Chapter 25 The Skin
The Skin: More Than a Mechanical Barrier
Little more than a century ago, the noted pathologist Rudolph Virchow considered the skin as a mere protective covering for more delicate and functionally sophisticated internal viscera.1 Then, and for the century that followed, the skin was considered primarily a passive protective barrier to both fluid loss and mechanical injury. Over the last few decades, however, studies have demonstrated the skin to be a complex organ—the largest in the body—in which precisely regulated cellular and molecular interactions govern many crucial responses to our environment.
Like other complex organs, the skin is composed of several interdependent cell types and structures that are functionally cooperative (Fig. 25-1).
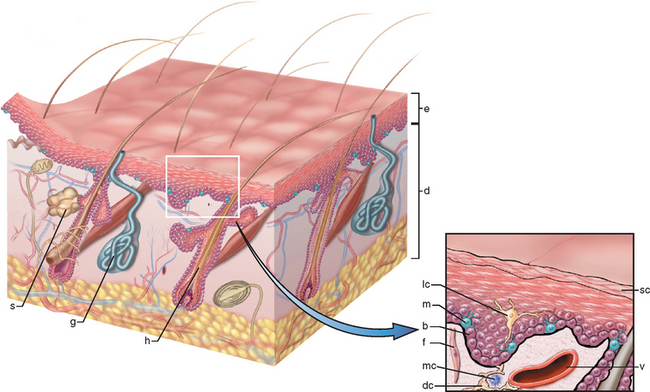
FIGURE 25-1 A, The skin is composed of an epidermal layer (e) from which specialized adnexa (hair follicles, h; sweat glands, g; and sebaceous glands, s) descend into the underlying dermis (d). B, This projection of the epidermal layer (e) and underlying superficial dermis demonstrates the progressive upward maturation of basal cells (b) into cornified squamous epithelial cells of the stratum corneum (sc). Melanin-containing dendritic melanocytes (m) (and rare Merkel cells containing neurosecretory granules) and midepidermal dendritic Langerhans cells (lc) are also present. The underlying dermis contains small vessels (v), fibroblasts (f), perivascular mast cells (mc), and dendrocytes (dc), potentially important in dermal immunity and repair.
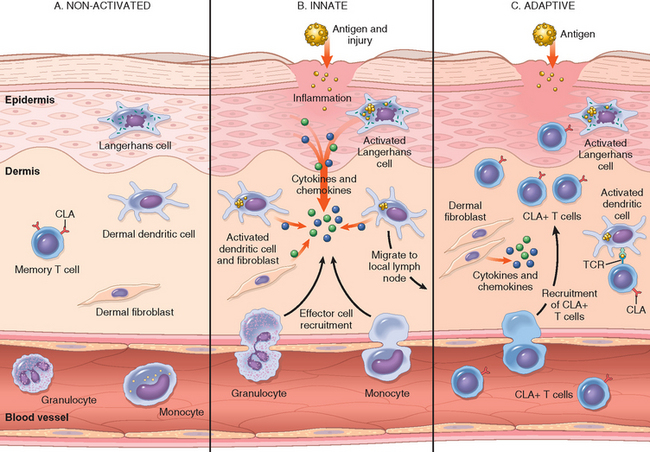
FIGURE 25-2 The cutaneous immune system. A, In the absence of inflammation, skin is populated by several immune cells, some stationary and others transitory, that survey the environment and are primed for response. B, The innate response to epithelial injury or antigen presentation activates the resident immune cells that then recruit nonspecific effector cells such as neutrophils and eosinophils. C, The adaptive response occurs when antigen presented in the context of the major histocompatibility complex is specifically recognized by T cells and as a result, additional antigen-specific skin-homing (CLA+) T cells are recruited. TCR, T-cell receptor.
Although the human integument may appear drab compared with the skin and pelage of certain other members of the animal kingdom, it is indeed extraordinarily vibrant with regard to the diversity and complexity of protective functions that it serves.
Imbalances in factors affecting the delicate homeostasis that exists among skin cells may result in conditions as diverse as wrinkles and hair loss, blisters and rashes, and life-threatening cancers and disorders of immune regulation. For example, chronic exposure to sunlight fosters premature cutaneous aging, blunting of immunological responses to environmental antigens, and the development of a variety of premalignant and malignant cutaneous neoplasms. Ingested agents, such as therapeutic drugs, can cause an enormous number of rashes or exanthems. Systemic disorders, such as diabetes mellitus, amyloidosis, and lupus erythematosus, may also have important manifestations in the skin.
Skin conditions are very common, affecting about one third of the United States population each year. Since skin is uniquely accessible to visual examination, for the experienced observer it can yield numerous insights into the functional state of the body, if not the soul, of a patient. Accurate description of the appearance of skin lesions is critical, since the gross appearance is often essential in formulating diagnoses and in understanding pathogenesis. To underscore the point, the appearance of skin lesions will be emphasized under each of the specific entities.
There are literally thousands of specific skin diseases. Only those that are common or that illustrate important pathologic mechanisms are described here. But first a brief primer of important clinical and histologic terms used in describing diseases of the skin is presented below.
DEFINITIONS OF MACROSCOPIC TERMS
| Excoriation | Traumatic lesion breaking the epidermis and causing a raw linear area (i.e., deep scratch); often self-induced |
| Lichenification | Thickened and rough skin characterized by prominent skin markings (as lichen on a tree trunk); usually the result of repeated rubbing |
| Macule | Circumscribed lesion, 5 mm or smaller in diameter, characterized by flatness and distinguished by coloration (patch is greater than 5 mm) |
| Onycholysis | Separation of nail plate from nail bed |
| Papule | Elevated dome-shaped or flat-topped lesion 5 mm or less across (nodule is greater than 5 mm) |
| Plaque | Elevated flat-topped lesion, usually greater than 5 mm across (may be caused by coalescent papules) |
| Pustule | Discrete, pus-filled, raised lesion |
| Scale | Dry, horny, platelike excrescence; usually the result of imperfect cornification |
| Vesicle | Fluid-filled raised lesion 5 mm or less across (Bulla is greater than 5 mm. Blister is the common term for either.) |
| Wheal | Itchy, transient, elevated lesion with variable blanching and erythema formed as the result of dermal edema |
DEFINITIONS OF MICROSCOPIC TERMS
| Acantholysis | Loss of intercellular cohesion between keratinocytes |
| Acanthosis | Diffuse epidermal hyperplasia |
| Dyskeratosis | Abnormal, premature keratinization within cells below the stratum granulosum |
| Erosion | Discontinuity of the skin showing incomplete loss of the epidermis |
| Exocytosis | Infiltration of the epidermis by inflammatory cells |
| Hydropic swelling (ballooning) | Intracellular edema of keratinocytes, often seen in viral infections |
| Hypergranulosis | Hyperplasia of the stratum granulosum, often due to intense rubbing |
| Hyperkeratosis | Thickening of the stratum corneum, often associated with a qualitative abnormality of the keratin |
| Lentiginous | A linear pattern of melanocyte proliferation within the epidermal basal cell layer |
| Papillomatosis | Surface elevation caused by hyperplasia and enlargement of contiguous dermal papillae |
| Parakeratosis | Keratinization with retained nuclei in the stratum corneum. On mucous membranes, parakeratosis is normal. |
| Spongiosis | Intercellular edema of the epidermis |
| Ulceration | Discontinuity of the skin showing complete loss of the epidermis revealing dermis or subcutis |
| Vacuolization | Formation of vacuoles within or adjacent to cells; often refers to basal cell–basement membrane zone area |
Disorders of Pigmentation and Melanocytes
Skin pigmentation has historically had major societal implications. Cosmetic desire for increased pigmentation (tanning) among whites and for fairness among normally dark-skinned people have both resulted in deleterious practices. Focal or widespread loss of normal protective pigmentation can render individuals targets of unwanted attention and extraordinarily vulnerable to the harmful effects of sunlight (as in albinism). Change in preexisting skin pigmentation may signify important primary events in the skin (e.g., malignant transformation of a mole) or disorders of internal viscera (e.g., in Addison disease, see Chapter 24).
FRECKLE (EPHELIS)
Freckles are the most common pigmented lesions of childhood in lightly pigmented individuals. They are generally small (1 to several mm in diameter), tan-red or light brown macules that appear after sun exposure. Once present, freckles fade and darken in a cyclic fashion during winter and summer, respectively. This is not because of changes in the number of melanocytes, but in the degree of pigmentation.
Morphology. Hyperpigmentation of freckles results from increased amounts of melanin pigment within basal keratinocytes; melanocytes may be slightly enlarged but are normal in density. It is unclear whether the freckle represents: (1) a focal abnormality in pigment production by a discrete field of melanocytes, (2) enhanced pigment donation to adjacent basal keratinocytes, or (3) both. The café au lait spots seen in neurofibromatosis (Chapter 5) are histologically indistinguishable from freckles, but the former evolve independently from sun exposure and can contain aggregated melanosomes (macromelanosomes) within the cytoplasm of melanocytes.
LENTIGO
The term lentigo (plural, lentigines) refers to a common benign localized hyperplasia of melanocytes occurring at all ages, but often initiated in infancy and childhood. There is no sex or racial predilection, and the cause and pathogenesis are unknown. These lesions may involve mucous membranes as well as the skin, and consist of small (5–10 mm across), oval, tan-brown macules or patches. Unlike freckles, lentigines do not darken when exposed to sunlight.
Morphology. The essential histologic feature of a lentigo is linear (non-nested) melanocytic hyperplasia restricted to the cell layer immediately above the basement membrane that produces a hyperpigmented basal cell layer. So characteristic is this pattern that the term lentiginous is used to describe similar cellular proliferations within the basal cell layer in melanocytic tumors, such as in lentiginous nevi and in certain melanomas (termed acral lentiginous melanomas). Elongation and thinning of the rete ridges are also commonly seen in a lentigo.
MELANOCYTIC NEVUS (PIGMENTED NEVUS, MOLE)
Most of us have at least a few moles and probably regard them as mundane and uninteresting. However, in truth moles (or nevi) are diverse, dynamic, and biologically intriguing. Strictly speaking, the term nevus denotes any congenital skin lesion (e.g., a birthmark). Melanocytic nevus refers specifically to any neoplasm of melanocytes and hence is somewhat of a misnomer, since most melanocytic nevi are acquired.
Common acquired melanocytic nevi are tan to brown, uniformly pigmented, small (usually <6 mm across), solid regions of relatively flat (macules) to elevated skin (papules) with well-defined, rounded borders (Figs. 25-3A and 25-4A). There are numerous clinical and histologic types of melanocytic nevi, with variable clinical appearance. Table 25-1 provides a comparative summary of salient features of some commonly encountered forms of melanocytic nevi. Acquired melanocytic nevi (Figs. 25-3 and 25-4) are probably the most common type and virtually universal. They may become more prominent during pregnancy, indicating a degree of hormone sensitivity.
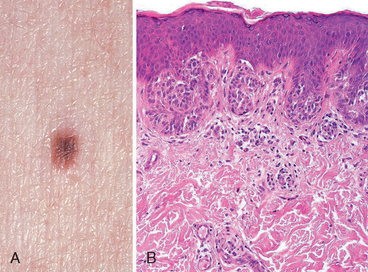
FIGURE 25-3 Melanocytic nevus, junctional type. A, In clinical appearance, lesions are small, relatively flat, symmetric, and uniform. B, On histologic examination, junctional nevi are characterized by rounded nests of nevus cells originating at the tips of rete ridges along the dermoepidermal junction.
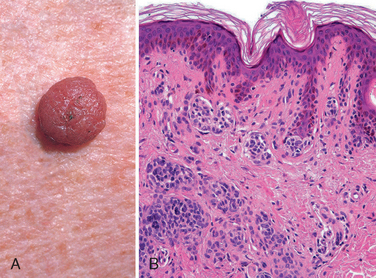
FIGURE 25-4 Melanocytic nevus, compound type. In contrast to the junctional nevus, the compound nevus (A) is more raised and dome-shaped. The symmetry and uniform pigment distribution suggest a benign process. Histologically (B), compound nevi combine the features of junctional nevi (intraepidermal nevus cell nests) with nests and cords of nevus cells in the underlying dermis.

Morphology. Melanocytic nevi are thought to progress through a series of morphologic changes over time. The earliest lesions are believed to be junctional nevi, which consist of aggregates or nests of round cells that grow along the dermoepidermal junction (see Fig. 25-3B). Nuclei of nevus cells are uniform and rounded in contour, contain inconspicuous nucleoli, and show little or no mitotic activity. Eventually, most junctional nevi grow into the underlying dermis as nests or cords of cells to form compound nevi (see Fig. 25-4B). In older lesions the epidermal nests may be lost entirely to form pure intradermal nevi. Clinically, compound and dermal nevi are often more elevated than junctional nevi.
Progressive growth of nevus cells from the dermoepidermal junction into the underlying dermis is accompanied by a process termed maturation (Fig. 25-5). Whereas superficial nevus cells are larger, tend to produce melanin, and grow in nests, deeper nevus cells are smaller, produce little or no pigment, and appear as cords and single cells. The most “mature” nevus cells may be found at the deepest extent of lesions, where they often acquire fusiform contours and grow in fascicles resembling neural tissue (neurotization, see Fig. 25-5E). This striking metamorphosis correlates with enzymatic changes (progressive loss of tyrosinase activity and acquisition of cholinesterase activity in deeper, nonpigmented, nervelike nevus cells). This sequence of maturation of individual nevus cells is of diagnostic importance in distinguishing some benign nevi from melanomas, which usually show little or no maturation.
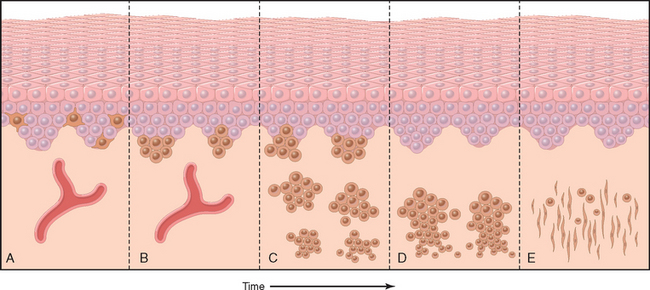
FIGURE 25-5 Maturation sequence of nondysplastic melanocytic nevi. A, Normal skin shows only scattered dendritic melanocytes within the epidermal basal cell layer. B, Junctional nevus. C, Compound nevus. D, Dermal nevus. E, Dermal nevus with neurotization (extreme maturation). Nevi may exist at any stage in this sequence for variable periods of time, although many are believed to progress through this sequence.
Although melanocytic nevi are common, their clinical and histologic diversity necessitates thorough knowledge of their appearance and natural evolution, lest they be confused with other skin conditions, most notably melanoma. The biologic importance of some nevi, however, resides in their possible transformation to melanoma (see below).
Pathogenesis.
Proof that nevi are neoplasms has come from studies showing that many have acquired mutations in either BRAF or NRAS. BRAF encodes a serine/threonine kinase that is a positive mediator of RAS signals, and the mutations in BRAF and NRAS found in nevi both cause constitutive activation of the RAS/BRAF signaling pathway (described in more detail under melanoma).6 Given that RAS lies in a potent transforming signaling pathway, it is reasonable to ask why nevi only rarely give rise to malignant melanomas. The answer appears to lie in the phenomenon referred to as oncogene-induced senescence. Expression of either activated RAS or BRAF in normal human melanocytes causes only a limited period of proliferation that is followed by a permanent growth arrest mediated by the accumulation of p16/INK4a,7 which you will recall is a potent inhibitor of several cyclin-dependent kinases, including CDK4 and CDK6 (Chapter 7). This protective response is disrupted in melanoma and some melanoma precursor lesions.
DYSPLASTIC NEVI
The association of melanocytic nevi with melanoma was made more than 185 years ago,8 but a precursor of melanoma was not clearly identified until 1978 when Clark and colleagues described the lesions that are now referred to as dysplastic nevi.9 Several lines of evidence support the concept that dysplastic nevi are precursors of melanoma. One of the most compelling pieces of evidence involves studies of families affected by dysplastic nevus syndrome, 10 in which a tendency to develop multiple dysplastic nevi and melanoma are co-inherited. The probability that a person with dysplastic nevus syndrome will develop melanoma is over 50% by age 60,11 and at-risk individuals sometimes develop several melanomas at different sites. Even more directly, transformation of dysplastic nevi to melanoma has been documented histologically.
Although dysplastic nevi can give rise to melanoma, the vast majority of such lesions are clinically stable and never progress. Conversely, not all melanomas in individuals with dysplastic nevus syndrome arise from dysplastic nevi, suggesting that these lesions are best viewed as indicators or markers of increased melanoma risk. Dysplastic nevi may also occur as isolated lesions in otherwise normal individuals, in which case the risk of malignant change is very low.
Dysplastic nevi are larger than most acquired nevi (often >5 mm across) and may number in the hundreds in those with the dysplastic nevus syndorme (Fig. 25-6A). They are flat macules, slightly raised plaques with a “pebbly” surface, or target-like lesions with a darker raised center and irregular flat periphery. They can be recognized by their size, variability in pigmentation (variegation) and irregular borders, and seem to be acquired rather than congenital in most instances. Unlike ordinary moles, dysplastic nevi occur on both sun-exposed and protected body surfaces.
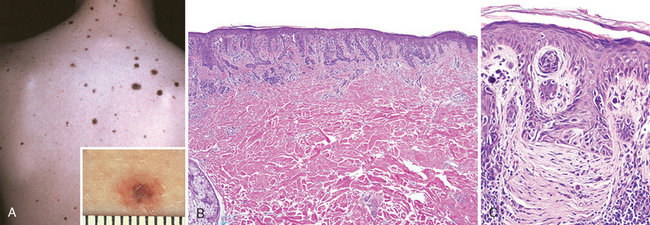
FIGURE 25-6 Dysplastic nevus. A, Numerous clinically atypical nevi on the back. B, One such lesion (inset A) has a compound nevus component (left side of scanning field) and an asymmetric junctional nevus component (right side of scanning field). The former correlates grossly with the more pigmented and raised central zone and the latter with the less pigmented, flat peripheral rim. C, An important feature is the presence of cytologic atypia (irregularly shaped, dark-staining nuclei). The dermis underlying the atypical cells characteristically shows linear, or lamellar, fibrosis.
Morphology. Microscopically, dysplastic nevi are usually compound and exhibit both architectural and cytologic atypia (Fig. 25-6A, B). Nevus cell nests within the epidermis may be enlarged and often fuse or coalescence with adjacent nests. As part of this process, single nevus cells begin to replace the normal basal cell layer along the dermoepidermal junction, producing lentiginous hyperplasia. Cytologic atypia takes the form of nuclear enlargement, irregular, often angulated, nuclear contours, and hyperchromasia (Fig. 25-6C). Associated alterations in the superficial dermis include lymphocytic infiltrates (usually sparse); release of melanin from dead nevus cells into the dermis (melanin incontinence), where it is phagocytosed by dermal macrophages; and a peculiar linear fibrosis surrounding the epidermal rete ridges that are involved by the nevus. This constellation of features assists in the histologic diagnosis.
Pathogenesis.
