Chapter 26 Bones, Joints, and Soft-Tissue Tumors

BONES
Recognized by the bard for its persistence after death (Alas, poor Yorick!), the skeletal system is vital during life. It has an essential role in mineral homeostasis, houses the hematopoietic elements, provides mechanical support for movement, protects viscera, and determines body size and shape. As is well known, bones are largely made up of an organic matrix (osteoid) and the mineral calcium hydroxyapatite, which gives the bones strength and hardness. What is not obvious is that, despite its stony hard structure, bone is a dynamic tissue that is continuously resorbed, renewed, and remodeled. These processes are carried out by several different types of bone cells that are regulated by a number of transcription factors, cytokines, and growth factors1-3 (Fig. 26-1).
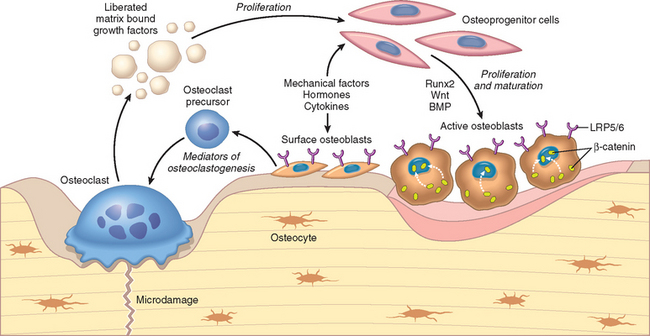
FIGURE 26-1 Bone cells and their interrelated activities. Hormones, cytokines, growth factors, and signal-transducing molecules are key in their formation and maturation, and allow communication between osteoblasts and osteoclasts. Bone resorption and formation in remodeling are coupled processes that are controlled by systemic factors and local cytokines, some of which are deposited in the bone matrix. BMP, bone morphogenic protein; LRP5/6, LDL receptor related proteins 5 and 6.
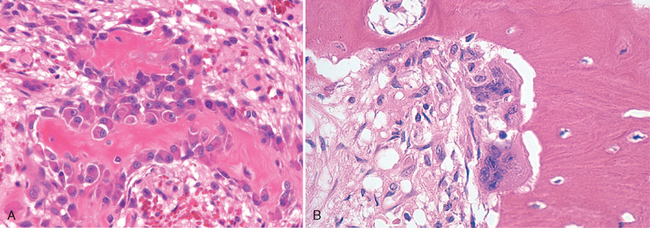
FIGURE 26-2 A, Active osteoblasts synthesizing bone matrix. The surrounding spindle cells represent osteoprogenitor cells. B, Two osteoclasts resorbing bone.
These cells and locally produced factors work together to regulate bone homeostasis. The control mechanisms are not known completely, but several signaling pathways of particular importance have emerged (Fig. 26-3). One such pathway involves three factors: (1) the transmembrane receptor RANK (receptor activator for NF-κB), which is expressed on osteoclast precursors; (2) RANK ligand, (RANKL) which is expressed on osteoblasts and marrow stromal cells; and (3) osteoprotegrin (OPG), a secreted “decoy” receptor made by osteoblasts and several other types of cells that can bind RANKL and thus short-circuit its interaction with RANK.9 When stimulated by RANKL, RANK signaling activates the transcription factor NF-κB, which is essential for the generation and survival of osteoclasts. A second important pathway involves M-CSF produced by osteoblasts and the M-CSF receptor, which is expressed by osteoclast progenitors. Activation of the M-CSF receptor stimulates a tyrosine kinase activity that is also crucial for the generation of osteoclasts. The other notable pathway is the WNT/β-catenin pathway. WNT proteins produced by marrow stromal cells bind to the LRP5 and LRP6 receptors on osteoblasts (see Fig. 26-1) and thereby trigger the activation of β-catenin and the production of OPG. The importance of these pathways is proven by rare but informative germline mutations in the OPG, RANK, RANKL, and LRP5 genes, which cause severe disturbances of bone metabolism (described later).
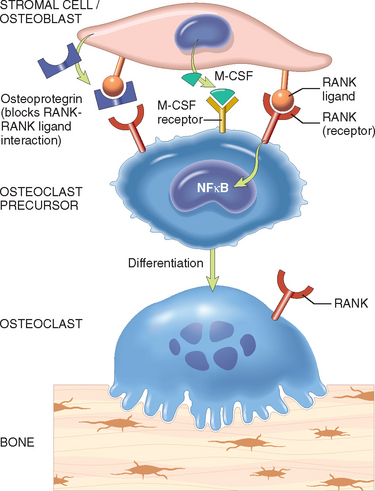
FIGURE 26-3 Paracrine molecular mechanisms that regulate osteoclast formation and function. Osteoclasts are derived from the same stem cells that produce macrophages. Osteoblast/stromal cell membrane–associated RANKL binds to its receptor RANK located on the cell surface of osteoclast precursors. This interaction in the background of macrophage colony-stimulating factor (M-CSF) causes the precursor cells to produce functional osteoclasts. Stromal cells also secrete osteoprotegrin (OPG), which acts as a decay receptor for RANKL, preventing it from binding the RANK receptor on osteoclast precursors. Consequently, OPG prevents bone resorption by inhibiting osteoclast differentiation.
Bone formation and resorption are tightly coupled and subject to fine-tuning at several levels. For example, because OPG and RANKL oppose one another, either bone resorption or bone formation can be favored by simply tipping the RANKL : OPG ratio one way or the other. Systemic factors that affect RANKL and OPG expression include hormones (parathyroid hormone, estrogen, testosterone, and glucocorticoids), vitamin D, inflammatory cytokines (e.g., IL-1), and growth factors (such as bone morphogenetic factors); each presumably acts by altering the levels of NF-κB and WNT/β-catenin signaling in osteoblasts. Another level of control involves paracrine crosstalk between osteoblasts and osteoclasts, and possibly osteocytes as well. We have seen that osteoblasts can enhance or inhibit osteoclast development and function by expressing OPG and RANKL in various proportions. As osteoclasts disassemble matrix proteins deposited by osteoblasts, growth factors, cytokines, and enzymes (such as collagenase) bound to the matrix are liberated and activated, including some that stimulate osteoblasts. Thus, as bone is broken down to its elemental units, substances are released into the microenvironment that initiate its renewal (see Fig. 26-1).
The proteins of bone include type 1 collagen and many noncollagenous proteins derived mainly from osteoblasts. Osteoblasts deposit collagen either in a random weave known as woven bone or in an orderly layered manner designated lamellar bone (Fig. 26-4). Normally, woven bone is seen in sites of rapid bone formation such as the fetal skeleton and the base of growth plates. It is produced quickly and resists forces equally from all directions. The presence of woven bone in the adult is always abnormal; however, it is not diagnostic of a particular disease. We will see examples of this later in this chapter. Lamellar bone, which gradually replaces woven bone during growth, is deposited much more slowly and is stronger than woven bone.
The noncollagenous proteins of bone are bound to the matrix and grouped according to their function (Table 26-1).8,10 Of these, only osteocalcin is unique to bone. It is measurable in the serum and used as a sensitive and specific marker for osteoblast activity. Cytokines and growth factors control bone cell proliferation, maturation, and metabolism, thereby playing a crucial role translating mechanical and metabolic signals into local bone cell activity and eventual skeletal adaptation.8,11 In this fashion the skeleton is uniquely able to change its structure in response to new physical forces; witness the repositioning of teeth by braces.
TABLE 26-1 Proteins of Bone Matrix
| OSTEOBLAST-DERIVED PROTEINS |
| PROTEINS CONCENTRATED FROM SERUM |
IGF, insulin-like growth factor; TGF, transforming growth factor; PDGF, platelet-derived growth factor; IL, interleukin; RANKL, RANK ligand.
Bone Modeling, Remodeling, and Peak Bone Mass
Local collections of osteocytes, osteoblasts, and osteoclasts work together to control bone formation and resorption, creating a functional unit referred to as the basic multicellular unit (BMU).12,13 Early in life, as the skeleton grows and enlarges (modeling), bone formation predominates. Once the skeleton has reached maturity, the breakdown and renewal of bone that constitutes skeletal maintenance is called remodeling and is probably initiated at sites experiencing fatigue and microdamage. In adults, BMUs remodel or replace 10% of the skeleton annually.
Peak bone mass is achieved in early adulthood after the cessation of growth, and it is determined by a variety of factors, including polymorphisms in the receptors for vitamin D and LRP5/6, nutrition, physical activity, age, and hormonal status. Beginning in the fourth decade, however, the amount of bone resorbed by the BMUs exceeds that formed, so that there is a steady decrement in skeletal mass.
Bone Growth and Development
Skeletal morphogenesis is determined by the homeobox genes, which encode transcription factors essential for the normal development of the skeleton.12,14 Most bones are first formed as a cartilage model or anlage. Subsequently, around the eighth week of gestation the process of enchondral ossification begins, and the cartilage is removed by osteoclast-type cells forming the medullary canal. This process progresses along the length of the bone, while concurrently the periosteum in the midshaft generates osteoblasts that deposit the beginnings of the cortex; this region is known as the primary center of ossification. A similar sequence of events occurs in the epiphysis, resulting in the removal of cartilage and deposition of bone in a centrifugal fashion (secondary center of ossification), such that a plate of the cartilage anlage becomes entrapped between the expanding centers of ossification forming the physis or growth plate (Fig. 26-5). The chondrocytes within the growth plate are responsible for longitudinal growth as they undergo a series of changes, including proliferation, growth, maturation, and apoptosis—controlled by a number of signaling pathways, including those involving FGF receptors and bone morphogenetic protein, hedgehog protein, and parathyroid-hormone (PTH)-related protein.15 In the region of apoptosis the matrix mineralizes and is resorbed by osteoclasts; however, remnant struts persist and act as scaffolding for the deposition of bone on their surfaces. These structures are known as primary spongiosa and are the first bony trabeculae. A similar process occurs at the base of articular cartilage, and by this mechanism bones increase in length, and articular surfaces increase in diameter. In contrast, bones derived from intramembranous formation, such as the cranium and lateral portions of the clavicles, are formed by osteoblasts directly from a fibrous layer of tissue that is derived from mesenchyme. Because bone tissue is made only by osteoblasts, the enlargement of bones is achieved by the deposition of new bone on a preexisting surface. This mechanism of appositional growth is key to understanding bone growth and modeling.
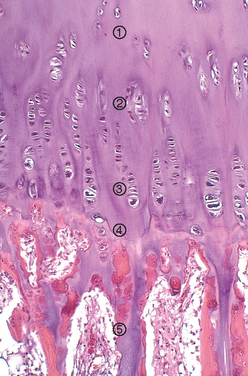
FIGURE 26-5 Active growth plate with ongoing enchondral ossification. 1, Reserve zone. 2, Zone of proliferation. 3, Zone of hypertrophy. 4, Zone of mineralization. 5, Primary spongiosa.
The complexity of the skeleton’s growth, development, maintenance, and relationships with other organ systems makes it unusually vulnerable to adverse influences. Not surprisingly, then, primary and secondary diseases of bone are varied and numerous. The spectrum of bone disorders is broad and the classification system is not standardized; here we will categorize the various disorders according to their perceived pathogenesis.
Developmental Abnormalities in Bone Cells, Matrix, and Structure
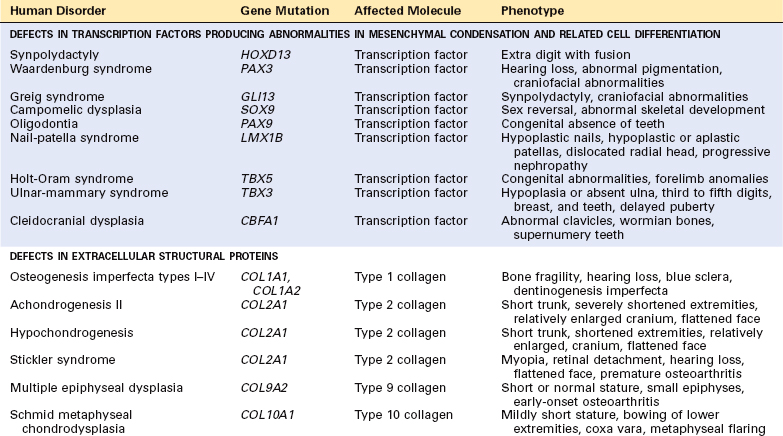
Developmental abnormalities of the skeleton are frequently genetically based; they first become manifest during the earliest stages of bone formation. In contrast, acquired diseases are usually detected in adulthood.11 Developmental anomalies resulting from localized problems in the migration of the mesenchymal cells and the formation of the condensations are known as dysostoses. They are usually limited to defined embryologic structures and may result from mutations in certain transcription factors (e.g., homeobox genes). In contrast, mutations in the regulators of skeletal organogenesis, such as signaling molecules (e.g., growth factors and their receptors) and matrix components (e.g., types 1 and 2 collagen) affect cartilage and bone tissues globally; these disorders are known as dysplasias.8,11,12,15-17 Table 26-2 shows a classification of developmental abnormalities of the bone based on the nature of the genetic abnormality. The classification of developmental and acquired abnormalities that follows is based on identified genetic defects and the skeletal manifestations of the disease processes. Many of the conditions can be classified in more than one category.

MALFORMATIONS AND DISEASES CAUSED BY DEFECTS IN NUCLEAR PROTEINS AND TRANSCRIPTION FACTORS
Congenital malformations or dysostoses of bone are relatively uncommon. The simplest anomalies include failure of a bone to develop (e.g., congenital absence of a phalanx, rib, or clavicle); the formation of extra bones (supernumerary ribs or digits); the fusion of two adjacent digits (syndactylism); or the development of long, spider-like digits. Some of these result from defects in the formation of the mesenchymal condensations and their differentiation into the cartilage anlage. They are caused by genetic alterations that affect transcription factors, especially those encoded by the homeobox genes, and certain cytokines.8,11 One example of a defect in mesenchymal condensation is caused by a mutation in the homeobox HOXD13 gene, which produces an extra digit between the third and fourth fingers as well as some degree of syndactyly.12 Loss of function mutations in the RUNX2 gene, which produces transcription factors important in osteoblastogenesis and some chondrocyte cell activity, results in cleidocranial dysplasia, an autosomal dominant disorder characterized by patent fontanelles, delayed closure of cranial sutures, Wormian bones, delayed eruption of secondary teeth, primitive clavicles, and short height.
DISEASES CAUSED BY DEFECTS IN HORMONES AND SIGNAL TRANSDUCTION MECHANISMS
Achondroplasia is the most common disease of the growth plate and is a major cause of dwarfism. It is caused by a mutation in the FGF receptor 3 (FGFR3).11 Normally, FGFmediated activation of FGFR3 inhibits cartilage proliferation; in achondroplasia, the mutations cause constitutive activation of FGFR3 and thereby suppress growth.
Achondroplasia is an autosomal dominant disorder; curiously approximately 80% of cases stem from new mutations, almost all of which occur in the paternal allele. Affected individuals have shortened proximal extremities, a trunk of relative normal length, and an enlarged head with bulging forehead and conspicuous depression of the root of the nose. The skeletal abnormalities are usually not associated with changes in longevity, intelligence, or reproductive status.
Thanatophoric dwarfism is the most common lethal form of dwarfism, affecting about one in every 20,000 live births. It is also caused by gain-of-function mutations in FGFR3 that differ from those in achondroplasia.11 Affected individuals have micromelic shortening of the limbs, frontal bossing, relative macrocephaly, a small chest cavity, and a bell-shaped abdomen. The underdeveloped thoracic cavity leads to respiratory insufficiency, and these individuals frequently die at birth or soon after. The histologic changes in the growth plate show diminished proliferation of chondrocytes and poor columnization in the zone of proliferation.
Increased bone mass is a manifestation of a variety of diseases. Several are caused by caused by gain-of-function mutations in the gene that encodes LPR5, a cell surface receptor that is essential for the activation of the WNT/β-catenin pathway in osteoblasts. These diseases, namely endosteal hyperostosis, Van Buchem disease, and autosomal dominant osteopetrosis type 1, are characterized by increased bone mass including cortical thickening, enlarged and elongated mandible, and increased density and enlargement of the cranial vault; some affected individuals may develop torus palatinus.
Conversely, inactivating mutations in LPR5 cause osteoporosis pseudoglioma syndrome. In this disorder the skeleton is severely osteoporotic, resulting in fractures due to insufficient bone formation.
DISEASES ASSOCIATED WITH DEFECTS IN EXTRACELLULAR STRUCTURAL PROTEINS
The interaction of the organic components of bone matrix is complex and a focus of intense scientific investigation. The importance of the structural bone proteins is exemplified by the diseases associated with deranged metabolism of the collagens important in bone and cartilage formation (types 1, 2, 9, 10, and 11). Their clinical manifestations are highly variable, ranging from lethal disease to premature osteoarthritis.
Type 1 Collagen Diseases (Osteogenesis Imperfecta)
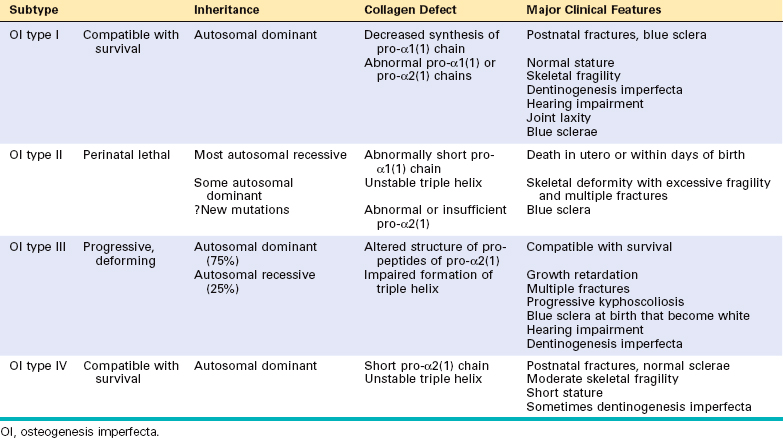
Osteogenesis imperfecta, or brittle bone disease, is a phenotypically diverse disorder caused by deficiencies in the synthesis of type 1 collagen. It is the most common inherited disorder of connective tissue. It principally affects bone, but also impacts other tissues rich in type 1 collagen (joints, eyes, ears, skin, and teeth). Osteogenesis imperfecta usually results from autosomal dominant mutations (over 800 have been identified) in the genes that enco de theα1 and α2 chains of collagen.19 Many of these mutations involve the substitution of glycine residues in the triple-helical domain. The genotypephenotype relationship underlying osteogenesis imperfecta is based on the location of the mutation within the protein. Mutations resulting in decreased synthesis of qualitatively normal collagen are associated with mild skeletal abnormalities. More severe or lethal phenotypes have abnormal polypeptide chains that cannot be arranged in the triple helix. Recently, mutations in the genes for cartilage-associated protein (CRTAP) and leucine proline-enriched proteoglycan 1 (LEPRE1) have been shown to be responsible for three rare variants of the disease.20
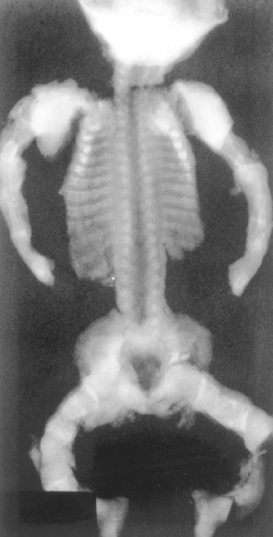
Clinically, osteogenesis imperfecta is separated into four major subtypes that vary widely in severity (Table 26-3). The type II variant is at one end of the spectrum and is uniformly fatal in utero or during the perinatal period. It is characterized by extraordinary bone fragility with multiple intrauterine fractures (Fig. 26-6). In contrast, individuals with the type I form have a normal life span but experience childhood fractures that decrease in frequency following puberty. Other findings include blue sclerae caused by decreased collagen content, making the sclera translucent and allowing partial visualization of the underlying choroid; hearing loss related to both a sensorineural deficit and impeded conduction due to abnormalities in the bones of the middle and inner ear; and dental imperfections (small, misshapen, and blue-yellow teeth) secondary to a deficiency in dentin. The basic abnormality in all forms of osteogenesis imperfecta is too little bone, thus constituting a type of osteoporosis with marked cortical thinning and attenuation of trabeculae.
Diseases Associated with Mutations of Types 2, 9, 10, and 11 Collagen
Types 2, 9, 10, and 11 collagens are important structural components of hyaline cartilage. Although uncommon, mutations in the genes encoding them produce an array of disorders ranging from the fatal to those compatible with life but associated with early destruction of joints (see Table 26-2). In the severe disorders, the type 2 collagen molecules are not secreted by the chondrocytes, and insufficient bone formation occurs. In the milder disorders there is reduced synthesis of normal type 2 collagen.
DISEASES ASSOCIATED WITH DEFECTS IN FOLDING AND DEGRADATION OF MACROMOLECULES
Mucopolysaccharidoses
The mucopolysaccharidoses, as discussed earlier (Chapter 5), are a group of lysosomal storage diseases that are caused by deficiencies in the enzymes that degrade dermatan sulfate, heparan sulfate, and keratan sulfate. The affected enzymes are mainly acid hydrolases. Mesenchymal cells, especially chondrocytes, normally metabolize extracellular matrix mucopolysaccharides; hence, cartilage formation is severely affected. Consequently, many of the skeletal manifestations of the mucopolysaccharidoses result from abnormalities in hyaline cartilage, including the cartilage anlage, growth plates, costal cartilages, and articular surfaces. It is not surprising, therefore, that affected individuals are frequently of short stature and have chest wall abnormalities, and malformed bones.
DISEASES ASSOCIATED WITH DEFECTS IN METABOLIC PATHWAYS (ENZYMES, ION CHANNELS, AND TRANSPORTERS)
Osteopetrosis
Osteopetrosis, also known as marble bone disease and Albers-Schönberg disease, refers to a group of rare genetic diseases that are characterized by reduced bone resorption and diffuse symmetric skeletal sclerosis due to impaired formation or function of osteoclasts (Fig. 26-7). The term osteopetrosis reflects the stonelike quality of the bones; however, the bones are abnormally brittle and fracture easily, like a piece of chalk. Osteopetrosis is classified into variants based on both the mode of inheritance and the clinical findings. The two major groups include autosomal recessive and dominant forms. The autosomal recessive type is further divided into mild and severe variants. The autosomal recessive severe type and the autosomal dominant mild type are the most common variants.
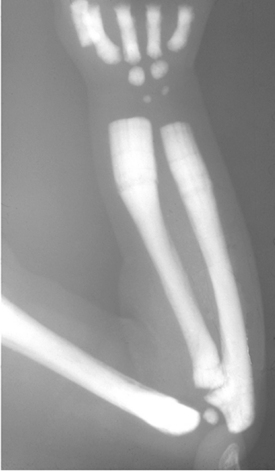
FIGURE 26-7 Radiogram of the upper extremity in an individual with osteopetrosis. The bones are diffusely sclerotic, and the distal metaphyses of the ulna and radius are poorly formed
(Erlenmeyer flask deformity).
Pathogenesis.
