Chapter 27 Peripheral Nerve and Skeletal Muscle
The functions of the neuromuscular system depend on the motor units (Fig. 27-1), each consisting of (1) a lower motor neuron in the anterior horn of the spinal cord or cranial nerve motor nucleus in the brain stem, (2) the axon of that neuron, and (3) the multiple muscle fibers it innervates. The principal structural component of peripheral nerve is the nerve fiber (an axon with its Schwann cells and myelin sheath). A nerve consists of numerous fibers that are grouped into fascicles by connective tissue sheaths. Myelinated and unmyelinated nerve fibers are intermingled within the fascicle. Peripheral nervous system axons are myelinated in segments (internodes) separated by nodes of Ranvier. A single Schwann cell supplies the myelin sheath for each internode. Unmyelinated axons are far more numerous than myelinated axons, and the cytoplasm of one Schwann cell envelops a variable number of unmyelinated fibers (5 to 20 axons in humans). The three major connective tissue components of peripheral nerve are the epineurium, which encloses the entire nerve; the perineurium, a multilayered concentric connective tissue sheath that encloses each fascicle; and the endoneurium, which surrounds individual nerve fibers. The motor and sensory fibers, which are separated within anterior and posterior roots, intermingle within the mixed sensorimotor nerve that exits the spinal canal.
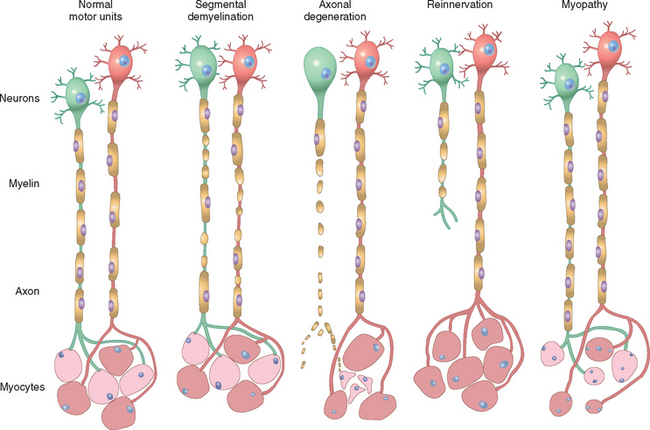
FIGURE 27-1 Normal and abnormal motor units. Normal motor units: Two adjacent motor units are shown (red and green neurons, red and pale-pink myocytes). Segmental demyelination: Random internodes of myelin are injured and are remyelinated by multiple Schwann cells, while the axon and myocytes remain intact. Axonal degeneration: The axon and its myelin sheath undergo anterograde degeneration (shown for the green neuron), with resulting denervation atrophy of the myocytes within its motor unit (pale-pink myocytes). Reinnervation of muscle: Sprouting of adjacent (red) uninjured motor axons leads to fiber type grouping of myocytes, while the injured axon attempts axonal sprouting. Myopathy: Scattered myocytes of adjacent motor units are small (degenerated or regenerated), whereas the neurons and nerve fibers are normal.
General Reactions of the Motor Unit
Neuromuscular diseases are accompanied by weakness and are often due to disorders of the motor unit—either the motor neuron and axon, or the muscle fibers. The two main responses of peripheral nerve to injury are determined by the target of the injury: either the Schwann cell or the axon. Diseases that affect primarily the Schwann cell lead to a loss of myelin, referred to as segmental demyelination. In contrast, primary involvement of the neuron and its axon leads to axonal degeneration. In some diseases axonal degeneration may be followed by axonal regeneration and reinnervation of muscle.1 The two principal pathologic processes seen in skeletal muscle are denervation atrophy, which follows loss of axons, and those due to a primary abnormality of the muscle fiber itself, referred to as myopathy.
SEGMENTAL DEMYELINATION
Segmental demyelination occurs when there is dysfunction of the Schwann cell (as in hereditary motor and sensory neuropathy) or damage to the myelin sheath (e.g., in Guillain-Barré syndrome); there is no primary abnormality of the axon. The process may affect some Schwann cells and their corresponding internodes while sparing others (see Fig. 27-1). The disintegrating myelin is engulfed initially by Schwann cells and later by macrophages. The denuded axon provides a stimulus for remyelination. A population of precursor cells within the endoneurium has the capacity to replace injured Schwann cells. These cells proliferate and encircle the axon and, in time, remyelinate the denuded portion.2 Newly formed myelinated internodes are shorter than normal, however, and several are required to bridge the demyelinated region (see Fig. 27-1). The new myelin sheath is also thin in proportion to the diameter of the axon.
With sequential episodes of demyelination and remyelination, there is an accumulation of tiers of Schwann cell processes that, on transverse section, appear as concentric layers of Schwann cell cytoplasm and redundant basement membrane surrounding a thinly myelinated axon (called onion bulbs) (Fig. 27-2). In time, many chronic demyelinating neuropathies give way to axonal injury. The specific conditions giving rise to demyelination are described later.
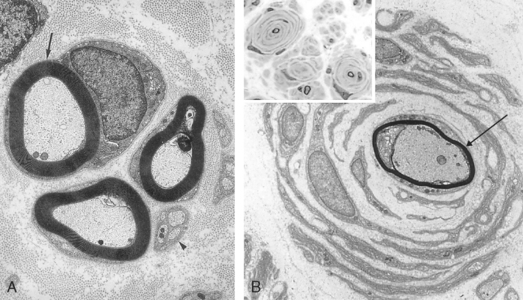
FIGURE 27-2 Compared with the normal ultrastructure of nerve (A), an “onion bulb” (B) is composed of a thinly myelinated axon (arrow) surrounded by concentrically arranged Schwann cells. Inset, Light-microscopic appearance of an onion bulb neuropathy, characterized by “onion bulbs” surrounding axons.
(B, Courtesy of G. Richard Dickersin, MD, from Diagnostic Electron Microscopy: A Text Atlas. New York, Igaku-Shoin Medical Publishers, 2000, p 984.)
AXONAL DEGENERATION AND MUSCLE FIBER ATROPHY
Axonal degeneration is the result of primary destruction of the axon, with secondary disintegration of its myelin sheath (see Fig. 27-1). Damage to the axon may be due to a focal event occurring at some point along the length of the nerve (such as trauma or ischemia) or to a more generalized abnormality affecting the neuron cell body (neuronopathy) or its axon (axonopathy). When axonal injury occurs as the result of a focal lesion, such as traumatic transection of a nerve, the distal portion of the fiber undergoes Wallerian degeneration (Fig. 27-3). Within a day the axon begins to break down, and the affected Schwann cells begin to catabolize myelin and later engulf axon fragments, forming small oval compartments (myelin ovoids).2 Macrophages are recruited into the area and participate in the phagocytosis of axonal and myelin-derived debris. The stump of the proximal portion of the severed nerve shows degenerative changes involving only the most distal two or three internodes and then undergoes regenerative activity. In the slowly evolving neuronopathies or axonopathies, evidence of axonal degeneration is scant because only a few fibers are actively degenerating at any given time.
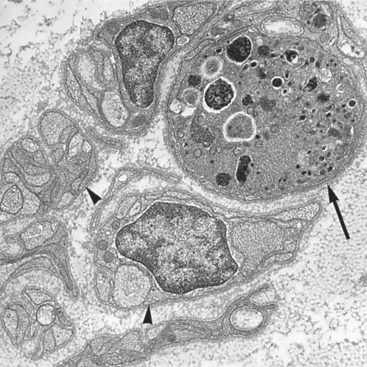
FIGURE 27-3 Electron micrograph of a degenerating axon (arrow) adjacent to several intact unmyelinated fibers (arrowheads). The axon is markedly distended and contains numerous degenerating organelles and dense bodies.
When axonal degeneration occurs, the muscle fibers within the affected motor unit lose their neural input and undergo denervation atrophy. Denervation of muscle leads to breakdown of myosin and actin, with a decrease in cell size and resorption of myofibrils, but cells remain viable. In cross-section, the atrophic fibers are smaller than normal and have a roughly triangular (“angulated”) shape. There is also cytoskeletal reorganization of some muscle cells, which results in a rounded zone of disorganized myofibers in the center of the fiber (target fiber).
NERVE REGENERATION AND REINNERVATION OF MUSCLE
The proximal stumps of degenerated axons sprout and elongate, and they may develop new growth cones during the process of axonal regeneration. These growth cones use the Schwann cells vacated by the degenerated axons to guide them. The presence of multiple closely aggregated, thinly myelinated small-caliber axons is evidence of regeneration (regenerating cluster). This regrowth of axons is apparently limited by the rate of the slow component of axonal transport, and the movement of tubulin, actin, and intermediate filaments, proceeding at about 1 mm per day. Despite its slow pace, axonal regeneration accounts for some of the functional recovery after nerve injury and can be accelerated with marrow stromal cell transplants.4
The reinnervation of skeletal muscle changes its composition, altering the distribution of the two major types of fibers, type 1 and type 2. The fiber types, defined on the basis of histochemistry and physiology (Table 27-1), are determined by the neuron of the motor unit, and their properties are imparted through innervation. Type 1 fibers are high in myoglobin and oxidative enzymes and have many mitochondria, in keeping with their ability to perform tonic contraction; operationally, they are most often defined by their dark staining for adenosine triphosphatase (ATPase) at pH 4.2 but light staining at pH 9.4. Type 2 fibers are rich in glycolytic enzymes and are involved in rapid phasic contractions; they are dark staining on ATPase stain performed at pH 9.4 but light staining at pH 4.2. Since the motor neuron determines fiber type, all muscle fibers of a single unit are of the same type. The fibers of a single motor unit are distributed across the muscle, giving rise to a checkerboard pattern of alternating fiber types, as is demonstrated especially well with staining for ATPase (Fig. 27-4A). Normally, there is some variability in the relative abundance of type 1 and type 2 fibers among different muscles. The mnemonic “one (type 1 fiber) slow (twitch) fat (lipid-rich) red (appearance) ox (oxidative)” is useful to keep the physiology and histochemistry of the fiber types in mind.
| Type 1 | Type 2 | |
|---|---|---|
| Action | Sustained force | Sudden movements |
| Strength | Weight-bearing | Purposeful motion |
| Enzyme content | NADH-TR dark staining | NADH light staining |
| ATPase at pH 4.2, dark staining | ATPase at pH 4.2, light staining | |
| ATPase at pH 9.4, light staining | ATPase at pH 9.4, dark staining | |
| Lipids | Abundant | Scant |
| Glycogen | Scant | Abundant |
| Ultrastructure | Many mitochondria | Few mitochondria |
| Wide Z-band | Narrow-Z-band | |
| Physiology | Slow-twitch | Fast-twitch |
| Color | Red | White |
| Prototype | Soleus (pigeon) | Pectoral (pigeon) |
ATPase, adenosine triphosphatase; NADH-TR, nicotinamide adenine dinucleotide, reduced form, tetrazolium reductase.
