:17.
32 Hou X, et al. Canonical Wnt signaling is critical to estrogen-mediated uterine growth. Mol Endocrinol. 2004;18:3035.
33 Groothuis PG, et al. Estrogen and the endometrium: lessons learned from gene expression profiling in rodents and human. Hum Reprod Update. 2007;13:405.
34 Kiviat NB, et al. Endometrial histopathology in patients with culture-proved upper genital tract infection and laparoscopically diagnosed acute salpingitis. Am J Surg Pathol. 1990;14:167.
35 Bulun SE. Mechanisms of disease: Endometriosis. New Engl J Med. 2009;360:268.
36 Noble LS, et al. Prostaglandin E2 stimulates aromatase expression in endometriosis-derived stromal cells. J Clin Endocrinol Metab. 1997;82:600.
37 Burney RO, et al. Gene expression analysis of endometrium reveals progesterone resistance and candidate susceptibility genes in women with endometriosis. Endocrinology. 2007;148:3814.
38 Nabeshima H, et al. Analysis of the clonality of ectopic glands in peritoneal endometriosis using laser microdissection. Fertil Steril. 2003;80:1144.
39 Wu Y, et al. Resolution of clonal origins for endometriotic lesions using laser capture microdissection and the human androgen receptor (HUMARA) assay. Fertil Steril. 2003;79(Suppl 1):710.
40 Wells M. Recent advances in endometriosis with emphasis on pathogenesis, molecular pathology, and neoplastic transformation. Int J Gynecol Pathol. 2004;23:316.
41 Corley D, et al. Postmenopausal bleeding from unusual endometrial polyps in women on chronic tamoxifen therapy. Obstet Gynecol. 1992;79:111.
42 Kazmerczak B, et al. HMGIY is the target of 6p21.3 rearrangements in various benign mesenchymal tumors. Genes Chromosome Cancer. 1998;23:279.
43 Kurman RJ, et al. The behavior of endometrial hyperplasia. A long-term study of “untreated” hyperplasia in 170 patients. Cancer. 1985;56:403.
44 Di Cristofano A, Ellenson LH. Endometrial carcinoma. Annu Rev Pathol. 2007;2:57.
45 Tashiro H, et al. Mutations in PTEN are frequent in endometrial carcinoma but rare in other common gynecological malignancies. Cancer Res. 1997;57:3935.
46 Maxwell GL, et al. Mutation of the PTEN tumor suppressor gene in endometrial hyperplasias. Cancer Res. 1998;58:2500.
47 Vilgelm A, et al. Akt-mediated phosphorylation and activation of estrogen receptor alpha is required for endometrial neoplastic transformation in Pten+/− mice. Cancer Res. 2006;66:3375.
48 Silverberg SG. Problems in the differential diagnosis of endometrial hyperplasia and carcinoma. Mod Pathol. 2000;13:309.
49 Trimble CL, et al. Concurrent endometrial carcinoma in women with a biopsy diagnosis of atypical endometrial hyperplasia: a Gynecologic Oncology Group study. Cancer. 2006;106:812.
50 Ferenczy A, Gelfand M. The biologic significance of cytologic atypia in progestogen-treated endometrial hyperplasia. Am J Obstet Gynecol. 1989;160:126.
51 O’Connell JT, et al. Identification of a basal/reserve cell immunophenotype in benign and neoplastic endometrium: a study with the p53 homologue p63. Gynecol Oncol. 2001;80:30.
52 Sherman ME. Theories of endometrial carcinogenesis: a multidisciplinary approach. Mod Pathol. 2000;13:295.
53 Mutter GL, et al. Allelotype mapping of unstable microsatellites establishes direct lineage continuity between endometrial precancers and cancer. Cancer Res. 1996;56:4483.
54 Levine RL, et al. PTEN mutations and microsatellite instability in complex atypical hyperplasia, a precursor lesion to uterine endometrioid carcinoma. Cancer Res. 1998;58:3254.
55 Oda K, et al. High frequency of coexistent mutations of PIK3CA and PTEN genes in endometrial carcinoma. Cancer Res. 2005;65:10669.
56 Hayes MP, et al. PIK3CA and PTEN mutations in uterine endometrioid carcinoma and complex atypical hyperplasia. Clin Cancer Res. 2006;12:5932.