Clark and associates have proposed stages in the development of dysplastic nevi and their eventual progression to melanoma (Fig. 25-7),12 presumably through stepwise acquisition of mutations or epigenetic changes. Dysplastic nevus syndrome is inherited in an autosomal dominant pattern. Several mutated genes have been discovered in affected families, including CDKN2A on chromosome 9p21 and CDK4 (cyclin-dependent kinase 4) on chromosome 12q14, both of which are also associated with familial forms of melanoma (discussed later). However, linkage of these mutations to the dysplastic nevus phenotype is not straightforward, as not all patients with germline CDKN2A and CDK4 mutations have dysplastic nevi, and not all familial dysplastic nevi are associated with mutations in these genes. It is suspected that other genetic modifiers determine whether CDKN2A and CDK4 mutations cause dysplastic nevi in a particular individual; the identities of these modifier genes, as well as the other genes that are responsible for the syndrome, are being sought. Like conventional nevi, dysplastic nevi also frequently have acquired activating mutations in NRAS and BRAF.
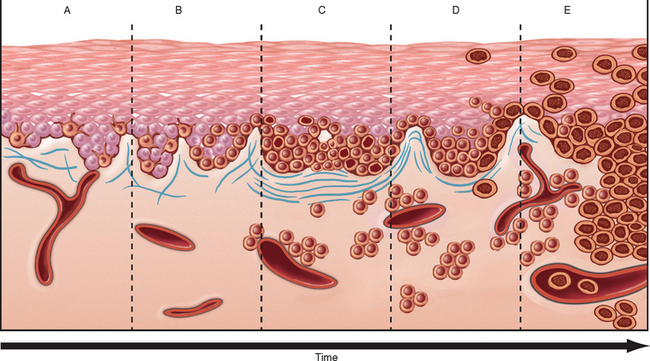
FIGURE 25-7 Potential steps of tumor progression in dysplastic nevi. A, Lentiginous melanocytic hyperplasia. B, Lentiginous junctional nevus. C, Lentiginous compound nevus with abnormal architectural and cytologic features (dysplastic nevus). D, Early melanoma, or melanoma in radial growth phase (large dark cells in epidermis). E, Advanced melanoma (vertical growth phase) with malignant spread into the dermis and vessels. The risk of malignant transformation of any single dysplastic nevus is extremely low but can occur.
MELANOMA
Melanoma is a relatively common neoplasm that remains deadly if not caught at its earliest stages. The great preponderance of melanomas arises in the skin; other sites of origin include the oral and anogenital mucosal surfaces, esophagus, meninges, and the eye (see Chapter 29). The following comments apply to cutaneous melanomas.
Today, as a result of increased public awareness of the signs of cutaneous melanoma, most are cured surgically.13 Nevertheless, the reported incidence of melanoma is increasing; more than 60,000 cases and more than 8000 deaths are expected in the United States in 2008.14
Clinical Features.
Because melanomas evolve over time from localized skin lesions to aggressive tumors that metastasize and are resistant to therapy, early recognition and complete excision are critical. Melanoma of the skin is usually asymptomatic, although itching or pain may be early manifestations. The majority of lesions are greater than 10 mm in diameter at diagnosis. The most consistent clinical signs are changes in the color, size, or shape of a pigmented lesion. Unlike benign nevi, melanomas show striking variations in color, appearing in shades of black, brown, red, dark blue, and gray (Fig. 25-8A). On occasion, zones of white or flesh-colored hypopigmentation also appear, sometimes due to focal regression of the tumor. The borders of melanomas are irregular and often notched, not smooth, round, and uniform as in melanocytic nevi. To reiterate, the most important warning signs, sometimes called the ABCs of melanoma, are (1) asymmetry; (2) irregular borders; and (3) variegated color. Other features of pigmented lesions that should raise concern are a diameter greater than 6 mm, any change in appearance, and new onset of itching or pain.
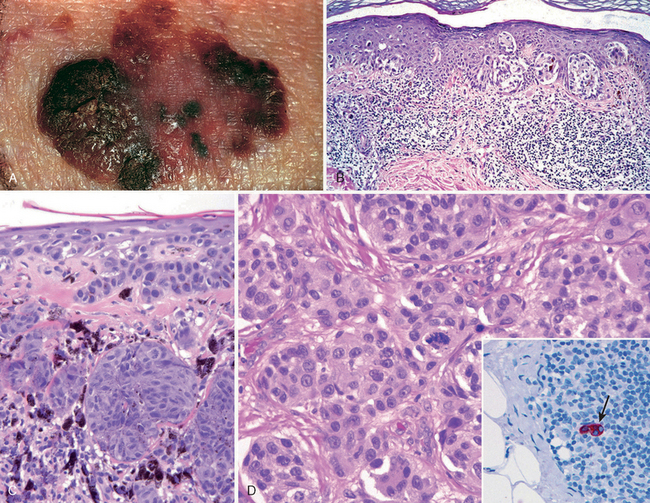
FIGURE 25-8 Melanoma. A, Typically, lesions are irregular in contour and pigmentation. Macular areas correlate with the radial growth phase, while raised areas usually correspond to nodular aggregates of malignant cells in the vertical phase of growth. B, Radial growth phase, showing irregular nested and single-cell growth of melanoma cells within the epidermis and an underlying inflammatory response within the dermis. C, Vertical growth phase, demonstrating nodular aggregates of infiltrating cells. D, High-power view of melanoma cells. The inset shows a sentinel lymph node with a tiny cluster of melanoma cells (arrow) staining for the melanocytic marker HMB-45. Even small numbers of malignant cells in a draining lymph node may confer a worse prognosis.
Morphology. Central to understanding the progression of melanoma is the concept of radial and vertical growth phases.15 Radial growth describes the horizontal spread of melanoma within the epidermis and superficial dermis (Fig. 25-8B). During this initial stage the tumor cells seem to lack the capacity to metastasize. Tumors in radial growth phase fall into several clinicopathologic classes, including: lentigo maligna, usually presenting as an indolent lesion on the face of older men that may remain in the radial growth phase for several decades; superficial spreading, the most common type of melanoma, usually involving sun-exposed skin; and acral/mucosal lentiginous melanoma that is unrelated to sun exposure.
After a variable (and unpredictable) period of time, melanoma shifts from the radial phase to a vertical growth phase, during which the tumor cells invade downward into the deeper dermal layers as an expansile mass (Fig. 25-8C). The vertical growth phase is often heralded by the appearance of a nodule and correlates with the emergence of a clone of cells with metastatic potential. Unlike melanocytic nevi, maturation is absent from the deep invasive portion of melanoma. The probability of metastasis in such lesions correlates with the depth of invasion, which by convention is the distance from the superficial epidermal granular cell layer to the deepest intradermal tumor cells; this measurement is known as the Breslow thickness.16 Other histologic features that correlate with outcome include the number of mitoses and the presence of ulceration;17,18 these and other prognostic factors are discussed further below.
Individual melanoma cells are usually considerably larger than normal melanocytes or cells found in melanocytic nevi. They contain large nuclei with irregular contours, chromatin that is characteristically clumped at the periphery of the nuclear membrane, and prominent red (eosinophilic) nucleoli (Fig. 25-8D). The appearance of the tumor cells is similar in the radial and vertical phases of growth. While most nevi and melanomas are easily distinguished based on their appearance, a minority of “atypical” lesions occupy a histologic gray zone and have been termed melanocytic tumors of uncertain malignant potential;13,19 such lesions require complete excision and close clinical follow-up.
Prognostic Factors.
Once a melanoma is excised, a number of clinical and pathologic features are used to gauge the probability of metastatic spread and prognosis. One model predicts outcome based on the following variables:20 (1) tumor depth (the Breslow thickness); (2) number of mitoses; (3) evidence of tumor regression (presumably due to the host immune response); (4) the presence and number of tumor infiltrating lymphocytes (TILs); (5) gender; and (6) location (central body or extremity). Determinants of a more favorable prognosis in this model include tumor depth of less than 1.7 mm, no or very few mitoses, a brisk TIL response, absence of regression, female gender, and location on an extremity. In a retrospective multivariate study by the American Joint Committee on Cancer (AJCC), tumor thickness and presence or absence of ulceration had prognostic significance independent of clinical stage.21,22 Because most melanomas initially metastasize to regional lymph nodes, additional prognostic information may be obtained by performing a sentinel lymph node biopsy; as in breast cancer (Chapter 23), this involves the identification, removal, and careful examination of the lymph node (or nodes) that is the initial site of drainage of intratumoral lymphatic vessels. Microscopic involvement of a sentinel node by even a small number of melanoma cells (micrometastases) confers a worse prognosis (Fig. 25-8D, inset).23 The degree of involvement and the total number of lymph nodes involved correlate well with overall survival.
Pathogenesis.
The two most important predisposing factors are inherited genes and sun exposure. Melanomas most commonly arise on sun-exposed surfaces, particularly the upper back in men and the back and legs in women, and lightly pigmented individuals are at higher risk than are darkly pigmented individuals. However, the relationship between sun exposure and melanoma is not as straightforward as with other skin cancers (discussed later); some studies suggest that severe sunburns early in life are the most important risk factor. Since melanomas occur in dark-skinned individuals and at body sites that are not sun-exposed, sunlight is clearly not the only predisposing factor, and other environmental factors may also contribute to risk.24
It is estimated that 10% to 15% of melanomas are familial, and many (but not all) of those with familial melanoma also have dysplastic nevi. Several of the genes responsible for familial melanoma encode well-characterized tumor suppressors and are also mutated in sporadic tumors (discussed below). Other genetic variants linked to melanoma risk in fair-skinned populations control melanin production; these pigmentation genes have weak effects, conferring a slightly elevated risk. They include MC1R, which encodes the melanocortin-1 receptor; ASIP (agouti signaling protein), which encodes a regulator of melanocortin receptor signaling, and TYR, which encodes tyrosinase, a melanocyte-specific enzyme that is required for melanin synthesis.25
Mutations that diminish the activity of the retinoblastoma (RB) tumor suppressor proteins are common in both familial and sporadic melanomas. The CDKN2A gene (discussed earlier under dysplastic nevi) is mutated in approximately 40% of pedigrees with autosomal dominant familial melanoma. CDKN2A is a complex locus that encodes three different tumor suppressors, p15/INK4b, p16/INK4a, and p14/ARF. Of these, loss of p16/INK4a is clearly implicated in human melanoma, and experimental evidence also strongly supports a role for loss of p14/ARF. You will recall from Chapter 7 that p16/INK4a enhances the activity of tumor suppressor proteins of the RB family by inhibiting cyclin-dependent kinase 4 (CDK4) and cyclin-dependent kinase 6 (CDK6), while p14/ARF enhances the activity of the p53 tumor suppressor by inhibiting the activity of the MDM2 oncoprotein. CDKN2A is mutated in approximately 10% of sporadic melanomas,26 and these mutations uniformly abolish the production of p16/INK4a and more variably affect p14/ARF. However, it is suspected that these mutations are the tip of the “oncogenic iceberg” with respect to molecular lesions affecting the function of RB proteins. For example, as many as 30% to 70% of melanomas show loss of p16/INK4a expression though varied mechanisms,27 and other uncommon familial and sporadic melanomas have mutations in CDK4 that prevent its inhibition by p16/INK4a. The net effect of all of these alterations is the same; increased melanocytic proliferation and escape from oncogene-induced cellular senescence.
The second common group of molecular lesions in sporadic melanoma leads to aberrant increases in RAS and PI-3K/AKT signaling (Fig. 25-9), which you will recall are pathways that promote cell growth and survival (Chapter 7). Activating mutations in BRAF, which encodes a serine/threonine kinase that is downstream of RAS, are seen in 60% to 70% of melanomas, while activating mutations in NRAS (which is upstream of BRAF) occur in additional 10% to 15% of tumors. For reasons that are unclear, melanomas arising in non-sun exposed sites are much more likely to have activating mutations in the c-KIT receptor tyrosine kinase,28,29 which sits upstream of both RAS and PI-3K/AKT, than in NRAS or BRAF. PTEN, a tumor suppressor that acts by downregulating PI-3K/AKT signaling, is epigenetically silenced in another 20% of melanomas.30,31
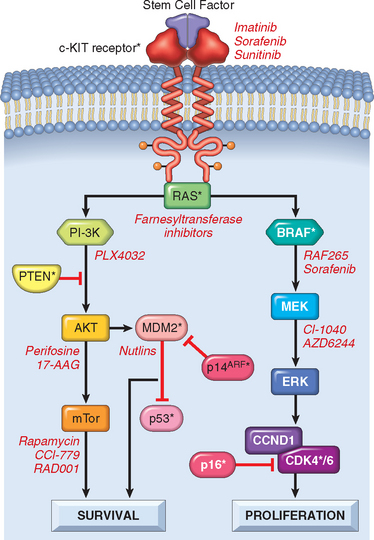
FIGURE 25-9 Pathways important in melanoma. The PI-3K/PTEN/AKT and BRAF/ERK pathways regulate cell survival and proliferation. Proteins altered in melanoma are indicated by asterisks. Inhibitors of these pathways are being studied as therapeutic agents; some specific examples are listed in red.
Molecular lesions of these two kinds are probably necessary, but not sufficient, for the development of melanoma. As discussed earlier, melanocytic nevi have the same activating mutations in NRAS and BRAF that are found in melanomas, yet become malignant very rarely, probably because unbridled RAS signaling leads to cellular senescence. p16/INK4a appears to be essential for the oncogene-induced cellular senescence,31 and in its absence proliferation persists, placing melanocytes at risk for transformation into a melanoma. However, dysplastic nevi sometimes have lesions in both p16/INK4a and RAS or BRAF, yet transform into full-blown melanomas only somewhat more frequently than conventional nevi. Clearly, other pathways yet to be identified also contribute to the transformation process.
These molecular insights have spawned attempts to treat melanoma with new therapeutic agents that target the RAS and PI-3K/AKT pathways (Fig. 25-9). Such approaches are urgently needed, as metastatic melanoma is resistant to both conventional chemotherapy and radiation treatment. Ultimately, it is likely that these types of targeted therapies will be used in combinations tailored to the oncogenic leions found in individual tumors.32 For example, a tumor with an activating mutation in c-KIT will require treatment with different inhibitors than a tumor with an activating mutation in RAS or BRAF. Immunotherapeutic approaches for melanoma in which host lymphocytes are “trained” to recognize and kill melanoma cells have also generated considerable interest, sparked in part by the recognition that spontaneous remissions of metastatic melanoma occur sporadically that are presumably mediated by the host immune response. Excellent responses are obtained on occasion,33 but the general applicability of such treatments has yet to be demonstrated.
Benign Epithelial Tumors
Benign epithelial neoplasms are common and usually biologically inconsequential, although they may cause significant psychological discomfort for the affected individual. These tumors, derived from the keratinizing stratified squamous epithelium of the epidermis and hair follicles and the ductular epithelium of cutaneous glands, often recapitulate the structures from which they arise. They are sometimes confused clinically with malignancy, particularly when they are pigmented or inflamed, and histologic examination of a biopsy is frequently required to establish a definitive diagnosis. In rare instances they are a telltale sign of syndromes associated with potentially life-threatening visceral malignancies, such as multiple trichilemmomas in Cowden syndrome or multiple sebaceous neoplasms in Muir-Torre syndrome. Diagnosis of epithelial tumors in these instances may facilitate recognition of the underlying syndrome and implementation of appropriate clinical interventions.
SEBORRHEIC KERATOSES
These common epidermal tumors occur most frequently in middle-aged or older individuals. They arise spontaneously and are particularly numerous on the trunk, although the extremities, head, and neck may also be involved. In people of color, multiple small lesions on the face are termed dermatosis papulosa nigra.
Seborrheic keratoses characteristically appear as round, flat, coin-like, waxy plaques that vary in diameter from millimeters to several centimeters (Fig. 25-10, inset). They are uniformly tan to dark brown and usually have a velvety to granular surface. Inspection with a hand lens usually reveals small, round, porelike ostia impacted with keratin, a feature helpful in differentiating these pigmented lesions from melanomas.
FIGURE 25-10 Seborrheic keratosis. A well-demarcated coinlike pigmented lesion containing dark keratin-filled surface plugs (inset) is composed histologically of benign basaloid cells associated with prominent keratin-filled “horn” cysts, some of which communicate with the surface (pseudo-horn cysts).
Morphology. On histologic examination, these neoplasms are exophytic and sharply demarcated from the adjacent epidermis. They are composed of sheets of small cells that most resemble basal cells of the normal epidermis (Fig. 25-10). Variable melanin pigmentation is present within these basaloid cells, accounting for the brown coloration. Exuberant keratin production (hyperkeratosis) occurs at the surface, and small keratin-filled cysts (horn cysts) and invaginations of keratin into the main mass (invagination cysts) are characteristic features. Interestingly, when seborrheic keratoses become irritated and inflamed, they develop whirling foci of squamous differentiation resembling eddy currents in a stream.27
Pathogenesis.
Activating mutations in the fibroblast growth factor receptor-3 (FGFR3) gene are found in many sporadic seborrheic keratoses and are thought to drive the growth of the tumor.34,35 Seborrheic keratoses may occur explosively in large numbers, as part of a paraneoplastic syndrome (Leser-Trélat sign), possibly under the stimulation of transforming growth factor-α produced by tumor cells, most commonly carcinomas of the gastrointestinal tract.36
ACANTHOSIS NIGRICANS
Acanthosis nigricans is a condition marked by thickened, hyperpigmented skin with a “velvet-like” texture that most commonly appears in the flexural areas (axillae, skin folds of the neck, groin, and anogenital regions). It can be an important cutaneous marker of benign and malignant conditions and, accordingly, is divided into two types.37 The benign type, which constitutes about 80% of all cases, develops gradually and usually occurs in childhood or during puberty. It may occur (1) as an autosomal dominant trait with variable penetrance, (2) in association with obesity or endocrine abnormalities (particularly with pituitary or pineal tumors and diabetes), and (3) as part of several rare congenital syndromes. As with seborrheic keratoses, acanthosis nigricans sometimes occurs as a paraneoplastic process resulting from the production of growth factors by a variety of tumors. The malignant type refers to lesions arising in middle-aged and older individuals in association with underlying cancers, most commonly gastrointestinal adenocarcinomas.
Morphology. All forms of acanthosis nigricans have similar histologic features. The epidermis and underlying enlarged dermal papillae undulate sharply to form numerous repeating peaks and valleys. Variable hyperplasia may be seen, along with hyperkeratosis and slight basal cell layer hyperpigmentation (but no melanocytic hyperplasia).
Pathogenesis.
Because lesions of acanthosis nigricans may precede clinical symptoms and signs of the underlying disorder, recognition of this entity may lead to early detection of covert systemic disease. The familial form is associated with germline activating mutations in FGFR3; depending on the mutation, acanthosis may be an isolated finding or be seen together with skeletal deformities, including achondroplasia and thanatophoric dysplasia.38
FIBROEPITHELIAL POLYP
The fibroepithelial polyp has many names (acrochordon, squamous papilloma, skin tag) and is one of the most common cutaneous lesions. It is generally detected as an incidental finding in middle-aged and older individuals on the neck, trunk, face, and intertriginous areas as a soft, flesh-colored, bag-like tumor often attached to the surrounding skin by a slender stalk. Rarely, fibroepithelial polyps and tumors of perifollicular mesenchyme (specialized fibroblasts associated with the hair bulb) are seen together in Birt-Hogg-Dubé syndrome, but the vast majority of polyps are sporadic.39
Morphology. On histologic examination these tumors consist of fibrovascular cores covered by benign squamous epithelium. It is not uncommon for the polyps to undergo ischemic necrosis due to torsion, which may cause pain and precipitate their removal.
