Chapter 18 Liver and Biliary Tract
 THE LIVER
THE LIVER
THE LIVER
The normal adult liver weighs 1400 to 1600 gm, constituting approximately 2.5% of body weight. The liver has a dual blood supply: the portal vein provides 60% to 70% of hepatic blood flow, and the hepatic artery supplies 30% to 40%. The portal vein and the hepatic artery enter the liver through the hilum, also called porta hepatis, which is a transverse fissure in the inferior surface of the liver. Within the liver, the branches of the portal veins, hepatic arteries, and bile ducts travel in parallel in portal tracts, ramifying variably through 17 to 20 orders of branches.
Terminology of the hepatic microarchitecture is based on two different concepts: the hepatic lobule and the hepatic acinus. According to the lobular model, the liver is divided into 1- to 2-mm diameter hexagonal lobules oriented around the terminal tributaries of the hepatic vein (terminal hepatic veins), with portal tracts at the periphery of the lobule. The hepatocytes in the vicinity of the terminal hepatic vein are called “centrilobular”; those near the portal tract are “periportal” (Fig. 18-1). In the acinar model the hepatocytes near the terminal hepatic veins are the distal apices of roughly triangular acini, whose bases are formed by the penetrating septal venules from the portal vein extending out from the portal tracts.1 In the acinus the parenchyma is divided into three zones, zone 1 being closest to the vascular supply, zone 3 abutting the terminal hepatic venule and most remote from the afferent blood supply, and zone 2 being intermediate. Regardless of the model used, zonation of the parenchyma is an important concept because of the gradient of activity displayed by many hepatic enzymes, and the zonal distribution of certain types of hepatic injury. While the acinar model best describes the physiologic relationships between hepatocytes and their vascular supply, the histopathology of the liver is usually discussed on the basis of a lobular architecture.
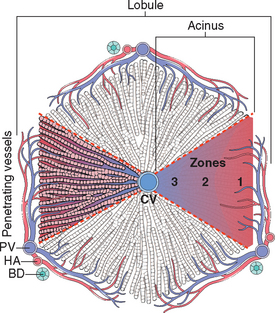
FIGURE 18-1 Microscopic anatomy of the liver; the two models, hepatic lobular model and acinar model, are illustrated. In the lobular model the terminal hepatic vein (CV) is at the center of a “lobule,” while the portal tracts (PV) are at the periphery. Pathologists refer to the regions of the parenchyma as “periportal and centrilobular.” In the acinar model, on the basis of blood flow, three zones can be defined, zone 1 being the closest to the blood supply and zone 3 being the farthest. BD, bile duct; HA, hepatic artery.
Hepatocytes are organized into cribriform, anastomosing sheets or “plates” extending from portal tracts to the terminal hepatic veins. Between the plates of hepatocytes are vascular sinusoids. Blood traverses the sinusoids and exits into the terminal hepatic veins through numerous orifices in the vein wall. Hepatocytes are thus bathed on two sides by well-mixed portal venous and hepatic arterial blood, making hepatocytes among the most richly perfused cells in the body. The sinusoids are lined by fenestrated and discontinuous endothelial cells. Deep to the endothelial cells lies the space of Disse, into which protrude abundant microvilli of hepatocytes. Scattered Kupffer cells of the mononuclear phagocyte system are attached to the luminal face of endothelial cells, and fat-containing hepatic stellate cells (HSCs) are found in the space of Disse. Between abutting hepatocytes are bile canaliculi, which are channels 1 to 2 μm in diameter, formed by grooves in the plasma membranes of facing hepatocytes and separated from the vascular space by tight junctions. These channels drain into the canals of Hering, ductular structures that connect the bile canaliculi to bile ductules in the periportal region. The ductules empty into the terminal bile ducts within the portal tracts.2 The liver also contains lymphocytes, including relatively large numbers of natural killer cells, and NK-T cells (Chapter 6).
General Features of Hepatic Disease
The liver is vulnerable to a wide variety of metabolic, toxic, microbial, circulatory, and neoplastic insults. The major primary diseases of the liver are viral hepatitis, alcoholic liver disease, nonalcoholic fatty liver disease (NAFLD), and hepatocellular carcinoma (HCC). Hepatic damage also occurs secondary to some of the most common diseases in humans, such as cardiac decompensation, disseminated cancer, and extrahepatic infections. The enormous functional reserve of the liver masks the clinical impact of mild liver damage, but with progression of diffuse disease or disruption of bile flow, the consequences of deranged liver function may become life-threatening.
With the rare exception of fulminant hepatic failure, liver disease is an insidious process in which clinical detection and symptoms of hepatic decompensation may occur weeks, months, or many years after the onset of injury. The ebb and flow of hepatic injury may be imperceptible to the patient and detectable only by abnormal laboratory tests (Table 18-1), and liver injury and healing may also occur without clinical detection. Hence, individuals with hepatic abnormalities who are referred to hepatologists most frequently have chronic liver disease. Surveillance studies in the United States document an annual incidence of newly diagnosed chronic liver disease of 72 per 100,000 population.3 Liver disease accounts for over 27,000 deaths per year in the United States (1.1% of all deaths).
TABLE 18-1 Laboratory Evaluation of Liver Disease
| Test Category | Serum Measurement* |
|---|---|
| Hepatocyte integrity | |
| Biliary | |
| Hepatocyte function |
* The most common tests are in italics.
† An elevation implicates liver disease.
‡ A decrease implicates liver disease.
PATTERNS OF HEPATIC INJURY
The liver has a relatively limited repertoire of cellular and tissue responses to injury, regardless of cause. The most common are:
Clinically, a few common syndromes occur that are a consequence of many different diseases. Before considering specific diseases, we will discuss some of these syndromes, which include hepatic failure, cirrhosis, portal hypertension, and disturbances of bilirubin metabolism causing jaundice and cholestasis.
HEPATIC FAILURE
The most severe clinical consequence of liver disease is hepatic failure. It may be the result of sudden and massive hepatic destruction (fulminant hepatic failure), which accounts for about 2000 cases per year in the United States, or, more often, represents the end stage of progressive chronic damage to the liver. End-stage liver disease may occur by insidious destruction of hepatocytes or by repetitive discrete waves of parenchymal damage. In cases of severe hepatic dysfunction, hepatic failure is often triggered by intercurrent diseases. Whatever the sequence, 80% to 90% of hepatic functional capacity must be lost before hepatic failure ensues. When the liver can no longer maintain homeostasis, transplantation offers the best hope for survival; the mortality of hepatic failure without liver transplantation is about 80%.
The alterations that cause liver failure fall into three categories:4
Clinical Features.
The clinical signs of hepatic failure are much the same regardless of cause, and are the result of hepatocytes failing to perform their homeostatic functions. Jaundice is an almost invariable finding. Hypoalbuminemia, which predisposes to peripheral edema, and hyperammonemia, which plays a major role in cerebral dysfunction, are worrisome developments. Fetor hepaticus is a characteristic body odor that is variously described as “musty” or “sweet and sour.” It is related to the formation of mercaptans by the action of gastrointestinal bacteria on the sulfurcontaining amino acid methionine, and shunting of splanchnic blood from the portal into the systemic circulation (portosystemic shunting). Impaired estrogen metabolism and consequent hyperestrogenemia are the putative causes of palmar erythema (a reflection of local vasodilatation) and spider angiomas of the skin. Each angioma is a central, pulsating, dilated arteriole from which small vessels radiate. In the male, hyperestrogenemia also leads to hypogonadism and gynecomastia.
Hepatic failure is life-threatening, because with severely impaired liver function, patients are highly susceptible to encephalopathy and failure of multiple organ systems. Respiratory failure with pneumonia, and sepsis combined with renal failure, claim the lives of many individuals with hepatic failure. A coagulopathy develops, attributable to impaired hepatic synthesis of several blood clotting factors. These defects can lead to massive gastrointestinal bleeding. Intestinal absorption of blood places a further metabolic load on the liver, which worsens the extent of hepatic failure. A rapid downhill course is usual, death occurring within weeks to a few months. A fortunate few survive acute episodes of hepatic failure, and hepatic function can be restored by hepatocellular regeneration if the liver does not have advanced fibrosis. As noted, liver transplantation may be life-saving.
Three particular complications associated with hepatic failure merit separate consideration, since they have grave implications.
CIRRHOSIS
Cirrhosis is the twelfth most common cause of death in the United States, accounting for most liver-related deaths. The chief worldwide causes of cirrhosis are alcohol abuse, viral hepatitis, and non-alcoholic steatohepatitis (NASH). Other etiologies include biliary disease and iron overload. Cirrhosis, as the end stage of chronic liver disease, is defined by three main morphologic characteristics:
Pathogenesis.
The central pathogenic processes in cirrhosis are death of hepatocytes, extracellular matrix (ECM) deposition, and vascular reorganization.10 In the normal liver, interstitial collagens (types I and III) are concentrated in portal tracts and around central veins, and thin strands of type IV collagen are present in the space of Disse. In cirrhosis, types I and III collagen are deposited in the space of Disse, creating fibrotic septal tracts. The vascular architecture of the liver is disrupted by the parenchymal damage and scarring, with the formation of new vascular channels in the fibrotic septa that connect the vessels in the portal region (hepatic arteries and portal veins) to terminal hepatic veins, shunting blood from the parenchyma. The deposition of collagen in the space of Disse is accompanied by the loss of fenestrations of sinusoidal endothelial cells (capillarization of sinusoids), impairing the function of sinusoids as channels that permit the exchange of solutes between hepatocytes and plasma (Fig. 18-2).
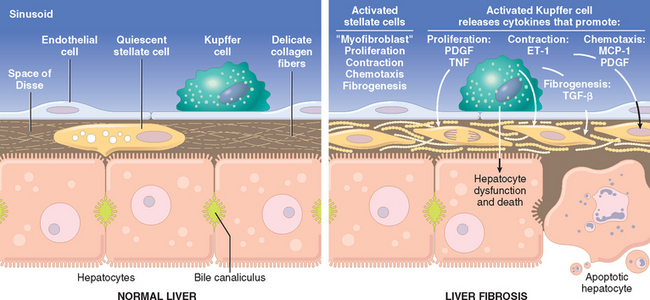
FIGURE 18-2 Stellate cell activation and liver fibrosis. Kupffer cell activation leads to secretion of multiple cytokines. Platelet-derived growth factor (PDGF) and tumor necrosis factor (TNF) activate stellate cells, and contraction of the activated stellate cells is stimulated by endothelin-1 (ET-1). Fibrogenesis is stimulated by transforming growth factor β (TGF-β). Chemotaxis of activated stellate cells to areas of injury is promoted by PDGF and monocyte chemotactic protein-1 (MCP-1). See text for details.
The predominant mechanism of fibrosis is the proliferation of hepatic stellate cells and their activation into highly fibrogenic cells, but other cell types, such as portal fibroblasts, fibrocytes, and cells derived from epithelium-mesenchymal transitions may also produce collagen. Proliferation of hepatic stellate cells and their activation into myofibroblasts is initiated by a series of changes that include an increase in the expression of platelet-derived growth factor receptor β (PDGFR-β) in the stellate cells. At the same time, Kupffer cells and lymphocytes release cytokines and chemokines that modulate the expression of genes in stellate cells that are involved in fibrogenesis. These include transforming growth factor β (TGF-β) and its receptors, metalloproteinase 2 (MMP−2), and tissue inhibitors of metalloproteinases 1 and 2 (TIMP-1 and −2). As they are converted into myofibroblasts, the cells release chemotactic and vasoactive factors, cytokines, and growth factors. Myofibroblasts are contractile cells, capable of constricting sinusoidal vascular channels and increasing vascular resistance within the liver parenchyma; their contraction is stimulated by endothelin-1 (ET-1). The stimuli for stellate cell activation may originate from several sources (Fig 18-2): (a) chronic inflammation, with production of inflammatory cytokines such as tumor necrosis factor (TNF), lymphotoxin, and interleukin 1β (IL-1β), and lipid peroxidation products; (b) cytokine and chemokine production by Kupffer cells, endothelial cells, hepatocytes, and bile duct epithelial cells; in response to (c) disruption of the ECM; and (d) direct stimulation of stellate cells by toxins.
Throughout the process of liver damage and fibrosis in the development of cirrhosis, the surviving hepatocytes are stimulated to regenerate and proliferate as spherical nodules within the confines of the fibrous septa. The net outcome is a fibrotic, nodular liver in which delivery of blood to hepatocytes is severely compromised, as is the ability of hepatocytes to secrete substances into plasma. Disruption of the interface between the parenchyma and portal tracts may also obliterate biliary channels, leading to the development of jaundice.
Clinical Features.
About 40% of individuals with cirrhosis are asymptomatic until late in the course of the disease. When symptomatic, they present with nonspecific clinical manifestations: anorexia, weight loss, weakness, and, in advanced disease, symptoms and signs of hepatic failure discussed earlier. Incipient or overt hepatic failure may develop, usually precipitated by a superimposed metabolic load on the liver, usually from systemic infection or gastrointestinal hemorrhage. Imbalances of pulmonary blood flow may lead to severely impaired oxygenation (hepatopulmonary syndrome, already discussed under liver failure), further stressing the patient. The ultimate mechanism of deaths in most cirrhotic patients is (1) progressive liver failure, (2) a complication related to portal hypertension, or (3) the development of hepatocellular carcinoma. In a small number of cases, cessation of liver injury may give the necessary time for resorption of the fibrous tissue and “reversal” of the cirrhosis.11 Even in such instances, the portal hypertension and risk of hepatocellular carcinoma remain.
PORTAL HYPERTENSION
Increased resistance to portal blood flow may develop in a variety of circumstances, which can be divided into prehepatic, intrahepatic, and posthepatic causes. The major prehepatic conditions are obstructive thrombosis, narrowing of the portal vein before it ramifies within the liver, or massive splenomegaly with increased splenic vein blood flow. The main post-hepatic causes are severe right-sided heart failure, constrictive pericarditis, and hepatic vein outflow obstruction. The dominant intrahepatic cause is cirrhosis, accounting for most cases of portal hypertension. Far less frequent intrahepatic causes are schistosomiasis, massive fatty change, diffuse fibrosing granulomatous disease such as sarcoidosis, and diseases affecting the portal microcirculation such as nodular regenerative hyperplasia (discussed later).
The pathophysiology of portal hypertension is complex and involves the resistance to portal flow at the level of sinusoids and the increase in portal flow caused by hyperdynamic circulation.
The four major clinical consequences of portal hypertension are (1) ascites, (2) the formation of portosystemic venous shunts, (3) congestive splenomegaly, and (4) hepatic encephalopathy (discussed earlier). These are illustrated in Figure 18-3.

FIGURE 18-3 The major clinical consequences of portal hypertension in the setting of cirrhosis, shown for the male. In women, oligomenorrhea, amenorrhea, and sterility are frequent, as a result of hypogonadism.
Ascites.
Ascites is the accumulation of excess fluid in the peritoneal cavity. In 85% of cases, ascites is caused by cirrhosis. Ascites usually becomes clinically detectable when at least 500 mL have accumulated. The fluid is generally serous, having less than 3 gm/dL of protein (largely albumin), and a serum to ascites albumin gradient of ≥1.1 gm/dL. The concentration of solutes such as glucose, sodium, and potassium are similar to that in the blood. The fluid may contain a scant number of mesothelial cells and mononuclear leukocytes. Influx of neutrophils suggests secondary infection, whereas the presence of blood cells points to possible disseminated intra-abdominal cancer. With long-standing ascites, seepage of peritoneal fluid through transdiaphragmatic lymphatics may produce hydrothorax, more often on the right side.
The pathogenesis of ascites is complex, involving the following mechanisms:11,12
Portosystemic Shunts.
With the rise in portal system pressure, the flow is reversed from portal to systemic circulation by dilation of collateral vessels and development of new vessels. Venous bypasses develop wherever the systemic and portal circulation share common capillary beds (see Fig. 18-3). Principal sites are veins around and within the rectum (manifest as hemorrhoids), the esophagogastric junction (producing varices), the retroperitoneum, and the falciform ligament of the liver (involving periumbilical and abdominal wall collaterals). Although hemorrhoidal bleeding may occur, it is rarely massive or life-threatening. Much more important are the esophagogastric varices that appear in about 40% of individuals with advanced cirrhosis of the liver and cause massive hematemesis and death in about half of them. Each episode of bleeding is associated with a 30% mortality. Abdominal wall collaterals appear as dilated subcutaneous veins extending from the umbilicus toward the rib margins (caput medusae) and constitute an important clinical hallmark of portal hypertension.
Splenomegaly.
Long-standing congestion may cause congestive splenomegaly. The degree of splenic enlargement varies widely and may reach as much as 1000 gm, but it is not necessarily correlated with other features of portal hypertension. The massive splenomegaly may secondarily induce hematologic abnormalities attributable to hypersplenism, such as thrombocytopenia or even pancytopenia.
JAUNDICE AND CHOLESTASIS
The common causes of jaundice are bilirubin overproduction, hepatitis, and obstruction of the flow of bile. Hepatic bile serves two major functions: (1) the emulsification of dietary fat in the lumen of the gut through the detergent action of bile salts, and (2) the elimination of bilirubin, excess cholesterol, xenobiotics, and other waste products that are insufficiently water-soluble to be excreted into urine. Alterations of bile formation become clinically evident as yellow discoloration of the skin and sclera (jaundice and icterus, respectively) due to retention of bilirubin, and as cholestasis, characterized by systemic retention of not only bilirubin but also other solutes eliminated in bile. To understand the pathophysiology of jaundice it is important first to become familiar with the major aspects of bile formation and metabolism. The metabolism of bilirubin by the liver consists of four separate but interrelated events: uptake from the circulation; intracellular storage; conjugation with glucoronic acid; and biliary excretion. These are described next.
Bilirubin and Bile Formation
Bilirubin is the end product of heme degradation (Fig. 18-4). The majority of daily production (0.2 to 0.3 gm, 85%) is derived from breakdown of senescent red cells by the mononuclear phagocytic system, especially in the spleen, liver, and bone marrow. Most of the remainder (15%) of bilirubin is derived from the turnover of hepatic heme or hemoproteins (e.g., the P-450 cytochromes) and from premature destruction of red cell precursors in the bone marrow (Chapter 13). Whatever the source, intracellular heme oxygenase oxidizes heme to biliverdin (step 1 in Fig. 18-4), which is immediately reduced to bilirubin by biliverdin reductase. Bilirubin thus formed outside the liver is released and bound to serum albumin (step 2). Albumin binding is necessary to transport bilirubin because bilirubin is virtually insoluble in aqueous solutions at physiologic pH. Hepatic processing of bilirubin involves carrier-mediated uptake at the sinusoidal membrane (step 3), conjugation with one or two molecules of glucuronic acid by bilirubin uridine diphosphate (UDP)–glucuronyltransferase (UGT1A1, step 4) in the endoplasmic reticulum, and excretion of the water-soluble, nontoxic bilirubin glucuronides into bile. Most bilirubin glucuronides are deconjugated in the gut lumen by bacterial β-glucuronidases and degraded to colorless urobilinogens (step 5). The urobilinogens and the residue of intact pigment are largely excreted in feces. Approximately 20% of the urobilinogens formed are reabsorbed in the ileum and colon, returned to the liver, and re-excreted into bile. A small amount of reabsorbed urobilinogen is excreted in the urine.