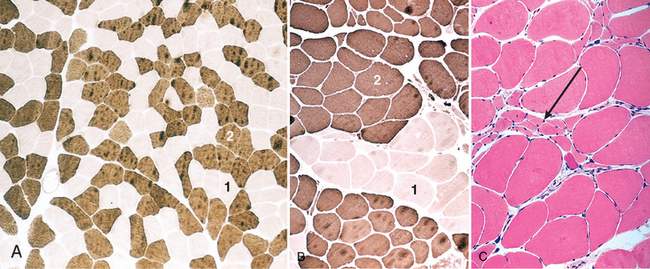
FIGURE 27-4 A, ATPase histochemical staining, at pH 9.4, of normal muscle showing checkerboard distribution of intermingled type 1 (light) and type 2 (dark) fibers. B, In contrast, fibers of both histochemical types are grouped together after reinnervation of muscle. C, A cluster of atrophic fibers (group atrophy) in the center (arrow).
Reinnervation of the atrophic muscle fibers within an injured motor unit occurs when the axons belonging to an unaffected neighboring motor unit extend sprouts to reinnervate the denervated myocytes and incorporate them into the healthy motor unit. The number of muscle fibers within the healthy reinnervating motor unit can thus be increased. Furthermore, since muscle fiber type is imparted by the innervating neuron, the newly adopted reinnervated fibers assume the fiber type of their neighboring new siblings. The result of reinnervation is the loss of the checkerboard pattern and the occurrence of a patch of contiguous myocytes having the same histochemical type (type grouping) (Fig. 27-4B). Group atrophy ensues when a type group in turn becomes denervated, because it is affected in the course of disease progression (Fig. 27-4C).
Type-specific atrophy is characteristic of some disease states. Type 2 fiber atrophy is a relatively common finding and is associated with inactivity or disuse. This type of “disuse atrophy” may occur after fracture of a limb and application of a plaster cast, in pyramidal tract degeneration, or in neurodegenerative diseases. Type 2 fiber atrophy may also occur during therapy with glucocorticoids and is characteristic of “steroid myopathy.”
REACTIONS OF THE MUSCLE FIBER
Although a wide spectrum of diseases may affect muscle, the pathologic reactions of myocytes are relatively limited. Pathologic changes may be seen in myopathies as well as in diseases that secondarily involve the muscle cells. The most common forms of reaction include the following:
Diseases of Peripheral Nerve
Peripheral nerve is susceptible to the same wide range of categories of disease (inflammatory, traumatic, metabolic, toxic, genetic, neoplastic) as are other tissues. The pattern of disease, however, reflects the unique structure and function of nerves.
INFLAMMATORY NEUROPATHIES
These diseases are characterized by inflammatory cell infiltrates in peripheral nerves, roots, and sensory and autonomic ganglia. In some, an infectious agent elicits the inflammatory responses; in others, immune mechanisms are presumed to be the primary cause of the inflammation.
Immune-Mediated Neuropathies
Guillain-Barré Syndrome (Acute Inflammatory Demyelinating Polyradiculoneuropathy)
Guillain-Barré syndrome is a life-threatening disease of the peripheral nervous system, with an overall annual incidence of one to three cases per 100,000 persons throughout the world.5 The disease is characterized clinically by weakness beginning in the distal limbs but rapidly advancing to affect proximal muscle function (“ascending paralysis”), and histologically by inflammation and demyelination of spinal nerve roots and peripheral nerves (radiculoneuropathy).
Pathogenesis.
Guillain-Barré syndrome is thought to be an acute-onset immune-mediated demyelinating neuropathy. Approximately two thirds of cases are preceded by an acute, influenza-like illness from which the affected individual has recovered by the time the neuropathy becomes symptomatic. Infections with Campylobacter jejuni, cytomegalovirus, Epstein-Barr virus, and Mycoplasma pneumoniae, or prior vaccination, have been shown to have a significant epidemiologic association with Guillain-Barré syndrome.6 There has been no consistent demonstration of an infectious agent in peripheral nerves in these patients, and an immunological reaction is generally favored as the underlying cause. A similar inflammatory disease of peripheral nerves can be induced in experimental animals by immunization with a peripheral nerve myelin protein. A T cell–mediated immune response ensues, accompanied by segmental demyelination induced by activated macrophages. Transfer of these T cells to a naive animal results in comparable lesions. Moreover, lymphocytes from individuals with Guillain-Barré syndrome have been shown to produce demyelination in tissue cultures of myelinated nerve fibers. Circulating antibodies may also play a part, and plasmapheresis can be an effective treatment.6
Morphology. The dominant histopathologic finding is inflammation of peripheral nerve, manifested as perivenular and endoneurial infiltration by lymphocytes, macrophages, and a few plasma cells. Inflammatory foci and demyelination are widely distributed throughout the peripheral nervous system, although their intensity is variable. The most intense inflammatory reaction is often localized in spinal and cranial motor roots and the adjacent parts of the spinal and cranial nerves. Segmental demyelination affecting peripheral nerves is the primary lesion, but damage to axons is also characteristic, particularly when the disease is severe. Electron microscopy has identified an early effect on myelin sheaths. The cytoplasmic processes of macrophages penetrate the basement membrane of Schwann cells, particularly in the vicinity of the nodes of Ranvier, and extend between the myelin lamellae, stripping away the myelin sheath from the axon. Ultimately, the remnants of the myelin sheath are engulfed by the macrophages. Remyelination follows the demyelination.
Clinical Course.
The clinical picture is dominated by the ascending paralysis. Deep tendon reflexes disappear early in the process; although sensory involvement can often be detected, it is less troublesome than the weakness. The nerve conduction velocity is slowed because of the multifocal destruction of myelin segments involving many axons within a nerve. There is elevation of the CSF protein due to inflammation and altered permeability of the microcirculation within the spinal roots as they traverse the subarachnoid space. Inflammatory cells are contained within the roots, however, and there is little to no CSF pleocytosis. Many patients spend weeks in hospital intensive-care units before recovering normal function. With improved respiratory care and support, the mortality rate has fallen from 25% in the past but is still considerable; some 2% to 5% die of respiratory paralysis, autonomic instability, cardiac arrest, or the complications of treatment. Up to 20% of hospitalized patients have long-term disability.5,6
Chronic Inflammatory Demyelinating Polyradiculoneuropathy
In some patients, inflammatory demyelinating polyradiculoneuropathy follows a subacute or chronic course, usually with relapses and remissions over a period of several years, rather than the acute course of Guillain-Barré syndrome.7 In these cases there is often a symmetric, mixed sensorimotor polyneuropathy, although some patients have predominantly sensory or motor impairment. Clinical remissions may occur with steroid treatment and plasmapheresis. Biopsies of sural nerves show evidence of recurrent demyelination and remyelination associated with well-developed onion bulb structures.8
INFECTIOUS POLYNEUROPATHIES
Many infectious processes affect peripheral nerve. Leprosy, diphtheria, and varicella-zoster cause unique and specific pathologic changes in nerves and are also discussed as systemic infectious diseases in Chapter 8.
Leprosy (Hansen Disease)
Peripheral nerves are involved in both lepromatous and tuberculoid leprosy (discussed in Chapter 8).9 In lepromatous leprosy, Schwann cells are invaded by Mycobacterium leprae, which proliferate and eventually infect other cells. There is evidence of segmental demyelination and remyelination and loss of both myelinated and unmyelinated axons. As the infection advances, endoneurial fibrosis and multilayered thickening of the perineurial sheaths occur. Affected individuals develop a symmetric polyneuropathy affecting the cool extremities (due to lower temperatures favoring mycobacterial growth). The infection prominently involves pain fibers, and the resulting loss of sensation contributes to injury, since the patient is rendered unaware of injurious stimuli and damaged tissues. Thus, large traumatic ulcers may develop in the extremities. Tuberculoid leprosy shows evidence of active cell-mediated immune response to M. leprae, with nodules of granulomatous inflammation situated in the dermis. The inflammation injures cutaneous nerves in the vicinity; axons, Schwann cells, and myelin are lost, and there is fibrosis of the perineurium and endoneurium. In tuberculoid leprosy, affected individuals have much more localized nerve involvement.
Diphtheria
Peripheral nerve involvement results from the effects of the diphtheria exotoxin and begins with paresthesias and weakness10; early loss of proprioception and vibratory sensation is common. The earliest changes are seen in the sensory ganglia, where the incomplete blood-nerve barrier allows entry of the toxin. There is selective demyelination of axons that extends into adjacent anterior and posterior roots as well as into mixed sensorimotor nerves. The mechanism of action of diphtheria toxin is described in Chapter 8.
Varicella-Zoster Virus
Varicella-zoster virus is one of the most common viral infections of the peripheral nervous system.11 Following chickenpox, a latent infection persists within neurons in the sensory ganglia of the spinal cord and brain stem, and reactivation leads to a painful, vesicular skin eruption in the distribution of sensory dermatomes (shingles), most frequently thoracic or trigeminal. The virus may be transported along the sensory nerves to the skin, where it establishes an active infection of epidermal cells. In a small proportion of patients, weakness is also apparent in the same distribution. Although the factors that give rise to reactivation are not fully understood, decreased cell-mediated immunity is of major importance in some cases.
Affected ganglia show neuronal destruction and loss, usually accompanied by abundant mononuclear inflammatory infiltrates; regional necrosis with hemorrhage may also be found. Peripheral nerve shows axonal degeneration after the death of the sensory neurons. Focal destruction of the large motor neurons of the anterior horns or cranial nerve motor nuclei may be seen at the corresponding levels. Intranuclear inclusions generally are not found in the peripheral nervous system.