Most of the mutations underlying osteopetrosis interfere with the process of acidification of the osteoclast resorption pit, which is required for the dissolution of the calcium hydroxyapatite within the matrix. Examples include autosomal recessive defects in the gene CA2, which encodes the enzyme carbonic anhydrase II.21 Carbonic anhydrase II is required by osteoclasts and renal tubular cells to generate protons from carbon dioxide and water. Absence of CAII prevents osteoclasts from acidifying the resorption pit and solubilizing hydroxyapatite, and also blocks the acidification of urine by the renal tubular cells. In an autosomal recessive severe form of the disease, a mutation in the chloride channel gene CLCN7 interferes with the function of the H+-ATPase proton pump located on the osteoclast ruffled border.21 Another severe autosomal recessive form is caused by a mutation in the gene TCIRG1, which encodes a component of the proton pump. A less severe autosomal recessive variant results from a mutation in the gene that encodes RANKL. Not surprisingly, these individuals have fewer osteoclasts than normal. In animals, osteopetrosis can also be caused by mutations in a large number of other genes, including M-CSF, RANK, and OPG, which you will recall regulate osteoclast formation and function.21
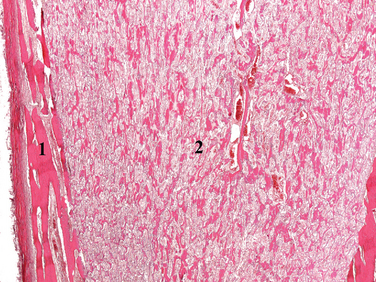
Morphology. The morphologic changes of osteopetrosis are explained by deficient osteoclast activity. The bones lack a medullary canal, and the ends of long bones are bulbous (Erlenmeyer flask deformity) and misshapen. The neural foramina are small and compress exiting nerves. The primary spongiosa, which is normally removed during growth, persists and fills the medullary cavity, leaving no room for the hematopoietic marrow and preventing the formation of mature trabeculae (Fig. 26-8). Deposited bone is not remodeled and tends to be woven in architecture. In the end, these intrinsic abnormalities cause the bone to be brittle and predisposed to fracture. Histologically, the number of osteoclasts may be normal, increased, or decreased depending on the underlying genetic defect.
Clinical Features.
Severe infantile malignant osteopetrosis is autosomal recessive and usually becomes evident in utero or soon after birth. Fracture, anemia, and hydrocephaly are often seen, resulting in postpartum mortality. Affected individuals who survive into their infancy have cranial nerve defects (optic atrophy, deafness, and facial paralysis) and repeated—often fatal—infections because of inadequacies of the marrow produced in extramedullary sites, which also causes prominent hepatosplenomegaly. The mild autosomal dominant benign form may not be detected until adolescence or adulthood, when it is discovered on x-rays performed because of repeated fractures. These individuals may also have mild cranial nerve deficits and anemia.
Osteopetrosis was the first genetic disease treated with bone marrow transplantation, since osteoclasts are derived from marrow monocyte precursors. The donor progenitor cells produce normal functioning osteoclasts, which reverse many of the skeletal abnormalities.
DISEASES ASSOCIATED WITH DECREASED BONE MASS
Osteoporosis
Osteoporosis is a disease characterized by porous bones and a reduced bone mass. The associated structural changes predispose the bone to fracture. The disorder may be localized to a certain bone or region, as in disuse osteoporosis of a limb, or may involve the entire skeleton, as a manifestation of a metabolic bone disease. Generalized osteoporosis, in turn, may be primary or secondary to a large variety of conditions (Table 26-4).
TABLE 26-4 Categories of Generalized Osteoporosis
| PRIMARY |
| SECONDARY |
| Endocrine Disorders |
| Neoplasia |
| Gastrointestinal |
| Drugs |
| Miscellaneous |
When the term osteoporosis is used in an unqualified manner, it usually refers to the most common forms, senile and postmenopausal osteoporosis, in which the loss of bone mass makes the skeleton vulnerable to fractures. It is estimated that one million Americans experience a fracture related to osteoporosis each year, at a cost of over 14 billion dollars. Effective treatment and prevention are imperative. The following discussion relates largely to these dominant forms of osteoporosis.
Pathogenesis.
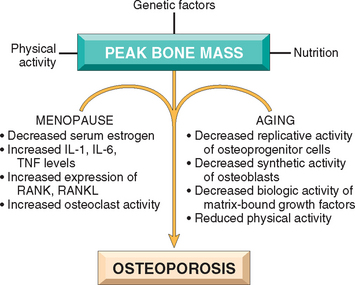
Peak bone mass is achieved during young adulthood. Its magnitude is determined largely by hereditary factors, especially polymorphisms in the genes that influence bone metabolism (discussed later).22 Physical activity, muscle strength, diet, and hormonal state also make important contributions. Once maximal skeletal mass is attained, a small deficit in bone formation accrues with every resorption and formation cycle of each basic multicellular unit. Accordingly, age-related bone loss, which may average 0.7% per year, is a normal and predictable biologic phenomenon. Both sexes are affected equally and whites more so than blacks. Differences in the peak skeletal mass in men versus women and in blacks versus whites may partially explain why certain populations are prone to develop this disorder.
Although much remains unknown, discoveries in the molecular biology of bone have provided intriguing new hypotheses about the pathogenesis of osteoporosis (Fig. 26-9):
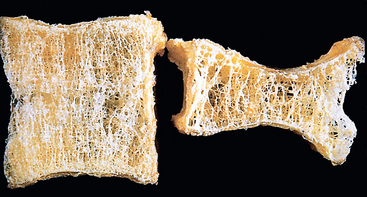
Morphology. The entire skeleton is affected in postmenopausal and senile osteoporosis (Fig. 26-10), but certain regions tend to be more severely involved than others. In postmenopausal osteoporosis the increase in osteoclast activity affects mainly bones or portions of bones that have increased surface area, such as the cancellous compartment of vertebral bodies. The trabecular plates become perforated, thinned, and lose their interconnections, leading to progressive microfractures and eventual vertebral collapse. In senile osteoporosis the cortex is thinned by subperiosteal and endosteal resorption, and the haversian systems are widened. In severe cases the haversian systems are so enlarged that the cortex mimics cancellous bone. The bone that remains is of normal composition.
Clinical Course.
The clinical manifestations of osteoporosis depend on which bones are involved. Vertebral fractures that frequently occur in the thoracic and lumbar regions are painful, and when multiple can cause significant loss of height and various deformities, including lumbar lordosis and kyphoscoliosis. Complications of fractures of the femoral neck, pelvis, or spine, such as pulmonary embolism and pneumonia, are frequent and result in 40,000 to 50,000 deaths per year.
Osteoporosis cannot be reliably detected in plain radiograms until 30% to 40% of the bone mass is lost, and measurement of blood levels of calcium, phosphorus, and alkaline phosphatase are not diagnostic. Osteoporosis is thus a difficult condition to diagnose accurately, since it remains asymptomatic until skeletal fragility is well advanced. The best procedures to accurately estimate the amount of bone loss, aside from biopsy, are specialized radiographic imaging techniques, such as dual-energy X-ray absorptiometry and quantitative computed tomography, which measure bone density.
The prevention and treatment of senile and postmenopausal osteoporosis includes exercise, appropriate calcium and vitamin D intake, and pharmacologic agents, most commonly bisphosphonates, which bind to bone and inhibit osteoclasts.
DISEASES CAUSED BY OSTEOCLAST DYSFUNCTION
Paget Disease (Osteitis Deformans)
This unique skeletal disease can be divided into three phases; (1) an initial osteolytic stage, followed by (2) a mixed osteoclastic-osteoblastic stage, which ends with a predominance of osteoblastic activity and evolves ultimately into (3) a burnt-out quiescent osteosclerotic stage (Fig. 26-11). The net effect is a gain in bone mass; however, the newly formed bone is disordered and architecturally unsound.
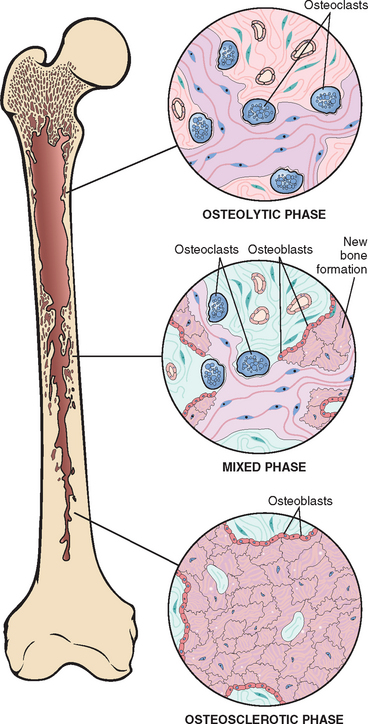
FIGURE 26-11 Diagrammatic representation of Paget disease of bone demonstrating the three phases in the evolution of the disease.
Paget disease usually begins in late adulthood (average age at diagnosis, 70 years) and becomes progressively more common thereafter. An intriguing aspect is the striking variation in its prevalence, both within certain countries and throughout the world. Paget disease is relatively common in whites in England, France, Austria, regions of Germany, Australia, New Zealand, and the United States. In contrast, Paget disease is rare in the native populations of Scandinavia, China, Japan, and Africa. The exact incidence is hard to determine because many affected individuals are asymptomatic; it is estimated that 1% of the US population over the age of 40 is affected and the prevalence rate in England is 2.5% for men and 1.6% for women 55 years or older. Recent surveys show that there has been a fall in new cases over the last 25–30 years, and a decline in its clinical severity.
Pathogenesis.
The cause of Paget disease remains uncertain, and current evidence suggests both environmental and genetic factors. The risk of developing the disorder is approximately seven times greater in first-degree relatives of affected individuals than it is in normal controls,25 and 15% to 40% of individuals with Paget disease have a family history that shows an autosomal dominant pattern of inheritance. Mutations in the SQSTM1 gene are present in 40% to 50% of cases of familial Paget disease, and in 5% to 10% of patients without a family history. The SQSTM mutations enhance NF-κB activation by RANK signaling, leading to increased osteoclast activity and an increased susceptibility to the disease. Mutations in RANKL and RANK/OPG have also been found in genetic diseases that have some phenotypic overlap with Paget disease; including familial expansile osteolysis, expansile skeletal hyperphosphatasia, early-onset Paget disease, juvenile Paget disease, and the syndrome of hereditary inclusion body myopathy, and frontotemporal dementia.
When Sir James Paget first described this condition in 1876, he attributed the skeletal changes to an inflammatory process, hence the term osteitis deformans. Support for this idea over the years has centered on a possible role for infection by a paramyxovirus, but this hypothesis remains unproven.
Morphology. Paget disease is a focal process that shows remarkable histologic variation over time and from site to site. The hallmark is the mosaic pattern of lamellar bone. This pattern, which is likened to a jigsaw puzzle, is produced by prominent cement lines that anneal haphazardly oriented units of lamellar bone (Fig. 26-12). In the initial lytic phase there are waves of osteoclastic activity and numerous resorption pits. The osteoclasts are abnormally large and have many more than the normal 10 to 12 nuclei; sometimes 100 nuclei are present. Osteoclasts persist in the mixed phase, but now many of the bone surfaces are lined by prominent osteoblasts. The marrow adjacent to the bone-forming surface is replaced by loose connective tissue that contains osteoprogenitor cells and numerous blood vessels, which transport nutrients and catabolites to and from these metabolically active sites. The newly formed bone may be woven or lamellar, but eventually all of it is remodeled into lamellar bone. As the mosaic pattern unfolds and the cell activity decreases, the periosseous fibrovascular tissue recedes and is replaced by normal marrow. In the end, the bone becomes a caricature of itself: larger than normal and composed of coarsely thickened trabeculae and cortices that are soft and porous and lack structural stability. These aspects make the bone vulnerable to deformation under stress; consequently, it fractures easily.
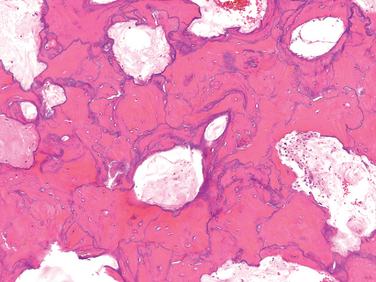
Clinical Course.
Clinical findings are extremely variable and depend on the extent and site of the disease. Most cases are mild and are discovered as an incidental radiographic finding. Paget disease is monostotic in about 15% of cases and polyostotic in the remainder. The axial skeleton or proximal femur is involved in up to 80% of cases. Even though no bone is immune, involvement of the ribs, fibula, and small bones of the hands and feet is unusual.
Pain localized to the affected bone is common. It is caused by microfractures or by bone overgrowth that compresses spinal and cranial nerve roots. Enlargement of the craniofacial skeleton may produce leontiasis ossea and a cranium so heavy that is difficult for the person to hold the head erect. The weakened pagetic bone may lead to invagination of the skull base (platybasia) and compression of the posterior fossa structures. Weight bearing causes anterior bowing of the femurs and tibiae and distorts the femoral heads, resulting in the development of severe secondary osteoarthritis. Chalkstick-type fractures are another frequent complication and usually occur in the long bones of the lower extremities. Compression fractures of the spine result in spinal cord injury and the development of kyphoses. The hypervascularity of pagetic bone warms the overlying skin, and in severe polyostotic disease the increased blood flow acts like an arteriovenous shunt, leading to high-output heart failure or exacerbation of underlying cardiac disease.
A variety of tumor and tumor-like conditions develop in pagetic bone. The benign lesions include giant-cell tumor, giant-cell reparative granuloma, and extra-osseous masses of hematopoiesis. The most dreaded complication is sarcoma, which occurs in 0.7% to 0.9% of all individuals with Paget disease, and in 5% to 10% of those with severe polyostotic disease. The sarcomas are usually osteosarcoma or fibrosarcoma, and they arise in Paget lesions in the long bones, pelvis, skull, and spine.
The diagnosis can frequently be made from the radiographic findings. Pagetic bone is typically enlarged with thick, coarsened cortices and cancellous bone(Fig. 26-13). Active disease has a wedge-shaped lytic leading edge that may progress along the length of the bone at a rate of 1 cm per year.26 Many affected individuals have elevated serum alkaline phosphatase levels and increased urinary excretion of hydroxyproline.
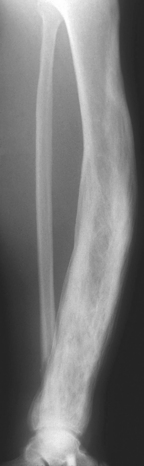
FIGURE 26-13 Severe Paget disease. The tibia is bowed and the affected portion is enlarged, sclerotic, and exhibits irregular thickening of both the cortical and cancellous bone.
In the absence of malignant transformation, Paget disease is usually not a serious or life-threatening disease. Most affected individuals have mild symptoms that are readily suppressed by calcitonin and bisphosphonates.
DISEASES ASSOCIATED WITH ABNORMAL MINERAL HOMEOSTASIS
Rickets and Osteomalacia
Rickets and osteomalacia are disorders characterized by a defect in matrix mineralization, most often related to a lack of vitamin D or some disturbance in its metabolism. The term rickets refers to the disorder in children in which deranged bone growth produces distinctive skeletal deformities. In the adult the disorder is called osteomalacia, because the bone that forms during the remodeling process is inadequately mineralized. This results in osteopenia and predisposition to insufficiency fractures. Both rickets and osteomalacia are discussed in Chapter 9.
Hyperparathyroidism
Hyperparathyroidism is classified into primary and secondary types as discussed in Chapter 24. Primary hyperparathyroidism results from autonomous hyperplasia or a tumor, usually an adenoma, of the parathyroid gland, whereas secondary hyperparathyroidism is commonly caused by prolonged states of hypocalcemia resulting in compensatory hypersecretion of PTH. Whatever the basis, the increased PTH concentrations are detected by receptors on osteoblasts, which then release factors that stimulate osteoclast activity. Thus, through a chain of signals, the skeletal manifestations of hyperparathyroidism are caused by unabated osteoclastic bone resorption. The following points should be noted:
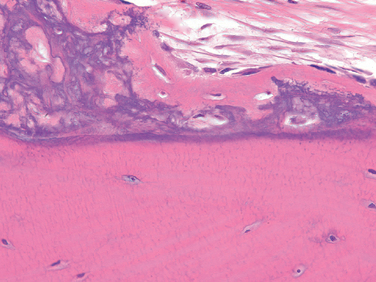
Morphology. For unknown reasons, the increased osteoclast activity in hyperparathyroidism affects cortical bone (subperiosteal, osteonal, and endosteal surfaces) more severely than cancellous bone. Subperiosteal resorption produces thinned cortices and the loss of the lamina dura around the teeth. X-rays reveal a pattern of radiolucency that is virtually diagnostic of hyperparathyroidism. In cancellous bone, osteoclasts tunnel into and dissect centrally along the length of the trabeculae, creating the appearance of railroad tracks and producing what is known as dissecting osteitis (Fig. 26-14). The correlative radiographic finding is a decrease in bone density or osteopenia. Since bone resorption and formation are coupled processes, it is not surprising that osteoblast activity is also increased in hyperparathyroidism. The marrow spaces around the affected surfaces are replaced by fibrovascular tissue.
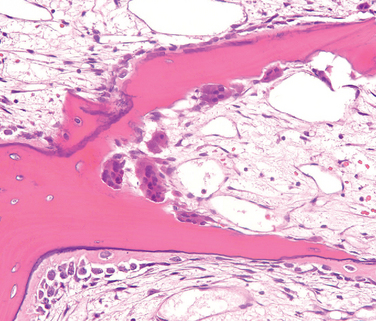
FIGURE 26-14 Hyperparathyroidism with osteoclasts boring into the center of the trabeculum (dissecting osteitis).
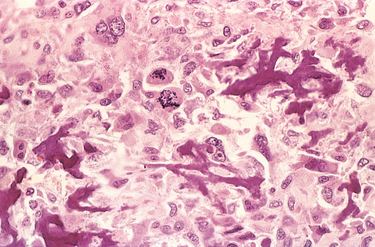
The bone loss predisposes to microfractures and secondary hemorrhages that elicit an influx of macrophages and an ingrowth of reparative fibrous tissue, creating a mass of reactive tissue, known as a brown tumor (Fig. 26-15). The brown color is the result of the vascularity, hemorrhage, and hemosiderin deposition, and it is not uncommon for the lesions to undergo cystic degeneration. The combined picture of increased bone cell activity, peritrabecular fibrosis, and cystic brown tumors is the hallmark of severe hyperparathyroidism and is known as generalized osteitis fibrosa cystica (von Recklinghausen disease of bone).
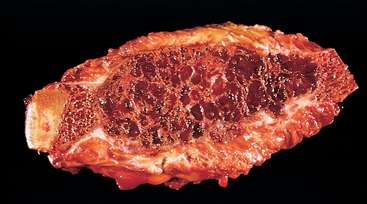
The decrease in bone mass predisposes to fractures, deformities caused by the stress of weight bearing, and joint pain and dysfunction as the lines of normal weight bearing are altered. Control of the hyperparathyroidism allows the bony changes to regress significantly or disappear completely.
Renal Osteodystrophy
The term renal osteodystrophy is used to describe collectively all of the skeletal changes of chronic renal disease, including (1) increased osteoclastic bone resorption mimicking osteitis fibrosa cystica, (2) delayed matrix mineralization (osteomalacia), (3) osteosclerosis, (4) growth retardation, and (5) osteoporosis. As advances in medical technology have prolonged the lives of individuals with renal disease, its impact on skeletal homeostasis has assumed greater clinical importance.
The various histologic bone changes in individuals with end-stage renal failure can be divided into three major types of disorders.27 High-turnover osteodystrophy is characterized by increased bone resorption and bone formation, with the former predominating. In contrast, low-turnover or aplastic disease is manifested by adynamic bone (little osteoclastic and osteoblastic activity) and, less commonly, osteomalacia. Many affected individuals have the third type, which is a mixed pattern of disease.
Pathogenesis.
The pathogenesis of the various skeletal lesions can be summarized as follows:
Fractures
Traumatic and nontraumatic fractures are some of the most common pathologic conditions affecting bone. Fractures are classified as complete or incomplete; closed (simple) when the overlying tissue is intact; compound when the fracture site communicates with the skin surface; comminuted when the bone is splintered; or displaced when the ends of the bone at the fracture site are not aligned. If the break occurs in bone already altered by a disease process, it is described as a pathologic fracture. A stress fracture is a slowly developing fracture that follows a period of increased physical activity in which the bone is subjected to new repetitive loads—as in sports training or marching in military boot camp.
Bone is unique in its ability to repair itself; it can completely reconstitute itself by reactivating processes that normally occur during embryogenesis. This process involves regulated expression of a multitude of genes and can be separated into overlapping stages with particular molecular, biochemical, histologic, and biomechanical features, described next.
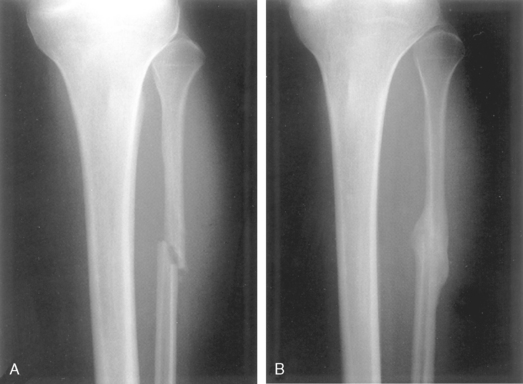
FIGURE 26-16 A, Recent fracture of the fibula. B, Marked callus formation 6 weeks later.
(Courtesy of Dr. Barbara Weissman, Brigham and Women’s Hospital, Boston, MA.)
The sequence of events in the healing of a fracture can be easily impeded or even blocked. For example, displaced and comminuted fractures frequently result in some deformity, and inadequate immobilization permits constant movement at the fracture site, so that the normal constituents of callus do not form, resulting in delayed union and nonunion. If a nonunion allows too much motion along the fracture gap, the central portion of the callus undergoes cystic degeneration, and the luminal surface can actually become lined by synovial-like cells, creating a false joint or pseudoarthrosis. A serious obstacle to healing is infection of the fracture site, which is a risk in comminuted and open fractures. The infection must be eradicated before bony union can be achieved.
Generally, with children and young adults, in whom most uncomplicated fractures are found, near perfect reconstitution is the norm. In older age groups in whom fractures tend to occur on a background of some other disease (e.g., osteoporosis and osteomalacia), repair is more often imperfect and may require mechanical immobilization (e.g., placement of stabilizing pins).
Osteonecrosis (Avascular Necrosis)
Infarction of bone and marrow is a relatively common event that can occur in the medullary cavity of the metaphysis or diaphysis and the subchondral region of the epiphysis. Ischemia underlies all forms of bone necrosis, which can occur in the setting of diverse predisposing conditions (Table 26-5) or as an isolated, idiopathic event. Aside from fracture, most cases of bone necrosis either are idiopathic or follow corticosteroid administration.
TABLE 26-5 Conditions Associated with Osteonecrosis
| Trauma |
| Corticosteroid administration |
| Infection |
| Dysbarism (e.g., the “bends”) |
| Radiation therapy |
| Connective tissue disorders |
| Pregnancy |
| Gaucher disease |
| Sickle cell and other anemias |
| Alcohol abuse |
| Chronic pancreatitis |
| Tumors |
| Epiphyseal disorders |
Morphology. Medullary infarcts are geographic and involve the cancellous bone and marrow. The cortex is usually not affected because of its collateral blood flow. In subchondral infarcts, a triangular or wedge-shaped segment of tissue that has the subchondral bone plate as its base undergoes necrosis. The overlying articular cartilage remains viable becauseit receives nutrition from the synovial fluid. The dead bone, recognized by its empty lacunae, is surrounded by necrotic adipocytes that frequently rupture, releasing their fatty acids, which bind calcium and form insoluble calcium soaps that may persist for life. In the healing response, osteoclasts resorb the necrotic trabeculae; however, those that remain act as scaffolding for the deposition of new bone in a process known as creeping substitution. In subchondral infarcts the pace of this substitution is too slow to be effective, so there is eventual collapse of the necrotic cancellous bone and distortion, fracture, and even sloughing of the articular cartilage (Fig. 26-17).