Fibroepithelial polyps are usually inconsequential, but can occasionally be associated with diabetes, obesity, and intestinal polyposis. Of interest, like melanocytic nevi and hemangiomas they often become more numerous or prominent during pregnancy, presumably related to hormonal stimulation.
EPITHELIAL CYST (WEN)
Epithelial cysts are common lesions formed by the invagination and cystic expansion of the epidermis or a hair follicle. The lay term, wen, derives from the Anglo-Saxon wenn, meaning a lump or tumor. These cysts are filled with keratin and lipid-containing debris derived from sebaceous secretions. Clinically, they are dermal or subcutaneous, well-circumscribed, firm, and often moveable nodules. When large, they may be subject to traumatic rupture, which can lead to inflammation and pain.
Morphology. Epithelial cysts are divided into several histologic types. The epidermal inclusion cyst has a wall resembling normal epidermis and is filled with laminated strands of keratin. Pilar or trichilemmal cysts have a wall that resembles follicular epithelium, without a granular cell layer and filled by a more homogeneous mixture of keratin and lipid. The dermoid cyst is similar to the epidermal inclusion cyst, but also contains multiple appendages (such as small hair follicles) budding outward from its wall. Finally, steatocystoma simplex is a cyst with a wall resembling the sebaceous gland duct, and from which numerous compressed sebaceous lobules originate. The importance of recognition of this cyst derives from the often dominantly heritable nature of the lesion (steatocystoma multiplex), which can be caused by missense mutations in the gene encoding keratin 17. Interestingly, the same mutations are also associated with a syndrome called pachyonychia congenital type 2, which is associated with focal defects in skin, nails, and pilosebaceous cysts.40,41 These phenotypes are consistent with the pattern of keratin 17 expression, which is restricted to nail bed and epidermal appendages.
ADNEXAL (APPENDAGE) TUMORS
There are literally hundreds of neoplasms arising from or showing differentiation toward cutaneous appendages. Their significance varies according to type and clinical context. Some are entirely benign, but may be confused with cutaneous cancers such as basal cell carcinoma. Other appendage tumors are associated with mendelian patterns of inheritance and occur as multiple disfiguring lesions. In some instances, these lesions warn of a predisposition for internal malignancy; such is the reationship between multiple trichilemmomas and Cowden syndrome, a disorder caused by germline mutations in the tumor suppressor gene PTEN that is associated with an increased risk of breast cancer and many other tumors.42 Selected examples are provided here to illustrate neoplasms of hair follicles and sebaceous, eccrine, and apocrine glands.
Appendage tumors are often nondescript, flesh-colored solitary or multiple papules and nodules. Some have a predisposition for occurrence on specific body surfaces. For example, the eccrine poroma occurs predominantly on the palms and soles. Cylindroma, an appendage tumor with ductal (apocrine or eccrine) differentiation, usually occurs on the forehead and scalp (Fig. 25-11A), where coalescence of nodules with time may produce a hat-like growth, hence the name turban tumor. These lesions may be dominantly inherited; in such cases they appear early in life and are associated with inactivating mutations in the tumor suppressor gene CYLD, which encodes a deubiquitinating enzyme that regulates NF-κB and the cell cycle.43,44 Germline mutations in this gene are associated with several related genetic syndromes including familial cylindromatosis, multiple familial trichoepithelioma (a follicular tumor), and Brooke-Spiegler syndrome (showing both tumor types).45 Syringomas, lesions with eccrine differentiation, usually occur as multiple, small, tan papules in the vicinity of the lower eyelids. Sebaceous adenomas can be associated with internal malignancy in the Muir-Torre syndrome, a subset of the hereditary nonpolyposis colorectal carcinoma syndrome (Chapter 17) associated with germline deficits in DNA mismatch repair proteins; some of these have mutations in genes involved in WNT signaling.46,47 Pilomatricomas, showing follicular differentiation, are associated with activating mutations in CTNNB1, the gene encoding β-catenin.48 Mutations in this gene are seen in numerous neoplasms but are of interest here since WNT signaling through β-catenin is critical for early hair development and regulates the hair cycle. Adnexal tumors can also show primarily apocrine differentiation; these usually arise in body areas where apocrine glands are most prevalent, such as the axilla and scalp. Some skin adnexal tumors may arise from multipotent cutaneous stem cells, which are believed to reside in a specialized niche associated with hair follicles.49
FIGURE 25-11 Adnexal tumors. A, Multiple cylindromas (papules) on the forehead are composed of islands of (B) basaloid cells containing occasional ducts that fit together like pieces of a jigsaw puzzle. C, Perinasal papules and small nodules of trichoepithelioma are composed of (D) buds of basaloid cells that resemble primitive hair follicles.
Morphology. The cylindroma is composed of islands of cells resembling those of the normal epidermal or adnexal basal cell layer (basaloid cells). These islands fit together like pieces of a jigsaw puzzle within a fibrous dermal matrix (Fig. 25-11B). Trichoepithelioma is a proliferation of basaloid cells that forms primitive structures resembling hair follicles (Fig. 25-11C, D). Sebaceous adenoma shows a lobular proliferation of sebocytes with increased peripheral basaloid cells and more mature sebocytes in the central portion, characterized by frothy or bubbly cytoplasm due to lipid vesicle content (Fig. 25-12A). Pilomatrixomas are composed of basaloid cells that show trichilemmal or hairlike differentiation similar to that seen in the germinal portion of the normal hair bulb in the anagen growth phase (Fig. 25-12B). Apocrine carcinoma shows ductal differentiation with prominent decapitation secretion similar to that seen in the normal apocrine gland (Fig. 25-12C). The infiltrative growth pattern is a hint of malignancy in this otherwise well-differentiated tumor.
FIGURE 25-12 Adnexal tumors. A, Sebaceous adenoma; inset demonstrates sebaceous differentiation. B, Pilomatrixoma; inset shows hair matrical differentiation with characteristic maturation to anucleate “ghost cells.” C, Apocrine carcinoma (well-differentiated); inset shows apocrine differentiation with characteristic luminal decapitation secretion.
Although most appendage tumors are benign, malignant variants do exist. Apocrine tumors are unusual in that malignant forms seem to be more common than benign forms. Sebaceous carcinoma, for example, arises from the meibomian glands of the eyelid and may follow an aggressive course replete with systemic metastases. Eccrine and apocrine carcinomas are often confused with metastatic adenocarcinomas to the skin because of their tendency for abortive gland formation.
Premalignant and Malignant Epidermal Tumors
ACTINIC KERATOSIS
The development of epidermal malignancy is typically preceded by a period of progressively worsening dysplastic changes, which are analogous to the precursor lesions that give rise to carcinoma of the squamous mucosa of the uterine cervix (Chapter 22). In the skin, these precursor lesions are called actinic keratoses; as the name implies, these usually occur in sun-damaged skin and exhibit hyperkeratosis. As would be expected, they occur with particularly high incidence in lightly pigmented individuals. Exposure to ionizing radiation, industrial hydrocarbons and arsenicals may induce similar lesions.
Actinic keratoses are usually less than 1 cm in diameter; are tan-brown, red, or skin-colored; and have a rough, sandpaper-like consistency. Some lesions may produce so much keratin that a “cutaneous horn” develops (Fig. 25-13A). Such horns may become so prominent that they actually resemble the horns of animals! Sun-exposed sites (face, arms, dorsum of hands) are most frequently affected. The lips may also develop similar lesions (termed actinic cheilitis).
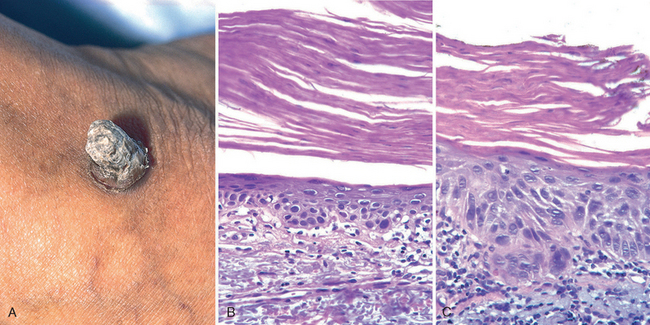
FIGURE 25-13 Actinic keratosis. A, Excessive scale formation in this lesion has produced a “cutaneous horn.” B, Basal cell layer atypia (dysplasia) is associated with marked hyperkeratosis and parakeratosis. C, Progression to full-thickness nuclear atypia, with or without the presence of superficial epidermal maturation, heralds the development of squamous cell carcinoma in situ.
Morphology. Cytologic atypia is seen in the lowermost layers of the epidermis and may be associated with hyperplasia of basal cells (Fig. 25-13B) or, alternatively, with atrophy that results in thinning of the epidermis. The atypical basal cells usually have pink or reddish cytoplasm due to dyskeratosis. Intercellular bridges are present, in contrast to basal cell carcinoma, in which they are not visible. The superficial dermis contains thickened, blue-gray elastic fibers (elastosis), a probable result of abnormal elastic fiber synthesis by sun-damaged fibroblasts34. The stratum corneum is thickened, and unlike normal skin, the cells in this layer often retain their nuclei (parakeratosis).
Whether all actinic keratoses would inexorably lead to skin cancer (usually squamous cell carcinoma), if given enough time, is conjectural. Studies indicate that lesions may regress or remain stable during a normal life span. However, enough become malignant to warrant local eradication. This can usually be accomplished by gentle curettage, freezing, or topical application of chemotherapeutic agents. More recently, a reagent called imiquimod, which activates the innate immune system through stimulation of Toll-like receptors (TLRs), has been used to eradicate the abnormal cells that compose this tumor.50
SQUAMOUS CELL CARCINOMA
Squamous cell carcinoma is the second most common tumor arising on sun-exposed sites in older people, exceeded only by basal cell carcinoma. Except for lesions on the lower legs, these tumors have a higher incidence in men than in women. Invasive squamous cell carcinomas are usually discovered while they are small and resectable. Less than 5% of these tumors metastasize to regional nodes; these lesions are generally deeply invasive and involve the subcutis.
Morphology. Squamous cell carcinomas that have not invaded through the basement membrane of the dermoepidermal junction (termed in situ carcinoma) appear as sharply defined, red, scaling plaques. More advanced, invasive lesions are nodular, show variable keratin production (appreciated grossly as hyperkeratotic scale), and may ulcerate (Fig. 25-14A).

FIGURE 25-14 Invasive squamous cell carcinoma. A, Lesions are often nodular and ulcerated as seen in this scalp tumor. B, Tongues of atypical squamous epithelium have transgressed the basement membrane, invading deeply into the dermis. C, A magnified image reveals invasive tumor cells showing enlarged nuclei with angulated contours and prominent nucleoli.
Unlike actinic keratoses, in squamous cell carcinoma in situ, cells with atypical (enlarged and hyperchromatic) nuclei involve all levels of the epidermis (Fig. 25-13C). Invasive squamous cell carcinoma (Fig. 25-14B, C) shows variable degrees of differentiation, ranging from tumors composed of polygonal cells arranged in orderly lobules and having numerous large areas of keratinization, to neoplasms associated with geographic necrosis consisting of highly anaplastic cells that exhibit only abortive, single-cell keratinization (dyskeratosis). The latter tumors may be so poorly differentiated that immunohistochemical stains for keratins are needed to confirm the diagnosis.
Keratoacanthoma is a controversial lesion; some regard it as a variant of well-differentiated squamous cell carcinoma, while others consider it to be a distinct entity. It differs from conventional squamous cell carcinomas in that after a period of rapid growth, it usually regresses spontaneously. Grossly, it is a symmetric cup-shaped tumor with a central depression filled with keratin debris. Histologically, the tumor is composed of lobules of squamous cells with glassy cytoplasm that undergo kertinization without an intervening granular layer. Once established, keratoacanthomas elicit a brisk lymphocytic and eosinophilic host response.
Pathogenesis.
The most important cause of cutaneous squamous cell carcinoma is DNA damage induced by exposure to UV light. Tumor incidence is proportional to the degree of lifetime sun exposure. A second common association is with immunosuppression, most notably chronic immunosuppression as a result of chemotherapy or organ transplantation.51 Immunosuppression may contribute to carcinogenesis by reducing host surveillance and increasing the susceptibility of keratinocytes to infection and transformation by oncogenic viruses, particularly human papilloma virus (HPV) subtypes 5 and 8.52 These same HPVs have been implicated in tumors arising in patients with a rare autosomal recessive condition, epidermodysplasia verruciformis, that is marked by a high susceptibility to cutaneous squamous cell carcinomas.53 Sunlight, in addition to its damaging effect on DNA, seems to cause a defect in cutaneous immunity by dampening the immune surveillance function of epidermal Langerhans cells.54 Other risk factors for squamous cell carcinoma include industrial carcinogens (tars and oils), chronic ulcers and draining osteomyelitis, old burn scars, ingestion of arsenicals, ionizing radiation, and (in the oral cavity) tobacco and betel nut chewing.
Most studies on the genetics of squamous cell carcinoma have focused on acquired defects in sporadic tumors and their precursors (actinic keratoses), and the relationships between these defects and sun-exposure. The incidence of p53 mutations in actinic keratoses found in Caucasians is high, suggesting that p53 dysfunction is an early event in the development of tumors induced by sunlight. Normally, DNA damaged by UV light is sensed by checkpoint kinases such as ATM and ATR, which send out signals that upregulate the expression and stability of p53. p53 in turn arrests cells in the G1 phase of the cell cycle and promotes either “high-fidelity” DNA repair or the elimination of cells that are damaged beyond repair by apoptosis (Chapter 7). When these protective functions of p53 are lost, DNA damage induced by UV light is more likely to be “repaired” by error-prone mechanisms, creating mutations that are passed down to daughter cells. Of note, the mutations that are seen in p53 often occur at pyrimidine dimers, indicating that they, too, stem from damage caused by UV light. A similar story underlies the remarkable susceptibility of patients with xeroderma pigmentosum to squamous cell carcinoma. This disorder is caused by inherited mutations in genes in the nucleotide excision repair pathway, which is required for accurate repair of pyrimidine dimers; when this pathway is defective, error-prone repair pathways take over, leading to the rapid accumulation of mutations and eventual carcinogenesis.
As with all other forms of cancer, cutaneous squamous cell carcinoma stems from molecular lesions in multiple genes. In addition to defects in p53, studies of explanted human keratinocytes suggest that dysregulated RAS signaling plays an important role in the transformation process.55
BASAL CELL CARCINOMA
Basal cell carcinoma is the most common invasive cancer in humans, with nearly 1 million estimated cases per year in the United States.56 These are slow-growing tumors that rarely metastasize. They have a tendency to occur at sun-exposed sites and in lightly pigmented people. As with squamous cell carcinoma, the incidence of basal cell carcinoma rises sharply with immunosuppression and in people with inherited defects in DNA repair such as xeroderma pigmentosum (Chapter 7).
These tumors present clinically as pearly papules often containing prominent, dilated subepidermal blood vessels (telangiectasias) (Fig. 25-15A). Some tumors contain melanin and thus appear similar to melanocytic nevi or melanomas. Advanced lesions may ulcerate, and extensive local invasion of bone or facial sinuses may occur after many years of neglect or in unusually aggressive tumors, explaining the archaic designation rodent ulcers. One common and important variant, the superficial basal cell carcinoma, presents as an erythematous, occasionally pigmented plaque that may resemble early forms of melanoma.
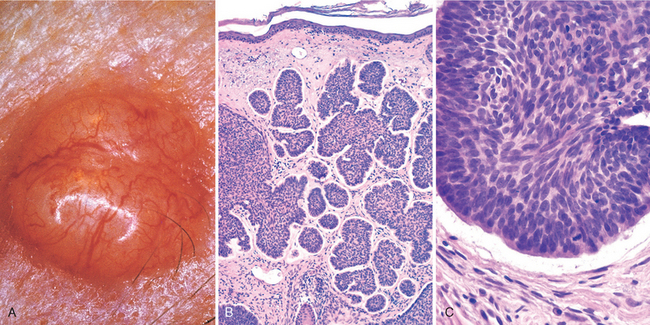
FIGURE 25-15 Basal cell carcinoma. Pearly, telangiectatic nodules (A) are composed of nests of uniformly atypical basaloid cells within the dermis (B) that are often separated from the adjacent stroma by thin clefts (C), an artifact of sectioning.
Morphology. Histologically, the tumor cells resemble those in the normal basal cell layer of the epidermis. They arise from the epidermis or follicular epithelium and do not occur on mucosal surfaces. Two patterns are seen: multifocal growths originating from the epidermis and sometimes extending over several square centimeters or more of skin surface (multifocal superficial type) and nodular lesions growing downward deeply into the dermis as cords and islands of variably basophilic cells with hyperchromatic nuclei, embedded in a mucinous matrix, and often surrounded by many fibroblasts and lymphocytes (Fig. 25-15B). The cells at the periphery of the tumor cell islands tend to be arranged radially with their long axes in parallel alignment (palisading). In sections, the stroma retracts away from the carcinoma (Fig. 25-15C), creating clefts or separation artifacts that assist in differentiating basal cell carcinomas from certain appendage tumors that are also characterized by proliferation of basaloid cells, such as trichoepithelioma.
Pathogenesis.
Nevoid basal cell carcinoma syndrome (NBCCS; also known as basal cell nevus or Gorlin syndrome) is an autosomal dominant disorder characterized by multiple basal cell carcinomas.57 Most of these tumors develop before age 20 and are accompanied by various other conditions including tumors (especially medulloblastomas and ovarian fibromas), odontogenic keratocysts, and pits of the palms and soles. Multiple systemic manifestations such as intracranial calcification, cleft lip and palate, abnormal segmentation of the vertebra, and rib anomalies (bifid, fused, missing, splayed ribs) may also be present.58 Though quite rare (incidence of 1 in 56,000), this syndrome has helped to elucidate the molecular genetics of basal cell carcinoma, including the common sporadic type. Examples of other genetic syndromes associated with skin tumors are listed in Table 25-2.
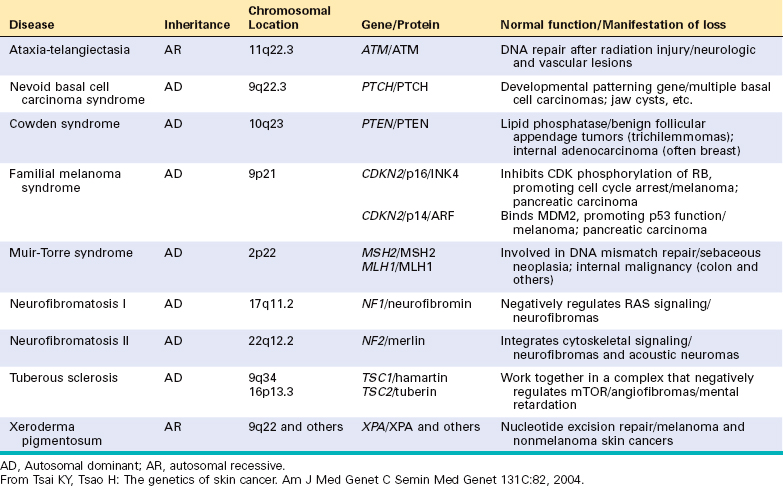
The gene associated with NBCCS, located on chromosome 9q22.3, is PTCH, the human homologue of the Drosophila developmental gene patched.59 The pathogenesis of basal cell carcinomas in NBCCS fits the classical Knudson “two-hit” hypothesis for familial retinoblastoma (Chapter 7). Individuals with NBCCS are born with a germline mutation in one of the PTCH alleles; the second normal allele is inactivated in tumors by a mutation acquired by chance or due to exposure to mutagens (such as UV light).
