Chapter 8 Infectious Diseases
General Principles of Microbial Pathogenesis
Despite the availability and use of effective vaccines and antibiotics, infectious diseases remain an important health problem in the United States and worldwide. In the United States, 2 of the top 10 leading causes of death are infectious diseases (pneumonia and influenza, and septicemia).1 Infectious diseases are particularly important causes of death among the elderly, people with acquired immunodeficiency syndrome (AIDS), those with chronic diseases, and those receiving immunosuppressive drugs. In developing countries, unsanitary living conditions and malnutrition contribute to a massive burden of infectious diseases that kills more than 10 million people each year. Most of these deaths are among children, especially from respiratory and diarrheal infections.2
CATEGORIES OF INFECTIOUS AGENTS
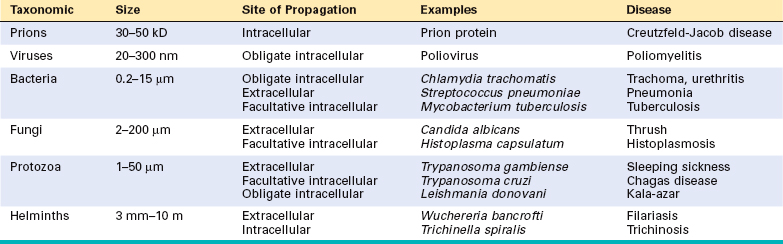
Infectious agents belong to a wide range of classes and vary in size from the approximately 27-kD nucleic acid–free prion to 20-nm poliovirus to 10-m tapeworms (Table 8-1).
Prions
Prions are composed of abnormal forms of a host protein, termed prion protein (PrP).3 These agents cause transmissible spongiform encephalopathies, including kuru (associated with human cannibalism), Creutzfeldt-Jakob disease (CJD), bovine spongiform encephalopathy (BSE; better known as mad cow disease), and variant Creutzfeldt-Jakob disease (vCJD; probably transmitted to humans from BSE-infected cattle).4 PrP is normally found in neurons. Diseases occur when the PrP undergoes a conformational change that confers resistance to proteases. The protease-resistant PrP promotes conversion of the normal protease-sensitive PrP to the abnormal form, explaining the infectious nature of these diseases. Accumulation of abnormal PrP leads to neuronal damage and distinctive spongiform pathologic changes in the brain. Spontaneous or inherited mutations in PrP, which make PrP resistant to proteases, have been observed in the sporadic and familial forms of CJD, respectively. CJD can be transmitted from person to person iatrogenically, by surgery, organ transplant, or blood transfusion. These diseases are discussed in detail in Chapter 28.
Viruses
Viruses are obligate intracellular parasites that depend on the host cell’s metabolic machinery for their replication. They consist of a nucleic acid genome surrounded by a protein coat (called a capsid) that is sometimes encased in a lipid membrane. Viruses are classified by their nucleic acid genome (DNA or RNA but not both), the shape of the capsid (icosahedral or helical), the presence or absence of a lipid envelope, their mode of replication, the preferred cell type for replication (called tropism), or the type of pathology. Because viruses are only 20 to 300 nm in size, they are best visualized with the electron microscope (Fig. 8-1). However, some viral particles aggregate within the cells they infect and form characteristic inclusion bodies, which may be seen with the light microscope and are useful for diagnosis. For example, cytomegalovirus (CMV)-infected cells are enlarged and show a large eosinophilic nuclear inclusion and smaller basophilic cytoplasmic inclusions; herpesviruses form a large nuclear inclusion surrounded by a clear halo; and both smallpox and rabies viruses form characteristic cytoplasmic inclusions. Many viruses do not produce inclusions (e.g., Epstein-Barr virus [EBV]).
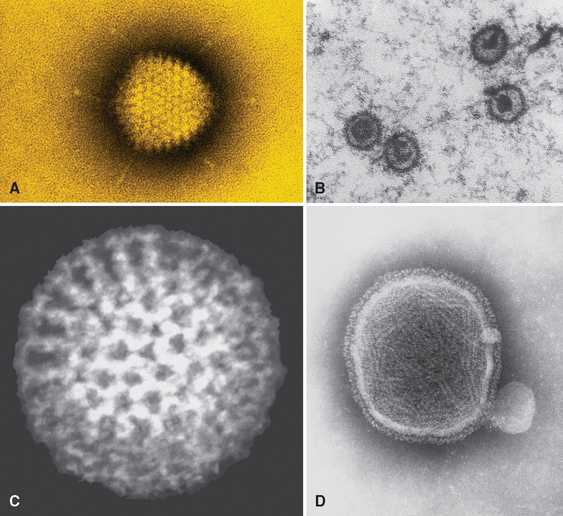
FIGURE 8-1 The variety of viral structures, as seen by electron microscopy. A, Adenovirus, an icosahedral nonenveloped DNA virus with fibers. B, Epstein-Barr virus, an icosahedral enveloped DNA virus. C, Rotavirus, a nonenveloped, wheel-like, RNA virus. D, Paramyxovirus, a spherical enveloped RNA virus. RNA is seen spilling out of the disrupted virus.
(Photos courtesy of Science Source; © Photo Researchers, Inc., New York, NY.)
Viruses account for a large share of human infections. Many viruses cause transient illnesses (e.g., colds, influenza). Other viruses are not eliminated from the body and persist within cells of the host for years, either continuing to multiply (e.g., chronic infection with hepatitis B virus [HBV]) or surviving in some nonreplicating form (termed latent infection) with the potential to be reactivated later. For example, herpes zoster virus, the cause of chickenpox, can enter dorsal root ganglia and establish latency there and later be periodically activated to cause shingles, a painful skin condition. Some viruses are involved in transformation of a host cell into a benign or malignant tumor (e.g., human papillomavirus [HPV]-induced benign warts and cervical carcinoma). Different species of viruses can produce the same clinical picture (e.g., upper respiratory infection); conversely, a single virus can cause different clinical manifestations depending on host age or immune status (e.g., CMV).
Bacteria
Bacteria are prokaryotes, meaning that they have a cell membrane but lack membrane-bound nuclei and other membrane-enclosed organelles. Most bacteria are bound by a cell wall consisting of peptidoglycan, a polymer of long sugar chains linked by peptide bridges. There are two forms of cell wall structures: a thick wall surrounding the cell membrane that retains crystal-violet stain (gram-positive bacteria) or a thin cell wall sandwiched between two phospholipid bilayer membranes (gram-negative bacteria) (Fig. 8-2). Bacteria are classified by Gram staining (positive or negative), shape (spherical ones are cocci; rod-shaped ones are bacilli) (Fig. 8-3), and need for oxygen (aerobic or anaerobic). Many bacteria have flagella, long helical filaments extending from the cell surface that enable bacteria to move. Some bacteria possess pili, another kind of surface projection that can attach bacteria to host cells or extracellular matrix. Most bacteria synthesize their own DNA, RNA, and proteins, but they depend on the host for favorable growth conditions.
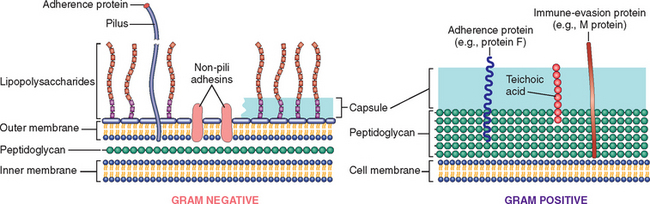
FIGURE 8-2 Molecules on the surface of gram-negative and gram-positive bacteria involved in pathogenesis. Not shown is the type 3 secretory apparatus of gram-negative bacteria (see text).
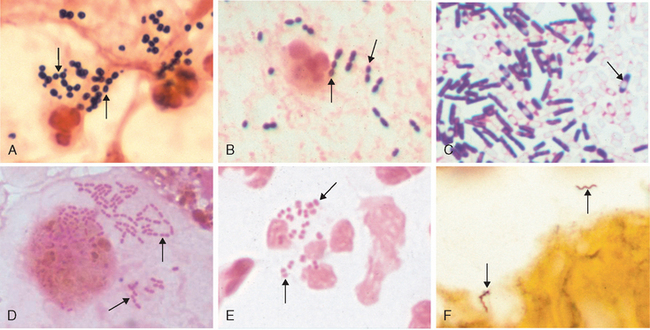
FIGURE 8-3 The variety of bacterial morphology. The bacteria are indicated by arrows in each panel. A, Gram stain of sputum from a patient with pneumonia. There are gram-positive cocci in clusters (Staphylococcus aureus) with degenerating neutrophils. B, Gram stain of sputum from a patient with pneumonia. Gram-positive, elongated cocci in pairs and short chains (Streptococcus pneumoniae) and a neutrophil are seen. C, Gram stain of Clostridium sordellii grown in culture. A mixture of gram-positive and gram-negative rods, many of which have subterminal spores (clear areas), are present. Clostridia species often stain as both gram-positive and gram-negative, although they are true gram-positive bacteria. D, Gram stain of a bronchoalveolar lavage specimen showing gram-negative intracellular rods typical of Enterobacteriaceae such as Klebsiella pneumoniae or Escherichia coli. E, Gram stain of urethral discharge from a patient with gonorrhea. Many gram-negative diplococci (Neisseria gonorrhoeae) are present within a neutrophil. F, Silver stain of brain tissue from a patient with Lyme disease meningoencephalitis. Two helical spirochetes (Borrelia burgdorferi) are indicated by arrows. The panels are at different magnifications.
(D, Courtesy of Dr. Karen Krisher, Clinical Microbiology Institute, Wilsonville, OR. Other panels courtesy of Dr. Kenneth Van Horn, Focus Diagnostics.)
Normal healthy people can be colonized by as many as 1012 bacteria on the skin, 1010 bacteria in the mouth, and 1014 bacteria in the gastrointestinal tract. Bacteria colonizing the skin include Staphylococcus epidermidis and Propionibacterium acnes, the cause of acne. Aerobic and anaerobic bacteria in the mouth, particularly Streptococcus mutans, contribute to dental plaque, a major cause of tooth decay. High-throughput sequencing methods have recently allowed detailed analysis of the diversity of intestinal bacterial flora. There are at least 395 species of bacteria in the normal intestinal flora, but only a small subset, mainly anaerobes, account for the great majority. In-depth analysis of the collective genome (called the “microbiome”) of the intestinal flora may yield insights into the evolutionary pressures that have selected the organisms that have succeeded in making humans their home, as well as disturbances in this symbiotic relationship, as in inflammatory bowel diseases.5 Many bacteria remain extracellular when they invade the body, while others can survive and replicate either outside or inside of host cells (facultative intracellular bacteria) and some grow only inside host cells (obligate intracellular bacteria).
Obligate intracellular bacteria include Chlamydia and Rickettsia, which replicate inside membrane-bound vacuoles in epithelial and endothelial cells, respectively. These bacteria get most or all of their energy source, ATP, from the host cell. Chlamydia trachomatis is the most frequent infectious cause of female sterility (by scarring and narrowing of the fallopian tubes) and blindness (by chronic inflammation of the conjunctiva that eventually causes scarring and opacification of the cornea). Rickettsiae injure the endothelial cells in which they grow, and so cause a hemorrhagic vasculitis, often visible as a rash, but they may also injure the central nervous system (CNS) and cause death (Rocky Mountain spotted fever [RMSF] and epidemic typhus). Rickettsiae are transmitted by arthropod vectors, including lice (epidemic typhus), ticks (RMSF and ehrlichiosis), and mites (scrub typhus).6
Mycoplasma organisms and those belonging to the related genus Ureaplasma are unique among extracellular bacterial pathogens, because they do not have a cell wall. These are the tiniest free-living organisms known (125–300 nm).
Fungi
Fungi are eukaryotes that possess thick chitin-containing cell walls and ergosterol-containing cell membranes. Fungi can grow either as rounded yeast cells or as slender filamentous hyphae. Hyphae may be septate (with cell walls separating individual cells) or aseptate, which is an important distinguishing characteristic in clinical material. Some of the most important pathogenic fungi exhibit thermal dimorphism; that is, they grow as hyphal forms at room temperature but as yeast forms at body temperature. Fungi may produce sexual spores or, more commonly, asexual spores referred to as conidia. The latter are produced on specialized structures or fruiting bodies arising along the hyphal filament. Fungi may cause superficial or deep infections. Superficial infections involve the skin, hair, and nails. Fungal species that are confined to superficial layers of the human skin are known as dermatophytes. These infections are commonly referred to by the term “tinea” followed by the area of the body affected (e.g., tinea pedis, “athlete’s foot”; tinea capitis, “ringworm of the scalp”). Certain fungal species invade the subcutaneous tissue, causing abscesses or granulomas (e.g., sporotrichosis and tropical mycoses).
Deep fungal infections can spread systemically and invade tissues, destroying vital organs in immunocompromised hosts, but usually heal or remain latent in otherwise normal hosts. Some deep fungal species are limited to a particular geographic region (e.g., Coccidioides in the southwestern United States and Histoplasma in the Ohio River Valley). Opportunistic fungi (e.g., Candida, Aspergillus, Mucor, and Cryptococcus), by contrast, are ubiquitous organisms that either colonize individuals or are encountered from environmental sources. In immunodeficient individuals, opportunistic fungi give rise to life-threatening infections characterized by tissue necrosis, hemorrhage, and vascular occlusion, with little or no inflammatory response. AIDS patients are often infected by the opportunistic fungus Pneumocystis jiroveci (previously called Pneumocystis carinii).
Protozoa
Parasitic protozoa are single-celled eukaryotes that are major causes of disease and death in developing countries. Protozoa can replicate intracellularly within a variety of cells (e.g., Plasmodium in red blood cells, Leishmania in macrophages) or extracellularly in the urogenital system, intestine, or blood. Trichomonas vaginalis are flagellated protozoal parasites that are sexually transmitted and can colonize the vagina and male urethra. The most prevalent intestinal protozoans, Entamoeba histolytica and Giardia lamblia, have two forms: (1) motile trophozoites that attach to the intestinal epithelial wall and may invade, and (2) immobile cysts that are resistant to stomach acids and are infectious when ingested. Blood-borne protozoa (e.g., Plasmodium, Trypanosoma, and Leishmania) are transmitted by insect vectors, in which they replicate before being passed to new human hosts. Intestinal protozoa are acquired by ingestion of cysts from contaminated food or water. Toxoplasma gondii is acquired either by contact with oocyst-shedding kittens or by eating cyst-ridden, undercooked meat.
Helminths
Parasitic worms are highly differentiated multicellular organisms. Their life cycles are complex; most alternate between sexual reproduction in the definitive host and asexual multiplication in an intermediary host or vector. Thus, depending on parasite species, humans could harbor adult worms (e.g., Ascaris lumbricoides) or immature stages (e.g., Toxocara canis) or asexual larval forms (e.g., Echinococcus species). Once adult worms take up residence in humans, they do not multiply but they produce eggs or larvae that are usually passed out in stool. Often, the severity of disease is in proportion to the number of organisms that have infected the individual (e.g., 10 hookworms cause little disease, whereas 1000 hookworms cause severe anemia by consuming 100 mL of blood per day). In some helminthic infections, disease is caused by inflammatory responses to the eggs or larvae rather than to the adults (e.g., schistosomiasis).
Ectoparasites
Ectoparasites are insects (lice, bedbugs, fleas) or arachnids (mites, ticks, spiders) that attach to and live on or in the skin. Arthropods may produce disease directly by damaging the human host or indirectly by serving as the vectors for transmission of an infectious agent into a human host. Some arthropods cause itching and excoriations (e.g., pediculosis caused by lice attached to hair shafts, or scabies caused by mites burrowing into the stratum corneum). At the site of the bite, mouth parts may be found associated with a mixed infiltrate of lymphocytes, macrophages, and eosinophils. In addition, attached arthropods can be vectors for other pathogens. For example, deer ticks transmit the Lyme disease spirochete Borrelia burgdorferi.
SPECIAL TECHNIQUES FOR DIAGNOSING INFECTIOUS AGENTS
Some infectious agents or their products can be directly observed in hematoxylin and eosin–stained sections (e.g., the inclusion bodies formed by CMV and herpes simplex virus (HSV); bacterial clumps, which usually stain blue; Candida and Mucor among the fungi; most protozoans; and all helminths). Many infectious agents, however, are best visualized by special stains that identify organisms on the basis of particular characteristics of their cell wall or coat—Gram, acid-fast, silver, mucicarmine, and Giemsa stains—or after labeling with specific antibody probes (Table 8-2). Regardless of the staining technique, organisms are usually best visualized at the advancing edge of a lesion rather than at its center, particularly if there is necrosis.