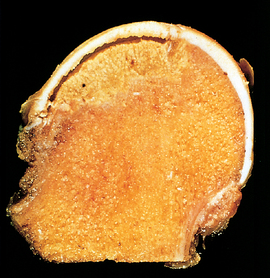
Clinical Course.
The symptoms depend on the location and extent of infarction. Typically, subchondral infarcts cause chronic pain that is initially associated only with activity but then becomes progressively more constant as secondary changes supervene. In contrast, medullary infarcts are clinically silent except for large ones occurring in Gaucher disease, dysbarism, and sickle cell anemia. Medullary infarcts usually remain stable over time. Subchondral infarcts, however, often collapse and may predispose to severe, secondary osteoarthritis. More than 10% of the 500,000 joint replacements performed annually in the United States are for treatment of the complications of osteonecrosis.
Infections—Osteomyelitis
Osteomyelitis denotes inflammation of bone and marrow, and the common use of the term virtually always implies infection. Osteomyelitis may be a complication of any systemic infection but frequently manifests as a primary solitary focus of disease. All types of organisms, including viruses, parasites, fungi, and bacteria, can produce osteomyelitis, but infections caused by certain pyogenic bacteria and mycobacteria are the most common. Currently in the United States, exotic infections in third world immigrants and opportunistic infections in immunosuppressed individuals have made the diagnosis and treatment of osteomyelitis quite challenging.
PYOGENIC OSTEOMYELITIS
Pyogenic osteomyelitis is almost always caused by bacteria. Organisms may reach the bone by (1) hematogenous spread, (2) extension from a contiguous site, and (3) direct implantation. In otherwise healthy children, most cases of osteomyelitis are hematogenous in origin and develop in the long bones.30 The initiating bacteremia may stem from seemingly trivial injuries to the mucosa, such as may occur during defecation or vigorous chewing of hard foods, or minor infections of the skin. In adults, however, osteomyelitis more often occurs as a complication of open fractures, surgical procedures, and diabetic infections of the feet.31
Staphylococcus aureus is responsible for 80% to 90% of the cases of pyogenic osteomyelitis in which an organism is recovered. These organisms express receptors for bone matrix components such as collagen, which facilitates their adherence to bone tissue. Escherichia coli, Pseudomonas, and Klebsiella are more frequently isolated from individuals with genitourinary tract infections or who are intravenous drug abusers. Mixed bacterial infections are seen in the setting of direct spread or inoculation of organisms during surgery or open fractures. In the neonatal period, Haemophilus influenzae and group B streptococci are frequent pathogens, and individuals with sickle cell disease are predisposed to Salmonella infection. In almost 50% of cases, no organisms can be isolated.
The location of the infection within a bone is influenced by the osseous vascular circulation, which varies with age. In the neonate the metaphyseal vessels penetrate the growth plate, resulting in frequent infection of the metaphysis, epiphysis, or both. In children, localization of microorganisms in the metaphysis is typical. After growth plate closure, the metaphyseal vessels reunite with their epiphyseal counterparts and provide a route for the bacteria to seed the epiphyses and subchondral regions in the adult.
Morphology. The morphologic changes of osteomyelitis depend on the stage (acute, subacute, or chronic) and location of the infection. Once in bone, the bacteria proliferate and induce an acute inflammatory reaction. The entrapped bone undergoes necrosis within the first 48 hours, and the bacteria and inflammation spread within the shaft of the bone and may percolate throughout the haversian systems to reach the periosteum. In children the periosteum is loosely attached to the cortex; sizable subperiosteal abscesses may form that can track for long distances along the bone surface. Lifting of the periosteum further impairs the blood supply to the affected region, and both the suppurative and the ischemic injury may cause segmental bone necrosis; the dead piece of bone is known as a sequestrum. Rupture of the periosteum leads to a soft-tissue abscess and the eventual formation of a draining sinus. Sometimes the sequestrum crumbles and forms free foreign bodies that pass through the sinus tract.
In infants, but uncommonly in adults, epiphyseal infection spreads through the articular surface or along capsular and tendoligamentous insertions into a joint, producing septic or suppurative arthritis, which can cause destruction of the articular cartilage and permanent disability. An analogous process involves the vertebrae, in which the infection destroys the hyaline cartilage end plate and intervertebral disc and spreads into adjacent vertebrae.
After the first week chronic inflammatory cells become more numerous and their release of cytokines stimulates osteoclastic bone resorption, ingrowth of fibrous tissue, and the deposition of reactive bone in the periphery. When the newly deposited bone forms a sleeve of living tissue around the segment of devitalized infected bone, it is known as an involucrum (Fig. 26-18). Several morphologic variants of osteomyelitis have eponyms: Brodie abscess is a small intraosseous abscess that frequently involves the cortex and is walled off by reactive bone; sclerosing osteomyelitis of Garré typically develops in the jaw and is associated with extensive new bone formation that obscures much of the underlying osseous structure.
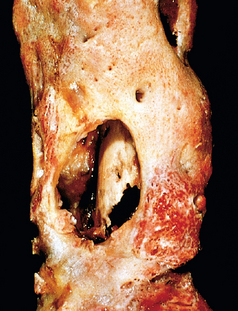
Clinical Course.
Clinically, hematogenous osteomyelitis may manifest as an acute systemic illness with malaise, fever, chills, leukocytosis, and marked-to-intense throbbing pain over the affected region. The presentation may be subtler with only unexplained fever, particularly in infants, or only localized pain in the absence of fever in the adult. The diagnosis can be strongly suggested by the characteristic radiographic findings of a lytic focus of bone destruction surrounded by a zone of sclerosis. In many untreated cases blood cultures are positive, but biopsy and bone cultures are required to identify the pathogen in most instances. The combination of antibiotics and surgical drainage is usually curative.In 5% to 25% of cases, acute osteomyelitis fails to resolve and persists as chronic infection. Chronicity may develop when there is delay in diagnosis, extensive bone necrosis, inadequate antibiotic therapy or surgical debridement, and weakened host defenses. Acute flare-ups may mark the clinical course of chronic infection and are usually spontaneous, have no obvious cause, and may occur after years of dormancy. Other complications of chronic osteomyelitis include pathologic fracture, secondary amyloidosis, endocarditis, sepsis, development of squamous cell carcinoma in the sinus tract, and rarely sarcoma in the infected bone.
TUBERCULOUS OSTEOMYELITIS
A resurgence of tuberculous osteomyelitis is occurring in developed countries, attributed to the influx of immigrants from countries where tuberculosis is endemic, and the greater numbers of immunosuppressed people (Chapter 8). In developing countries the affected individuals are usually adolescents or young adults, whereas in the indigenous population of the United States they tend to be older, except for those who are immunosuppressed. Approximately 1% to 3% of individuals with pulmonary or extrapulmonary tuberculosis have osseous infection.
The organisms are usually blood borne and originate from a focus of active visceral disease during the initial stages of primary infection. Direct extension (e.g., from a pulmonary focus into a rib or from tracheobronchial nodes into adjacent vertebrae) or spread via draining lymphatics may also occur. The bony infection is usually solitary and in some cases may be the only manifestation of the disease that may fester for years before being recognized. Individuals with acquired immunodeficiency syndrome frequently have multifocal bone involvement.
The spine (40% of cases, especially the thoracic and lumbar vertebrae) followed by the knees and hips are the most common sites of skeletal involvement.32 Tuberculous osteomyelitis tends to be more destructive and resistant to control than pyogenic osteomyelitis. In the spine (Pott disease) the infection breaks through intervertebral discs to involve multiple vertebrae and extends into the soft tissues forming abscesses. The histologic findings are typical of tuberculosis elsewhere (Chapter 8).
Typically, affected individuals present with pain on motion, localized tenderness, low-grade fevers, chills, and weight loss. Severe destruction of vertebrae frequently results in permanent compression fractures that produce severe scoliotic or kyphotic deformities and neurologic deficits secondary to spinal cord and nerve compression. Other complications of tuberculous osteomyelitis include tuberculous arthritis, sinus tract formation, psoas abscess, and amyloidosis.
SKELETAL SYPHILIS
Syphilis (Treponema pallidum) and yaws (Treponema pertenue) both can involve bone. Currently, syphilis is experiencing a resurgence; however, bone involvement remains infrequent because the disease is usually diagnosed and treated before this complication develops.
In congenital syphilis the bone lesions begin to appear about the fifth month of gestation and are fully developed at birth. The spirochetes tend to localize in areas of active enchondral ossification (osteochondritis) and in the periosteum (periostitis). In acquired syphilis bone disease may begin early in the tertiary stage, which is usually 2 to 5 years after the initial infection. The bones most frequently involved are those of the nose, palate, skull, and extremities, especially the long tubular bones such as the tibia. The syphilitic saber shin is produced by massive reactive periosteal bone deposition on the medial and anterior surfaces of the tibia.
Morphology. Syphilitic bone infection is characterized by edematous granulation tissue containing numerous plasma cells and necrotic bone. Typical gummas may also form in both congenital and acquired syphilis (Chapter 8). The spirochetes can be demonstrated in the inflammatory tissue with special silver stains.
Bone Tumors and Tumor-Like Lesions
Bone tumors are diverse in their gross and morphologic features, and vary in their natural history from innocuous to the rapidly fatal. It is critical to diagnose these tumors correctly, stage them accurately, and treat them appropriately, so that affected patients not only survive, but also maintain optimal function of the affected body parts.
Most bone tumors are classified according to the normal cell or tissue type they recapitulate. Lesions that do not have normal tissue counterparts are grouped according to their distinct clinicopathologic features (Table 26-6). Overall, matrix-producing and fibrous tumors are the most common, and among the benign tumors, osteochondroma and fibrous cortical defect are most frequent. Excluding malignant neoplasms of marrow origin (myeloma, lymphoma, and leukemia), osteosarcoma is the most common primary cancer of bone, followed by chondrosarcoma and Ewing sarcoma.
TABLE 26-6 Classification of Major Primary Tumors Involving Bones
| Histologic Type | Benign | Malignant |
|---|---|---|
| Hematopoietic (40%) | ||
| Chondrogenic (22%) | ||
| Osteogenic (19%) | Osteosarcoma | |
| Fibrogenic | Fibrosarcoma | |
| Unknown origin (10%) | ||
| Neuroectodermal | Ewing sarcoma | |
| Notochordal | Benign notochordal cell tumor | Chordoma |
Data on percentage of each type from Unni KK: Dahlin’s Bone Tumors, 5th ed. Philadelphia, Lippincott-Raven, 1996, p 4; by permission of Mayo Foundation.
The precise incidence of specific bone tumors is not known, because many benign lesions are not biopsied. Benign tumors greatly outnumber their malignant counterparts and occur with greatest frequency within the first three decades of life, whereas in the elderly a bone tumor is likely to be malignant. In the United States about 2400 new cases of bone sarcoma are diagnosed annually, and approximately 1300 deaths from bone sarcoma occur each year.
As a group these neoplasms affect all ages and arise in virtually every bone, but most develop during the first several decades of life and have a propensity to originate in the long bones of the extremities. However, specific types of tumors target certain age groups and anatomic sites. Thus, the location of a tumor provides important diagnostic information.
Although the cause of most bone tumors is unknown, genetic alterations similar to those that occur in other tumors clearly play a role. For instance, bone sarcomas occur in the Li-Fraumeni and hereditary retinoblastoma cancer syndromes, which are linked to mutations in the genes encoding p53 and RB (Chapter 7). Bone infarcts, chronic osteomyelitis, Paget disease, radiation, and metal prostheses are also associated with a bone neoplasia. Such secondary neoplasms, however, account for only a small fraction of skeletal tumors.
Clinically, bone tumors present in various ways. The more common benign lesions are frequently asymptomatic and are detected as incidental findings. Many tumors, however, produce pain or are noticed as a slow-growing mass. In some circumstances the first hint of a tumor’s presence is a sudden pathologic fracture. Radiologic imaging studies have an important role in diagnosing these lesions. In addition to providing the exact location and extent of the tumor, imaging studies can detect features that help limit diagnostic possibilities and give clues to the aggressiveness of the tumor. Ultimately, in most instances, biopsy and histologic study are necessary.
BONE-FORMING TUMORS
Common to all these neoplasms is the production of bone by the neoplastic cells. The tumor bone is usually deposited as woven trabeculae (except in osteomas) and is variably mineralized.
Osteoma
Osteomas are bosselated, round-to-oval sessile tumors that project from the subperiosteal surface of the cortex. They most often arise on or inside the skull and facial bones. They are usually solitary and are detected in middle age. Multiple osteomas are seen in the setting of Gardner syndrome (Chapter 17). They consist of a composite of woven and lamellar bone that is frequently deposited in a cortical pattern with haversian-like systems. Some variants contain a component of trabecular bone in which the intertrabecular spaces are filled with hematopoietic marrow.
Osteomas are generally slow-growing tumors of little clinical significance except when they cause obstruction of a sinus cavity, impinge on the brain or eye, interfere with function of the oral cavity, or produce cosmetic problems.
Osteoid Osteoma and Osteoblastoma
Osteoid osteoma and osteoblastoma are terms used to describe benign bone tumors that have identical histologic features but differ in size, sites of origin, and symptoms. Osteoid osteomas are by definition less than 2 cm in greatest dimension and usually occur in the teens and 20s. Seventy-five percent of affected individuals are younger than 25 years old, and men outnumber women 2 : 1. They can arise in any bone but have a predilection for the appendicular skeleton and posterior elements of the spine. In 50% of cases the femur or tibia is involved, wherein they commonly arise in the cortex and less frequently within the medullary cavity. Osteoid osteomas produce severe nocturnal pain that is relieved by aspirin.33 The pain is probably caused by excess prostaglandin E2 (PGE2) production by the proliferating osteoblasts. Osteoblastoma is larger than 2 cm and involves the spine more frequently; the pain is dull, achy, and unresponsive to salicylates, and the tumor usually does not induce a marked bony reaction.
Morphology. Osteoid osteoma and osteoblastoma are round-to-oval masses of hemorrhagic gritty tan tissue. They are well circumscribed and composed of randomly interconnecting trabeculae of woven bone that are prominently rimmed by osteoblasts (Fig. 26-19). The stroma surrounding the neoplastic bone consists of loose connective tissue that contains many dilated and congested capillaries. The relatively small size, well-defined margins, and benign cytologic features of the neoplastic osteoblasts help distinguish these tumors from osteosarcoma. Osteoid osteomas, especially those that arise beneath the periosteum, usually elicit a tremendous amount of reactive bone formation that encircles the lesion. The actual tumor, known as the nidus, manifests radiographically as a small round lucency that may be centrally mineralized (Fig. 26-20).
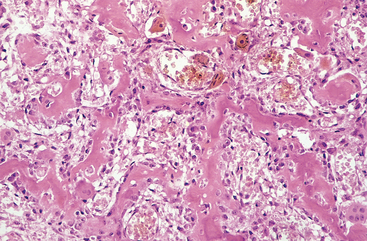
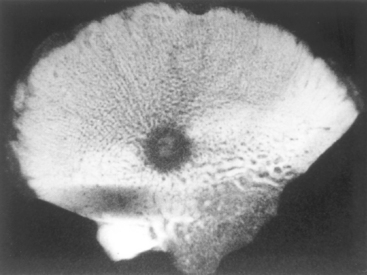
Osteoid osteoma is frequently treated by radioablation. Osteoblastoma is usually curetted or excised en bloc in a conservative fashion. The possibility of malignant transformation is remote except when osteoblastoma is treated with radiation (large tumors in the base of the skull and spine), which may promote this dreaded complication.
Osteosarcoma
Osteosarcoma is a malignant mesenchymal tumor in which the cancerous cells produce bone matrix. It is the most common primary malignant tumor of bone, exclusive of myeloma and lymphoma, and accounts for approximately 20% of primary bone cancers. Osteosarcoma occurs in all age groups but has a bimodal age distribution; 75% occur in persons younger than 20 years of age.34 The smaller second peak occurs in the elderly, who frequently suffer from conditions known to predispose to osteosarcoma—Paget disease, bone infarcts, and prior irradiation. Overall, men are more commonly affected than women (1.6 : 1). The tumors usually arise in the metaphyseal region of the long bones of the extremities, and almost 50% occur about the knee (Fig. 26-21). Any bone can be involved, however, and in persons beyond the age of 25, the incidence in flat bones and long bones is almost equal.
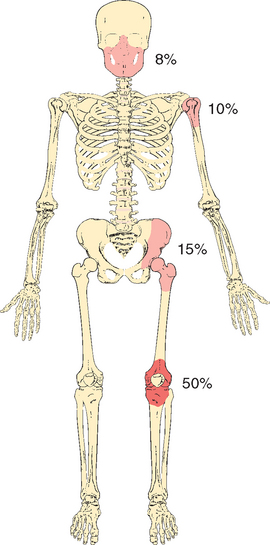
FIGURE 26-21 Major sites of origin of osteosarcomas. The numbers are approximate percentages arising at each site.
Pathogenesis.
Approximately 70% of osteosarcomas have acquired genetic abnormalities such as ploidy changes and chromosomal aberrations, none of which are specific to this tumor. More telling is the presence of very frequent mutations that interfere with function of two genes: (1) RB, the retinoblastoma gene, a critical cell cycle regulator; and (2) p53, a gene whose product regulates DNA repair and certain aspects of cellular metabolism (Chapter 7). Although the basic mechanisms that cause the development of osteosarcoma are still unknown, it is clear that defects in RB and p53 play important roles in the process. This association is emphasized by rare patients with germline mutations in RB, who have a roughly 1000-fold increased risk of osteosarcoma; and similarly by patients with Li-Fraumeni syndrome (germline p53 mutations), who also have a greatly elevated incidence of this tumor. Abnormalities in INK4a, which encodes p16 (a cell cycle regulator) and p14 (which aids and abets p53 function), also are seen in osteosarcoma. It is also noteworthy that osteosarcomas tend to occur at sites of bone growth, presumably because proliferation makes osteoblastic cells prone to acquire mutations that could lead to transformation. The association may contribute to the high incidence of osteosarcoma in large dog breeds, such as St. Bernards and Great Danes.
Morphology. Several subtypes of osteosarcoma are recognized and are grouped according to
The most common subtype arises in the metaphysis of long bones and is primary, solitary, intramedullary, and poorly differentiated.
Grossly, osteosarcomas are big bulky tumors that are gritty, gray-white, and often contain areas of hemorrhage and cystic degeneration (Fig. 26-22). The tumors frequently destroy the surrounding cortices and produce soft-tissue masses. They spread extensively in the medullary canal, infiltrating and replacing the marrow surrounding the preexisting bone trabeculae. Infrequently, they penetrate the epiphyseal plate or enter the joint. When joint invasion occurs, the tumor grows into it along tendoligamentous structures or through the attachment site of the joint capsule. The tumor cells vary in size and shape and frequently have large hyperchromatic nuclei. Bizarre tumor giant cells are common, as are mitoses. The formation of bone by the tumor cells is characteristic (Fig. 26-23). The neoplastic bone usually hasa coarse, lace-like architecture but also may be deposited in broad sheets or as primitive trabeculae. Other matrices, including cartilage or fibrous tissue, may be present in varying amounts. When malignant cartilage is abundant, the tumor is called chondroblastic osteosarcoma. Vascular invasion is usually conspicuous, and up to 50% to 60% of an individual tumor may be necrotic.
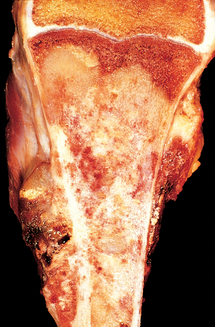
Clinical Course.
Osteosarcomas typically present as painful, progressively enlarging masses. Sometimes a sudden fracture of the bone is the first symptom. Radiograms of the primary tumor usually show a large destructive, mixed lytic and blastic mass with infiltrative margins (Fig. 26-24). The tumor frequently breaks through the cortex and lifts the periosteum, resulting in reactive periosteal bone formation. The triangular shadow between the cortex and raised ends of periosteum is known radiographically as Codman triangle and is characteristic but not diagnostic of this tumor. These aggressive neoplasms spread hematogenously, and at the time of diagnosis approximately 10% to 20% of affected individuals have demonstrable pulmonary metastases, and it is likely that many more have occult metastases. In those who die of the neoplasm, 90% have metastases to the lungs, bones, brain, and elsewhere.
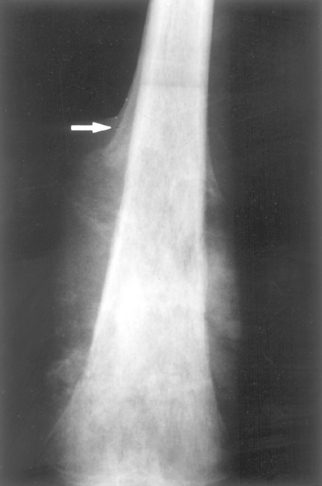
FIGURE 26-24 Distal femoral osteosarcoma with prominent bone formation extending into the soft tissues. The periosteum, which has been lifted, has laid down a proximal triangular shell of reactive bone known as a Codman triangle (arrow).
Osteosarcoma is treated with a multimodality approach that includes chemotherapy, which is given under the assumption that all patients at the time of diagnosis have metastases, which are usually too small to detect by imaging. The prognosis of patients without detectable metastases has improved substantially, with 5-year survival rates reaching 60% to 70% with aggressive chemotherapy and limb salvaging surgery. Unfortunately, the outcome for patients with overt metastases or recurrent disease is still poor (approximately 20% 5-year survival rate).
CARTILAGE-FORMING TUMORS
Cartilage tumors account for the majority of primary bone tumors and are characterized by the formation of hyaline or myxoid cartilage; fibrocartilage and elastic cartilage are rare components. As in most types of bone tumors, benign cartilage tumors are much more common than malignant ones.
Osteochondroma
Osteochondroma, also known as an exostosis, is a benign cartilage-capped tumor that is attached to the underlying skeleton by a bony stalk. It is the most common benign bone tumor; about 85% are solitary. The remainder are seen as part of the multiple hereditary exostosis syndrome, which is an autosomal dominant hereditary disease. Hereditary exostoses are caused by germline loss-of-function mutations in either the EXT1 or EXT2 genes, whereas inactivation of only EXT1 has been detected in sporadic tumors. These genes encode proteins that function in the biosynthesis of heparin sulfate proteoglycans (Chapter 3). Reduced expression of EXT1 and EXT2 results in defective endochondral ossification, which somehow sets the stage for abnormal growth. Solitary osteochondromas are usually first diagnosed in late adolescence and early adulthood, but multiple osteochondromas become apparent during childhood. Men are affected three times more often than women. Osteochondromas develop only in bones of endochondral origin and arise from the metaphysis near the growth plate of long tubular bones, especially about the knee. Occasionally, they develop from bones of the pelvis, scapula, and ribs, and in these sites they are frequently sessile and have short stalks. Rarely, they involve the short tubular bones of the hands and feet.