57 Tashiro H, et al. p53 gene mutations are common in uterine serous carcinoma and occur early in their pathogenesis. Am J Pathol. 1997;150:177.
58 Grice J, et al. Uterine papillary serous carcinoma: evaluation of long-term survival in surgically staged patients. Gynecol Oncol. 1998;69:69.
59 Tay EH, Ward BG. The treatment of uterine papillary serous carcinoma (UPSC): are we doing the right thing? Int J Gynecol Cancer. 1999;9:463.
60 Lim P, et al. Early stage uterine papillary serous carcinoma of the endometrium: effect of adjuvant whole abdominal radiotherapy and pathologic parameters on outcome. Cancer. 2001;91:752.
61 Silverberg SG, et al. Carcinosarcoma (malignant mixed mesodermal tumor) of the uterus. A Gynecologic Oncology Group pathologic study of 203 cases. Int J Gynecol Pathol. 1990;9:1.
62 Abeln EC, et al. Molecular genetic evidence for the conversion hypothesis of the origin of malignant mixed müllerian tumours. J Pathol. 1997;183:424.
63 Clement PB, Scully RE. Müllerian adenosarcoma of the uterus: a clinicopathologic analysis of 100 cases with a review of the literature. Hum Pathol. 1990;21:363.
64 Chang KL, et al. Primary uterine endometrial stromal neoplasms. A clinicopathologic study of 117 cases. Am J Surg Pathol. 1990;14:415.
65 Li H, et al. Gene fusion and RNA trans-splicing in normal and neoplastic cells. Cell Cycle. 2009;8:218.
66 Ligon AH, Morton CC. Leiomyomata: treatability and cytogenetic studies. Human Reproduction update. 2008;7:8.
67 Ligon AH, Morton CC. Genetics of uterine leiomyomata. Genes Chromosomes Cancer. 2000;28:235.
68 Quade BJ, et al. Frequent loss of heterozygosity for chromosome 10 in uterine leiomyosarcoma in contrast to leiomyoma. Am J Pathol. 1999;154:945.
69 Bell SW, et al. Problematic uterine smooth muscle neoplasms. A clinicopathologic study of 213 cases. Am J Surg Pathol. 1994;18:535.
70 Obermair A, et al. Primary fallopian tube carcinoma: the Queensland experience. Int J Gynecol Cancer. 2001;11:69.
71 Aziz S, et al. A genetic epidemiological study of carcinoma of the fallopian tube. Gynecol Oncol. 2001;80:341.
72 Ovarian pathology in infertility. In: Young RHS, Scully RE, Krausz FT, editors. Pathology of Reproductive Failure. Baltimore: Williams and Wilkins; 1991:104-139.
73 Homburg R. Polycystic ovary syndrome—from gynaecological curiosity to multisystem endocrinopathy. Hum Reprod. 1996;11:29.
74 Ovalle F, Azziz R. Insulin resistance, polycystic ovary syndrome, and type 2 diabetes mellitus. Fertil Steril. 2002;77:1095.
75 The Ovary. In: Young RH, Steinberg S, et al, editors. Diagnostic Surgical Pathology. New York: Raven Press, 1994.
76 Scully RE. Pathology of ovarian cancer precursors. J Cell Biochem Suppl. 1995;23:208.
77 Narod SA, Boyd J. Current understanding of the epidemiology and clinical implications of BRCA1 and BRCA2 mutations for ovarian cancer. Curr Opin Obstet Gynecol. 2002;14:19.
78 Narod SA, et al. Tubal ligation and risk of ovarian cancer in carriers of BRCA1 or BRCA2 mutations: a case-control study. Lancet. 2001;357:1467.
79 Ness RB, et al. Oral contraceptives, other methods of contraception, and risk reduction for ovarian cancer. Epidemiology. 2001;12:307.
80 Malpica A, et al. Interobserver and intraobserver variability of a two-tier system for grading ovarian serous carcinoma. Am J Surg Pathol. 2007;31:1168.