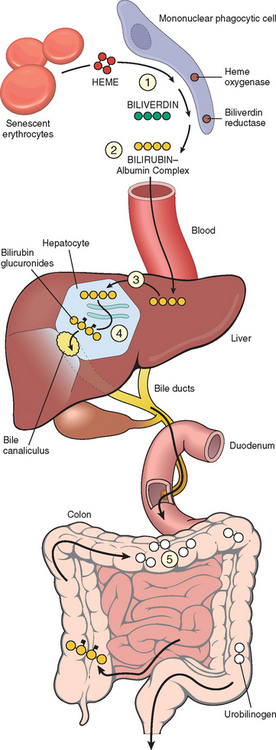
FIGURE 18-4 Bilirubin metabolism and elimination. (1) Normal bilirubin production from heme (0.2–0.3 gm/day) is derived primarily from the breakdown of senescent circulating erythrocytes. (2) Extrahepatic bilirubin is bound to serum albumin and delivered to the liver. (3) Hepatocellular uptake and (4) glucuronidation in the endoplasmic reticulum generate bilirubin monoglucuronides and diglucuronides, which are water soluble and readily excreted into bile. (5) Gut bacteria deconjugate the bilirubin and degrade it to colorless urobilinogens. The urobilinogens and the residue of intact pigments are excreted in the feces, with some reabsorption and excretion into urine.
The hepatic conjugating enzyme UGT1A1 is a product of the UGT1 gene located on chromosome 2q37. It is a member of a family of enzymes that catalyze the glucuronidation of an array of substrates such as steroid hormones, carcinogens, and drugs. In humans, UGT1A1, generated from the exon 1A of the UGT1 gene, is the only isoform responsible for bilirubin glucuronidation. Mutations of UGT1A1 cause hereditary unconjugated hyperbilirubinemias: Crigler-Najjar syndrome types I and II, and Gilbert syndrome.
Two thirds of the organic materials in bile are bile salts, which are formed by the conjugation of bile acids with taurine or glycine. Bile acids, the major catabolic products of cholesterol, are a family of water-soluble sterols with carboxylated side chains. The primary human bile acids are cholic acid and chenodeoxycholic acid. Bile acids in bile salts act as highly effective detergents. Their primary physiologic role is to solubilize water-insoluble lipids secreted by hepatocytes into bile, and also to solubilize dietary lipids in the gut lumen. Ninety-five percent of secreted bile acids, conjugated or unconjugated, are reabsorbed from the gut lumen and recirculate the liver (enterohepatic circulation), thus helping to maintain a large endogenous pool of bile acids for digestive and excretory purposes.
Pathophysiology of Jaundice
Both unconjugated bilirubin and conjugated bilirubin (bilirubin glucuronides) may accumulate systemically. There are two important pathophysiologic differences between the two forms of bilirubin. Unconjugated bilirubin is virtually insoluble in water at physiologic pH and exists in tight complexes with serum albumin. This form cannot be excreted in the urine even when blood levels are high. Normally, a very small amount of unconjugated bilirubin is present as an albumin-free anion in plasma. This fraction of unbound bilirubin may diffuse into tissues, particularly the brain in infants, and produce toxic injury. The unbound plasma fraction may increase in severe hemolytic disease or when protein-binding drugs displace bilirubin from albumin. Hence, hemolytic disease of the newborn (erythroblastosis fetalis) may lead to accumulation of unconjugated bilirubin in the brain, which can cause severe neurologic damage, referred to as kernicterus (Chapter 10). In contrast, conjugated bilirubin is water-soluble, nontoxic, and only loosely bound to albumin. Because of its solubility and weak association with albumin, excess conjugated bilirubin in plasma can be excreted in urine. With prolonged conjugated hyperbilirubinemia, a portion of circulating pigment may become covalently bound to albumin; this is termed the bilirubin delta fraction.
Serum bilirubin levels in the normal adult vary between 0.3 and 1.2 mg/dL, and the rate of systemic bilirubin production is equal to the rates of hepatic uptake, conjugation, and biliary excretion. Jaundice becomes evident when the serum bilirubin levels rise above 2.0 to 2.5 mg/dL; levels as high as 30 to 40 mg/dL can occur with severe disease. Jaundice occurs when the equilibrium between bilirubin production and clearance is disturbed by one or more of the following mechanisms (Table 18-2): (1) excessive extrahepatic production of bilirubin; (2) reduced hepatocyte uptake; (3) impaired conjugation; (4) decreased hepatocellular excretion; and (5) impaired bile flow. The first three mechanisms produce unconjugated hyperbilirubinemia, and the latter two produce predominantly conjugated hyperbilirubinemia. Although more than one mechanism may be operative, generally one mechanism predominates, so knowledge of the major form of plasma bilirubin is of value in evaluating possible causes of hyperbilirubinemia.
| PREDOMINANTLY UNCONJUGATED HYPERBILIRUBINEMIA |
| PREDOMINANTLY CONJUGATED HYPERBILIRUBINEMIA |
UGT, uridine diphosphate–glucuronyltransferase.
Two conditions result from specific defects in hepatocellular bilirubin metabolism.
Neonatal Jaundice.
Because the hepatic machinery for conjugating and excreting bilirubin does not fully mature until about 2 weeks of age, almost every newborn develops transient and mild unconjugated hyperbilirubinemia, termed neonatal jaundice or physiologic jaundice of the newborn. This may be exacerbated by breastfeeding, as a result of the presence of bilirubin-deconjugating enzymes in breast milk. Nevertheless, sustained jaundice in the newborn is abnormal, discussed later under neonatal hepatitis.
Hereditary Hyperbilirubinemias.
Multiple genetic mutations can cause hereditary hyperbilirubinemia15 (Table 18-3). In Crigler-Najjar syndrome type I hepatic UGT1A1 (described earlier) is completely absent, and the colorless bile contains only trace amounts of unconjugated bilirubin. The liver is morphologically normal by light and electron microscopy. However, serum unconjugated bilirubin reaches very high levels, producing severe jaundice and icterus. Without liver transplantation, this condition is invariably fatal, causing death secondary to kernicterus within 18 months of birth.
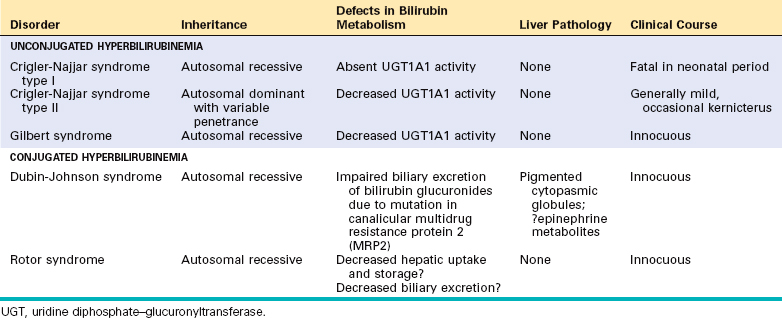
Crigler-Najjar syndrome type II is a less severe, nonfatal disorder in which UGT1A1 enzyme activity is greatly reduced, and the enzyme is capable of forming only monoglucuronidated bilirubin. Unlike Crigler-Najjar syndrome type I, the only major consequence is extraordinarily yellow skin. Phenobarbital treatment can improve bilirubin glucuronidation by inducing hypertrophy of the hepatocellular endoplasmic reticulum.
Gilbert syndrome is a relatively common, benign, inherited condition presenting with mild, fluctuating hyperbilirubinemia, in the absence of hemolysis or liver disease. It affects 3% to 10% of the U.S. population. In Gilbert syndrome, hepatic bilirubin-glucuronidating activity is about 30% of normal, a less severe reduction than in Crigler-Najjar syndromes. It is caused in most patients by the homozygous insertion of two extra bases in the 5′ promoter region of the UGT1 gene, leading to reduced transcription. The mild hyperbilirubinemia may go undiscovered for years and is not associated with functional derangements. When detected in adolescence or adult life it is typically in association with stress, such as an intercurrent illness, strenuous exercise, or fasting. Gilbert syndrome itself has no clinical consequence except for the anxiety that a jaundiced sufferer might justifiably experience with this otherwise innocuous condition. However, individuals who have Gilbert syndrome may be more susceptible to adverse effects of drugs that are metabolized by UGT1A1.
Dubin-Johnson syndrome is an autosomal recessive disorder characterized by chronic conjugated hyperbilirubinemia. It is caused by a defect in hepatocellular excretion of bilirubin glucuronides across the canalicular membrane. The molecular basis for this syndrome is absence of the canalicular protein, multidrug resistance protein 2, which is responsible for transport of bilirubin glucuronides and related organic anions into bile.16 The liver is darkly pigmented because of coarse pigmented granules within the cytoplasm of hepatocytes (Fig. 18-5). Electron microscopy reveals that the pigment is located in lysosomes: it appears to be composed of polymers of epinephrine metabolites. The liver is otherwise normal. Apart from chronic or recurrent jaundice of fluctuating intensity, most patients are asymptomatic and have a normal life expectancy.
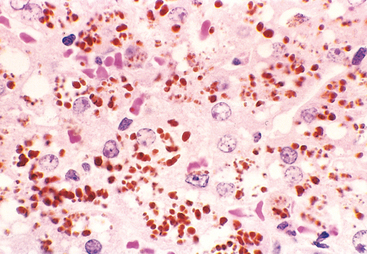
FIGURE 18-5 Dubin-Johnson syndrome, showing abundant pigment inclusions in otherwise normal hepatocytes.
Rotor syndrome is a rare form of asymptomatic conjugated hyperbilirubinemia associated with multiple defects in hepatocellular uptake and excretion of bilirubin pigments. The precise molecular basis for this syndrome is unknown. The liver is morphologically normal. As with Dubin-Johnson syndrome, patients with Rotor syndrome have jaundice but otherwise have normal lives.
Cholestasis
Cholestasis denotes a pathologic condition of impaired bile formation and bile flow, leading to accumulation of bile pigment in the hepatic parenchyma.17 It can be caused by extrahepatic or intrahepatic obstruction of bile channels, or by defects in hepatocyte bile secretion. Patients may have jaundice, pruritus, skin xanthomas (focal accumulation of cholesterol), or symptoms related to intestinal malabsorption, including nutritional deficiencies of the fat-soluble vitamins A, D, or K. A characteristic laboratory finding is elevated serum alkaline phosphatase and γ-glutamyl transpeptidase (GGT), enzymes present on the apical membranes of hepatocytes and bile duct epithelial cells.
Morphology. The morphologic features of cholestasis depend on its severity, duration, and underlying cause. Common to both obstructive and nonobstructive cholestasis is the accumulation of bile pigment within the hepatic parenchyma (Figs. 18-6 and 18-7). Elongated green-brown plugs of bile are visible in dilated bile canaliculi (Fig. 18-7B). Rupture of canaliculi leads to extravasation of bile, which is quickly phagocytosed by Kupffer cells. Droplets of bile pigment also accumulate within hepatocytes, which can take on a fine, foamy appearance (feathery degeneration).
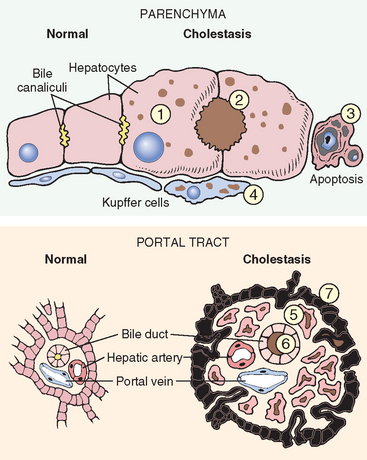
FIGURE 18-6 Morphologic features of cholestasis (right) and comparison with normal liver (left). In the parenchyma (upper panel) cholestatic hepatocytes (1) are enlarged with dilated canalicular spaces (2). Apoptotic cells (3) may be seen, and Kupffer cells (4) frequently contain regurgitated bile pigments. In the portal tracts of obstructed liver (lower panel) there is also bile ductular proliferation (5), edema, bile pigment retention (6), and eventually neutrophilic inflammation (not shown). Surrounding hepatocytes (7) are swollen and undergoing degeneration.
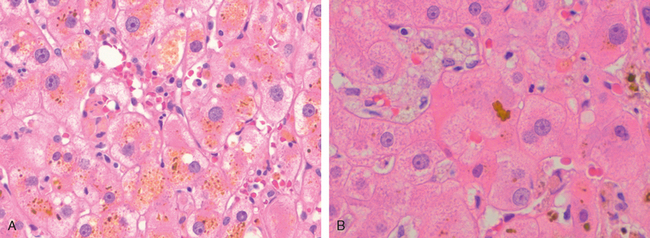
FIGURE 18-7 Histology of cholestasis. A, Intracellular cholestasis showing the bile pigments in the cytoplasm; B, bile plug showing the expansion of bile canaliculus by bile.
Obstruction of the biliary tree, either intrahepatic or extrahepatic, causes distention of upstream bile ducts and ductules by bile. The bile stasis and back-pressure induce proliferation of the duct epithelial cells, and looping and reduplication of ducts and ductules in the portal tracts. The labyrinthine ductules reabsorb secreted bile salts, serving to protect the downstream obstructed bile ducts from the toxic detergent action of bile salts. Associated histologic findings include portal tract edema and periductular infiltrates of neutrophils. Prolonged obstructive cholestasis leads not only to feathery change of hepatocytes but also to focal dissolution of hepatocytes by detergents, giving rise to bile lakes filled with cellular debris and pigment. Unrelieved obstruction leads to portal tract fibrosis, and ultimately, to biliary cirrhosis.
Since extrahepatic biliary obstruction is frequently amenable to surgical alleviation, correct and prompt diagnosis is imperative. In contrast, cholestasis due to diseases of the intrahepatic biliary tree or hepatocellular secretory failure (collectively termed intrahepatic cholestasis) is not benefited by surgery (short of transplantation), and the patient’s condition may be worsened by an operative procedure. There is thus some urgency in making a correct diagnosis of the cause of jaundice and cholestasis.
Progressive Familial Intrahepatic Cholestasis (PFIC).
Here we discuss a striking but heterogeneous group of autosomal-recessively inherited cholestatic conditions known as PFICs.17 PFIC-1 (also known as Byler disease as it was first identified in the descendents of Jacob Byler, an Amish patient), PFIC-2, and PFIC-3 are caused by mutations of three different genes. PFIC-1 and PFIC-2 share a similar phenotype, which includes normal or near normal GGT activity, and lack of bile ductular proliferation in portal tracts.
Progressive familial intrahepatic cholestasis 1 (PFIC-1) is characterized by cholestasis beginning in infancy, with severe pruritus due to high serum bile acid levels, and relentlessly progresses to liver failure before adulthood. The genetic defect is usually a mutation in the ATP8B1 gene on chromosome 18q21 that causes impaired bile secretion, through mechanisms that are not yet fully elucidated.18 In the mild form of PFIC-1 called benign recurrent intrahepatic cholestasis, there are intermittent attacks of cholestasis over life without progression to chronic liver disease.
Progressive familial intrahepatic cholestasis 2 (PFIC-2) is caused by mutations in the hepatocyte canalicular bile salt export pump (BSEP), encoded by the ABCB11 gene. BSEP is a member of the adenosine triphosphate–binding cassette (ABC) family of transporters.19 Mutations of the ABCB11 gene cause severely impaired bile salt secretion into bile. Patients suffer extreme pruritus, growth failure, and progression to cirrhosis in the first decade of life. These patients also have higher risk for cholangiocarcinoma.
Progressive familial intrahepatic cholestasis 3 (PFIC-3) is caused by mutations in the ABCB4 gene, and is characterized by cholestasis with a high serum GGT.20 The ABCB4-encoded protein, MDR3, is a liver-specific canalicular transport protein. In individuals with PFIC-3, there is absence of secreted phosphatidylcholine in bile, which leaves the apical surfaces of the biliary tree epithelia subject to the full detergent action of secreted bile salts, with resultant toxic destruction of these epithelia and release of GGT into the circulation.
Infectious Disorders
Inflammatory disorders of the liver dominate the clinical practice of hepatology. This is partly because virtually any insult to the liver can kill hepatocytes and recruit inflammatory cells, but also because inflammatory diseases are frequently long-term chronic conditions. Among inflammatory disorders, viral infection is by far the most frequent.
VIRAL HEPATITIS
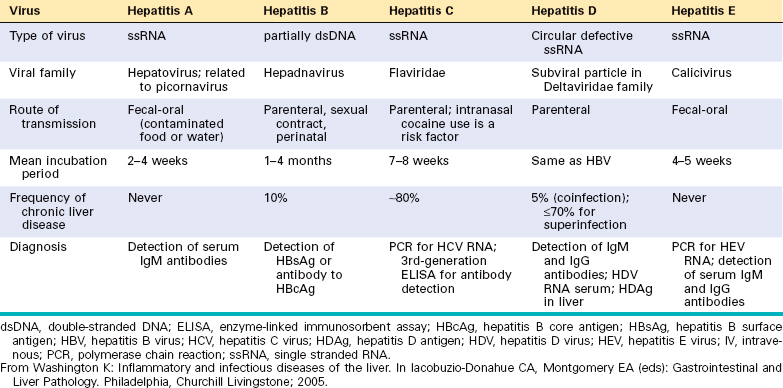
Systemic viral infections can involve the liver as in (1) infectious mononucleosis (Epstein-Barr virus), which may cause a mild hepatitis during the acute phase; (2) cytomegalovirus infection, particularly in the newborn or immunosuppressed patient; and (3) yellow fever (yellow fever virus), which has been a major and serious cause of hepatitis in tropical countries. Infrequently, in children and immunosuppressed patients, the liver is affected in the course of rubella, adenovirus, herpesvirus, or enterovirus infections. However, unless otherwise specified, the term viral hepatitis is applied for hepatic infections caused by a group of viruses known as hepatotropic virus (hepatitis viruses A, B, C, D, and E) that have a particular affinity for the liver (Table 18-4). We first present the main features of each hepatotropic virus, followed by a discussion of the clinicopathologic characteristics of acute and chronic viral hepatitis.
Hepatitis A Virus
Hepatitis A virus (HAV), the scourge of military campaigns since antiquity, is a benign, self-limited disease with an incubation period of 3 to 6 weeks. HAV does not cause chronic hepatitis or a carrier state and only rarely causes fulminant hepatitis, so the fatality rate associated with HAV is about 0.1%. HAV occurs throughout the world and is endemic in countries with substandard hygiene and sanitation, where populations may have detectable antibodies to HAV by the age of 10 years. Clinical disease tends to be mild or asymptomatic, and is rare after childhood. In developed countries, the prevalence of seropositivity (indicative of previous exposure) increases gradually with age, reaching 50% by age 50 years in the United States. In this population acute HAV tends to be a sporadic febrile illness. Affected individuals have nonspecific symptoms such as fatigue and loss of appetite, and often develop jaundice. Overall, HAV accounts for about 25% of clinically evident acute hepatitis worldwide and an estimated 30,000 to 50,000 new cases per year in the United States.
HAV, discovered in 1973, is a small, nonenveloped, positive-strand RNA picornavirus that occupies its own genus, Hepatovirus. Ultrastructurally, HAV is an icosahedral capsid 27 nm in diameter and can be cultured in vitro. The receptor for HAV is HAVcr-1, a 451–amino acid class I integralmembrane mucin-like glycoprotein of unknown normal function.21 HAV is spread by ingestion of contaminated water and foods and is shed in the stool for 2 to 3 weeks before and 1 week after the onset of jaundice. Thus, close personal contact with an infected individual or fecal-oral contamination during this period accounts for most cases and explains the outbreaks in institutional settings such as schools and nurseries, and the water-borne epidemics in places where people live in overcrowded, unsanitary conditions. HAV can also be detected in serum and saliva. Because HAV viremia is transient, blood-borne transmission of HAV occurs only rarely; therefore, donated blood is not specifically screened for this virus. In developed countries, sporadic infections may be contracted by the consumption of raw or steamed shellfish (oysters, mussels, clams), which concentrate the virus from seawater contaminated with human sewage. Infected workers in the food industry may also be the source of outbreaks. HAV itself does not seem to be cytopathic. Cellular immunity, particularly CD8+ T cells, plays a key role in hepatocellular injury during HAV infection.22
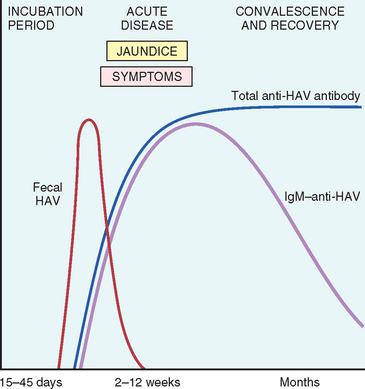
Specific IgM antibody against HAV appears in blood at the onset of symptoms, constituting a reliable marker of acute infection (Fig. 18-8). Fecal shedding of the virus ends as the IgM titer rises. The IgM response usually begins to decline in a few months and is followed by the appearance of IgG anti-HAV. The latter persists for years, perhaps conferring lifelong immunity against reinfection by all strains of HAV. However, there are no routinely available tests for IgG anti-HAV. The presence of this antibody is inferred from the difference between total and IgM anti-HAV. HAV vaccine, available since 1992, is effective in preventing infection.23
Hepatitis B Virus (HBV)
HBV can produce (1) acute hepatitis with recovery and clearance of the virus, (2) nonprogressive chronic hepatitis, (3) progressive chronic disease ending in cirrhosis, (4) fulminant hepatitis with massive liver necrosis, and (5) an asymptomatic carrier state. HBV-induced chronic liver disease is an important precursor for the development of hepatocellular carcinoma.24 The approximate frequencies of clinical outcomes of HBV infection are depicted in Figure 18-9.