Morphology. Osteochondromas are sessile or mushroom shaped, and range in size from 1 to 20 cm. The cap is composed of benign hyaline cartilage varying in thickness (Fig. 26-25) and is covered peripherally by perichondrium. The cartilage has the appearance of disorganized growth plate and undergoes enchondral ossification, with the newly made bone forming the inner portion of the head and stalk. The cortex of the stalk merges with the cortex of the host bone, so that the medullary cavity of the osteochondroma and bone are in continuity.
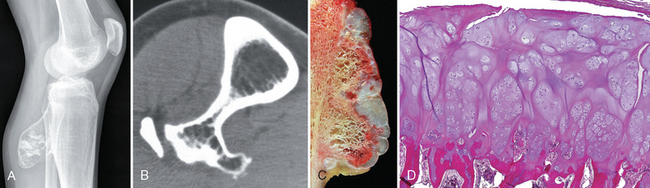
FIGURE 26-25 Osteochondroma. A, X-ray of an osteochondroma arising off the posterior surface of the tibia. B, Axial CT scan shows continuity of the cortex of the bone and the center of the osteochondroma. The fibula is adjacent to the mass. C, Gross specimen of sessile osteochondroma composed of a cap of hyaline cartilage undergoing enchondral ossification. D, The cartilage cap has the histologic appearance of disorganized growth plate-like cartilage.
Clinically, osteochondromas present as slow-growing masses, which can be painful if they impinge on a nerve or if the stalk is fractured. In many cases they are detected as an incidental finding. In multiple hereditary exostosis the underlying bones may be bowed and shortened, reflecting an associated disturbance in epiphyseal growth. Osteochondromas usually stop growing at the time of growth plate closure. Rarely in sporadic cases, but more commonly in those with multiple hereditary exostosis, they give rise to a chondrosarcoma or some other type of sarcoma.
Chondromas
Chondromas are benign tumors of hyaline cartilage that usually occur in bones of enchondral origin. They can arise within the medullary cavity, where they are known as enchondromas, or on the surface of bone, where they are called subperiosteal or juxtacortical chondromas. Enchondromas are the most common of the intraosseous cartilage tumors and are usually diagnosed in individuals who are in their 20s to 40s. They are usually solitary metaphyseal lesions of tubular bones; the favored sites are the short tubular bones of the hands and feet. A syndrome of multiple enchondromas or enchondromatosis is known as Ollier disease. If the enchondromatosis is associated with soft-tissue hemangiomas, the disorder is called Maffucci syndrome.
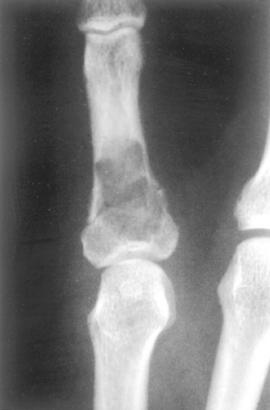
Morphology. Enchondromas are usually smaller than 3 cm and grossly are gray-blue and translucent. They are composed of well-circumscribed nodules of cyto logically benign hyaline cartilage (Fig. 26-26). The peripheral portion of the nodules may undergo enchondral ossification, and the center can calcify and die. The chondromas in Ollier disease and Maffucci syndrome are sometimes more cellular and exhibit cytologic atypia, making it difficult to distinguish them from chondrosarcoma.

Clinical Features.
Most enchondromas are asymptomatic and are detected incidentally. Occasionally they are painful and cause pathologic fracture. The tumors in enchondromatosis may be numerous and large, producing severe deformities. The radiographic features are characteristic; the unmineralized nodules of cartilage produce well-circumscribed oval lucencies that are surrounded by a thin rim of radiodense bone (C or O ring sign). If the matrix calcifies it is detected as irregular opacities. The nodules scallop the endosteum, but usually leave the cortex intact (Fig. 26-27). The growth potential of chondromas is limited, and most remain stable. Treatment depends on the clinical situation and is usually observation or curettage. Solitary chondromas rarely undergo sarcomatous transformation, but those associated with enchondromatoses do so more frequently. Individuals with Maffucci syndrome are also at risk of developing other types of malignancies, including ovarian carcinomas and brain gliomas.
Chondroblastoma
Chondroblastoma is a rare benign tumor that accounts for less than 1% of primary bone tumors. It usually occurs in young patients in their teens and has a male-to-female ratio of 2 : 1. Most arise about the knee; less common sites such as the pelvis and ribs are affected in older patients. Chondroblastoma has a striking predilection for epiphyses and apophyses (epiphyseal equivalents, i.e., iliac crest).36
Morphology. The tumor is composed of sheets of compact polyhedral chondroblasts that have well-defined cytoplasmic borders, moderate amounts of pink cytoplasm, and nuclei that are hyperlobulatedwith longitudinal grooves (Fig. 26-28). Mitotic activity and necrosis are frequently present. The tumor cells are surrounded by scant amounts of hyaline matrix that is deposited in a lace-like configuration; nodules of well-formed hyaline cartilage are distinctly uncommon. When the matrix calcifies it produces a characteristic chicken-wire pattern of mineralization (see Fig. 26-28). Scattered through the lesion are non-neoplastic osteoclast-type giant cells. Occasionally the tumors undergo prominent hemorrhagic cystic degeneration.
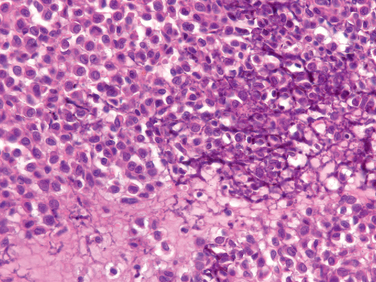
Chondroblastomas are usually painful, and because of their location near a joint they also cause effusions and restrict joint mobility. Radiographically, they produce a well-defined geographic lucency that commonly has spotty calcifications. Recurrences are not uncommon after curettage. Pulmonary metastases occur rarely in lesions that have undergone prior pathologic fracture or repeated curettage. Apparently in these circumstances the tumor cells are pushed into ruptured vessels, giving them access to the systemic circulation.
Chondromyxoid Fibroma
Chondromyxoid fibroma is the rarest of cartilage tumors and because of its varied morphology can be mistaken for sarcoma. It affects individuals in their teens and 20s and has a male preponderance. The tumors most frequently arise in the metaphysis of long tubular bones, but can involve virtually any bone of the body.
Morphology. The tumors range from 3 to 8 cm in greatest dimension and are well-circumscribed, solid, and glistening tan-gray. Microscopically, there are nodules of poorly formed hyaline cartilage and myxoid tissue delineated by fibrous septae. The cellularity varies; the areas of greatest cellularity are at the periphery of the nodules. In the cartilaginous regions the tumor cells are situated in lacunae; however, in the myxoid areas, the cells are stellate, and their delicate cell processes extend through the mucinous ground substance and approach or contact neighboring cells (Fig. 26-29). In contrast to other benign cartilage tumors, the neoplastic cells in chondromyxoid fibroma show varying degrees of cytologic atypia, including the presence of large hyperchromatic nuclei. Other findings include small foci of calcification of the cartilaginous matrix and scattered non-neoplastic, osteoclast-type giant cells.
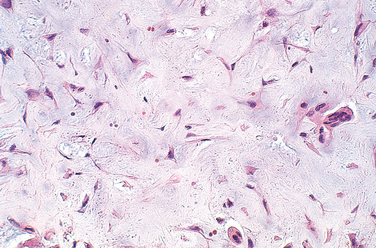
Individuals with chondromyxoid fibroma usually complain of localized dull, achy pain. In most instances, radiograms demonstrate an eccentric geographic lucency that is well delineated from the adjacent bone by a rim of sclerosis. Occasionally the tumor expands the overlying cortex. The treatment of choice is simple curettage, and even though they may recur, they do not pose a threat for malignant transformation or metastasis.
Chondrosarcoma
Chondrosarcomas are a group of tumors that span a broad spectrum of clinical and pathologic findings. The feature common to all of them is the production of neoplastic cartilage. Chondrosarcoma is subclassified according to site as central (intramedullary) and peripheral (juxtacortical and surface). Histologically, they include conventional (hyaline and/or myxoid), clear cell, dedifferentiated, and mesenchymal variants. Conventional central tumors constitute about 90% of chondrosarcomas.
Chondrosarcoma of the skeleton is about half as frequent as osteosarcoma and is the second most common malignant matrix-producing tumor of bone. Individuals with chondrosarcoma are usually in their 40s or older. The clear cell and especially the mesenchymal variants occur in younger patients, in their teens or 20s. The tumor affects men twice as frequently as women. About 15% of conventional chondrosarcomas (usually peripheral tumors) arise from a preexisting enchondroma or osteochondroma.
Morphology. Conventional chondrosarcoma is composed of malignant hyaline and myxoid cartilage. The large bulky tumors are made up of nodules of gray-white, somewhat translucent glistening tissue (Fig. 26-30). In predominantly myxoid variants, the tumors are viscous and gelatinous and the matrix oozes from the cut surface. Spotty calcifications are typically present, and central necrosis may create cystic spaces. The adjacent cortex is thickened or eroded, and the tumor grows with broad pushing fronts into the surrounding soft tissue. The malignant cartilage infiltrates the marrow space and surrounds pre-existing bony trabeculae. The tumors vary in degree of cellularity, cytologic atypia, and mitotic activity (Fig. 26-31). Low-grade or grade 1 lesions demonstrate mild hypercellularity, and the chondrocytes have plump vesicular nuclei with small nucleoli. Binucleate cells are sparse, and mitotic figures are difficult to find. Portions of the matrix frequently mineralize, and the cartilage may undergo endochondral ossification. By contrast, grade 3 chondrosarcomas are characterized by marked hypercellularity, extreme pleomorphism with bizarre tumor giant cells, and mitoses. Pure grade 3 chondrosarcomas are uncommon. Such malignant cartilage is more frequently a component of chondroblastic osteosarcoma (see earlier).
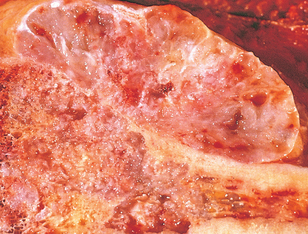
FIGURE 26-30 Chondrosarcoma with lobules of hyaline and myxoid cartilage permeating throughout the medullary cavity, growing through the cortex, and forming a relatively well-circumscribed soft-tissue mass.
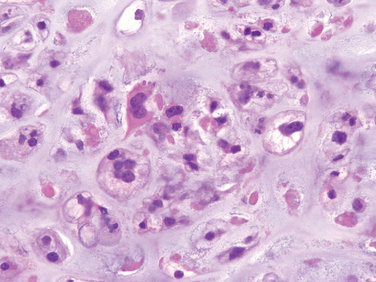
Approximately 10% of conventional low-grade chondrosarcomas have a second high-grade component that has the morphology of a poorly differentiated sarcoma; this combination defines dedifferentiated chondrosarcomas. The hallmark of clear cell chondrosarcoma is sheets of large malignant chondrocytes that have abundant clear cytoplasm, numerous osteoclast-type giant cells, and intralesional reactive bone formation, which often causes confusion with osteosarcoma. Mesenchymal chondrosarcoma is composed of islands of well-differentiated hyaline cartilage surrounded by sheets of small round cells, which can mimic Ewing sarcoma.
Chondrosarcomas commonly arise in the central portions of the skeleton, including the pelvis, shoulder, and ribs. The clear cell variant is unique in that it originates in the epiphyses of long tubular bones. In contrast to enchondroma, chondrosarcoma rarely involves the distal extremities. These tumors usually present as painful, progressively enlarging masses. The nodular growth pattern of the cartilage produces prominent endosteal scalloping radiographically. The calcified matrix appears as foci of flocculent densities. A slow-growing, low-grade tumor causes reactive thickening of the cortex, whereas a more aggressive high-grade neoplasm destroys the cortex and forms a soft-tissue mass. There is a direct correlation between the grade and the biologic behavior of the tumor.37 Fortunately, most conventional chondrosarcomas are indolent and fall into the range of grade 1 and grade 2. In one analysis, the 5-year survival rates were 90%, 81%, and 43% for grades 1 through 3, respectively. None of the grade 1 tumors metastasized, whereas 70% of the grade 3 tumors disseminated. Another prognostic feature is size; tumors greater than 10 cm are more aggressive than smaller tumors. When chondrosarcomas metastasize, they spread preferentially to the lungs and skeleton. The treatment of conventional chondrosarcoma is wide surgical excision. The mesenchymal and dedifferentiated tumors are also treated with chemotherapy, because of their aggressive clinical course.
FIBROUS AND FIBRO-OSSEOUS TUMORS
Tumors composed solely or predominantly of fibrous elements are diverse and include some of the most common lesions of the skeleton.
Fibrous Cortical Defect and Non-Ossifying Fibroma
Fibrous cortical defects are extremely common, being found in 30% to 50% of children older than 2 years. They are believed to be developmental defects rather than neoplasms. The vast majority arise eccentrically in the metaphysis of the distal femur and proximal tibia, and almost half are bilateral or multiple. Often they are small, about 0.5 cm in diameter. Those that grow to 5 or 6 cm in size develop into non-ossifying fibromas, which are usually not detected until adolescence.
Morphology. Both fibrous cortical defects and non-ossifying fibromas produce elongated, sharply demarcated radiolucencies that are surrounded by a thin rim of sclerosis (Fig. 26-32). They consist of gray to yellow-brown cellular lesions containing fibroblasts and macrophages (histiocytes). The cytologically bland fibroblasts are frequently arranged in a storiform (pinwheel) pattern, and the histiocytes are either multinucleated giant cells or clusters of foamy macrophages (Fig. 26-33).
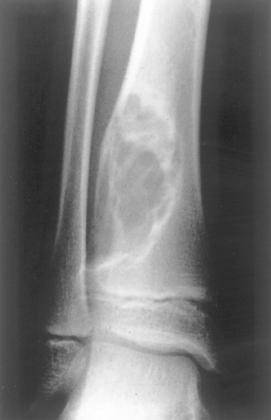
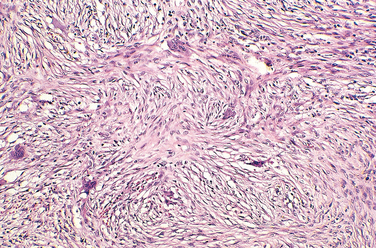
Fibrous cortical defects are asymptomatic and are usually detected on radiography as an incidental finding. Most have limited growth potential and undergo spontaneous resolution within several years, being replaced by normal cortical bone. The few that progressively enlarge into non-ossifying fibromas may present with pathologic fracture or require biopsy and curettage to exclude other types of tumors.
Fibrous Dysplasia
Fibrous dysplasia is a benign tumor that has been likened to a localized developmental arrest; all of the components of normal bone are present, but they do not differentiate into their mature structures. The lesions arise during skeletal growth and development, and appear in three distinctive but sometimes overlapping clinical patterns: (1) involvement of a single bone (monostotic); (2) involvement of multiple bones (polyostotic); and (3) polyostotic disease, associated with café-au-lait skin pigmentations and endocrine abnormalities, especially precocious puberty. The skeletal, skin, and endocrine lesions result from a somatic gain-of-function mutation occurring during embryogenesis in the GNAS gene, which you will recall is also mutated in pituitary adenomas (Chapter 24). The result of the mutations in both types of tumors is the same—the production of a hyperactive guanyl nucleotide binding protein, encoded by the GNAS gene, that drives abnormal growth.38
Monostotic fibrous dysplasia accounts for 70% of all cases. It occurs equally in boys and girls, usually in early adolescence, and often stops enlarging at the time of growth plate closure. The femur, tibia, ribs, jawbones, calvaria, and humerus are most commonly affected. The lesion is frequently asymptomatic and usually discovered incidentally but it may cause pain, fracture, and discrepancies in limb length. Fibrous dysplasia can cause marked enlargement and distortion of bone, so that if the craniofacial skeleton is involved, disfigurement, sometimes severe, can occur. Monostotic disease does not evolve into the polyostotic form.
Polyostotic fibrous dysplasia without endocrine dysfunction accounts for 27% of all cases. It manifests at a slightly earlier age than the monostotic type and may continue to cause problems into adulthood. The bones affected, in descending order of frequency, are the femur, skull, tibia, humerus, ribs, fibula, radius, ulna, mandible, and vertebrae. Craniofacial involvement is present in 50% of those who have a moderate number of bones affected and in 100% of those with extensive skeletal disease. Polyostotic disease has a propensity to involve the shoulder and pelvic girdles, resulting in severe, sometimes crippling deformities (e.g., shepherd-crook deformity of the proximal femur) and spontaneous and often recurrent fractures.
Polyostotic fibrous dysplasia associated with café-au-lait skin pigmentation and endocrinopathies is known as the McCune-Albright syndrome and accounts for 3% of all cases. The endocrinopathies include sexual precocity, hyperthyroidism, pituitary adenomas that secrete growth hormone, and primary adrenal hyperplasia. The severity of manifestations in McCune-Albright syndrome depends on the number and cell types that harbor the mutation in the GNAS gene. The most common clinical presentation is precocious sexual development, which occurs most often in girls. The bone lesions are often unilateral but can be bilateral, and the skin pigmentation is usually limited to the same side of the body. The cutaneous macules are classically large; are dark to café-au-lait; have irregular serpiginous borders (coastline of Maine); and are found primarily on the neck, chest, back, shoulder, and pelvic region.
Morphology. The lesions of fibrous dysplasia are well circumscribed, intramedullary, and vary greatly in size. Larger lesions expand and distort the bone. The lesional tissue is tan-white and gritty and is composed of curvilinear trabeculae of woven bone surrounded by a moderately cellular fibroblastic proliferation. The shapes of the trabeculae mimic Chinese letters, and the bone lacks prominent osteoblastic rimming (Fig. 26-34). Nodules of hyaline cartilage with the appearance of disorganized growth plate are also present in approximately 20% of cases. Cystic degeneration, hemorrhage, and foamy macrophages are other common findings.
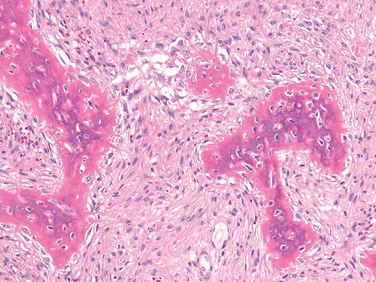
Clinical Course.
The natural history of fibrous dysplasia is variable and depends on the extent of skeletal involvement. Individuals with monostotic disease usually have minimal symptoms, except if the tumor is strategically located, such as in the femoral neck. The lesion is readily diagnosed by radiology because of its typical ground-glass appearance and well-defined margination. Lesions that fracture or cause significant symptoms are cured by conservative surgery. Polyostotic involvement is frequently associated with progressive disease. Those diagnosed at an earlier age are more likely to have severe skeletal complications, such as recurring fractures, long-bone deformities and persistent pain, and involvement and distortion of the craniofacial bones. These patients may require multiple corrective orthopedic surgical procedures. Bisphosphonates can be used to reduce the severity of the bone pain. A rare complication, usually in the setting of polyostotic involvement, is malignant transformation of a lesion into a sarcoma.
Fibrosarcoma Variants
Collagen-producing sarcomas with a fibroblastic phenotype occur at any age, but most affect the middle-aged and elderly. They have a nearly equal sex distribution and usually arise de novo; however, a few develop in preexisting benign tumors, bone infarcts, pagetic bone, and previously irradiated tissue.
Morphology. Grossly these tumors are large, hemorrhagic, tan-white masses that destroy the underlying bone and frequently extend into the soft tissues. They are composed of cytologically malignant fibroblasts arranged in a herringbone storiform pattern. The level of differentiation determines the amount of collagen produced and degree of cytologic atypia. In the past, some of these tumors were called malignant fibrous histiocytoma because the pleomorphic cells resembled histiocytes (activated tissue macrophages).
Fibrosarcoma presents as an enlarging painful mass that usually arises in the metaphysis of long bones and pelvic flat bones. Pathologic fracture is a frequent complication. Radiographically it is permeative and lytic and often extends into the adjacent soft tissue. The prognosis depends on the size, location, stage, and grade of the tumor; large, high-grade tumors that are difficult to resect have a very poor prognosis.
Miscellaneous Tumors
EWING SARCOMA/PRIMITIVE NEUROECTODERMAL TUMOR
The Ewing sarcoma family of tumors encompasses Ewing sarcoma and primitive neuroectodermal tumor (PNET), which are primary malignant small round-cell tumors of bone and soft tissue (Chapter 10). Both Ewing sarcoma and PNET have a similar neural phenotype, and because they share an identical chromosome translocation they should be viewed as two variants of the same tumor that differ only in their degree of neural differentiation. Tumors that demonstrate neural differentiation are labeled PNETs, and those that are undifferentiated are diagnosed as Ewing sarcoma. This distinction has no clinical significance.
Ewing sarcoma and PNET together account for approximately 6% to 10% of primary malignant bone tumors and follow osteosarcoma as the second most common group of bone sarcomas in children. Of all bone sarcomas, Ewing sarcoma/PNET has the youngest average age at presentation, since most affected individuals are 10 to 15 years old, and approximately 80% are younger than 20 years. Boys are affected slightly more frequently than girls, and there is a striking predilection for whites; blacks are rarely afflicted. Most Ewing sarcoma/PNET have a translocation involving the EWS gene on chromosome 22 and a gene encoding an ETS family transcription factor; the most commonly involved ETS gene is FLI1, as part of a (11;22) (q24;q12) translocation. The fusion genes generated by these translocations produce chimeric transcription factors that alter the expression of a network of target genes, resulting in abnormal cell proliferation and survival.39 Other recent evidence suggests that the precursor cell of Ewing sarcoma/PNET is a multipotent mesenchymal stem cell.40
Morphology. Arising in the medullary cavity, Ewing sarcoma and PNET usually invade the cortex, periosteum, and soft tissue. The tumor is soft, tan-white, and frequently contains areas of hemorrhage and necrosis. It is composed of sheets of uniform small, round cells that are slightly larger than lymphocytes (Fig. 26-35). They have scant cytoplasm, which may appear clear because it is rich in glycogen. The presence of Homer-Wright rosettes (tumor cells arranged in a circle about a central fibrillary space) is indicative of neural differentiation. Although the tumor contains fibrous septae, there is generally little stroma. Necrosis may be prominent, and there are relatively few mitotic figures in relation to the dense cellularity of the tumor.
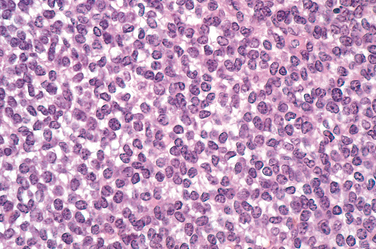
Clinical Features.
Ewing sarcoma and PNET usually arise in the diaphysis of long tubular bones, especially the femur and the flat bones of the pelvis. They present as painful enlarging masses, and the affected site is frequently tender, warm, and swollen. Some affected individuals have systemic findings, including fever, elevated sedimentation rate, anemia, and leukocytosis, which mimic infection. Plain radiograms show a destructive lytic tumor that has permeative margins and extension into the surrounding soft tissues. The characteristic periosteal reaction produces layers of reactive bone deposited in an onion-skin fashion.