HEREDITARY NEUROPATHIES
This is a group of heterogeneous, typically progressive, and often disabling syndromes that affect peripheral nerves. The genetic and molecular basis of many of the hereditary peripheral neuropathies is being elucidated,12,13 and as they are further defined, adjustments in the current classification scheme can be anticipated. They can be divided into several groups:
TABLE 27-2 Hereditary Sensory and Autonomic Neuropathies (HSANs)
| Disease and Inheritance | Gene and Locus | Clinical and Pathologic Findings |
|---|---|---|
| HSAN I; autosomal dominant | Serine palmitoyl transferase, long-chain base, subunit 1 (SPTLC1) gene; 9q22.1–q22.3 | Predominantly sensory neuropathy, presenting in young adults; axonal degeneration (mostly myelinated fibers) |
| HSAN II; autosomal recessive (some cases are sporadic) | HSN2 gene; 12q13.3 | Predominantly sensory neuropathy, presenting in childhood; axonal degeneration (mostly myelinated fibers) |
| HSAN III; (Riley-Day syndrome; familial dysautonomia; most often in Jewish children); autosomal recessive | IKAP histone acetyltransferase (IKAP) gene; 9q31 | Predominantly autonomic neuropathy, presenting in infancy; axonal degeneration (mostly unmyelinated fibers); atrophy and loss of sensory and autonomic ganglion cells |
| HSAN IV; autosomal recessive dysautonomia, type II; | Neurotrophic tyrosine kinase receptor, type 1, or NTRK1 gene; 1q21–q22 | Congenital insensitivity to pain and anhidrosis; presentation in infancy; nearly complete loss of small myelinated and unmyelinated fibers |
| HSAN V; autosomal recessive | Nerve growth factor β subunit (NGFB) gene; 1p13.1 | Congenital insensitivity to pain and temperature; presentation in infancy; nearly complete loss of small myelinated fibers |
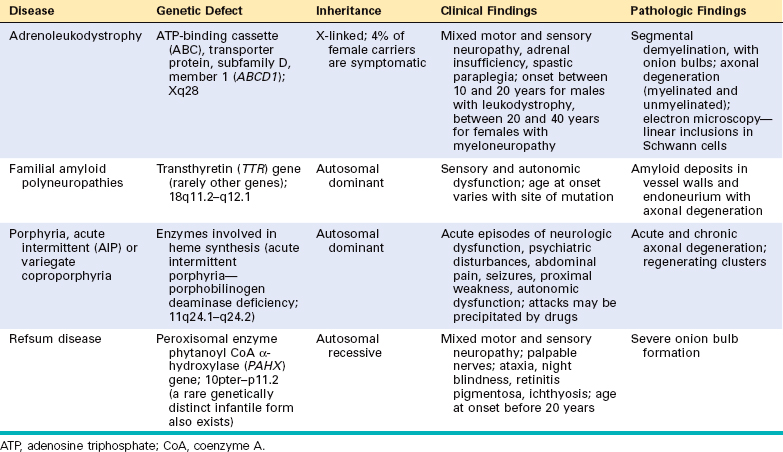
The most common hereditary neuropathy, HMSN I, results in demyelination of peripheral nerve and slowing of the velocity of axonal conduction. The other hereditary neuropathies are axonal neuropathies that have fiber loss as their most prominent pathologic finding.
Hereditary Motor and Sensory Neuropathy Type I
HMSN I, also referred to as Charcot-Marie-Tooth (CMT) disease, demyelinating type, usually presents in childhood or early adulthood. A characteristic progressive muscular atrophy of the leg below the knee seen in these patients gives rise to the common clinical term peroneal muscular atrophy. Affected individuals may be asymptomatic, but when they present, it is often with symptoms such as distal muscle weakness, atrophy of the leg below the knee, or secondary orthopedic problems of the foot (such as pes cavus).
Pathogenesis and Molecular Genetics.
The disease is genetically heterogeneous. The most common subtype (known as HMSN IA or CMT1A) has a duplication of a large region of chromosome 17p11.2, resulting in “segmental trisomy” of this region. The duplicated segment includes the gene that encodes peripheral myelin protein 22 (PMP22), a transmembrane protein expressed in compacted myelin. PMP22 and a set of related proteins are involved in the compaction of myelin in the peripheral nervous system (Fig. 27-5). Mutations affecting these myelin-associated genes result in demyelinating neuropathies of the HMSN I phenotype.14 Mutations in another gene, on chromosome 1, which codes for myelin protein zero (MPZ), produce an identical clinical phenotype (HMSN IB). Additional pedigrees with hereditary demyelinating neuropathy show mutations in genes encoding structural proteins (connexin-32), protein degradation pathways (LITAF), and myelination induction genes (early growth response 2, EGR2).12,14
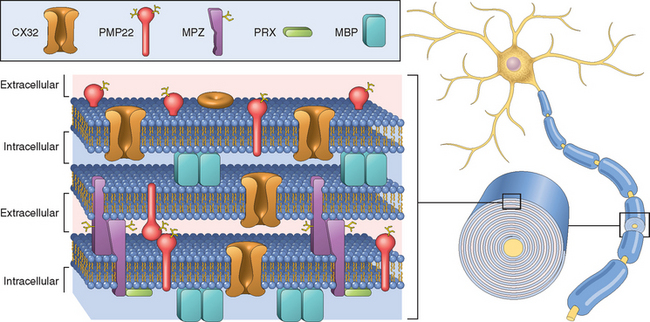
FIGURE 27-5 Relationship between the proteins of compacted myelin and the lipid bilayers. Myelin basic protein (MBP) is an intracellular protein that has a role in myelin compaction. Mutant forms of myelin protein zero (MPZ), peripheral myelin protein 22 (PMP22), and periaxin (PRX) cause hereditary demyelinating neuropathies of the Charcot-Marie-Tooth, type 1 category.
Morphology. HMSN I is a demyelinating neuropathy. Histologic examination shows the consequences of repetitive demyelination and remyelination, with multiple onion bulbs, more pronounced in distal nerves than in proximal nerves (see Fig. 27-2). The axon is often present in the center of the onion bulb, and the myelin sheath is usually thin or absent. The redundant layers of Schwann cell hyperplasia surrounding individual axons are associated with enlargement of involved peripheral nerves that may become palpable, which has led to the term hypertrophic neuropathy. In the longitudinal plane, the axon may show evidence of segmental demyelination. Autopsy studies of affected individuals have shown degeneration of the posterior columns of the spinal cord.
Other Hereditary Motor and Sensory Neuropathies
HMSN II
The axonal form of autosomal dominant CMT HMSN II (CMT-2) presents with signs and symptoms similar to those of HMSN I, although nerve enlargement is not seen and the disease presents at a slightly later age. This form is less common than HMSN I. Some cases (designated HMSN IIA1 or CMT2A1) are caused by mutations in a gene encoding a kinesin family member, KIF1B.15 Additional, less common mutations in other genes have been identified; these are classified as CMT2B to 2L.23 Nerve biopsy specimens in HMSN II show loss of myelinated axons as the predominant finding. Segmental demyelination of internodes is infrequent. These findings suggest that the site of primary cellular dysfunction is the axon or neuron.
Dejerine-Sottas Neuropathy (HMSN III)
Dejerine-Sottas neuropathy is a slowly progressive, autosomal recessive disorder that begins in early childhood, and is manifested by delay in developmental milestones, such as the acquisition of motor skills. In contrast to HMSN I and HMSN II, in which muscular atrophy is limited to the leg, both trunk and limb muscles are involved in Dejerine-Sottas disease. On physical examination, enlarged peripheral nerves can be detected by inspection and palpation. The deep tendon reflexes are depressed or absent, and nerve conduction velocity is slowed. HMSN III is genetically heterogeneous, and arises from mutations in the same myelin-associated genes that are mutated in HMSN I. These include genes encoding PMP22, MPZ, periaxin (PRX), and EGR2.16 Morphologically, the size of individual peripheral nerve fascicles is increased, often markedly, with abundant onion bulb formation as well as segmental demyelination. There is usually evidence of axonal loss, and the axons that remain are often of diminished caliber. Distal portions of peripheral nerves are most severely affected; however, autopsy studies have shown that the spinal roots are also involved.
ACQUIRED METABOLIC AND TOXIC NEUROPATHIES
Functional and structural changes in peripheral nerves develop in response to various metabolic alterations—either from endogenous disorders or from exogenous agents. The most common of these processes are discussed here.
Peripheral Neuropathy in Adult-Onset Diabetes Mellitus
The prevalence of peripheral neuropathy in individuals with diabetes mellitus depends on the duration of the disease; up to 50% of diabetics overall have peripheral neuropathy clinically, and up to 80% of those who have had the disease for more than 15 years. Several distinct clinicopathologic patterns of diabetes-related peripheral nerve abnormalities have been recognized (Chapter 24). They are categorized as distal symmetric sensory or sensorimotor neuropathy, autonomic neuropathy, and focal or multifocal asymmetric neuropathy. Individuals may develop any combination of these lesions; in fact, the first two (sensorimotor and autonomic) are often found together. The mechanism of diabetic neuropathy is not completely resolved, but there is evidence for involvement of both the polyol pathway and the nonenzymatic glycation of proteins.17
Morphology. In individuals with a distal symmetric sensorimotor neuropathy, the predominant pathologic finding is an axonal neuropathy. As with other chronic axonal neuropathies, there is often some segmental demyelination. There is a relative loss of small myelinated fibers and of unmyelinated fibers, but large fibers are also affected. Endoneurial arterioles show thickening, hyalinization, and intense periodic acid–Schiff positivity in their walls and extensive reduplication of the basement membrane18 (Fig. 27-6).
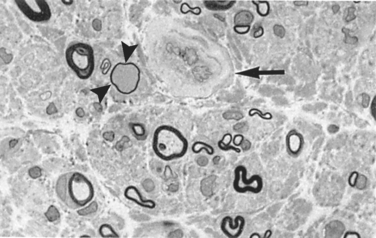
Clinical Course.
The most common peripheral neuropathy in type 2 diabetes mellitus is the symmetric neuropathy that involves distal sensory and motor nerves. Individuals with the neuropathy develop decreased sensation in the distal extremities with less evident motor abnormalities. The loss of pain sensation can result in the development of ulcers, which heal poorly because of the diffuse vascular injury in diabetes and are a major cause of morbidity. Another manifestation of diabetic neuropathy is dysfunction of the autonomic nervous system; this affects 20% to 40% of individuals with diabetes mellitus, nearly always in association with a distal sensorimotor neuropathy.17 Diabetic autonomic neuropathy has protean manifestations, including postural hypotension, incomplete emptying of the bladder resulting in recurrent infections, and sexual dysfuction. Some affected individuals, especially elderly adults with a long history of diabetes, develop a peripheral neuropathy that manifests itself as a disorder of single individual peripheral or cranial (oculomotor nerve) nerves (mononeuropathy), or of several individual nerves in an asymmetric distribution (multiple mononeuropathy or mononeuropathy multiplex). The pathogenesis of mononeuropathies in adult-onset diabetes is thought to involve vascular insufficiency, and ischemia of the affected peripheral nerve.17
Metabolic and Nutritional Peripheral Neuropathies
Most individuals with renal failure have a peripheral neuropathy (uremic neuropathy).19 This is typically a distal, symmetric neuropathy that may be asymptomatic or may be associated with muscle cramps, distal dysesthesias, and diminished deep tendon reflexes. In these patients axonal degeneration is the primary event; occasionally there is secondary demyelination. Regeneration and recovery are common after dialysis.