The PTCH gene encodes a receptor for the protein product of the sonic hedgehog gene (SHH), a member of the hedgehog (HH) family of genes that determine polarity during embryonic development.60 The pathway is also involved in hair follicle formation and the hair cycle in skin.5 The PTCH protein forms a receptor complex with another transmembrane protein, known as SMO (for “smoothened”). In the absence of its ligand, SHH, PTCH inactivates SMO and sequesters it from transducing a down-stream signal. Binding of SHH to PTCH releases the suppression of SMO, causing the up-regulation of hedgehog target genes through a signal cascade that involves the GLI1 transcription factor (Fig. 25-16). Experimental work in mice has shown that animals with defects in the PTCH signaling pathway, including overexpression of GLI1, develop skin tumors resembling basal cell carcinomas.61,62 In NBCCS the absence of PTCH causes constitutive activation of SMO, leading to the development of basal cell carcinoma.
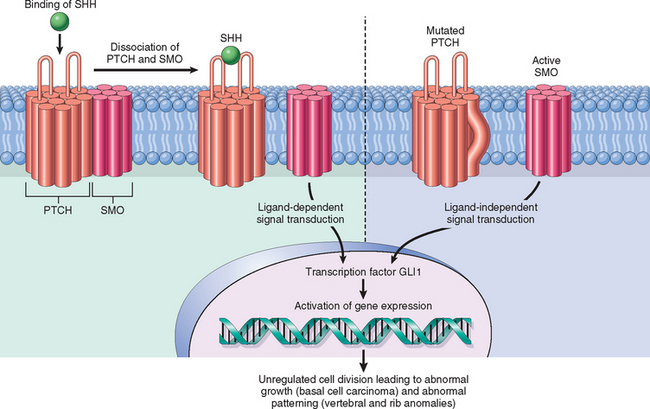
FIGURE 25-16 Normal and oncogenic hedgehog signaling. Left, Normally, PTCH and SMO form a receptor complex that binds sonic hedgehog (SHH). In the absence of SHH, PTCH blocks SMO activity. When SHH binds PTCH, SMO is released to trigger a signal transduction cascade that leads to activation of GLI1 and other transcription factors. Right, Mutations in PTCH, and less often in SMO, allow SMO to signal without ligand binding and underlie the nevoid basal cell carcinoma (Gorlin) syndrome.
Mutations of genes belonging to the PTCH signaling pathway are also important for the development of the common sporadic form of basal cell carcinoma, as documented by the inactivation of PTCH and the presence of SMO activating mutations in these skin neoplasms.63 PTCH mutations are found in approximately 30% of sporadic basal cell carcinomas, and of these about one third have mutations (C→T transitions) that are considered hallmarks of UV damage. Mutations in p53 occur in 40% to 60% of basal cell carcinomas, and 60% of these have “UV signature.”64 Xeroderma pigmentosum, a disorder of DNA repair, presents a striking example of the connection between sun exposure and defects in PTCH and p53.65 In these tumors the frequency of mutations in PTCH and p53 are, respectively, 90% and 40%, and the majority of these bear the UV signature.
Tumors of the Dermis
The dermis contains a variety of elements such as smooth muscle, pericytes, fibroblasts, neural tissue, and endothelium. Neoplasms with differentiation toward all of these tissues occur not just in the skin, but also in soft tissues and viscera. In this section we consider two representative dermal neoplasms—one benign, one malignant—that arise primarily in the skin.
BENIGN FIBROUS HISTIOCYTOMA (DERMATOFIBROMA)
Benign fibrous histiocytoma refers to a heterogeneous family of morphologically and histogenetically related benign dermal neoplasms of uncertain lineage. These tumors are usually seen in adults and often occur on the legs of young to middle-aged women. Their biologic behavior is indolent.
These neoplasms appear as firm, tan to brown papules (Fig. 25-17A). Lesions are asymptomatic or tender and may increase and decrease slightly in size over time. Most are less than 1 cm in diameter, but actively growing lesions may reach several centimeters in diameter; with time they often become flattened. The tendency for fibrous histiocytomas to dimple inward on lateral compression can be helpful in distinguishing them from nodular melanomas, which protrude when squeezed.
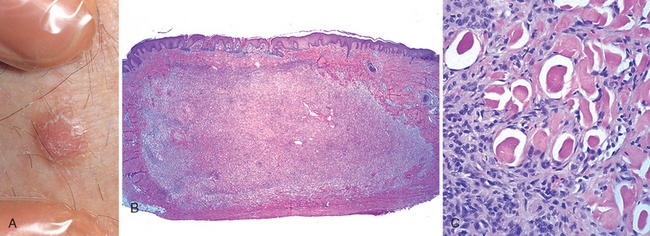
FIGURE 25-17 Benign fibrous histiocytoma (dermatofibroma). This firm, tan papule on the leg (A) shows a localized proliferation of benign-appearing spindle cells within the dermis (B). C, Note the characteristic overlying epidermal hyperplasia and the tendency of fibroblasts to surround individual collagen bundles.
The cause of fibrous histiocytomas remains a mystery. Many cases have a history of antecedent trauma, suggesting an abnormal response to injury and inflammation,66 perhaps analogous to the deposition of increased amounts of altered collagen in a hypertrophic scar or keloid. These common yet curious tumors appear to be composed at least partially of factor XIIIa–positive dermal dendrocytes.
Morphology. The most common form of fibrous histiocytoma is referred to as a dermatofibroma. These tumors are formed by benign, spindle-shaped cells arranged in a well-defined, nonencapsulated mass within the mid-dermis (Fig. 25-17B, C). Extension of these cells into the subcutaneous fat is sometimes observed. Many cases demonstrate a peculiar form of overlying epidermal hyperplasia, characterized by downward elongation of hyperpigmented rete ridges (a pseudo-epitheliomatous pattern). Numerous histologic variants are noted, such as more cellular forms or tumors with pools of extravascular blood and hemosiderin (aneurysmal).
DERMATOFIBROSARCOMA PROTUBERANS
Dermatofibrosarcoma protuberans is best regarded as a well-differentiated, primary fibrosarcoma of the skin. These tumors are slow growing, and although they are locally aggressive and can recur, they rarely metastasize. Clinically they are firm, solid nodules that arise most frequently on the trunk. They often develop as aggregated “protuberant” tumors within a firm (indurated) plaque or nodule that may sometimes ulcerate.
Morphology. On microscopic examination, these neoplasms are cellular, composed of fibroblasts arranged radially, reminiscent of blades of a pinwheel, a pattern referred to as storiform. Mitoses are rare. In contrast to that in dermatofibroma, the overlying epidermis is generally thinned. Deep extension from the dermis into subcutaneous fat, producing a characteristic “honeycomb” pattern, is frequently present (Fig. 25-18B, C). These tumors may extend down fibrous septae in the subcutis and thus require wider excision than would appear to be necessary to prevent local recurrence.
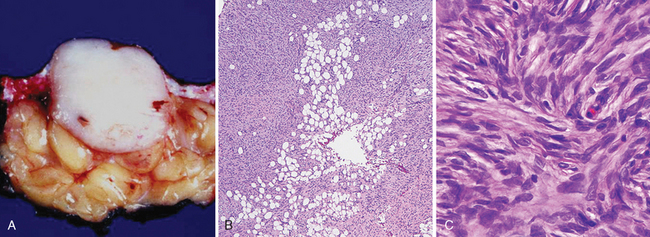
FIGURE 25-18 Dermatofibrosarcoma protuberans. A, The tumor usually presents as a flesh-colored to erythematous nodule, and has a fibrotic appearance on sectioning. B, C, Characteristic storiform cellularity is noted histologically, and the lesion often infiltrates the subcutis in a manner that resembles “Swiss cheese” to some afficianados.
Pathogenesis.
The molecular hallmark of dermatofibrosarcoma protuberans is a balanced translocation between genes encoding collagen 1A1 (COL1A1) and the platelet-derived growth factor-β (PDGFB). It results in the juxtaposition of COL1A1 promoter sequences and the coding region of PDGFβ; this in turn leads to over-expression and secretion of PDGFβ, which drives tumor cell growth through an autocrine loop.67,68 Such fusion genes are increasingly being recognized in a wide spectrum of tumors, including carcinomas.69 While the primary mode of treatment is local wide excision, in cases not amenable to this approach the targeted tyrosine kinase inhibitor imatinib mesylate is used. This inhibits PDGF-β receptor activation and has marked antitumor activity.70 Withdrawal of the drug allows tumor regrowth even after long-term treatment, so use of this agent is lifelong.
Tumors of Cellular Migrants to the Skin
Aside from tumors that arise directly from epidermal and dermal cells, several proliferative disorders of the skin involve cells whose progenitors arise elsewhere and home to the cutaneous microenvironment. The two examples discussed in this section—namely, cutaneous T-cell lymphoma and mastocytosis—are primary cutaneous disorders that arise from lymphocytes and mast cells, respectively.
MYCOSIS FUNGOIDES (CUTANEOUS T-CELL LYMPHOMA)
Cutaneous T cell lymphoma (CTCL) represents a spectrum of lymphoproliferative disorders affecting the skin (see also Chapter 13).71 Two different clinical types of malignant T-cell disorders were originally recognized: mycosis fungoides, a chronic proliferative process; and a more aggressive nodular eruptive variant, mycosis fungoides d’emblée. It is now known that a variety of presentations of T-cell lymphoma occur in the skin, but this section will focus on mycosis fungoides.
Mycosis fungoides is a T-cell lymphoma that presents in the skin and may evolve into generalized lymphoma.72 Most affected individuals have disease that remains localized to the skin for many years; a minority have rapid systemic dissemination. This condition may occur at any age, but most commonly it afflicts persons older than age 40.
Lesions of mycosis fungoides usually involve truncal areas and include scaly, red-brown patches; raised, scaling plaques that may even be confused with psoriasis; and fungating nodules. Prognosis is related to the percentage of body surface involved and progression from patch to plaque to nodular forms. Eczema-like lesions typify early stages of disease when obvious visceral or nodal spread has not occurred. Raised, indurated, irregularly outlined, erythematous plaques may then supervene. Development of multiple, large (≤10 cm or more in diameter), red-brown nodules correlates with systemic spread. Sometimes plaques and nodules ulcerate (Fig. 25-19A). Ultimately, lesions may affect numerous body surfaces, including the trunk, extremities, face, and scalp. In some individuals, seeding of the blood by malignant T cells is accompanied by diffuse erythema and scaling of the entire body surface (erythroderma), a condition known as Sézary syndrome (Chapter 13).
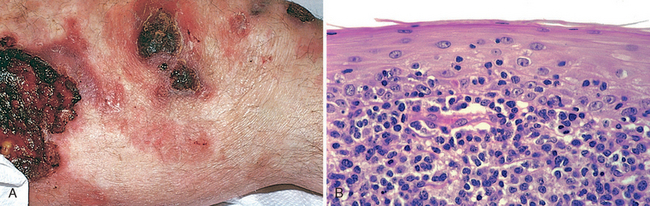
FIGURE 25-19 Cutaneous T-cell lymphoma. A, Several ill-defined, erythematous, often scaling, and occasionally ulcerated plaques. B, Microscopically, there is an infiltrate of atypical lymphocytes that show a tendency to accumulate beneath the epidermal layer and to invade the epidermis.
The proliferating cells in CTCL are clonal populations of lymphocytes of the CD4 subset.2,73 The neoplastic cells are targeted to the skin by expression of CLA (see Fig. 25-2) and often express aberrant cell surface antigens as well as clonal T-cell receptor gene rearrangements. Detection of these features may be of diagnostic assistance in difficult cases. Topical therapy with steroids or UV light is often used for early lesions of CTCL, whereas more aggressive systemic chemotherapy is indicated for advanced disease.
Morphology. The histologic hallmark of CTCL of the mycosis fungoides type is the presence of the Sézary-Lutzner cells. These are T-helper cells (CD4+) that characteristically form band-like aggregates within the superficial dermis (Fig. 25-19B) and invade the epidermis as single cells and small clusters (Pautrier microabscesses). These cells have markedly infolded nuclear membranes, imparting a hyperconvoluted or cerebriform contour. Although patches and plaques show pronounced epidermal infiltration by Sézary-Lutzner cells (epidermotropism), in more advanced nodular lesions the malignant T cells often lose this epidermotropic tendency, grow deeply into the dermis, and eventually spread systemically.
MASTOCYTOSIS
The term mastocytosis refers to a spectrum of rare disorders characterized by increased numbers of mast cells in the skin and, in some instances, in other organs. A localized cutaneous form of the disease that affects predominantly children and accounts for more than 50% of all cases is termed urticaria pigmentosa. These lesions are multiple, although solitary mastocytomas may also occur, usually shortly after birth. About 10% of individuals with mast cell disease have systemic disease, with mast cell infiltration of many organs. These individuals are often adults, and unlike localized cutaneous disease, the prognosis may be poor.
The pathologic findings in mastocytosis are highly variable. In urticaria pigmentosa, lesions are multiple and widely distributed, consisting of round to oval, red-brown, nonscaling papules and small plaques. Solitary mastocytomas present as one or several pink to tan-brown nodules that may be pruritic or show blister formation (Fig. 25-20A). In systemic mastocytosis skin lesions similar to those of urticaria pigmentosa are accompanied by mast cell infiltration of bone marrow, liver, spleen, and lymph nodes. Many of the signs and symptoms of mastocytosis are due to the effects of histamine, heparin, and other substances released as a result of mast cell degranulation. Darier sign refers to a localized area of dermal edema and erythema (wheal) that occurs when lesion skin is rubbed. Dermatographism refers to an area of dermal edema resembling a hive that occurs in normal skin as a result of localized stroking with a pointed instrument. In systemic disease, all of the following may be seen: pruritus and flushing triggered by certain foods, temperature changes, alcohol, and certain drugs (morphine, codeine, aspirin); watery nasal discharge (rhinorrhea); rarely, gastrointestinal or nasal bleeding, possibly due to the anticoagulant effects of heparin; and bone pain as a result of osteoblastic and osteoclastic involvement.
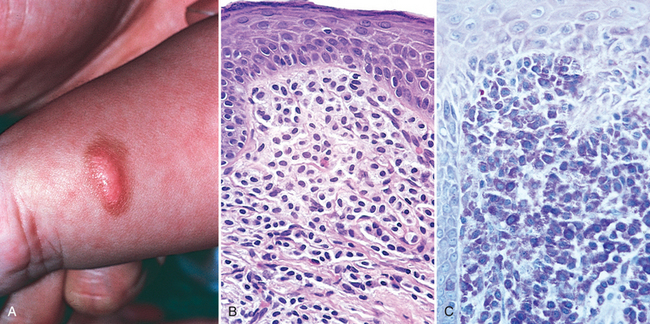
FIGURE 25-20 Mastocytosis. A, Solitary mastocytoma in a 1-year-old child. B, By routine histology, numerous ovoid cells with uniform, centrally located nuclei are observed in the dermis. C, Giemsa staining reveals purple, “metachromatic” granules within the cytoplasm of the cells.
Morphology. The histologic picture in urticaria pigmentosa or solitary mastocytoma varies from a subtle increase in the numbers of spindle-shaped and stellate mast cells around superficial dermal blood vessels, to large numbers of tightly packed, round to oval mast cells in the upper to mid-dermis (Fig. 25-20B). Variable fibrosis, edema, and small numbers of eosinophils may also be present. Mast cells may be difficult to differentiate from lymphocytes in routine, hematoxylin and eosin–stained sections, and special metachromatic stains (toluidine blue or Giemsa) must be used to visualize their granules (Fig. 25-20C). Even with these stains, extensive degranulation may result in failure to recognize these cells by light microscopy, but their identity can be readily confirmed with immunohistochemical stains for mast cell markers, such as mast cell tryptase.
Pathogenesis.
The pathogenesis of many cases of mastocytosis involves acquired activating point mutations in the c-KIT receptor tyrosine kinase. The resulting increase in c-KIT signaling drives mast cell growth and survival.74,75 Recognition of this etiology has led to attempts to treat this disorder with c-KIT inhibitors.
Disorders of Epidermal Maturation
ICHTHYOSIS
Of the numerous disorders that impair epidermal maturation, ichthyosis is perhaps one of the most striking. The term is derived from the Greek root ichthy-, meaning “fishy,” and accordingly, this group of genetically inherited disorders is associated with chronic, excessive keratin buildup (hyperkeratosis) that results clinically in fish-like scales (Fig. 25-21A). Most ichthyoses become apparent either at or around the time of birth. Acquired (noninherited) variants also exist; in the acquired ichthyosis vulgaris in adults, there can be an association with lymphoid and visceral malignancies. The clinical types of ichthyosis vary according to the mode of inheritance, histology, and clinical features; the primary categories include ichthyosis vulgaris (autosomal dominant or acquired), congenital ichthyosiform erythroderma (autosomal recessive), lamellar ichthyosis (autosomal recessive), and X-linked ichthyosis.76
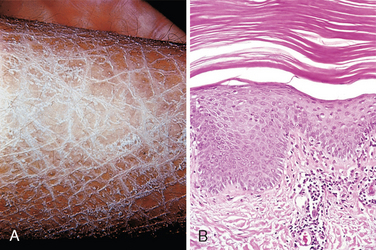
FIGURE 25-21 Ichthyosis. Note prominent fishlike scales (A) and compacted, thickened stratum corneum (B).
Morphology. All forms of ichthyosis exhibit a buildup of compacted stratum corneum that is associated with loss of the normal basket-weave pattern (Fig. 25-21B). There is generally little or no inflammation. Variations in the thickness of the epidermis and the stratum granulosum and the gross appearance and distribution of lesions are used to subclassify these disorders.
Pathogenesis.
The primary abnormality in some forms of ichthyosis is defective desquamation, leading to retention of abnormally formed scale. For example, in X-linked ichthyosis, affected individuals have a deficiency of steroid sulfatase, an enzyme important for the removal of proadhesive cholesterol sulfate secreted into the intercellular spaces. Accumulation of cholesterol sulfate results in persistent cell-to-cell adhesion within the stratum corneum, hindering the desquamation process.
Acute Inflammatory Dermatoses
Literally thousands of inflammatory dermatoses have been described. In general, acute lesions last from days to weeks and are characterized by inflammatory infiltrates (usually composed of lymphocytes and macrophages rather than neutrophils), edema, and variable degrees of epidermal, vascular, or subcutaneous injury. Chronic lesions, on the other hand, persist for months to years and are often associated with changes in epidermal growth (atrophy or hyperplasia) or dermal fibrosis. The lesions discussed here are selected as examples of the more commonly encountered acute dermatoses.