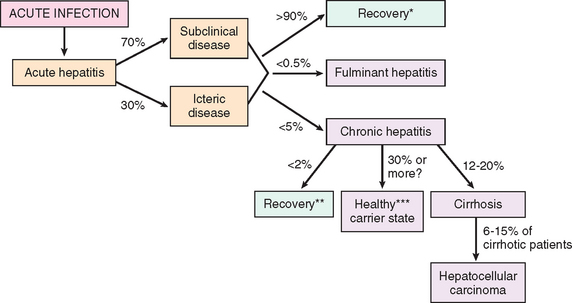
FIGURE 18-9 Potential outcomes of hepatitis B infection in adults, with their approximate frequencies in the United States.
*Recovery from acute hepatitis refers to complete recovery as well as latent infections with maintenance of T cell response.
**Recovery from chronic hepatitis is indicated by negative test for HBsAg.
***Healthy carrier state is indicated by positive HBsAg >6 months; HBeAg negative; serum HBV DNA <105 copies/mL; persistently normal AST and ALT levels; absence of significant inflammation and necrosis on liver biopsy.
Liver disease due to HBV is an enormous global health problem. One third of the world population (2 billion people) have been infected with HBV, and 400 million people have chronic infection. Seventy-five percent of all chronic carriers live in Asia and the Western Pacific rim. The global prevalence of chronic hepatitis B infection varies widely, from high (>8%) in Africa, Asia, and the Western Pacific to intermediate (2% to 7%) in southern and eastern Europe to low (<2%) in western Europe, North America, and Australia. As will be discussed later, the carrier rate is largely dictated by the age at infection, being the highest when infection occurs in children perinatally and the lowest when adults are infected. In the United States, incidence of HBV infection has dramatically decreased; there are now an estimated 46,000 new infections per year with about 5,000 acute symptomatic cases.
The mode of transmission of HBV varies with geographical areas. In high prevalence regions of the world, perinatal transmission during childbirth accounts for 90% of cases. In areas with intermediate prevalence, horizontal transmission, especially in early childhood, is the dominant mode of transmission. Such spread occurs through minor cuts and breaks in the skin or mucous membranes among children with close bodily contact. In low prevalence areas such as the United States, unprotected heterosexual or homosexual intercourse and intravenous drug abuse (sharing of needles and syringes) are the chief modes of spread. The incidence of transfusion-related spread has dwindled greatly in recent years due to screening of donated blood and HBsAg and exclusion of paid blood donors.
HBV has a prolonged incubation period (4–26 weeks). Unlike HAV, HBV remains in the blood until and during active episodes of acute and chronic hepatitis. In the United States acute HBV infection mostly affects adults. Approximately 70% have mild or no symptoms and do not develop jaundice. The remaining 30% have nonspecific constitutional symptoms such as anorexia, fever, jaundice, and upper right quadrant pain. In almost all cases the infection is self-limited and resolves without treatment. Chronic disease rarely occurs in adults in non-endemic areas. Fulminant hepatitis is also rare, occurring in approximately 0.1 to 0.5% of cases.
HBV was first linked to hepatitis in the 1960s when Australia antigen (later known as HBV surface antigen) was identified.25 The virus is a member of the Hepadnaviridae, a family of DNA viruses that cause hepatitis in multiple animal species. There are eight HBV genotypes with geographic distribution around the globe. The mature HBV virion is a 42-nm, spherical double-layered “Dane particle” that has an outer surface envelope of protein, lipid, and carbohydrate enclosing an electron-dense, 28-nm, slightly hexagonal core. The genome of HBV is a partially double-stranded circular DNA molecule having 3200 nucleotides (Fig. 18-10). The HBV genome contains four open reading frames coding for:26
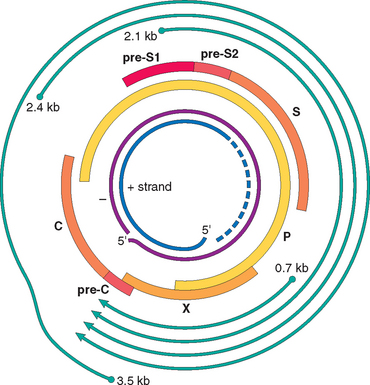
FIGURE 18-10 Diagrammatic representation of genomic structure and transcribed components of the hepatitis B virion. The innermost circles represent the DNA (+) strand and the DNA (−) strand of the virion. The thick bars labeled P, X, pro-C, C, pre-SI, pre-S2, and S denote the peptides derived from the virion. The outermost lines denote the mRNA transcripts of the virion.
The natural course of the disease can be followed by serum markers (18-11).
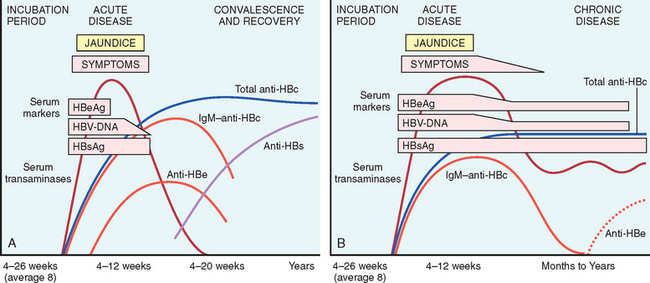
FIGURE 18-11 Sequence of serologic markers for hepatitis B viral hepatitis demonstrating (A) acute infection with resolution and (B) progression to chronic infection.
Occasionally, mutated strains of HBV emerge that do not produce HBeAg but are replication competent and express HBcAg. In such patients, the HBeAg may be low or undetectable despite the presence of HBV viral load. A second ominous development is the appearance of vaccine-induced escape mutants, which replicate in the presence of vaccine-induced immunity. For instance, in one such viral mutant, replacement of arginine at amino acid 145 of HBsAg with glycine significantly alters recognition of HBsAg by anti-HBsAg antibodies.
Despite the self-limited nature of acute HBV infection, recent studies show that very low levels of HBV DNA can be detected by PCR analysis in the blood of some individuals who may have anti-HBe antibodies. It is unclear at this time whether the viral material detected is composed of virus fragments, infectious virus, or non-infectious virus, but the material persists for many years.
The host immune response to the virus is the main determinant of the outcome of the infection.27 The mechanisms of innate immunity protect the host during the initial phases of the infection, and a strong response by virus-specific CD4+ and CD8+ interferon γ-producing cells is associated with the resolution of acute infection. There are several reasons to believe that HBV does not cause direct hepatocyte injury. Most importantly, many chronic carriers have virions in their hepatocytes with no evidence of cell injury. Hepatocyte damage is believed to result from damage to the virus-infected cells by CD8+ cytotoxic T cells.
Hepatitis B can prevented by vaccination and by the screening of donor blood, organs, and tissues. The vaccine is prepared from purified HbsAg produced in yeast. Vaccination induces a protective anti-HBs antibody response in 95% of infants, children, and adolescents. Universal vaccination has had notable success in Taiwan and Gambia, but unfortunately, it has not been adopted worldwide.
Hepatitis C Virus
Hepatitis C virus (HCV) is a major cause of liver disease worldwide, with approximately 170 million people affected. Approximately 4.1 million Americans, or 1.6% of the population, have chronic HCV infection. This makes HCV the most common chronic blood-borne infection and accounts for almost half of all US individuals with chronic liver disease. Notably, there has been a decrease in the annual incidence of infection from its mid-1980s peak of over 230,000 new infections per year to a current 19,000 new infections per year. This welcome decline resulted primarily from a marked reduction in transfusion-associated causes as a result of screening procedures. Nevertheless, the number of patients with chronic infection will continue to increase, as a result of potential lifelong persistence of HCV infection. In contrast to HBV, progression to chronic disease occurs in the majority of HCV-infected individuals, and cirrhosis eventually occurs in 20% to 30% of individuals with chronic HCV infection. Thus, HCV is the most common cause of chronic liver disease in the United States and the most common indication for liver transplantation.
According to the 2008 data from the USA Centers for Disease Control, the most common risk factors for HCV infection are:
Currently, transmission of HCV by blood transfusion is close to zero in the United States; the risk of acquiring HCV by needle sticks is about six times higher than that for HIV (1.8 vs 0.3%). For children, the major route of infection is perinatal, but is much lower than for HBV (6% vs. 20%). Note that patients may have multiple risk factors (the total of the listed risks above is >100%).
HCV, discovered in 1989, is a member of the Flaviviridae family. It is a small, enveloped, single-stranded RNA virus with a 9.6-kilobase (kb) genome that codes for a single polyprotein with one open reading frame, which is subsequently processed into functional proteins (Fig. 18-12). We review, briefly, the genomic structure of HCV because this has a bearing on the pathogenesis of hepatitis C. The 5′ end of the genome encodes a highly conserved nucleocapsid core protein, followed by envelope proteins E1 and E2. Two hypervariable regions (HVR 1 and 2) are present in the E2 sequence. A protein, p7, is believed to function as an ion channel. Toward the 3′ end are six less conserved nonstructural proteins: NS2, NS3, NS4A, NS4B, NS5A, and NS5B. NS5B is the viral RNA-dependent RNA polymerase. The 3′ sequences of both the positive- and negative-strand RNAs contribute cis-acting functions that are essential for viral replication. The secondary structure and protein-binding properties of these highly conserved nontranslated regions are thought to promote HCV RNA synthesis and genome stability through the binding of various host and viral proteins.
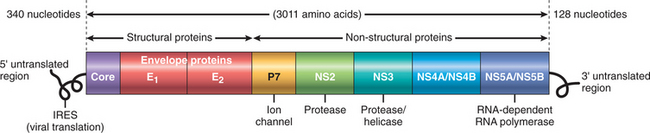
FIGURE 18-12 Diagrammatic representation of the hepatitis C viral (HCV) genomic structure. HCV is a (+) strand RNA virus containing two untranslated regions at the 5′ and the 3′ ends. The virus encodes a single polypeptide that is processed into multiple viral proteins. The potential function of each individual protein is highlighted.
Because of the poor fidelity of the HCV RNA polymerase (NS5B), the virus is inherently unstable, giving rise to multiple genotypes and subtypes. Indeed, within any given patient HCV circulates as a population of divergent but closely related variants known as quasispecies.28 Over time, dozens of quasispecies can be detected within one individual and mapped as derivative strains of the original HCV strain that infected the patient. The E2 protein of the envelope is the target of many anti-HCV antibodies but is also the most variable region of the entire viral genome, enabling emergent virus strains to escape from neutralizing antibodies. This genomic instability and antigenic variability have seriously hampered efforts to develop an HCV vaccine. In particular, elevated titers of anti-HCV IgG occurring after an active infection do not consistently confer effective immunity. A characteristic feature of HCV infection, therefore, is repeated bouts of hepatic damage, the result of reactivation of a preexisting infection or emergence of an endogenous, newly mutated strain.
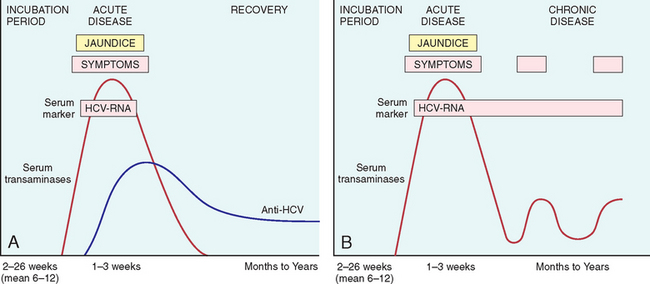
The incubation period for HCV hepatitis ranges from 2 to 26 weeks, with a mean between 6 and 12 weeks. In about 85% of individuals, the clinical course of the acute infection is asymptomatic and easily missed. HCV RNA is detectable in blood for 1 to 3 weeks, coincident with elevations in serum transaminases. In symptomatic acute HCV infection, anti-HCV antibodies are detected in only 50% to 70% of patients; in the remaining patients, the anti-HCV antibodies emerge after 3 to 6 weeks. The clinical course of acute HCV hepatitis is milder than that of HBV; rare cases may be severe and indistinguishable from HAV or HBV hepatitis. Strong immune responses involving CD4+ and CD8+ T cells are associated with self-limited HCV infections, but it is not known why only a small minority of individuals are capable of clearing HCV infection.
Persistent infection and chronic hepatitis are the hallmarks of HCV infection, despite the generally asymptomatic nature of the acute illness. This occurs in 80% to 85% of cases. Cirrhosis may develop over 5 to 20 years after acute infection in 20% to 30% of patients with persistent infection. The mechanisms that lead to the chronicity of HCV infection are not well understood, but it is clear that the virus has developed multiple strategies to evade host antiviral immunity.29 HCV is able to actively inhibit the interferon (IFN)-mediated cellular antiviral response at multiple steps, including Toll-like receptor signaling in response to viral RNA recognition and signaling downstream of IFN receptors that confers on cells an anti-viral state.
In chronic HCV infection, circulating HCV RNA persists in many patients despite the presence of neutralizing antibodies, including more than 90% of patients with chronic disease (Fig. 18-13). Hence, in persons with chronic hepatitis, HCV RNA testing must be performed to assess viral replication and to confirm the diagnosis of HCV infection. A clinical feature that is quite characteristic of chronic HCV infection is episodic elevations in serum aminotransferases, with intervening normal or near-normal periods. Fulminant hepatic failure rarely occurs.
Hepatitis D Virus
Also called “hepatitis D virus,” hepatitis D virus (HDV) is a unique RNA virus that is dependent for its life cycle on HBV. Infection with HDV arises in the following settings.30
Coinfection of HBV and HDV results in acute hepatitis B + D which is clinically indistinguishable from classical acute hepatitis B and is usually transient and self-limited. Elimination of hepatitis B leads to elimination of HDV. The rate of progression to chronic infection is not different from that observed after classical acute hepatitis B. However, a high incidence of liver failure has been reported among drug addicts.
Superinfection with HDV in a chronic HBsAg carrier may present as severe acute hepatitis in a previously unrecognized HBV carrier, or as an exacerbation of preexisting chronic hepatitis B. Chronic HDV infection occurs in 80% to 90% of such patients. The superinfection may have two phases; an acute phase with active HDV replication and suppression of HBV with high ALT levels; and a chronic phase in which HDV replication decreases, HBV replication increases, ALT levels fluctuate, and the disease progresses to cirrhosis and hepatocellular cancer (HCC).
Helper-independent latent infection may be seen in liver transplants. HDV can be detected in nuclei of the grafted liver within a few hours after transplantation without evidence of productive HDV infection or HBV reinfection. This likely occurs due to infection of the allograft by HDV alone, while concomitant infection by HBV is prevented by hepatitis B immunoglobulin administered to prevent HBV reinfection. During this latent phase, there is no evidence of liver disease. HD viremia and hepatitis ensues only when HBV escapes neutralization and coinfection of the graft with high levels of HBV replication occurs, leading to activation of the HDV by the helper virus.
Infection by HDV is worldwide with an estimate of 15 million affected individuals (about 5% of 300 million of HBV infected persons). The prevatence varies widely in different countries. It is high in the Amazonian basin, and in Africa, the Middle East and southern Italy. Twenty to forty percent of HbsAg carriers may have anti-HDV antibody, although there has been a definite decline in recent years. In the United States HDV has virtually disappeared from hemophiliacs and other individuals who receive blood transfusion, because of the HBV screening procedures. Surprisingly HDV infection is uncommon in the large population of HBsAg carriers in Southeast Asia and China.
HDV, discovered in 1977, is a 35-nm, double-shelled particle that by electron microscopy resembles the “Dane particle” of HBV. The external coat antigen of HBsAg surrounds an internal polypeptide assembly, designated delta antigen (HDAg), the only protein produced by the virus. Associated with HDAg is a small circular molecule of single-stranded RNA, whose length is smaller than the genome of any known animal virus. Replication of the virus is through RNA-directed RNA synthesis by host RNA polymerase, mainly Pol II.
Diagnosis.
HDV RNA is detectable in the blood and liver just before and in the early days of acute symptomatic disease. IgM anti-HDV is the most reliable indicator of recent HDV exposure, although its appearance is late and frequently short-lived. Nevertheless, acute co-infection by HDV and HBV is best indicated by detection of IgM against both HDAg and HBcAg (denoting new infection with hepatitis B). With chronic delta hepatitis arising from HDV superinfection, HBsAg is present in serum, and anti-HDV antibodies (IgG and IgM) persist for months or longer. Treatment of HDV infection is limited to IFN-α.30 Other antiviral agents for HBV have not shown effectiveness. Vaccination for HBV can also prevent HDV infection.
Hepatitis E Virus
Hepatitis E virus (HEV) hepatitis is an enterically trans-mitted, water-borne infection that occurs primarily in young to middle-aged adults; sporadic infection and overt illness in children are rare. HEV is a zoonotic disease with animal reservoirs, such as monkeys, cats, pigs, and dogs.31 Epidemics have been reported in Asia and the Indian subcontinent, sub-Saharan Africa, and Mexico. Sporadic infection may occur in travelers to these regions, but, most importantly, HEV infection accounts for more than 30% to 60% of cases of sporadic acute hepatitis in India, exceeding the frequency of HAV. A characteristic feature of HEV infection is the high mortality rate among pregnant women, approaching 20%. In most cases the disease is self-limiting; HEV is not associated with chronic liver disease or persistent viremia. The average incubation period following exposure is 6 weeks.
Discovered in 1983, HEV is an unenveloped, positive-stranded RNA virus in the Hepevirus genus.31 Viral particles are 32 to 34 nm in diameter, and the RNA genome is approximately 7.3 kb in size. A specific antigen (HEV Ag) can be identified in the cytoplasm of hepatocytes during active infection, and virions are shed in stool during the acute illness.
Diagnosis.
Before the onset of clinical illness, HEV RNA and HEV virions can be detected by PCR in stool and serum. The onset of rising serum aminotransferases, clinical illness, and elevated IgM anti-HEV titers are virtually simultaneous. Symptoms resolve in 2 to 4 weeks, during which time the IgM is replaced with a persistent IgG anti-HEV titer.
Hepatitis G Virus
A flavivirus bearing similarities to HCV was cloned in 1995 and designated hepatitis G virus (HGV, also referred to as GBV-C). HGV is transmitted by contaminated blood or blood products, and via sexual contact. However, HGV is inappropriately named: it is not hepatotropic and does not cause elevations in serum aminotransferases. Instead, the virus appears to replicate in the bone marrow and spleen. The prevalence of HGV RNA in American blood donors ranges from 1% to 4%; but since the virus does not cause known human disease, blood donors do not need screening. HGV commonly co-infects individuals infected with the human immunodeficiency virus (HIV; prevalence 35%); curiously this dual infection is somewhat protective against HIV disease.32
Clinicopathologic Syndromes of Viral Hepatitis
Several clinical syndromes may develop following exposure to hepatitis viruses: (1) acute asymptomatic infection with recovery (serologic evidence only); (2) acute symptomatic hepatitis with recovery, anicteric or icteric; (3) chronic hepatitis, without or with progression to cirrhosis; and (4) fulminant hepatitis with massive to submassive hepatic necrosis.