Peripheral neuropathy can also develop in individuals with chronic liver disease, chronic respiratory insufficiency, and thyroid dysfunction. Thiamine deficiency is associated with axonal neuropathy, a clinical condition termed neuropathic beriberi. Axonal neuropathies also occur with deficiencies of vitamins B12 (cobalamin), B6 (pyridoxine), and E (α-tocopherol). Excessive chronic consumption of ethyl alcohol often leads to axonal neuropathy. There is a strong contribution of associated dietary deficiency, and affected individuals often have signs of thiamine deficiency. However, ethyl alcohol may have a direct toxic effect on peripheral nerve, since some affected individuals have adequate thiamine intake.20
Neuropathies Associated with Malignancy
Direct infiltration or compression of peripheral nerves by tumor is a common cause of mononeuropathy and may be the presenting symptom of cancer. These neuropathies include brachial plexopathy from neoplasms of the apex of the lung, obturator palsy from pelvic malignant neoplasms, and cranial nerve palsies from intracranial tumors and tumors of the base of the skull. A polyradiculopathy involving the lower extremity may develop when the cauda equina is involved by meningeal carcinomatosis.
In contrast, a diffuse, symmetric peripheral neuropathy may occur in individuals with a distant carcinoma as a paraneoplastic effect (Chapters 7 and 28. The most common type is a sensorimotor neuropathy characterized by weakness and sensory deficits that are often more pronounced in the lower extremities and that progress during months to years.21 The neuropathy is most frequently associated with small-cell carcinoma of the lung; as many as 2% to 5% of people with lung cancer have clinical evidence of peripheral neuropathy. Patients with the less frequent pure sensory neuropathy present with numbness and paresthesias that may precede the diagnosis of the malignancy by 6 to 15 months. An immunological mechanism for the neuropathy has been suggested based on the presence of inflammatory infiltrates within the dorsal root ganglia and the identification of IgG antibodies that bind a 35- to 38-kD RNA-binding protein expressed by neurons and the tumor.22 The severity of clinical symptoms correlates with antibody titer, suggesting a causative relationship.
Paraneoplastic neuropathy may also develop in individuals with plasma cell neoplasms in one of two ways. The first is through the deposition of light-chain (AL type) amyloid in peripheral nerves (Chapter 6). The second is related to the production of monoclonal immunoglobulin that recognizes a major protein of myelin, myelin-associated glycoprotein.23
Toxic Neuropathies
Peripheral neuropathies can occur after exposure to industrial or environmental chemicals, biologic toxins, or therapeutic drugs.24 Prominent among the environmental chemicals are heavy metals, including lead and arsenic (Chapter 9). In addition, many organic compounds are known to be toxic to the peripheral nervous system, leading to peripheral neuropathy.
TRAUMATIC NEUROPATHIES
Peripheral nerves are commonly injured in the course of trauma. Lacerations result from cutting injuries and can complicate fractures when a sharp fragment of bone lacerates the nerve. Avulsions occur when tension is applied to a peripheral nerve, often as the result of a force applied to one of the limbs. Regeneration of peripheral nerve axons following these types of injuries does occur, albeit slowly. Regrowth may be complicated by discontinuity between the proximal and distal portions of the nerve sheath as well as by the misalignment of individual fascicles. Axons, even in the absence of correctly positioned distal segments, may continue to grow, resulting in a mass of tangled axonal processes known as a traumatic neuroma (pseudoneuroma or amputation neuroma). Within this mass, small bundles of axons appear randomly oriented; each, however, is surrounded by organized layers containing Schwann cells, fibroblasts, and perineurial cells (Fig. 27-7).
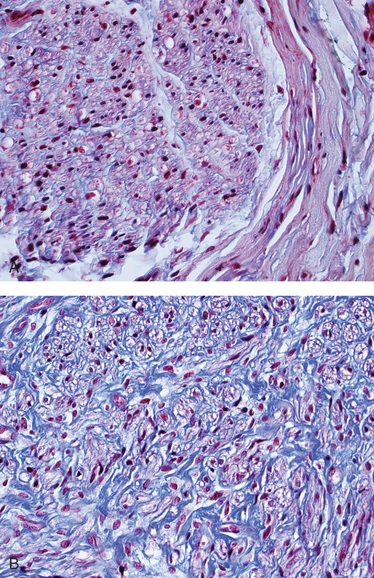
FIGURE 27-7 Normal appearance of peripheral nerve (A), with all the axons aligned in a single plane with sheaths of connective tissue, as compared with traumatic neuroma (B) showing disordered orientation of axons (pale purple) intermixed with connective tissue (blue).
Compression neuropathy (entrapment neuropathy) occurs when a peripheral nerve is compressed, often within an anatomic compartment. Carpal tunnel syndrome, the most common entrapment neuropathy, results from compression of the median nerve at the level of the wrist within the compartment delimited by the transverse carpal ligament.25 Women are more commonly affected than men, and the problem is frequently bilateral. The disorder may be observed in association with many conditions including tissue edema, pregnancy, inflammatory arthritis, hypothyroidism, amyloidosis (especially that related to β2-microglobulin deposition in individuals on renal dialysis), acromegaly, diabetes mellitus, and excessive repetitive motions of the wrist. Symptoms are limited to dysfunction of the median nerve, including numbness and paresthesias of the tips of the thumb and first two digits. Other nerves prone to compression neuropathies include the ulnar nerve at the level of the elbow, the peroneal nerve at the level of the knee, and the radial nerve in the upper arm; the latter occurs from sleeping with the arm in an awkward position (“Saturday night palsy”). Another form of compression neuropathy is found in the foot, affecting the interdigital nerve at intermetatarsal sites. This problem, which occurs more often in women than in men, leads to foot pain (metatarsalgia). The histologic findings of the lesion (Morton neuroma) include evidence of chronic compression injury.
TUMORS OF PERIPHERAL NERVE
Both benign and malignant tumors can be derived from elements of the nerve sheath. These are discussed with tumors of the central nervous system (Chapter 28).
Diseases of Skeletal Muscle
DENERVATION ATROPHY
Neurogenic atrophy of muscle is caused by disorders that affect motor neurons (see Fig. 27-1). The response of muscle to denervation and the histologic changes associated with reinnervation have been described earlier.
Spinal Muscular Atrophy (Infantile Motor Neuron Disease)
Motor neuron diseases are progressive neurologic illnesses that selectively destroy the anterior horn cells in the spinal cord and cranial nerve motor neurons. Motor neuron diseases in adults are discussed in Chapter 28. Spinal muscular atrophy (SMA) is a distinctive group of autosomal recessive motor neuron diseases that begin in childhood or adolescence. SMA is discussed here because the disease is commonly considered with the childhood myopathies and because the pathologic findings in skeletal muscle are characteristic.
Genetics.
All forms of SMA are associated with mutations affecting survival motor neuron 1 (SMN1), a gene on chromosome 5 that is required for motor neuron survival. This region of chromosome 5 also contains variable numbers of copies of a second highly homologous gene, SMN2. Homozygous deletions of SMN1 (or less commonly, intragenic mutations) cause SMA.26 The number of copies of the homologous SMN2 modifies the clinical phenotype, with more copies being associated with milder neurologic phenotype. SMN genes are expressed in all tissues, so why mutations or deletions of these genes cause only neuronal loss is not clear. It is postulated that the SMN protein is critical for normal axonal transport and integrity of neuromuscular junctions, and thus promotes survival of motor neurons.
Morphology. The typical histologic finding in muscle is large numbers of atrophic fibers, often only a few micrometers in diameter (Fig. 27-8). This is unlike the groups of angulated atrophic fibers seen in denervation atrophy of muscle in adults. In SMA the muscle fiber atrophy often involves an entire fascicle, a feature known as panfascicular atrophy. There are also scattered large fibers that are two to four times normal size.
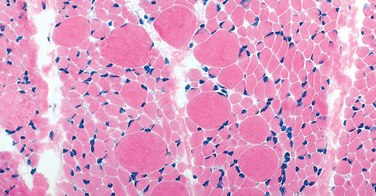
Clinical Course.
The most common form of SMA, Werdnig-Hoffmann disease (SMA type 1), has its onset at birth or within the first 4 months of life with severe hypotonia (lack of muscle tone and “floppiness”). It usually leads to death within the first 3 years of life. The other two forms (SMA 2 and SMA 3) present at later ages, either in early childhood (between 3 and 15 months of age in SMA 2) or in later childhood (after 2 years of age in SMA 3). Those with SMA 2 usually die in childhood after age 4, whereas those with SMA 3 often survive into adulthood.
MUSCULAR DYSTROPHIES
The muscular dystrophies are a heterogeneous group of inherited disorders of muscle, often beginning in childhood, that lead to progressive weakness and muscle wasting. Histologically, in advanced cases muscle fibers undergo degeneration and are replaced by fibrofatty tissue and collagen. This feature distinguishes dystrophies from myopathies (described later), which also present with muscle weakness.
X-Linked Muscular Dystrophy (Duchenne Muscular Dystrophy and Becker Muscular Dystrophy)
The two most common forms of muscular dystrophy are X linked: Duchenne muscular dystrophy (DMD) and Becker muscular dystrophy (BMD). DMD is the most severe and common form of muscular dystrophy, with an incidence of about 1 per 3500 live male births. DMD becomes clinically manifest by the age of 5 years. It leads to wheelchair dependence by 10 to 12 years of age, and thereafter progresses relentlessly. Although BMD involves the same genetic locus, it is less common and much less severe than DMD.
Pathogenesis and Molecular Genetics.