TABLE 8-2 Special Techniques for Diagnosing Infectious Agents
| Techniques | Infectious Agents |
|---|---|
| Gram stain | Most bacteria |
| Acid-fast stain | Mycobacteria, nocardiae (modified) |
| Silver stains | Fungi, legionellae, pneumocystis |
| Periodic acid–Schiff | Fungi, amebae |
| Mucicarmine | Cryptococci |
| Giemsa | Campylobacteria, leishmaniae, malaria parasites |
| Antibody probes | All classes |
| Culture | All classes |
| DNA probes | All classes |
Acute infections can be diagnosed serologically by detecting pathogen-specific antibodies in the serum. The presence of specific IgM antibody shortly after the onset of symptoms is often diagnostic. Alternatively, specific antibody titers can be measured early (“acute”) and 4–6 weeks (“convalescent”) after infection; a four-fold rise in titer is usually considered diagnostic.
Nucleic acid–based tests, collectively called molecular diagnostics, have become routine methods for detecting and quantifying several pathogens. For instance, in people infected with the human immunodeficiency virus (HIV), quantification of HIV RNA is an important guide to antiretroviral therapy.7 The management of HBV and HCV infections is similarly guided by nucleic acid–based viral quantification or typing to predict resistance to antiviral drugs.
Nucleic acid amplification tests, such as polymerase chain reaction (PCR) and transcription-mediated amplification, have become routine for diagnosis of gonorrhea, chlamydial infection, tuberculosis, and herpes encephalitis. In some cases, molecular assays are much more sensitive than conventional testing.8,9 PCR testing of cerebrospinal fluid (CSF) for herpes simplex virus (HSV) encephalitis has a sensitivity of about 80%, while viral culture of CSF has a sensitivity of less than 10%. Similarly, nucleic acid tests for genital chlamydia detect 10% to 30% more infections than does conventional chlamydia culture. In other cases, such as gonorrhea, the sensitivity of nucleic acid testing is similar to that of culture.
NEW AND EMERGING INFECTIOUS DISEASES
Although infectious diseases such as leprosy have been known since biblical times, and parasitic schistosomes and mycobacteria have been demonstrated in Egyptian mummies, a surprising number of new infectious agents continue to be discovered (Table 8-3). The infectious causes of some diseases with significant morbidity and mortality were previously unrecognized, because some of the infectious agents are difficult to culture; examples include Helicobacter pylori gastritis, HBV and HCV, and Legionnaires pneumonia. Some infectious agents are genuinely new to humans, e.g., HIV, which causes AIDS, and B. burgdorferi, which causes Lyme disease. Other infections have become much more common because of immunosuppression caused by AIDS or therapy for transplant rejection and some cancers (e.g., CMV, Kaposi sarcoma herpesvirus, Mycobacterium avium-intracellulare, P. jiroveci, and Cryptosporidium parvum).10,11 Finally, infectious diseases that are common in one area may be introduced into a new area. For example, West Nile virus has been common in Europe, Asia, and Africa for years but was first described in the United States in 1999.
TABLE 8-3 Some Recently Recognized Infectious Agents and Manifestations
| Date Recognized | Infectious Agent | Manifestations |
|---|---|---|
| 1977 | Ebola virus | Epidemic Ebola hemorrhagic fever |
| Hantaan virus | Hemorrhagic fever with renal syndrome | |
| Legionella pneumophila | Legionnaires disease | |
| Campylobacter jejuni | Enteritis | |
| 1980 | HTLV-I | T-cell lymphoma or leukemia, HTLV-associated myelopathy |
| 1981 | Staphylococcus aureus | Toxic shock syndrome |
| 1982 | Escherichia coli O157:H7 | Hemorrhagic colitis, hemolytic-uremic syndrome |
| Borrelia burgdorferi | Lyme disease | |
| 1983 | HIV | AIDS |
| Helicobacter pylori | Gastric ulcers | |
| 1988 | Hepatitis E | Enterically transmitted hepatitis |
| 1989 | Hepatitis C | Hepatitis C |
| 1992 | Vibrio cholerae O139 | New epidemic cholera strain |
| Bartonella henselae | Cat-scratch disease | |
| 1995 | KSHV (HHV-8) | Kaposi sarcoma in AIDS |
| 1999 | West Nile virus | West Nile fever, neuroinvasive disease |
| 2003 | SARS coronavirus | Severe acute respiratory syndrome |
Human demographics and behavior are among the many variables that contribute to the emergence of infectious diseases. AIDS was first recognized in the United States as predominantly a disease of homosexuals and drug abusers, but heterosexual transmission is now common. In sub-Saharan Africa, the area of the world with the highest number of AIDS cases, it is predominantly a heterosexual disease.12 Changes in the environment occasionally drive rates of infectious diseases. Reforestation of the eastern United States has led to massive increases in the populations of deer and mice, which carry the ticks that transmit Lyme disease, babesiosis, and ehrlichiosis.13 Failure of DDT to control the mosquitoes that transmit malaria and the development of drug-resistant parasites have dramatically increased the morbidity and mortality of Plasmodium falciparum in Asia, Africa, and Latin America. Microbial adaptation to widespread antibiotic use contributed to the emergence of drug resistance in many species of bacteria, including Mycobacterium tuberculosis, Neisseria gonorrhoeae, Staphylococcus aureus, and Enterococcus faecium. Infections with antibiotic-resistant bacteria are becoming a serious problem, such as methicillin-resistant staphylococcus (discussed later).
AGENTS OF BIOTERRORISM
Sadly, the anthrax attacks in the United States in 2001 transformed the theoretical threat of bioterrorism into reality. The Centers for Disease Control and Prevention (CDC) have evaluated the microorganisms that pose the greatest danger as weapons on the basis of the efficiency with which disease can be transmitted, how difficult the microorganisms are to produce and distribute, what can be done to defend against them, and the extent to which they are likely to alarm the public and produce widespread fear. The CDC has ranked bioweapons into three categories, A, B, and C, based on these criteria (Table 8-4).14
TABLE 8-4 Potential Agents of Bioterrorism
| Category A Diseases/Agents |
| Category B Diseases/Agents |
| Category C Diseases/Agents |
| Emerging infectious disease threats such as Nipah virus and Hantavirus |
Adapted from Centers for Disease Control Information.
Category A agents pose the highest risk and can be readily disseminated or transmitted from person to person, can cause high mortality with potential for major public health impact, might cause public panic and social disruption, and might require special action for public health preparedness. For example, smallpox is a category A agent because of its high transmissibility in any climate or season, case mortality rate of 30% or greater, and lack of effective antiviral therapy. This agent can be easily disseminated because of the stability of the virus in aerosol form and the very small dose needed for infection. Smallpox naturally spreads from person to person mainly by direct contact with virus in skin lesions or contaminated clothing or bedding. Symptoms appear after 7 to 17 days. Initially there is high fever, headache, and backache, followed by the appearance of the rash, which first appears on the mucosa of the mouth and pharynx, face, and forearms and later spreads to the trunk and legs and becomes vesicular and later pustular. Because people can be contagious during the incubation period, this virus has the potential to continue to spread throughout an unprotected population. Since vaccination ended in the United States in 1972 and vaccination immunity has waned, the population is highly susceptible to smallpox. Recent concern that smallpox could be used for bioterrorism has led to a return of vaccination for selected groups in the United States and Israel.
Category B agents are moderately easy to disseminate, produce moderate morbidity but low mortality, and require specific diagnostic and disease surveillance. Many of these agents are food-borne or water-borne. Category C agents include emerging pathogens that could be engineered for mass dissemination because of availability, ease of production and dissemination, potential for high morbidity and mortality, and great impact on health.
TRANSMISSION AND DISSEMINATION OF MICROBES
Routes of Entry of Microbes
Microbes can enter the host by inhalation, ingestion, sexual transmission, insect or animal bites, or injection. The first defenses against infection are intact skin and mucosal surfaces, which provide physical barriers and produce antimicrobial substances. In general, respiratory, gastrointestinal, or genitourinary tract infections that occur in healthy persons are caused by relatively virulent microorganisms that are capable of damaging or penetrating intact epithelial barriers. In contrast, most skin infections in healthy persons are caused by less virulent organisms entering the skin through damaged sites (cuts and burns).
Skin.
The dense, keratinized outer layer of skin is a natural barrier to infection, and the low pH of the skin (∼5.5) and the presence of fatty acids inhibit growth of microorganisms other than residents of the normal flora. Human skin is normally inhabited by a variety of bacterial and fungal species, including some potential opportunists, such as S. epidermidis and Candida albicans. Although skin is usually an effective barrier, certain types of fungi (dermatophytes) can infect the stratum corneum, hair, and nails, and a few microorganisms are able to traverse the unbroken skin. For example, Schistosoma larvae released from freshwater snails penetrate swimmers’ skin by releasing collagenase, elastase, and other enzymes that dissolve the extracellular matrix. Most microorganisms, however, penetrate through breaks in the skin, including superficial pricks (fungal infections), wounds (staphylococci), burns (Pseudomonas aeruginosa), and diabetic and pressure-related foot sores (multibacterial infections). Intravenous catheters in hospitalized patients can produce local or systemic infection (bacteremia). Needle sticks can expose the recipient to potentially infected blood and may transmit HBV, hepatitis C virus (HCV), or HIV. Some pathogens penetrate the skin via an insect or animal bite. For instance, bites by fleas, ticks, mosquitoes, mites, and lice break the skin and transmit arboviruses (causes of yellow fever and encephalitis), rickettsiae (Rocky Mountain spotted fever [RMSF]), bacteria (plague, Lyme disease), protozoa (malaria, leishmaniasis), and helminths (filariasis). Animal bites can lead to infections with bacteria or certain viruses such as rabies.
Gastrointestinal Tract.
Most gastrointestinal pathogens are transmitted by food or drink contaminated with fecal material. Where hygiene fails, diarrheal disease becomes rampant.
Acidic gastric secretions are important defenses within the gastrointestinal tract and are lethal for many gastrointestinal pathogens.14 Healthy volunteers do not become infected by Vibrio cholerae unless they are fed 1011 organisms, whereas volunteers given V. cholerae and sodium bicarbonate have a 10,000-fold increase in susceptibility to cholera. In contrast, some ingested agents, such as Shigella and Giardia cysts, are relatively resistant to gastric acid; hence, as few as 100 organisms of each are sufficient to cause illness.
Other normal defenses within the gastrointestinal tract include (1) the layer of viscous mucus covering the intestinal epithelium, (2) lytic pancreatic enzymes and bile detergents, (3) mucosal antimicrobial peptides called defensins, (4) normal flora, and (5) secreted IgA antibodies. IgA antibodies are made by plasma cells located in mucosa-associated lymphoid tissues (MALT). These lymphoid aggregates are covered by a single layer of specialized epithelial cells called M cells. M cells are important for transport of antigens to mucosa-associated lymphoid tissues and for binding and uptake of numerous gut pathogens, including poliovirus, enteropathic Escherichia coli, V. cholerae, Salmonella typhi, and Shigella flexneri.15
Infections via the gastrointestinal tract occur when local defenses are weakened or the organisms develop strategies to overcome these defenses. Host defenses are weakened by low gastric acidity, by antibiotics that alter the normal bacterial flora (e.g., in pseudomembranous colitis), or when there is stalled peristalsis or mechanical obstruction (e.g., in blind-loop syndrome). Most enveloped viruses are inactivated by bile and digestive enzymes, but nonenveloped viruses may be resistant (e.g., the hepatitis A virus, rotaviruses, reoviruses, and norovirus).
Enteropathogenic bacteria elicit gastrointestinal disease by a variety of mechanisms:
Fungal infection of the gastrointestinal tract occurs mainly in immunologically compromised people. Candida, part of the normal gastrointestinal flora, shows a predilection for stratified squamous epithelium, causing oral thrush or membranous esophagitis, but may also spread to the stomach, lower gastrointestinal tract, and systemic organs.
The cyst forms of intestinal protozoa are essential for their transmission, because cysts resist stomach acid. In the gut, cysts convert to motile trophozoites and attach to sugars on the intestinal epithelia through surface lectins. What happens next differs among pathogens. Giardia lamblia attaches to the epithelial brush border, whereas cryptosporidia are taken up by enterocytes, in which they form gametes and spores. E. histolytica causes contact-mediated cytolysis through a channel-forming pore protein and thereby ulcerates and invades the colonic mucosa. Intestinal helminths, as a rule, cause disease only when they are present in large numbers or in ectopic sites, for example, by obstructing the gut or invading and damaging the bile ducts (Ascaris lumbricoides). Hookworms may cause iron deficiency anemia by chronic loss of blood sucked from intestinal villi; the fish tapeworm Diphyllobothrium latum can deplete its host of vitamin B12, giving rise to an illness resembling pernicious anemia. Finally, the larvae of several helminths pass through the gut briefly on their way to another organ; for example, Trichinella spiralis larvae preferentially encyst in muscle, Echinococcus species larvae in the liver or lung.
Respiratory Tract.
A large number of microorganisms, including viruses, bacteria, and fungi, are inhaled daily by every city inhabitant. In many cases, the microbes are inhaled in dust or aerosol particles. The distance these particles travel into the respiratory system is inversely proportional to their size. Large particles are trapped in the mucociliary blanket that lines the nose and the upper respiratory tract. Inhaled microorganisms are trapped in the mucus secreted by goblet cells and are then transported by ciliary action to the back of the throat, where they are swallowed and cleared. Particles smaller than 5 μm travel directly to the alveoli, where they are phagocytosed by alveolar macrophages or by neutrophils recruited to the lung by cytokines.
Microorganisms that invade the normal healthy respiratory tract have developed specific mechanisms to overcome the mucociliary defenses or to avoid destruction by alveolar macrophages. Some successful respiratory pathogens evade these defenses by attaching to epithelial cells in the lower respiratory tract and pharynx. For example, influenza viruses possess hemagglutinin proteins that project from the surface of the virus and bind to sialic acid on the surface of epithelial cells. This attachment induces the host cell to engulf the virus, leading to viral entry and replication within the host cell. However, sialic acid also interferes with shedding of newly synthesized viruses from the host cell. Influenza viruses have another cell surface protein, neuraminidase, which cleaves sialic acid and allows virus to release from the host cell. Neuraminidase also lowers the viscosity of mucus and facilitates viral transit within the respiratory tract. Interestingly, some anti-influenza drugs are sialic acid analogues that inhibit neuraminidase and prevent viral release from host cells.
Certain bacterial respiratory pathogens can impair ciliary activity. For instance, Haemophilus influenzae and Bordetella pertussis elaborate toxins that paralyze mucosal cilia; P. aeruginosa, a cause of severe respiratory infection in persons with cystic fibrosis, and M. pneumoniae produce ciliostatic substances. Some bacteria such as Streptococcus pneumoniae or Staphylococcus species lack specific adherence factors and often gain access after viral infection causes loss of ciliated epithelium, making individuals with a viral respiratory infection more susceptible to these secondary bacterial superinfections. Chronic damage to mucociliary defense mechanisms occurs in smokers and people with cystic fibrosis, while acute injury occurs in intubated patients and in those who aspirate gastric acid.
Some respiratory pathogens avoid phagocytosis or destruction after phagocytosis. M. tuberculosis, for example, gains its foothold in normal alveoli because it is able to escape killing within the phagolysosomes of macrophages. Opportunistic fungi infect the lungs when cellular immunity is depressed or when leukocytes are reduced in number (e.g., P. jiroveci in AIDS patients and Aspergillus species following chemotherapy).
Urogenital Tract.
The urinary tract is almost always invaded from the exterior via the urethra. The regular flushing of the urinary tract with urine serves as a defense against invading microorganisms. Urine in the bladder is normally sterile, and successful pathogens (e.g., N. gonorrhoeae, E. coli) adhere to the urinary epithelium. Anatomy plays an important role in infection. Women have more than 10 times as many urinary tract infections as men, because the distance between the urinary bladder and skin (i.e., the length of the urethra) is 5 cm in women, in contrast to 20 cm in men. Obstruction of urinary flow and/or reflux can compromise normal defenses and increase susceptibility to urinary tract infections. Urinary tract infections can spread retrogradely from the bladder to the kidney and cause acute and chronic pyelonephritis, which is the major preventable cause of renal failure.
From puberty until menopause the vagina is protected from pathogens by a low pH resulting from catabolism of glycogen in the normal epithelium by lactobacilli. Antibiotics can kill the lactobacilli and make the vagina susceptible to infection. Sexually transmitted pathogens have developed specific mechanisms for attaching to the vaginal or cervical mucosa, or they enter via local breaks in the mucosa during sexual intercourse (HIV, HPV, Treponema pallidum).