Each of the hepatotropic viruses can cause acute asymptomatic or symptomatic infection. A small number of HBV-infected adult patients develop chronic hepatitis. In contrast, HCV is notorious for chronic infection. HAV and HEV do not cause chronic hepatitis. Fulminant hepatitis is unusual and is seen primarily with HBV. Although HBV and HCV are responsible for most cases of chronic hepatitis, there are many other causes of chronic hepatitis (described later), including chronic alcoholism, drugs (e.g., isoniazid, α-methyldopa, methotrexate), toxins, Wilson disease, α1-antitrypsin deficiency, and autoimmunity. Therefore, serologic and molecular studies are essential for the diagnosis of viral hepatitis, and for distinguishing between the various types.
Acute Asymptomatic Infection with Recovery.
Patients in this group are identified only incidentally on the basis of minimally elevated serum transaminases or, after the fact, by the presence of antiviral antibodies. Worldwide, HAV and HBV infection are frequently subclinical events in childhood, verified only in adulthood by the presence of anti-HAV or anti-HBV antibodies.
Acute Symptomatic Infection with Recovery.
Any one of the hepatotropic viruses can cause symptomatic acute viral hepatitis. Whatever the agent, the disease is more or less the same and can be divided into four phases: (1) an incubation period, (2) a symptomatic preicteric phase, (3) a symptomatic icteric phase, and (4) convalescence. The incubation period for the different viruses is given in Table 18-4. Peak infectivity occurs during the last asymptomatic days of the incubation period and the early days of acute symptoms.
Chronic Hepatitis.
Chronic hepatitis is defined as symptomatic, biochemical, or serologic evidence of continuing or relapsing hepatic disease for more than 6 months. As mentioned earlier, HCV infection causes chronic hepatitis at a high frequency while only a small number of patients with HBV infection develop chronic disease. The clinical features of chronic hepatitis are extremely variable and are not predictive of outcome. In some patients the only signs of chronic disease are persistent elevations of serum transaminases. The most common symptom is fatigue; less common symptoms are malaise, loss of appetite, and occasional bouts of mild jaundice. Physical findings are few, the most common being spider angiomas, palmar erythema, mild hepatomegaly, hepatic tenderness, and mild splenomegaly. Laboratory studies may reveal prolongation of the prothrombin time and, in some instances, hyperglobulinemia, hyperbilirubinemia, and mild elevations in alkaline phosphatase levels. Occasionally, in cases of HBV and HCV, immune complex disease may develop secondary to the presence of circulating antibody-antigen complexes, in the form of vasculitis (subcutaneous or visceral, Chapter 11) and glomerulonephritis (Chapter 20). Cryoglobulinemia is found in about 35% of individuals with chronic hepatitis C.
The development of chronic infection after exposure to HBV is an important clinical problem. Age at the time of infection is the best determinant of chronicity. The younger the age at the time of infection, the higher the probability of chronicity. In many endemic areas, maternal-to-infant transmission is a major risk factor for chronic HBV infection. Though uncommon, patients can recover completely from chronic HBV infection.33 Despite progress in the treatment of chronic HBV infection, complete cure is extremely difficult to achieve. Thus, the goal of the treatment of chronic hepatitis B is to slow disease progression, reduce liver damage, and prevent liver cirrhosis or liver cancer. The major problems associated with the current treatment regimens are viral resistance and side effects.
HCV is by far the most common cause of chronic viral hepatitis. The clinical diagnosis may not be apparent because patients with chronic HCV infection often have mild or no symptoms. However, even patients with normal transaminases are at high risk of developing permanent liver damage. Therefore, any individual with detectable HCV RNA in the serum needs medical attention.
HCV infection is potentially curable. Treatment is currently based on combination of pegylated IFN-α and ribavirin. The response to therapy depends on the viral genotype; patients with genotype 2 or 3 infection generally have the best responses. Several new drugs targeting viral protease and polymerase are under investigation.
The Carrier State.
A “carrier” is an individual who harbors and can transmit an organism, but has no manifest symptoms. In the case of hepatotropic virus this definition is somewhat confusing, as it can be interpreted to mean: (1) individuals who carry one of the viruses but have no liver disease; (2) those who harbor one of the viruses and have non-progressive liver damage, but are essentially free of symptoms or disability. In both cases, particularly the latter, these individuals constitute reservoirs for infection. In the case of HBV infection a “healthy carrier” is often defined as an individual without HBeAg, but with presence of anti-HBe, normal aminotransferases, low or undetectable serum HBV DNA, and a liver biopsy showing a lack of significant inflammation and necrosis (Fig. 18-9). In non-endemic areas such as the United States, less than 1% of HBV infections acquired by adults produces a carrier state. This frequency is larger in those who have chronic hepatitis B (Fig. 18-9). In contrast, HBV infection acquired early in life in endemic areas (such as Southeast Asia, China, and Sub-Saharan Africa) gives rise to a carrier state of the two categories described above, in more than 90% of cases. It has been estimated that HCV infection in the United States may yield a carrier state in 10% to 40% of cases, but in most of the studies, absence of liver disease was assessed by persistent normal levels of aminotransferases, rather than liver biopsy. This is a limitation of such studies.
HIV and Chronic Viral Hepatitis.
Because of the similar transmission mode and the similar high-risk patient population, co-infection of HIV and hepatitis viruses is becoming a common clinical problem. Among HIV patients, 10% are infected with HBV and 30% with HCV. Chronic HBV and HCV infection is now a leading cause of morbidity and mortality for HIV-infected patients, and liver disease is the second most common cause of death in individuals with acquired immunodeficiency syndrome (AIDS).34 It is clear that HIV infection significantly exacerbates the severity of liver disease caused by HBV or HCV. Less clear is the impact of HBV or HCV on the course of HIV infection. In addition, anti-HIV agents may cause hepatotoxicity in some patients with HBV or HCV co-infection.
Morphology of Acute and Chronic Hepatitis. The general morphologic features of viral hepatitis are depicted schematically in Figure 18-14. The morphologic changes in acute and chronic viral hepatitis are shared among the hepatotropic viruses and can be mimicked by drug reactions or autoimmune liver disease. Tissue alterations caused by acute infection with HAV, HBV, HCV, and HEV are generally similar, as is the chronic hepatitis caused by HBV, HCV, and HBV + HDV. A few histologic changes may be indicative of a particular type of virus. HBV-infected hepatocytes may show a cytoplasm packed with spheres and tubules of HBsAg, producing a finely granular cytoplasm (“ground-glass hepatocytes,” Fig. 18-15). HCV-infected livers frequently show lymphoid aggregates within portal tracts and focal lobular regions of hepatocyte macrovesicular steatosis, which are to be distinguished from the extensive panlobular microvesicular and macrovesicular steatosis seen in many forms of toxic hepatitis (e.g., alcohol-induced).
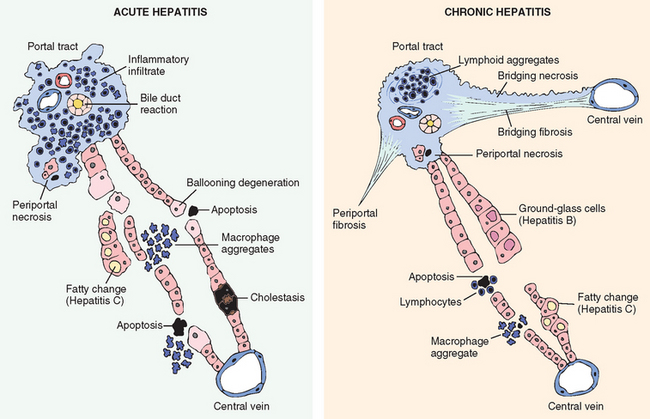
FIGURE 18-14 Diagrammatic representation of the morphologic features of acute and chronic hepatitis. Bridging necrosis (and fibrosis) is shown only for chronic hepatitis but may also occur in acute hepatitis (not shown).
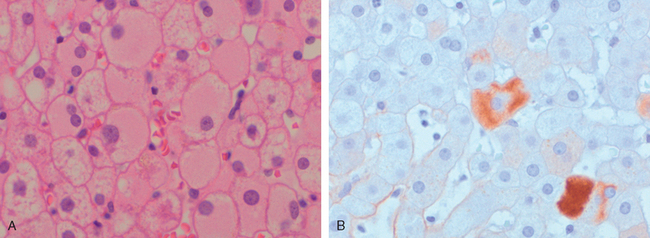
FIGURE 18-15 Chronic HBV infection. A, Showing the diffuse granular cytoplasm, so-called ground-glass hepatocytes. B, Immunoperoxidase stain for HBsAg from the same case, showing cytoplasmic inclusions of viral particles.
Acute Hepatitis. With acute hepatitis (Fig. 18-16) hepatocyte injury takes the form of diffuse swelling (“ballooning degeneration”;), so the cytoplasm looks empty and contains only scattered eosinophilic remnants of cytoplasmic organelles. An inconstant finding is cholestasis, with bile plugs in canaliculi and brown pigmentation of hepatocytes. The canalicular bile plugs result from cessation of the contractile activity of the hepatocyte pericanalicular actin microfilament web. Several patterns of hepatocyte cell death are seen.

FIGURE 18-16 Acute viral hepatitis showing disruption of lobular architecture, inflammatory cells in the sinusoids, and hepatocyte apoptosis (arrow).
Inflammation is a characteristic and usually prominent feature of acute hepatitis. Kupffer cells undergo hypertrophy and hyperplasia and are often laden with lipofuscin pigment as a result of phagocytosis of hepatocellular debris. The portal tracts are usually infiltrated with a mixture of inflammatory cells. The inflammatory infiltrate may spill over into the adjacent parenchyma, causing apoptosis of periportal hepatocytes. This is known as interface hepatitis, which can occur in acute and chronic hepatitis. Cells in the canals of Hering proliferate, forming ductular structures at the parenchymal interface (ductular reaction).
Chronic Hepatitis. The histologic features of chronic hepatitis range from exceedingly mild to severe (Fig. 18-17). In the mildest forms, inflammation is limited to portal tracts and consists of lymphocytes, macrophages, occasional plasma cells, and rare neutrophils or eosinophils. Liver architecture is usually well preserved, but smoldering hepatocyte apoptosis throughout the lobule may occur in all forms of chronic hepatitis. In chronic HCV infection, common findings (occurring in 55% of HCV infections) are lymphoid aggregates and bile duct reactive changes in the portal tracts, and focal mild to moderate macrovesicular steatosis. The steatosis is more prevalent and prominent in HCV genotype 3 infections. In all forms of chronic hepatitis, continued interface hepatitis and bridging necrosis between portal tracts and portal tracts-to-terminal hepatic veins, are harbingers of progressive liver damage.
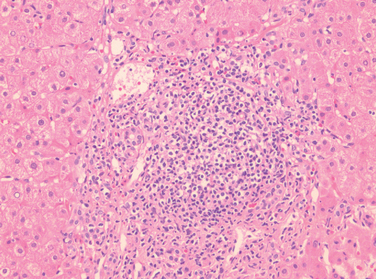
FIGURE 18-17 Chronic viral hepatitis due to HCV, showing portal tract expansion with inflammatory cells and fibrous tissue and interface hepatitis with spillover of inflammation into the adjacent parenchyma. A lymphoid aggregate is present.
The hallmark of chronic liver damage is the deposition of fibrous tissue. At first, only portal tracts show increased fibrosis, but with time periportal septal fibrosis occurs, followed by linking of fibrous septa (bridging fibrosis), especially between portal tracts. In clinical practice, several systems have been used to score the severity and progression of liver damage due to HBV and HCV infection.35 In each system the key elements are inflammation and hepatocyte destruction (grade), and the severity of fibrosis (stage)
Continued loss of hepatocytes and fibrosis results in cirrhosis. It is characterized by irregularly sized nodules separated by variable but mostly broad scars, and is often referred to as post-necrotic cirrhosis (Fig. 18-18). However, this term is not specific to viral etiology, and is applied to all forms of cirrhosis in which the liver shows large, irregular-sized nodules with broad scars. In addition to viral hepatitis, autoimmune hepatitis, hepatotoxins (carbon tetrachloride, mushroom poisoning), pharmaceutical drugs (acetaminophen, α-methyldopa), and even alcohol (discussed later) can give rise to cirrhotic livers with irregular-sized large nodules. In about 20% of cases the inciting cause of the cirrhosis cannot be determined, and these are labeled as cryptogenic cirrhosis. Thus, the morphology of the end-stage cirrhotic liver is often not helpful in determining the basis of the liver injury.
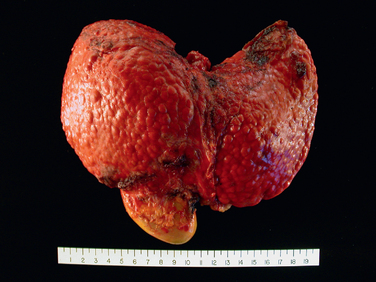
The clinical course of viral hepatitis is unpredictable. Patients may experience spontaneous remission or may have indolent disease without progression for many years. Conversely, some patients have rapidly progressive disease and develop cirrhosis within a few years. The major causes of death from cirrhosis are: liver failure and hepatic encephalopathy, massive hematemesis from esophageal varices, and HCC in those with long-standing HBV (particularly neonatal) or HCV infection.
Fulminant Hepatic Failure.
Hepatic insufficiency that progresses from onset of symptoms to hepatic encephalopathy within 2 to 3 weeks in individuals who do not have chronic liver disease is termed fulminant hepatic failure. Viral hepatitis is responsible for about 12% of cases of fulminant hepatic failure; of these 8% are caused by HBV infection and the rest by HAV. Occasionally, HCV, herpesvirus infection, and Dengue virus cause fulminant hepatitis. Noninfectious causes, such as acetaminophen toxicity, were mentioned earlier. In about 15% of cases, the cause of fulminant hepatic failure is unknown.
The pathogenesis of fulminant hepatic failure varies depending on etiology. In the case of HBV-induced fulminant hepatitis, there is massive apoptosis.36
Morphology of Fulminant Hepatitis. Viral hepatitis and all other causative agents produce essentially identical morphologic changes that vary with the severity of the necrotizing process. The distribution of liver destruction is extremely capricious, since the entire liver or only random areas may be involved. With massive loss of mass, the liver may shrink to as little as 500 to 700 gm, and becomes a limp, red organ covered by a wrinkled, too-large capsule. On transection (Fig. 18-19A), necrotic areas have a muddy red, mushy appearance with hemorrhage. Microscopically, complete destruction of hepatocytes in contiguous lobules leaves only a collapsed reticulin framework and preserved portal tracts. There may be surprisingly little inflammatory reaction. Alternatively, with survival for several days, there is a massive influx of inflammatory cells to begin the phagocytic cleanup process (Fig. 18-19B).
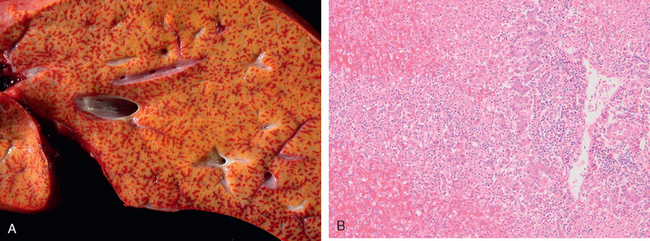
FIGURE 18-19 Massive necrosis. A, Cut section of liver. The liver is small (700 gm), bile-stained, and soft. The capsule is wrinkled. B, Microscopic section. Portal tracts and terminal hepatic veins are closer together than normal, as a result of necrosis and collapse of the intervening parenchyma. The rudimentary ductal structures are the result of early ductular regeneration. An infiltrate of mononuclear inflammatory cells is present.
Survival for more than a week may permit the replication of residual hepatocytes. The proliferation and differentiation of a quiescent stem/progenitor cell population in the canals of Hering, known as oval cells (Chapter 3), creates a ductular reaction. Maturation of these proliferating cells can generate both hepatocytes and bile duct cells. If the parenchymal framework is preserved, regeneration resulting mostly from hepatocyte replication can completely restore the liver architecture. With more massive destruction of confluent lobules, regeneration is disorderly, yielding nodular masses of liver cells that produce a more irregular liver on healing. Fibrous scarring may occur in patients with a protracted course of submassive or patchy necrosis, leading to cirrhosis.
The treatment for fulminant hepatic failure is to correct the underlying liver abnormality and provide supportive care. Liver transplantation is the only option for patients whose disease does not resolve before secondary infection and other organ failure develop. The mortality of fulminant hepatic failure is approximately 80% without liver transplantation, and about 35% with transplantation.
BACTERIAL, PARASITIC, AND HELMINTHIC INFECTIONS
Extrahepatic bacterial infections, particularly sepsis, can induce mild hepatic inflammation and varying degrees of hepatocellular cholestasis. The latter effect is attributable to the effects of pro-inflammatory cytokines released by Kupffer cells and endothelial cells, in response to circulating endotoxin. Several bacteria can infect the liver directly, including Staphylococcus aureus in the setting of toxic shock syndrome, Salmonella typhi in the setting of typhoid fever, and T. pallidum in secondary or tertiary syphilis. Alternatively, bacteria may proliferate in a biliary tree especially when outflow is compromised by partial or complete obstruction. The intra-biliary bacterial composition reflects the gut flora, and the severe acute inflammatory response within the intrahepatic biliary tree is called ascending cholangitis.
Parasitic and helminthic infections are major causes of morbidity worldwide, and the liver is frequently involved (Chapter 8). These diseases include malaria, schistosomiasis, strongyloidiasis, cryptosporidiosis, leishmaniasis, echinococcosis, and infections by the liver flukes Fasciola hepatica and Clonorchis sinensis.
Liver abscesses, a form of liver infection that is common in developing countries, deserve special mention. They are usually caused by echinococcal and amebic infections (Chapter 8), and less commonly, by other protozoal and helminthic organisms. In developed countries liver abscesses are uncommon; the incidence of amebic infections is low and is usually present in immigrants from endemic regions. Most such abscesses are pyogenic, representing a complication of a bacterial infection elsewhere. The organisms reach the liver by (1) the portal vein, (2) arterial supply, (3) ascending infection in the biliary tract (ascending cholangitis), (4) direct invasion of the liver from a nearby source, or (5) a penetrating injury. The majority of hepatic abscesses used to result from portal spread of intra-abdominal infections (e.g., appendicitis, diverticulitis, colitis). With improved management of these conditions, spread now occurs primarily through the biliary tree or the arterial supply in patients suffering from some form of immune deficiency (e.g., old age with debilitating disease, immunosuppression, or cancer chemotherapy with marrow failure). In these settings, abscesses may develop without a primary focus elsewhere.
Morphology. Liver abscesses may occur as solitary or multiple lesions, ranging in size from millimeters to massive lesions many centimeters in diameter. Bacteremic spread through the arterial or portal system tends to produce multiple small abscesses, whereas direct extension and trauma usually cause solitary large abscesses. Biliary abscesses, which are usually multiple, may contain purulent material from adjacent bile ducts. Gross and microscopic features are similar to those seen in any abscess. The causative organism can occasionally be identified in the case of fungal or parasitic abscesses. On rare occasions, abscesses located in the subdiaphragmatic region, particularly amebic, may burrow into the thoracic cavity to produce empyema or a lung abscess. Rupture of subcapsular liver abscesses can lead to peritonitis or localized peritoneal abscesses. Echinococcal infection has a characteristic cystic structure; the wall is laminated, and hooklets and intact organisms can be identified (Fig. 18-20). Calcification in the cystic wall is common.