DMD and BMD are caused by abnormalities in DMD, a gene that is located in the Xp21 region. DMD is one of the largest human genes, spanning 2.3 million base pairs, and 79 exons. It encodes a 427-kD protein named dystrophin. A large proportion of the genetic abnormalities are deletions, with frameshift and point mutations accounting for the rest.27 Approximately two thirds of the cases are familial, and the remainder represent new mutations. In the affected families females are carriers; they are clinically asymptomatic but often have elevated serum creatine kinase and show minimal histologic abnormalities on muscle biopsy. Female carriers and affected males who survive into adulthood are also at risk for developing dilated cardiomyopathy.27
Dystrophin is a cytoplasmic protein located adjacent to the sarcolemmal membrane in myocytes (Fig. 27-9). It is concentrated at the plasma membrane over Z-bands, where it forms a strong mechanical link to cytoplasmic actin. Thus, dystrophin and the dystrophin-associated protein complex form an interface between the intracellular contractile apparatus and the extracellular connective tissue matrix. The role of this complex of proteins in transferring the force of contraction to connective tissue has been proposed to be the basis for the myocyte degeneration that occurs in the absence of dystrophin28 or various other proteins that interact with dystrophin (see later). Muscle biopsy specimens from individuals with DMD show little or no dystrophin by both staining and western blot analysis (Fig. 27-10). People with BMD, who also have mutations in the dystrophin gene, have diminished amounts of dystrophin, usually of an abnormal molecular weight, reflecting mutations that allow synthesis of an abnormal protein of smaller size (Fig. 27-10B).
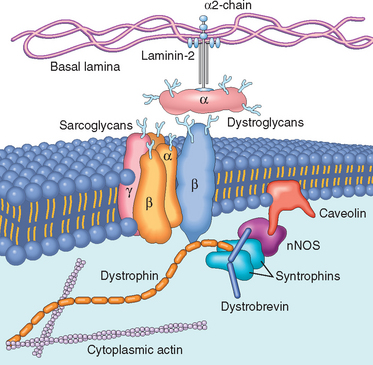
FIGURE 27-9 Relationship between the cell membrane (sarcolemma) and the sarcolemmal associated proteins. Dystrophin, an intracellular protein, forms an interface between the cytoskeletal proteins and a group of transmembrane proteins, the dystroglycans and the sarcoglycans. These transmembrane proteins have interactions with the extracellular matrix, including the laminin proteins. Dystrophin also interacts with dystrobrevin and the syntrophins, which form a link with neuronal-type nitric oxide synthetase (nNOS) and caveolin. Mutations in dystrophin are associated with the X-linked muscular dystrophies; mutations in caveolin and the sarcoglycan proteins with the limb-girdle muscular dystrophies, which can be autosomal dominant or recessive disorders; and mutations in the α2-laminin (merosin) with autosomal recessive congenital muscular dystrophy.
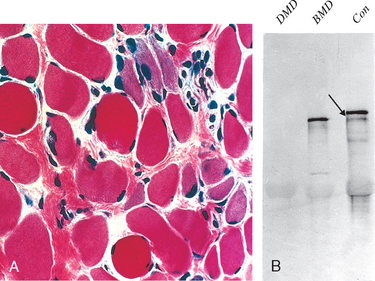
FIGURE 27-10 A, Duchenne muscular dystrophy (DMD) showing variation in muscle fiber size, increased endomysial connective tissue, and regenerating fibers (blue hue). B, Western blot showing absence of dystrophin in DMD and altered dystrophin size in Becker muscular dystrophy (BMD) compared with control
(arrow) (Con). (Courtesy of Dr. L. Kunkel, Children’s Hospital, Boston, MA.)
Morphology. Histopathologic abnormalities common to DMD and BMD include (1) variation in fiber size (diameter) due to the presence of both small and enlarged fibers, sometimes with fiber splitting; (2) increased numbers of internalized nuclei (beyond the normal range of 3% to 5%); (3) degeneration, necrosis, and phagocytosis of muscle fibers; (4) regeneration of muscle fibers; and (5) proliferation of endomysial connective tissue (see Fig. 27-10A). DMD cases also often show enlarged, rounded, hyaline fibers that have lost their normal cross-striations; such fibers, believed to be hypercontracted, are rare in BMD. Both type 1 and type 2 fibers are involved, and no alterations in the proportion or distribution of fiber types are evident. Histochemical reactions sometimes fail to identify distinct fiber types in DMD. In later stages the muscles eventually become almost totally replaced by fat and connective tissue. Cardiac involvement, when present, consists of interstitial fibrosis, which is more prominent in the subendocardium.
Clinical Course.
Boys with DMD are normal at birth, and early motor milestones are met on time. Walking, however, is often delayed, and the first indications of muscle weakness are clumsiness and inability to keep up with peers. Weakness begins in the pelvic girdle muscles and then extends to the shoulder girdle. Enlargement of the muscles of the lower leg associated with weakness, a phenomenon termed pseudohypertrophy, is an important clinical finding. The increased muscle bulk is caused initially by an increase in the size of the muscle fibers and then, as the muscle atrophies, by an increase in fat and connective tissue. Pathologic changes are also found in the heart, and patients may develop heart failure or arrhythmias.27 Although there are no well-established structural abnormalities of the central nervous system, cognitive impairment is a component of the disease and is sometimes severe enough to be considered a form of mental retardation. Serum creatine kinase is elevated during the first decade of life but returns to normal as muscle mass decreases. Death results from respiratory insufficiency, pulmonary infection, and cardiac decompensation. Gene therapy has received a great deal of attention but has been hampered by the size of the DMD gene. There has been success in experimental animals with two delivery systems: wild-type stem cell injections directly into muscle; and systemic injection of adenoassociated viruses carrying genes engineered to produce small, but functionally competent, portions of the dystrophin protein.29 Induction of specific exon skipping using antisense RNA has been shown to restore the open reading in certain mutated DMD genes, and increase dystrophin expression in muscle biopsies.30
Boys with BMD develop symptoms at a later age than those with DMD. The onset occurs in later childhood or in adolescence, and is followed by a slower and more variable rate of progression, although there is considerable variation between pedigrees. Many patients have a nearly normal life span. Cardiac disease is frequently seen in these patients.
Other Muscular Dystrophies
Other less common forms of muscular dystrophy share many features of DMD and BMD, but have distinct clinical and pathologic characteristics. Some of these muscular dystrophies affect specific muscle groups, and the diagnosis is based largely on the pattern of muscle weakness (Table 27-4). Several autosomal muscular dystrophies, however, affect the proximal musculature of the trunk and limbs, similar to the X-linked muscular dystrophies, and are termed limb girdle muscular dystrophies.
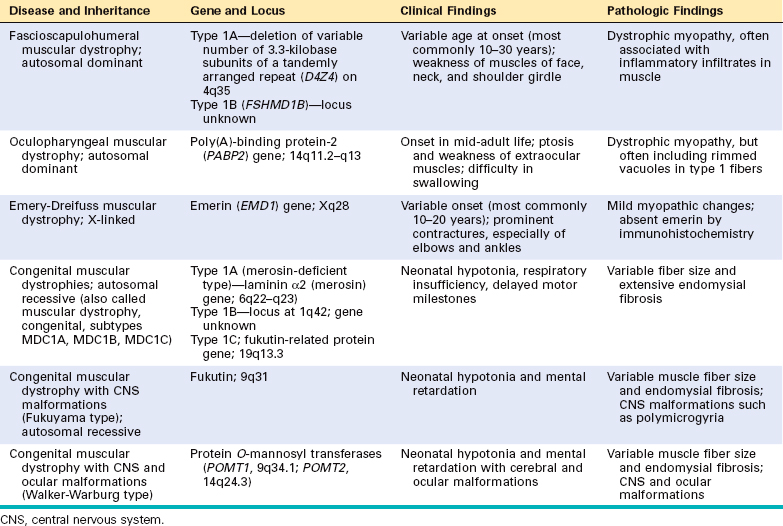
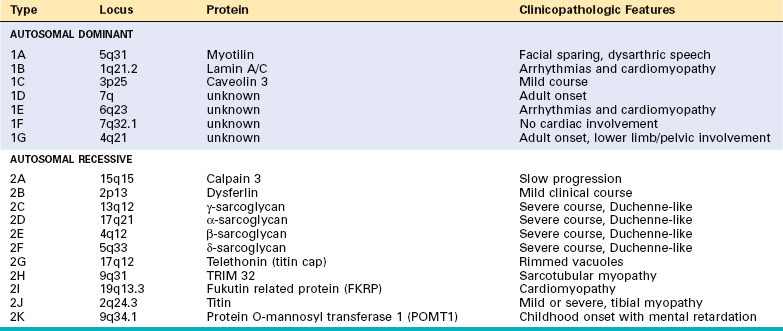
Limb girdle muscular dystrophies (LGMDs) are inherited in either an autosomal dominant (type 1) or autosomal recessive (type 2) pattern (Table 27-5). Six subtypes of the dominant LGMDs (1A to 1F) and eleven subtypes of the recessive LGMDs (2A to 2K) have been identified. Mutations of the sarcoglycan complex of proteins have been identified in four of the limb girdle muscular dystrophies (2C, 2D, 2E, and 2F).31,32 These membrane proteins interact with dystrophin through another transmembrane protein, β-dystroglycan (see Fig. 27-9).
Myotonic Dystrophy
Myotonia, the sustained involuntary contraction of a group of muscles, is the cardinal symptom in this disease. Patients often complain of “stiffness” and have difficulty in releasing their grip, for instance, after a handshake. Myotonia can often be elicited by percussion of the thenar eminence.
Pathogenesis.
Inherited as an autosomal dominant trait, myotonic dystrophy is associated with a CTG trinucleotide repeat expansion on chromosome 19q13.2–q13.3. This expansion affects the mRNA for the dystrophia myotonia protein kinase (DMPK).33 In normal subjects, fewer than 30 repeats are present; disease develops with expansion of this repeat, and several thousand repeats may be present in severely affected individuals. The mutation is not stable within a pedigree; with each generation more repeats accumulate, and the disease becomes more severe, a phenomenon called anticipation (Chapter 5). Expansion of the trinucleotide repeat influences the eventual concentration of protein product. However, it is not established if the disease is caused by abnormality of the protein affected by the trinucleotide repeat, or if this alters splicing of other RNAs.
Morphology. Skeletal muscle may show variation in fiber size. In addition, there is a striking increase in the number of internal nuclei, which on longitudinal section may form conspicuous chains. Another well-recognized abnormality is the ring fiber, with a subsarcolemmal band of cytoplasm that appears distinct from the center of the fiber. The rim contains myofibrils that are oriented circumferentially around the longitudinally oriented fibrils in the rest of the fiber. The ring fiber may be associated with an irregular mass of sarcoplasm (sarcoplasmic mass) extending outward from the ring. These sarcoplasmic masses stain blue with hematoxylin and eosin, red with Gomori trichrome, and intensely blue with the NAD–tetrazolium reductase histochemical reaction. Histochemical techniques have demonstrated a relative atrophy of type 1 fibers early in the course in some cases. Of all the dystrophies, only myotonic dystrophy shows pathologic changes in the intrafusal fibers of muscle spindles, with fiber splitting, necrosis, and regeneration.