81 Bell DA. Origins and molecular pathology of ovarian cancer. Mod Pathol. 2005;18:S19.
82 Shih IeM, Kurman RJ. Ovarian tumorigenesis: a proposed model based on morphological and molecular genetic analysis. Am J Pathol. 2004;164:1511.
83 Werness BA, et al. Histopathology of familial ovarian tumors in women from families with and without germline BRCA1 mutations. Hum Pathol. 2000;31:1420.
84 Szych C, et al. Molecular genetic evidence supporting the clonality and appendiceal origin of pseudomyxoma peritonei in women. Am J Pathol. 1999;154:1849.
85 Cuatrecasas M, et al. K-RAS mutations in mucinous ovarian tumors: a clinicopathologic and molecular study of 95 cases. Cancer. 1997;79:1581.
86 Lee KR, Scully RE. Mucinous tumors of the ovary: a clinicopathologic study of 196 borderline tumors (of intestinal type) and carcinomas, including an evaluation of 11 cases with “pseudomyxoma peritonei.”. Am J Surg Pathol. 2000;24:1447.
87 Watkin W, et al. Mucinous carcinoma of the ovary. Pathologic prognostic factors. Cancer. 1992;69:208.
88 Ronnett BM, et al. Immunohistochemical evidence supporting the appendiceal origin of pseudomyxoma peritonei in women. Int J Gynecol Pathol. 1997;16:1.
89 Eifel P, et al. Simultaneous presentation of carcinoma involving the ovary and the uterine corpus. Cancer. 1982;50:163.
90 Catasus L, et al. Molecular genetic alterations in endometrioid carcinomas of the ovary: similar frequency of β-catenin abnormalities but lower rate of microsatellite instability and PTEN alterations than in uterine endometrioid carcinomas. Hum Pathol. 2004;35:1360.
91 Sato N, et al. Loss of heterozygosity on 10q23.3 and mutation of the tumor suppressor gene PTEN in benign endometrial cyst of the ovary: possible sequence progression from benign endometrial cyst to endometrioid carcinoma and clear cell carcinoma of the ovary. Cancer Res. 2000;60:7052.
92 Berek JS, Bast RCJr. Ovarian cancer screening. The use of serial complementary tumor markers to improve sensitivity and specificity for early detection. Cancer. 1995;76(10 Suppl):2092.
93 Kim JH, et al. Osteopontin as a potential diagnostic biomarker for ovarian cancer. JAMA. 2002;287:1671.
94 Petricoin EF, et al. Use of proteomic patterns in serum to identify ovarian cancer. Lancet. 2002;359:572.
95 Hankinson SE, et al. Tubal ligation, hysterectomy, and risk of ovarian cancer. A prospective study. JAMA. 1993;270:2813.
96 Linder D, et al. Pathenogenic origin of benign ovarian teratomas. N Engl J Med. 1975;292:63.
97 Mutter GL. Teratoma genetics and stem cells: a review. Obstet Gynecol Surv. 1987;42:661.
98 O’Connor DM, Norris HJ. The influence of grade on the outcome of stage I ovarian immature (malignant) teratomas and the reproducibility of grading. Int J Gynecol Pathol. 1994;13:283.
99 Hole-Hansen CE, et al. Ovarian dysgerminomas are characterized by frequent KIT mutations and abundant expression of pluripotency markers. Mol Cancer. 2007;6:12.
100 Sever M, et al. Expression of CD117 (C-KIT) receptor in dysgerminoma of the ovary: diagnostic and therapeutic implications. Mod Pathol. 2005;18:1411.
101 Williams S, et al. Adjuvant therapy of ovarian germ cell tumors with cisplatin, etoposide, and bleomycin: a trial of the Gynecologic Oncology Group. J Clin Oncol. 1994;12:701.
102 Young RH, Scully RE. Ovarian sex cord–stromal tumors: recent progress. Int J Gynecol Pathol. 1982;1:101.
103 Robertson DM, et al. Inhibins/activins as diagnostic markers for ovarian cancer. Mol Cell Endocrinol. 2002;191:97.
104 Prat J, Scully RE. Cellular fibromas and fibrosarcomas of the ovary: a comparative clinicopathologic analysis of seventeen cases. Cancer. 1981;47:2663.