Spread and Dissemination of Microbes
Some microorganisms proliferate locally, at the site of infection, whereas others penetrate the epithelial barrier and spread to distant sites via the lymphatics, the blood, or nerves (Fig. 8-4). Pathogens that cause superficial infections stay confined to the lumen of hollow viscera (e.g., Vibrio cholera), or adhere to or proliferate exclusively in or on epithelial cells (e.g., papillomaviruses, dermatophytes). A variety of pathogenic bacteria, fungi, and helminths are invasive by virtue of their motility or ability to secrete lytic enzymes (e.g., streptococci and staphylococci secrete hyaluronidase, which degrades the extracellular matrix between host cells). Microbial spread initially follows tissue planes of least resistance and to sites drained by regional lymphatics. For example, staphylococcal infections may progress from a localized abscess or furuncle to regional lymph nodes. This can sometimes leads to bacteremia and colonization of distant organs (heart, liver, brain, kidney, bone). Within the blood, microorganisms may be transported free or within host cells. Some viruses (e.g., poliovirus and HBV), most bacteria and fungi, some protozoa (e.g., African trypanosomes), and all helminths are transported free in the plasma. Leukocytes can carry herpesviruses, HIV, mycobacteria, and Leishmania and Toxoplasma organisms. Certain viruses (e.g., Colorado tick fever virus) and parasites (Plasmodium and Babesia) are carried by red blood cells. Most viruses spread from cell to cell by replication and release of infectious virions, but others may propagate from cell to cell by fusion or transport within nerves (e.g., rabies virus). Infectious foci seeded by blood are called secondary foci. They can be single and large (a solitary abscess or tuberculoma) or multiple and tiny (e.g., miliary tuberculosis or Candida microabscesses in many tissues). Sporadic bloodstream invasion by low-virulence or nonvirulent microbes (e.g., during brushing of teeth) is common but is quickly controlled by normal host defenses. By contrast, disseminated viremia, bacteremia, fungemia, or parasitemia by virulent pathogens is a serious insult and manifests itself by fever, low blood pressure, and multiple other systemic signs and symptoms of sepsis. Massive bloodstream invasion by bacteria or their endotoxins can rapidly become fatal, even for previously healthy individuals.
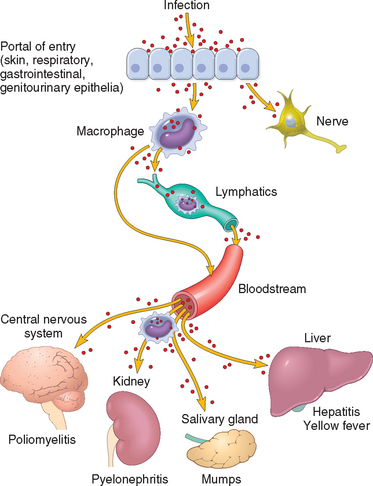
FIGURE 8-4 Routes of entry and dissemination of microbes. To enter the body microbes penetrate the epithelial or mucosal barriers. Infection may remain localized at the site of entry or spread to other sites in the body. Most common microbes (selected examples are shown) spread through the lymphatics or bloodstream (either freely or within inflammatory cells). However, certain viruses and bacterial toxins may also travel through nerves.
(Adapted from Mims CA: The Pathogenesis of Infectious Disease, 4th ed. San Diego, Academic Press, 1996.)
The major manifestations of infectious disease may appear at sites distant from the point of microbe entry. For example, chickenpox and measles viruses enter through the airways but cause rashes in the skin; poliovirus enters through the intestine but kills motor neurons. Schistosoma mansoni parasites penetrate the skin but eventually localize in blood vessels of the portal system and mesentery, damaging the liver and intestine. Schistosoma hematobium, on the other hand, localizes to the urinary bladder and causes cystitis. The rabies virus travels to the brain in a retrograde fashion within nerves, while the varicella zoster virus (VZV) hides in dorsal root ganglia, and on reactivation, travels along nerves to cause shingles.
The placental-fetal route is an important mode of transmission (Chapter 10). When infectious organisms reach the pregnant uterus through the cervical orifice or the bloodstream and are able to traverse the placenta, severe damage to the fetus can result. Bacterial or mycoplasmal placentitis can cause premature delivery or stillbirth. Viral infections can cause maldevelopment of the fetus, with infection early in pregnancy resulting in the most severe disease. Rubella infection during the first trimester can cause congenital heart disease, mental retardation, cataracts, or deafness in the infant, while little damage is caused by rubella infection during the third trimester. Transmission of treponemes leads to congenital syphilis only when T. pallidum infects the mother late in the second trimester but then causes severe fetal osteochondritis and periostitis that leads to multiple bony lesions. Infection also can occur during passage through the birth canal (e.g., gonococcal or chlamydial conjunctivitis) or through maternal milk (e.g., CMV, HBV, human T-cell leukemia virus-1 [HTLV-1]). Maternal transmission of HIV is the major cause of AIDS in children. Maternal transmission of HBV can later cause chronic hepatitis or liver cancer.
Release of Microbes from the Body
For transmission of disease, the mode of exit of a microorganism from the host’s body is as important as entry into it. Depending on the location of infection, release may be accomplished by skin shedding, coughing, sneezing, voiding of urine or feces, during sexual contact, or through insect vectors. Some microbes are hardy and can survive for extended periods in dust, food, or water. Bacterial spores, protozoan cysts, and thick-shelled helminth eggs can survive in a cool and dry environment. Some enteric pathogens are shed for long periods by asymptomatic carrier hosts (e.g., S. typhi). Less hardy microorganisms must be quickly passed from person to person, often by direct contact.
Transmission from person to person can occur by respiratory, fecal-oral, or sexual routes (discussed below). Viruses and bacteria transmitted by the respiratory route (e.g., M. tuberculosis) are infectious only when lesions are open to the airways. Many pathogens, ranging from viruses to helminths, can be transmitted by the fecal-oral route, that is, by ingestion of stool-contaminated food or water. Water-borne viruses involved in epidemic outbreaks include hepatitis A and E viruses, poliovirus, and rotavirus. Some parasitic helminths (e.g., hookworms, schistosomes) shed eggs in stool that gain access to new hosts by larval penetration of the skin rather than by oral intake. Protozoa and helminths have evolved complex transmission cycles involving a chain of intermediate and vector hosts bearing successive developmental stages of the parasites. Viruses infecting the oropharynx (e.g., EBV, CMV, mumps viruses) are transmitted principally through saliva. Other pathogens are spread mainly by prolonged intimate or mucosal contact (as occurs during sexual transmission) including viruses (HPV, HSV, HBV, HIV), bacteria (T. pallidum, N. gonorrhoeae, Chlamydia trachomatis), fungi (Candida species), protozoa (Trichomonas species), and arthropods (Phthirus pubis, or crab lice). Transmission of HBV, HCV, and HIV infections through blood and blood products may be caused by human behavior, e.g., needle sharing by drug abusers, cuts, and needle sticks.
Microbes can be transmitted from animal to human (termed zoonotic infections), either by direct contact or consumption of animal products or indirectly via an invertebrate vector. Invertebrate vectors (insects, ticks, mites) can spread infection passively in some cases or serve as necessary hosts for replication and development of the pathogen.
Sexually Transmitted Infections
A number of organisms can be transmitted through sexual contact (Table 8-5). Some, such as C. trachomatis and N. gonorrhoeae, are usually spread by sexual intercourse, while others, such as Shigella species and E. histolytica, are typically spread by other means but are also occasionally spread by oral-anal sex. Groups that are at greater risk for some sexually transmitted infections (STIs) include adolescents, men who have sex with men, and people who use illegal drugs. While the increased risk among these groups is partially due to unsafe sexual practices, limited access to health care is also often a contributing factor. The presence of an STI in young children, unless acquired during birth, strongly suggests sexual abuse.
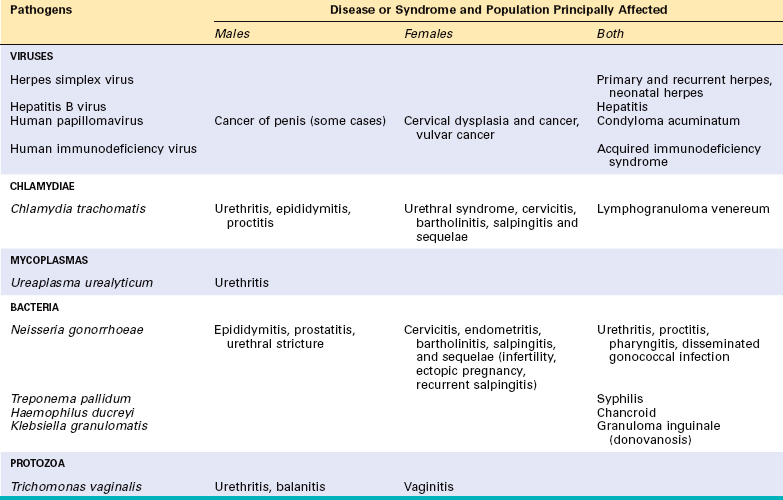
The initial site of an STI may be the urethra, vagina, cervix, rectum, or oral pharynx. The organisms that cause these infections tend to be short-lived outside of the host, so they usually depend on direct person-to-person spread. Most of these agents can be infectious in the absence of symptoms, so transmission often occurs from people who do not realize that they have an infection. To reduce the spread of STIs, these infections are often reported to public health authorities so that people who have had intimate contact with the person may be tested and treated.
Although the various pathogens that cause STIs differ in many ways, some general features should be noted.
Syphilis is discussed later in this chapter, and other STIs are described in Chapters 21 and 22.
Healthcare-Associated Infections
An increasingly important source of infections is the healthcare setting. “Nosocomial” infections are those acquired in the hospital. It is estimated that about 1.7 million patients each year get nosocomial infections in the United States.15 These infections can be transmitted in many ways (e.g. blood transfusion or organ transplant), but perhaps the most common, and readily preventable, means of transmission include spread from the hands of healthcare workers or from contaminated surfaces such as bedrails. Proper attention to hygiene and cleansing (e.g., hand washing) can greatly reduce the transmission of important pathogens such as methicillin-resistant S. aureus and vancomycin-resistant enterococci.
Host Defenses Against Infections
The outcome of infection is determined by the ability of the microbe to infect, colonize, and damage host tissues and the ability of host defense mechanisms to eradicate the infection. Host barriers to infection prevent microbes from entering the body and consist of innate and adaptive immune defenses. Innate immune defense mechanisms exist before infection and respond rapidly to microbes. These mechanisms include physical barriers to infection, phagocytic cells, natural killer (NK) cells, and plasma proteins, including the complement system proteins and other mediators of inflammatory responses (cytokines, collectins, acute-phase reactants). Adaptive immune responses are stimulated by exposure to microbes and increase in magnitude, speed, and effectiveness with successive exposures to microbes. Adaptive immunity is mediated by T and B lymphocytes and their products (Chapter 6).
Microbes and the immune system have been engaged in an evolutionary battle, in which each tries to outwit the other. The immune system is capable of effectively combating many infections, but different microbes have developed ways of evading host defenses (discussed later). In some infections, a balance is reached between the microbe and the host such that the infection persists in a state of latency but does not cause significant pathology. In such situations, decline of immune responses can result in rapid reactivation of the infection and severe pathologic manifestations. Such reactivation is seen in latent viral infections (e.g., EBV) and some bacterial infections (e.g., tuberculosis).
HOW MICROORGANISMS CAUSE DISEASE
Infectious agents establish infection and damage tissues by three mechanisms:
Here we describe some of the mechanisms whereby viruses and bacteria damage host tissues.
Mechanisms of Viral Injury
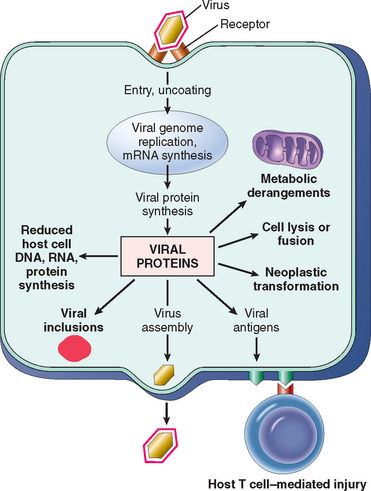
Viruses can directly damage host cells by entering them and replicating at the host’s expense. The predilection for viruses to infect certain cells and not others is called tropism and is determined by several factors, including (1) expression of host cell receptors for the virus, (2) presence of cellular transcription factors that recognize viral enhancer and promoter sequences, (3) anatomic barriers, and (4) local temperature, pH, and host defenses.17 Each of these is described briefly.
A major determinant of tissue tropism is the presence of viral receptors on host cells. Viruses possess specific cell-surface proteins that bind to particular host cell-surface proteins. Many viruses use normal cellular receptors of the host to enter cells. For example, HIV glycoprotein gp120 binds to CD4 on T cells and to the chemokine receptors CXCR4 (mainly on T cells) and CCR5 (mainly on macrophages) (Chapter 6). EBV envelope glycoprotein gp350 binds to complement receptor 2 (CR2/CD21) on B cells. In some cases, host proteases are needed to enable binding of virus to host cells; for instance, a host protease cleaves and activates the influenza virus hemagglutinin. Another determinant of viral tropism is the ability of the virus to replicate inside some cells but not in others, and this is related to the presence of cell type–specific transcription factors. For example, the JC virus, which causes leukoencephalopathy (Chapter 28), is restricted to oligodendroglia in the CNS, because the promoter and enhancer DNA sequences upstream from the viral genes are active in glial cells but not in neurons or endothelial cells. Physical barriers also can contribute to tissue tropism. For example, enteroviruses replicate in the intestine in part because they can resist inactivation by acids, bile, and digestive enzymes. Rhinoviruses infect only within the upper respiratory tract because they replicate optimally at the lower temperature of the upper respiratory tract.
Once viruses are inside host cells, they can damage or kill the cells by a number of mechanisms (Fig. 8-5):
Mechanisms of Bacterial Injury
Bacterial Virulence.
Bacterial damage to host tissues depends on the ability of the bacteria to adhere to host cells, invade cells and tissues, or deliver toxins. Pathogenic bacteria have virulence genes that encode proteins that confer these properties. Virulence genes are frequently found grouped together in clusters called pathogenicity islands. All of the Salmonella strains that infect humans are closely related enough to form a single species, meaning that they share many “housekeeping” genes.16 Differences in a relatively small number of pathogenicity genes determine whether an isolate of Salmonella can cause life-threatening typhoid fever or self-limited enteritis.
Plasmids and bacteriophages (viruses) are mobile genetic elements that spread between bacteria and can encode virulence factors (e.g., toxins, or enzymes that confer antibiotic resistance). Bacteriophages or plasmids can convert otherwise nonpathogenic bacteria into virulent ones. Exchange of these elements between bacteria can endow the recipient with a survival advantage, with the capacity to cause disease, or both. Plasmids or transposons encoding antibiotic resistance can convert an antibiotic-susceptible bacterium into a resistant one, making therapy difficult (e.g., vancomycin-resistant enterococci and methicillin-resistant staphylococci are endemic in many hospitals).
Many bacteria coordinately regulate gene expression within a large population of organisms by quorum sensing. For example, bacteria can induce expression of virulence factors as their concentration in the tissues increases. This may allow bacteria growing in discrete host sites, such as an abscess or consolidated pneumonia, to overcome host defenses. S. aureus coordinately regulates virulence factors by secreting autoinducer peptides.17 As the bacteria grow to increasing concentrations, the level of the autoinducer peptide increases, stimulating toxin production. Within the population, some bacteria produce the autoinducer peptide and others respond to it by secreting toxins. Thus, because of quorum sensing, unicellular bacteria acquire some of the more complex properties of multicellular organisms, in which different cells perform different functions.
Communities of bacteria can form biofilms in which the organisms live within a viscous layer of extracellular polysaccharides that adhere to host tissues or devices such as intravascular catheters and artificial joints. In addition to enhancing adherence to host tissues, biofilms increase the virulence of bacteria by making them inaccessible to immune effector mechanisms and increasing their resistance to antimicrobial drugs. Biofilm formation seems to be important in the persistence and relapse of infections such as bacterial endocarditis, artificial joint infections, and respiratory infections in people with cystic fibrosis.
Bacterial Adherence to Host Cells.
Adhesins are bacterial surface molecules that bind to host cells or extracellular matrix. Bacterial adhesins that bind bacteria to host cells are limited in structural type but have a broad range of host cell specificity. Streptococcus pyogenes is a gram-positive bacterium that adheres to host tissues by virtue of protein F and teichoic acid projecting from the bacterial cell wall and binding to fibronectin on the surface of host cells and in the extracellular matrix.
Pili are filamentous proteins on the surface of bacteria. The stalks of pili are composed of conserved repeating subunits, while the variable amino acids on the tips of the pili determine the binding specificity of the bacteria. Strains of E. coli that cause urinary tract infections uniquely express a specific P pilus, which binds to a gal(α1–4)gal moiety expressed on uroepithelial cells.18 Pili on N. gonorrhoeae bacteria mediate adherence of the bacteria to host cells and also are targets of the antibody response against N. gonorrhoeae. Variation in the type of pili expressed is an important mechanism by which N. gonorrhoeae escapes the immune response.19
Virulence of Intracellular Bacteria.
Facultative intracellular bacteria infect either epithelial cells (Shigella and enteroinvasive E. coli), macrophages (M. tuberculosis, M. leprae), or both (S. typhi). The growth of bacteria in cells may allow the bacteria to escape from certain effector mechanisms of the immune response (e.g., antibodies), or it may facilitate spread of the bacteria, as migration of macrophages carries M. tuberculosis bacteria from the lung to other sites.