Treatment includes chemotherapy and surgical excision with or without irradiation. The advent of effective chemotherapy has markedly improved the prognosis from a dismal 5% to 15% to an approximately 75% 5-year survival; at least 50% have long-term cures. The amount of chemotherapy-induced necrosis is an important prognostic finding. Gene expression arrays of tumors appear to identify individuals with the most aggressive tumors.41
GIANT-CELL TUMOR
Giant-cell tumor is so named because it contains a mixture of mononuclear cells and a profusion of multinucleated osteoclast-type giant cells, giving rise to the synonym osteoclastoma. This tumor is a relatively uncommon benign but locally aggressive neoplasm. It usually arises in individuals in their 20s to 40s. The mononuclear cells in giant-cell tumors express RANKL, and the giant osteoclast-like cells are believed to form via the RANK/RANKL signaling pathway.42
Morphology. These are large, red-brown tumors that frequently undergo cystic degeneration. They are mostly composed of uniform oval mononuclear cells that constitute the proliferating component of the tumor. Scattered within this background are numerous osteoclast-type giant cells having 100 or more nuclei that resemble those of the mononuclear cells (Fig. 26-36). Necrosis, hemorrhage, hemosiderin deposition, and reactive bone formation are common secondary features.
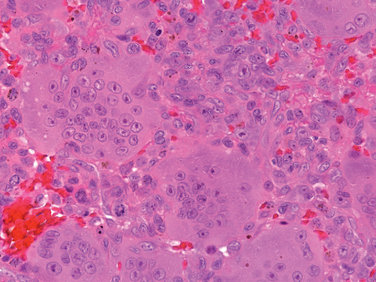
Clinical Course.
Giant-cell tumors in adults involve both the epiphyses and the metaphyses, but in adolescents they are confined proximally by the growth plate and are limited to the metaphysis. The majority arise around the knee (distal femur and proximal tibia), but virtually any bone can be involved. The typical location of these tumors near joints frequently causes arthritis-like symptoms. Occasionally, they present with pathologic fractures. Most are solitary; however, multiple or multicentric tumors do occur, especially in the distal extremities. Giant-cell tumors often erode into the subchondral bone plate (Fig. 26-37) and destroy the overlying cortex, producing a bulging soft-tissue mass delineated by a thin shell of reactive bone. The margins with the adjacent bone are fairly circumscribed but seldom sclerotic. The biologic unpredictability of these neoplasms complicates their management. Conservative surgery such as curettage is associated with a 40% to 60% recurrence rate, and up to 4% metastasize to the lungs.

ANEURYSMAL BONE CYST
Aneurysmal bone cyst is a benign tumor of bone characterized by multiloculated blood-filled cystic spaces that may present as a rapidly growing expansile tumor. Despite its aggressive radiographic appearance, aneurysmal bone cyst behaves in a benign fashion. This tumor is associated with distinctive 17p13 translocations that result in up-regulation of USP6, a deubiquitinating enzyme.43
Morphology. Grossly, aneurysmal bone cyst consists of multiple blood-filled cystic spaces separated by thin, tan-white septa (Fig. 26-38). The walls are composed of plump uniform fibroblasts (which may be mitotically active), multinucleated osteoclast-like giant cells, and reactive woven bone. The bone is lined by osteoblasts, and its deposition typically follows the contours of the fibrous septa. Approximately one third of cases contain an unusual cartilage-like matrix, called “blue bone.” Necrosis is uncommon unless there has been a previous pathologic fracture.
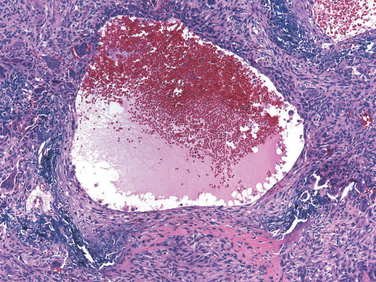
Clinical Course.
Aneurysmal bone cyst affects all age groups but generally occurs during the first 2 decades of life and has no sex predilection. It most frequently develops in the metaphyses of long bones and the posterior elements of vertebral bodies. The most common signs and symptoms are pain and swelling. When an aneurysmal bone cyst involves the vertebrae, it can compress nerves and cause neurologic symptoms. Rarely, pathologic fractures occur.
Radiographically, aneurysmal bone cyst is usually an eccentric, expansile lesion with well-defined margins (Fig. 26-39A). Most lesions are completely lytic and often contain a thin shell of reactive bone at the periphery. Computed tomography and magnetic resonance imaging may demonstrate internal septa and characteristic fluid-fluid levels (Fig. 26-39B).
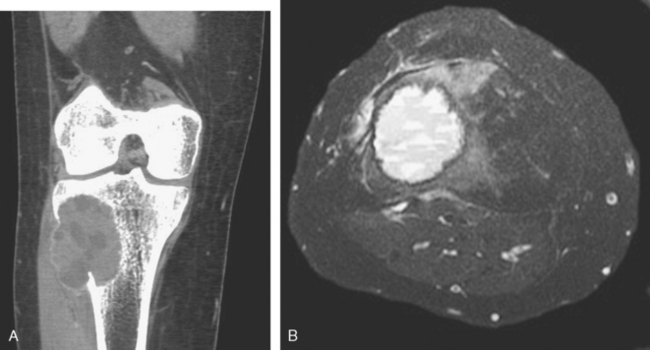
FIGURE 26-39 A, Coronal computed axial tomography scan showing eccentric aneurysmal bone cyst of tibia. The soft-tissue component is delineated by a thin rim of reactive subperiosteal bone. B, Axial magnetic resonance image demonstrating characteristic fluid-fluid levels.
The treatment of aneurysmal bone cyst is surgical, usually in the form of curettage or, in certain situations, en bloc resection. The recurrence rate is low, and spontaneous regression may occur following incomplete removal.
METASTATIC DISEASE
Metastatic tumors are the most common form of skeletal malignancy. They usually develop in later stages of tumor progression. The pathways of spread include (1) direct extension, (2) lymphatic or hematogenous dissemination, and (3) intraspinal seeding (via the Batson plexus of veins). Any cancer can spread to bone, but in adults more than 75% of skeletal metastases originate from cancers of the prostate, breast, kidney, and lung. In children, metastases to bone originate from neuroblastoma, Wilms tumor, osteosarcoma, Ewing sarcoma, and rhabdomyosarcoma.
Skeletal metastases are typically multifocal; however, carcinomas of the kidney and thyroid are notorious for producing solitary lesions. The metastases may occur in any bone, but most involve the axial skeleton (vertebral column, pelvis, ribs, skull, sternum), proximal femur, and humerus in descending order of frequency. The red marrow in these areas, with its rich capillary network and slow blood flow, facilitates implantation and growth of the tumor cells. Metastases to the small bones of the hands and feet are uncommon and usually originate from cancers of the lung, kidney, or colon.
The radiographic manifestations of metastases may be purely lytic, purely blastic, or mixed lytic and blastic. In lytic lesions, the metastatic cells secrete substances such as prostaglandins, cytokines, and PTH-related protein that stimulate osteoclastic bone resorption; the tumor cells themselves do not directly resorb bone. Lysis of bone tissue rich in growth factors such as TGF-β, IGF-1, FGF, PDGF, and bone morphogenetic proteins, in turn helps create an environment conducive to tumor cell growth. Carcinomas of the kidney, lung, and gastrointestinal tract and malignant melanoma produce lytic bone destruction. Other metastases elicit a sclerotic response, particularly prostate adenocarcinoma, which may do so by secreting WNT proteins that stimulate osteoblastic bone formation. Most metastases induce a mixed lytic and blastic reaction.
JOINTS
Joints are constructed to provide both movement and mechanical stability. They are classified as solid (nonsynovial) and cavitated (synovial). The solid joints, known as synarthroses, provide structural integrity and allow for minimal movement. They lack a joint space and are grouped according to the type of connective tissue (fibrous tissue or cartilage) that bridges the ends of the bones; fibrous synarthroses include the cranial sutures and the bonds between roots of teeth and the jawbones; cartilaginous synarthroses (synchondroses) are represented by the symphyses (manubriosternalis and pubic). Synovial joints, in contrast, have a joint space that allows for a wide range of motion. Situated between the ends of bones formed via enchondral ossification, they are strengthened by a dense fibrous capsule reinforced by ligaments and muscles. The boundary of the joint space consists of the synovial membrane, which is firmly anchored to the underlying capsule and does not cover the articular surface. Its contour is smooth except near the osseous insertion, where it is thrown into numerous villous folds. Synovial membranes are lined by synoviocytes, cuboidal connective cells that are arranged one to four cell layers deep. Synoviocytes synthesize hyaluronic acid and various proteins. The synovial lining lacks a basement membrane, which allows for quick exchange between blood and synovial fluid. Synovial fluid is clear and viscous, and is a filtrate of plasma containing hyaluronic acid that acts as a lubricant and provides nutrition for the articular hyaline cartilage.
Hyaline cartilage is a unique connective tissue ideally suited to serve as an elastic shock absorber and wear-resistant surface. It lacks a blood supply and does not have lymphatic drainage or innervation. Hyaline cartilage is composed of type 2 collagen, water, proteoglycans, and chondrocytes, each of which has specific functions. The collagen fibers enable the cartilage to resist tensile stresses and transmit vertical loads. The water and proteoglycans give hyaline cartilage its turgor and elasticity and have an important role in limiting friction. The chondrocytes synthesize the matrix as well as enzymatically digest it, with the half-life of the different components ranging from weeks (proteoglycans) to years (type 2 collagen). Chondrocytes secrete the degradative enzymes in an inactive form and enrich the matrix with enzyme inhibitors. Diseases that destroy articular cartilage do so by activating the catabolic enzymes and decreasing the production of inhibitors, thereby accelerating the rate of matrix breakdown. Cytokines such as IL-1 and TNF trigger the degradative process; their sources include chondrocytes, synoviocytes, fibroblasts, and inflammatory cells. Destruction of articular cartilage by indigenous cells is an important mechanism in many joint diseases.
Arthritis
OSTEOARTHRITIS
Osteoarthritis, also called degenerative joint disease, is the most common type of joint disease and is one of the 10 most disabling conditions in developed nations. It is characterized by the progressive erosion of articular cartilage. It is estimated that more than 33 billion dollars are spent annually in the United States for its treatment and for lost days of work. The term osteoarthritis implies an inflammatory disease; however, even though inflammatory cells may be present (usually in small numbers), osteoarthritis is considered to be an intrinsic disease of cartilage in which biochemical and metabolic alterations in individuals with genetic susceptibility result in its breakdown.
In most instances osteoarthritis appears insidiously, without apparent initiating cause, as an aging phenomenon (idiopathic or primary osteoarthritis). In these cases the disease is usually oligoarticular (affects few joints) but may be generalized. In about 5% of cases, osteoarthritis may appear in younger individuals having some predisposing condition, such as previous injuries to a joint; a congenital developmental deformity of a joint(s); or some underlying systemic disease such as diabetes, ochronosis, hemochromatosis, or marked obesity. In these settings the disease is called secondary osteoarthritis and often involves one or several predisposed joints; witness the shoulder or elbow involvements in baseball players and knees in basketball players. Gender has some influence on distribution. The knees and hands are more commonly affected in women and the hips in men.
Pathogenesis.
Osteoarthritis (OA) is a multifactorial disease that has genetic and environmental components. Studies of families and twins have suggested that the risk of OA is related to the net impact of multiple genes, each with a small effect. Genome-wide association studies (GWAS) are underway and it is likely that many risk-associated genes will be identified and validated shortly; a number of candidates have been identified, including genes involved in prostaglandin metabolism and WNT signaling.44 The major environmental factors relate to aging and biomechanical stress, which is influenced by obesity, muscle strength, and joint stability, structure, and alignment. The association with aging is strong; the prevalence of OA increases exponentially beyond the age of 50, and about 80% to 90% of individuals have evidence of the disease by age 65. Thus, OA joins heart disease and cancer as one of the unfortunate dividends of growing older. However, it is an oversimplification to consider OA an inevitable consequence of cartilage wear and tear. The mechanisms leading to OA are complex and not yet clear, but chondrocytes are at the center of the process, which can be divided into several phases: (1) chondrocyte injury, which is related to aging and genetic and biochemical factors; (2) early OA, in which chondrocytes proliferate (cloning) and secrete inflammatory mediators, collagens, proteoglycans, and proteases, which act together to remodel the cartilaginous matrix and initiate secondary inflammatory changes in the synovium and subchondral bone; and (3) late OA, in which repetitive injury and chronic inflammation lead to chondrocyte drop out, marked loss of cartilage, and extensive subchondral bone changes.45
Morphology. In the early stages of osteoarthritis the chondrocytes proliferate, forming clusters. Concurrently, the water content of the matrix increases and the concentration of proteoglycans decreases. Subsequently, vertical and horizontal fibrillation and cracking of the matrix occur as the superficial layers of the cartilage and type 2 collagen molecules are degraded. Grossly this manifests as a granular soft articular surface. Eventually, chondrocytes die and full-thickness portions of the cartilage are sloughed. The dislodged pieces of cartilage and subchondral bone tumble into the joint, forming loose bodies (joint mice). The exposed subchondral bone plate becomes the new articular surface, and friction with the opposing degenerated articular surface smooths and burnishes the exposed bone, giving it the appearance of polished ivory (bone eburnation) (Fig. 26-40). Concurrently there is rebuttressing and sclerosis of the underlying cancellous bone. Small fractures through the articulating bone are common, and the fracture gaps allow synovial fluid to be forced into the subchondral regions in a one-way, ball valve–like mechanism. The loculated fluid collection increases in size, forming fibrous-walled cysts. Mushroom-shaped osteophytes (bony outgrowths) develop at the margins of the articular surface and are capped by fibrocartilage and hyaline cartilage that gradually ossify. The synovium is usually only mildly congested and fibrotic, and may have scattered chronic inflammatory cells.
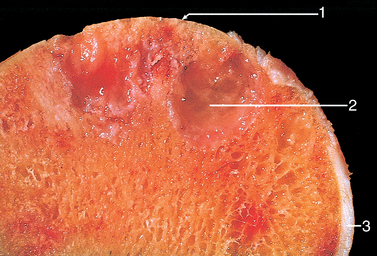
Clinical Course.
Osteoarthritis is an insidious disease. Patients with primary disease are usually asymptomatic until they are in their 50s. If a young person has significant manifestations of osteoarthritis, a search for some underlying cause should be made. Characteristic symptoms include deep, achy pain that worsens with use, morning stiffness, crepitus, and limitation of range of movement. Impingement on spinal foramina by osteophytes results in cervical and lumbar nerve root compression and radicular pain, muscle spasms, muscle atrophy, and neurologic deficits. Typically, only one or a few joints are involved except in the uncommon generalized variant. The joints commonly involved include the hips, knees, lower lumbar and cervical vertebrae, proximal and distal interphalangeal joints of the fingers, first carpometacarpal joints, and first tarsometatarsal joints of the feet (Fig. 26-41). Heberden nodes, prominent osteophytes at the distal interphalangeal joints, are common in women (but not men). The wrists, elbows, and shoulders are usually spared. There are still no satisfactory means of preventing primary osteoarthritis, and there are no effective methods of halting its progression. The disease may stabilize for years but more often is slowly progressive, and it is second only to cardiovascular diseases in causing long-term disability.
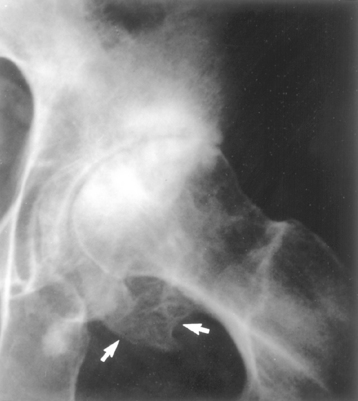
RHEUMATOID ARTHRITIS
Rheumatoid arthritis is a chronic systemic inflammatory disorder that may affect many tissues and organs—skin, blood vessels, heart, lungs, and muscles—but principally attacks the joints, producing a nonsuppurative proliferative and inflammatory synovitis that often progresses to destruction of the articular cartilage and ankylosis of the joints. Although the cause of rheumatoid arthritis remains unknown, genetic predisposition, environment, and autoimmunity have pivotal roles in the development, progression, and chronicity of the disease.
About 1% of the world’s population is afflicted by rheumatoid arthritis, women three to five times more often than men. It is most common in those 40 to 70 years old, but no age is immune. We first consider the morphology as a background to discuss pathogenesis.
Joints. Rheumatoid arthritis causes a broad spectrum of morphologic alterations; the most severe are manifested in the joints. Initially the synovium becomes grossly edematous, thickened, and hyperplastic, transforming its smooth contour to one covered by delicate and bulbous fronds (Fig. 26-42). The characteristic histologic features include (1) infiltration of synovial stroma by a dense perivascular inflammatory infiltrate composed of lymphoid aggregates (mostly CD4+ helper T cells), B cells, plasma cells, dendritic cells, and macrophages (Fig. 26-42C); (2) increased vascularity due to vasodilation and angiogenesis, with superficial hemosiderin deposits; (3) aggregation of organizing fibrin covering portions of the synovium and floating in the joint space as rice bodies; (4) accumulation of neutrophils in the synovial fluid and along the surface of synovium but usually not deep in the synovial stroma; (5) osteoclastic activity in underlying bone, allowing the synovium to penetrate into the bone and cause juxta-articular erosions, subchondral cysts, and osteoporosis; and (6) pannus formation. The pannus is a mass of synovium and synovial stroma consisting of inflammatory cells, granulation tissue, and synovial fibroblasts, which grows over the articular cartilage and causes its erosion. In time, after the cartilage has been destroyed, the pannus bridges the apposing bones to form a fibrous ankylosis, which eventually ossifies and results in bony ankylosis. Inflammation in the tendons, ligaments, and occasionally the adjacent skeletal muscle frequently accompanies the arthritis.
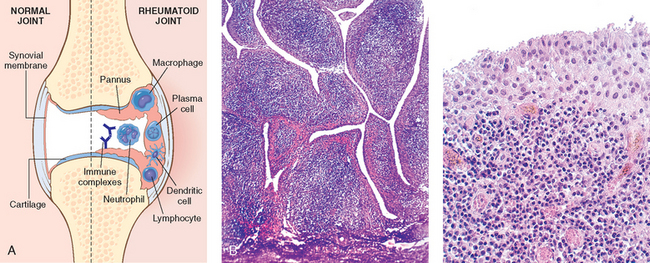
FIGURE 26-42 Rheumatoid arthritis. A, Schematic view of the joint lesion. B, Low magnification reveals marked synovial hypertrophy with formation of villi. C, At higher magnification, subsynovial tissue containing a dense lymphoidaggregate is seen.
(Modified from Feldmann M: Development of anti-TNF therapy for rheumatoid arthritis. Nat Rev Immunol 2:364, 2002.)
Skin. Rheumatoid nodules are the most common cutaneous lesion. They occur in approximately 25% of affected individuals, usually those with severe disease, and arise in regions of the skin that are subjected to pressure, including the ulnar aspect of the forearm, elbows, occiput, and lumbosacral area. Less commonly they form in the lungs, spleen, pericardium, myocardium, heart valves, aorta, and other viscera. Rheumatoid nodules are firm, nontender, and round to oval, and in the skin arise in the subcutaneous tissue. Microscopically they have a central zone of fibrinoid necrosis surrounded by a prominent rim of epithelioid histiocytes (activated macrophages) and numerous lymphocytes and plasma cells (Fig. 26-43).
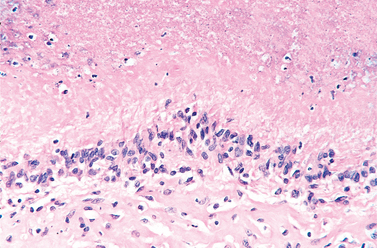
FIGURE 26-43 Subcutaneous rheumatoid nodule with an area of necrosis (top) surrounded by a palisade of macrophages and scattered chronic inflammatory cells.
Blood Vessels. Affected individuals with severe erosive disease, rheumatoid nodules, and high titers of rheumatoid factor are at risk of developing vasculitic syndromes (Chapter 11). Rheumatoid vasculitis is a potentially catastrophic complication of rheumatoid arthritis, particularly when it affects vital organs. The involvement of medium- to small-size arteries is similar to that occurring in polyarteritis nodosa, except that in rheumatoid arthritis the kidneys are not involved. Frequently, segments of small arteries such as vasa nervorum and digital arteries are obstructed by an obliterating endarteritis resulting in peripheral neuropathy, ulcers, and gangrene. Leukocytoclastic venulitis produces purpura, cutaneous ulcers, and nail bed infarction.
Pathogenesis.
Although much remains uncertain, it is currently believed that rheumatoid arthritis is triggered by exposure of a genetically susceptible host to an arthritogenic antigen resulting in a breakdown of immunological self-tolerance and a chronic inflammatory reaction. In this manner, an acute arthritis is initiated, but it is the continuing autoimmune reaction, the activation of CD4+ helper T cells, and the local release of inflammatory mediators and cytokines that ultimately destroys the joint (Fig. 26-44).
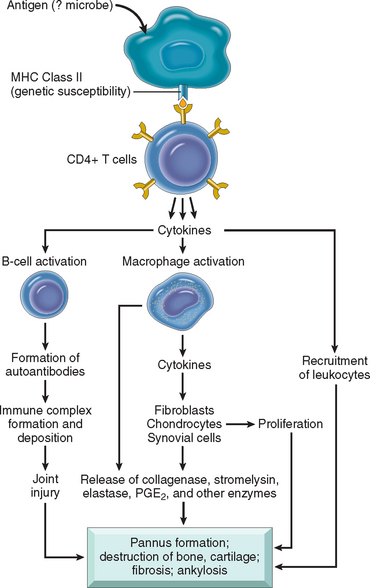
What mediators then bring about the destructive proliferative synovitis? These represent the “usual suspects.” The cytokines secreted by the T cells, such as interferon-γ and IL-17, act upon and stimulate synoviocytes and macrophages, which produce pro-inflammatory molecules such as IL-1, IL-6, IL-23, TNF, PGE2, nitric oxide, and the growth factors granulocyte-macrophage colony-stimulating factor and TGF-β. The inflammatory mediators activate endothelial cells in the synovium and thus facilitate leukocyte binding and transmigration. They also cause an increased production of cartilage matrix metalloproteinases, which along with antigen-antibody complexes, are important in the destruction of the articular cartilage. Additionally, they are potent stimulators of osteoclastogenesis and osteoclast activity by up-regulating the production of RANKL. RANKL is also expressed by the T cells and activated synoviocytes. Consequently the edematous, hyperplastic, and sticky (synoviocytes up-regulate vascular cell adhesion molecule) synovium rich in inflammatory cells becomes adherent to and grows over the articular surface, forming a pannus, and stimulates resorption of the adjacent bone. In the end, the pannus produces sustained, irreversible cartilage destruction and erosion of subchondral bone.
From this alphabet soup of mediators and cytokines, only one has been firmly implicated in the pathogenesis of rheumatoid arthritis—TNF. Happily, proof of its involvement has come from trials of specific TNF antagonists, which relieve swelling and pain, and appear to arrest disease progression (see below).
Clinical Course.
The clinical course of rheumatoid arthritis is extremely variable. The disease begins slowly and insidiously in more than half of affected individuals. Initially there is malaise, fatigue, and generalized musculoskeletal pain, and only after several weeks to months do the joints become involved. The pattern of joint involvement varies, but it is generally symmetrical and the small joints are affected before the larger ones. Symptoms usually develop in the hands (metacarpophalangeal and proximal interphalangeal joints) and feet, followed by the wrists, ankles, elbows, and knees. Uncommonly the upper spine is involved, but the lumbosacral region and hips are usually spared.