URTICARIA
Urticaria (hives) is a common disorder of the skin characterized by localized mast cell degranulation and resultant dermal microvascular hyperpermeability. This gives rise to pruritic edematous plaques called wheals. Angioedema is closely related to urticaria and is characterized by deeper edema of both the dermis and the subcutaneous fat.
Urticaria most often occurs between ages 20 and 40, although all age groups are susceptible. Individual lesions develop and fade within hours (usually <24 hours), and episodes may last for days or persist for months. Lesions vary from small, pruritic papules to large edematous plaques (Fig. 25-22A). Individual lesions may coalesce to form annular, linear, or arciform configurations. Sites of predilection for urticarial eruptions include any area exposed to pressure, such as the trunk, distal extremities, and ears. Persistent episodes of urticaria may herald an underlying disease (e.g., collagen vascular disorders, Hodgkin lymphoma), but in the majority of cases no underlying cause can be identified.
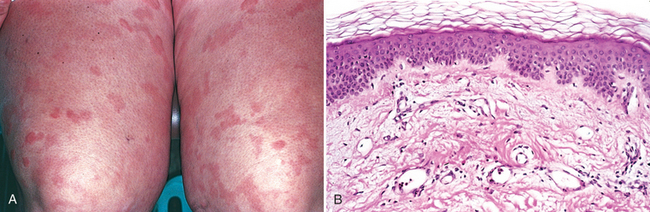
FIGURE 25-22 Urticaria. A, Erythematous, edematous, often circular plaques are characteristic. B, Histologically, there is superficial dermal edema, manifested by spaces between collagen bundles, and dilated lymphatic and blood-filled vascular spaces; the epithelium is normal.
Morphology. The histologic features of urticaria may be very subtle. There is usually a sparse superficial perivenular infiltrate consisting of mononuclear cells and rare neutrophils. Eosinophils may also be present. Collagen bundles are more widely spaced than in normal skin, a result of superficial dermal edema (Fig. 25-22B). Superficial lymphatic channels are dilated due to increased absorption of edema fluid. Epidermal changes are typically absent.
Pathogenesis.
In most cases, urticaria results from antigen-induced release of vasoactive mediators from mast cell granules through sensitization with specific IgE antibodies. This IgE-dependent reaction can follow exposure to many different antigens (pollens, foods, drugs, insect venom) (Chapter 6). IgE-independent urticaria may also result from substances that in certain individuals directly incite the degranulation of mast cells, such as opiates, certain antibiotics, curare, and radiographic contrast media. Another cause of IgE-independent urticaria is exposure to chemicals, such as aspirin, that suppress prostaglandin synthesis from arachidonic acid. Complement-mediated urticaria is seen in hereditary angioneurotic edema (Chapter 6), caused by an inherited deficiency of C1 inhibitor that results in uncontrolled activation of the early components of the complement system and production of vasoactive mediators.77
ACUTE ECZEMATOUS DERMATITIS
The Greek word eczema, meaning “to boil over,” vividly describes the appearance of acute eczematous dermatitis. All forms of eczema are characterized by red, papulovesicular, oozing, and crusted lesions that, if persistent, develop into raised, scaling plaques due to reactive acanthosis and hyperkeratosis (Fig. 25-23). Based on initiating factors, eczematous dermatitis can be subdivided into the following categories: (1) allergic contact dermatitis, (2) atopic dermatitis, (3) drug-related eczematous dermatitis, (4) photoeczematous dermatitis, and (5) primary irritant dermatitis.
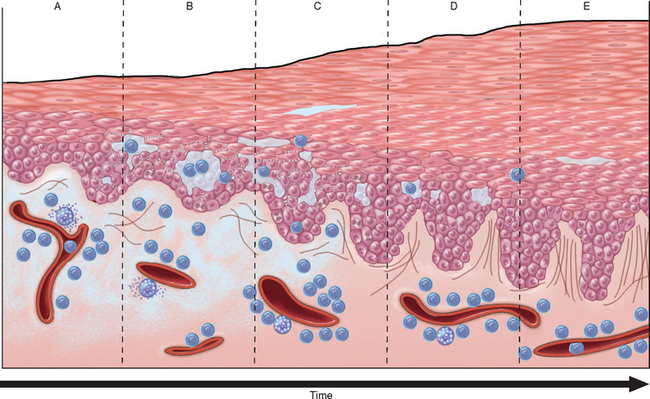
FIGURE 25-23 Stages of eczema development. A, Initial dermal edema and perivascular infiltration by inflammatory cells is followed within 24 to 48 hours by epidermal spongiosis and microvesicle formation (B). C, Abnormal scale, including parakeratosis, follows, along with progressive acanthosis (D) and hyperkeratosis (E) as the lesion becomes chronic.
A striking example of eczema is an acute contact reaction to topical antigens such as poison ivy/oak (Rhus toxicodendron), characterized by pruritic, edematous, oozing plaques, often containing small and large blisters (vesicles and bullae) (Fig. 25-24A). Such lesions are prone to bacterial superinfection, which produces a yellow crust (impetiginization). With time, persistent lesions become less “wet” (fail to ooze or form vesicles) and become progressively scaly (hyperkeratotic) as the epidermis thickens (acanthosis). The clinical causes of eczema are sometimes broadly divided into “inside” and “outside” types: disease resulting from external application of antigen (such as poison ivy) or reaction to an internal circulating antigen (such as ingested food or drug).
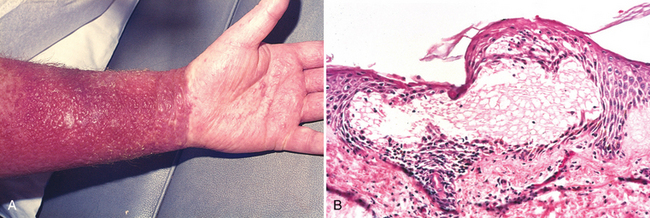
FIGURE 25-24 Eczematous dermatitis. A, Acute allergic contact dermatitis, with numerous vesicles on erythematous skin due to antigen exposure (in this case, laundry detergent in clothing). B, Histologically, intercellular edema within the epidermis creates small, fluid-filled intraepidermal vesicles.
Treatment of eczema involves a search for offending substances that can be removed from the environment. Topical steroids can also be used that nonspecifically block the inflammatory response. Such treatments are palliative and not curative, but are helpful in interrupting acute exacerbations of eczema that can become self perpetuating if unchecked.
Morphology. Spongiosis characterizes acute eczematous dermatitis, hence the histologic synonym spongiotic dermatitis. Unlike urticaria, in which edema is restricted to the superficial dermis, edema seeps into the intercellular spaces of the epidermis, splaying apart keratinocytes, particularly in the stratum spinosum. Mechanical shearing of intercellular attachment sites (desmosomes) and cell membranes by progressive accumulation of intercellular fluid may result in the formation of intraepidermal vesicles (Fig. 25-24B).
During the earliest stages of eczematous dermatitis, there is a superficial, perivascular, lymphocytic infiltrate associated with papillary dermal edema and mast cell degranulation. The pattern and composition of this infiltrate may provide clues to the underlying cause. For example, eczema resulting from certain ingested drugs is marked by a lymphocytic infiltrate, often containing eosinophils, around deep as well as superficial dermal vessels. By contrast, eczematous dermatitis resulting from contact antigens tends to produce an inflammatory reaction that preferentially affects the superficial dermal layer.
Pathogenesis.
This has been well studied in dermatitis triggered by contact antigens (e.g., poison ivy). Initially, antigens at the epidermal surface are taken up by dendritic Langerhans cells, which then migrate by way of dermal lymphatics to draining lymph nodes. Here, antigens, now processed by the Langerhans cell, are presented to naive CD4+ T cells, which are activated and develop into effector and memory cells (Chapter 6). On antigen re-exposure, these memory T cells migrate to affected skin sites of antigen localization, where they adhere to post-capillary venules, extravasate into tissues, and release cytokines and chemokines that recruit the numerous inflammatory cells characteristic of eczema. This process occurs within 24 hours and accounts for the initial erythema and pruritus that characterize cutaneous delayed hypersensitivity in the acute, spongiotic phase.2
Langerhans cells within the epidermis play a central role in contact dermatitis, and understandably factors that affect Langerhans cell function impact the inflammatory reaction. Chronic exposure to UV light is injurious to epidermal Langerhans cells and can prevent sensitization to contact antigens.78 Other factors that modulate Langerhans cell function include neuropeptides released from nerve endings that terminate near the Langerhans cell body.79
ERYTHEMA MULTIFORME
Erythema multiforme is an uncommon, self-limited disorder that seems to be a hypersensitivity reaction to certain infections and drugs. This disorder affects individuals of any age and is associated with the following conditions: (1) infections such as herpes simplex, mycoplasmal infections, histoplasmosis, coccidioidomycosis, typhoid, and leprosy, among others; (2) administration of certain drugs (sulfonamides, penicillin, barbiturates, salicylates, hydantoins, and antimalarials); (3) malignant disease (carcinomas and lymphomas); and (4) collagen vascular diseases (lupus erythematosus, dermatomyositis, and polyarteritis nodosa).80
Affected individuals present with an array of “multiform” lesions, including macules, papules, vesicles, and bullae as well as the characteristic target lesion consisting of a red macule or papule with a pale, vesicular, or eroded center (Fig. 25-25A). Although lesions may be widely distributed, symmetric involvement of the extremities frequently occurs. An extensive and symptomatic febrile form of the disease, which is often but not exclusively seen in children, is called Stevens-Johnson syndrome. Typically, erosions and hemorrhagic crusts involve the lips and oral mucosa, although the conjunctiva, urethra, and genital and perianal areas may also be affected. Secondary infection of involved areas due to loss of skin integrity may result in life-threatening sepsis. Another variant, termed toxic epidermal necrolysis, results in diffuse necrosis and sloughing of cutaneous and mucosal epithelial surfaces, producing a clinical situation analogous to an extensive burn when both infection and fluid loss are clinical concerns.81
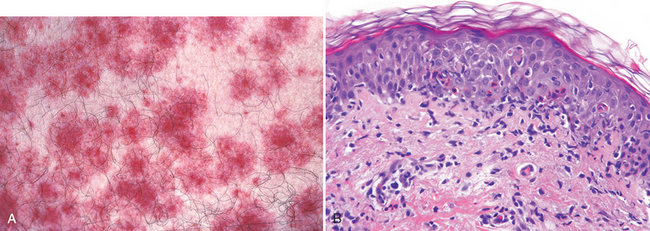
FIGURE 25-25 Erythema multiforme. A, The target-like clinical lesions consist of a central blister or zone of epidermal necrosis surrounded by macular erythema. B, Early lesions show lymphocytes collecting along the dermoepidermal junction where basal keratinocytes have begun to become vacuolated. With time, necrotic/apoptotic keratinocytes accumulate in the overlying epithelium.
Morphology. On histologic examination, early lesions show a superficial perivascular, lymphocytic infiltrate associated with dermal edema and accumulation of lymphocytes along the dermoepidermal junction, where they are intimately associated with degenerating and necrotic keratinocytes, a pattern termed interface dermatitis (Fig. 25-25B). With time there is upward migration of lymphocytes into the epidermis. Discrete and confluent zones of epidermal necrosis occur with concomitant blister formation. Epidermal sloughing leads to shallow erosions. The clinical targetoid (target-like) lesion shows central necrosis surrounded by a rim of perivenular inflammation (see Fig. 25-25A).
Pathogenesis.
Erythema multiforme has similarities to other conditions characterized by immunologically mediated epidermal cell injury such as acute graft-versus-host disease, skin allograft rejection, and fixed drug eruptions.82 In all these conditions epithelial cells are killed by skin-homing (CLA+) CD8+ cytotoxic T lymphocytes (CTLs). In erythema multiforme these cytotoxic cells are more prominent in the central portion of the lesions while CD4+ helper and Langerhans cells are more common in the raised, erythematous periphery. The precise target antigens within the epidermis that are recognized by CTLs in erythema multiforme remain unknown, but there is growing evidence that the basal cells that reside at the very tips of epidermal rete ridges may preferentially undergo apoptosis.83
Chronic Inflammatory Dermatoses
This category includes inflammatory skin disorders that persist for many months to years. The skin surface in some chronic inflammatory dermatoses is roughened as a result of excessive or abnormal scale formation and shedding. However, not all scaling lesions are inflammatory; witness the here-ditary ichthyoses, described above, with extensive scale due to defects in desquamation.
PSORIASIS
Psoriasis is a common chronic inflammatory dermatosis affecting as many as 1% to 2% of people in the United States. Persons of all ages may develop the disease. Psoriasis is sometimes associated with arthritis, myopathy, enteropathy, spondylitic joint disease, or the acquired immunodeficiency syndrome. Psoriatic arthritis may be mild or may produce severe deformities resembling the joint changes seen in rheumatoid arthritis.
Psoriasis most frequently affects the skin of the elbows, knees, scalp, lumbosacral areas, intergluteal cleft, and glans penis. The typical lesion is a well-demarcated, pink to salmon-colored plaque covered by loosely adherent scale that is characteristically silver-white in color (Fig. 25-26A). Variations exist, with some lesions occurring in annular, linear, gyrate, or serpiginous configurations. Psoriasis is one cause of total body erythema and scaling known as erythroderma. Nail changes occur in 30% of cases of psoriasis and consist of yellow-brown discoloration (often likened to an oil slick), with pitting, dimpling, separation of the nail plate from the underlying bed (onycholysis), thickening, and crumbling. In the rare variant called pustular psoriasis multiple small pustules form on erythematous plaques. This type of psoriasis is either benign and localized (hands and feet) or generalized and life-threatening, with associated fever, leukocytosis, arthralgia, diffuse cutaneous and mucosal pustules, secondary infection, and electrolyte disturbances.84
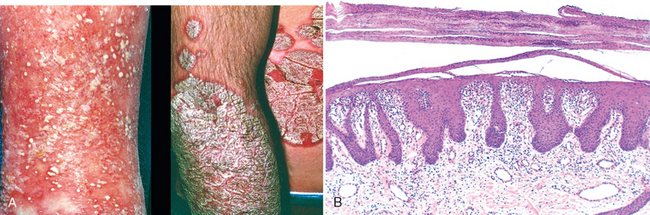
FIGURE 25-26 Clinical evolution of psoriasis. A, Early and eruptive lesions may be dominated by signs of inflammation, including small pustules and erythema (left). Established, chronic lesions demonstrate erythema surmounted by characteristic silver-white scale (right). B, Histologically, established lesions demonstrate marked epidermal hyperplasia, parakeratotic scale, and neutrophils within the superficial epidermal layers.
Morphology. Established lesions of psoriasis have a characteristic histologic picture. Increased epidermal cell turnover results in marked epidermal thickening (acanthosis), with regular downward elongation of the rete ridges sometimes described as appearing like test tubes in a rack (Fig. 25-26B). Mitotic figures are easily identified well above the basal cell layer, where mitotic activity is confined in normal skin. The stratum granulosum is thinned or absent, and extensive overlying parakeratotic scale is seen. Typical of psoriatic plaques is thinning of the portion of the epidermal cell layer that overlies the tips of dermal papillae (suprapapillary plates) and dilated, tortuous blood vessels within these papillae. This constellation of changes results in abnormal proximity of vessels within the dermal papillae to the overlying parakeratotic scale, and accounts for the characteristic clinical phenomenon of multiple, minute, bleeding points when the scale is lifted from the plaque (Auspitz sign). Neutrophils form small aggregates within slightly spongiotic foci of the superficial epidermis (spongiform pustules) and within the parakeratotic stratum corneum (Munro microabscesses). In pustular psoriasis, larger abscess-like accumulations of neutrophils are present directly beneath the stratum corneum.
Pathogenesis.
The pathogenesis of psoriasis seems to be multifactorial, with contributions from genetic and environmental factors. There is a strong association between psoriasis and HLA-C, particularly with the HLA-Cw*0602 allele. About two thirds of affected individuals carry this allele; homozygotes for HLA-Cw*0602 have a 2.5-fold higher risk of developing psoriasis than heterozygotes. Nevertheless, only 10% of individuals with HLA-Cw*0602 develop psoriasis, indicating that other factors interact with this MHC molecule in causing disease susceptibility.85 Sensitized populations of CD4+ TH1 and TH17 cells and activated CD8+ effector T cells enter the skin and accumulate in the epidermis. The culprit antigens remain elusive. T cell homing to the skin may create an abnormal microenvironment by inducing the secretion of cytokines and growth factors that induce keratinocyte proliferation, resulting in the characteristic lesions. The interactions between CD4+ T cells, CD8+ T cells, dendritic cells, and keratinocytes give rise to a cytokine “soup” dominated by TH1-type and TH17-type cytokines such as IL-12, interferon-γ, tumor necrosis factor (TNF), and IL-17.2 The lymphocytes also produce growth factors for keratinocytes. Psoriatic lesions can be induced in susceptible individuals by local trauma, a process known as the Koebner phenomenon. The trauma may induce a local inflammatory response that promotes lesion development. There is much evidence that, as with rheumatoid arthritis, TNF is a major mediator in the pathogenesis of psoriasis. Indeed, anti-TNF therapies provide benefit and are currently in use.
SEBORRHEIC DERMATITIS
Seborrheic dermatitis is a chronic inflammatory dermatosis even more common than psoriasis, affecting 1% to 3% of the general population.84 It classically involves regions with a high density of sebaceous glands, such as the scalp, forehead (especially the glabella), external auditory canal, retroauricular area, nasolabial folds, and the presternal area. However, it is important to note that seborrheic dermatitis is not a disease of the sebaceous glands. The individual lesions are macules and papules on an erythematous-yellow, often greasy base, typically in association with extensive scaling and crusting. Fissures may also be present, particularly behind the ears. Dandruff is the common clinical expression of seborrheic dermatitis of the scalp. In infants, seborrheic dermatitis presents as cradle cap but can also be part of a disorder known as Leiner disease, in which the condition is generalized and associated with diarrhea and failure to thrive. A severe form of seborrheic dermatitis that is difficult to treat is seen in many HIV–infected individuals with low CD4 counts.86
Morphology. Lesions of seborrheic dermatitis share histologic features with both spongiotic dermatitis and psoriasis, with earlier lesions being more spongiotic and later ones more acanthotic. Typically, mounds of parakeratosis containing neutrophils and serum are present at the ostia of hair follicles (so-called follicular lipping). A superficial perivascular inflammatory infiltrate generally consists of an admixture of lymphocytes and neutrophils. With human immunodeficiency virus infection, apoptotic keratinocytes and plasma cells may also be present.
Pathogenesis.
The etiology of seborrheic dermatitis is unknown. However, therapeutic studies with ketoconazole, an antifungal agent, suggest that the lipophilic yeast Malassezia furfur, which is associated with tinea versicolor, may have a role in some cases.87 Involvement of sebum is supported by clinical observations of people with Parkinson disease, who typically show increased sebum production secondary to dopamine deficiency and have a markedly increased incidence of seborrheic dermatitis.88 Indeed, once treated with levodopa, the oiliness of the skin decreases and the seborrheic dermatitis improves. However, because other conditions associated with increased sebaceous activity such as acne (discussed below) do not show this association, sebum production is unlikely to be the sole or primary factor in the pathogenesis of seborrheic dermatitis.