Bacteria have a number of mechanisms for entering host cells. Some bacteria use the host immune response to gain entry into macrophages. Coating of bacteria with antibodies or the complement protein C3b (opsonization) normally results in phagocytosis of bacteria by macrophages. Like many bacteria, M. tuberculosis activates the alternative complement pathway, resulting in opsonization with C3b. Once coated with C3b, M. tuberculosis binds to the CR3 complement receptor on macrophages and is endocytosed into the cell.20 Gram-negative bacteria use a complex secretion system to enter epithelial cells.21 This system consists of needle-like structures projecting from the bacterial surface that bind to host cells, form pores in the host cell membrane, and then inject proteins that mediate rearrangement of the cell cytoskeleton, facilitating bacterial entry. Bacteria such as Listeria monocytogenes can manipulate the cell cytoskeleton to spread directly from cell to cell, perhaps allowing the bacteria to evade immune effector mechanisms.22
Once in the cytoplasm, bacteria have different strategies for interacting with the host cell. Shigella and E. coli inhibit host protein synthesis, replicate rapidly, and lyse the host cell within 6 hours. Within macrophages, most bacteria are killed when the phagosome fuses with an acidic lysosome to form a phagolysosome, but certain bacteria elude this host defense. For example, M. tuberculosis blocks fusion of the lysosome with the phagosome,23 allowing it to proliferate unchecked within the macrophage. Other bacteria avoid destruction in macrophages by escaping from the phagosome. L. monocytogenes produces a pore-forming protein called listeriolysin O and two phospholipases that degrade the phagosome membrane, allowing the bacteria to escape into the cytoplasm.22
Bacterial Toxins.
Any bacterial substance that contributes to illness can be considered a toxin. Toxins are classified as endotoxins, which are components of the bacterial cell, and exotoxins, which are proteins that are secreted by the bacterium.
Bacterial endotoxin is a lipopolysaccharide (LPS) that is a large component of the outer membrane of gram-negative bacteria. LPS is composed of a long-chain fatty acid anchor (lipid A) connected to a core sugar chain, both of which are very similar in all gram-negative bacteria. Attached to the core sugar is a variable carbohydrate chain (O antigen), which is used diagnostically to serotype and discriminate between different strains of bacteria. The response to bacterial LPS can be both beneficial and harmful to the host. The response is beneficial in that LPS activates protective immunity in several ways, including induction of important cytokines and chemoattractants (chemokines) of the immune system as well as increased expression of costimulatory molecules, which enhance T-lymphocyte activation. However, high levels of LPS are thought to play an important role in septic shock, disseminated intravascular coagulation (DIC), and adult respiratory distress syndrome, mainly through induction of excessive levels of cytokines such as TNF, IL-1, and IL-12 (Chapter 4). LPS binds to the cell-surface receptor CD14, and the complex then binds to Toll-like receptor 4 (TLR4), which is a pattern recognition receptor of the innate immune system and transmits signals that lead to the cellular response.24
Exotoxins are secreted proteins that cause cellular injury and disease. They can be classified into broad categories by their site and mechanism of action. These are briefly described next and discussed in more detail in the specific sections about each type of bacteria.
Injurious Effects of Host Immunity
As was mentioned earlier, the host immune response to microbes can sometimes be the cause of tissue injury. The granulomatous inflammatory reaction to M. tuberculosis is a delayed hypersensitivity response that sequesters the bacilli and prevents spread but also can produce tissue damage and fibrosis. Similarly, the liver damage following HBV and HCV infection of hepatocytes is mainly due to the immune response to the infected liver cells and not to cytopathic effects of the virus. The humoral immune response to microbes also can have pathologic consequences. For example, following infection with S. pyogenes, antibodies produced against the streptococcal M protein can cross-react with cardiac proteins and damage the heart, leading to rheumatic heart disease. Poststreptococcal glomerulonephritis, which also can develop following infection with S. pyogenes, is caused by antistreptococcal antibodies that bind to streptococcal antigens and form immune complexes, which deposit in renal glomeruli and produce nephritis. Thus, antimicrobial immune responses can have beneficial and pathologic consequences.
The examples provided above are relatively infrequent situations in which specific immune responses to microbes have been identified as causative agents of various diseases. Recent clinical, epidemiologic, and experimental studies suggest that infections may be associated with a wide variety of chronic inflammatory disorders as well as cancer.28 In some chronic inflammatory diseases, such as inflammatory bowel disease (Chapter 17), an important early event may be compromise of the intestinal epithelial barrier, which enables the entry of both pathogenic and commensal microbes and their interactions with local immune cells, resulting in inflammation. The cycle of inflammation and epithelial injury may be the basis of the disease, with microbes playing the central role. Viruses (HBV, HCV) and bacteria (H. pylori) that are not known to carry or activate oncogenes are associated with cancers, presumably because these microbes trigger chronic inflammation, which provides fertile ground for the development of cancer (Chapter 7).
IMMUNE EVASION BY MICROBES
Humoral and cellular immune responses that protect the host from most infections were discussed in Chapter 6. Not surprisingly, microorganisms have developed many means to resist and evade the immune system (Fig. 8-6).29 These mechanisms are important determinants of microbial virulence and pathogenicity. They include (1) growth in niches that are inaccessible to the host immune system, (2) antigenic variation, (3) resistance to innate immune defenses, and (4) impairment of effective T-cell antimicrobial responses by specific or nonspecific immunosuppression.
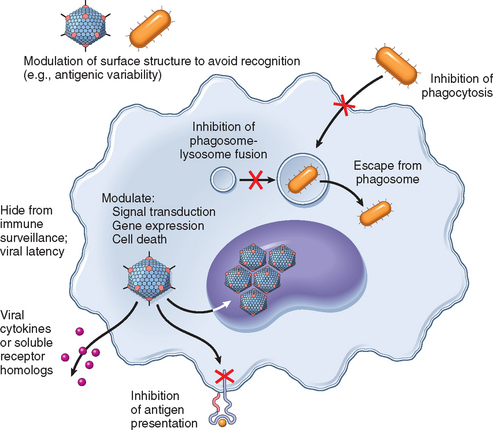
FIGURE 8-6 An overview of mechanisms used by viral and bacterial pathogens to evade innate and adaptive immunity.
(Modified with permission from Finlay B, McFadden G: Anti-immunology: evasion of the host immune system by bacterial and viral pathogens. Cell 124:767–782, 2006.)
Some microorganisms replicate in sites that are inaccessible to the host immune response. Microbes that propagate in the lumen of the intestine (e.g., toxin-producing Clostridium difficile) or gallbladder (e.g., S. typhi) are concealed from cell-mediated immune defenses. Some organisms establish infections by rapidly invading host cells before the host humoral response becomes effective (e.g., malaria sporozoites entering liver cells, Trichinella and Trypanosoma cruzi entering skeletal or cardiac muscle cells). Some larger parasites (e.g., the larvae of tapeworms) form cysts in host tissues that are covered by a dense capsule and are thus inaccessible to host immune cells and antibodies. Viral latency is the ultimate strategy for hiding antigens from the immune system. During the latent state, many viral genes are not expressed.
Some microbes can evade immune responses by varying the antigens they express. Neutralizing antibodies block the ability of microbes to infect cells and recruit effector mechanisms to kill pathogens. To escape recognition, microbes use many strategies that involve genetic mechanisms for generating antigenic variation. The low fidelity of viral RNA polymerases (in HIV and many respiratory viruses including influenza virus) and reassortment of viral genomes (influenza viruses) create viral antigenic variation (Table 8-6). The spirochete Borrelia recurrentis repeatedly switches its surface antigens, and the Lyme disease Borrelia organisms use similar mechanisms to vary their outer membrane proteins.30 Trypanosoma species have many genes for their major surface antigen, VSG, and can vary the expression of this surface protein. There are at least 80 different serotypes of S. pneumoniae, each with different capsular polysaccharides.
TABLE 8-6 Mechanisms of Antigenic Variation
| Type | Example | Disease |
|---|---|---|
| High mutation rate | HIV | AIDS |
| Influenza virus | Influenza | |
| Genetic reassortment | Influenza virus | Influenza |
| Rotavirus | Diarrhea | |
| Genetic rearrangement (e.g., gene recombination, gene conversion, site-specific inversion) | Borrelia burgdorferi | Lyme disease |
| Neisseria gonorrhoeae | Gonorrhea | |
| Trypanosoma sp. | African sleeping sickness | |
| Plasmodium sp. | Malaria | |
| Large diversity of serotypes | Rhinoviruses | Colds |
| Streptococcus pneumoniae | Pneumonia | |
| Meningitis |
Some microbes have devised methods for evading innate immune defenses, such as escaping killing by phagocytic cells and complement.31 Cationic antimicrobial peptides, including defensins, cathelicidins, and thrombocidins, provide important initial defense against invading microbes. Resistance to these antimicrobial peptides is key to the virulence of a number of bacterial pathogens, enabling them to avoid killing by neutrophils and macrophages.32 The carbohydrate capsule on the surface of all the major bacteria that cause pneumonia or meningitis (pneumococcus, meningococcus, H. influenzae) makes them more virulent by shielding bacterial antigens and by preventing phagocytosis of the organisms by neutrophils. For example, E. coli with the sialic acid–containing K1 capsule causes meningitis in newborns. Sialic acid will not bind C3b, which is critical for activation of the alternative complement pathway, so the bacteria escape from complement-mediated lysis and opsonization-directed phagocytosis. Many bacteria make toxic proteins that kill phagocytes, prevent their migration, or diminish their oxidative burst. Bacteria also can circumvent immune defenses by covering themselves with host proteins. Some bacterial pathogens including Salmonella can modify the lipid moiety of LPS to reduce Toll-like receptor (TLR) activation. S. aureus are covered by protein A molecules that bind the Fc portion of antibodies and so inhibit phagocytosis. Neisseria, Haemophilus, and Streptococcus all secrete proteases that degrade antibodies. As already mentioned, another successful strategy for circumventing defense mechanisms is to replicate within phagocytic cells. A number of viruses, some intracellular bacteria (including mycobacteria, Listeria, and Legionella), fungi (e.g., Cryptococcus neoformans), and protozoa (e.g., leishmania, trypanosomes, toxoplasmas) can multiply within phagocytes.
Viruses can produce molecules that inhibit innate immunity.29,33 Some viruses (e.g., herpesviruses and poxviruses) produce proteins that block complement activation. Viruses have developed a large number of strategies to combat interferons (IFNs), an early host defense against viruses. Some viruses produce soluble homologues of IFN-α/β or IFN-γ receptors that bind to and inhibit actions of secreted IFNs, or produce proteins that inhibit intracellular JAK/STAT signaling downstream of IFN receptors. They may also inactivate or inhibit double-stranded RNA–dependent protein kinase (PKR), a key mediator of the antiviral effects of IFN. Some viruses encode within their genomes homologues of other cytokines and chemokines, or their receptors, which act in various ways to inhibit immune responses. Many viruses have developed strategies to block apoptosis in the host cell, which may give the viruses time to complete replication, assembly, and exit, promote viral persistence, and contribute to cell transformation.
Some microbes produce factors that decrease recognition of infected cells by CD4+ helper T cells and CD8+ cytotoxic T cells. For example, several DNA viruses (e.g., herpesviruses, including HSV, CMV, and EBV) can bind to or alter localization of major histocompatibility complex (MHC) class I proteins, impairing peptide presentation to CD8+ T cells.34,35 Downregulation of MHC class I molecules might make it likely that virus-infected cells would be targets for NK cells. However, herpesviruses also express MHC class I homologues that act as effective inhibitors of NK cells by engaging killer inhibitory receptors (Chapter 6). Herpesviruses can target MHC class II molecules for degradation, impairing antigen presentation to CD4+ T helper cells. Viruses also can infect leukocytes and directly compromise their function. HIV infects CD4+ T cells, macrophages, and dendritic cells, and EBV infects B lymphocytes.
INFECTIONS IN IMMUNOSUPPRESSED HOSTS
Inherited or acquired defects in immunity (Chapter 6) often impair only part of the immune system, rendering the affected individual susceptible to specific types of infections. Patients with antibody deficiency, as in X-linked agammaglobulinemia, contract severe bacterial infections by organisms including S. pneumoniae, H. influenzae, and S. aureus, as well as a few viral infections (rotavirus and enteroviruses). Patients with T-cell defects are especially susceptible to infections with intracellular pathogens, notably viruses and some parasites. Patients with deficiencies in complement proteins are particularly susceptible to infections by S. pneumoniae, H. influenzae, and N. meningitidis. Some children have deficiencies in neutrophil function, leading to increased infections with S. aureus as well as some gram-negative bacteria and fungi.
Acquired immunodeficiencies have a variety of causes, the most important being infection with HIV, which causes AIDS. HIV infects and eventually kills CD4+ helper T lymphocytes. As discussed in Chapter 6, this leads to profound immunosuppression and a multitude of infections. While most organisms that infect people with AIDS were common pathogens before the era of HIV, others were uncommon (cryptococcus, pneumocystis), and one, Kaposi sarcoma herpesvirus (KSHV), also called human herpesvirus-8 (HHV-8), was discovered as a result of research in AIDS patients.36
Diseases that impair production of leukocytes, such as leukemia, which fills the bone marrow with cancerous cells, make patients vulnerable to opportunistic infections. Iatrogenic causes of immunosuppression include immunosuppressive drugs used to treat patients with autoimmune diseases and organ transplant recipients, as well as drugs used to treat cancer. Therapy to prevent rejection of organ transplants leads to severe immunosuppression, making transplant recipients very susceptible to infectious diseases. Patients receiving bone marrow transplants have profound defects in innate and adaptive immunity during the long time that it takes for the donated bone marrow to engraft, and become susceptible to infection with almost any organism, including opportunistic organisms that seldom cause disease in healthy people (e.g., Aspergillus species and Pseudomonas species).
Diseases of organ systems other than the immune system can also make patients susceptible to specific microorganisms. People with cystic fibrosis commonly get respiratory infections with P. aeruginosa, S. aureus, and Burkholdaria cepacia.37 The lack of splenic function in individuals with sickle cell disease makes them susceptible to infection with encapsulated bacteria such as S. pneumoniae, which are normally opsonized and phagocytosed by splenic macrophages. Burns destroy skin, removing this barrier to microbes, allowing infection with pathogens such as P. aeruginosa. Finally, malnutrition may impair the host defenses.
SPECTRUM OF INFLAMMATORY RESPONSES TO INFECTION
In contrast to the vast molecular diversity of microbes, the morphologic patterns of tissue responses to microbes are limited, as are the mechanisms directing these responses. Therefore, many pathogens produce similar reaction patterns, and few features are unique or pathognomonic for a particular microorganism. Moreover, it is the interaction between the microorganism and the host that determines the histologic features of the inflammatory response. Thus, pyogenic bacteria, which normally evoke vigorous leukocyte responses, may cause rapid tissue necrosis with little leukocyte exudation in a profoundly neutropenic host. Similarly, in a normal patient, M. tuberculosis causes well-formed granulomas with few mycobacteria present, whereas in an AIDS patient the same mycobacteria multiply profusely in macrophages, which fail to coalesce into granulomas.
There are five major histologic patterns of tissue reaction in infections.
Suppurative (Purulent) Inflammation
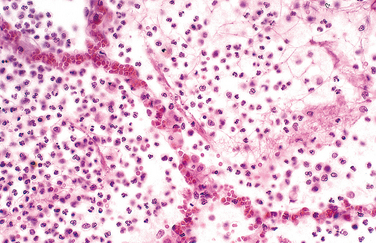
This pattern is the reaction to acute tissue damage, described in Chapter 2, characterized by increased vascular permeability and leukocytic infiltration, predominantly of neutrophils (Fig. 8-7). The neutrophils are attracted to the site of infection by release of chemoattractants from the “pyogenic” (pus-forming) bacteria that evoke this response, mostly extracellular gram-positive cocci and gram-negative rods. Massing of neutrophils and liquefactive necrosis of the tissue form pus. The sizes of exudative lesions range from tiny microabscesses formed in multiple organs during bacterial sepsis secondary to a colonized heart valve to diffuse involvement of entire lobes of the lung in pneumonia. How destructive the lesions are depends on their location and the organism involved. For example, pneumococci usually spare alveolar walls and cause lobar pneumonia that resolves completely, whereas staphylococci and Klebsiella species destroy alveolar walls and form abscesses that heal with scar formation. Bacterial pharyngitis resolves without sequelae, whereas untreated acute bacterial inflammation of a joint can destroy it in a few days.