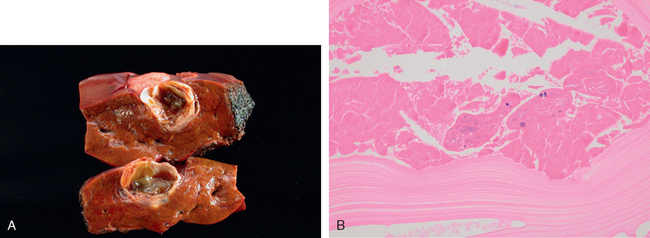
Liver abscesses are associated with fever and, in many instances, right upper quadrant pain and tender hepatomegaly. Jaundice may result from extrahepatic biliary obstruction. Although antibiotic therapy may control smaller lesions, surgical drainage is often necessary for the larger lesions. Because diagnosis is frequently delayed since the patients are elderly and have serious coexistent disease, the mortality rate of patients with large liver abscesses ranges from 30% to 90%. With early recognition and management as many as 80% of patients can survive. In the case of echinococcal cysts, rupture of this cyst has severe clinical consequences, including systemic spread of the organism and resultant shock from massive immune response.
Autoimmune Hepatitis
Autoimmune hepatitis is a chronic and progressive hepatitis of unknown etiology.37 The pathogenesis is attributed to T cell–mediated autoimmunity, in which hepatocyte injury is caused by IFN-γ produced by CD4+ and CD8+ T cells and by CD8+ T-cell–mediated cytotoxicity. A defect in regulatory T-cells may underlie the uncontrolled activation of pathogenic, self-reactive lymphocytes. Genetic factors likely play a role in the autoimmunity (Chapter 6). The injurious immune reaction may be triggered by viral infections, certain drugs such as minocycline, atorvastatin, simvastatin, methyldopa, interferons, nitrofurantoin, and pemoline, and herbal products (such as black cohosh). Autoimmune hepatitis commonly occurs concurrently with other autoimmune disorders, such as celiac disease, systemic lupus erythematosus, rheumatoid arthritis, thyroiditis, Sjögren syndrome, and ulcerative colitis.
Clinicopathologic Features.
The disease may run an indolent or severe course (including fulminant hepatitis). There is a female predominance (78%), particularly in young and perimenopausal women. The annual incidence is highest among white northern Europeans at 1.9 per 100,000, but all ethnic groups are susceptible. The salient features38 include the absence of serologic markers of viral infection, elevated serum IgG and γ-globulin levels (1.2 to 3.0 times normal), and high serum titers of autoantibodies. Autoimmune hepatitis is classified into types 1 and 2, based on the patterns of circulating antibodies. Type 1 is characterized by the presence of antinuclear (ANA), anti–smooth muscle (SMA), anti–actin (AAA), and anti–soluble liver antigen/liver-pancreas antigen (anti-SLA/LP) antibodies. The main antibodies detected in Type 2 autoimmune hepatitis are anti–liver kidney microsome-1 (ALKM-1) antibodies, which are mostly directed against CYP2D6, and anti–liver cytosol-1 (ACL-1). Type 1 is much more common than Type 2 in the United States and is associated with the HLA-DR3 serotype. There is a female predominance, but the disease occurs in children and adults of both sexes.
The entire histologic spectrum of chronic hepatitis may be seen in autoimmune hepatitis, but it is marked by prominent inflammatory infiltrates of lymphocytes and plasma cells. Clusters of plasma cells in the interface of portal tracts and hepatic lobules are fairly characteristic for autoimmune hepatitis (Fig. 18-21). Symptomatic patients with autoimmune hepatitis tend to show substantial liver destruction and scarring at the time of diagnosis. Alternatively, autoimmune hepatitis may present in an atypical fashion with symptoms primarily from involvement of other organ systems, or may be asymptomatic and progress to cirrhosis without clinical diagnosis. An acute appearance of clinical illness is common (40%), and a fulminant presentation with onset of hepatic encephalopathy within 8 weeks of disease onset is possible. In a small subset of patients, autoimmune hepatitis diagnosed clinically may show histologic destruction of bile ducts (“autoimmune cholangitis”), making distinction from primary biliary cirrhosis (PBC) or primary sclerosing cholangitis (PSC) quite difficult. In some cases there is overlap of the clinical and histologic features of autoimmune hepatitis with those of PBC or PSC.
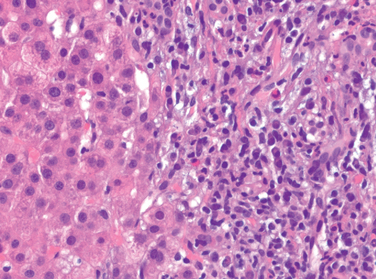
FIGURE 18-21 Autoimmune hepatitis. The photograph shows the interface hepatitis with prominent plasma cells.
The mortality of patients with severe untreated autoimmune hepatitis is approximately 40% within 6 months of diagnosis, and cirrhosis develops in at least 40% of survivors. Hence, diagnosis and intervention are clinical imperatives. Prednisone alone or in combination with azathioprine is the mainstay of therapy. Other immunosuppressants such as cyclosporine, tacrolimus, azathioprine, mycophenolate mofetil, and rapamycin are also used in various clinical settings. Liver transplantation is indicated for patients with end-stage liver disease. The ten-year survival rate after transplantation is 75%, but the disease recurs in 22% to 42% of transplanted patients.
Drug- and Toxin-Induced Liver Disease
The liver is subject to potential damage from an enormous array of pharmaceutical and environmental chemicals.39 Drug-induced liver injury accounts for about 10% of adverse drug reactions, and is the most common cause of fulminant hepatitis in the United States. The incidence of liver injury induced by prescribed drugs is estimated to be between 14 and 40 per 100,000 patients. Genetic variability is a critical factor that influences the susceptibility to drug-induced injury. Injury may result (1) from direct toxicity to hepatocytes or biliary epithelial cells, causing necrosis, apoptosis, or disruption of cellular function; (2) through hepatic conversion of a xenobiotic to an active toxin; or (3) through immune mechanisms, usually by a drug or a metabolite acting as a hapten to convert a cellular protein into an immunogen.40
The main principles of drug and toxic injury are discussed in Chapter 9. Here it suffices to recall that drug reactions may be predictable (intrinsic) or unpredictable (idiosyncratic). Predictable drug reactions can occur in anyone who receives a sufficient dose of an agent. Unpredictable reactions depend on idiosyncracies of the host, particularly the rate at which the host metabolizes the agent, and the intensity of the immune response. Idiosyncratic drug reaction should be considered in any patient receiving a therapeutic drug who develops evidence of liver damage. Generally, adults are more susceptible than children, and women are affected more than men. Important examples include chlorpromazine, an agent that causes cholestasis in patients who are slow to metabolize it to an innocuous byproduct, and halothane, which can cause a fatal immune-mediated hepatitis in some patients who are exposed to this anesthetic on multiple occasions. Table 18-5 lists more common offending agents, grouped according to the type of morphologic injury. It should be noted that the injury may be immediate or may take weeks to months to develop, presenting only after severe liver damage has developed. It may take the form of hepatocyte necrosis, cholestasis, or insidious onset of liver dysfunction. Drug-induced chronic hepatitis is clinically and histologically indistinguishable from chronic viral hepatitis; hence, serologic markers of viral infection are critical for making the distinction.
TABLE 18-5 Patterns of Injury in Drug- and Toxin-Induced Hepatic Injury
| Pattern of Injury | Morphologic Findings | Examples of Associated Agents |
|---|---|---|
| Cholestatic | Bland hepatocellular cholestasis, without inflammation | Contraceptive and anabolic steroids; estrogen replacement therapy |
| Cholestatic hepatitis | Cholestasis with lobular necroinflammatory activity; may show bile duct destruction | Numerous antibiotics; phenothiazines |
| Hepatocellular necrosis | Spotty hepatocyte necrosis | Methyldopa, phenytoin |
| Submassive necrosis, zone 3 | Acetaminophen, halothane | |
| Massive necrosis | Isoniazid, phenytoin | |
| Steatosis | Macrovesicular | Ethanol, methotrexate, corticosteroids, total parenteral nutrition |
| Steatohepatitis | Microvesicular, Mallory bodies | Amiodarone, ethanol |
| Fibrosis and cirrhosis | Periportal and pericellular fibrosis | Methotrexate, isoniazid, enalapril |
| Granulomas | Noncaseating epithelioid granulomas | Sulfonamides, numerous other agents |
| Vascular lesions | Sinusoidal obstruction syndrome (veno-occlusive disease): obliteration of central veins | High-dose chemotherapy, bush teas |
| Budd-Chiari syndrome | Oral contraceptives | |
| Sinusoidal dilatation | Oral contraceptives, numerous other agents | |
| Peliosis hepatis: blood-filled cavities, not lined by endothelial cells | Anabolic steroids, tamoxifen | |
| Neoplasms | Hepatic adenoma | Oral contraceptives, anabolic steroids |
| Hepatocellular carcinoma | Thorotrast | |
| Cholangiocarcinoma | Thorotrast | |
| Angiosarcoma | Thorotrast, vinyl chloride |
From Washington K: Metabolic and toxic conditions of the liver. In Iacobuzio-Donahue CA, Montgomery EA (eds): Gastrointestinal and Liver Pathology. Philadelphia, Churchill Livingstone; 2005.
Among the agents listed in Table 18-5, hepatic injury is considered predictable with overdoses of acetaminophen, exposure to Amanita phalloides toxin, carbon tetrachloride, and, to a certain extent, alcohol. However, individual genetic differences in the hepatic metabolism of xenobiotics through activating and detoxification pathways play a major role in individual susceptibility to even “predictable” hepatotoxins. Many other xenobiotics, such as sulfonamides, α-methyldopa, and allopurinol, cause idiosyncratic reactions. As already mentioned (in this chapter and in Chapter 9), acetaminophen is the leading cause of drug-induced acute liver failure. The most common prescription drugs causing idiosyncratic injury (that is, drug toxicity unrelated to drug dosage) include antibiotics and, in particular, isonazid, nonsteroidal analgesics, and anti-seizure medications. Idiosyncratic reactions evolve with a subacute course and are usually characterized by high bilirubin levels. Herbal preparations can be responsible for both predictable and idiosyncratic liver damage. Reye syndrome, a rare and potentially fatal syndrome of mitochondrial dysfunction in liver, brain, and elsewhere, occurs predominantly in children and is characterized morphologically by extensive accumulation of fat droplets within hepatocytes (microvesicular steatosis). Its development has been associated with the administration of acetylsalicylic acid (aspirin) for the relief of fever, but a causal relationship between aspirin and Reye syndrome has not been established. Nevertheless, aspirin should be avoided in children with febrile illness. Long-term methotrexate administration, an effective treatment for moderate to severe psoriasis, can cause liver injury, including hepatic steatosis and fibrosis.41
Drug-induced liver disease is usually followed by recovery upon removal of the drug. Exposure to a toxin or therapeutic agent should always be included in the differential diagnosis of liver disease.
ALCOHOLIC LIVER DISEASE
Excessive alcohol (ethanol) consumption is the leading cause of liver disease in most Western countries. In the United States, 50% of the population 18 years of age or older drink alcohol. A subset of these individuals suffer serious health consequences associated with alcoholism (Chapter 9). Of greatest impact is alcoholic liver disease, which affects more than 2 million Americans and causes 27,000 deaths a year. There are three distinctive, albeit overlapping, forms of alcoholic liver disease: (1) hepatic steatosis (fatty liver disease), (2) alcoholic hepatitis, and (3) cirrhosis (Fig. 18-22). The morphology of the three forms of alcoholic liver disease is presented first, followed by a discussion of their pathogenesis.

FIGURE 18-22 Alcoholic liver disease. The interrelationships among hepatic steatosis, hepatitis, and cirrhosis are shown, depicting key morphologic features.
Hepatic Steatosis (Fatty Liver). After even moderate intake of alcohol, microvesicular lipid droplets accumulate in hepatocytes. With chronic intake of alcohol, lipid accumulates creating large, clear macrovesicular globules that compress and displace the hepatocyte nucleus to the periphery of the cell. Macroscopically, the fatty liver of chronic alcoholism is a large (as heavy as 4 to 6 kg), soft organ that is yellow and greasy. Although there is little or no fibrosis at the outset, with continued alcohol intake fibrous tissue develops around the terminal hepatic veins and extends into the adjacent sinusoids. The fatty change is completely reversible if there is abstention from further intake of alcohol.
Alcoholic Hepatitis (Alcoholic Steatohepatitis). Alcoholic hepatitis is characterized by:
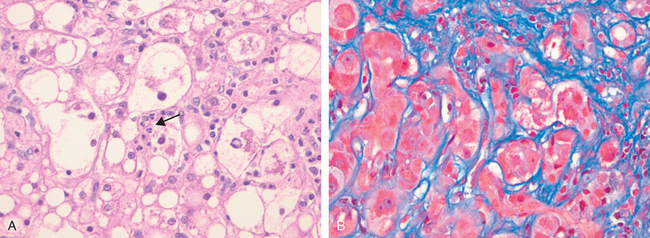
FIGURE 18-23 Alcoholic hepatitis. A, The cluster of inflammatory cells marks the site of a necrotic hepatocyte (arrow). B, Eosinophilic Mallory bodies are seen in hepatocytes, which are surrounded by fibrous (Masson stain) tissue.
Cirrhosis. The final and irreversible form of alcoholic liver disease usually evolves slowly and insidiously but may develop in 1 or 2 years in some cases. At first the cirrhotic liver is yellowtan, fatty, and enlarged, usually weighing over 2 kg. Over the span of years, it is transformed into a brown, shrunken, nonfatty organ, sometimes less than 1 kg in weight. Initially the developing fibrous septa are delicate and extend through sinusoids from central to portal regions as well as from portal tract to portal tract. Regenerative activity of entrapped parenchymal hepatocytes generates uniform micronodules. With time the nodularity becomes more prominent; scattered larger nodules create a “hobnail” appearance on the surface of the liver (Fig. 18-24A). As fibrous septa dissect and surround nodules, the liver becomes more fibrotic, loses fat, and shrinks progressively in size. Parenchymal islands are engulfed by wider bands of fibrous tissue, and the liver is converted into a mixed micronodular and macronodular pattern (Fig. 18-24B). Ischemic necrosis and fibrous obliteration of nodules eventually create broad expanses of tough, pale scar tissue (“Laennec cirrhosis”). Bile stasis often develops; Mallory bodies are only rarely evident at this stage. Thus, end-stage alcoholic cirrhosis comes to resemble, both macroscopically and microscopically, the cirrhosis developing from viral hepatitis and other causes.
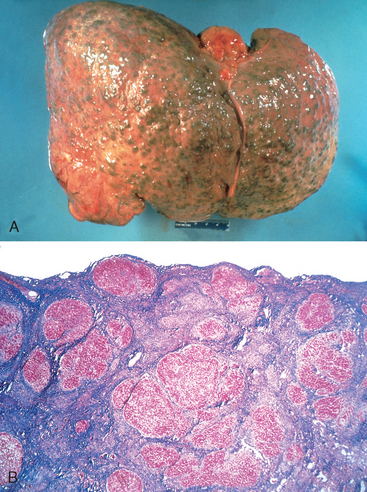
FIGURE 18-24 Alcoholic cirrhosis. A, The characteristic diffuse nodularity of the surface reflects the processes of nodular regeneration and scarring. The greenish tint of some nodules is due to bile stasis. A hepatocellular carcinoma is present as a budding mass at the lower edge of the right lobe (lower left). B, The microscopic view shows nodules of varying sizes entrapped in blue-staining fibrous tissue. The liver capsule is at the top (Masson trichrome).
Pathogenesis.
Short-term ingestion of as much as 80 gm of alcohol (six beers or 8 ounces of 80-proof liquor) over one to several days generally produces mild, reversible hepatic steatosis. Daily intake of 80 gm or more of ethanol generates significant risk for severe hepatic injury, and daily ingestion of 160 gm or more for 10 to 20 years is associated more consistently with severe injury. Only 10% to 15% of alcoholics, however, develop cirrhosis. Thus, other factors must also influence the development and severity of alcoholic liver disease. These factors include:
The pharmacokinetics and metabolism of alcohol were described in Chapter 9. Pertinent to our discussion here are the detrimental effects of alcohol and its byproducts on hepatocellular function. Exposure to alcohol causes steatosis, dysfunction of mitochondrial and cellular membranes, hypoxia, and oxidative stress. At millimolar concentrations, alcohol directly affects microtubular and mitochondrial function and membrane fluidity.
Hepatocellular steatosis results from (1) shunting of normal substrates away from catabolism and toward lipid biosynthesis, as a result of generation of excess reduced nicotinamide adenine dinucleotide (NADH + H+) by the two major enzymes of alcohol metabolism, alcohol dehydrogenase and acetaldehyde dehydrogenase; (2) impaired assembly and secretion of lipoproteins; and (3) increased peripheral catabolism of fat.
The causes of alcoholic hepatitis are uncertain but some of the factors in its pathogenesis are discussed next. Acetaldehyde (the major intermediate metabolite of alcohol) induces lipid peroxidation and acetaldehyde-protein adduct formation, further disrupting cytoskeletal and membrane function. Cytochrome P-450 metabolism produces reactive oxygen species (ROS) that react with cellular proteins, damage membranes, and alter hepatocellular function. In addition, alcohol-induced impaired hepatic metabolism of methionine leads to decreased intrahepatic glutathione levels, thereby sensitizing the liver to oxidative injury. The induction of CYP2E1 and other cytochrome P-450 enzymes in the liver by alcohol increases alcohol catabolism in the endoplasmic reticulum and enhances the conversion of other drugs (e.g., acetaminophen) to toxic metabolites.
Alcohol can become a major source of calories in the diet of an alcoholic, displacing other nutrients and leading to malnutrition and deficiencies of vitamins (such as thiamine). This is compounded by impaired digestive function, primarily related to chronic gastric and intestinal mucosal damage, and pancreatititis.
Alcohol causes the release of bacterial endotoxin from the gut into the portal circulation, inducing inflammatory responses in the liver, such as the activation of NF-κB, and release of TNF, IL-6, and TGF-α. In addition, alcohol stimulates the release of endothelins from sinusoidal endothelial cells, causing vasoconstriction and the contraction of activated stellate cells (“myofibroblasts”), leading to a decrease in hepatic sinusoidal perfusion (already discussed under “Portal Hypertension”).
In summary, alcoholic liver disease is a chronic disorder featuring steatosis, hepatitis, progressive fibrosis, cirrhosis, and marked derangement of vascular perfusion. It can be regarded as a maladaptive state in which cells in the liver respond in an increasingly pathologic manner to a stimulus (alcohol) that originally was only marginally harmful. For some unknown reason, cirrhosis develops in only a small fraction of chronic alcoholics.
Clinical Features.
Hepatic steatosis (fatty liver) may become evident as hepatomegaly, with mild elevation of serum bilirubin and alkaline phosphatase levels. Severe hepatic dysfunction is unusual. Alcohol withdrawal and the provision of an adequate diet are sufficient treatment. In contrast, alcoholic hepatitis tends to appear acutely, usually following a bout of heavy drinking. Symptoms and laboratory manifestations may range from minimal to fulminant hepatic failure. Between these two extremes are the nonspecific symptoms of malaise, anorexia, weight loss, upper abdominal discomfort, tender hepatomegaly, and the laboratory findings of hyperbilirubinemia, elevated alkaline phosphatase, and often a neutrophilic leukocytosis. An acute cholestatic syndrome may appear, resembling large bile duct obstruction. The outlook is unpredictable; each bout of hepatitis incurs about a 10% to 20% risk of death. With repeated bouts, cirrhosis appears in about one third of patients within a few years. Alcoholic hepatitis also may be superimposed on established cirrhosis. With proper nutrition and total cessation of alcohol consumption, the alcoholic hepatitis may clear slowly. However, in some patients, the hepatitis persists, despite abstinence, and progresses to cirrhosis.