LICHEN PLANUS
“Pruritic, purple, polygonal, planar papules, and plaques” are the tongue-twisting “six p’s” of this disorder of skin and mucosa. Lichen planus is usually self-limited and most commonly resolves spontaneously 1 to 2 years after onset, often leaving zones of postinflammatory hyperpigmentation. Oral lesions may persist for years. Squamous cell carcinoma has been noted to occur in chronic mucosal and paramucosal lesions of lichen planus, but a direct pathogenetic relationship has not been established.89,90
Cutaneous lesions consist of itchy, violaceous, flat-topped papules that may coalesce focally to form plaques (Fig. 25-27A). These papules are often highlighted by white dots or lines called Wickham striae, which are created by areas of hypergranulosis. In darkly pigmented individuals, lesions may acquire a dark-brown color due to release of melanin into the dermis as the basal cell layer is destroyed. Multiple lesions are characteristic and are symmetrically distributed, particularly on the extremities, often about the wrists and elbows, and on the glans penis. In 70% of cases, oral lesions are present as white, reticulated, or netlike areas involving the mucosa. As in psoriasis, the Koebner phenomenon may be seen in lichen planus.91
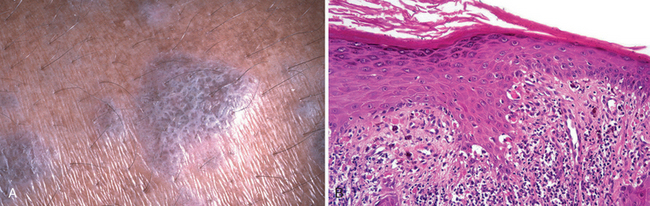
FIGURE 25-27 Lichen planus. A, This flat-topped pink-purple, polygonal papule has a white lacelike pattern that is referred to as Wickham stria. B, Biopsy specimen demonstrating a bandlike infiltrate of lymphocytes at the dermoepidermal junction, hyperkeratosis, hypergranulosis and pointed rete ridges (sawtoothing) as a result of chronic basal cell layer injury.
Morphology. Lichen planus is characterized histologically by a dense, continuous infiltrate of lymphocytes along the dermoepidermal junction, a prototypic example of interface dermatitis (Fig. 25-27B). The lymphocytes are intimately associated with basal keratinocytes, which show degeneration, necrosis, and a resemblance in size and contour to more mature cells of the stratum spinosum (squamatization). A consequence of this destructive infiltration of lymphocytes is a redefinition of the normal, smoothly undulating configuration of the dermoepidermal interface to a more angulated zigzag contour (sawtoothing). Anucleate, necrotic basal cells may become incorporated into the inflamed papillary dermis, where they are referred to as colloid or Civatte bodies. Though characteristic of lichen planus, these bodies may be detected in any chronic dermatitis in which basal keratinocytes are injured and destroyed. Although the lesions bear some similarities to those in erythema multiforme, lichen planus shows changes of chronicity, namely, epidermal hyperplasia (or rarely atrophy) and thickening of the granular cell layer and stratum corneum (hypergranulosis and hyperkeratosis, respectively). Lichen planus preferentially affecting the epithelium of hair follicles is referred to as lichen planopilaris.
Pathogenesis.
The pathogenesis of lichen planus is not known. It is plausible that expression of altered antigens at the level of the basal cell layer and the dermoepidermal junction elicit a cell-mediated cytotoxic (CD8+/CLA+ T cell) immune response. In support of this notion, T lymphocyte infiltrates and hyperplasia of Langerhans cells are characteristic features of this disorder.92
Blistering (Bullous) Diseases
Although vesicles and bullae (blisters) occur in several unrelated conditions such as herpesvirus infection, spongiotic dermatitis, erythema multiforme, and thermal burns, there exists a group of disorders in which blisters are the primary and most distinctive features. These bullous diseases, as they are called, produce dramatic lesions and in some instances are fatal if untreated. Blisters in the various disorders occur at different levels within the skin (Fig. 25-28); histologic assessment is essential for accurate diagnosis and provides insight into the pathogenic mechanisms. Knowledge of the molecular structure of the intercellular and cell-to-matrix attachments that provide the skin with mechamical stability is helpful in understanding these diseases (Fig. 25-29).
FIGURE 25-28 Schematic representation of histologic levels of blister formation. A, In a subcorneal blister the stratum corneum forms the roof of the bulla (as in pemphigus foliaceus). B, In a suprabasal blister a portion of the epidermis, including the stratum corneum, forms the roof (as in pemphigus vulgaris). C, In a subepidermal blister the entire epidermis separates from the dermis (as in bullous pemphigoid).
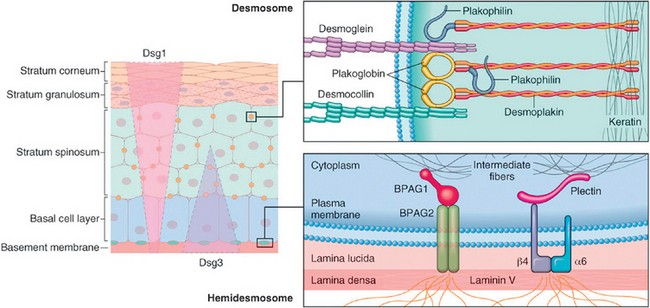
FIGURE 25-29 Squamous cell adhesion molecules. Knowledge of the proteins composing desmosomes and hemodesmosomes is key to understanding blistering disorders. Desmogleins 1 and 3 (Dsg1, Dsg3) are functionally interchangeable components of desmosomes, but have different distributions within the epidermis (left panel). In pemphigus vulgaris, autoantibodies against Dsg1 and Dsg3 cause blisters in the deep suprabasal epidermis, whereas in pemphigus foliaceus, the autoantibodies are against Dsg1 alone, leading to superficial, subcorneal blisters. In bullous phemphigoid, autoantibodies bind BPAG2, a component of the hemidesomes, leading to blister formation at the level of the lamina lucida of the basement membrane. Dermatitis herpetiformis is caused by lgA autoantibodies to the fibrils that anchor hemidesmosomes to the dermis. The various forms of epidermolysis bullosa are caused by genetic defects in genes encoding proteins that either form or stabilize desmosomes or hemidesmosomes. α6/β4, α6/β4 integrin.
INFLAMMATORY BLISTERING DISORDERS
Pemphigus
Pemphigus is a blistering disorder caused by autoantibodies that result in the dissolution of intercellular attachments within the epidermis and mucosal epithelium. Though rare, without treatment it may be life-threatening, and its pathobiology provides important insight into the molecular mechanisms of keratinocyte adhesion. The majority of individuals who develop pemphigus are in the fourth to sixth decades of life, and men and women are affected equally. There are multiple variants: (1) pemphigus vulgaris, (2) pemphigus vegetans, (3) pemphigus foliaceus, (4) pemphigus erythematosus, and (5) paraneoplastic pemphigus.93
Pemphigus vulgaris, by far the most common type (accounting for more than 80% of cases worldwide), involves the mucosa and skin, especially on the scalp, face, axilla, groin, trunk, and points of pressure. It may present as oral ulcers that may persist for months before skin involvement appears. Primary lesions are superficial vesicles and bullae that rupture easily, leaving shallow erosions covered with dried serum and crust (Fig. 25-30A). Pemphigus vegetans is a rare form that usually presents not with blisters but with large, moist, verrucous (wart-like), vegetating plaques studded with pustules on the groin, axillae, and flexural surfaces. Pemphigus foliaceus is a more benign form that is endemic in Brazil (where it is called fogo selvagem) and occurs sporadically in other geographic regions. Sites of predilection are the scalp, face, chest, and back, and the mucous membranes are only rarely affected. Bullae are so superficial that only zones of erythema and crusting, sites of previous blister rupture resulting in superficial erosions, are usually present on physical examination (Fig. 25-31A). Pemphigus erythematosus is considered to be a localized, less severe form of pemphigus foliaceus that may selectively involve the malar area of the face in a lupus erythematosus–like fashion. Paraneoplastic pemphigus occurs in association with various malignancies, most commonly non-Hodgkin lymphoma.
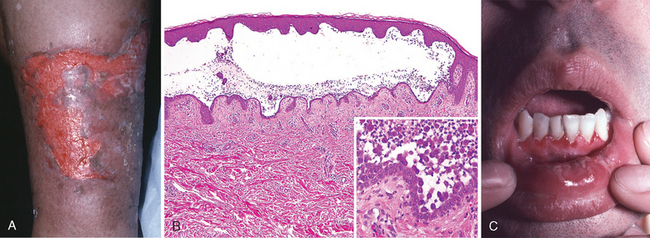
FIGURE 25-30 Pemphigus vulgaris. A, Eroded plaques are formed on rupture of confluent, thin-roofed bullae, here affecting axillary skin. B, Suprabasal acantholysis results in an intraepidermal blister in which rounded (acantholytic) epidermal cells are identified (inset). C, Ulcerated blisters in the oral mucosa are also common as seen here on the mucosal portion of the lip.
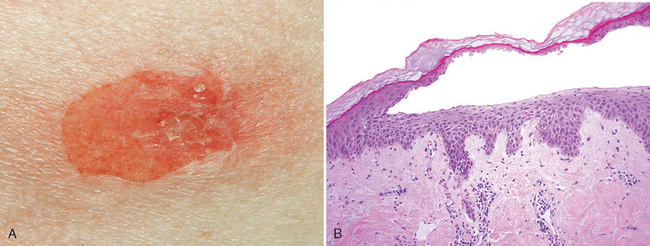
FIGURE 25-31 Pemphigus foliaceus. A, The delicate, superficial (subcorneal) blisters are much less erosive than seen in pemphigus vulgaris. B, Subcorneal separation of the epithelium is seen.
The mainstay of treatment is immunosuppressive agents, which decrease the titers of the pathogenic antibodies. In those with the most severe form of the disease, pemphigus vulgaris, this lowers but does not entirely prevent mortality.
Morphology. The common histologic denominator in all forms of pemphigus is acantholysis, the dissolution, or lysis, of the intercellular adhesions that connect squamous epithelial cells. Acantholytic cells dissociate from one another, lose their polyhedral shape and become rounded. In pemphigus vulgaris and pemphigus vegetans, acantholysis selectively involves the cells immediately above the basal cell layer. In the vegetans variant, there is also overlying epidermal hyperplasia. The suprabasal acantholytic blister that forms is characteristic of pemphigus vulgaris (see Fig. 25-30B). The single layer of intact basal cells that forms the blister base has been likened to a row of tombstones. In pemphigus foliaceus, a blister forms by similar mechanisms but, unlike the case with pemphigus vulgaris, selectively involves the superficial epidermis at the level of the stratum granulosum (Fig. 25-31B). Variable superficial dermal infiltration by lymphocytes, histiocytes, and eosinophils accompanies all forms of pemphigus.
Pathogenesis.
All forms of pemphigus are caused by IgG autoantibodies against desmogleins, and show linkage to specific HLA types.94 By direct immunofluorescence, lesional sites show a characteristic net-like pattern of intercellular IgG deposits. IgG is usually seen at all levels of the epithelium in pemphigus vulgaris, but tends to be more superficial in pemphigus foliaceus (Fig. 25-32). The distribution of desmoglein 1 and 3 in the epidermis and the presence of autoantibodies to one or both proteins appear to explain the position and severity of the blisters (Fig. 25-29). The antibodies seem to function primarily by directly disrupting the intercellular adhesive function of the desmosomes and may activate intercellular proteases as well.95 Paraneoplastic pemphigus most often arises in the setting of lymphoid neoplasms, and is caused by autoantibodies that may recognize desmogleins of other proteins involved in intercellular adhesion.96
FIGURE 25-32 Direct immunofluorescence of pemphigus. A, In pemphigus vulgaris there is deposition of immunoglobulin along the plasma membranes of epidermal keratinocytes in a reticular or fishnet-like pattern. Also note the early suprabasal separation due to loss of cell-to-cell adhesion (acantholysis). B, In pemphigus foliaceus the immunoglobulin deposits are more superficial.
Bullous Pemphigoid
Generally affecting elderly individuals, bullous pemphigoid shows a wide range of clinical presentations. It may involve only the skin, either locally or generally, or the skin and mucosal surfaces. Clinically, lesions are tense bullae, filled with clear fluid, on normal or erythematous skin (Fig. 25-33A). Lesions are usually up to 2 cm in diameter but occasionally may reach 4 to 8 cm in diameter. The bullae do not rupture as easily as do the blisters seen in pemphigus and, if they are not secondarily infected, heal without scarring. Sites of involvement include the inner aspects of the thighs, flexor surfaces of the forearms, axillae, groin, and lower abdomen. Oral lesions are present in 10% to 15% of affected individuals, usually appearing after the cutaneous lesions.97 Some patients may present with urticarial plaques, with extreme associated pruritus.
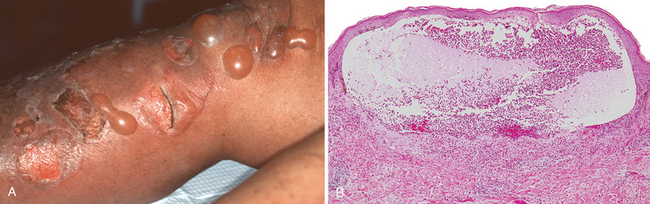
FIGURE 25-33 Bullous pemphigoid. A, Clinical bullae result from basal cell layer vacuolization, producing tense, intact subepidermal blisters that are difficult to rupture given the roof formed by the full thickness of the epidermis. Ulceration results upon rupture as the blisters are subepidermal. B, Histopathology shows an intact blister with eosinophils, as well as lymphocytes and occasional neutrophils, that may be intimately associated with basal cell layer destruction and the creation of the subepidermal cleft.
Morphology. The separation of bullous pemphigoid from pemphigus is based on the identification of subepidermal, nonacantholytic blisters. Early lesions show a superficial and sometimes deep perivascular infiltrate of lymphocytes and variable numbers of eosinophils, occasional neutrophils, superficial dermal edema, and associated basal cell layer vacuolization (Fig. 25-33B). Eosinophils showing degranulation are typically detected directly beneath the epidermal basal cell layer. The vacuolated basal cell layer eventually gives rise to a fluid-filled blister.
Pathogenesis.
Bullous pemphigoid features linear deposition of immunoglobulin and complement in the basement membrane zone (Fig. 25-34A). Reactivity also occurs in the basal cell–basement membrane attachment plaques (hemidesmosomes), where most of the bullous pemphigoid antigen (BPAG) is located. This protein is involved normally in dermoepidermal bonding. There are two forms of the antigen recognized by auto-antibodies—BPAG1, a 230-kD plakin protein, and BPAG2, a 180-kD protein also known as collagen type XVIII. Both BPAG1 and BPAG2 are recognized as normal constituents of the hemidesmosomes that bind basal cells at the dermoepidermal junction (Figs. 25-34B and 25-29). Only antibodies to BPAG2 are proven to cause blistering.98 Generation of auto-antibodies to this hemidesmosome component results in continuous and linear deposition of IgG along the basement membrane and the fixation of complement with subsequent tissue injury by means of locally recruited neutrophils and eosinophils.
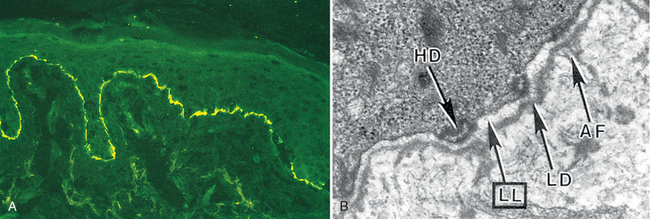
FIGURE 25-34 A, Linear deposition of complement along the dermoepidermal junction in bullous pemphigoid; the pattern has been likened to ribbon candy. B, Bullous pemphigoid antigen is located in the lowermost portion of the basal cell cytoplasm in association with hemidesmosomes (HD), with blister formation affecting the lamina lucida (LL) of the basement membrane zone. AF, anchoring fibrils; LD, lamina densa. (See also Fig. 25-31.)
DERMATITIS HERPETIFORMIS
Dermatitis herpetiformis is a rare disorder characterized by urticaria and grouped vesicles. The disease affects predominantly males, often in the third and fourth decades. In some cases it occurs in association with intestinal celiac disease and responds to a gluten-free diet.
Pathogenesis.
The association of dermatitis herpetiformis with celiac disease provides a clue to its pathogenesis. Genetically predisposed individuals develop lgA antibodies to dietary gluten (derived from the wheat protein gliadin). The antibodies cross-react with reticulin, a component of the anchoring fibrils that tether the epidermal basement membrane to the superficial dermis. The resultant injury and inflammation produce a subepidermal blister. Some people with dermatitis herpetiformis and gluten-sensitive enteropathy respond to a gluten-free diet.
Morphology. As an early event, fibrin and neutrophils accumulate selectively at the tips of dermal papillae, forming small microabscesses (Fig. 25-35A). The basal cells overlying these microabscesses show vacuolization and focal dermoepidermal separation that ultimately coalesce to form a true subepidermal blister. By direct immunofluorescence, dermatitis herpetiformis shows discontinuous, granular deposits of lgA selectively localized in the tips of dermal papillae (Fig. 25-35B).
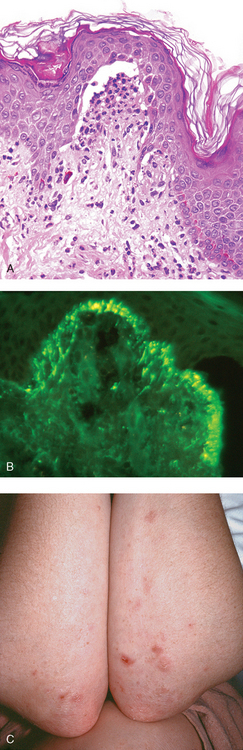
FIGURE 25-35 Dermatitis herpetiformis. A, The blisters are associated with basal cell layer injury initially caused by accumulation of neutrophils (microabscesses) at the tips of dermal papillae. B, Selective deposition of IgA autoantibody at the tips of dermal papillae is characteristic. C, Lesions consist of intact and eroded (usually scratched) erythematous blisters, often grouped (seen here on elbows and arms).
(B, Courtesy of Dr. Victor G. Prieto, Houston, Texas.)
Clinical Features.
The urticarial plaques and vesicles of dermatitis herpetiformis are extremely pruritic. The lesions are bilateral, symmetric and grouped, involving preferentially the extensor surfaces, elbows, knees, upper back, and buttocks (Fig. 25-35C).
NONINFLAMMATORY BLISTERING DISORDERS
Epidermolysis Bullosa and Porphyria
Some primary disorders characterized by vesicles and bullae are mediated by non-inflammatory mechanisms. Two such diseases are epidermolysis bullosa and porphyria.