Mononuclear and Granulomatous Inflammation
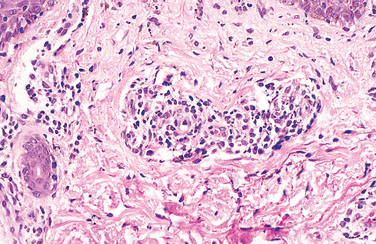
Diffuse, predominantly mononuclear, interstitial infiltrates are a common feature of all chronic inflammatory processes, but when they develop acutely, they often are a response to viruses, intracellular bacteria, or intracellular parasites. In addition, spirochetes and helminths provoke chronic inflammatory responses. Which mononuclear cell predominates within the inflammatory lesion depends on the host immune response to the organism. For example, plasma cells are abundant in the primary and secondary lesions of syphilis (Fig. 8-8), whereas lymphocytes predominate in HBV infection or viral infections of the brain. The presence of these lymphocytes reflects cell-mediated immune responses against the pathogen or pathogen-infected cells. At the other extreme, macrophages may become filled with organisms, as occurs in M. avium-intracellulare infections in AIDS patients, who cannot mount an effective immune response to the organisms. Granulomatous inflammation is a distinctive form of mononuclear inflammation usually evoked by infectious agents that resist eradication and are capable of stimulating strong T cell–mediated immunity (e.g., M. tuberculosis, Histoplasma capsulatum, schistosome eggs). Granulomatous inflammation is characterized by accumulation of activated macrophages called “epithelioid” cells, which may fuse to form giant cells. In some cases there is a central area of caseous necrosis (see Chapter 2 and “Tuberculosis” in this chapter).
Cytopathic-Cytoproliferative Reaction
These reactions are usually produced by viruses. The lesions are characterized by cell necrosis or cellular proliferation, usually with sparse inflammatory cells. Some viruses replicate within cells and make viral aggregates that are visible as inclusion bodies (e.g., herpesviruses or adenovirus) or induce cells to fuse and form multinucleated cells called polykaryons (e.g., measles virus or herpesviruses). Focal cell damage in the skin may cause epithelial cells to become detached, forming blisters (Fig. 8-9). Some viruses can cause epithelial cells to proliferate (e.g., venereal warts caused by HPV or the umbilicated papules of molluscum contagiosum caused by poxviruses). Finally, viruses can contribute to the development of malignant neoplasms (Chapter 7).

Tissue Necrosis
Clostridium perfringens and other organisms that secrete powerful toxins can cause such rapid and severe necrosis (gangrenous necrosis) that tissue damage is the dominant feature. Because few inflammatory cells are present, these lesions resemble infarcts with disruption or loss of basophilic nuclear staining and preservation of cellular outlines. Clostridia are often opportunistic pathogens that are introduced into muscle tissue by penetrating trauma or infection of the bowel in a neutropenic host. Similarly, the parasite E. histolytica causes colonic ulcers and liver abscesses characterized by extensive tissue destruction with liquefactive necrosis and without a prominent inflammatory infiltrate. By entirely different mechanisms, viruses can cause widespread and severe necrosis of host cells associated with inflammation, as exemplified by total destruction of the temporal lobes of the brain by herpesvirus or the liver by HBV.
Chronic Inflammation and Scarring
Many infections elicit chronic inflammation, which can lead either to complete healing or to extensive scarring. For example, chronic HBV infection may cause cirrhosis of the liver, in which dense fibrous septae surround nodules of regenerating hepatocytes. Sometimes the exuberant scarring response is the major cause of dysfunction (e.g., the “pipe-stem” fibrosis of the liver or fibrosis of the bladder wall caused by schistosomal eggs [Fig. 8-10] or the constrictive fibrous pericarditis in tuberculosis).
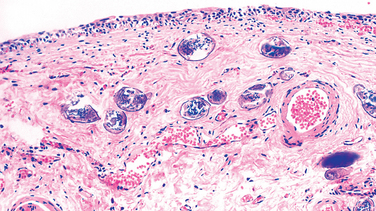
FIGURE 8-10 Schistosoma haematobium infection of the bladder with numerous calcified eggs and extensive scarring.
These patterns of tissue reaction are useful guidelines for analyzing microscopic features of infectious processes, but they rarely appear in pure form because different types of host reactions often occur at the same time. For example, the lung of an AIDS patient may be infected with CMV, which causes cytolytic changes, and at the same time by Pneumocystis, which causes interstitial inflammation. Similar patterns of inflammation also can be seen in tissue responses to physical or chemical agents and in inflammatory diseases of unknown cause (Chapter 2).
This concludes our discussion of the general principles of the pathogenesis and pathology of infectious disease. We now turn to descriptions of specific infections caused by viruses, bacteria, fungi, and parasites. In this discussion we emphasize pathogenic mechanisms and pathologic changes, rather than details of clinical features, which are available in clinical textbooks. Infections that typically involve a specific organ are discussed in other chapters.
Viral Infections
Viruses are the cause of many clinically important acute and chronic infections affecting virtually every organ system (Table 8-7).
TABLE 8-7 Selected Human Viruses and Viral Diseases
| Organ System | Species | Disease |
|---|---|---|
| Respiratory | Adenovirus | Upper and lower respiratory tract infections, conjunctivitis, diarrhea |
| Rhinovirus | Upper respiratory tract infection | |
| Influenza viruses A, B | Influenza | |
| Respiratory syncytial virus | Bronchiolitis, pneumonia | |
| Digestive | Mumps virus | Mumps, pancreatitis, orchitis |
| Rotavirus | Childhood gastroenteritis | |
| Norovirus | Gastroenteritis | |
| Hepatitis A virus | Acute viral hepatitis | |
| Hepatitis B virus | Acute or chronic hepatitis | |
| Hepatitis D virus | With HBV, acute or chronic hepatitis | |
| Hepatitis C virus | Acute or chronic hepatitis | |
| Hepatitis E virus | Enterically transmitted hepatitis | |
| Systemic with Skin Eruptions | Measles virus | Measles (rubeola) |
| Rubella virus | German measles (rubella) | |
| Varicella-zoster virus | Chickenpox, shingles | |
| Herpes simplex virus 1 | Oral herpes (“cold sore”) | |
| Herpes simplex virus 2 | Genital herpes | |
| Systemic with Hematopoietic Disorders | Cytomegalovirus | Cytomegalic inclusion disease |
| Epstein-Barr virus | Infectious mononucleosis | |
| HIV-1 and HIV-2 | AIDS | |
| Arboviral and Hemorrhagic Fevers | Dengue virus 1–4 | Dengue hemorrhagic fever |
| Yellow fever virus | Yellow fever | |
| Skin/Genital Warts | Papillomavirus | Condyloma; cervical carcinoma |
| Central Nervous System | Poliovirus | Poliomyelitis |
| JC virus | Progressive multifocal leukoencephalopathy (opportunistic) |
ACUTE (TRANSIENT) INFECTIONS
The viruses that cause transient infections are structurally heterogeneous, but each elicits an effective immune response that eliminates the organism and may or may not confer lifelong protection. The mumps virus, for example, has only one serotype and infects people only once, whereas other transient viruses, such as influenza viruses, can repeatedly infect the same individual because of antigenic variation. The immune response to some transient viruses wanes with time, allowing even the same serotype of virus to infect repeatedly (e.g., respiratory syncytial virus).
Measles
Measles (rubeola) virus is a leading cause of vaccine-preventable death and illness worldwide. More than 20 million people are affected by measles each year. In 2005, there were an estimated 345,000 deaths globally, the majority of them in children in developing countries. Because of poor nutrition, children in developing countries are 10 to 1000 times more likely to die of measles pneumonia than are children in developed countries.38 Epidemics of measles occur among unvaccinated individuals. Measles can produce severe disease in people with defects in cellular immunity (such as HIV-infected people or people with hematologic malignancy). In the United States the incidence of measles has decreased dramatically since 1963, when a measles vaccine was licensed. The diagnosis is usually made clinically, or by serology or detection of viral antigen in nasal exudate or urinary sediment.
Pathogenesis.
Measles virus is a single-stranded RNA virus of the paramyxovirus family that includes mumps, respiratory syncytial virus (the major cause of lower respiratory infections in infants), parainfluenza virus (a cause of croup), and human metapneumovirus. There is only one serotype of measles virus. Two cell-surface receptors have been identified for the virus: CD46, a complement-regulatory protein that inactivates C3 convertases, and signaling lymphocytic activation molecule (SLAM), a molecule involved in T-cell activation.39 CD46 is expressed on all nucleated cells, while SLAM is expressed on cells of the immune system. Both these receptors bind the viral hemagglutinin protein. Measles virus is transmitted by respiratory droplets. The virus initially multiplies within upper respiratory epithelial cells and then spreads to local lymphoid tissue. Measles can replicate in epithelial cells, endothelial cells, monocytes, macrophages, dendritic cells, and lymphocytes. Replication of the virus in lymphatic tissue is followed by viremia and systemic dissemination of the virus to many tissues, including the conjunctiva, respiratory tract, urinary tract, small blood vessels, lymphatic system, and CNS. Measles may cause croup, pneumonia, diarrhea with protein-losing enteropathy, keratitis with scarring and blindness, encephalitis, and hemorrhagic rashes (“black measles”) in malnourished children with poor medical care. Most children develop T cell–mediated immunity to measles virus that controls the viral infection and produces the measles rash, a hypersensitivity reaction to measles-infected cells in the skin. The rash is less frequent in people with deficiencies in cell-mediated immunity but does occur in agammaglobulinemic people. Antibody-mediated immunity to measles virus protects against reinfection. Measles also can cause transient but profound immunosuppression, resulting in secondary bacterial and viral infection, which are responsible for much of measles-related morbidity and mortality. Alterations of both innate and adaptive immune responses occur following measles infection, including defects in dendritic cell and lymphocyte function.40 Subacute sclerosing panencephalitis (described in Chapter 28) and measles inclusion body encephalitis (in immunocompromised individuals) are rare late complications of measles. The pathogenesis of subacute sclerosing panencephalitis is not well understood, but a replication-defective variant of measles may be involved in this persistent viral infection.41
Morphology. The blotchy, reddish brown rash of measles virus infection on the face, trunk, and proximal extremities is produced by dilated skin vessels, edema, and a moderate, nonspecific, mononuclear perivascular infiltrate. Ulcerated mucosal lesions in the oral cavity near the opening of Stensen ducts (the pathognomonic Koplik spots) are marked by necrosis, neutrophilic exudate, and neovascularization. The lymphoid organs typically have marked follicular hyperplasia, large germinal centers, and randomly distributed multinucleate giant cells, called Warthin-Finkeldey cells, which have eosinophilic nuclear and cytoplasmic inclusion bodies. These are pathognomonic of measles and are also found in the lung and sputum (Fig. 8-11). The milder forms of measles pneumonia show the same peribronchial and interstitial mononuclear cell infiltration that is seen in other nonlethal viral infections. In severe or neglected cases, bacterial superinfection may be a cause of death.
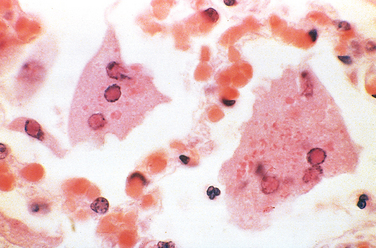
Mumps
Like measles virus, mumps virus is a member of the paramyxovirus family. Mumps virus has two types of surface glycoproteins, one with hemagglutinin and neuraminidase activities and the other with cell fusion and cytolytic activities. Mumps viruses enter the upper respiratory tract through inhalation of respiratory droplets, spread to draining lymph nodes where they replicate in lymphocytes (preferentially in activated T cells), and then spread through the blood to the salivary and other glands. Mumps virus infects salivary gland ductal epithelial cells, resulting in desquamation of involved cells, edema, and inflammation that leads to the classic salivary gland pain and swelling of mumps. Mumps virus also can spread to other sites, including the CNS, testis and ovary, and pancreas. Aseptic meningitis is the most common extrasalivary gland complication of mumps infection, occurring in about 10% of cases. The mumps vaccine has reduced the incidence of mumps by 99% in the United States. The diagnosis is usually made clinically, but serology or viral culture can be used for definitive diagnosis.
Morphology. In mumps parotitis, which is bilateral in 70% of cases, affected glands are enlarged, have a doughy consistency, and are moist, glistening, and reddish brown on cross-section. On microscopic examination the gland interstitium is edematous and diffusely infiltrated by macrophages, lymphocytes, and plasma cells, which compress acini and ducts. Neutrophils and necrotic debris may fill the ductal lumen and cause focal damage to the ductal epithelium.
In mumps orchitis testicular swelling may be marked, caused by edema, mononuclear cell infiltration, and focal hemorrhages. Because the testis is tightly contained within the tunica albuginea, parenchymal swelling may compromise the blood supply and cause areas of infarction. Sterility, when it occurs, is caused by scars and atrophy of the testis after resolution of viral infection.
In the enzyme-rich pancreas, lesions may be destructive, causing parenchymal and fat necrosis and neutrophil-rich inflammation. Mumps encephalitis causes perivenous demyelination and perivascular mononuclear cuffing.
Poliovirus Infection
Poliovirus is a spherical, unencapsulated RNA virus of the enterovirus genus. Other enteroviruses cause childhood diarrhea as well as rashes (coxsackievirus A), conjunctivitis (enterovirus 70), viral meningitis (coxsackieviruses and echovirus), and myopericarditis (coxsackievirus B). There are three major strains of poliovirus, each of which is included in the Salk formalin-fixed (killed) vaccine and the Sabin oral, attenuated (live) vaccine.42 These vaccines have nearly eliminated poliovirus from the Western hemisphere, because the poliovirus, like smallpox virus, infects people but not other animals, is only briefly shed, does not undergo antigenic variation, and is effectively prevented by immunization.43 Poliovirus still persists in parts of Africa.
Poliovirus, like other enteroviruses, is transmitted by the fecal-oral route. It first infects tissues in the oropharynx, then is secreted into the saliva and swallowed, and subsequently multiplies in the intestinal mucosa and lymph nodes, causing a transient viremia and fever. The virus infects only humans because it uses human CD155 to gain entry into cells but does not bind to cells in other species.44 Although most polio infections are asymptomatic, in about 1 of 100 infected persons poliovirus invades the CNS and replicates in motor neurons of the spinal cord (spinal poliomyelitis) or brain stem (bulbar poliomyelitis). Antiviral antibodies control the disease in most cases, and it is not known why they fail to contain the virus in some individuals. Viral spread to the nervous system may be secondary to viremia or occur by retrograde transport of the virus along axons of motor neurons.45 Rare cases of poliomyelitis that occur after vaccination are caused by mutations of the attenuated viruses to wild-type forms. The diagnosis can be made by viral culture of throat secretions or stool, or by serology. The neurologic features and neuropathology of poliovirus infection are described in Chapter 28.
West Nile Virus
West Nile virus is an arthropod-borne virus (arbovirus) of the flavivirus group, which also includes viruses that cause dengue fever and yellow fever. West Nile virus has a broad geographic distribution in the Old World, with outbreaks in Africa, the Middle East, Europe, Southeast Asia, and Australia. This virus was first detected in the United States in 1999 during an outbreak in New York City.46 West Nile virus is transmitted by mosquitoes to birds and to mammals. Wild birds develop prolonged viremia and are the major reservoir for the virus. Humans are usually incidental hosts. However, West Nile virus has been transmitted by blood transfusion, transplanted organs, breast milk, and transplacentally.47
After inoculation by a mosquito, West Nile virus replicates in skin dendritic cells, which then migrate to lymph nodes, where virus replicates further, enters the bloodstream, and, in some individuals, crosses the blood-brain barrier. In the CNS the virus infects neurons. Chemokines have essential roles in directing leukocytes to the CNS for viral clearance. In humans and in mice, the chemokine receptor CCR5 functions as an essential host factor to resist neuroinvasive infection. The CCR5Δ32 allele, which contains a 32–base pair deletion in the coding sequence and results in a complete loss of function in a homozygous individual, is associated with symptomatic and lethal West Nile virus infection. Thus, the loss of CCR5 receptor increases the risk of fatal West Nile virus infection but is protective against HIV-1 infection because HIV uses the receptor to infect host cells (Chapter 6).48
West Nile virus infection is usually asymptomatic, but in 20% of infected individuals it gives rise to a mild, short-lived febrile illness associated with headache and myalgia. A maculopapular rash is seen in approximately half the cases. CNS complications (meningitis, encephalitis, meningoencephalitis) are not frequent, occurring in about 1 in 150 clinically apparent infections. There is a mortality of about 10% in people with meningoencephalitis and long-term cognitive and neurologic impairment in many survivors. Perivascular and leptomeningeal chronic inflammation, microglial nodules (Chapter 28), and neuronophagia predominantly involving the temporal lobes and brain stem have been observed in the brains of patients who died of West Nile virus infection. Immunosuppressed persons and the elderly appear to be at the greatest risk for severe disease. Rare complications include hepatitis, myocarditis, and pancreatitis. The diagnosis is usually made by serology, but viral culture and PCR-based tests are also used.