The manifestations of alcoholic cirrhosis are similar to those of other forms of cirrhosis. Laboratory findings reflect the hepatic dysfunction, with elevated serum aminotransferase, hyperbilirubinemia, variable elevation of serum alkaline phosphatase, hypoproteinemia (globulins, albumin, and clotting factors), and anemia. In some instances, liver biopsy may be indicated, since in about 10% to 20% of cases of presumed alcoholic cirrhosis, another disease process is found. Finally, cirrhosis may be clinically silent, discovered only at autopsy or when stress such as infection or trauma tips the balance toward hepatic insufficiency.
The long-term outlook for alcoholics with liver disease is variable. Five-year survival approaches 90% in abstainers who are free of jaundice, ascites, or hematemesis; it drops to 50% to 60% in those who continue to imbibe. In the end-stage alcoholic the proximate causes of death are (1) hepatic coma, (2) massive gastrointestinal hemorrhage, (3) intercurrent infection (to which these patients are predisposed), (4) hepatorenal syndrome following a bout of alcoholic hepatitis, and (5) hepatocellular carcinoma (the risk of developing this tumor in alcoholic cirrhosis is 1% to 6% of cases annually).
Metabolic Liver Disease
A distinct group of liver diseases is attributable to disorders of metabolism, either acquired or inherited. The most common acquired metabolic disorder is non-alcoholic fatty liver disease. Among inherited metabolic diseases, hemochromatosis, Wilson disease, and α1-antitrypsin deficiency are most prominent. Also included among liver metabolic diseases is neonatal hepatitis, a broad disease category encompassing rare inherited diseases and neonatal infections.
NONALCOHOLIC FATTY LIVER DISEASE (NAFLD)
NAFLD is a group of conditions that have in common the presence of hepatic steatosis (fatty liver), in individuals who do not consume alcohol, or do so in very small quantities (less than 20 g of ethanol/week). It has become the most common cause of chronic liver disease in the United States, and in its various forms, probably affects more than 30% of the population. However, these estimates are approximate, because fatty liver without other complications may not be detected clinically. NAFLD includes simple hepatic steatosis, steatosis accompanied by minor, non-specific inflammation, and non-alcoholic steatohepatitis (NASH).43 Steatosis with or without non-specific inflammation is generally a stable condition without significant clinical problems. In contrast, NASH is a condition in which there is hepatocyte injury that may progress to cirrhosis in 10% to 20% of cases. The main components of NASH are hepatocyte ballooning, lobular inflammation, and steatosis.44 With progressive disease fibrosis occurs. NASH affects men and women equally and the condition is strongly associated with obesity and the other components of the metabolic syndrome, such as dyslipidemia, hyperinsulinemia and insulin resistance. It is estimated that more than 70% of obese individuals have some form of NAFLD. It is the most common cause of so-called cryptogenic cirrhosis, namely cirrhosis of “unknown” origin. NAFLD contributes to the progression of other liver diseases such as HCV infection and HCC. The epidemic of obesity in the United States heightens concern that NAFLD will increase in prevalence.
Pathogenesis.
The precise mechanisms of steatosis and hepatocellular damage in NAFLD are not entirely known, but genetics and environment play a role in the pathogenesis.44 A “two-hit” model of pathogenesis has been proposed, encompassing two sequential events: (1) hepatic fat accumulation and, (2) hepatic oxidative stress.45 The oxidative stress acts upon the accumulated hepatic lipids, resulting in lipid peroxidation and the release of lipid peroxides, which can produce reactive oxygen species.
Clinical Features.
Individuals with simple steatosis are generally asymptomatic. Clinical presentation is often related to other metabolic derangements, such as obesity, insulin resistance, and diabetes.46 Imaging studies may reveal fat accumulation in the liver. However, liver biopsy is the most reliable diagnostic tool for NASH and helps determine the extent of steatosis, presence of steatohepatitis, and degree of fibrosis. Serum AST and ALT are elevated in about 90% of patients with NASH. The AST/ALT ratio is usually less than 1, in contrast to alcoholic steatohepatitis in which the ratio is generally above 2.0 to 2.5. Despite the enzyme elevations, patients may be asymptomatic. Others have general symptoms such as fatigue and right-sided abdominal discomfort caused by hepatomegaly. Because of the association between NASH and the metabolic syndrome, cardiovascular disease is a frequent cause of death in patients with NASH. The goal of treating individuals with NASH is to reverse the steatosis and prevent cirrhosis. The current management strategy seeks to correct the underlying risk factors, such as obesity and hyperlipidemia, and to treat insulin resistance.
Morphology. Steatosis usually involves more than 5% of the hepatocytes and sometimes more than 90%. Large (macrovesicular) and small (microvesicular) droplets of fat, predominantly triglycerides, accumulate within hepatocytes (Fig. 18-25A). At the most clinically benign end of the spectrum, there is no appreciable hepatic inflammation, hepatocyte death, or scarring, despite persistent elevation of serum liver enzymes. Steatohepatitis (NASH) is characterized by steatosis and multifocal parenchymal inflammation, mainly neutrophils, Mallory bodies, hepatocyte death (both ballooning degeneration and apoptosis), and sinusoidal fibrosis. Fibrosis also occurs within portal tracts and around terminal hepatic venules (Fig. 18-25B). These histological changes are similar to those of alcoholic steatohepatitis. Cirrhosis may develop, presumably the result of years of subclinical progression of the necroinflammatory and fibrotic processes. When cirrhosis is established, the steatosis or steatohepatitis tends to be reduced and sometimes is not identifiable.
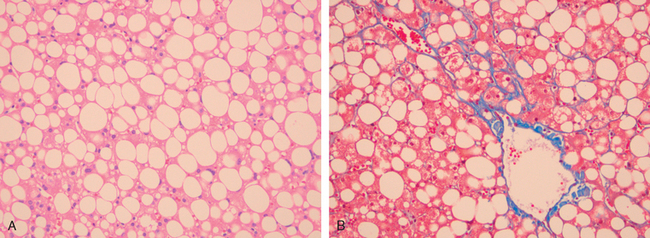
HEMOCHROMATOSIS
Hemochromatosis was first described by von Recklinghausen in 1889. It is characterized by the excessive accumulation of body iron, most of which is deposited in parenchymal organs such as the liver and pancreas. Iron can also accumulate in the heart, joints, or endocrine organs. Hemochromatosis (also known as primary or hereditary hemochromatosis) is a homozygous-recessive inherited disorder47 that is caused by excessive iron absorption. Accumulation of iron in tissues, which may occur as a consequence of parenteral administration of iron, usually in the form of transfusions, or other causes (Table 18-6), is variably known as secondary hemochromatosis, acquired hemochromatosis, or hemosiderosis. We will use the terms hemochromatosis for the hereditary disease and hemosiderosis for the acquired deposition of iron in some tissues.
TABLE 18-6 Classification of Iron Overload
* Neonatal hemochromatosis develops in utero and does not appear to be a hereditary condition.
As was discussed in Chapter 14, the total body iron pool ranges from 2 to 6 gm in normal adults; about 0.5 gm is stored in the liver, 98% of which is in hepatocytes. In hemochromatosis, total iron accumulation may exceed 50 gm, over one third of which accumulates in the liver. The following features characterize this disease:
Pathogenesis.
Because there is no regulated iron excretion from the body, the total body content of iron is tightly regulated by intestinal absorption as described below. In hemochromatosis, regulation of intestinal absorption of dietary iron is abnormal, leading to net iron accumulation of 0.5 to 1.0 gm/year, mainly in the liver. The disease manifests itself typically after 20 gm of stored iron have accumulated. Excessive iron appears to be directly toxic to host tissues, by the following mechanisms: (1) lipid peroxidation via iron-catalyzed free radical reactions, (2) stimulation of collagen formation by activation of hepatic stellate cells, and (3) interaction of reactive oxygen species and of iron itself with DNA, leading to lethal cell injury or predisposition to hepatocellular carcinoma. The actions of iron are reversible in cells that are not fatally injured, and removal of excess iron with therapy promotes recovery of tissue function.
The main regulator of iron absorption is the protein hepcidin (also known as liver expressed antimicrobial peptide or LEAP1), encoded by the HAMP gene. Hepcidin, which also has antibacterial activity, is produced in hepatocytes as an 84 amino acid propeptide that is cleaved into a mature form of 25 amino acids and smaller circulating forms of 20 and 23 amino acids. Transcription of hepcidin is increased by inflammatory cytokines and iron, and decreased by iron deficiency, hypoxia and ineffective erythropoiesis. Hepcidin binds to the cellular iron efflux channel ferroportin (FPN), causing internalization and proteolysis of the channel. This prevents the release of iron from intestinal cells and macrophages; thus hepcidin lowers plasma iron levels. Conversely, a deficiency in hepcidin causes iron overload.
Other proteins involved in iron metabolism, do so by regulating hepcidin levels. These include: (1) hemojuvelin (HJV), which is expressed in the liver, heart, and skeletal muscle; (2) transferrin receptor 2 (TfR2), which is highly expressed in hepatocytes, where it mediates the uptake of transferrin-bound iron, and (3) HFE, the product of the hemochromatosis gene. Lack of hepcidin expression caused by mutations in hepcidin, HJV, TfR2, and HFE cause hemochromatosis. Of these mutations, those in HFE are the most common, as discussed below. Mutations of HAMP and HJV cause a severe form of hereditary hemochromatosis, known as juvenile hemochromatosis. Mutations of HFE and TfR2 cause the classic form of hereditary adult hemochromatosis, a milder disease than the juvenile form. Mutations of ferroportin cause a distinctive iron storage disease that is different from hereditary hemochromatosis. The precise mechanisms by which HFE, HJV, and TfR2 regulate hepcidin and ferroportin remain to be determined. A serine protease (TMPRSS6) was recently identified as an iron sensor that suppresses HAMP expression.48
The adult form of hemochromatosis is almost always caused by mutations of HFE, a gene located on the short arm of chromosome 6 at 6p21.3, close to the HLA gene locus. It encodes an HLA class I-like molecule that regulates intestinal absorption of dietary iron. The most common HFE mutation is a cysteine-to-tyrosine substitution at amino acid 282 (called C282Y), due to a single G to A transition at nucleotide 845 (G845A). This mutation, which causes inactivation of the protein, is present in 70% to 100% of the patients diagnosed with hereditary hemochromatosis. The other common mutation is H63D (histidine at position 63 to aspartate). The H63D homozygous state and C282Y/H63D compound heterozygous mutations often cause only mild iron accumulation.
The C282Y mutation is largely confined to white populations of European origin, while the H63D has a worldwide distribution. The frequency of C282Y homozygosity is 0.45% (1 of every 220 persons), and the heterozygous frequency is 11%, making hereditary hemochromatosis one of the most common genetic disorders in humans. However, the penetrance of this disorder is low in patients with the homozygous C282Y mutation, so the genetic condition does not lead to clinical disease in all individuals.
Morphology. The morphologic changes in hereditary hemochromatosis are characterized principally by: (1) deposition of hemosiderin in the following organs (in decreasing order of severity): liver, pancreas, myocardium, pituitary gland, adrenal gland, thyroid and parathyroid glands, joints, and skin (detected by the Prussian blue histologic reaction or by atomic absorption analysis of tissue); (2) cirrhosis; and (3) pancreatic fibrosis. In the liver, iron becomes evident first as golden-yellow hemosiderin granules in the cytoplasm of periportal hepatocytes, which stain blue with the Prussian blue stain (Fig. 18-26). With increasing iron load, there is progressive involvement of the rest of the lobule, along with bile duct epithelium and Kupffer cell pigmentation. Iron is a direct hepatotoxin, and inflammation is characteristically absent. At this stage, the liver is typically slightly larger than normal, dense, and chocolate brown. Fibrous septa develop slowly, leading ultimately to a micronodular pattern of cirrhosis in an intensely pigmented liver.
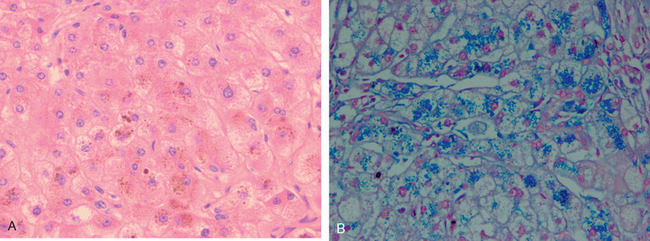
FIGURE 18-26 Histologic appearance of hereditary hemochromatosis. Hepatocellular iron deposition is dark-brown in H&E stain (A) and blue in Prussian blue–stained section (B). This is a section from an early stage of the disease, in which parenchymal architecture is normal.
Biochemical determination of hepatic tissue iron concentration is the standard for quantitating hepatic iron content. In normal individuals, the iron content of liver tissue is less than 1000 μg per gram dry weight of liver. Adult patients with hereditary hemochromatosis exhibit over 10,000 μg iron per gram dry weight; hepatic iron concentrations in excess of 22,000 μg per gram dry weight are associated with the development of fibrosis and cirrhosis.
The pancreas becomes intensely pigmented, has diffuse interstitial fibrosis, and may exhibit some parenchymal atrophy. Hemosiderin is found in both the acinar and the islet cells, and sometimes in the interstitial fibrous stroma. The heart is often enlarged and has hemosiderin granules within the myocardial fibers, producing a striking brown coloration to the myocardium. A delicate interstitial fibrosis may appear. Although skin pigmentation is partially attributable to hemosiderin deposition in dermal macrophages and fibroblasts, most of the pigmentation results from increased epidermal melanin production. The combination of these pigments imparts a characteristic slate-gray color to the skin. With hemosiderin deposition in the joint synovial linings, an acute synovitis may develop. Excessive deposition of calcium pyrophosphate damages the articular cartilage, producing a disabling polyarthritis referred to as pseudo-gout. The testes may be small and atrophic but are not usually significantly pigmented. It is thought that the atrophy is secondary to a derangement in the hypothalamic-pituitary axis resulting in reduced gonadotropin and testosterone levels.
Clinical Features.
Classical hemochromatosis is more often a disease of males and rarely becomes evident before age 40. The principal manifestations include hepatomegaly, abdominal pain, skin pigmentation (particularly in sun-exposed areas), deranged glucose homeostasis or frank diabetes mellitus due to destruction of pancreatic islets, cardiac dysfunction (arrhythmias, cardiomyopathy), and atypical arthritis. In some patients, the presenting complaint is hypogonadism (e.g., amenorrhea in the female, impotence and loss of libido in the male). The classic triad of pigment cirrhosis with hepatomegaly, skin pigmentation, and diabetes mellitus might not develop until late in the course of the disease. Death may result from cirrhosis or cardiac disease. A significant cause of death is hepatocellular carcinoma; the risk is 200-fold greater than in the general population, and treatment for iron overload does not remove the risk for this tumor.
Fortunately, hemochromatosis can be diagnosed long before irreversible tissue damage has occurred. Screening involves demonstration of very high levels of serum iron and ferritin, exclusion of secondary causes of iron overload, and liver biopsy if indicated. Screening of family members of probands is important. Heterozygotes also accumulate excessive iron, but not to a level that causes significant tissue damage. Currently most patients with hemochromatosis are diagnosed in the subclinical, precirrhotic stage due to routine serum iron measurements (as part of other diagnostic workup). They are treated by regular phlebotomy and have a normal life expectancy.
Neonatal hemochromatosis (also called congenital hemochromatosis) is a disease of unknown origin manifested by severe liver disease and extrahepatic hemosiderin deposition.49 Neonatal hemochromatosis is not an inherited disease; liver injury, leading to hemosiderin accumulation, occurs in utero, and might be related to maternal alloimmune injury to the fetal liver. Extrahepatic hemosiderin deposition, detected by buccal biopsy, needs to be documented for the correct diagnosis. There is no specific treatment, except for supportive care, and liver transplantation in severe cases.
The most common causes of hemosiderosis (secondary or acquired hemochromatosis) are disorders associated with ineffective erythropoiesis, such as severe forms of thalassemia (Chapter 14) and myelodysplastic syndromes (Chapter 13). In these disorders, the excess iron results not only from transfusions, but also from increased absorption. Transfusions alone, when given repeatedly over a period of years (such as in patients with chronic hemolytic anemias), can also lead to systemic hemosiderosis and parenchymal organ injury. Alcoholic cirrhosis is often associated with a modest increase in stainable iron within liver cells. However, this represents alcohol-induced redistribution of iron, since total body iron is not significantly increased. A rather unusual form of iron overload resembling hereditary hemochromatosis occurs in sub-Saharan Africa, the result of ingesting large quantities of alcoholic beverages fermented in iron utensils (Bantu siderosis). Home brewing in steel drums continues to this day, and genetic susceptibility to this disease, such as mutations of ferroportin has been proposed in these populations.50 Lastly, chronic HBV and HCV infection may increase iron storage within hepatocytes.
WILSON DISEASE
Wilson disease is an autosomal recessive disorder caused by mutation of the ATP7B gene, resulting in impaired copper excretion into bile and a failure to incorporate copper into ceruloplasmin.51 This disorder is marked by the accumulation of toxic levels of copper in many tissues and organs, principally the liver, brain, and eye. Normally, 40% to 60% of ingested copper (2 to 5 mg/day) is absorbed in the duodenum and proximal small intestine, and is transported to the portal circulation complexed with albumin and histidine. Free copper dissociates and is taken up by hepatocytes. Copper is incorporated into enzymes and also binds to a α2-globulin (apoceruloplasmin) to form ceruloplasmin, which is secreted into the blood. Excess copper is transported into the bile. Ceruloplasmin accounts for 90% to 95% of plasma copper. Circulating ceruloplasmin is eventually desialylated, endocytosed by the liver, and degraded within lysosomes, after which the released copper is excreted into bile. This degradation/excretion pathway is the primary route for copper elimination. The estimated total body copper is only 50 to 150 mg.
The ATP7B gene, located on chromosome 13, encodes a transmembrane copper-transporting ATPase, expressed on the hepatocyte canalicular membrane. More than 300 mutations in the ATP7B gene have been identified, but not all genes cause the disease. The overwhelming majority of patients are compound heterozygotes containing different mutations on each ATP7B allele. The overall frequency of mutated alleles is 1 : 100, and the prevalence of the disease of approximately 1 : 30,000 to 1 : 50,000 (approximately 9000 patients in the United States). Deficiency in the ATP7B protein causes a decrease in copper transport into bile, impairs its incorporation into ceruloplasmin, and inhibits ceruloplasmin secretion into the blood. These changes cause copper accumulation in the liver and a decrease in circulating ceruloplasmin. The copper causes toxic liver injury, through the production of ROS by the Fenton reaction (Chapter 1). Although there is a latent period of variable duration for the disease, once the hepatic capacity for incorporating copper into ceruloplasmin is exceeded, there may be sudden onset of critical systemic illness. Specifically, non-ceruloplasmin–bound copper spills over from the liver into the circulation, causing hemolysis and pathologic changes at other sites such as the brain, corneas, kidneys, bones, joints, and parathyroids. Concomitantly, urinary excretion of copper markedly increases from its normal minuscule levels.
Morphology. The liver often bears the brunt of injury, but the disease may also present as a neurologic disorder. The hepatic changes are variable, ranging from relatively minor to massive damage. Fatty change (steatosis) may be mild to moderate, with vacuolated nuclei (glycogen or water) and occasionally, focal hepatocyte necrosis. An acute hepatitis can show features mimicking acute viral hepatitis, except possibly for the accompanying fatty change. The chronic hepatitis of Wilson disease exhibits moderate to severe inflammation and hepatocyte necrosis, with the particular features of macrovesicular steatosis, vacuolated hepatocellular nuclei, and Mallory bodies. With progression of chronic hepatitis, cirrhosis will develop. Massive liver necrosis is a rare manifestation that is indistinguishable from that caused by viruses or drugs. Excess copper deposition can often be demonstrated by special stains (rhodamine stain for copper, orcein stain for copper-associated protein). Because copper also accumulates in chronic obstructive cholestasis and because histology cannot reliably distinguish Wilson disease from viral- and drug-induced hepatitis, demonstration of hepatic copper content in excess of 250 μg per gram dry weight is most helpful for making a diagnosis.