Epidermolysis bullosa constitutes a group of disorders caused by inherited defects in structural proteins that lend mechanical stability to the skin. The common feature is a proclivity to form blisters at sites of pressure, rubbing, or trauma, at or soon after birth.99 In the simplex type, defects of the basal cell layer of the epidermis result from mutations in the genes encoding keratins 14 or 5. These two proteins normally pair with one another to make a functional keratin fiber, thus explaining the similar phenotype resulting from mutation of either gene. In the junctional type, blisters occur in otherwise histologically normal skin at precisely the level of the lamina lucida (Figs. 25-36 and 25-29). In the scarring dystrophic types, blisters develop beneath the lamina densa, in association with rudimentary or defective anchoring fibrils. Dystrophic epidermolysis bullosa is an inherited disease resulting from mutations in the COL7A1 gene that encodes type VII collagen (Chapter 3). Squamous cell carcinoma can sometime arise in these chronic blisters.100 The histologic changes are so subtle that electron microscopy may be required to differentiate among these types in clinically ambiguous settings. Overall, approximately ten genes encoding structural proteins at the dermoepidermal junction have been implicated in the various forms of this disease.101 One form, termed non-Herlitz junctional epidermolysis bullosa is caused by a genetic defect (usually a point mutation) in the LAMB3 gene that encodes laminin Vβ3. Successful gene therapy has been carried out in one individual with this disease by inserting a normal LAMB3 gene into keratinocytes.
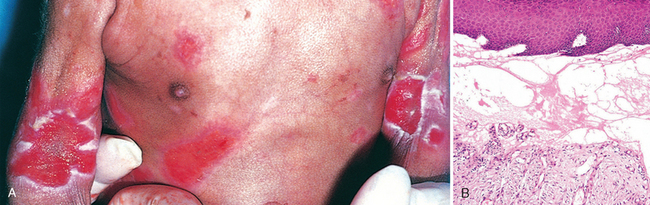
FIGURE 25-36 Epidermolysis bullosa. A, Junctional epidermolysis bullosa showing typical erosions in flexural creases. B, A noninflammatory subepidermal blister has formed at the level of the lamina lucida.
Porphyria refers to a group of uncommon inborn or acquired disturbances of porphyrin metabolism. Porphyrins are pigments normally present in hemoglobin, myoglobin, and cytochromes. The classification of porphyrias is based on both clinical and biochemical features. The five major types are (1) congenital erythropoietic porphyria, (2) erythrohepatic protoporphyria, (3) acute intermittent porphyria, (4) porphyria cutanea tarda, and (5) mixed porphyria. Cutaneous manifestations consist of urticaria and vesicles that heal with scarring and that are exacerbated by exposure to sunlight. The primary alterations by light microscopy are a subepidermal vesicle (Fig. 25-37) with associated marked thickening of the walls of superficial dermal vessels. The pathogenesis of these alterations is not well understood, although serum proteins, including immunoglobulins, typically form glassy deposits in the walls of superficial dermal microvessels.
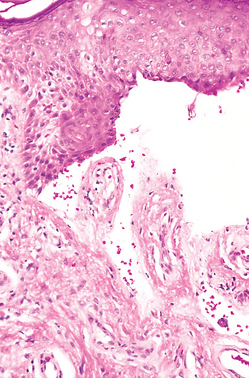
Disorders of Epidermal Appendages
ACNE VULGARIS
Virtually universal in the middle to late teenage years, acne vulgaris affects both males and females, although males tend to have more severe disease. Acne is seen in all races but is usually milder in people of Asian descent. Acne vulgaris in adolescents is believed to occur as a result of physiologic hormonal variations and alterations in hair follicles, particularly the sebaceous gland. The clinical features of acne may be induced or exacerbated by drugs (corticosteroids, adrenocorticotropic hormone, testosterone, gonadotropins, contraceptives, trimethadione, iodides, and bromides), occupational contactants (cutting oils, chlorinated hydrocarbons, and coal tars), and occlusive conditions such as heavy clothing, cosmetics, and tropical climates. Some families seem to be particularly affected by acne, suggesting a hereditary component.
Acne is divided into noninflammatory and inflammatory types, although the types may coexist. The former consists of open and closed comedones. Open comedones are small follicular papules containing a central black keratin plug. This color is the result of oxidation of melanin pigment (not dirt). Closed comedones are follicular papules without a visible central plug. Because the keratin plug is trapped beneath the epidermal surface, these lesions are potential sources of follicular rupture and inflammation. Inflammatory acne is characterized by erythematous papules, nodules, and pustules (Fig. 25-38A). Severe variants (e.g., acne conglobata) result in sinus tract formation and physical scarring.
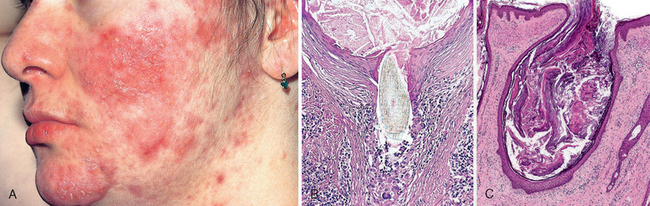
FIGURE 25-38 Acne. A, Inflammatory acne associated with erythematous papules and pustules. B, A hair shaft pierces the follicular epithelium, eliciting inflammation and fibrosis. C, Open comedone.
Morphology. Four key components contribute to the development of acne: (1) changes in keratinization of the lower portion of the follicular infundibulum with the development of a keratin plug blocking outflow of sebum to the skin surface; (2) hypertrophy of sebaceous glands with puberty or increased activity due to hormonal stimulation; (3) lipase-synthesizing bacteria (Propionibacterium acnes) colonizing the upper and midportion of the hair follicle, converting lipids within sebum to pro-inflammatory fatty acids; and (4) inflammation of the follicle associated with release of cytotoxic and chemotactic factors.103 Depending on the stage of the disease, open or closed comedones, papules, pustules, or deep inflammatory nodules may develop. Open comedones have large, patulous orifices, whereas those of closed comedones are identifiable only microscopically (Fig. 25-38B, C). Variable lymphohistiocytic infiltrates are present in and around affected follicles, and extensive acute and chronic inflammation accompanies follicular rupture. Dermal abscesses may form in association with rupture (see Fig. 25-38C), and gradual resolution, often with scarring, ensues.
Pathogenesis.
The pathogenesis of acne is incompletely understood. Endocrine factors have been implicated (especially androgens) because, historically, young castrated males generally do not develop the condition (a questionable tradeoff). It has been postulated that bacterial lipases of Propionibacterium acnes break down sebaceous oils, liberating highly irritating fatty acids and resulting in the earliest inflammatory phases of acne.104 Inhibition of lipase production is a rationale for administration of antibiotics to individuals with inflammatory acne. The synthetic vitamin A derivative 13-cisretinoic acid (isotretinoin) has brought about remarkable clinical improvement in some cases of severe acne through its strong anti-sebaceous action.105,106
ROSACEA
Rosacea is a common disease of middle age and beyond, affecting up to 3% of the US population, with a predilection for females. Four stages are recognized: (1) flushing episodes (pre-rosacea), (2) persistent erythema and telangiectasia, (3) pustules and papules, and (4) rhinophyma—permanent thickening of the nasal skin by confluent erythemaous papules and follicular prominence.107
Morphology. Rosacea is characterized by a nonspecific perifollicular infiltrate composed of lymphocytes surrounded by dermal edema and telangiectasia. In the pustular phase neutrophils may colonize the follicles, and follicular rupture may cause a granulomatous dermal response. The development of rhinophyma is associated with hypertrophy of sebaceous glands and follicular plugging by keratotic debris.
Pathogenesis.
Individuals with rosacea have high cutaneous levels of the endogenous antimicrobial peptide cathelicidin, an important mediator of the innate immune response. The cathelicidin peptides present are qualitatively distinct from those seen in individuals without rosacea as a result of alternative processing by serine proteases such as stratum corneum tryptic enzyme.108 While more study is needed, it seems that inappropriate activation of the innate immune system may play a role in this disorder.
Panniculitis
ERYTHEMA NODOSUM AND ERYTHEMA INDURATUM
Panniculitis is an inflammatory reaction in the subcutaneous adipose tissue that may preferentially affect (1) the connective tissue septa separating lobules of fat, or (2) the lobules of fat themselves. Panniculitis often involves the lower legs and usually has a subacute to chronic course. Erythema nodosum is the most common form and usually has an acute presentation. Its occurrence is often associated with infections (β-hemolytic streptococcal infection, tuberculosis and, less commonly, coccidioidomycosis, histoplasmosis, and leprosy), drug administration (sulfonamides, oral contraceptives), sarcoidosis, inflammatory bowel disease, and certain malignant neoplasms, but many times a cause cannot be identified.109
Erythema nodosum presents as poorly defined, exquisitely tender, erythematous plaques and nodules that may be more readily palpated than seen. Fever and malaise may accompany the cutaneous signs. Over the course of weeks, lesions usually flatten and become bruiselike, leaving no residual clinical scars, while new lesions develop. Biopsy of a deep wedge of tissue to generously sample the subcutis is usually required for histologic diagnosis.
Erythema induratum is an uncommon type of panniculitis that affects primarily adolescents and menopausal women. Although the cause is not known, most observers regard this as a primary vasculitis affecting deep vessels supplying lobules of the subcutis, with subsequent necrosis and inflammation within the fat. Erythema induratum presents as an erythematous, slightly tender nodule that usually goes on to ulcerate. Originally considered a hypersensitivity response to tuberculosis, erythema induratum today most commonly occurs without an associated underlying disease.
Morphology. The histopathology of erythema nodosum is distinctive. In early lesions, widening of the connective tissue septa is due to edema, fibrin exudation, and neutrophilic infiltration. Later, infiltration by lymphocytes, histiocytes, multinucleated giant cells, and occasional eosinophils is associated with septal fibrosis. Vasculitis is not present. In erythema induratum, on the other hand, granulomatous inflammation and zones of caseous necrosis involve the fat lobule. Early lesions show necrotizing vasculitis affecting small- to medium-sized arteries and veins in the deep dermis and subcutis.
Erythema nodosum and erythema induratum are but two of the many types of panniculitis. Weber-Christian disease (relapsing febrile nodular panniculitis) is a rare form of lobular, nonvasculitic panniculitis seen in children and adults. It is marked by crops of erythematous plaques or nodules, predominantly on the lower extremities, created by deep-seated foci of inflammation with aggregates of foamy histiocytes admixed with lymphocytes, neutrophils, and giant cells. Factitial panniculitis is a form of secondary panniculitis caused by self-inflicted trauma or injection of foreign or toxic substances. Rarely, T cell lymphomas (panniculitic T cell lymphoma) home to fat lobules, producing fat necrosis and superimposed inflammation that mimics panniculitis. Finally, disorders such as lupus erythematosus may occasionally have deep inflammatory components with associated panniculitis.110 The etiologies for all of these except the self-inflicted form and the rare T cell lymphomas are not understood.
Infection
The skin frequently succumbs to the attack of microorganisms, parasites, and insects. We have already discussed the possible role of bacteria in the pathogenesis of common acne, and the dermatoses resulting from viruses are too numerous to list. In the setting of the immunocompromised individual, ordinarily trivial cutaneous infections may become life-threatening. Many disorders, such as herpes simplex and herpes zoster, the viral exanthems, deep fungal infections, and immune reactions in skin provoked by infectious agents, are discussed in Chapter 8. Here we cover a representative sampling of common infections whose primary clinical manifestations are in the skin.
VERRUCAE (WARTS)
Verrucae are common lesions of children and adolescents, although they may be encountered at any age. They are caused by human papillomaviruses. Transmission of disease usually involves direct contact between individuals or auto-inoculation. Verrucae are generally self-limited, regressing spontaneously within 6 months to 2 years.
The classification of verrucae is based largely on appearance and location.111 Verruca vulgaris is the most common type of wart. The lesions of verruca vulgaris occur anywhere but most frequently on the hands, particularly on the dorsal surfaces and periungual areas, where they appear as gray-white to tan, flat to convex, 0.1- to 1-cm papules with a rough, pebble-like surface (Fig. 25-39A). Verruca plana, or flat wart, is common on the face or the dorsal surfaces of the hands. The warts are slightly elevated, flat, smooth, tan papules that are generally smaller than verruca vulgaris. Verruca plantaris and verruca palmaris occur on the soles and palms, respectively. Rough, scaly lesions may reach 1 to 2 cm in diameter, coalesce, and be confused with ordinary calluses. Condyloma acuminatum (venereal wart) occurs on the penis, female genitalia, urethra, perianal areas, and rectum. Venereal warts appear as soft, tan, cauliflower-like masses that occasionally reach many centimeters in diameter.
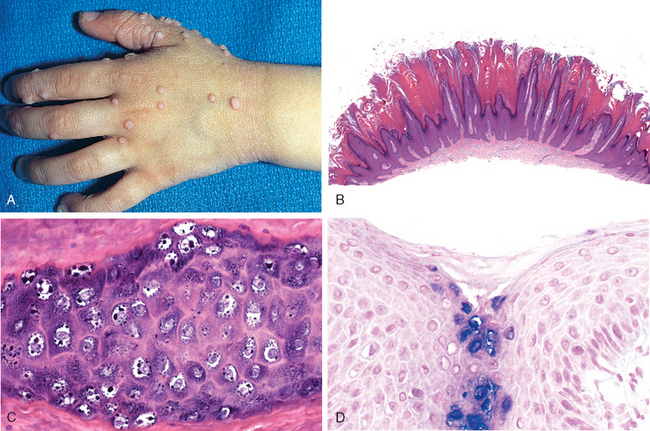
FIGURE 25-39 Verruca vulgaris. A, Multiple papules with rough pebble-like surfaces. B (low power) and C, (high power) histology of the lesions showing papillomatous epidermal hyperplasia and cytopathic alterations that include nuclear pallor and prominent keratohyaline granules. D, In situ hybridization demonstrating viral DNA within epidermal cells.
Morphology. Histologic features common to verrucae include epidermal hyperplasia that is often undulant in character, termed verrucous or papillomatous epidermal hyperplasia (Fig. 25-39B); and cytoplasmic vacuolization (koilocytosis) involving the more superficial epidermal layers, producing haloes of pallor surrounding infected nuclei. Electron microscopy of these zones reveals numerous viral particles within nuclei. Infected cells may also demonstrate prominent and apparently condensed keratohyaline granules and jagged eosinophilic intracytoplasmic keratin aggregates as a result of viral cytopathic effects (Fig. 25-39C). These cellular alterations are not as prominent in condylomas; hence, their diagnosis is based primarily on hyperplastic papillary architecture containing wedge-shaped zones of koilocytosis.
Pathogenesis.
More than 150 types of papillomavirus have been identified, many of them capable of producing warts in humans. The clinical variants of warts are often associated with distinct HPV subtypes. For example, anogenital warts are caused predominantly by HPV types 6 and 11. In contrast, there is a tendency for lesions induced by HPV type 16 to show some degree of dysplasia.112 HPV type 16 has also been associated with in situ squamous cell carcinoma of the genitalia and with bowenoid papulosis (genital lesions of young adults with the histologic appearance of carcinoma in situ, but which usually regress spontaneously).111 The relationship of HPV subtypes 5 and 8 to squamous cell carcinomas, particularly in individuals affected by the rare condition epidermodysplasia verruciformis, was mentioned earlier. These patients develop multiple flat warts that contain HPV genomes, some of which progress to carcinoma. Viral typing can be accomplished by either in situ hybridization (Fig. 25-39D) or polymerase chain reaction.
MOLLUSCUM CONTAGIOSUM
Molluscum contagiosum is a common, self-limited viral disease of the skin caused by a poxvirus. The virus is characteristically brick shaped, has a dumbbell-shaped DNA core, and measures 300 nm in maximal dimension, and thus represents the largest pathogenic poxvirus in humans and one of the largest viruses in nature. Infection is usually spread by direct contact, particularly among children and young adults.
Clinically, multiple lesions may occur on the skin and mucous membranes, with a predilection for the trunk and anogenital areas. Individual lesions are firm, often pruritic, pink to skin-colored umbilicated papules generally ranging in diameter from 0.2 to 0.4 cm. Rarely, “giant” forms occur measuring up to 2 cm in diameter. A curd-like material can be expressed from the central umbilication. Smearing this material onto a glass slide and staining with Giemsa reagent often shows diagnostic molluscum bodies.
Morphology. On microscopic examination, lesions show cuplike verrucous epidermal hyperplasia. The diagnostically specific structure is the molluscum body, which occurs as a large (up to 35 μm), ellipsoid, homogeneous, cytoplasmic inclusion in cells of the stratum granulosum and the stratum corneum (Fig. 25-40). In the H&E stain, these inclusions are eosinophilic in the blue-purple stratum granulosum and acquire a pale blue hue in the red stratum corneum. Numerous virions are present within molluscum bodies.
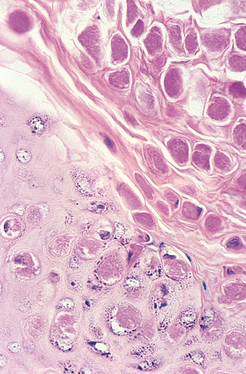
IMPETIGO
Impetigo is a common superficial bacterial infection of skin. It is highly contagious and is frequently seen in otherwise healthy children as well as occasionally in adults in poor health. Two forms exist, classically referred to as impetigo contagiosa and impetigo bullosa; they differ from each other simply by the size of the pustules. Over the past decade a remarkable shift in etiology has been observed. Whereas in the past impetigo contagiosa was almost exclusively caused by group A β-hemolytic streptococci and impetigo bullosa by Staphylococcus aureus, both are now usually caused by Staphylococcus aureus.114
The infection usually involves exposed skin, particularly that of the face and hands. It presents as an erythematous macule, but multiple small pustules rapidly supervene. As pustules break, shallow erosions form, covered with drying serum, giving the characteristic clinical appearance of honey-colored crust. If the crust is not removed, new lesions form about the periphery and extensive epidermal damage may ensue. A bullous form of impetigo mainly occurs in children.
Morphology. The characteristic microscopic feature of impetigo is accumulation of neutrophils beneath the stratum corneum, often producing a subcorneal pustule. Special stains reveal the presence of bacteria in these foci. Nonspecific, reactive epidermal alterations and superficial dermal inflammation accompany these findings. Rupture of pustules results in superficial layering of serum, neutrophils, and cellular debris to form the characteristic crust. Blistering lesions may show accumulation of fluid and neutrophils beneath the stratum corneum.
Pathogenesis.
Bacterial species in the epidermis evoke an innate immune response that tends to be destructive within the epidermis leading to local serous exudate with formation of a scale crust (scab). The pathogenesis of blister formation in impetigo is related to bacterial production of a toxin that specifically cleaves desmoglein 1, the protein responsible for cell-to-cell adhesion within the uppermost epidermal layers.115 Recall that in pemphigus foliaceus, which has a similar plane of blister formation, desmoglein 1 is compromised not by a toxin but by an autoantibody (see Fig. 25-29). Because there is virtually no involvement of the dermis, once the bacteria are eliminated the lesions heal without scarring.