Viral Hemorrhagic Fevers
Viral hemorrhagic fevers (VHFs) are systemic infections. They are caused by enveloped RNA viruses in four different families: arenaviruses, filoviruses, bunyaviruses, and flaviviruses. Although structurally distinct, these viruses all depend on an animal or insect host for survival and transmission. VHF viruses are restricted geographically to areas in which their hosts reside. Humans are infected when they come into contact with infected hosts or insect vectors, but humans are not the natural reservoir for any of these viruses. Some viruses that cause hemorrhagic fever (Ebola, Marburg, Lassa) also can spread from person to person. VHF viruses produce a spectrum of illnesses, ranging from relatively mild acute disease characterized by fever, headache, myalgia, rash, neutropenia, and thrombocytopenia to severe, life-threatening disease in which there is sudden hemodynamic deterioration and shock. These viruses are potential biologic weapons because of their infectious properties, morbidity and mortality, and the absence of therapy and vaccines.
The pathogenesis of viral hemorrhagic fevers is not well understood. The hemorrhagic manifestations are due to thrombocytopenia or severe platelet or endothelial dysfunction. Typically there is increased vascular permeability. There may be necrosis and hemorrhage in many organs, particularly the liver. Although the viruses that cause hemorrhagic fever can replicate in endothelial cells and direct cytopathic effects may contribute to disease, most disease manifestations are related to activation of innate immune responses.49 Viral infection of macrophages and dendritic cells leads to release of mediators that modify vascular function and have procoagulant activity.
CHRONIC LATENT INFECTIONS (HERPESVIRUS INFECTIONS)
Herpesviruses are large encapsulated viruses that have a double-stranded DNA genome that encodes approximately 70 proteins. Herpesviruses cause acute infection followed by latent infection in which the viruses persist in a noninfectious form with periodic reactivation and shedding of infectious virus. Latency is operationally defined as the inability to recover infectious particles from cells that harbor the virus. There are eight types of human herpesviruses, belonging to three subgroups defined by the type of cell most frequently infected and the site of latency: α-group viruses, including HSV-1, HSV-2, and VZV, which infect epithelial cells and produce latent infection in neurons; lymphotropic β-group viruses, including CMV, human herpesvirus-6 (which causes exanthem subitum, also known as roseola infantum and sixth disease, a benign rash of infants), and human herpesvirus-7 (a virus without a known disease association), which infect and produce latent infection in a variety of cell types; and the γ-group viruses EBV and KSHV/HHV-8, the cause of Kaposi sarcoma,58 which produce latent infection mainly in lymphoid cells. In addition, herpesvirus simiae is an Old World monkey virus that resembles HSV-1 and can cause fatal neurologic disease in animal handlers, usually resulting from an animal bite.
Herpes Simplex Virus (HSV)
HSV-1 and HSV-2 differ serologically but are genetically similar and cause a similar set of primary and recurrent infections.50 These viruses produce acute and latent infections. Both viruses replicate in the skin and the mucous membranes at the site of entrance of the virus (usually oropharynx or genitals), where they produce infectious virions and cause vesicular lesions of the epidermis. The viruses spread to sensory neurons that innervate these primary sites of replication. Viral nucleocapsids are transported along axons to the neuronal cell bodies, where the viruses establish latent infection. In immunocompetent hosts, primary HSV infection resolves in a few weeks, although the virus remains latent in nerve cells. During latency the viral DNA remains within the nucleus of the neuron, and only latency-associated viral RNA transcripts (LATs) are synthesized.51 In this state, no viral proteins appear to be produced. Recent data suggest that some LATs may be microRNAs that confer resistance to apoptosis and thus contribute to virus persistence in sensory neurons.52 Reactivation of HSV-1 and HSV-2 may occur repeatedly with or without symptoms, and results in the spread of virus from the neurons to the skin or to mucous membranes. Reactivation occurs in the presence of host immunity, because herpesviruses have developed ways to avoid immune recognition. For example, HSVs can evade antiviral CTLs by inhibiting the MHC class I recognition pathway, and elude humoral immune defenses by producing receptors for the Fc domain of immunoglobulin and inhibitors of complement.34,35
In addition to causing cutaneous lesions, HSV-1 is the major infectious cause of corneal blindness in the United States; corneal epithelial disease is thought to be due to direct viral damage, while corneal stromal disease appears to be immune mediated. HSV-1 is also the major cause of fatal sporadic encephalitis in the United States, when the virus spreads to the brain, particularly the temporal lobes and orbital gyri of the frontal lobes. In addition, neonates and individuals with compromised cellular immunity (e.g., secondary to HIV infection or chemotherapy) may suffer disseminated herpesvirus infections.
Morphology. HSV-infected cells contain large, pink to purple intranuclear inclusions (Cowdry type A) that consist of intact and disrupted virions with the stained host cell chromatin pushed to the edges of the nucleus (Fig. 8-12). Due to cell fusion, HSV also produces inclusion-bearing multinucleated syncytia.
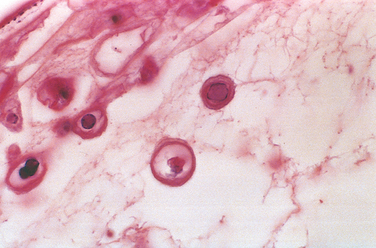
FIGURE 8-12 High-power view of cells from the blister in Figure 8-9 showing glassy intranuclear herpes simplex inclusion bodies.
HSV-1 and HSV-2 cause lesions ranging from self-limited cold sores and gingivostomatitis to life-threatening disseminated visceral infections and encephalitis. Fever blisters or cold sores favor the facial skin around mucosal orifices (lips, nose), where their distribution is frequently bilateral and independent of skin dermatomes. Intraepithelial vesicles (blisters), which are formed by intracellular edema and ballooning degeneration of epidermal cells, frequently burst and crust over, but some may result in superficial ulcerations.
Gingivostomatitis, which is usually encountered in children, is caused by HSV-1. It is a vesicular eruption extending from the tongue to the retropharynx and causing cervical lymphadenopathy. Swollen, erythematous HSV lesions of the fingers or palm (herpetic whitlow) occur in infants and, occasionally, in health care workers.
Genital herpes is more often caused by HSV-2 than by HSV-1. It is characterized by vesicles on the genital mucous membranes as well as on the external genitalia that are rapidly converted into superficial ulcerations, rimmed by an inflammatory infiltrate (Chapter 22). Herpesvirus (usually HSV-2) can be transmitted to neonates during passage through the birth canal of infected mothers. Although HSV-2 infection in the neonate may be mild, more commonly it is fulminating with generalized lymphadenopathy, splenomegaly, and necrotic foci throughout the lungs, liver, adrenals, and CNS.
Two forms of corneal lesions are caused by HSV (Chapter 29). Herpes epithelial keratitis shows typical virus-induced cytolysis of the superficial epithelium. In contrast, herpes stromal keratitis is characterized by infiltrates of mononuclear cells around keratinocytes and endothelial cells, leading to neovascularization, scarring, opacification of the cornea, and eventual blindness. This is an immunological reaction to the HSV infection.
Herpes simplex encephalitis is described in Chapter 28.
Disseminated skin and visceral herpes infections are usually encountered in hospitalized patients with some form of underlying cancer or immunosuppression. Kaposi varicelliform eruption is a generalized vesiculating involvement of the skin, whereas eczema herpeticum is characterized by confluent, pustular, or hemorrhagic blisters, often with bacterial superinfection and viral dissemination to internal viscera. Herpes esophagitis is frequently complicated by superinfection with bacteria or fungi. Herpes bronchopneumonia, which may be introduced by intubation of a patient with active oral lesions, is often necrotizing, and herpes hepatitis may cause liver failure.
Varicella-Zoster Virus (VZV)
Two conditions—chickenpox and shingles—are caused by VZV. Acute infection with VZV causes chickenpox; reactivation of latent VZV causes shingles (also called herpes zoster). Chickenpox is mild in children but more severe in adults and in immunocompromised people. Shingles is a source of morbidity in elderly and immunosuppressed persons.53 Like HSV, VZV infects mucous membranes, skin, and neurons and causes a self-limited primary infection in immunocompetent individuals. Also like HSV, VZV evades immune responses and establishes a latent infection in sensory ganglia.51 In contrast to HSV, VZV is transmitted in epidemic fashion by aerosols, disseminates hematogenously, and causes widespread vesicular skin lesions. VZV infects neurons and/or satellite cells around neurons in the dorsal root ganglia and may recur many years after the primary infection, causing shingles. Localized recurrence of VZV is most frequent and painful in dermatomes innervated by the trigeminal ganglia, where it is most likely to exist in a state of latency. In contrast to numerous recurrences of HSV, most people do not have a recurrence of VZV. VZV usually recurs only once in immunocompetent individuals, but immunosuppressed or elderly persons can have multiple recurrences of VZV. VZV infection is diagnosed by viral culture or detection of viral antigens in cells scraped from superficial lesions.
Morphology. The chickenpox rash occurs approximately 2 weeks after respiratory infection. Lesions appear in multiple waves centrifugally from the torso to the head and extremities. Each lesion progresses rapidly from a macule to a vesicle, which resembles a dewdrop on a rose petal. On histologic examination, chickenpox vesicles contain intranuclear inclusions in the epithelial cells like those of HSV-1 (Fig. 8-13). After a few days most chickenpox vesicles rupture, crust over, and heal by regeneration, leaving no scars. However, bacterial superinfection of vesicles that are ruptured by trauma may lead to destruction of the basal epidermal layer and residual scarring.
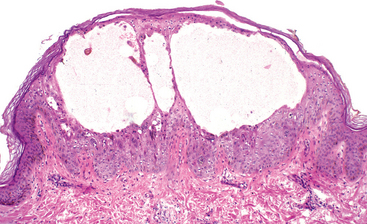
Shingles occurs when VZV that has long remained latent in the dorsal root ganglia after a previous chickenpox infection is reactivated and infects sensory nerves that carry it to one or more dermatomes. There, the virus infects keratinocytes and causes vesicular lesions, which, unlike chickenpox, are often associated with intense itching, burning, or sharp pain because of the simultaneous radiculoneuritis. This pain is especially severe when the trigeminal nerves are involved; rarely, the geniculate nucleus is involved, causing facial paralysis (Ramsay Hunt syndrome). The sensory ganglia contain a dense, predominantly mononuclear infiltrate, with herpetic intranuclear inclusions within neurons and their supporting cells (Fig. 8-14). VZV can also cause interstitial pneumonia, encephalitis, transverse myelitis, and necrotizing visceral lesions, particularly in immunosuppressed people.
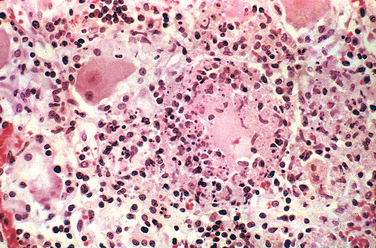
Cytomegalovirus (CMV)
Cytomegalovirus (CMV), a β-group herpesvirus, can produce a variety of disease manifestations, depending on the age of the host, and, more important, on the host’s immune status. CMV latently infects monocytes and their bone marrow progenitors and can be reactivated when cellular immunity is depressed. CMV causes an asymptomatic or mononucleosis-like infection in healthy individuals but devastating systemic infections in neonates and in immunocompromised people. As its name implies, CMV-infected cells exhibit gigantism of both the entire cell and its nucleus. Within the nucleus is a large inclusion surrounded by a clear halo (owl’s eye).
Transmission of CMV can occur by several mechanisms, depending on the age group affected.54,55 These include the following:
Acute CMV infection induces transient but severe immunosuppression. CMV can infect dendritic cells and impair their maturation and ability to stimulate T cells.55 Similar to other herpesviruses, CMV can elude immune responses by downmodulating MHC class I and II molecules and producing homologues of TNF receptor, IL-10, and MHC class I molecules.29,34,35 Interestingly, CMV can both activate and evade NK cells by inducing ligands for activating receptors and class I–like proteins that engage inhibitory receptors. Thus, CMV can both hide from immune defenses and actively suppress immune responses.
Morphology. The characteristic enlargement of infected cells can be appreciated histologically. Prominent intranuclear basophilic inclusions spanning half the nuclear diameter are usually set off from the nuclear membrane by a clear halo (Fig. 8-15). Within the cytoplasm of these cells, smaller basophilic inclusions can also be seen. In the glandular organs, the parenchymal epithelial cells are affected; in the brain, the neurons; in the lungs, the alveolar macrophages and epithelial and endothelial cells; and in the kidneys, the tubular epithelial and glomerular endothelial cells. Affected cells are strikingly enlarged, often to a diameter of 40 μm, and they show cellular and nuclear pleomorphism. Disseminated CMV causes focal necrosis with minimal inflammation in virtually any organ.
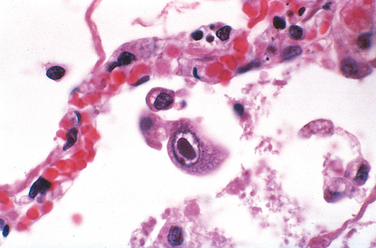
Congenital Infections.
Infection acquired in utero may take many forms. In approximately 95% of cases it is asymptomatic. However, sometimes when the virus is acquired from a mother with primary infection (who does not have protective antibodies), classic cytomegalic inclusion disease develops. Cytomegalic inclusion disease resembles erythroblastosis fetalis. Affected infants may suffer intrauterine growth retardation, be profoundly ill, and manifest jaundice, hepatosplenomegaly, anemia, bleeding due to thrombocytopenia, and encephalitis. In fatal cases the brain is often smaller than normal (microcephaly) and may show foci of calcification. Diagnosis of neonatal CMV is made by shell-virus culture of urine or oral secretions.
The infants who survive usually have permanent deficits, including mental retardation, hearing loss, and other neurologic impairments. The congenital infection is not always devastating, however, and may take the form of interstitial pneumonitis, hepatitis, or a hematologic disorder. Most infants with this milder form of cytomegalic inclusion disease recover, although a few develop mental retardation later. Uncommonly, a totally asymptomatic infection may be followed months to years later by neurologic sequelae, including delayed-onset mental retardation and deafneas.
Perinatal Infections.
Infection acquired during passage through the birth canal or from breast milk is asymptomatic in the vast majority of cases, although, uncommonly, infants may develop an interstitial pneumonitis, failure to thrive, skin rash, or hepatitis. These children have acquired maternal antibodies against CMV, which reduce the severity of disease. Despite the lack of symptoms, many of these people continue to excrete CMV in their urine or saliva for months to years. Subtle effects on hearing and intelligence later in life have been reported in some studies.
Cytomegalovirus Mononucleosis.
In healthy young children and adults the disease is nearly always asymptomatic. In surveys around the world, 50% to 100% of adults demonstrate antibodies to CMV in the serum, indicating previous exposure. The most common clinical manifestation of CMV infection in immunocompetent hosts beyond the neonatal period is an infectious mononucleosis–like illness, with fever, atypical lymphocytosis, lymphadenopathy, and hepatomegaly accompanied by abnormal liver function test results, suggesting mild hepatitis. The diagnosis is made by serology. Most people recover without any sequelae, although excretion of the virus may occur in body fluids for months to years.
Irrespective of the presence or absence of symptoms, once infected a person becomes seropositive for life. The virus remains latent within leukocytes.
CMV in Immunosuppressed Individuals.
Immunocompromised individuals (e.g., transplant recipients, HIV-infected individuals) are susceptible to severe CMV infection. This can be either primary infection or reactivation of latent CMV. CMV is the most common opportunistic viral pathogen in AIDS. Recipients of solid-organ transplants (heart, liver, kidney) also may contract CMV from the donor organ.
In all these settings, serious, life-threatening disseminated CMV infections primarily affect the lungs (pneumonitis) and gastrointestinal tract (colitis). In the pulmonary infection an interstitial mononuclear infiltrate with foci of necrosis develops, accompanied by the typical enlarged cells with inclusions. The pneumonitis can progress to full-blown acute respiratory distress syndrome. Intestinal necrosis and ulceration can develop and be extensive, leading to the formation of pseudomembranes and debilitating diarrhea. Diagnosis of CMV infections is made by demonstration of characteristic morphologic alterations in tissue sections, viral culture, rising antiviral antibody titer, detection of CMV antigens, and PCR-based detection of CMV DNA. The antigen- and PCR-based methods have revolutionized the approach to monitoring CMV infection in people after transplantation.
CHRONIC PRODUCTIVE INFECTIONS
In some infections the immune system is unable to eliminate the virus, and continued viral replication leads to persistent viremia. The high mutation rate of viruses such as HIV and HBV may allow them to escape control by the immune system.
Hepatitis B Virus
HBV is a significant cause of acute and chronic liver disease worldwide. Here we will briefly discuss HBV as an example of a chronic productive viral infection; viral hepatitis is discussed in detail in Chapter 18. HBV, a member of the hepadnavirus family, is a DNA virus that can be transmitted percutaneously (e.g., intravenous drug use or blood transfusion), perinatally, and sexually. HBV infects hepatocytes, and cellular injury occurs mainly from the immune response to infected liver cells and not to cytopathic effects of the virus.56 The effectiveness of the cytotoxic T-lymphocyte (CTL) response is a major determinant of whether a person clears the virus or becomes a chronic carrier. When infected hepatocytes are destroyed by CTLs, replicating virus is also eliminated and the infection is cleared. However, if the rate of infection of hepatocytes outpaces the ability of CTLs to eliminate infected cells, a chronic infection is established. This may happen in about 5% of adults and up to 90% of children infected perinatally. In this setting the liver develops a chronic hepatitis, with lymphocytic inflammation, apoptotic hepatocytes resulting from CTL-mediated killing, and progressive destruction of the liver parenchyma. Long-term viral replication and recurrent immune-mediated liver injury can lead to cirrhosis of the liver and an increased risk for hepatocellular carcinoma. The morphology and pathogenesis of hepatocellular carcinoma and the role of HBV are discussed in Chapter 18. In some infected individuals, hepatocytes are infected but the CTL response is dormant, resulting in the establishment of a “carrier” state, without progressive liver damage.