Clinical Course.
The disease often presents in late childhood with abnormalities in gait secondary to weakness of foot dorsiflexors and subsequently progresses to weakness of the hand intrinsic muscles and wrist extensors. Atrophy of muscles of the face and ptosis ensue, leading to the typical facial appearance. Cataracts, which are present in virtually every patient, may be detected early in the course by slit-lamp examination. Other associated abnormalities include frontal balding, gonadal atrophy, cardiomyopathy, smooth muscle involvement, decreased plasma IgG, and abnormal glucose tolerance. Dementia has been reported in some cases.
ION CHANNEL MYOPATHIES (CHANNELOPATHIES)
The ion channel myopathies, or channelopathies, are a group of familial diseases featuring myotonia, relapsing episodes of hypotonic paralysis (induced by vigorous exercise, cold, or a high-carbohydrate meal), or both. Hypotonia variants associated with elevated, depressed, or normal serum potassium levels at the time of the attack are called hyperkalemic, hypokalemic, and normokalemic periodic paralysis, respectively.
Pathogenesis.
As their name indicates, these diseases are caused by mutations in genes that encode ion channels.34 Hyperkalemic periodic paralysis results from mutations in the gene that encodes a skeletal muscle sodium channel protein (SCN4A), which regulates the entry of sodium into muscle during contraction. The gene for hypokalemic periodic paralysis encodes a voltage-gated L-type calcium channel.
Malignant hyperpyrexia (malignant hyperthermia) is a rare clinical syndrome characterized by a marked hypermetabolic state (tachycardia, tachypnea, muscle spasms, and later hyperpyrexia) triggered by anesthetics, most commonly halogenated inhalational agents and succinylcholine. The clinical syndrome may occur in predisposed individuals with hereditary muscle diseases, including congenital myopathies, dystrophinopathies, and metabolic myopathies. Mutations in several genes have been identified in families with susceptibility to malignant hyperthermia, including genes encoding L-type voltage-dependent calcium channel, notably the rynodine receptor (RyR1).35 Upon exposure to anesthetic, the mutant receptor allows uncontrolled efflux of calcium from the sarcoplasm. This leads to tetany, increased muscle metabolism, and excessive heat production. Diagnosis can be made either by identifying the genetic mutation or by exposing biopsied muscle to the anesthetic agent and observing contraction.
CONGENITAL MYOPATHIES
The congenital myopathies are a group of disorders defined largely on the basis of the pathologic findings within muscle.36 Most of these conditions share common clinical features, including onset in early life, nonprogressive or slowly progressive course, proximal or generalized muscle weakness, and hypotonia. Those affected at birth or in early infancy may present as “floppy infants” because of hypotonia or may have severe joint contractures (arthrogryposis); however, both hypotonia and arthrogryposis may also be caused by other neuromuscular dysfunction.
The best-characterized congenital myopathies are listed in Table 27-6. Figure 27-11 shows the structural characteristics of nemaline myopathy, one of the most distinctive types.
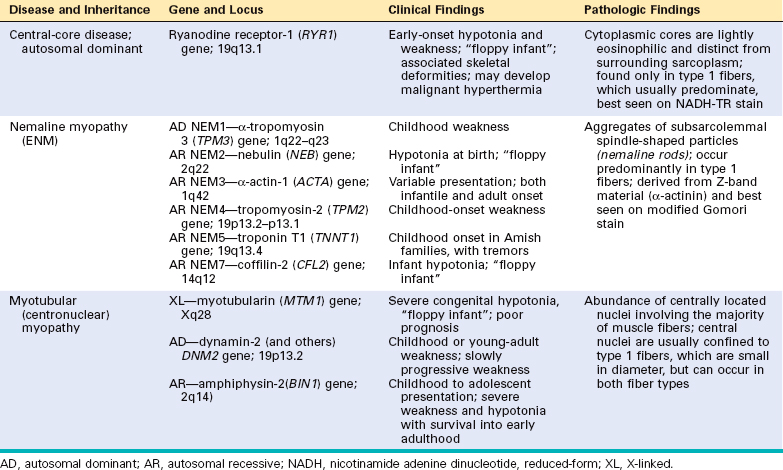
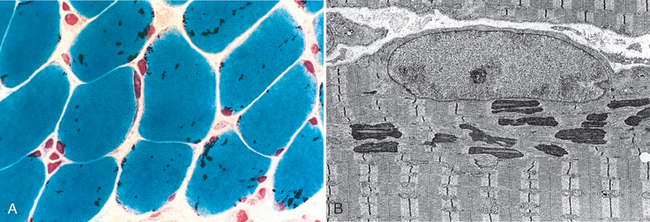
FIGURE 27-11 A, Nemaline myopathy with numerous rod-shaped, intracytoplasmic inclusions (dark purple structures). B, Electron micrograph of subsarcolemmal nemaline bodies, showing material of Z-band density located adjacent to nucleus, with normal sarcomeres (Z-band to Z-band) creating the typical cross-striation pattern of skeletal muscle.
MYOPATHIES ASSOCIATED WITH INBORN ERRORS OF METABOLISM
Many of the myopathies associated with metabolic disease involve disorders of glycogen synthesis and degradation (Chapter 5). Combinations of clinical, pathologic, and molecular information are used to arrive at a specific diagnosis.37 Myopathies can also result from disorders of mitochondrial function.
Lipid Myopathies
Abnormalities of carnitine transport or deficiencies of the mitochondrial dehydrogenase enzyme systems can lead to blocks in fatty acid oxidation and accumulation of lipid droplets within muscle (lipid myopathies).38,39 Patients with these disorders develop muscle pain, tightness, and myoglobinuria following prolonged exercise or exercise during fasting states. Fatty acids provide energy for muscle contraction, especially when glycogen stores are depleted (as in fasting). With a metabolic block in fatty acid oxidation, the required energy is not available, resulting in symptoms. Concomitant cardiomyopathies and fatty liver may also occur.
Mitochondrial Myopathies (Oxidative Phosphorylation Diseases)
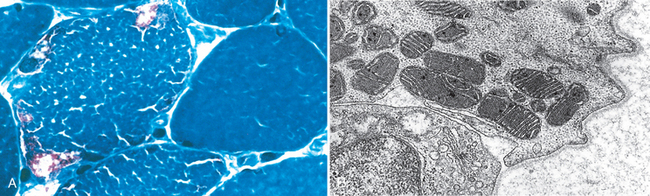
Approximately one fifth of the proteins involved in oxidative phosphorylation are encoded by the mitochondrial genome (mtDNA); additionally, this circular genome encodes 22 mitochondrial-specific transfer RNAs and 2 ribosomal RNA species. The remainder of the mitochondrial enzyme complexes are encoded in the nuclear genome. Mutations in both nuclear and mitochondrial genes cause the so-called mitochondrial myopathies.40 Diseases that involve the mtDNA show maternal inheritance, since only the oocyte contributes mitochondria to the embryo (Chapter 5). There is a high mutation rate for mtDNA compared with nuclear DNA. The mitochondrial diseases may present in young adulthood and manifest with proximal muscle weakness, sometimes with severe involvement of the extraocular muscles involved in eye movements (external ophthalmoplegia). The weakness may be accompanied by other neurologic symptoms, lactic acidosis, and cardiomyopathy, so this group of disorders is sometimes classified as mitochondrial encephalomyopathies (Chapter 28).41
Morphology. The most consistent pathologic finding in skeletal muscle is aggregates of abnormal mitochondria that are demonstrable only by special techniques.42 These occur under the sarcolemma in early stages, but with severe involvement, they may extend throughout the fiber. Since they are also associated with distortion of the myofibrils, the muscle fiber contour becomes irregular on cross-section, and the descriptive term ragged red fibers has been applied to them (Fig. 27-12A). Electron microscopy shows increased numbers of mitochondria with irregular shapes. Some contain paracrystalline parking lot inclusions or alterations in the structure of cristae (Fig. 27-12B). Cytochrome oxidase–negative fibers may be present in several mitochondrial myopathies, as assessed by histochemistry.
Clinical Course and Genetics.
The relationship between clinical course and the genetic alterations in the mitochondrial disorders is not straightforward; however, three general types of mutations have been defined. One consists of point mutations in mtDNA. Disorders associated with these show a maternal pattern of inheritance; some examples include myoclonic epilepsy with ragged red fibers, Leber hereditary optic neuropathy, and mitochondrial encephalomyopathy with lactic acidosis and strokelike episodes. As in other diseases caused by mutations in mtDNA, the expression of the disease is quite variable due to unequal distribution of mtDNA in affected cells (Chapter 5). A second set of mutations involves genes encoded by nuclear DNA and shows autosomal dominant or autosomal recessive inheritance. Some cases of subacute necrotizing encephalopathy (Leigh syndrome), exertional myoglobinuria, and infantile X-linked cardioskeletal myopathy (Barth syndrome) are due to mutations in nuclear DNA. The final subset of mitochondrial myopathies is caused by deletions or duplications of mtDNA. Examples include chronic progressive external ophthalmoplegia, characterized by a myopathy with prominent weakness of external ocular movements. Kearns-Sayre syndrome, another myopathy in this group, is also characterized by ophthalmoplegia but, in addition, includes pigmentary degeneration of the retina and complete heart block.
INFLAMMATORY MYOPATHIES
There are three subgroups of inflammatory muscle diseases: infectious, noninfectious inflammatory, and systemic inflammatory diseases that involve muscle along with other organs. Infectious myositis (Chapter 8) and systemic inflammatory diseases (Chapter 6) are discussed elsewhere.
Noninfectious Inflammatory Myopathies
Noninfectious inflammatory myopathies are a heterogeneous group of disorders that are most likely immune mediated and are characterized by injury and inflammation of skeletal muscle. Three relatively distinct disorders, dermatomyositis, polymyositis, and inclusion body myositis, are included in this category.43 These may occur as an isolated myopathy or as one component of an immune-mediated systemic disease, particularly systemic sclerosis (Chapter 6). The clinical features of each disorder are presented first to facilitate discussion of pathogenesis and morphologic changes.
Dermatomyositis.