SUPERFICIAL FUNGAL INFECTIONS
As opposed to deep fungal infections of the skin, where the dermis or subcutis is primarily involved, superficial fungal infections of the skin are confined to the stratum corneum, and are caused primarily by dermatophytes. These organisms grow in the soil and on animals and produce a number of diverse and characteristic clinical lesions.116
Tinea capitis usually occurs in children and is only rarely seen in infants and adults. It is a dermatophytosis of the scalp characterized by asymptomatic, often hairless patches of skin associated with mild erythema, crust formation, and scaling. Tinea barbae is a dermatophyte infection of the beard area that affects adult men; it is a relatively uncommon disorder. Tinea corporis, on the other hand, is a common superficial fungal infection of the body surface that affects persons of all ages, but particularly children. Predisposing factors include excessive heat and humidity, exposure to infected animals, and chronic dermatophytosis of the feet or nails. The most common type of tinea corporis is an expanding, round, slightly erythematous plaque with an elevated scaling border (Fig. 25-41A). Tinea cruris occurs most frequently in the inguinal areas of obese men during warm weather. Heat, friction, and maceration all predispose to its development. The infection usually first appears on the upper inner thighs, with gradual extension of moist, red patches that have raised, scaling borders. Tinea pedis (athlete’s foot) affects 30% to 40% of the population at some time in their lives. There is diffuse erythema and scaling, often initially localized to the web spaces. Most of the inflammatory tissue reaction, however, has recently been shown to be the result of bacterial superinfection and not directly related to the primary dermatophytosis.117 Spread to or primary infection of the nails is referred to as onychomycosis. This produces discoloration, thickening, and deformity of the nail plate. Tinea versicolor usually occurs on the upper trunk and is highly distinctive in appearance. Caused by Malassezia furfur, a yeast rather than a dermatophyte, the lesions consist of groups of macules of all sizes, lighter or darker than surrounding skin, with a fine peripheral scale.87
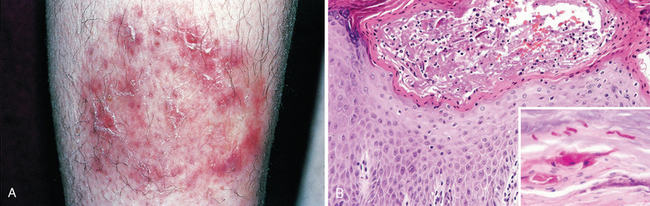
FIGURE 25-41 Tinea. A, Characteristic plaque of tinea corporis. Routine histology (B) shows a mild eczematous (spongiotic) dermatitis and focal neutrophilic abscesses. A periodic acid–Schiff stain (inset) reveals deep red hyphae within the stratum corneum.
Morphology. The histologic features of dermatophytoses are variable, depending on the properties of the organism, the host response, and the degree of bacterial superinfection. There may be mild eczematous dermatitis associated with intraepidermal neutrophils (Fig. 25-41B). Fungal cell walls, rich in mucopolysaccharides, stain bright pink to red with periodic acid–Schiff stain. They are present in the anucleate cornified layer of lesional skin, hair, or nails (Fig. 25-41B, inset). Culture of material scraped from these areas will usually permit the identification of the offending species.
1 Virchow R. Cellular Pathology. London: John Churchill, 1860;33.
2 Kupper TS, Fuhlbrigge RC. Immune surveillance in the skin: mechanisms and clinical consequences. Nat Rev Immunol. 2004;4:211.
3 Murphy GF. The secret of “NIN,” a novel neural immunological network potentially integral to immunologic function in human skin. In: Nickoloff BJ, editor. Dermal Immune System. Boca Raton, FL: CRC Press; 1993:227.
4 Johnson KO. The roles and functions of cutaneous mechanoreceptors. Curr Opin Neurobiol. 2001;11:455.
5 Fuchs E. Scratching the surface of skin development. Nature. 2007;445:834.
6 Pollock PM, et al. High frequency of BRAF mutations in nevi. Nat Genet. 2003;33:19.
7 Michaloglou C, et al. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. 2005;436:720.
8 Norris W. A case of fungoid disease. Edinburgh Med Surg J. 1820;16:562.
9 Clark WHJr., et al. Origin of familial malignant melanomas from heritable melanocytic lesions. “The B-K mole syndrome.”. Arch Dermatol. 1978;114:732.
10 Tsai KY, Tsao H. The genetics of skin cancer. Am J Med Genet C Semin Med Genet. 2004;131C:82.
11 Greene MH, et al. High risk of malignant melanoma in melanoma-prone families with dysplastic nevi. Ann Intern Med. 1985;102:458.
12 Clark WHJr., et al. A study of tumor progression: the precursor lesions of superficial spreading and nodular melanoma. Hum Pathol. 1984;15:1147.
13 Kim JC, Murphy GF. Dysplastic melanocytic nevi and prognostically indeterminate nevomelanomatoid proliferations. Clin Lab Med. 2000;20:691.
14 Geller AC, et al. Screening, early detection, and trends for melanoma: current status (2000–2006) and future directions. J Am Acad Dermatol. 2007;57:555.
15 Elder DE. Melanocytic tumors of the skin. In: Atlas of Tumor Pathology, Third Series, Fascicle 1. Washington, DC: Armed Forces Institute of Pathology; 1991:183.
16 Breslow A. Prognosis in cutaneous melanoma: tumor thickness as a guide to treatment. Pathol Annu. 1980;15:1.
17 Kruper LL, et al. Predicting sentinel node status in AJCC stage I/II primary cutaneous melanoma. Cancer. 2006;107:2436.
18 Barnhill RL, et al. The importance of mitotic rate as a prognostic factor for localized cutaneous melanoma. J Cutan Pathol. 2005;32:268.
19 Elder DE, Xu X. The approach to the patient with a difficult melanocytic lesion. Pathology. 2004;36:428.
20 Clark WHJr., et al. Model predicting survival in stage I melanoma based on tumor progression. J Natl Cancer Inst. 1989;81:1893.
21 Balch CM, et al. Prognostic factors analysis of 17,600 melanoma patients: validation of the American Joint Committee on Cancer melanoma staging system. J Clin Oncol. 2001;19:3622.
22 Balch CM, et al. The new melanoma staging system. Semin Cutan Med Surg. 2003;22:42.
23 Rousseau DLJr., Gershenwald JE. The new staging system for cutaneous melanoma in the era of lymphatic mapping. Semin Oncol. 2004;31:415.
24 Berwick M, et al. Sun exposure and mortality from melanoma. J Natl Cancer Inst. 2005;97:195.
25 Gudbjartsson DF, et al. ASIP and TYR pigmentation variants associated with cutaneous melanoma and basal cell carcinoma. Nat Genet. 2008;40:886.
26 Chin L. The genetics of malignant melanoma: lessons from mouse and man. Nat Rev Cancer. 2003;3:559.
27 Peters G. Tumor suppression for ARFicionados: the relative contributions of p16/INK4a and p14/ARF in melanoma. J Natl Cancer Inst. 2008;100:757.
28 Curtin JA, et al. Somatic activation of KIT in distinct subtypes of melanoma. J Clin Oncol. 2006;24:4340.
29 Curtin JA, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med. 2005;353:2135.
30 Ibrahim N, Haluska FG. Molecular pathogenesis of cutaneous melanocytic neoplasms. Annu Rev Pathol Mech Dis. 2009;4:551.
31 Goel VK, et al. Examinations of mutations in BRAF, NRAS, and PTEN in primary cutaneous melanoma. J Invest Dermatol. 2006;126:154.
32 Chudnovsky Y, et al. Melanoma genetics and the development of rational therapeutics. J Clin Invest. 2005;115:813.
33 Hunder NN, et al. Treatment of metastatic melanoma with autologous CD4+ T Cells against NY-ESO-1. N Engl J Med. 2008;358:2698.
34 Hafner C, et al. High frequency of FGFR3 mutations in adenoid seborrheic keratoses. J Invest Dermatol. 2006;126:2404.
35 Ellis DL, et al. Melanoma, growth factors, acanthosis nigricans, the sign of Leser-Trelat, and multiple acrochordons. A possible role for alpha-transforming growth factor in cutaneous paraneoplastic syndromes. N Engl J Med. 1987;317:1582.
36 Logie A, et al. Activating mutations of the tyrosine kinase receptor FGFR3 are associated with benign skin tumors in mice and humans. Hum Mol Genet. 2005;14:1153.
37 Torley D, et al. Genes, growth factors and acanthosis nigricans. Br J Dermatol. 2002;147:1096.
38 Hafner C, et al. FGFR3 mutations in benign skin tumors. Cell Cycle. 2006;5:2723.
39 Leter EM, et al. Birt-Hogg-Dube syndrome: Clinical and genetic studies of 20 families. J Invest Dermatol. 2007;128:45.
40 Covello SP, et al. Keratin 17 mutations cause either steatocystoma multiplex or pachyonychia congenita type 2. Br J Dermatol. 1998;139:475.
41 McLean WH, et al. Keratin 16 and keratin 17 mutations cause pachyonychia congenita. Nat Genet. 1995;9:273.
42 Liaw D, et al. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet. 1997;16:64.
43 Bignell GR, et al. Identification of the familial cylindromatosis tumour-suppressor gene. Nat Genet. 2000;25:160.
44 Trompouki E, et al. CYLD is a deubiquitinating enzyme that negatively regulates NF-kappaB activation by TNFR family members. Nature. 2003;424:793.
45 Young AL, et al. CYLD mutations underlie Brooke-Spiegler, familial cylindromatosis, and multiple familial trichoepithelioma syndromes. Clin Genet. 2006;70:246.
46 Barana D, et al. Spectrum of genetic alterations in Muir-Torre syndrome is the same as in HNPCC. Am J Med Genet A. 2004;125:318.
47 Takeda H, et al. Human sebaceous tumors harbor inactivating mutations in LEF1. Nat Med. 2006;12:395.
48 Chan EF, et al. A common human skin tumour is caused by activating mutations in β-catenin. Nat Genet. 1999;21:410.
49 Owens DM, Watt FM. Contribution of stem cells and differentiated cells to epidermal tumours. Nat Rev Cancer. 2003;3:444.
50 Jorizzo JL. Current and novel treatment options for actinic keratosis. J Cutan Med Surg. 2004;8(Suppl 3):13.
51 Alam M, Ratner D. Cutaneous squamous-cell carcinoma. N Engl J Med. 2001;344:975.
52 Harwood CA, Proby CM. Human papillomaviruses and non-melanoma skin cancer. Curr Opin Infect Dis. 2002;15:101.
53 Ramoz N, et al. Mutations in two adjacent novel genes are associated with epidermodysplasia verruciformis. Nat Genet. 2002;32:579.
54 Beissert S, Schwarz T. Mechanisms involved in ultraviolet light-induced immunosuppression. J Investig Dermatol Symp Proc. 1999;4:61.
55 Khavari PA. Modelling cancer in human skin tissue. Nat Rev Cancer. 2006;6:270.
56 Rubin AI, et al. Basal-cell carcinoma. N Engl J Med. 2005;353:2262.
57 Gorlin RJ, Goltz RW. Multiple nevoid basal-cell epithelioma, jaw cysts and bifid rib. A syndrome. N Engl J Med. 1960;262:908.
58 Mirowski GW, et al. Nevoid basal cell carcinoma syndrome. J Am Acad Dermatol. 2000;43:1092.
59 Hahn H, et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell. 1996;85:841.
60 Riobo NA, Manning DR. Pathways of signal transduction employed by vertebrate Hedgehogs. Biochem J. 2007;403:369.
61 Fan H, et al. Induction of basal cell carcinoma features in transgenic human skin expressing Sonic Hedgehog. Nat Med. 1997;3:788.
62 Nilsson M, et al. Induction of basal cell carcinomas and trichoepitheliomas in mice overexpressing GLI-1. Proc Natl Acad Sci U S A. 2000;97:3438.
63 Bale AE. The nevoid basal cell carcinoma syndrome: genetics and mechanism of carcinogenesis. Cancer Invest. 1997;15:180.
64 Kim MY, et al. Mutations of the p53 and PTCH gene in basal cell carcinomas: UV mutation signature and strand bias. J Dermatol Sci. 2002;29:1.
65 D’Errico M, et al. UV mutation signature in tumor suppressor genes involved in skin carcinogenesis in xeroderma pigmentosum patients. Oncogene. 2000;19:463.
66 Calonje E. Is cutaneous benign fibrous histiocytoma (dermatofibroma) a reactive inflammatory process or a neoplasm? Histopathology. 2000;37:278.
67 Simon MP, et al. Deregulation of the platelet-derived growth factor B-chain gene via fusion with collagen gene COL1A1 in dermatofibrosarcoma protuberans and giant-cell fibroblastoma. Nat Genet. 1997;15:95.
68 Patel KU, et al. Dermatofibrosarcoma protuberans COL1A1-PDGFB fusion is identified in virtually all dermatofibrosarcoma protuberans cases when investigated by newly developed multiplex reverse transcription polymerase chain reaction and fluorescence in situ hybridization assays. Hum Pathol. 2008;39:184.
69 Mitelman F, et al. The impact of translocations and gene fusions on cancer causation. Nat Rev Cancer. 2007;7:233.
70 Rubin BP, et al. Molecular targeting of platelet-derived growth factor B by imatinib mesylate in a patient with metastatic dermatofibrosarcoma protuberans. J Clin Oncol. 2002;20:3586.
71 Willemze R, Meijer CJ. Classification of cutaneous T-cell lymphoma: from Alibert to WHO-EORTC. J Cutan Pathol. 2006;33(Suppl 1):18.
72 Duvic M, Edelson R. Cutaneous T-cell lymphoma. J Am Acad Dermatol. 2004;51:S43.
73 Zinzani PL, et al. Mycosis fungoides. Crit Rev Oncol Hematol. 2008;65:172.
74 Longley BJ, et al. Somatic c-KIT activating mutation in urticaria pigmentosa and aggressive mastocytosis: establishment of clonality in a human mast cell neoplasm. Nat Genet. 1996;12:312.
75 Longley BJ, et al. Chronically KIT-stimulated clonally-derived human mast cells show heterogeneity in different tissue microenvironments. J Invest Dermatol. 1997;108:792.
76 Oji V, Traupe H. Ichthyoses: differential diagnosis and molecular genetics. Eur J Dermatol. 2006;16:349.
77 Nettis E, et al. Clinical and aetiological aspects in urticaria and angio-oedema. Br J Dermatol. 2003;148:501.
78 Murphy GF, et al. Topical tretinoin replenishes CD1a-positive epidermal Langerhans cells in chronically photodamaged human skin. J Cutan Pathol. 1998;25:30.
79 Steinhoff M, et al. Modern aspects of cutaneous neurogenic inflammation. Arch Dermatol. 2003;139:1479.
80 Lamoreux MR, et al. Erythema multiforme. Am Fam Physician. 2006;74:1883.
81 Parrillo SJ. Stevens-Johnson syndrome and toxic epidermal necrolysis. Curr Allergy Asthma Rep. 2007;7:243.
82 Murphy GF, et al. Cytotoxic T lymphocytes and phenotypically abnormal epidermal dendritic cells in fixed cutaneous eruptions. Hum Pathol. 1985;16:1264.
83 Gilliam AC, et al. Apoptosis is the predominant form of epithelial target cell injury in acute experimental graft-versus-host disease. J Invest Dermatol. 1996;107:377.
84 Schon MP, Reich K. Tumor necrosis factor antagonists in the therapy of psoriasis. Clin Dermatol. 2008;26:486.
85 Bowcock AM, Cookson WO. The genetics of psoriasis, psoriatic arthritis and atopic dermatitis. Hum Mol Genet. 2004;13(Spec No 1):R43.
86 Schwartz RA, et al. Seborrheic dermatitis: an overview. Am Fam Physician. 2006;74:125.
87 Gupta AK, et al. Skin diseases associated with Malassezia species. J Am Acad Dermatol. 2004;51:785.
88 Fischer M, et al. Skin function and skin disorders in Parkinson’s disease. J Neural Transm. 2001;108:205.
89 Mithani SK, et al. Molecular genetics of premalignant oral lesions. Oral Dis. 2007;13:126.
90 Patel GK, et al. Cutaneous lichen planus and squamous cell carcinoma. J Eur Acad Dermatol Venereol. 2003;17:98.
91 Katta R. Lichen planus. Am Fam Physician. 2000;61:3319.
92 Iijima W, et al. Infiltrating CD8+ T cells in oral lichen planus predominantly express CCR5 and CXCR3 and carry respective chemokine ligands RANTES/CCL5 and IP-10/CXCL10 in their cytolytic granules: a potential self-recruiting mechanism. Am J Pathol. 2003;163:261.
93 Edelson RL. Pemphigus—decoding the cellular language of cutaneous autoimmunity. N Engl J Med. 2000;343:60.
94 Tron F, et al. Immunogenetics of pemphigus: an update. Autoimmunity. 2006;39:531.
95 Gniadecki R. Desmoglein autoimmunity in the pathogenesis of pemphigus. Autoimmunity. 2006;39:541.
96 Wade MS, Black MM. Paraneoplastic pemphigus: a brief update. Australas J Dermatol. 2005;46:1.
97 Walsh SR, et al. Bullous pemphigoid: from bench to bedside. Drugs. 2005;65:905.
98 Blank M, et al. New insights into the autoantibody-mediated mechanisms of autoimmune bullous diseases and urticaria. Clin Exp Rheumatol. 2006;24:S20.
99 Fine JD, et al. Revised classification system for inherited epidermolysis bullosa: report of the Second International Consensus Meeting on diagnosis and classification of epidermolysis bullosa. J Am Acad Dermatol. 2000;42:1051.
100 Mallipeddi R, et al. Increased risk of squamous cell carcinoma in junctional epidermolysis bullosa. J Eur Acad Dermatol Venereol. 2004;18:521.
101 McGrath JA, Mellerio JE. Epidermolysis bullosa. Br J Hosp Med (Lond). 2006;67:188.
102 Mavilio F, et al. Correction of junctional epidermolysis bullosa by transplantation of genetically modified epidermal stem cells. Nat Med. 2006;12:1397.
103 Harper JC, Thiboutot DM. Pathogenesis of acne: recent research advances. Adv Dermatol. 2003;19:1.
104 Miskin JE, et al. Propionibacterium acnes, a resident of lipid-rich human skin, produces a 33 kDa extracellular lipase encoded by gehA. Microbiology. 1997;143(Pt 5):1745.
105 Goldsmith LA, et al. American Academy of Dermatology Consensus Conference on the safe and optimal use of isotretinoin: summary and recommendations. J Am Acad Dermatol. 2004;50:900.
106 Clarke SB, et al. Pharmacologic modulation of sebaceous gland activity: mechanisms and clinical applications. Dermatol Clin. 2007;25:137.
107 Diamantis S, Waldorf HA. Rosacea: clinical presentation and pathophysiology. J Drugs Dermatol. 2006;5:8.
108 Yamasaki K, et al. Increased serine protease activity and cathelicidin promotes skin inflammation in rosacea. Nat Med. 2007;13:975.
109 Requena L, Sanchez Yus E. Erythema nodosum. Semin Cutan Med Surg. 2007;26:114.
110 Ter Poorten MC, Thiers BH. Panniculitis. Dermatol Clin. 2002;20:421.
111 Lio P. Warts, molluscum and things that go bump on the skin: a practical guide. Arch Dis Child Educ Pract Ed. 2007;92:ep119.
112 de Villiers EM. Papillomavirus and HPV typing. Clin Dermatol. 1997;15:199.
113 Orth G. Genetics of epidermodysplasia verruciformis: insights into host defense against papillomaviruses. Semin Immunol. 2006;18:362.
114 Sladden MJ, Johnston GA. Current options for the treatment of impetigo in children. Expert Opin Pharmacother. 2005;6:2245.
115 Payne AS, et al. Desmosomes and disease: pemphigus and bullous impetigo. Curr Opin Cell Biol. 2004;16:536.
116 Schwartz RA. Superficial fungal infections. Lancet. 2004;364:1173.
117 Leyden JJ, Aly R. Tinea pedis. Semin Dermatol. 1993;12:280.