TRANSFORMING INFECTIONS
This group includes several viruses that have been implicated in the causation of human cancer: EBV, HPV, HBV, and HTLV-1. EBV is discussed here; others are discussed in later chapters.
Epstein-Barr Virus (EBV)
EBV causes infectious mononucleosis, a benign, self-limited lymphoproliferative disorder, and is associated with the development of a number of neoplasms, most notably certain lymphomas and nasopharyngeal carcinoma.57 Infectious mononucleosis is characterized by fever, generalized lymphadenopathy, splenomegaly, sore throat, and the appearance in the blood of atypical activated T lymphocytes (mononucleosis cells). Some people develop hepatitis, meningoencephalitis, and pneumonitis. Infectious mononucleosis occurs principally in late adolescents or young adults among upper socioeconomic classes in developed nations. In the rest of the world, primary infection with EBV occurs in childhood and is usually asymptomatic.
Pathogenesis.
EBV is transmitted by close human contact, frequently with the saliva during kissing. An EBV envelope glycoprotein binds to CD21 (CR2), the receptor for the C3d component of complement (Chapter 2), present on B cells.58 The viral infection begins in nasopharyngeal and oropharyngeal lymphoid tissues, particularly the tonsils (Fig. 8-16). Either through transient infection of epithelium or transcytosis into the submucosa, EBV gains access to submucosal lymphoid tissues. Here, infection of B cells may take one of two forms. In a minority of B cells there is productive infection with lysis of infected cells and release of virions, which may infect other B cells. In most B cells, EBV establishes latent infection. Of note, people with X-linked agammaglobulinemia, who lack B cells, do not become latently infected with EBV or shed virus, suggesting B cells are the main reservoir of latent infection. During latent infection, a small number of EBV genes are expressed and they are involved in the establishment of latency. The gene products include EBNA1, which binds the EBV genome to chromosomes, mediating episomal persistence and maintenance, and EBNA2 and latent membrane protein 1 (LMP1), which drive B-cell activation and proliferation.59 LMP1 appears to act by binding to TNF receptor–associated factors, and activates signaling pathways that mimic B-cell activation by CD40, which is involved in normal B-cell responses (Chapter 6). EBNA2 stimulates transcription of many host cell genes, including genes that drive cell cycle entry. The activated B cells then disseminate in the circulation and secrete antibodies with several specificities, including the heterophile anti–sheep red blood cell antibodies used for the diagnosis of infectious mononucleosis. Heterophile antibodies bind to antigens that differ from the antigens that induced them. Thus, people with mononucleosis make antibodies that agglutinate sheep or horse red blood cells in the laboratory, but these antibodies do not react with EBV.
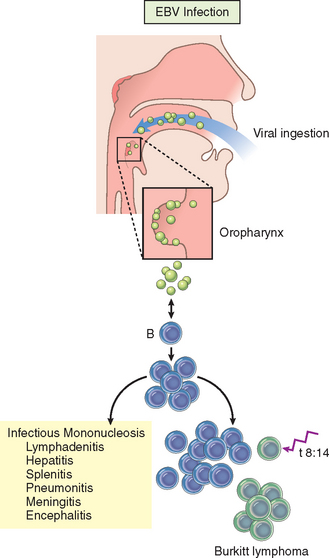
FIGURE 8-16 Outcome of Epstein-Barr virus (EBV) infection. In an individual with normal immune function, infection is usually either asymptomatic or leads to mononucleosis. In the setting of cellular immunodeficiency, the proliferation of infected B cells may be uncontrolled, leading to the development of B-cell neoplasms. In other instances, persons without overt evidence of immunodeficiency develop EBV-positive tumors, which are usually (but not always) also derived from B cells. One secondary genetic event that collaborates with EBV to cause B-cell transformation is a balanced 8;14 chromosomal translocation, which is seen in Burkitt lymphoma. EBV is also implicated in the pathogenesis of nasopharyngeal carcinoma, Hodgkin lymphoma, and certain other rare non-Hodgkin lymphomas.
The symptoms of infectious mononucleosis appear upon initiation of the host immune response. Cellular immunity mediated by CD8+ cytotoxic T cells and NK cells is the most important component of this response. The atypical lymphocytes seen in the blood, so characteristic of this disease, are mainly EBV-specific CD8+ cytotoxic T cells, but also include CD16+ NK cells. The reactive proliferation of T cells is largely centered in lymphoid tissues, which accounts for the lymphadenopathy and splenomegaly. Early in the course of the infection, IgM antibodies are formed against viral capsid antigens; later, IgG antibodies are formed that persist for life. In otherwise healthy persons, the fully developed humoral and cellular responses to EBV act as brakes on viral shedding, resulting in the elimination of B cells expressing the full complement of EBV latency–associated genes. However, EBV persists throughout life in a small population of resting B cells in which expression of EBV genes is limited to EBNA1 and LMP2. Cells within this pool are thought to occasionally reactivate expression of the other latency-associated genes, such as EBNA2 and LMP1, causing them to proliferate. In hosts with acquired defects in cellular immunity (e.g., AIDS, organ transplantation), this proliferation can progress through a multi-step process to EBV-associated B-cell lymphomas. EBV also contributes to the development of some cases of Burkitt lymphoma (Chapter 13), in which a chromosomal translocation (most commonly an 8:14 translocation) involving the c-myc oncogene is the critical oncogenic event (see Fig. 8-16).
Morphology. The major alterations involve the blood, lymph nodes, spleen, liver, CNS, and, occasionally, other organs. The peripheral blood shows absolute lymphocytosis; more than 60% of white blood cells are lymphocytes. Between 5% and 80% of these are large, atypical lymphocytes, 12 to 16 μm in diameter, characterized by an abundant cytoplasm containing multiple clear vacuolations, an oval, indented, or folded nucleus, and scattered cytoplasmic azurophilic granules (Fig. 8-17). These atypical lymphocytes, most of which express CD8, are sufficiently distinctive to strongly suggest the diagnosis.

The lymph nodes are typically discrete and enlarged throughout the body, principally in the posterior cervical, axillary, and groin regions. On histologic examination the most striking feature is the expansion of paracortical areas by activated T cells (immunoblasts). A minor population of EBV-infected B cells expressing EBNA2, LMP1, and other latency-specific genes can also be detected in the paracortex using specific antibodies. Occasionally, EBV-infected B cells resembling Reed-Sternberg cells (the malignant cells of Hodgkin lymphoma, Chapter 13) may be found. B-cell areas (follicles) may also be hyperplastic, but this is usually mild. The T-cell proliferation is sometimes so exuberant that it is difficult to distinguish the nodal morphology from that seen in malignant lymphomas. Similar changes commonly occur in the tonsils and lymphoid tissue of the oropharynx.
The spleen is enlarged in most cases, weighing between 300 and 500 gm. It is usually soft and fleshy, with a hyperemic cut surface. The histologic changes are analogous to those of the lymph nodes, showing an expansion of white pulp follicles and red pulp sinusoids due to the presence of numerous activated T cells. These spleens are especially vulnerable to rupture, possibly in part because the rapid increase in size produces a tense, fragile splenic capsule.
The liver is usually involved to some degree, although hepatomegaly is at most moderate. On histologic examination, atypical lymphocytes are seen in the portal areas and sinusoids, and scattered, isolated cells or foci of parenchymal necrosis may be present. This histologic picture is similar to that of other forms of viral hepatitis.
Clinical Features.
Infectious mononucleosis classically presents with fever, sore throat, lymphadenitis, and the other features mentioned earlier. However, atypical presentations are common, and include malaise, fatigue, and lymphadenopathy, raising the specter of leukemia or lymphoma; a fever of unknown origin without significant lymphadenopathy or other localized findings; hepatitis resembling one of the hepatotropic viral syndromes; or a febrile rash resembling rubella. The diagnosis depends on the following findings (in increasing order of specificity): (1) lymphocytosis with the characteristic atypical lymphocytes in the peripheral blood, (2) a positive heterophile antibody reaction (monospot test), and (3) specific antibodies for EBV antigens (viral capsid antigens, early antigens, or Epstein-Barr nuclear antigen). In most patients, infectious mononucleosis resolves within 4 to 6 weeks, but sometimes the fatigue lasts longer. One or more complications occasionally supervene. Perhaps most common is marked hepatic dysfunction with jaundice, elevated hepatic enzyme levels, disturbed appetite, and rarely even liver failure. Other complications involve the nervous system, kidneys, bone marrow, lungs, eyes, heart, and spleen. Splenic rupture can occur even with minor trauma, leading to hemorrhage that may be fatal. A more serious complication in those suffering from some form of immunodeficiency, such as AIDS, or receiving immunosuppressive therapy (e.g., bone marrow or solid-organ transplant recipients) is B-cell lymphomas. As detailed in Chapter 13, EBV also causes another distinctive form of lymphoma, called Burkitt lymphoma, particularly in certain geographic locales.
Unfortunate consequences also occur in individuals suffering from the X-linked lymphoproliferation syndrome (also known as Duncan disease), a disorder caused by a defect in a gene, SH2D1A, that is expressed primarily in cytotoxic T cells and NK cells.60 SH2D1A (also called SAP) participates in a signaling pathway critical for an effective cellular response to EBV-infected B cells. Patients are often normal until they are acutely infected with EBV, often during adolescence. The failure to control EBV infection variously leads to chronic infectious mononucleosis, agammaglobulinemia, and B-cell lymphoma, each of which proves fatal in about a third of patients.
Bacterial Infections
Different classes of bacteria are responsible for diverse infections (Table 8-8).
TABLE 8-8 Selected Human Bacterial Diseases and Their Pathogens
| Clinical or Microbiologic Category | Species | Frequent Disease Presentations |
|---|---|---|
| Infections by pyogenic cocci | Staphylococcus aureus, S. epidermidis | Abscess, cellulitis, pneumonia, sepsis |
| Streptococcus pyogenes | Pharyngitis, erysipelas, scarlet fever | |
| Streptococcus pneumoniae (pneumococcus) | Lobar pneumonia, meningitis | |
| Neisseria meningitidis (meningococcus) | Meningitis | |
| Neisseria gonorrhoea (gonococcus) | Gonorrhea | |
| Gram-negative infections | Escherichia coli,* Klebsiella pneumoniae* | Urinary tract infection, wound infection, abscess, pneumonia, sepsis, shock, endocarditis |
| Enterobacter (Aerobacter) aerogenes* | ||
| Proteus spp. (P. mirabilis, P. morgagni)* | ||
| Serratia marcescens,* Pseudomonas spp. (P. aeruginosa)* | ||
| Bacteroides spp. (B. fragilis) | Anaerobic infection | |
| Legionella spp. (L. pneumophila) | Legionnaires disease | |
| Contagious childhood bacterial diseases | Haemophilus influenzae | Meningitis, upper and lower respiratory tract infections |
| Bordetella pertussis | Whooping cough | |
| Corynebacterium diphtheriae | Diphtheria | |
| Enteric infections | Enteropathogenic E. coli, Shigella spp. | Invasive or noninvasive gastroenterocolitis |
| Vibrio cholerae, Campylobacter jejuni, C. coli | ||
| Yersinia enterocolitica, Salmonella spp. (1000 strains) | ||
| Salmonella typhi | Typhoid fever | |
| Clostridial infections | Clostridium tetani | Tetanus (lockjaw) |
| Clostridium botulinum | Botulism (paralytic food poisoning) | |
| Clostridium perfringens, C. septicum | Gas gangrene, necrotizing cellulitis | |
| Clostridium difficile* | Pseudomembranous colitis | |
| Zoonotic bacterial infections | Bacillus anthracis | Anthrax |
| Yersinia pestis | Bubonic plague | |
| Francisella tularensis | Tularemia | |
| Brucella melitensis, B. suis, B. abortus | Brucellosis (undulant fever) | |
| Borrelia recurrentis | Relapsing fever | |
| Borrelia burgdorferi | Lyme disease | |
| Human treponemal infections | Treponema pallidum | Syphilis |
| Mycobacterial infections | Mycobacterium tuberculosis,* M. bovis | Tuberculosis |
| M. leprae | Leprosy | |
| M. kansasii,* M. avium, M. intracellulare | Atypical mycobacterial infections | |
| Actinomycetaceae | Nocardia asteroides* | Nocardiosis |
| Actinomyces israelii | Actinomycosis |
* Important opportunistic infections.
GRAM-POSITIVE BACTERIAL INFECTIONS
Common gram-positive pathogens include Staphylococcus, Streptococcus, and Enterococcus, each of which causes many types of infections. Four less common diseases caused by gram-positive rod-shaped organisms are also discussed here: diphtheria, listeriosis, anthrax, and nocardiosis. Clostridia, which are gram-positive, are discussed with the anaerobes. All these infections are diagnosed by culture and some special tests mentioned below.
Staphylococcal Infections
Staphylococcus aureus are pyogenic gram-positive cocci that form clusters like bunches of grapes. These bacteria cause a myriad of skin lesions (boils, carbuncles, impetigo, and scalded-skin syndrome) as well as abscesses, sepsis, osteomyelitis, pneumonia, endocarditis, food poisoning, and toxic shock syndrome (TSS) (Fig. 8-18). Here we review the general characteristics of S. aureus infection. Specific organ infections are described in other chapters. S. epidermidis, a species that is related to S. aureus, causes opportunistic infections in catheterized patients, patients with prosthetic cardiac valves, and drug addicts. S. saprophyticus is a common cause of urinary tract infections in young women.
Pathogenesis.
S. aureus possess a multitude of virulence factors, which include surface proteins involved in adherence, secreted enzymes that degrade proteins, and secreted toxins that damage host cells.
S. aureus expresses surface receptors for fibrinogen (called clumping factor), fibronectin, and vitronectin, and uses these molecules as a bridge to bind to host endothelial cells.61 Staphylococci infecting prosthetic valves and catheters have a polysaccharide capsule that allows them to attach to the artificial materials and to resist host cell phagocytosis. The lipase of S. aureus degrades lipids on the skin surface, and its expression is correlated with the ability of the bacteria to produce skin abscesses. Staphylococci also have protein A on their surface, which binds the Fc portion of immunoglobulins, allowing the organism to escape antibody-mediated killing.
S. aureus produces multiple membrane-damaging (hemolytic) toxins, including α-toxin, a pore-forming protein that intercalates into the plasma membrane of host cells and depolarizes them62; β-toxin, a sphingomyelinase; and δ-toxin, which is a detergent-like peptide. Staphylococcal γ-toxin and leukocidin lyse erythrocytes and phagocytic cells, respectively.
The exfoliative A and B toxins produced by S. aureus are serine proteases that cleave the protein desmoglein 1, which is part of the desmosomes that hold epidermal cells tightly together.25 This causes keratinocytes to detach from one another and the underlying skin, resulting in a loss of barrier function that often leads to secondary skin infections. Exfoliation can occur locally at the site of infection (bullous impetigo) or can be widespread, when secreted toxin causes disseminated loss of the superficial epidermis (staphylococcal scalded-skin syndrome).
Superantigens produced by S. aureus cause food poisoning and TSS. TSS came to public attention because of its association with the use of hyperabsorbent tampons, which became colonized with S. aureus during use. It is now clear that TSS can be caused by growth of S. aureus at many sites, most commonly the vagina and infected surgical sites. TSS is characterized by hypotension (shock), renal failure, coagulopathy, liver disease, respiratory distress, a generalized erythematous rash, and soft tissue necrosis at the site of infection. If not promptly treated, TSS can be fatal. TSS can also be caused by Streptococcus pyogenes. As mentioned earlier, bacterial superantigens bind to conserved portions of MHC molecules and to relatively conserved portions of T-cell receptor β chains. In this manner superantigens may stimulate up to 20% of T lymphocytes, leading to release of large amounts of cytokines such as TNF and IL-1, which can produce a condition resembling septic shock (Chapter 4). Superantigens produced by S. aureus also cause vomiting, presumably by affecting the CNS or the enteric nervous system.63
Morphology. Whether the lesion is located in the skin, lungs, bones, or heart valves, S. aureus causes pyogenic inflammation that is distinctive for its local destructiveness.