In the brain, toxic injury primarily affects the basal ganglia, particularly the putamen, which shows atrophy and even cavitation. Nearly all patients with neurologic involvement develop eye lesions called Kayser-Fleischer rings, green to brown deposits of copper in Desçemet’s membrane in the limbus of the cornea.
Clinical Features.
The age at onset and the clinical presentation of Wilson disease are extremely variable (average age is 11.4 years), but the disorder usually manifests in affected individuals between 6 and 40 years of age. The most common presentation is acute or chronic liver disease. Neuropsychiatric manifestations, including mild behavioral changes, frank psychosis, or a Parkinson disease–like syndrome (such as tremor), are the initial features in most of the remaining cases. The biochemical diagnosis of Wilson disease is based on a decrease in serum ceruloplasmin, an increase in hepatic copper content (the most sensitive and accurate test), and increased urinary excretion of copper (the most specific screening test). Serum copper levels are of no diagnostic value, since they may be low, normal, or elevated, depending on the stage of evolution of the disease. Demonstration of Kayser-Fleischer rings further favors the diagnosis. Early recognition and long-term copper chelation therapy (as with D-penicillamine, or Trientine) or zinc-based therapy has dramatically altered the usual progressive downhill course. Individuals with hepatitis or unmanageable cirrhosis require liver transplantation for survival, which can also lead to eventual cure.
α1-ANTITRYPSIN DEFICIENCY
α1-Antitrypsin deficiency is an autosomal recessive disorder marked by very low levels of α1-antitrypsin. The major function of this protein is the inhibition of proteases, particularly neutrophil elastase, cathepsin G, and proteinase 3, which are normally released from neutrophils at sites of inflammation. α1-Antitrypsin deficiency leads to the development of pulmonary emphysema, because the activity of destructive proteases is not inhibited (discussed in Chapter 15). It also causes liver disease, as a consequence of the accumulation of this protein in hepatocytes.52 In addition, cutaneous panniculitis, arterial aneurysm, bronchiectasis, and Wegener’s granulomatosis can occur in α1-antitrypsin deficiency.
α1-Antitrypsin is a small 394–amino acid plasma glycoprotein synthesized predominantly by hepatocytes. It is a member of the serine protease inhibitor (serpin) family. The gene, located on chromosome 14, is very polymorphic, and at least 75 α1-antitrypsin forms have been identified, denoted alphabetically by their relative migration on an isoelectric gel. The general notation is “Pi” for “protease inhibitor” and an alphabetic letter for the position on the gel; two letters denote the genotype of the two alleles. The most common genotype is PiMM, occurring in 90% of individuals (in the traditional sense, this would be the wild-type genotype). Most allelic variants show substitutions in the polypeptide chain but produce normal levels of functional α1-antitrypsin. Some deficiency variants, including the PiS variant, result in a moderate reduction in serum concentrations of α1-antitrypsin without clinical manifestations. Rare variants termed Pi-null have no detectable serum α1-antitrypsin. The most common clinically significant mutation is PiZ; homozygotes for the PiZZ protein have circulating α1-antitrypsin levels that are only 10% of normal. These individuals are at high risk for developing clinical disease. Expression of alleles is autosomal codominant, and consequently, PiMZ heterozygotes have intermediate plasma levels of α1-antitrypsin. Among people of northern European descent the PiS frequency is 6% and the PiZ frequency is 4%; the PiZZ state affects 1 in 1800 live births. Because of its occasionally early presentation for liver disease, α1-antitrypsin deficiency is the most commonly diagnosed genetic hepatic disorder in infants and children.
Pathogenesis.
With most allelic variants, the mRNA is transcribed, and the protein is synthesized and secreted normally. Deficiency variants show a selective defect in migration of this secretory protein from the endoplasmic reticulum to Golgi apparatus; this is most marked for the PiZ polypeptide, attributable to a single amino acid substitution of Glu342 to Lys342. The mutant polypeptide (α1AT-Z) is abnormally folded and polymerizes, creating endoplasmic reticulum stress and leading to apoptosis (Chapter 1; see Fig. 1-27). The precise mechanisms of liver disease with α1AT-Z are not well defined. The accumulated α1AT-Z in the endoplasmic reticulum triggers a series of events, including an autophagocytic response, mitochondrial dysfunction, and possible activation of pro-inflammatory NF-κB, causing hepatocyte damage.53 All individuals with the PiZZ genotype accumulate α1AT-Z in the endoplasmic reticulum of hepatocytes, but only 10% to 15% of PiZZ individuals develop overt clinical liver disease. Other genetic factors or environmental factors are thus posited to play a role in the development of liver disease.
Morphology. α1-Antitrypsin deficiency is characterized by the presence of round-to-oval cytoplasmic globular inclusions in hepatocytes, which in routine H&E stains are acidophilic and indistinctly demarcated from the surrounding cytoplasm. They are strongly periodic acid–Schiff (PAS)-positive and diastase-resistant (Fig. 18-27). The globules are also present but in diminished size and number in the PiMZ and PiSZ genotypes. For unknown reasons most of the globules are in hepatocytes surrounding the portal tracts. Moreover, the number of globule-containing hepatocytes in a patient’s liver is not correlated with the severity of pathologic findings. The hepatic pathology associated with PiZZ homozygosity is extremely varied, ranging from neonatal hepatitis (Fig. 18-28) without or with cholestasis and fibrosis (discussed below), to childhood cirrhosis, to a smoldering chronic inflammatory hepatitis or cirrhosis that becomes apparent only late in life. For the most part the only distinctive feature of the hepatic disease is the PAS-positive globules; infrequently, fatty change and Mallory bodies are present. The diagnostic α1-antitrypsin globules may be absent in the young infant; steatosis may be present as a tip-off to the possibility of α1-antitrypsin deficiency.
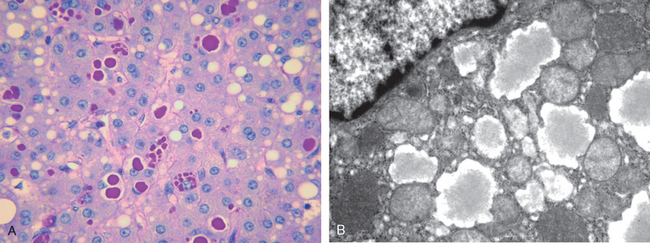
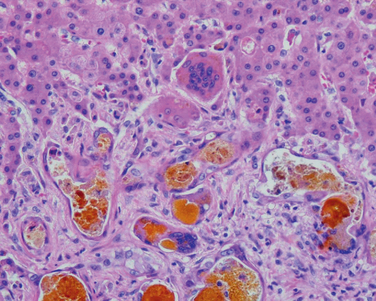
Clinical Features.
Neonatal hepatitis with cholestatic jaundice appears in 10% to 20% of newborns with the deficiency. In adolescence, presenting symptoms may be related to hepatitis or cirrhosis. Attacks of hepatitis may subside with apparent complete recovery, or they may become chronic and lead progressively to cirrhosis. Finally, the disease may remain silent until cirrhosis appears in middle to later life. HCC develops in 2% to 3% of PiZZ adults, usually but not always in the setting of cirrhosis. The treatment, and the cure, for severe hepatic disease is orthotopic liver transplantation. In patients with pulmonary disease the single most important treatment is avoidance of cigarette smoking, because smoking markedly accelerates emphysema and the destructive lung disease associated with α1-antitrypsin deficiency.
NEONATAL CHOLESTASIS
Prolonged conjugated hyperbilirubinemia in the neonate, termed neonatal cholestasis, affects approximately 1 in 2500 live births. The major conditions causing it are (1) cholangiopathies, primarily biliary atresia (discussed later), and (2) a variety of disorders causing conjugated hyperbilirubinemia in the neonate, collectively referred to as neonatal hepatitis. Neonatal cholestasis and hepatitis are not specific entities, nor are the disorders necessarily inflammatory. Instead, the finding of “neonatal cholestasis” should evoke a diligent search for recognizable toxic, metabolic, and infectious liver diseases, the more common of which are listed in Table 18-7. Once identifiable causes have been excluded, one is left with the syndrome of “idiopathic” neonatal hepatitis, which shows considerable clinical overlap with biliary atresia. Despite the long list of disorders associated with neonatal cholestasis, most are quite rare. “Idiopathic” neonatal hepatitis represents as many as 50% of cases, biliary atresia represents another 20%, and α1-antitrypsin deficiency represents 15%. Differentiation of biliary atresia from nonobstructive neonatal cholestasis assumes great importance, since definitive treatment of biliary atresia requires surgical intervention (Kasai procedure), whereas surgery may adversely affect the clinical course of a child with other disorders. Fortunately, discrimination can be made with clinical data in about 90% of cases, with or without liver biopsy. Affected infants have jaundice, dark urine, light or acholic stools, and hepatomegaly. Variable degrees of hepatic synthetic dysfunction may be identified, such as hypoprothrombinemia. Thus, liver biopsy is critical in distinguishing neonatal hepatitis from an identifiable cholangiopathy.
TABLE 18-7 Major Causes of Neonatal Cholestasis
| Idiopathic neonatal hepatitis |
Morphology. The morphologic features of neonatal hepatitis include lobular disarray with focal liver cell apoptosis and necrosis, panlobular giant-cell transformation of hepatocytes (Fig. 18-29), prominent hepatocellular and canalicular cholestasis, mild mononuclear infiltration of the portal areas, reactive changes in Kupffer cells, and extramedullary hematopoiesis. This predominantly parenchymal pattern of injury may blend imperceptibly into a ductal pattern of injury, with bile ductular proliferation and fibrosis of portal tracts. In these cases distinction from an obstructive biliary atresia may therefore be difficult.
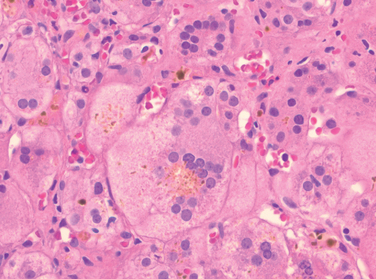
Intrahepatic Biliary Tract Disease
In this section we discuss three disorders of intrahepatic bile ducts: secondary biliary cirrhosis, primary biliary cirrhosis, and primary sclerosing cholangitis (summarized in Table 18-8). Secondary biliary cirrhosis is a condition resulting most often from uncorrected obstruction of the extrahepatic biliary tree. Primary biliary cirrhosis is a destructive disorder of the intrahepatic biliary tree. Primary selerosing cholangitis involves both the extrahepatic and intrahepatic biliary tree. It should also be noted that intrahepatic bile ducts are frequently damaged as part of more general liver diseases as in drug toxicity, viral hepatitis, liver transplantation, and graft-versus-host disease after bone marrow transplantation.
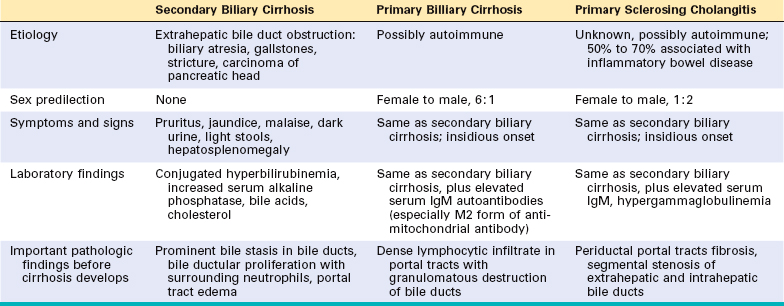
SECONDARY BILIARY CIRRHOSIS
Prolonged obstruction of the extrahepatic biliary tree results in profound hepatic alterations. The most common cause of obstruction in adults is extrahepatic cholelithiasis (gallstones, described later), followed by malignancies of the biliary tree or head of the pancreas, and strictures resulting from previous surgical procedures. Obstructive conditions in children include biliary atresia, cystic fibrosis, choledochal cysts (a cystic anomaly of the extrahepatic biliary tree, discussed later), and syndromes in which there are insufficient intrahepatic bile ducts (paucity of bile duct syndromes). The initial morphologic features of cholestasis were described earlier and are entirely reversible with correction of the obstruction. However, secondary inflammation resulting from biliary obstruction initiates periportal fibrosis, which eventually leads to hepatic scarring and nodule formation, generating secondary biliary cirrhosis. Subtotal obstruction may promote secondary bacterial infection of the biliary tree (ascending cholangitis), which aggravates the inflammatory injury. Enteric organisms such as coliforms and enterococci are common culprits.
Morphology. The end-stage obstructed liver shows yellow-green pigmentation that is accompanied by marked icteric discoloration of body tissues and fluids. On cut surface the liver is hard, with a finely granular appearance (Fig. 18-30). The histology is characterized by coarse fibrous septa that subdivide the liver in a jigsaw-like pattern. Embedded in the septa are distended small and large bile ducts, which frequently contain inspissated pigmented material. There is extensive proliferation of smaller bile ductules, particularly at the interface between septa in former portal tracts and the parenchyma. Cholestatic features in the parenchyma may be severe, with extensive feathery degeneration and formation of bile lakes. However, once regenerative nodules have formed, bile stasis may become less conspicuous. Ascending bacterial infection incites a robust neutrophilic infiltration of bile ducts; severe pylephlebitis and cholangitic abscesses may develop.

PRIMARY BILIARY CIRRHOSIS (PBC)
PBC is an inflammatory autoimmune disease mainly affecting the intrahepatic bile ducts. The primary feature of this disease is a nonsuppurative, inflammatory destruction of medium-sized intrahepatic bile ducts. It is accompanied by portal inflammation, scarring, and eventual development of cirrhosis and liver failure.54 Because cirrhosis develops only after many years, the disease name is somewhat misleading for patients diagnosed early at a pre-cirrhotic stage.
This is primarily a disease of middle-aged women, with a female predominance over males in excess of 6 : 1. It may occur between the ages of 20 and 80 years, with peak incidence between 40 and 50 years of age. The incidence of this disease in the United States is approximately 27 per million people (7 and 45 per million in males and females, respectively). Both the incidence and prevalence of PBC are increasing and geographic clustering has been reported, suggesting that genetic and environmental factors are important in the pathogenesis of the disease. Family members of PBC patients have an increased risk of developing the disease. The onset is insidious, usually presenting with fatigue and pruritus. Hepatomegaly is a typical finding, and eyelid xanthelasmas arise as a result of infiltration of the nasal area of the eyelid by cholesterol-rich macrophages. Hyperpigmentation due to melanin deposition and an inflammatory arthropathy are seen in 25% to 40% of cases. Signs and symptoms of chronic liver disease, such as spider nevi, are late features. Over a period of two or more decades, patients develop cirrhosis and complications that include portal hypertension with variceal bleeding, and hepatic encephalopathy.
Serum alkaline phosphatase and cholesterol are almost always elevated, even at onset; hyperbilirubinemia is a late development and usually signifies incipient hepatic decompensation. Antimitochondrial antibodies are present in 90% to 95% of patients. They are highly characteristic of PBC and an essential element for diagnosis, together with the elevation of alkaline phosphatase and γ-glutamyltransferase, which are markers of cholestasis.
Pathogenesis.
PBC is thought to be an autoimmune disorder, but its pathogenesis is still unknown. Many potential mechanisms have been proposed, including aberrant expression of MHC class II molecules on bile duct epithelial cells, accumulation of autoreactive T cells around bile ducts, reaction of antimitochondrial antibodies to hepatocytes, or of other antibodies against cellular components (nuclear pore proteins, and centromeric proteins, among others).55 The antimitochondrial antibodies, the characteristic autoantibodies in PBC, target the E2 component of the pyrurate dehydrogenase complex (PDC-E2). PDC-E2–specific T cells are also present in these patients, supporting the notion of immune-mediated pathogenesis.56
Morphology. PBC is the prototype of conditions leading to small-duct biliary fibrosis and cirrhosis. PBC is a focal and variable disease, showing different degrees of severity in different portions of the liver. During the pre-cirrhotic stage portal tracts are infiltrated by a dense accumulation of lymphocytes, macrophages, plasma cells, and occasional eosinophils. Interlobular bile ducts are infiltrated by lymphocytes and may show noncaseating granulomatous inflammation (Fig. 18-31) and undergo progressive destruction. With time the obstruction to intrahepatic bile flow leads to progressive secondary hepatic damage. Portal tracts upstream from damaged bile ducts show bile ductular proliferation, inflammation, and necrosis of the adjacent periportal hepatic parenchyma. The parenchyma develops generalized cholestasis. Over years to decades, relentless portal tract scarring and bridging fibrosis lead to cirrhosis.
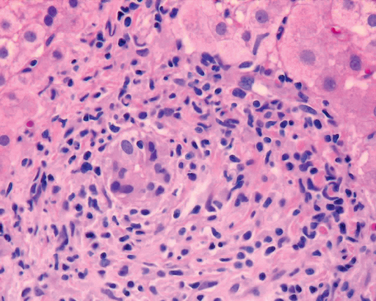
FIGURE 18-31 Primary biliary cirrhosis. A portal tract is markedly expanded by an infiltrate of lymphocytes and plasma cells. There is a granulomatous reaction to a bile duct undergoing destruction (florid duct lesion).
Macroscopically, the liver does not at first appear abnormal, but as the disease progresses bile stasis stains the liver green. The capsule remains smooth and glistening until a fine granularity appears, representing deposition of fibrous septa. This process culminates in a well-developed, uniform micronodular cirrhosis. Liver weight is at first normal to increased (because of inflammation) but is ultimately decreased. In most cases the end-stage picture is indistinguishable from secondary biliary cirrhosis or the cirrhosis that follows chronic hepatitis from other causes.
Clinical Features.
The onset is extremely insidious, and patients may be symptom-free for many years. Eventually, pruritus, fatigue, and abdominal discomfort develop, followed in time by secondary features: skin pigmentation, xanthelasmas, steatorrhea, and vitamin D malabsorption–related osteomalacia and/or osteoporosis. More general features of jaundice and hepatic decompensation, including portal hypertension and variceal bleeding, mark entry into the end stages of the disease. PBC patients have an increased risk to develop hepatocellular carcinomas. The major cause of death is liver failure, followed in order by massive variceal hemorrhage and intercurrent infection. Individuals with PBC may also have extrahepatic manifestations of autoimmunity, including the sicca complex of dry eyes and mouth (Sjögren syndrome; from the Latin sicca, meaning dryness), systemic sclerosis, thyroiditis, rheumatoid arthritis, Raynaud phenomenon, membranous glomerulonephritis, and celiac disease. There is no specific therapy for PBC but treatment with ursodeoxycholic acid, if started early, can provide complete remission and prolong survival is 25% to 30% of cases. Its mechanism of action is not well understood. Liver transplantation is the best form of treatment for persons with end-stage liver disease.
PRIMARY SCLEROSING CHOLANGITIS (PSC)
PSC is characterized by inflammation and obliterative fibrosis of intrahepatic and extrahepatic bile ducts, with dilation of preserved segments. Characteristic “beading” of a contrast medium in radiographs of the intrahepatic and extrahepatic biliary tree is attributable to the irregular strictures and dilations of affected bile ducts. PSC is commonly seen in association with inflammatory bowel disease (see Chapter 17), particularly chronic ulcerative colitis, which coexists in approximately 70% of individuals with primary sclerosing cholangitis. Conversely, the prevalence of PSC in persons with ulcerative colitis is about 4%. PSC tends to occur in the third through fifth decades of life, and males predominate 2 : 1. (See Table 18-8 for comparisons with primary and secondary biliary cirrhosis.)
Pathogenesis.