Involved joints are swollen, warm, painful, and particularly stiff on arising or following inactivity. Approximately 10% of affected individuals have an acute onset over several days with severe symptoms and polyarticular involvement. The typical patient has progressive joint involvement over a period of months to years, with initial minimal limitation of motion that steadily becomes more severe. The disease course may be slow or rapid, and fluctuates over the years, with the greatest damage occurring in the first 4 or 5 years. Approximately 20% of affected individuals enjoy periods of partial or complete remission, but the symptoms inevitably return and involve previously unaffected joints.
The radiographic hallmarks are joint effusions and juxta-articular osteopenia with erosions and narrowing of the joint space with loss of articular cartilage (Fig. 26-45). Destruction of tendons, ligaments, and joint capsules produces characteristic deformities, including radial deviation of the wrist, ulnar deviation of the fingers, and flexion-hyperextension abnormalities of the fingers (swan neck, boutonnière). The end result is deformed joints that have no stability and minimal or no range of motion. Large synovial cysts, like the Baker cyst in the posterior knee, may develop as the increased intra-articular pressure causes outpunching of the synovium.
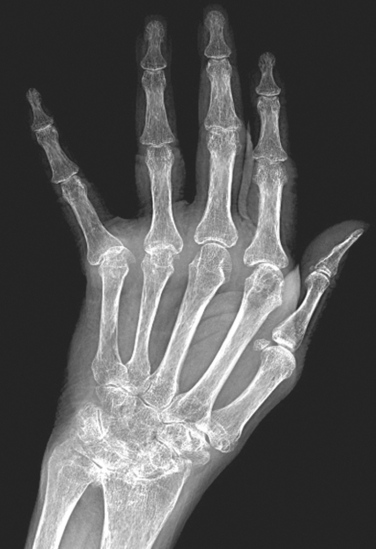
FIGURE 26-45 Rheumatoid arthritis of the hand. There is diffuse osteopenia, marked loss of the joint spaces of the carpal, metacarpal, phalangeal, and interphalangeal joints, periarticular bony erosions, and ulnar drift of the fingers.
The presence of rheumatoid factor and anti-CCP antibody are laboratory indicators that together are sensitive and fairly specific for rheumatoid arthritis. As pointed out, rheumatoid factor may not be present and also appears in many other conditions. Analysis of synovial fluid confirms an inflammatory arthritis with neutrophils, high protein content, and low mucin content, but is nonspecific. The diagnosis is based primarily on the clinical features and includes the presence of four of the following criteria: (1) morning stiffness, (2) arthritis in three or more joint areas, (3) arthritis of hand joints, (4) symmetric arthritis, (5) rheumatoid nodules, (6) serum rheumatoid factor, and (7) typical radiographic changes.
The treatment of rheumatoid arthritis is aimed at relieving the pain and inflammation, and slowing or arresting the relentless joint destruction. Therapies include corticosteroids, and synthetic and biologic disease-modifying drugs such as methotrexate and, most notably, antagonists of TNF. As mentioned earlier, these are remarkably effective and now constitute the mainstay of treatment. Most importantly, they prevent or slow joint destruction, which is the greatest source of disability, and have altered the natural history of the disease for the better. However, anti-TNF agents are not curative, and patients must be maintained on TNF antagonists or other immunosuppressive drugs to avoid disease flares. Also, inhibiting the activity of a key inflammatory mediator comes with a price; patients treated with anti-TNF agents are susceptible to certain infections, particularly M. tuberculosis.
JUVENILE IDIOPATHIC ARTHRITIS
Juvenile idiopathic arthritis (JIA), previously known as juvenile rheumatoid arthritis, encompasses all forms of arthritis that develop before 16 years of age and that persist for a minimum of 6 weeks. JIA is one of the more common connective tissue diseases; it affects 30,000 to 50,000 children in the United States and is an important cause of functional disability. The etiology of JIA is unknown. It is classified into seven discrete clinical subsets that may correspond to separate diseases and genetic backgrounds; (1) systemic arthritis, (2) oligoarthritis, (3) rheumatoid factor–positive polyarthritis, (4) rheumatoid factor–negative polyarthritis, (5) enthesitis (inflammation of a point of attachment of skeletal muscle to bone)-associated arthritis, (6) psoriatic arthritis, and (7) undifferentiated arthritis.
JIA differs from rheumatoid arthritis in adults in the following ways: (1) oligoarthritis is more common, (2) systemic disease is more frequent, (3) large joints are affected more often than small joints, (4) rheumatoid nodules and rheumatoid factor are usually absent, and (5) antinuclear antibody (ANA) seropositivity is common. As in rheumatoid arthritis, risk is associated with genetic susceptibility (with particular HLA alleles) and environmental factors; the heterogeneity of the disease indicates that different factors may be at play in different individuals. The inflammatory synovitis and morphologic changes are similar to those in rheumatoid arthritis. There is evidence of abnormal immunoregulation and a prevalence of activated CD4+ memory T cells within involved joints; cytokine production is prominent and helps drive the process.
Systemic arthritis may have a rather abrupt onset, is associated with remitting, high spiking fevers, migratory and transient skin rash, hepatosplenomegaly, and serositis. Long-term follow-up shows that affected individuals may experience recurrent flares or persistent disease that may be associated with significant morbidity and serious complications.
Arthritis affecting four or fewer joints during the first 6 months of disease in the absence of psoriasis and an HLA-B27 genotype defines the oligoarthritis variant. The arthritis is asymmetric, develops at an early age (younger than 6 years), and is commonly associated with iridocylitis and a positive ANA.
Rheumatoid factor–positive polyarthritis is similar to the adult form of the disease and is mainly seen in teenage girls. Rheumatoid factor–negative polyarthritis involves more than five joints within the first 6 months and consists of several subtypes that have features that overlap with oligoarthritis and rheumatoid factor–negative arthritis in adults, and a subset that shows stiffness and contractions, but little swelling.
Enthesitis-related arthritis mainly affects male children younger than 6 years, and most affected individuals are HLA-B27 positive. The enthesitis and arthritis affects tendoligamentous insertion sites and joints of the lower extremities.
Undifferentiated arthritis encompasses patients who do not fulfill inclusion criteria of the other groups or have overlapping features.
Long-term prognosis of JIA is very variable. Although many affected individuals may have sustained disease activity, only about 10% develop serious functional disability.
SERONEGATIVE SPONDYLOARTHROPATHIES
The seronegative spondyloarthropathies are a group of diseases that develop in genetically predisposed individuals and are initiated by ubiquitous environmental factors, especially infectious agents. The manifestations are immune mediated and are triggered by a T-cell response presumably directed against an undefined antigen that may cross-react with native molecules of the musculoskeletal system. Clinically, the diseases produce inflammatory peripheral or axial oligoarthritis and enthesopathies. The seronegative spondyloarthropathies include ankylosing spondylitis, reactive arthritis (Reiter syndrome and enteritis-associated arthritis), psoriatic arthritis, and arthritis associated with inflammatory bowel disease (ulcerative colitis, Crohn disease). Many are associated with the HLA-B27 allele and a triggering infection but without specific autoantibodies (hence the term “seronegative”). They all have inflammation of synovial joints and share overlapping clinical features, with extra-articular involvement of the eyes, skin, and cardiovascular system being relatively commonplace.
Ankylosing Spondyloarthritis
Also known as rheumatoid spondylitis and Marie-Strümpell disease, ankylosing spondyloarthritis is a chronic synovitis that causes destruction of articular cartilage and resultant bony ankylosis, especially of the sacroiliac and apophyseal joints (between tuberosities and processes). Inflammation of tendinoligamentous insertion sites eventuates in their ossification, producing squaring and fusion of the vertebral bodies, and bony outgrowths, which together result in severe spinal immobility. It usually becomes symptomatic in the second and third decades of life, and men are affected two to three times more frequently than women. Affected individuals characteristically present with low back pain, which frequently follows a chronic progressive course. Involvement of peripheral joints, such as the hips, knees, and shoulders, occurs in at least one third of affected individuals. Fracture of the spine, uveitis, aortitis, and amyloidosis are other recognized complications. Approximately 90% of the risk of developing the disease and the severity of the clinical manifestations is determined genetically. Although 90% of affected individuals are HLA-B27 positive, it has been suspected the other genes also contribute. Recent genome-wide association studies have shown associations with ARTS1, a gene that encodes a peptidase that trims antigens being processed for presentation by class I HLA molecules; and IL23R, the gene for the IL-23 receptor, suggesting that IL-23 (which promotes TH17 responses) may have a role in this disease.49
Reiter Syndrome
Reiter syndrome is a form of reactive arthritis and is defined by a triad of arthritis, nongonococcal urethritis or cervicitis, and conjunctivitis. Most affected individuals are men in their 20s or 30s, and more than 80% are HLA-B27 positive. This form of arthritis also affects individuals infected with the human immunodeficiency virus (HIV). The disease is probably caused by an autoimmune reaction initiated by prior infection of the gastrointestinal tract (Shigella, Salmonella, Yersinia, Campylobacter) and the genitourinary system (Chlamydia). Arthritic symptoms usually develop within several weeks of the inciting bout of urethritis or diarrhea. Joint stiffness and low back pain are common early symptoms. The ankles, knees, and feet are affected most often, frequently in an asymmetric pattern. Synovitis of a digital tendon sheath produces the sausage finger or toe, and ossification of tendoligamentous insertion sites leads to calcaneal spurs and bony outgrowths. Patients with severe chronic disease have involvement of the spine that is indistinguishable from ankylosing spondylitis. Extra-articular involvement manifests as inflammatory balanitis, conjunctivitis, cardiac conduction abnormalities, and aortic regurgitation. The natural behavior of Reiter syndrome is extremely variable. The episodes of arthritis usually wax and wane over a period of several weeks to 6 months. Almost 50% of affected individuals have recurrent arthritis, tendinitis, fasciitis, and lumbosacral pain that can cause significant functional disability.
Enteritis-Associated Arthritis
Enteritis-associated arthritis is caused by gastrointestinal infection by Yersinia, Salmonella, Shigella, and Campylobacter, among others. The outer cell membranes of these organisms have lipopolysaccharides as a major component, and they stimulate a host of immunological responses. The arthritis appears abruptly and tends to involve the knees and ankles but sometimes also the wrists, fingers, and toes. It lasts for about a year, then generally clears and only rarely is accompanied by ankylosing spondylitis.
Psoriatic Arthritis
Psoriatic arthritis is a chronic inflammatory arthropathy that affects peripheral and axial joints and entheses and is associated with psoriasis. Susceptibility to the disease is genetically determined and related to HLA-B27 and HLA-Cw6 alleles. It develops in more than 10% of the psoriatic population and has assorted phenotypic subtypes. Symptoms manifest between the ages of 30 and 50, and those involving the joints usually develop slowly but are acute in onset in one third of affected individuals. The patterns of joint involvement are diverse. The distal interphalangeal joints of the hands and feet are first affected in an asymmetric distribution in more than 50% of patients and may be associated with a sausage-like finger. The large joints such as the ankles, knees, hips, and wrists may be involved as well.50 Sacroiliac and spinal disease occurs in 20% to 40% of affected individuals. Aside from conjunctivitis and iritis, extra-articular manifestations are uncommon. Histologically, psoriatic arthritis is similar to rheumatoid arthritis. Psoriatic arthritis, however, is usually not as severe, remissions are more frequent, and joint destruction is less frequent.
INFECTIOUS ARTHRITIS
Microorganisms of all types can seed joints during hematogenous dissemination. Articular structures can also become infected by direct inoculation or from contiguous spread from a soft-tissue abscess or focus of osteomyelitis. Infectious arthritis is potentially serious, because it can cause rapid destruction of the joint and produce permanent deformities.
Bacterial Arthritis
Bacterial infections almost always cause an acute suppurative arthritis. The bacteria usually seed the joint during an episode of bacteremia; however, in neonates there is an increased incidence of contiguous spread from underlying epiphyseal osteomyelitis. The most common organisms are gonococcus, Staphylococcus, Streptococcus, Haemophilus influenzae, and gram-negative bacilli (E. coli, Salmonella, Pseudomonas, and others). H. influenzae arthritis predominates in children under 2 years of age, S. aureus is the main causative agent in older children and adults, and gonococcus is prevalent during late adolescence and young adulthood. Individuals with sickle cell disease are prone to infection with Salmonella at any age. These joint infections affect the sexes equally except for gonococcal arthritis, which is seen mainly in sexually active women. Predisposing conditions include immune deficiencies (congenital and acquired), debilitating illness, joint trauma, chronic arthritis of any cause, and intravenous drug abuse.
The classic presentation is the sudden development of an acutely painful and swollen infected joint that has a restricted range of motion. Systemic findings of fever, leukocytosis, and elevated sedimentation rate are common. In disseminated gonococcal infection the symptoms are more subacute. In 90% of nongonococcal cases, the infection involves only a single joint, usually the knee, followed in frequency by the hip, shoulder, elbow, wrist, and sternoclavicular joints. Axial articulations are more commonly involved in drug addicts. Prompt recognition and effective therapy prevent rapid joint destruction.
Tuberculous Arthritis
Tuberculous arthritis (Chapter 8) is a chronic progressive monoarticular disease that occurs in all age groups, especially adults. It usually develops as a complication of adjoining osteomyelitis or after hematogenous dissemination from a visceral (usually pulmonary) site of infection. Onset is insidious and causes gradual progressive pain. Systemic symptoms may or may not be present. Mycobacterial seeding of the joint induces the formation of confluent granulomas with central caseous necrosis. The affected synovium may grow as a pannus over the articular cartilage and erode the bone along the joint margins. Chronic disease results in severe destruction with fibrous ankylosis and obliteration of the joint space. The weight-bearing joints are usually affected, especially the hips, knees, and ankles in descending order of frequency.
Lyme Arthritis
As previously discussed (Chapter 8), Lyme arthritis is caused by infection with the spirochete Borrelia burgdorferi, which is transmitted by the ticks of the Ixodes ricinus complex. The initial infection of the skin is followed within several days or weeks by dissemination of the organism to other sites, especially the joints.
Approximately 60% to 80% of untreated individuals with Lyme disease develop joint symptoms within a few weeks to 2 years after the onset of the disease. The arthritis is the dominant feature of late disease; it tends to be remitting and migratory, and primarily involves large joints, especially the knees, shoulders, elbows, and ankles in descending order of frequency. Usually one or two joints are affected at a time, and the attacks last for a few weeks to months. Infected synovium exhibits a chronic papillary synovitis with synoviocyte hyperplasia, fibrin deposition, mononuclear cell infiltrates (especially CD4+ T cells), and onion-skin thickening of arterial walls. The morphology in severe cases can closely resemble that of rheumatoid arthritis. Silver stains may reveal small numbers of organisms in the vicinity of blood vessels in approximately 25% of cases. Chronic arthritis that is antibiotic refractory develops in approximately 10% of affected individuals and results from infection-induced autoimmunity. It is hypothesized that specific HLA-DR molecules bind an epitope of B. burgdorferi outer surface protein A, which initiates a T-cell reaction to this epitope. The T-cells may cross-react with an unknown self-antigen (an example of “molecular mimicry”). The joints in these patients have synovial pannus, which causes articular cartilage destruction and permanent deformities.51
Viral Arthritis
Arthritis can occur in the setting of a variety of viral infections, including alphavirus, parvovirus B19, rubella, Epstein-Barr virus, and hepatitis B and C virus. The clinical manifestations of the arthritis are variable and range from acute to subacute symptoms. It is unclear whether the joint symptoms are caused by direct infection of the joint by the virus, as seen in rubella and some alphavirus infections, or whether the viral infection generates an autoimmune reaction as seen in other forms of reactive or post-infectious arthritides.52 A variety of different rheumatic conditions, including reactive arthritis, psoriatic arthritis, and septic arthritis, have developed in individuals infected with HIV. The pathogenesis of some of these forms of HIV-associated chronic arthritis is probably autoimmune. The new effective antiretroviral therapies for HIV have ameliorated their severity.
CRYSTAL-INDUCED ARTHRITIS
Articular crystal deposits are associated with a variety of acute and chronic joint disorders. Endogenous crystals shown to be pathogenic include monosodium urate (gout), calcium pyrophosphate dihydrate, and basic calcium phosphate (hydroxyapatite). Exogenous crystals, such as corticosteroid ester crystals and talcum, and the biomaterials polyethylene and methyl methacrylate, may also induce joint disease. Silicone, polyethylene, and methyl methacrylate are used in prosthetic joints, and their debris that accumulates with long use and wear may result in local arthritis and failure of the prosthesis. Endogenous and exogenous crystals produce disease by triggering the cascade that results in cytokine-mediated cartilage destruction. Here we discuss the two most important crystal arthropathies: gout, caused by urates, and pseudo-gout, caused by calcium pyrophosphate.
Gout and Gouty Arthritis
Man is the only mammal to spontaneously develop hyperuricemia and gout, as only humans lack uricase, the enzyme responsible for the degradation of uric acid in other mammals. This, in combination with a high reabsorption rate of filtered urate, predisposes humans to hyperuricemia and gout, which is the common end point of a group of disorders that produce hyperuricemia.
Gout is marked by transient attacks of acute arthritis initiated by crystallization of urates within and about joints, leading eventually to chronic gouty arthritis and the appearance of tophi. Tophi represent large aggregates of urate crystals and the surrounding inflammatory reaction (see later). Most, but not all, individuals with chronic gout also develop urate nephropathy. Hyperuricemia (plasma urate level above 6.8 mg/dL) is necessary but not sufficient for the development of gout. More than 10% of the population of the Western hemisphere has hyperuricemia, and the prevalence is increasing; however, gout develops in fewer than 0.5% of these individuals. The various conditions producing hyperuricemia and gout (Table 26-7) are divided into those that produce primary gout (accounting for most idiopathic cases) and secondary gout (the cause of the hyperuricemia is known, and gout is not the main clinical expression of the disease). The role of hyperuricemia in the development of a variety of diseases, such as hypertension, chronic renal disease, cardiovascular disease, and the metabolic syndrome of hypertriglyceridemia, obesity, and insulin resistance, is controversial and remains a focus of investigation.
TABLE 26-7 Classification of Gout
| PRIMARY GOUT (90% OF CASES) |
| SECONDARY GOUT (10% OF CASES) |
Pathogenesis.
Uric acid is the end product of purine metabolism. Clinically, hyperuricemia develops from overproduction of urate in approximately 10% of cases (increased cell turnover—as in cancer, psoriasis, and during tumor lysis induced by chemotherapy) and reduced excretion in the remainder.
Plasma levels of uric acid are governed by a four-part renal transport system that involves glomerular filtration, reabsorption, secretion, and postsecretory reabsorption. Approximately 90% of the filtered urate is reabsorbed, and the urate transporter 1 gene (URAT1) has an important role in the reabsorption process. Decreased filtration and underexcretion of uric acid underlies most cases of primary gout. Two pathways are involved in purine synthesis: (1) a de novo pathway in which purines are synthesized from non-purine precursors and (2) a salvage pathway in which free purine bases derived from the breakdown of nucleic acids of endogenous or exogenous origin are recaptured (salvaged) (Fig. 26-46). The enzyme hypoxanthine guanine phosphoribosyl transferase (HGPRT) is involved in the salvage pathway. A deficiency of this enzyme leads to increased synthesis of purine nucleotides through the de novo pathway and hence increased production of uric acid. A complete lack of HGPRT occurs in the uncommon X-linked Lesch-Nyhan syndrome, seen only in males and characterized by hyperuricemia, severe neurologic deficits with mental retardation, self-mutilation, and in some cases gouty arthritis. Less severe deficiencies of the enzyme may also induce hyperuricemia and gouty arthritis with only mild neurologic deficits, but together these causes of gout are uncommon.
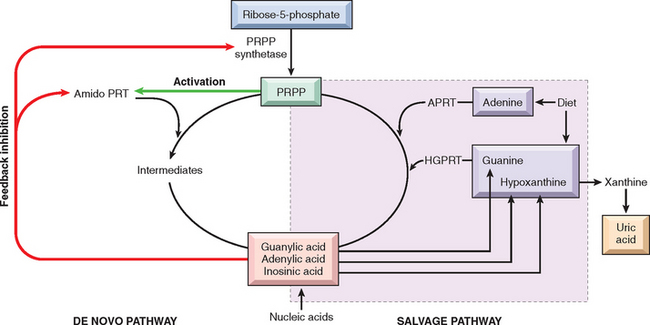
FIGURE 26-46 Purine metabolism. The conversion of PRPP to purine nucleotides is catalyzed by amido-PRT in the de novo pathway and by APRT and HGPRT in the salvage pathway. APRT, adenosine phosphoribosyltransferase; HGPRT, hypoxanthine-guanine phosporibosyltransferase; PRPP, phosphoribosyl pyrophosphate; PRT, phosphoribosyltransferase.
As stated earlier, hyperuricemia does not necessarily lead to gouty arthritis. Many factors contribute to the conversion of asymptomatic hyperuricemia into primary gout, including the following:
Central to the pathogenesis of the arthritis is precipitation of monosodium urate (MSU) crystals into the joints (Fig. 26-47). The solubility of MSU in a joint is modulated by temperature (the lower the less soluble) and by the intra-articular concentration of urate and cations. Crystallization is dependent on the presence of nucleating agents such as insoluble collagen fibers, chondroitin sulfate, proteoglycans, cartilage fragments, and other crystals. Since synovial fluid is a poorer solvent for monosodium urate than plasma, urates in the joint fluid become supersaturated more easily, particularly in the peripheral joints (ankles and toes), where temperatures are as low as 20°C. With prolonged hyperuricemia, crystals and microtophi of urates develop in the synovium and in the joint cartilage. Some unknown event, possibly trauma, causes the release of crystals into the synovial fluid, which begins a cascade of events that initiates, intensifies, and sustains a powerful inflammatory response that is the hallmark of the acute attack. MSU crystals are phagocytosed by macrophages and through an incompletely understood mechanism activate the NALP3 inflammasome, a multiprotein complex that includes the protease caspase 1. The inflammasome-activated caspase 1, in turn, cleaves and activates several cytokines, most notably IL-1β and IL-18. IL-1β induces the expression of adhesion molecules and the synthesis of the neurophil chemokine CXCL8, which is essential for the localization of neutrophils at the site of acute inflammation. The neutrophils pour fuel on the fire by releasing toxic free radicals, leukotrienes (leukotriene B4), and lysosomal enzymes. Thus comes about an acute arthritis, which typically remits spontaneously in days to weeks.53 A scheme of these events is shown in Figure 26-47.
Repeated attacks of acute arthritis lead eventually to chronic arthritis and the formation of tophi in the inflamed synovial membranes and periarticular tissue, as well as elsewhere. In time, severe damage to the cartilage develops and the function of the joints is compromised. It is not known why the chronic arthritis is asymptomatic for intervals of days to months, even though crystals are undoubtedly present in abundance in the joints.
Morphology. The distinctive morphologic changes in gout are (1) acute arthritis, (2) chronic tophaceous arthritis, (3) tophi in various sites, and sometimes (4) gouty nephropathy. Acute arthritis is characterized by a dense neutrophilic infiltrate that permeates the synovium and synovial fluid. The MSU crystals are frequently found in the cytoplasm of the neutrophils and are arranged in small clusters in the synovium. They are long, slender, and needle shaped, and are negatively birefringent. The synovium is edematous and congested, and also contains scattered lymphocytes, plasma cells, and macrophages. When the episode of crystallization abates and the crystals are resolubilized, the acute attack remits.