As the name implies, individuals with dermatomyositis have an inflammatory disorder of the skin as well as skeletal muscle. It is characterized by a distinctive skin rash that may accompany or precede the onset of muscle disease. The classic rash takes the form of a lilac or heliotrope discoloration of the upper eyelids associated with periorbital edema (Fig. 27-13A). It is often accompanied by a scaling erythematous eruption or dusky red patches over the knuckles, elbows, and knees (Grotton lesions). Muscle weakness is slow in onset, bilaterally symmetric, and often accompanied by myalgias. It typically affects the proximal muscles first. As a result, tasks such as getting up from a chair and climbing steps become increasingly difficult. Fine movements controlled by distal muscles are affected only late in the disease. Dysphagia resulting from involvement of oropharyngeal and esophageal muscles occurs in one third of the affected individuals. Extramuscular manifestations, including interstitial lung disease, vasculitis, and myocarditis, may be present in some cases. Compared with the control populations, adults with dermatomyositis have a higher risk of developing visceral cancers. According to several studies, 20% to 25% of adults with dermatomyositis have cancer.44
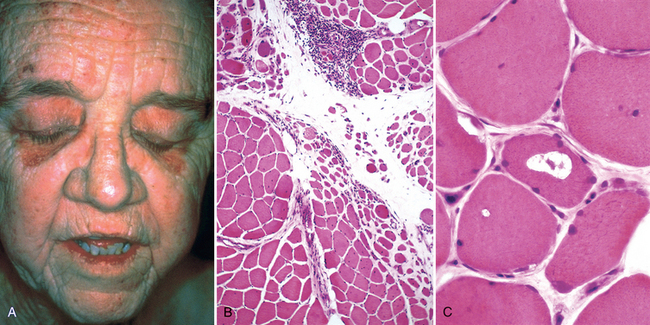
FIGURE 27-13 A, Dermatomyositis. Note the heliotrope rash affecting the eyelids. B, Dermatomyositis. The histologic appearance of muscle shows perifascicular atrophy of muscle fibers and inflammation. C, Inclusion body myositis showing a vacuole within a myocyte.
(Courtesy of Dr. Dennis Burns, Department of Pathology, University of Texas Southwestern Medical School, Dallas, TX.)
Juvenile dermatomyositis45 causes a similar onset of rash and muscle weakness but more often is accompanied by abdominal pain and involvement of the gastrointestinal tract. Mucosal ulceration, hemorrhage, and perforation may occur as the result of the dermatomyositis-associated vasculopathy. Calcinosis, which is uncommon in adult dermatomyositis, occurs in one third of youths with juvenile dermatomyositis.
Polymyositis.
This inflammatory myopathy is characterized by symmetric proximal muscle involvement, similar to that seen in dermatomyositis. It differs from dermatomyositis by the lack of cutaneous involvement and its occurrence mainly in adults. Similar to dermatomyositis, there may be inflammatory involvement of heart, lungs, and blood vessels.
Inclusion Body Myositis.
In contrast with the other two entities, inclusion body myositis begins with the involvement of distal muscles, especially extensors of the knee (quadriceps) and flexors of the wrists and fingers.46 Furthermore, the weakness may be asymmetric. It is an insidiously developing disorder that typically affects individuals over the age of 50 years. Most cases are sporadic, but familial cases have been recognized as “inclusion body myopathy.”
Etiology and Pathogenesis.
The cause of inflammatory myopathies is unknown, but the tissue injury is most likely mediated by immunological mechanisms. Capillaries seem to be the principal targets in dermatomyositis. Deposits of antibodies and complement are present in small blood vessels, and are associated with foci of myocyte necrosis. B cells and CD4+ T cells are present within the muscle, but there is a paucity of lymphocytes within the areas of myofiber injury. The perifascicular distribution of myocyte injury also suggests a vascular pathogenesis.
In contrast, polymyositis seems to be caused by cellmediated injury of myocytes. CD8+ cytotoxic T cells and macrophages are seen near damaged muscle fibers, and the expression of HLA class I and class II molecules is increased on the sarcolemma of normal fibers. Similar to other immune-mediated diseases, antinuclear antibodies are present in a variable number of cases, regardless of the clinical category (Chapter 6). The specificities of autoantibodies are quite varied, but those directed against transfer RNA synthetases seem to be more or less specific for inflammatory myopathies.47
The pathogenesis of inclusion body myositis is less clear. As in polymyositis, CD8+ cytotoxic T cells are found in the muscle, but in contrast to the other two forms of myositis, immunosuppressive therapy is not beneficial. Intracellular deposits of β-amyloid protein, amyloid β–pleated sheet fibrils, and hyperphosphorylated tau protein, features shared with Alzheimer disease, have drawn attention to a possible relationship to aging. The protein deposition may result from abnormal protein folding. The two hereditary forms of inclusion body myopathy have a similar morphology. The autosomal recessive form is caused by mutations in the GNE gene (encoding UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase), and the autosomal dominant form is caused by mutations in the gene encoding myosin heavy chain IIa.46,48
Morphology. The histologic features of the individual forms of myositis are quite distinctive and are described separately.
Dermatomyositis. The inflammatory infiltrates in dermatomyositis are located predominantly around small blood vessels and in the perimysial connective tissue. Typically, groups of atrophic fibers are particularly prominent at the periphery of fascicles. This “perifascicular atrophy” is sufficient for diagnosis, even if the inflammation is mild or absent (Fig. 27-13B), and is most likely related to a relative state of hypoperfusion of the periphery of muscle fascicles. Quantitative analyses reveal a marked reduction in the intramuscular capillaries, believed to result from vascular endothelial injury and fibrosis. Necrotic muscle fibers and regeneration may also be seen throughout the fascicle, as in polymyositis.
Polymyositis. In this condition, the inflammatory cells are found in the endomysium. CD8+ lymphocytes and other lymphoid cells surround and invade healthy muscle fibers. Both necrotic and regenerating muscle fibers are scattered throughout the fascicle, without the perifascicular atrophy seen in dermatomyositis. There is no evidence of vascular injury in polymyositis.
Inclusion Body Myositis. The diagnostic finding in inclusion body myositis is the presence of rimmed vacuoles (Fig. 27-13C). The vacuoles are present within myocytes, and they are highlighted by basophilic granules at their periphery. In addition, the vacuolated fibers may also contain amyloid deposits that reveal typical staining with Congo red. Under the electron microscope, tubular and filamentous inclusions are seen in the cytoplasm and the nucleus that are composed of β-amyloid or hyperphosphorylated tau. The inflammatory cell infiltrate is similar to that seen in polymyositis.
The diagnosis of myositis is based on clinical symptoms, electromyography, elevated creatinine kinase in serum, and biopsy. Electromyography (EMG) is particularly informative; mixed neurogenic and myopathic changes on EMG are suggestive of inflammatory myopathy. As might be expected, muscle injury is associated with elevated serum levels of creatine kinase. Biopsy is required for definitive diagnosis. Immunosuppressive therapy is beneficial in adult and juvenile dermatomyositis and in polymyositis but not in inclusion-body myositis.
TOXIC MYOPATHIES
Thyrotoxic Myopathy
Thyrotoxic myopathy presents most commonly as an acute or chronic proximal muscle weakness that may precede the onset of other signs of thyroid dysfunction. Exophthalmic ophthalmoplegia is characterized by swelling of the eyelids, edema of the conjunctiva, and diplopia. In hypothyroidism there may be cramping or aching of muscles, and movements and reflexes are slowed. Findings include fiber atrophy, an increased number of internal nuclei, glycogen aggregates, and, occasionally, deposition of mucopolysaccharides in the connective tissue.
In thyrotoxic myopathy, there is myofiber necrosis, regeneration, and interstitial lymphocytosis. In chronic thyrotoxic myopathy, there may be only slight variability of muscle fiber size, mitochondrial hypertrophy, and focal myofibril degeneration; fatty infiltration of muscle is seen in severe cases. Exophthalmic ophthalmoplegia is limited to the extraocular muscles, which may be edematous and enlarged.
Ethanol Myopathy
Binge drinking of alcohol produces an acute toxic syndrome of rhabdomyolysis with accompanying myoglobinuria, which may lead to renal failure. Clinically, the affected individual may develop acute pain that is either generalized or confined to a single muscle group. Some patients have a complicated clinicopathologic syndrome consisting of proximal muscle weakness and electrophysiologic evidence of myopathy superimposed on alcoholic neuropathy. On histologic examination there is swelling of myocytes, fiber necrosis, myophagocytosis, and regeneration. There may also be evidence of denervation.
Drug-Induced Myopathies
Proximal muscle weakness and atrophy can occur as a result of the deleterious effects of steroids on muscle, whether in Cushing syndrome or during therapeutic administration of steroids, a condition known as steroid myopathy. The severity of clinical disability is variable and not directly related to the steroid level or the therapeutic regimen. It is characterized by muscle fiber atrophy, predominantly of type 2 fibers. When the myopathy is severe, there may be a bimodal distribution of fibers due to the presence of type 1 fibers of nearly normal caliber and markedly atrophic type 2 fibers. Electron microscopy has shown dilation of the sarcoplasmic reticulum and thickening of the basal laminae.
Chloroquine, originally used in the treatment of malaria but subsequently used in other clinical settings, can produce a proximal myopathy in humans. The most prominent finding is the presence of vacuoles within myocytes. Two types of vacuoles have been described: (1) autophagic membrane-bound vacuoles containing membranous debris; and (2) curvilinear bodies with short curved membranous structures with alternating light and dark zones. Vacuoles can be seen in as many as 50% of the myocytes, most commonly type 1 fibers, and with progression, myocyte necrosis may develop. A similar vacuolar myopathy occurs in some individuals treated with hydrochloroquine.
Statins are very frequently prescribed in the United States to reduce cholesterol and the risks of acute ischemic cardiac events and stroke. Myopathy is the most common complication of statins.49 “Statin-induced myopathy” can occur with use of any of the statins (e.g., atorvastatin, simvastatin, pravastatin). The incidence is approximately 1.5% of users, and is unrelated to dose, cumulative dose, or statin subtype. Drug metabolism characteristics that can be predicted with pharmacogenetic predictors, on the other hand, may help to identify individuals at risk for myopathy.