Primary sclerosing cholangitis is a chronic cholestatic disorder characterized by non-specific inflammation, fibrosis and strictures of intra- and extrahepatic bile ducts.57 Several features of the disease suggest that it results from immunologically mediated injury to bile ducts. These include the detection of T cells in the periductal stroma, the presence of a plethora of circulating autoantibodies, and the association with ulcerative colitis. It has been proposed that T cells activated in the gut mucosa travel to the liver where they recognize a bile duct antigen that cross-reacts with gut antigens. Another proposed etiology is that the bile duct lesions are a consequence of cross-reaction of bile duct antigens with enteric bacteria or bacterial products. Antibodies commonly found in patients with primary sclerosing cholangitis include anti-smooth muscle antibodies, anti-nuclear antibodies (ANAs), rheumatoid factor, and an atypical p-ANCA which shows a perinuclear staining pattern but is directed against a nuclear envelope protein, instead of myeloperoxidase as is typical of p-ANCA antibodies. The atypical p-ANCA is found in up to 80% of patients, but its relationship with the pathogenesis of the disease is unknown. First degree relatives of patients with primary sclerosing cholangitis have an increased risk of developing the disease. As with many other immunologically mediated diseases, primary sclerosing cholangitis is associated with an increased prevalence of certain MHC class I and class II haplotypes.58
Morphology. PSC is a fibrosing cholangitis of bile ducts, with a lymphocytic infiltrate, progressive atrophy of the bile duct epithelium, and obliteration of the lumen (Fig. 18-32). The concentric periductal fibrosis around affected ducts (“onion-skin fibrosis”) is followed by their disappearance, leaving behind a solid, cordlike fibrous scar. In between areas of progressive stricture, bile ducts become ectatic and inflamed, presumably the result of downstream obstruction. As the disease progresses the liver becomes markedly cholestatic, culminating in biliary cirrhosis much like that seen with primary and secondary biliary cirrhosis.
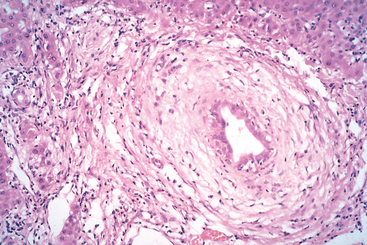
Clinical Features.
Asymptomatic patients may come to attention only because of persistent elevation of serum alkaline phosphatase. Alternatively, progressive fatigue, pruritus, and jaundice may develop. The disease follows a protracted course of 5 to 17 years, and the severely afflicted patients have the usual symptoms of chronic liver disease, including weight loss, ascites, variceal bleeding, and encephalopathy. Approximately 7% of individuals with PSC develop cholangiocarcinoma, a very high frequency relative to that of the general population. The incidence of chronic pancreatitis and hepatocellular carcinoma also seems to be increased in PSC patients. A distinctive type of sclerosing cholangitis, with elevated IgG4 and associated with autoimmune pancreatitis, has been recognized recently.59 There is no specific medical therapy for PSC. Cholestyramine has been used for pruritus, and endoscopic dilation with sphincterotomy or stenting is used for relieving symptoms. Liver transplantation is the definitive treatment for persons with end-stage liver disease.
ANOMALIES OF THE BILIARY TREES (INCLUDING LIVER CYSTS)
A heterogeneous group of lesions exist in which the primary abnormality is altered architecture or paucity of the intrahepatic biliary tree. Lesions may be found incidentally during radiographic studies, surgery, or at autopsy. Such conditions may become manifest as hepatosplenomegaly and portal hypertension in the absence of hepatic dysfunction, starting in late childhood or adolescence. There are five distinct conditions: von Meyenburg complexes, polycystic liver disease, congenital hepatic fibrosis, Caroli disease, and Alagille syndrome.
Von Meyenburg Complexes.
These are small clusters of modestly dilated bile ducts embedded in a fibrous, sometimes hyalinized, stroma located close to or within portal tracts. These lesions are often referred to as “bile duct hamartomas” (Fig. 18-33). Von Meyenburg complexes are common and without clinical significance except in the differential diagnosis of metastases to the liver.
Polycystic Liver Disease.
In this disease there are multiple diffuse cystic lesions in the liver, varying in number from a scattered few to hundreds (Fig. 18-34). The cysts, lined by cuboidal or flattened biliary epithelium, contain straw-colored fluid.

Congenital Hepatic Fibrosis.
In this condition portal tracts are enlarged by irregular, broad bands of collagenous tissue, forming septa that divide the liver into irregular islands. Variable numbers of abnormally shaped bile ducts are embedded in the fibrous tissue, and are in continuity with the biliary tree. This anomaly arises because of persistence of the embryonic form of the biliary tree, with ensuing portal tract fibrosis over the individual’s lifetime. Although individuals with congenital hepatic fibrosis rarely develop cirrhosis, they may still face complications of portal hypertension, particularly bleeding varices.
Caroli Disease.
In this disease the larger ducts of the intrahepatic biliary tree are segmentally dilated and may contain inspissated bile. Pure forms are rare; this disease is usually associated with portal tract fibrosis of the congenital hepatic fibrosis type. The disease is frequently complicated by intrahepatic cholelithiasis (described later), cholangitis, hepatic abscesses, and portal hypertension. Persons with Caroli disease and congenital hepatic fibrosis have an increased risk of developing cholangiocarcinomas.
Each of the four conditions discussed above can be associated with polycystic kidney disease. Single or multiple liver cysts are the most frequent extrarenal manifestation of autosomal-dominant polycystic kidney disease caused by a mutation in PKD1 (Chapter 20), and occur in 75% to 90% of patients with this type of kidney disease.60 A form of polycystic liver disease caused by mutations of the PRKCSH gene (which encodes a protein kinase C substrate 80K-H) does not coexist with polycystic kidney disease.61 Congenital hepatic fibrosis is strongly associated with the autosomal recessive form of polycystic kidney disease, which is caused by mutations of the PKHD1 (polycystic kidney and hepatic disease) gene.62 The exact pathogenesis of these biliary lesions and the basis of their association with polycystic kidney disorders remain unclear.
Alagille Syndrome (Syndromatic Paucity of Bile Ducts; Arteriohepatic Dysplasia).
This is a rare autosomal dominant multi-organ disorder, in which the liver pathology is characterized by absence of bile ducts in portal tracts. The syndrome is caused by mutations or deletion of the gene encoding Jagged1, which is located on chromosome 20p. Jagged1 is a cell surface protein that functions as a ligand for Notch receptors (Chapter 3). Mutations in Jagged can be detected in as many as 94% of individuals with a clinical diagnosis of Alagille syndrome, and some of the remaining patients have mutations in the Notch 2 receptor.63 The Jagged1-Notch signaling pathway regulates cell fate and is involved in the development of the organ systems affected in Alagille syndrome. Affected patients have five major clinical features: chronic cholestasis, peripheral stenosis of the pulmonary artery, butterfly-like vertebral arch defects, an eye defect known as posterior embryotoxon, and a peculiar hypertelic facies. Patients can survive into adulthood but are at risk for hepatic failure and hepatocellular carcinoma.
Circulatory Disorders
Given the enormous flow of blood through the liver, it is not surprising that circulatory disturbances have considerable impact on the liver. In most instances, however, clinically significant abnormalities of liver function do not develop, but hepatic morphology may be strikingly affected. These disorders can be grouped according to whether blood flow into, through, or from the liver is impaired (Fig. 18-35).
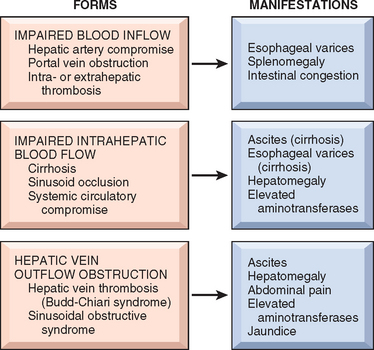
FIGURE 18-35 Hepatic circulatory disorders. Forms and clinical manifestations of compromised hepatic blood flow.
IMPAIRED BLOOD FLOW INTO THE LIVER
Hepatic Artery Compromise
Liver infarcts are rare, thanks to the double blood supply to the liver. Nonetheless, thrombosis or compression of an intrahepatic branch of the hepatic artery by embolism (Fig. 18-36), neoplasia, polyarteritis nodosa (Chapter 11), or sepsis may result in a localized infarct that is usually anemic and pale tan, or sometimes hemorrhagic, as a result of suffusion of portal blood. Interruption of the main hepatic artery does not always produce ischemic necrosis of the organ, particularly if the liver is otherwise normal. Retrograde arterial flow through accessory vessels, when coupled with the portal venous supply, is usually sufficient to sustain the liver parenchyma. The one exception is hepatic artery thrombosis in a transplanted liver, which generally leads to infarction of the major ducts of the biliary tree and loss of the organ.
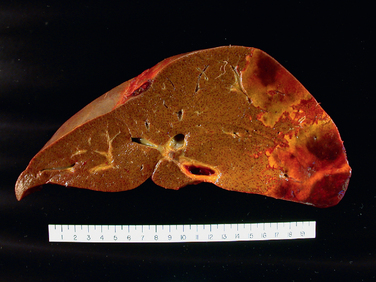
Portal Vein Obstruction and Thrombosis
Blockage of the extrahepatic portal vein may be insidious and well tolerated, or may be a catastrophic and potentially lethal event; most cases fall somewhere in between. Occlusive disease of the portal vein or its major radicles typically produces abdominal pain and, in most instances, other manifestations of portal hypertension, principally esophageal varices that are prone to rupture. Ascites is not common (because the block is presinusoidal), but when present, is often massive and intractable. As discussed earlier, ascites is common in cirrhosis due to sinusoidal block and hyperdynamic circulation. Acute impairment of visceral blood flow leads to profound congestion and bowel infarction.
Extrahepatic portal vein obstruction may arise from the following conditions, but in about one third of cases no cause can be implicated:
Intrahepatic portal vein radicles may be obstructed by acute thrombosis. The thrombosis does not cause ischemic infarction but instead results in a sharply demarcated area of red-blue discoloration called infarct of Zahn. There is no necrosis, only severe hepatocellular atrophy and marked hemostasis in distended sinusoids. Invasion of the portal vein system by primary or secondary cancer in the liver can progressively occlude portal inflow to the liver; tongues of HCC can even occlude the extrahepatic portal vein.
Noncirrhotic Portal Fibrosis and Idiopathic Portal Hypertension.
These conditions are similar and characterized by portal hypertension and a moderate degree of portal fibrosis without cirrhosis.64 Noncirrhotic portal fibrosis is common in India and generally presents with upper gastrointestinal bleeding. Idiopathic portal hypertension, described in Japan, has a female predominance, and usually presents with splenomegaly. The pathogenesis of these conditions is unknown. It has been proposed that they may result from bacterial infection of the gut causing septic embolization of the portal vein. Another proposed mechanism is the fibrosis of portal vein branches associated with the increased expression of vascular cell adhesion molecule-1 (VCAM-1). Histologically there is a variable involvement of portal tracts, only some of which have increased connective tissue deposition and fibrosis. In addition, there is obliteration of small branches of the portal veins. This histological picture is sometimes referred to as hepatic sclerosis or obliterative portal venopathy.
IMPAIRED BLOOD FLOW THROUGH THE LIVER
The most common intrahepatic cause of blood flow obstruction is cirrhosis, as described earlier. In addition, physical occlusion of the sinusoids occurs in a small but striking group of diseases. In sickle cell disease the hepatic sinusoids may become packed with sickled erythrocytes, free in the sinusoids or phagocytosed by Kupffer cells (Fig. 18-37), leading to panlobular parenchymal necrosis. Disseminated intravascular coagulation may occlude sinusoids. This is usually inconsequential except for the periportal sinusoidal occlusion and parenchymal necrosis that may arise in pregnancy as part of eclampsia (discussed later). Finally, metastatic tumor cells (e.g., breast carcinoma, lymphoma, malignant melanoma) may fill the hepatic sinusoids in the absence of a mass lesion. The attendant obstruction to blood flow and massive necrosis of hepatocytes can lead to fulminant hepatic failure.
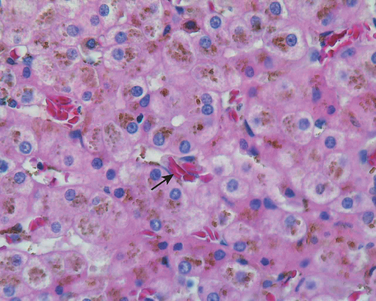
FIGURE 18-37 Sickle cell crisis in liver. The photomicrograph shows several aggregates of red blood cells, with some of them showing “sickle cell” appearance (arrow).
Passive Congestion and Centrilobular Necrosis
These hepatic manifestations of systemic circulatory compromise are considered together because they represent a morphologic continuum. Both changes are commonly seen at autopsy because there is an element of preterminal circulatory failure with virtually every nontraumatic death.
Right-sided cardiac decompensation leads to passive congestion of the liver. The liver is slightly enlarged, tense, and cyanotic, with rounded edges. Microscopically there is congestion of centrilobular sinusoids. With time, centrilobular hepatocytes become atrophic, resulting in markedly attenuated liver cell plates. Left-sided cardiac failure or shock may lead to hepatic hypoperfusion and hypoxia, causing ischemic coagulative necrosis of hepatocytes in the central region of the lobule (centrilobular necrosis). In most instances the only clinical evidence of centrilobular necrosis or its variants is transient elevation of serum aminotransferases, but the parenchymal damage may be sufficient to induce mild to moderate jaundice.
The combination of hypoperfusion and retrograde congestion acts synergistically to cause centrilobular hemorrhagic necrosis. The liver takes on a variegated mottled appearance, reflecting hemorrhage and necrosis in the centrilobular regions, known as the nutmeg liver (Fig. 18-38). By microscopy there is a sharp demarcation of viable periportal and necrotic pericentral hepatocytes, with suffusion of blood through the centrilobular region. An uncommon complication of sustained chronic severe congestive heart failure is so-called cardiac sclerosis. The pattern of liver fibrosis is distinctive, inasmuch as it is mostly centrilobular. The damage rarely fulfills the criteria for the diagnosis of cirrhosis, but the historically sanctified term cardiac cirrhosis cannot easily be dislodged.
Peliosis Hepatis
Sinusoidal dilation occurs in any condition in which efflux of hepatic blood is impeded. Peliosis hepatis is a rare condition in which the dilation is primary. The liver contains blood-filled cystic spaces, either unlined or lined with sinusoidal endothelial cells. The pathogenesis of peliosis hepatis is unknown. Focal apoptosis of hepatocytes or sinusoidal endothelial cells, and disruption of liver extracellular matrix seem to play a role in the pathogenesis. Bartonella species have been seen in the sinusoidal endothelial cells in AIDS-associated peliosis.65 Clinically, peliosis hepatis is associated with many diseases, including cancer, tuberculosis, AIDS, or post-transplantation immunodeficiency. It is also associated with exposure to anabolic steroids and, rarely, oral contraceptives and danazol. Clinical signs are generally absent even in advanced peliosis, but potentially fatal intra-abdominal hemorrhage or hepatic failure may occur. Peliotic lesions usually disappear after correction of the underlying causes.
HEPATIC VENOUS OUTFLOW OBSTRUCTION
Hepatic Vein Thrombosis and Inferior Vena Cava Thrombosis
Obstruction of a single main hepatic vein by thrombosis is clinically silent. The obstruction of two or more major hepatic veins produces liver enlargement, pain, and ascites, a condition known as Budd-Chiari syndrome. Hepatic damage is the consequence of increased intrahepatic blood pressure, and an inability of the massive hepatic blood flow to shunt around the blocked outflow tract. Hepatic vein thrombosis is associated with primary myeloproliferative disorders (including polycythemia vera), inherited disorders of coagulation (e.g., deficiencies in antithrombin, protein S, or protein C, or mutations of factor V; see Chapter 4), antiphospholipid syndrome, paroxysmal nocturnal hemoglobinuria, and intra-abdominal cancers, particularly HCC. The occurrence of hepatic vein thrombosis in the setting of pregnancy or oral contraceptive use is usually through interaction with an underlying thrombogenic disorder. About 10% of cases are idiopathic in origin, presumably unrecognized thrombogenic disorders.
A separate distinction is made for inferior vena cava obstruction at its hepatic portion (obliterative hepatocavopathy). This disorder is caused by inferior vena cava thrombosis or membranous obstruction of the inferior vena cava. It is endemic in Nepal, with a suspected association with infections.
Morphology. In the Budd-Chiari syndrome, acutely developing thrombosis of the major hepatic veins or the hepatic portion of the inferior vena cava, the liver is swollen and red-purple and has a tense capsule (Fig. 18-39). Microscopically the affected hepatic parenchyma reveals severe centrilobular congestion and necrosis. Centrilobular fibrosis develops in instances in which the thrombosis is more slowly developing. The major veins may contain totally occlusive fresh thrombi, subtotal occlusion, or, in chronic cases, organized adherent thrombi.
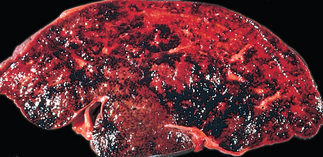
The mortality of untreated acute hepatic vein thrombosis is high. Prompt surgical creation of a portosystemic venous shunt permits reverse flow through the portal vein and considerably improves the prognosis. In the case of vena caval thrombosis, direct dilation of caval obstruction may be possible during angiography. The chronic forms of these thrombotic syndromes are far less lethal, and more than two thirds of patients are alive after 5 years.
Sinusoidal Obstruction Syndrome (Veno-Occlusive Disease)
Originally described in Jamaican drinkers of pyrrolizidine alkaloid–containing bush tea and named veno-occlusive disease, the disease is now called sinusoidal obstruction syndrome, and occurs primarily following allogeneic bone marrow transplantation, usually within the first 3 weeks. The incidence approaches 25% in recipients of allogeneic marrow transplants. Sinusoidal obstruction syndrome can occur in cancer patients receiving chemotherapy, especially with agents such as gemtuzumad and ozagamicin, used in the treatment of acute myeloid leukemia, actinomycin D in the treatment of Wilms’ tumors, dacarbazine (a drug activated by sinusoidal endothelial cells), and in patients who receive cytotoxic agents such as cyclophosphamide before bone marrow transplantation (discussed below). The mortality rates can be higher than 30%. Although histology is the gold standard for the diagnosis, a diagnosis of sinusoidal obstruction syndrome is frequently made on clinical grounds only (tender hepatomegaly, ascites, weight gain, and jaundice), because of the high risk of liver biopsy in these patients.
Morphology. Sinusoidal obstruction syndrome is characterized by obliteration of hepatic vein radicles by varying amounts of subendothelial swelling and finely reticulated collagen. In acute disease there is striking centrilobular congestion with hepatocellular necrosis and accumulation of hemosiderin-laden macrophages. As the disease progresses, obliteration of the lumen of the venule is easily identified with special stains for connective tissue (Fig. 18-40). In chronic or healed sinusoidal obstruction syndrome, dense perivenular fibrosis radiating out into the parenchyma may be present, frequently with total obliteration of the venule; hemosiderin deposition is evident in the scar tissue, and congestion is minimal.
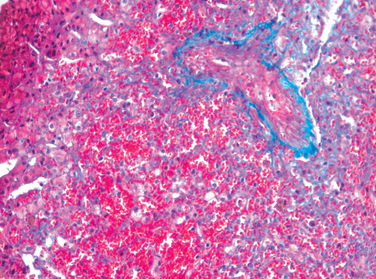
Sinusoidal obstruction syndrome arises from toxic injury to the sinusoidal endothelium.66 Endothelial lining cells round up and slough off the sinusoidal wall, embolizing downstream and obstructing sinusoidal blood flow. This is accompanied by entry of erythrocytes into the space of Disse, necrosis of perivenular hepatocytes, and downstream accumulation of cellular debris in the terminal hepatic vein. Proliferation of perisinusoidal stellate cells and subendothelial fibroblasts in the terminal hepatic vein follows, with fibrosis and deposition of extracellular matrix in the sinusoids.