Chronic tophaceous arthritis evolves from the repetitive precipitation of urate crystals during acute attacks. The urates may heavily encrust the articular surfaces and form visible deposits in the synovium (Fig. 26-48). The synovium becomes hyperplastic, fibrotic, and thickened by inflammatory cells and forms a pannus that destroys the underlying cartilage leading to juxta-articular bone erosions. In severe cases, fibrous or bony ankylosis ensues, resulting in partial to complete loss of joint function.
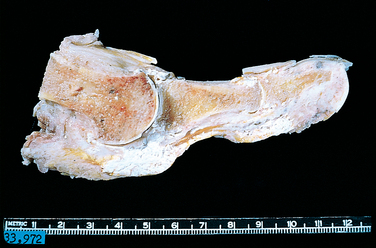
Tophi are the pathognomonic hallmark of gout. They are formed by large aggregations of urate crystals surrounded by an intense inflammatory reaction of macrophages, lymphocytes, and large foreign body giant cells, which may have completely or partially engulfed masses of crystals (Fig. 26-49). Tophi may appear in the articular cartilage of joints and in the periarticular ligaments, tendons, and soft tissues, including the olecranon and patellar bursae, Achilles tendons, and earlobes. Less frequently they may occur in the kidneys, nasal cartilages, skin of the fingertips, palms, soles, or elsewhere. Superficial tophi can ulcerate through the overlying skin.
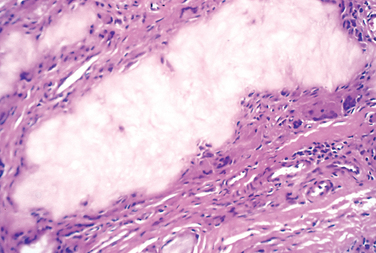
FIGURE 26-49 Photomicrograph of a gouty tophus. An aggregate of dissolved urate crystals is surrounded by reactive fibroblasts, mononuclear inflammatory cells, and giant cells.
Gouty nephropathy (Chapter 20) is associated with the deposition of MSU crystals in the renal medullary interstitium, sometimes forming tophi, intratubular precipitations, or free uric acid crystals, and the production of uric acid renal stones. Secondary complications, such as pyelonephritis, may ensue, particularly when the urates induce some urinary obstruction.
Clinical Course.
The natural history of gout is said to have four stages: (1) asymptomatic hyperuricemia, (2) acute gouty arthritis, (3) intercritical gout, and (4) chronic tophaceous gout. Asymptomatic hyperuricemia appears around puberty in males and after menopause in females. After many years acute arthritis appears in the form of the sudden onset of excruciating joint pain associated with localized hyperemia, warmth, and exquisite tenderness. Yet constitutional symptoms are uncommon except possibly mild fever. Most first attacks are monoarticular; 50% occur in the first metatarsophalangeal joint. Eventually, about 90% of affected individuals experience acute attacks in the following locations (in descending order of frequency): insteps, ankles, heels, knees, wrists, fingers, and elbows. Untreated, acute gouty arthritis may last for hours to weeks, but gradually there is complete resolution and the patient enters an asymptomatic intercritical period. Although some patients never have another attack, most experience a second acute episode within months to a few years. In the absence of appropriate therapy, the attacks recur at shorter intervals and frequently become polyarticular. Eventually, over the span of years, disabling chronic tophaceous gout develops. On average about 12 years pass between the initial acute attack and the appearance of chronic tophaceous arthritis. At this stage, radiograms show characteristic juxta-articular bone erosion caused by osteoclastic bone resorption and loss of the joint space. Progression leads to severe crippling disease.
Cardiovascular disease including atherosclerosis and hypertension is common in individuals with gout. Renal manifestations sometimes appear in the form of renal colic associated with the passage of gravel and stones and may proceed to chronic gouty nephropathy. About 20% of those with chronic gout die of renal failure. The diagnosis of gout should not be delayed, because numerous drugs are available to abort or prevent acute attacks of arthritis and mobilize tophaceous deposits. Their use is important, because many aspects of the disease are related to the duration and severity of the hyperuricemia. Generally, gout does not materially shorten the life span, but it may impair the quality of life.
Calcium Pyrophosphate Crystal Deposition Disease (Pseudo-Gout)
Calcium pyrophosphate crystal deposition disease (CPPD), also known as pseudo-gout and chondrocalcinosis, is one of the more common disorders associated with intra-articular crystal formation. It usually occurs in individuals over 50 years of age and becomes more common with increasing age, rising to a prevalence of 30% to 60% in those 85 years or older. The sexes and races are equally affected. CPPD is divided into sporadic (idiopathic), hereditary, and secondary types. In the hereditary variant the crystals develop relatively early in life and are associated with severe osteoarthritis. The autosomal dominant form of the disease is caused by germline mutations in the ANKH gene, which encodes a transmembrane pyrophosphate transport channel.54 The secondary form is associated with various disorders, including previous joint damage, hyperparathyroidism, hemochromatosis, hypomagnesemia, hypothyroidism, ochronosis, and diabetes. The basis for crystal formation is not known; altered activity of the matrix enzymes that produce and degrade pyrophosphate is suspected.
Morphology. The crystals first develop in the articular matrix, menisci, and intervertebral discs, and as the deposits enlarge they may rupture and seed the joint. Here, they are phagocytosed by macrophages, in which they activate the NALP3 inflammasome, eliciting a series of pro-inflammatory events similar or identical to those induced by urate crystals (Fig. 26-47). Neutrophils recruited by inflammatory mediators are thought to produce damage through the release of reactive oxygen species, catabolic enzymes, and cytokines, calling forth the more chronic reactions associated with macrophages and fibrosis. The crystals form chalky white friable deposits, which are seen histologically in stained preparations as oval blue-purple aggregates. Individual crystals are generally 0.5 to 5 μm in greatest dimension, are weakly birefringent, and have geometric shapes (Fig. 26-50). Rarely the crystals are deposited in masslike aggregates simulating tophi.
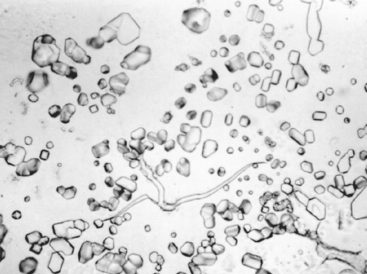
CPPD is frequently asymptomatic; however, it also can produce acute, subacute, or chronic arthritis that can be confused with osteoarthritis or rheumatoid arthritis. The joint involvement may last from several days to weeks and may be monoarticular or polyarticular; the knees, followed by the wrists, elbows, shoulders, and ankles, are most commonly affected. Ultimately, approximately 50% of affected individuals experience significant joint damage. Therapy is supportive. There is no known treatment that prevents or retards crystal formation.
Tumors and Tumor-Like Lesions
Reactive tumor-like lesions, such as ganglions, synovial cysts, and osteochondral loose bodies, commonly involve joints and tendon sheaths. They usually result from trauma or degenerative processes and are much more common than neoplasms. Primary neoplasms are unusual and tend to recapitulate the cells and tissue types (synovial membrane, fat, blood vessels, fibrous tissue, and cartilage) native to joints and related structures. Benign tumors are much more frequent than their malignant counterparts, which are rare and discussed with the soft-tissue tumors.
GANGLION AND SYNOVIAL CYST
A ganglion is a small (1–1.5 cm) cyst that is almost always located near a joint capsule or tendon sheath. A common location is around the joints of the wrist, where it appears as a firm, fluctuant, pea-sized translucent nodule. It arises as a result of cystic or myxoid degeneration of connective tissue; hence the cyst wall lacks a true cell lining. The lesion may be multilocular and enlarges through coalescence of adjacent areas of myxoid change. The fluid that fills the cyst is similar to synovial fluid; however, there is no communication with the joint space.
Herniation of synovium through a joint capsule or massive enlargement of a bursa may produce a synovial cyst. A well-recognized example is the synovial cyst that forms in the popliteal space in the setting of rheumatoid arthritis (Baker cyst). The synovial lining may be hyperplastic and contain inflammatory cells and fibrin but is otherwise unremarkable.
TENOSYNOVIAL GIANT-CELL TUMOR (LOCALIZED AND DIFFUSE)
Tenosynovial giant-cell tumor is the term for several closely related benign neoplasms that develop in the synovial lining of joints, tendon sheaths, and bursae. They harbor a consistent chromosomal translocation, t(1;2)(p13;q37), which fuses colony-stimulating factor 1 (CSF1) coding sequences to the promoter of the collagen type VI alpha-3 gene.55 As a result, the tumor cells overexpress CSF1, a chemoattractant for macrophages, which infiltrate the tumor in large numbers. Variants of tenosynovial giant-cell tumor include the diffuse type (previously known as pigmented villonodular synovitis), and the localized type (also known as giant-cell tumor of tendon sheath). Whereas the diffuse form tends to involve one or more joints, the localized kind usually occurs as a discrete nodule attached to a tendon sheath, commonly of the hand. Both variants usually are diagnosed in the 20s to 40s and affect the sexes equally.
Morphology. Grossly, tenosynovial giant-cell tumors are red-brown to mottled orange-yellow. In diffuse tumors the normally smooth joint synovium is converted into a tangled mat by red-brown folds, finger-like projections, and nodules (Fig. 26-51). In contrast, localized tumors are well circumscribed and resemble a small walnut. The neoplastic cells, which account for only 2% to 16% of the cells in the mass, are polyhedral, moderately sized, and resemble synoviocytes (Fig. 26-52). In the diffuse variant they spread along the surface and infiltrate the subsynovial issue. In nodular tumors the cells grow in a solid aggregate that may be attached to the synovium by a pedicle. Both variants are heavily infiltrated by macrophages, which may contain hemosiderin and lipid-filled vacuoles, or coalesce into multinucleated giant cells.
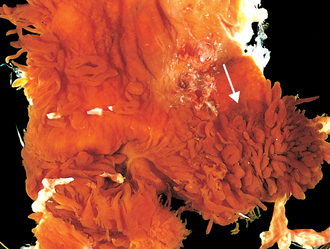
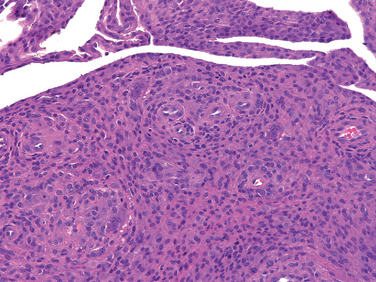
Diffuse tenosynovial giant-cell tumor usually presents in the knee in 80% of cases, followed in frequency by the hip, ankle, and calcaneocuboid joints. Affected individuals typically complain of pain, locking, and recurrent swelling. Tumor progression limits the range of movement of the joint and causes it to become stiff and firm. Sometimes a palpable mass is appreciated. Aggressive tumors erode into adjacent bones and soft tissues, causing confusion with other types of neoplasms. In contrast, the localized variant manifests as a solitary, slow-growing, painless mass that frequently involves the tendon sheaths along the wrists and fingers; it is the most common mesenchymal neoplasm of the hand. Cortical erosion of adjacent bone occurs in approximately 15% of cases. Surgery is the recommended treatment for both lesions; the diffuse tumors have a significant recurrence rate, because they are difficult to excise.
SOFT-TISSUE TUMORS AND TUMOR-LIKE LESIONS
Traditionally, soft-tissue tumors are defined as mesenchymal proliferations that occur in the extraskeletal, nonepithelial tissues of the body, excluding the viscera, coverings of the brain, and lymphoreticular system. They are classified according to the tissue they recapitulate (muscle, fat, fibrous tissue, vessels, and nerves) (Table 26-8), although there is little evidence that they actually arise from the normal differentiated counterpart. Some soft-tissue tumors have no normal tissue counterpart but have constant clinicopathologic features warranting their designation as distinct entities. The true frequency of soft-tissue tumors is difficult to estimate, because most benign lesions are not removed. A conservative estimate is that benign tumors outnumber their malignant counterparts (sarcomas) by a ratio of at least 100 : 1. In the United States, little more than 8000 sarcomas are diagnosed annually (0.8% of invasive malignancies), yet they are responsible for 2% of all cancer deaths, reflecting their lethal nature. In contrast to carcinomas, sarcomas usually metastasize via hematogenous routes, making the lung and skeleton common sites of dissemination.
| TUMORS OF ADIPOSE TISSUE |
| TUMORS AND TUMOR-LIKE LESIONS OF FIBROUS TISSUE |
| FIBROHISTIOCYTIC TUMORS |
| TUMORS OF SKELETAL MUSCLE |
| TUMORS OF SMOOTH MUSCLE |
| VASCULAR TUMORS |
| PERIPHERAL NERVE TUMORS |
| TUMORS OF UNCERTAIN HISTOGENESIS |
Pathogenesis and General Features
The cause of most soft-tissue tumors is unknown. There are documented associations, however, with radiation therapy, and rare instances in which chemical burns, thermal burns, or trauma were associated with subsequent development of a sarcoma. Exposure to phenoxyherbicides and chlorophenols has also been implicated in some cases. Kaposi sarcoma is causally associated with the human herpesvirus 8; however, viruses are probably not important in the pathogenesis of most human sarcomas. The majority of soft-tissue tumors occur sporadically, but a small minority are associated with genetic syndromes, the most notable of which are neurofibromatosis type 1 (neurofibroma, malignant peripheral nerve sheath tumor), Gardner syndrome (fibromatosis), Li-Fraumeni syndrome (soft-tissue sarcoma), and Osler-Weber-Rendu syndrome (telangiectasia). Current evidence suggests that soft-tissue tumors develop due to mutations in mesenchymal stem cells that are widely distributed in the body. Some of the abnormalities, such as specific chromosomal translocations, produce fusion genes that encode chimeric transcription factors. How these abnormal transcription factors drive neoplastic transformation is not clear. These genetic events can be specific enough to serve as diagnostic markers in some tumors56 (Table 26-9).
TABLE 26-9 Chromosomal and Genetic Abnormalities in Soft-Tissue Sarcomas
| Tumor | Cytogenetic Abnormality | Genetic Abnormality |
|---|---|---|
| Ewing sarcoma/Primitive neuroectodermal tumor | t(11;22)(q24;q12) | FLI1-EWS fusion gene |
| t(21;22)(q22;q12) | ERG-EWS fusion gene | |
| t(7;22)(q22;q12) | ETV1-EWS fusion gene | |
| Liposarcoma—myxoid and round-cell type | t(12;16)(q13;p11) | CHOP/TLS fusion gene |
| Synovial sarcoma | t(x;18)(p11;q11) | SYT-SSX fusion gene |
| Rhabdomyosarcoma—alveolar type | t(2;13)(q35;q14) | PAX3-FKHR fusion gene |
| t(1;13)(p36;q14) | PAX7-FKHR fusion gene | |
| Extraskeletal myxoid chondrosarcoma | t(9;22)(q22;q12) | CHN-EWS fusion gene |
| Desmoplastic small round-cell tumor | t(11;22)(p13;q12) | EWS-WT1 fusion gene |
| Clear-cell sarcoma | t(12;22)(q13;q12) | EWS-ATF1 fusion gene |
| Dermatofibrosarcoma protuberans | t(17;22)(q22;q15) | COLA1-PDGFB fusion gene |
| Alveolar soft-part sarcoma | t(X;17)(p11.2;q25) | TFE3-ASPL fusion gene |
| Congenital fibrosarcoma | t(12;15)(p13;q23) | ETV6-NTRK3 fusion gene |
Soft-tissue tumors may arise in any location; approximately 40% occur in the lower extremity, especially the thigh, 20% in the upper extremities, 10% in the head and neck, and 30% in the trunk and retroperitoneum. Regarding sarcomas, males are affected more frequently than females (1.4 : 1), and the incidence generally increases with age. Fifteen percent arise in children, and they constitute the fourth most common malignancy in this age group, following brain tumors, hematopoietic cancers, and Wilms tumor in frequency. Specific sarcomas tend to appear in certain age groups (e.g., rhabdomyosarcoma in children, synovial sarcoma in young adulthood, and liposarcoma and fibrosarcoma in middle to late adult life).
Several features of soft-tissue tumors influence their prognosis:
TABLE 26-10 Morphology of Cells in Soft Tissue Tumors
| Cell Type | Features | Tumor Type |
|---|---|---|
| Spindle cell | Rod-shaped, long axis twice as great as short axis | Fibrous, fibrohistiocytic, smooth muscle, Schwann cell |
| Small round cell | Size of a lymphocyte with little cytoplasm | Rhabdomyosarcoma, primitive neuroectodermal tumor |
| Epithelioid | Polyhedral with abundant cytoplasm, nucleus is centrally located | Smooth muscle, Schwann cell endothelial, epithelioid sarcoma |
TABLE 26-11 Architectural Patterns in Soft-Tissue Tumors
| Pattern | Tumor Type |
|---|---|
| Fascicles of eosinophilic spindle cells intersecting at right angles | Smooth muscle |
| Short fascicles of spindle cells radiating from a central point like spokes on a wheel—storiform | Fibrohistiocytic |
| Nuclei arranged in columns—palisading | Schwann cell |
| Herringbone | Fibrosarcoma |
| Mixture of fascicles of spindle cells and groups of epithelioid cells—biphasic | Synovial sarcoma |
With this brief background, we now turn to the individual tumors and tumor-like lesions. Some of the soft-tissuetumors are presented elsewhere: tumors of peripheral nerve (Chapter 28); tumors of vascular origin, including Kaposi sarcoma (Chapter 11); and uterine tumors of smooth muscle origin (Chapter 22).
Fatty Tumors
LIPOMAS
Benign tumors of fat, known as lipomas, are the most common soft-tissue tumor of adulthood. They are subclassified according to particular morphologic features as conventional lipoma, fibrolipoma, angiolipoma, spindle cell lipoma, myelolipoma, and pleomorphic lipoma. Some of the variants have characteristic chromosomal abnormalities; for example, conventional lipomas often show rearrangements of 12q14–q15, 6p, and 13q, and spindle cell and pleomorphic lipomas have rearrangements of 16q and 13q.
Morphology. The conventional lipoma, the most common subtype, is a well-encapsulated mass of mature adipocytes that varies considerably in size. It arises in the subcutis of the proximal extremities and trunk, most frequently during middle adulthood. Infrequently, lipomas are large, intramuscular, and poorly circumscribed.
Lipomas are soft, mobile, and painless (except angiolipoma) and are usually cured by simple excision.
LIPOSARCOMA
Liposarcomas are one of the most common sarcomas of adulthood and appear in the 40s to 60s; they are rare in children. They usually arise in the deep soft tissues of the proximal extremities and retroperitoneum, and are notorious for developing into large tumors.
Morphology. Liposarcomas are histologically divided into well-differentiated, myxoid/round cell, and pleomorphic variants. The cells in well-differentiated liposarcomas are readily recognized as lipocytes, and the tumor cells frequently contain supernumerary rings and giant rod chromosomes due to amplification of the 12q14–q15 region containing the MDM2 oncogene. This, you may recall, inhibits p53 (Chapter 7). In the other variants, most of the tumor cells are not obviously adipogenic, but some cells indicative of fatty differentiation are almost always present. These cells are known as lipoblasts; they mimic fetal fat cells and contain round clear cytoplasmic vacuoles of lipid that scallop the nucleus (Fig. 26-53). The myxoid/round cell variant of liposarcoma has a t(12;16)(q13;p11) chromosomal abnormality in most cases.
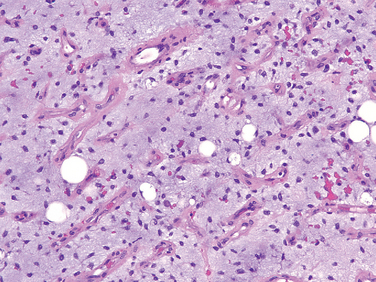
The well-differentiated variant is relatively indolent, the myxoid/round cell type is intermediate in its malignant behavior, and the pleomorphic variant usually is aggressive and frequently metastasizes. All types of liposarcoma recur locally and often repeatedly unless adequately excised.
Fibrous Tumors and Tumor-Like Lesions
REACTIVE PSEUDOSARCOMATOUS PROLIFERATIONS
Reactive pseudosarcomatous proliferations are nonneoplastic lesions that either develop in response to some form of local trauma (physical or ischemic) or are idiopathic. They are composed of plump reactive fibroblasts and related mesenchymal cells. Clinically they are alarming, because they develop suddenly and grow rapidly. Histologically they cause concern, because their hypercellularity, mitotic activity, and primitive appearance mimic sarcoma. Representative of this family of lesions are nodular fasciitis and myositis ossificans.
Nodular Fasciitis
Nodular fasciitis, also known as infiltrative or pseudosarcomatous fasciitis, is the most common of the reactive pseudosarcomas. It most often occurs in adults on the volar aspect of the forearm, followed in order of frequency by the chest and back. Affected individuals typically present with a several-week history of a solitary, rapidly growing, and sometimes painful mass. Preceding trauma is reported in only 10% to 15% of cases.
Morphology. Nodular fasciitis arises in the deep dermis, subcutis, or muscle. Grossly the lesion is several centimeters in greatest dimension, is nodular in configuration, and has poorly defined margins. The lesion is richly cellular and contains plump, immature-appearing fibroblasts and myofibroblasts arranged randomly or in short intersecting fascicles (Fig. 26-54). The cells vary in size and shape (spindle to stellate) and have conspicuous nucleoli; mitotic figures are abundant. Frequently the stroma is myxoid and contains lymphocytes and extravasated red blood cells. The histologic differential is extensive, but important lesions that must be excluded are fibromatosis and spindle cell sarcomas. Nodular fasciitis rarely recurs after excision.
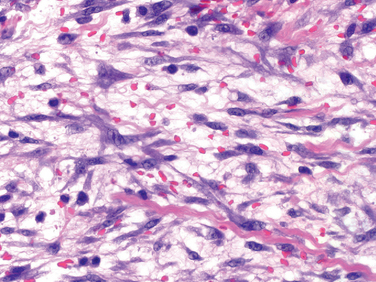
Myositis Ossificans
Myositis ossificans is distinguished from the other reactive fibroblastic proliferations by the presence of metaplastic bone. It usually develops in athletic adolescents and young adults and follows an episode of trauma in more than 50% of cases. The lesion typically arises in the musculature of the proximal extremities. The clinical findings are related to its stage of development; in the early phase the involved area is swollen and painful, and during the subsequent several weeks it becomes more circumscribed and firm. Eventually, it evolves into a painless, hard, well-demarcated mass.
Morphology. Grossly the usual lesion is 3 to 6 cm in greatest dimension and well-demarcated. Initially, the lesion is cellular and consists of plump, elongated fibroblast and myofibroblast-like cells simulating nodular fasciitis (see earlier). In due course these cells are surrounded by an intermediate zone that contains osteoblasts, which deposit ill-defined trabeculae of woven bone. The most peripheral zone contains well-formed, mineralized trabeculae that closely resemble cancellous bone. Eventually the entire lesion ossifies, and the intertrabecular spaces become filled with bone marrow. The mature lesion is completely ossified.
The radiographic findings parallel the morphologic progression. Initially, X-rays may show only soft-tissue fullness, but at about 3 weeks patchy flocculent radiodensities form in the periphery. The radiodensities become more extensive with time and slowly encroach on the radiolucent center (Fig. 26-55). Myositis ossificans must be distinguished from extraskeletal osteosarcoma. The latter usually occurs in elderly patients, the proliferating cells are cytologically malignant, and the tumor lacks the zonation of myositis ossificans. Simple excision of myositis ossificans is usually curative.