105 Roth LM, et al. Sertoli–Leydig cell tumors: a clinicopathologic study of 34 cases. Cancer. 1981;48:187.
106 Hart WR, Burkons DM. Germ cell neoplasms arising in gonadoblastomas. Cancer. 1979;43:669.
107 Wilcox AJ, et al. Incidence of early loss of pregnancy. N Engl J Med. 1988;319:189.
108 Rossing MA, et al. Past use of an intrauterine device and risk of tubal pregnancy. Epidemiology. 1993;4:245.
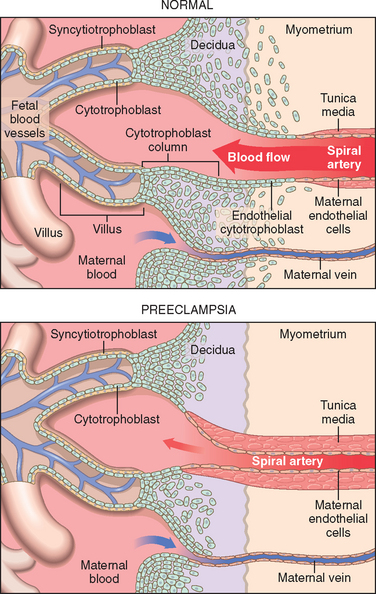
109 Schmidt M, et al. Altered angiogenesis in preeclampsia: evaluation of a new test system for measuring placental growth factor. Clin Chem Lab Med. 2007;45:1504.
110 Baumwell S, Karumanchi SA. Pre-eclampsia: clinical manifestations and molecular mechanisms. Nephron Clin Pract. 2007;106:c72.
111 Venkatesha S, et al. Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat Med. 2006;12:642.
112 Maynard S, Epstein FH, Karumanchi SA. Preeclampsia and angiogenic imbalance. Ann Rev Med. 2008;59:61.
113 Clark BA, et al. Urinary cyclic GMP, endothelin, and prostaglandin E2 in normal pregnancy and preeclampsia. Am J Perinatol. 1997;14:559.
114 Bracken MB, et al. Epidemiology of hydatidiform mole and choriocarcinoma. Epidemiol Rev. 1984;6:52.
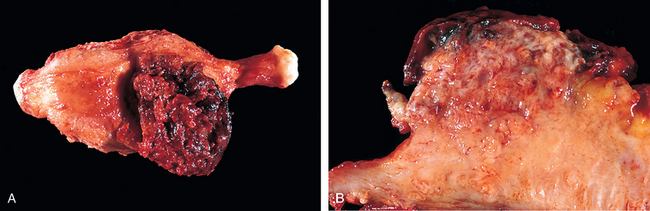
115 Lurain JR, et al. Natural history of hydatidiform mole after primary evacuation. Am J Obstet Gynecol. 1983;145:591.
116 Papadopoulos AJ, et al. Twenty-five years’ clinical experience with placental site trophoblastic tumors. J Reprod Med. 2002;47:460.
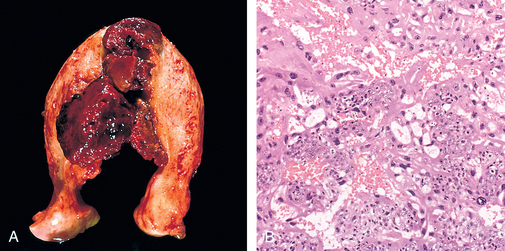
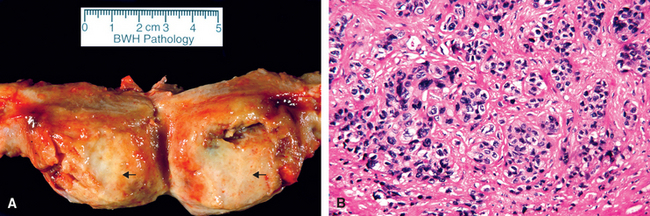
117 Chang YL, et al. Prognostic factors and treatment for placental site trophoblastic tumor—report of 3 cases and analysis of 88 cases. Gynecol Oncol. 1999;73:216.
118 Baergen RN, et al. Placental site trophoblastic tumor: a study of 55 cases and review of the literature emphasizing factors of prognostic significance. Gynecol Oncol. 2006;100:511.