DISEASES OF THE NEUROMUSCULAR JUNCTION
Myasthenia Gravis
Myasthenia gravis is a muscle disease caused by immune-mediated loss of acetylcholine receptor. It has a prevalence of about 30 in 100,000 persons.50 When arising before age 40 years it is most commonly seen in women, but it occurs equally in both sexes in older patients. Thymic hyperplasia is found in 65% and thymoma in 15% of affected patients. Analysis of neuromuscular transmission in myasthenia gravis shows a decrease in the number of muscle acetylcholine receptors (AChRs), and circulating antibodies to the AChR are present in nearly all cases. The disease can be passively transferred to animals with serum from affected individuals.
Pathogenesis.
In most instances the autoantibodies against the AChR lead to loss of functional AChRs at the neuromuscular junction by (1) fixing complement and causing direct injury to the postsynaptic membrane, (2) increasing the internalization and degradation of the receptors, and (3) inhibiting binding of acetylcholine. Electrophysiologic studies are notable for diminished motor responses after repeated stimulation; nerve conduction is normal. Sensory as well as autonomic functions are not affected. Despite the evidence that antibodies to AChR have a critical pathogenic role, there is not always a correlation between antibody titers and the neuromuscular deficit. Interestingly, in light of the immune-mediated etiology of the disease, thymic abnormalities are common in these patients, but the precise link between thymic function and autoimmunity is uncertain. Regardless of the type of thymic pathology, most affected individuals improve after thymectomy.
Morphology. By light microscopic examination, muscle biopsy specimens are usually unrevealing. In severe cases type 2 fiber atrophy due to disuse may be found. By electron microscopy the postsynaptic membrane is simplified, and there is loss of AChRs from the region of the synapse. Immune complexes and the complement membrane attack complex (C5–C9) can be found along the postsynaptic membrane as well.
Clinical Course.
Typically, weakness begins with the extraocular muscles; drooping eyelids (ptosis) and double vision (diplopia) cause the affected individual to seek medical attention. However, the initial symptoms may take the form of generalized weakness. The weakness fluctuates over days, hours, or even minutes, and intercurrent medical conditions can lead to exacerbations. Patients show improvement in strength in response to administration of anticholinesterase agents, which remains a useful clinical test. Respiratory compromise was a major cause of mortality in the past; 95% of affected individuals now survive more than 5 years after diagnosis because of improved methods of treatment and better ventilatory support. Effective forms of treatment include anticholinesterase drugs, prednisone, plasmapheresis, and thymectomy when thymic lesions are present.50
Lambert-Eaton Myasthenic Syndrome
Lambert-Eaton myasthenic syndrome is a disease of the neuromuscular junction that is distinct from myasthenia gravis. It is usually a paraneoplastic process, most commonly with small-cell carcinoma of the lung (60% of cases), but can occur in the absence of underlying malignant disease. Affected individuals develop proximal muscle weakness and autonomic dysfunction. Unlike myasthenia gravis, no clinical improvement is produced by anticholinesterase agents, and electrophysiologic studies show evidence of enhanced neurotransmission with repetitive stimulation. These clinical features allow this disorder to be distinguished from myasthenia gravis.
The content of anticholinesterase is normal in neuromuscular junction synaptic vesicles, and the postsynaptic membrane is normally responsive to anticholinesterase, but fewer vesicles are released in response to each presynaptic action potential. Some affected individuals have antibodies that recognize presynaptic PQ-type voltage-gated calcium channels, and a similar disease can be transferred to animals with these antibodies, suggesting that autoimmunity to the calcium channel causes the disease.
TUMORS OF SKELETAL MUSCLE
Skeletal muscle tumors are discussed with other soft-tissue tumors (Chapter 26).
1 Navarro X, et al. Neural plasticity after peripheral nerve injury and regeneration. Prog Neurobiol. 2007;82:163.
2 Chen Z-L, et al. Peripheral regeneration. Annu Rev Neurosci. 2007;30:209.
3 Raivich G, Makwana M. The making of successful axonal regeneration: genes, molecules and signal transduction pathways. Brain Res Rev. 2007;53:287.
4 Chen C-J, et al. Transplantation of bone marrow stromal cells for peripheral nerve repair. Exp Neurol. 2007;204:443.
5 Chio A, et al. Guillain-Barré syndrome: a prospective, population-based incidence and outcome survey. Neurology. 2003;60:1146.
6 Hartung H-P, et al. Acute immunoinflammatory neuropathy: update on Guillain-Barré syndrome. Curr Opin Neurol. 2002;15:571.
7 French CSG. Recommendations on diagnostic strategies for chronic inflammatory demyelinating polyradiculoneuropathy. J Neurol Neurosurg Psychiatr. 2008;79:115.
8 Rentzos M, et al. Chronic inflammatory demyelinating polyneuropathy: a 6-year retrospective clinical study of a hospital-based population. J Clin Neurosci. 2007;14:229.
9 de Freitas MRG. Infectious neuropathy. Curr Opin Neurol. 2007;20:548.
10 Piradov MA, et al. Diphtheritic polyneuropathy: clinical analysis of severe forms. Arch Neurol. 2001;58:1438.
11 Weinberg JM. Herpes zoster: epidemiology, natural history, and common complications. J Am Acad Dermatol. 2007;57:S130.
12 Klein CJ. The inherited neuropathies. Neurol Clin. 2007;25:173.
13 Suter U, Scherer SS. Disease mechanisms in inherited neuropathies. Nat Rev Neurosci. 2003;4:714.
14 Nave K-A, et al. Mechanisms of disease: inherited demyelinating neuropathies—from basic to clinical research. Nat Clin Pract Neurol. 2007;3:453.
15 Zhao C, et al. Charcot-Marie-Tooth disease type 2A caused by mutation in a microtubule motor KIF1Bbeta. Cell. 2001;105:587. Erratum in: Cell. 2001;106:127.
16 Szigeti K, et al. Molecular diagnostics of Charcot-Marie-Tooth disease and related peripheral neuropathies. Neuromolecular Med. 2006;8:243.
17 Said G. Diabetic neuropathy—a review. Nat Clin Pract Neurol. 2007;3:331.
18 Zochodne DW. Diabetes mellitus and the peripheral nervous system: manifestations and mechanisms. Muscle Nerve. 2007;36:144.
19 Krishnan AV, Kiernan MC. Uremic neuropathy: clinical features and new pathophysiological insights. Muscle Nerve. 2007;35:273.
20 Koike H, Sobue G. Alcoholic neuropathy. Curr Opin Neurol. 2006;19:481.
21 Rudnicki SA, Dalmau J. Paraneoplastic syndromes of the peripheral nerves. Curr Opin Neurol. 2005;18:598.
22 Aguirre-Cruz L, et al. Clinical relevance of non-neuronal auto-antibodies in patients with anti-Hu or anti-Yo paraneoplastic diseases. J Neuro-Oncol. 2005;71:39.
23 Steck AJ, et al. Anti-myelin-associated glycoprotein neuropathy. Curr Opin Neurol. 2006;19:458.
24 London Z, Albers JW. Toxic neuropathies associated with pharmaceutic and industrial agents. Neurol Clin. 2007;25:257.
25 Bland JDP. Carpal tunnel syndrome [see comment]. BMJ. 2007;335:343.
26 Lunn MR, Wang CH. Spinal muscular atrophy. Lancet. 2008;371:2120.
27 McNally EM. New approaches in the therapy of cardiomyopathy in muscular dystrophy. Annu Rev Med. 2007;58:75.
28 Ervasti JM. Dystrophin, its interactions with other proteins, and implications for muscular dystrophy. Biochim Biophys Acta. 2007;1772:108.
29 Rodino-Klapac LR, et al. Gene therapy for Duchenne muscular dystrophy: expectations and challenges. Arch Neurol. 2007;64:1236.
30 van Deutekom JC, et al. Local dystrophin restoration with antisense oligonucleotide PRO051 [see comment]. N Engl J Med. 2007;357:2677.
31 Bonnemann CG. Limb-girdle muscular dystrophy in childhood. Pediatr Ann. 2005;34:569.
32 Guglieri M, et al. Molecular etiopathogenesis of limb girdle muscular and congenital muscular dystrophies: boundaries and contiguities. Clin Chim Acta. 2005;361:54.
33 Schara U, Schoser BGH. Myotonic dystrophies type 1 and 2: a summary on current aspects. Semin Pediatr Neurol. 2006;13:71.
34 Vicart S, et al. Human skeletal muscle sodium channelopathies. Neurol Sci. 2005;26:194.
35 Brandom BW. The genetics of malignant hyperthermia. Anesthesiol Clin North America. 2005;23:615.
36 Bruno C, Minetti C. Congenital myopathies. Curr Neurol Neurosci Rep. 2004;4:68.
37 Di Mauro S. Muscle glycogenoses: an overview. Acta Myol. 2007;26:35.
38 Wieser T, et al. Carnitine palmitoyltransferase II deficiency: molecular and biochemical analysis of 32 patients. Neurology. 2003;60:1351.
39 Vladutiu GD, Slonim AE. Combined biochemical and molecular diagnosis in blood of a common lipid myopathy. Muscle Nerve. 2000;23:1773.
40 DiMauro S, Gurgel-Giannetti J. The expanding phenotype of mitochondrial myopathy. Curr Opin Neurol. 2005;18:538.
41 DiMauro S, Hirano M. Mitochondrial encephalomyopathies: an update. Neuromuscul Disord. 2005;15:276.
42 Sarnat HB, Marin-Garcia J. Pathology of mitochondrial encephalomyopathies. Can J Neurol Sci. 2005;32:152.
43 Dalakas MC. Mechanisms of disease: signaling pathways and immunobiology of inflammatory myopathies. Nat Clin Pract Rheumatol. 2006;2:219. Erratum in: Nat Clin Pract Rheumatol. 2006;2:398.
44 Callen JP, Wortmann RL. Dermatomyositis. Clin Dermatol. 2006;24:363.
45 Griffin TA, Reed AM. Pathogenesis of myositis in children. Curr Opin Rheumatol. 2007;19:487.
46 Askanas V, Engel WK. Inclusion-body myositis, a multifactorial muscle disease associated with aging: current concepts of pathogenesis. Curr Opin Rheumatol. 2007;19:550.
47 Mimori T, et al. Autoantibodies in idiopathic inflammatory myopathy: an update on clinical and pathophysiological significance. Curr Opin Rheumatol. 2007;19:523.
48 Needham M, et al. Genetics of inclusion-body myositis. Muscle Nerve. 2007;35:549.
49 Antons KA, et al. Clinical perspectives of statin-induced rhabdomyolysis. Am J Med. 2006;119:400.
50 Conti-Fine BM, et al. Myasthenia gravis: past, present, and future. J Clin Invest. 2006;116:2843.