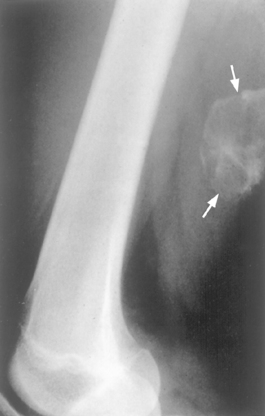
FIBROMATOSES
Superficial Fibromatosis (Palmar, Plantar, and Penile Fibromatoses)
Palmar, plantar, and penile fibromatoses, lesions that are more bothersome than serious, constitute a small group of superficial fibromatoses. They are characterized by nodular or poorly defined broad fascicles of fibroblasts and myofibroblasts surrounded by abundant dense collagen. The molecular mechanisms underlying superficial fibromatoses are unknown, but they are different from their deep-seated counterparts.
In the palmar variant (Dupuytren contracture) there is irregular or nodular thickening of the palmar fascia either unilaterally or bilaterally (50%). Over a span of years, attachment tothe overlying skin causes puckering and dimpling. At the same time a slowly progressive flexion contracture develops that mainly affects the fourth and fifth fingers of the hand. Essentially similar changes are seen with plantar fibromatosis except that flexion contractures are uncommon and bilateral involvement is infrequent. In penile fibromatosis (Peyronie disease) a palpable induration or mass appears, usually on the dorsolateral aspect of the penis. Eventually, it may cause abnormal curvature of the shaft, constriction of the urethra, or both.
All forms of superficial fibromatosis affect males more frequently than females. In about 20% to 25% of cases, the palmar and plantar fibromatoses stabilize and do not progress, in some instances resolving spontaneously. Some recur after excision, particularly the plantar variant.
Deep-Seated Fibromatosis (Desmoid Tumors)
Deep-seated fibromatoses lie in a gray zone between benign fibrous tumors and low-grade fibrosarcomas. On the one hand, they often present as large, infiltrative masses that frequently recur after incomplete excision; on the other, they are composed of banal well-differentiated fibroblasts that do not metastasize. They may occur at any age but are most frequent in the teens to 30s. Deep-seated fibromatosis is divided into extra-abdominal, abdominal, and intra-abdominal types, but all have similar gross and microscopic features. Extra-abdominal fibromatosis occurs in men and women with equal frequency and arises principally in the musculature of the shoulder, chest wall, back, and thigh. Abdominal fibromatosis generally arises in the musculoaponeurotic structures of the anterior abdominal wall in women during or after pregnancy. Intra-abdominal fibromatosis tends to occur in the mesentery or pelvic walls, often in individuals having familial adenomatous polyposis (Gardner syndrome) (Chapter 17). Mutations in the APC or β-catenin genes are present in the majority of these tumors (whether or not the affected individuals have underlying Gardner syndrome) and have an important role in their genesis.
Morphology. These tumors occur as gray-white, firm, poorly demarcated masses varying from 1 to 15 cm in greatest diameter. They are rubbery and tough, and infiltrate surrounding structures. Histologically deep-seated fibromatosis is composed of plump banal fibroblasts arranged in broad sweeping fascicles that infiltrate the adjacent tissue (Fig. 26-56). Mitoses may be frequent. Regenerative muscle cells when trapped within these lesions may take on the appearance of multinucleated giant cells.
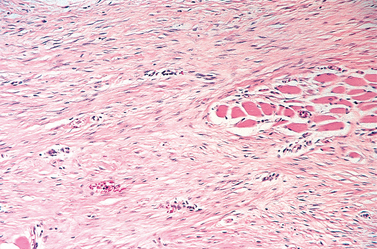
In addition to possibly being disfiguring or disabling, deep-seated fibromatosis is occasionally painful. Although curable by adequate excision, these lesions frequently recur locally and persistently when incompletely removed. Some tumors have responded to treatment with tamoxifen, and in other cases chemotherapy or irradiation has been effective. The rare reports of metastasis of fibromatosis must be interpreted as misdiagnosis of fibrosarcoma.
FIBROSARCOMA
Fibrosarcomas occur anywhere in the body, but are most common in the deep soft tissues of the extremities. Many tumors previously considered fibrosarcoma have been reclassified based on immunohistochemistry or cytogenetic findings as fibromatosis (desmoid), malignant peripheral nerve sheath tumors, or monophasic synovial sarcomas.
Morphology. Typically these neoplasms are unencapsulated, infiltrative, soft, fish-flesh masses often having areas of hemorrhage and necrosis. Better differentiated lesions may appear deceptivelyencapsulated. Histologic examination discloses all degrees of differentiation, from slowly growing tumors that closely resemble cellular fibromatosis and sometimes having spindled cells growing in a herringbone fashion (Fig. 26-57), to highly cellular neoplasms dominated by architectural disarray, pleomorphism, frequent mitoses, and areas of necrosis (Fig. 26-58).
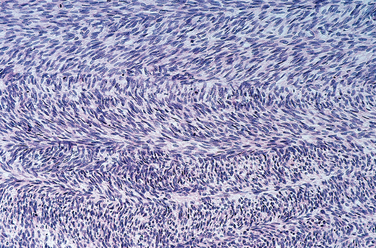
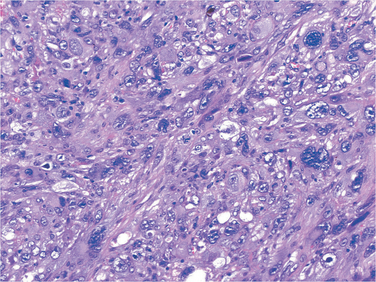
Fibrosarcomas are aggressive tumors, recurring in more than 50% of cases and metastasizing in more than 25%.
Fibrohistiocytic Tumors
Fibrohistiocytic tumors contain cellular elements that resemble both fibroblasts and histiocytes (macrophages). The phenotype of the neoplastic cells most closely resembles fibroblasts, and the term fibrohistiocytic should be viewed as descriptive in nature and not one that connotes the cell of origin.
BENIGN FIBROUS HISTIOCYTOMA (DERMATOFIBROMA)
Benign fibrous histiocytoma is a relatively common lesion that usually occurs in the dermis and subcutis. It is painless and slow growing, and most often presents in mid-adult life as a firm, small (≤1 cm) mobile nodule. Its morphologic features are described in Chapter 25.
MALIGNANT FIBROUS HISTIOCYTOMA
Once considered the most common sarcoma of adults, malignant fibrous histiocytoma referred to a group of soft-tissue tumors characterized by considerable cytologic pleomorphism, the presence of bizarre multinucleate cells, and storiform architecture. The phenotype of the neoplastic cell is now recognized to be fibroblastic and as a result malignant fibrous histiocytoma is being dropped as a diagnostic entity. Tumors previously diagnosed as malignant fibrous histiocytoma are currently classified as variants of fibrosarcoma (myxofibrosarcoma, pleomorphic fibrosarcoma, etc.) and other tumor types.
Tumors of Skeletal Muscle
Skeletal muscle neoplasms, in contrast to other groups of tumors, are almost all malignant. The benign variant, rhabdomyoma, is distinctly rare. The so-called cardiac rhabdomyoma is frequently seen in individuals with tuberous sclerosis and is discussed in Chapter 12.
RHABDOMYOSARCOMA
Rhabdomyosarcoma, the most common soft-tissue sarcoma of childhood and adolescence, usually appears before age 20. It may arise in any anatomic location, but most occur in the head and neck or genitourinary tract, where there is little if any skeletal muscle as a normal constituent. Only in the extremities do they appear in relation to skeletal muscle.
Morphology. Rhabdomyosarcoma is histologically subclassified into embryonal, alveolar, and pleomorphic variants. The rhabdomyoblast—the diagnostic cell in all types—contains eccentric eosinophilic granular cytoplasm rich in thick and thin filaments. Rhabdomyoblasts may be round or elongate; the latter are known as tadpole or strap cells, and may contain cross-striations visible by light microscopy (Fig. 26-59). Ultrastructurally, rhabdomyoblasts contain sarcomeres, and immunohistochemically they stain with antibodies to the myogenic markers desmin, MYOD1, and myogenin.
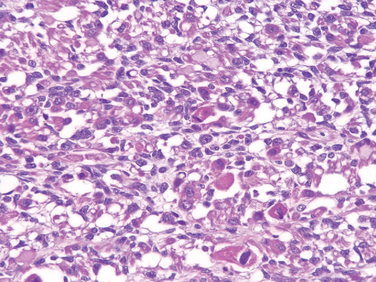
FIGURE 26-59 Rhabdomyosarcoma composed of malignant small round cells. The rhabdomyoblasts are large and round, and have abundant eosinophilic cytoplasm; no cross-striations are evident.
Embryonal rhabdomyosarcoma is the most common type, accounting for 60% of rhabdomyosarcomas. It includes the sarcoma botryoides, described in Chapter 22, as well as spindle cell and anaplastic variants. The tumor occurs in children younger than 10 years of age and typically arises in the nasal cavity, orbit, middle ear, prostate, and paratesticular region. This variant of rhabdomyosarcoma commonly has parental isodisomy of chromosome 11p15.5, which leads to overexpression of the imprinted IGFII gene.58 The sarcoma botryoides subtype develops in the walls of hollow, mucosal-lined structures, such as the nasopharynx, common bile duct, bladder, and vagina. Where the tumors abut the mucosa of an organ, they form a submucosal zone of hypercellularity called the cambium layer.
Most embryonal rhabdomyosarcomas present as a soft gray infiltrative mass. The tumor cells mimic skeletal muscle at various stages of embryogenesis and consist of sheets of both round and spindled cells in a myxoid stroma. Rhabdomyoblasts with visible cross-striations may be present.
Alveolar rhabdomyosarcoma tends to develop in early to middle adolescence, commonly arises in the deep musculature of the extremities, and represents approximately 20% of rhabdomyosarcomas. Histologically the tumor is traversed by a network of fibrous septae that divide the cells into clusters or aggregates that creates a crude resemblance to pulmonary alveolae (Fig. 26-60). The tumor cells are moderate in size, and many have little cytoplasm. Those in the center of the aggregates are dyscohesive, while those at the periphery adhere to the septae. Cells with cross-striations are identified in about 25% of cases. Cytogenetic studies have shown that this variant of rhabdomyosarcoma has a chromosomal translocation that either fuses the PAX3 to the FOXO1a gene, t(2,13)(q35;q14) or the PAX7 to the FOXO1a gene, t(1;13)(p36;q14).58 Tumors with the PAX3-FOXO1a fusion gene are more aggressive and associated with a worse prognosis.
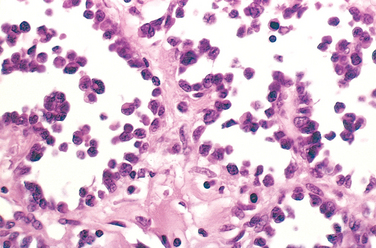
Pleomorphic rhabdomyosarcoma is characterized by numerous large, sometimes multinucleated, bizarre eosinophilic tumor cells. This variant is rare, has a tendency to arise in the deep soft tissue of adults, and can resemble other pleomorphic sarcomas histologically.
Rhabdomyosarcomas are aggressive neoplasms that are usually treated with surgery and chemotherapy with or without radiation therapy. The histologic type and location of the tumor influence survival. The botryoid subtype has the best prognosis, while the anaplastic embryonal, pleomorphic, and alveolar variants are often fatal.
Tumors of Smooth Muscle
LEIOMYOMAS
Leiomyomas, the benign smooth muscle tumors, often arise in the uterus; in fact, uterine leiomyomas are the most common neoplasm in women (Chapter 22). They develop in 77% of women and, depending on their number, size, and location, may cause a variety of symptoms including infertility. Leiomyomas may also arise from the arrector pili muscles found in the skin, nipples, scrotum, and labia and less frequently develop in the deep soft tissues and the wall of the gut. Those arising in the arrector muscles (pilar leiomyomas) may be multiple and painful. The phenotype of multiple cutaneous leiomyomas, in some individuals, is transmitted as an autosomal dominant trait and is associated with uterine leiomyomas and a predisposition to develop renal cell carcinoma—hereditary leiomyomatosis and renal cell cancer syndrome. This disorder is associated with a germline loss-of-function mutation in the fumarate hydratase gene located on chromosome 1q42.3.
Leiomyomas are usually not larger than 1 to 2 cm in greatest dimension and are composed of fascicles of spindle cells that tend to intersect each other at right angles. The tumor cells have blunt-ended, elongated nuclei and show minimal atypia and few mitotic figures. Solitary lesions are easily cured; however, multiple tumors may be so numerous that complete surgical removal is impractical.
LEIOMYOSARCOMA
Leiomyosarcomas account for 10% to 20% of soft-tissue sarcomas. They occur in adults and afflict women more frequently than men. Most develop in the skin and deep soft tissues of the extremities and retroperitoneum.
Morphology. Leiomyosarcomas present as painless firm masses. Retroperitoneal tumors may be large and bulky and cause abdominal symptoms. Histologically they consist of malignant spindle cells with cigar-shaped nuclei arranged in interweaving fascicles. Ultrastructurally, malignant smooth muscle cells contain bundles of thin filaments with dense bodies and pinocytic vesicles, and individual cells are surrounded by basal lamina. Immunohistochemically, they stain with antibodies to smooth muscle actin and desmin.
Treatment depends on the size, location, and grade. Superficial or cutaneous leiomyosarcomas are usually small and have a good prognosis, whereas those of the retroperitoneum are large, cannot be entirely excised, and cause death by both local extension and metastatic spread.
Synovial Sarcoma
Synovial sarcoma is so named because it was once believed to recapitulate synovium, but the cell of origin is still unclear. In addition, although the term synovial sarcoma implies an origin from the joint linings, less than 10% are intra-articular. Synovial sarcomas account for approximately 10% of all soft-tissue sarcomas and rank as the fourth most common sarcoma. Most occur in the 20s to 40s. The majority develop in the deep soft tissue and about 60% to 70% involve the lower extremity, especially around the knee and thigh. Patients usually present with a deep-seated mass that has been noted for several years. Uncommonly, these tumors occur in the head and neck or in viscera.
Morphology. Synovial sarcomas are morphologically biphasic or monophasic. The histologic hallmark of biphasic synovial sarcoma is dual lines of differentiation (i.e., epithelial-like and mesenchymal-like). The epithelial cells are cuboidal to columnar and form glands or grow in solid cords or aggregates. The spindle cells are arranged in densely cellular fascicles that surround the epithelial cells (Fig. 26-61). Many synovial sarcomas are monophasic, being composed of only spindled cells or, very rarely, epithelial cells. Lesions composed solely of spindled cells are easily mistaken for fibrosarcomas or malignant peripheral nerve sheath tumors. A characteristic feature when present is calcified concretions that can sometimes be detected radiographically. Immunohistochemistry is helpful in identifying these tumors, since the tumor cells yield positive reactions for keratin and epithelial membrane antigen, differentiating them from most other sarcomas. In addition, most synovial sarcomas show a characteristic chromosomal translocation t(x;18)(p11;q11) producing SS18-SSX1, SSX2, or SSX4 fusion genes that encode chimeric transcription factors.59 The specific type of translocation in synovial sarcoma has not been shown to be related to prognosis.
Synovial sarcomas are treated aggressively with limb-sparing therapy and frequently chemotherapy. The 5-year survival varies from 25% to 62%, and only 11% to 30% live for 10 years or longer. Common sites of metastases are the lung, skeleton, and occasionally the regional lymph nodes.
1 Glimcher M. Metabolic Bone Disease and Clinical Related Disorders, 2nd ed. Philadelphia: WB Saunders, 1990.
2 Baron R, Rawadi G. Targeting the Wnt/beta-catenin pathway to regulate bone formation in the adult skeleton. Endocrinology. 2007;148:2635.
3 Day TF, Yang Y. Wnt and hedgehog signaling pathways in bone development. J Bone Joint Surg Am. 2008;90(Suppl 1):19.
4 Cirmanova V, et al. The effect of leptin on bone—an evolving concept of action. Physiol Res. 2008;57(Suppl 1):S143.
5 Kogianni G, Noble BS. The biology of osteocytes. Curr Osteoporos Rep. 2007;5:81.
6 Robling AG, et al. Mechanical stimulation of bone in vivo reduces osteocyte expression of Sost/sclerostin. J Biol Chem. 2008;283:5866.
7 Asagiri M, Takayanagi H. The molecular understanding of osteoclast differentiation. Bone. 2007;40:251.
8 Cohen MM. The new bone biology: pathology, molecular, and clinical correlates. Am J Med Genet. 2006;140A:2646.
9 Boyce BF, Xing L. Biology of RANK, RANKL, and osteoprotegerin. Arthritis Res Ther. 2007;9(Suppl 1):S1.
10 Young MF, et al. Structure, expression, and regulation of the major noncollagenous matrix proteins of bone. Clin Orthop Relat Res. 1992;281:275.
11 Raisz LG. Physiology and pathophysiology of bone remodeling. Clin Chem. 1999;45:1353.
12 Olsen BR, et al. Bone development. Annu Rev Cell Dev Biol. 2000;16:191.
13 Zaidi M. Skeletal remodeling in health and disease. Nat Med. 2007;13:791.
14 Hartmann C. Skeletal development—Wnts are in control. Mol Cell. 2007;24:177.
15 Alman BA. Skeletal dysplasias and the growth plate. Clin Genet. 2008;73:24.
16 Mundlos S, Olsen BR. Heritable diseases of the skeleton. Part I: Molecular insights into skeletal development—transcription factors and signaling pathways. FASEB J. 1997;11:125.
17 Mundlos S, Olsen BR. Heritable diseases of the skeleton. Part II: Molecular insights into skeletal development—matrix components and their homeostasis. FASEB J. 1997;11:227.
18 Superti-Furga A, et al. Molecular-pathogenetic classification of genetic disorders of the skeleton. Am J Med Genet. 2001;106:282.
19 Marini JC, et al. Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans. Hum Mutat. 2007;28:209.
20 Martin E, Shapiro JR. Osteogenesis imperfecta:epidemiology and pathophysiology. Curr Osteoporos Rep. 2007;5:91.
21 Askmyr MK, et al. Towards a better understanding and new therapeutics of osteopetrosis. Br J Haematol. 2008;140:597.
22 Carbonell Sala S, Brandi ML. 2006 update on genetic determinants of osteoporosis. J Endocrinol Invest. 2007;30:2.
23 Mosekilde L. Mechanisms of age-related bone loss. Novartis Found Symp. 2001;235:150.
24 Styrkarsdottir U, et al. Multiple genetic loci for bone mineral density and fractures. N Engl J Med. 2008;358:2355.
25 Layfield R. The molecular pathogenesis of Paget disease of bone. Expert Rev Mol Med. 2007;9:1.
26 Whyte MP. Clinical practice. Paget’s disease of bone. N Engl J Med. 2006;355:593.
27 Schwarz C, et al. Diagnosis of renal osteodystrophy. Eur J Clin Invest. 2006;36(Suppl 2):13.
28 Giannoudis PV, et al. Fracture healing: the diamond concept. Injury. 2007;38(Suppl 4):S3.
29 Lafforgue P. Pathophysiology and natural history of avascular necrosis of bone. Joint Bone Spine. 2006;73:500.
30 Kaplan SL. Osteomyelitis in children. Infect Dis Clin North Am. 2005;19:787. vii,
31 Calhoun JH, Manring MM. Adult osteomyelitis. Infect Dis Clin North Am. 2005;19:765.
32 Gardam M, Lim S. Mycobacterial osteomyelitis and arthritis. Infect Dis Clin North Am. 2005;19:819.
33 Lee EH, et al. Osteoid osteoma: a current review. J Pediatr Orthop. 2006;26:695.
34 Klein MJ, Siegal GP. Osteosarcoma: anatomic and histologic variants. Am J Clin Pathol. 2006;125:555.
35 Kansara M, Thomas DM. Molecular pathogenesis of osteosarcoma. DNA Cell Biol. 2007;26:1.
36 Ramappa AJ, et al. Chondroblastoma of bone. J Bone Joint Surg Am. 2000;82A:1140.
37 Chow WA. Update on chondrosarcomas. Curr Opin Oncol. 2007;19:371.
38 Weinstein LS. G(s)alpha mutations in fibrous dysplasia and McCune-Albright syndrome. J Bone Miner Res. 2006;21(Suppl 2):P120.
39 Khoury JD. Ewing sarcoma family of tumors: a model for the new era of integrated laboratory diagnostics. Expert Rev Mol Diagn. 2008;8:97.
40 Ludwig JA. Ewing sarcoma: historical perspectives, current state-of-the-art, and opportunities for targeted therapy in the future. Curr Opin Oncol. 2008;20:412.
41 Nielson TO. Microarray analysis of sarcomas. Adv Anat Pathol. 2006;13:166.
42 Werner M. Giant cell tumour of bone: morphological, biological and histogenetical aspects. Int Orthop. 2006;30:484.
43 Oliveira AM, et al. Aneurysmal bone cyst variant translocations upregulate USP6 transcription by promoter swapping with the ZNF9, COL1A1, TRAP150, and OMD genes. Oncogene. 2005;24:3419.
44 Valdes AM, Spector TD. The contribution of genes to osteoarthritis. Med Clin North Am. 2009;93:45.
45 Goldring MB, Goldring SR. Osteoarthritis. J Cell Physiol. 2007;213:626.
46 Lundy SK, et al. Cells of the synovium in rheumatoid arthritis. T lymphocytes. Arthritis Res Ther. 2007;9:202.
47 Andersson AK, et al. Recent developments in the immunobiology of rheumatoid arthritis. Arthritis Res Ther. 2008;10:204.
48 Imboden JB. The immunopathogenesis of rheumatoid arthritis. Ann Rev Pathol Mech Dis. 2009;4:417.
49 Brown MA. Breakthroughs in genetic studies of ankylosing spondylitis. Rheumatology (Oxford). 2008;47:132.
50 Turkiewicz AM, Moreland LW. Psoriatic arthritis: current concepts on pathogenesis-oriented therapeutic options. Arthritis Rheum. 2007;56:1051.
51 Drouin EE, et al. Human homologues of a Borrelia T cell epitope associated with antibiotic-refractory Lyme arthritis. Mol Immunol. 2008;45:180.
52 Rulli NE, et al. The molecular and cellular aspects of arthritis due to alphavirus infections: lesson learned from Ross River virus. Ann N Y Acad Sci. 2007;1102:96.
53 Choi HK, et al. Pathogenesis of gout. Ann Intern Med. 2005;143:499.
54 Rosenthal AK. Update in calcium deposition diseases. Curr Opin Rheumatol. 2007;19:158.
55 Moller E, et al. Molecular identification of COL6A3-CSF1 fusion transcripts in tenosynovial giant cell tumors. Genes Chromosomes Cancer. 2008;47:21.
56 Riggi N, et al. Sarcomas: genetics, signalling, and cellular origins. Part 1: The fellowship of TET. J Pathol. 2007;213:4.
57 Rubin BP, Goldblum JR. Pathology of soft tissue sarcoma. J Natl Compr Canc Netw. 2007;5:411.
58 Paulino AC, Okcu MF. Rhabdomyosarcoma. Curr Probl Cancer. 2008;32:7.
59 de Bruijn DR, et al. The (epi)genetics of human synovial sarcoma. Genes Chromosomes Cancer. 2007;46:107.