Excluding impetigo, which is a staphylococcal or streptococcal infection restricted to the superficial epidermis, staphylococcal skin infections are centered around the hair follicles. A furuncle, or boil, is a focal suppurative inflammation of the skin and subcutaneous tissue, either solitary or multiple or recurrent in successive crops. Furuncles are most frequent in moist, hairy areas, such as the face, axillae, groin, legs, and submammary folds. Beginning in a single hair follicle, a boil develops into a growing and deepening abscess that eventually “comes to a head” by thinning and rupturing the overlying skin. A carbuncle is a deeper suppurative infection that spreads laterally beneath the deep subcutaneous fascia and then burrows superficially to erupt in multiple adjacent skin sinuses. Carbuncles typically appear beneath the skin of the upper back and posterior neck, where fascial planes favor their spread. Hidradenitis is a chronic suppurative infection of apocrine glands, most often in the axilla. Infections of the nail bed (paronychia) or on the palmar side of the fingertips (felons) are exquisitely painful. They may follow trauma or embedded splinters and, if deep enough, destroy the bone of the terminal phalanx or detach the fingernail.
Staphylococcal lung infections (Fig. 8-19) have a polymorphonuclear infiltrate similar to that of pneumococcus (Fig. 8-7) but cause much more tissue destruction. S. aureus lung infections usually occur in people with a hematogenous source, such as an infected thrombus, or a predisposing condition such as influenza.
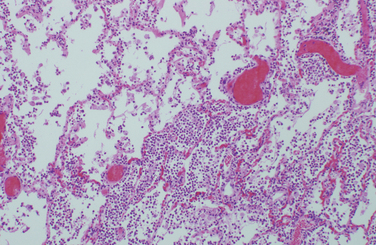
FIGURE 8-19 Staphylococcal abscess of the lung with extensive neutrophilic infiltrate and destruction of the alveoli (contrast with Fig. 8-7).
Staphylococcal scalded-skin syndrome, also called Ritter disease, most frequently occurs in children with staphylococcal infections of the nasopharynx or skin. There is a sunburn-like rash that spreads over the entire body and evolves into fragile bullae that lead to partial or total skin loss. The desquamation of the epidermis in staphylococcal scalded-skin syndrome occurs at the level of the granulosa layer, distinguishing it from toxic epidermal necrolysis, or Lyell’s disease, which is secondary to drug hypersensitivity and causes desquamation at the level of the epidermal-dermal junction (Chapter 25).
Antibiotic resistance is a growing problem in treatment of S. aureus infections. Methicillin-resistant S. aureus (MRSA) are resistant to all currently available beta-lactam cell-wall synthesis inhibitors (which include the penicillins and cephalosporins). Until recently, MRSA was mainly found in healthcare-associated infections, but community-acquired MRSA infections have now become common in many areas.64 As a result, empiric treatment of staphylococcal infections with beta-lactam antibiotics has become less effective. Community-acquired MRSA commonly produce a potent membrane damaging toxin, which kills leukocytes and may make these strains of S. aureus particularly virulent.
Streptococcal and Enterococcal Infections
Streptococci are gram-positive cocci that grow in pairs or chains and cause a myriad of suppurative infections of the skin, oropharynx, lungs, and heart valves. They are also responsible for a number of post-infectious syndromes, including rheumatic fever (Chapter 12), immune complex glomerulonephritis (Chapter 20), and erythema nodosum (Chapter 25). β-hemolytic streptococci are typed according to their surface carbohydrate (Lancefield) antigens. S. pyogenes (group A) causes pharyngitis, scarlet fever, erysipelas, impetigo, rheumatic fever, TSS, and glomerulonephritis. S. agalactiae (group B) colonizes the female genital tract and causes sepsis and meningitis in neonates and chorioamnionitis in pregnancy. S. pneumoniae, the most important α-hemolytic streptococcus, is a common cause of community-acquired pneumonia and meningitis in adults. The viridans group streptococci include several species of α-hemolytic and nonhemolytic streptococci that are normal oral flora and are also a common cause of endocarditis. Finally, S. mutans is the major cause of dental caries. Streptococcal infections are diagnosed by culture, and the rapid antigen test for pharyngitis.
Enterococci are also gram-positive cocci that grow in chains. Enterococci are often resistant to commonly used antibiotics and are a significant cause of endocarditis and urinary tract infections.
Pathogenesis.
The different species of streptococci produce many virulence factors and toxins. S. pyogenes, S. agalactiae, and S. pneumoniae have capsules that resist phagocytosis. S. pyogenes also expresses M protein, a surface protein that prevents bacteria from being phagocytosed, and a complement C5a peptidase, which degrades this chemotactic peptide.64 S. pyogenes secrete a phage-encoded pyrogenic exotoxin that causes fever and rash in scarlet fever. Poststreptococcal acute rheumatic fever is probably caused by antistreptococcal M protein antibodies and T cells that cross-react with cardiac proteins.66 Virulent S. pyogenes have been referred to as flesh-eating bacteria because they cause a rapidly progressive necrotizing fasciitis. Pneumolysin is a cytosolic bacterial protein released on disruption of S. pneumoniae.67 Pneumolysin inserts into host cell membranes and lyses them, greatly increasing tissue damage. This toxin also activates the classical pathway of complement, reducing complement available for opsonization of bacteria. S. mutans produces caries by metabolizing sucrose to lactic acid (which causes demineralization of tooth enamel) and by secreting high-molecular-weight glucans that promote aggregation of bacteria and plaque formation.
Enterococci have an antiphagocytic capsule and produce enzymes that cleave host tissues, but they are relatively low-virulence bacteria. The emergence of enterococci as pathogens is primarily due to their resistance to antibiotics, including the broad-spectrum antibiotic vancomycin.
Morphology. Streptococcal infections are characterized by diffuse interstitial neutrophilic infiltrates with minimal destruction of host tissues. The skin lesions caused by streptococci (furuncles, carbuncles, and impetigo) resemble those of staphylococci, although streptococci are less likely to cause the formation of discrete abscesses.
Erysipelas is most common among middle-aged persons in warm climates and is caused by exotoxins from superficial infection with S. pyogenes. It is characterized by rapidly spreading erythematous cutaneous swelling that may begin on the face or, less frequently, on the body or an extremity. The rash has a sharp, well-demarcated, serpiginous border and may form a “butterfly” distribution on the face (Fig. 8-20). On histologic examination there is a diffuse, edematous, neutrophilic inflammatory reaction in the dermis and epidermis extending into the subcutaneous tissues. Microabscesses may be formed, but tissue necrosis is usually minor.
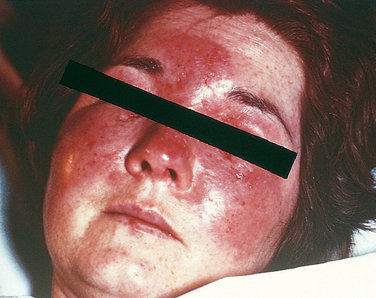
Streptococcal pharyngitis, which is the major antecedent of poststreptococcal glomerulonephritis (Chapter 20), is marked by edema, epiglottic swelling, and punctate abscesses of the tonsillar crypts, sometimes accompanied by cervical lymphadenopathy. Swelling associated with severe pharyngeal infection may encroach on the airways, especially if there is peritonsillar or retropharyngeal abscess formation.
Scarlet fever, associated with pharyngitis caused by S. pyogenes, is most common between the ages of 3 and 15 years. It is manifested by a punctate erythematous rash that is most prominent over the trunk and inner aspects of the arms and legs. The face is also involved, but usually a small area about the mouth remains relatively unaffected to produce a circumoral pallor. The inflammation of the skin usually leads to hyperkeratosis and scaling during defervescence.
S. pneumoniae is an important cause of lobar pneumonia (described in Chapter 15 and pictured in Fig. 8-7).
Diphtheria
Diphtheria is caused by Corynebacterium diphtheriae, a slender gram-positive rod with clubbed ends, that is passed from person to person through aerosols or skin exudate. C. diphtheriae may be carried asymptomatically or cause illnesses ranging from skin lesions in neglected wounds of combat troops in the tropics, and a life-threatening syndrome that includes formation of a tough pharyngeal membrane and toxin-mediated damage to the heart, nerves, and other organs. C. diphtheriae produces only one toxin, which is a phage-encoded A-B toxin that blocks host cell protein synthesis.68 The A fragment does this by catalyzing the covalent transfer of adenosine diphosphate (ADP)-ribose to elongation factor-2 (EF-2). This inhibits EF-2 function, which is essential for the translation of mRNA into protein. A single molecule of diphtheria toxin can kill a cell by ADP-ribosylating, and thus inactivating, more than a million EF-2 molecules. Immunization with diphtheria toxoid (formalin-fixed toxin) does not prevent colonization with C. diphtheriae but protects immunized people from the lethal effects of the toxin.
Morphology. Inhaled C. diphtheriae proliferate at the site of attachment on the mucosa of the nasopharynx, oropharynx, larynx, or trachea but also form satellite lesions in the esophagus or lower airways. Release of exotoxin causes necrosis of the epithelium, accompanied by an outpouring of a dense fibrinosuppurative exudate. The coagulation of this exudate on the ulcerated necrotic surface creates a tough, dirty gray to black, superficial membrane (Fig. 8-21). Neutrophilic infiltration in the underlying tissues is intense and is accompanied by marked vascular congestion, interstitial edema, and fibrin exudation. When the membrane sloughs off its inflamed and vascularized bed, bleeding and asphyxiation may occur. With control of the infection, the membrane is coughed up or removed by enzymatic digestion, and the inflammatory reaction subsides.
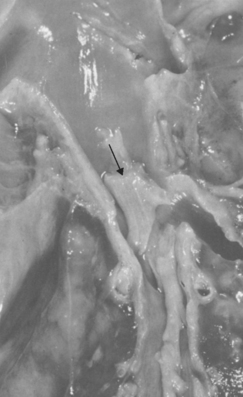
Although the bacterial invasion remains localized, generalized hyperplasia of the spleen and lymph nodes ensues as a result of the entry of soluble exotoxin into the blood. The exotoxin may cause fatty change in the myocardium with isolated myofiber necrosis, polyneuritis with degeneration of the myelin sheaths and axis cylinders, and (less commonly) fatty change and focal necroses of parenchymal cells in the liver, kidneys, and adrenals.
Listeriosis
Listeria monocytogenes is a gram-positive, facultative intracellular bacillus that causes severe food-borne infections. Mini-epidemics of L. monocytogenes infection have been linked to dairy products, chicken, and hot dogs. Pregnant women, their neonates, the elderly, and immunosuppressed persons (e.g., transplant recipients or AIDS patients) are particularly susceptible to severe L. monocytogenes infection. In pregnant women (and pregnant sheep and cattle), L. monocytogenes causes an amnionitis that may result in abortion, stillbirth, or neonatal sepsis. In neonates, L. monocytogenes may cause disseminated disease (granulomatosis infantiseptica) and an exudative meningitis, both of which are also seen in immunosuppressed adults.
Listeria monocytogenes has leucine-rich proteins on its surface called internalins, which bind to E-cadherin on host epithelial cells and induce internalization of the bacterium.69 Inside the cell, the bacteria escape from the membrane-bound phagolysosome by the action of a pore-forming protein, listeriolysin O, and two phospholipases.22 In the host cell cytoplasm, ACTA, a bacterial surface protein, binds to host cell cytoskeletal proteins and induces actin polymerization, which propels the bacteria into adjacent, uninfected host cells. Resting macrophages, which internalize L. monocytogenes through C3 activated on the bacterial surface, fail to kill the bacteria. In contrast, macrophages that are activated by IFN-γ phagocytose and kill the bacteria. Hence protection against L. monocytogenes is mediated largely by IFN-γ produced by NK cells and T cells.
Morphology. In acute human infections, L. monocytogenes evokes an exudative pattern of inflammation with numerous neutrophils. The meningitis it causes is macroscopically and microscopically indistinguishable from that caused by other pyogenic bacteria (Chapter 28). The finding of gram-positive, mostly intracellular, bacilli in the CSF is virtually diagnostic. More varied lesions may be encountered in neonates and immunosuppressed adults. Focal abscesses alternate with grayish or yellow nodules representing necrotic amorphous basophilic tissue debris. These can occur in any organ, including the lung, liver, spleen, and lymph nodes. In infections of longer duration, macrophages appear in large numbers, but granulomas are rare. Infants born with L. monocytogenes sepsis often have a papular red rash over the extremities, and listerial abscesses can be seen in the placenta. A smear of the meconium will disclose the gram-positive organisms.
Anthrax
Bacillus anthracis is a large, spore-forming gram-positive rod-shaped bacterium. These bacteria are common pathogens in farm and wild animals that have contact with soil contaminated with B. anthracis spores. Anthrax spores can be ground to a fine powder, making a potent biologic weapon. There are between 20,000 and 100,000 cases of anthrax each year, and recent use of the microbe as an agent of bioterrorism has heightened concern about this organism. In 1979, accidental release of B. anthracis spores at a military research institute in Russia killed 66 people. In 2001, 22 people in the United States were infected with B. anthracis, mostly through spores delivered in the mail.
B. anthracis is typically acquired through exposure to animals or animal products such as wool or hides.70 There are three major anthrax syndromes.
Pathogenesis.
Bacillus anthracis produces potent toxins and a polyglutamyl capsule that is antiphagocytic. The mode of action of anthrax toxin is well understood85 (Fig. 8-22). It has A and B subunits. The B subunit is also referred to as the protective antigen, because antibodies against this protein protect animals against the toxin. The protective antigen binds to a cell-surface protein, and then a host protease clips off a 20-kD fragment of the B subunit. The remaining 63-kD fragment self-associates to form a heptamer. Anthrax toxin has two alternate A subunits: edema factor (EF) and lethal factor (LF), each named for the effect of the toxin in experimental animals. Three A subunits bind to the B heptamer, and this complex is endocytosed into the host cell. The low pH of the endosome causes a conformational change in the B heptamer, which then forms a selective channel in the endosome membrane through which EF and LF move into the cytoplasm. In the cytoplasm, EF binds to calcium and calmodulin to form an adenylate cyclase. The active EF converts ATP to cyclic adenosine monophosphate (cAMP), an important signaling molecule that stimulates efflux of water from the cell, leading to interstitial edema. LF has a different mechanism of action. LF is a protease that destroys mitogen-activated protein kinase kinases (MAPKKs). These kinases regulate the activity of MAPKs, which are important regulators of cell growth and differentiation (Chapter 3). The mechanism of cell death due to dysregulation of MAPKs is not understood.
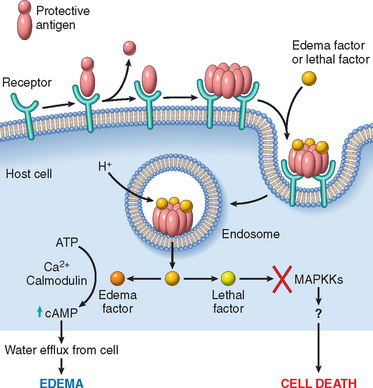
FIGURE 8-22 Mechanism of action of anthrax toxins.
(Adapted from Mourez M et al.: 2001: a year of major advances in anthrax toxin research. Trends Microbiol 10:287, 2002.)
Morphology. Anthrax lesions at any site are typified by necrosis and exudative inflammation with infiltration of neutrophils and macrophages. The presence of large, boxcar-shaped gram-positive extracellular bacteria in chains, seen histopathologically or recovered in culture, should suggest the diagnosis.
Inhalational anthrax causes numerous foci of hemorrhage in the mediastinum with hemorrhagic, enlarged hilar and peribronchial lymph nodes.72 Microscopic examination of the lungs typically shows a perihilar interstitial pneumonia with infiltration of macrophages and neutrophils and pulmonary vasculitis. Hemorrhagic lesions associated with vasculitis are also present in about half of cases. Mediastinal lymph nodes show lymphocytosis, macrophages with phagocytosed apoptotic lymphocytes, and a fibrin-rich edema (Fig. 8-23). B. anthracis is present predominantly in the alveolar capillaries and venules and, to a lesser degree, within the alveolar space. In fatal cases, B. anthracis is evident in multiple organs (spleen, liver, intestines, kidneys, adrenal glands, and meninges).
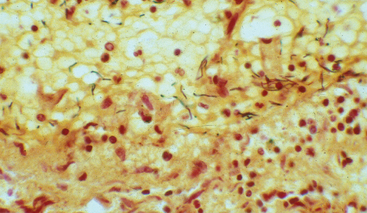
FIGURE 8-23 Bacillus anthracis in the subcapsular sinus of a hilar lymph node of a patient who died of inhalational anthrax.
(Courtesy of Dr. Lev Grinberg, Department of Pathology, Hospital 40, Ekaterinburg, Russia and Dr. David Walker, UTMB Center for Biodefense and Emerging Infectious Diseases, Galveston, TX.)
Nocardia
Nocardia are aerobic gram-positive bacteria that grow in distinctive branched chains. In culture, Nocardia form thin aerial filaments resembling hyphae. Despite this morphologic similarity to molds, Nocardia are true bacteria.
Nocardia are found in soil and cause opportunistic infections in immunocompromised people.73 Nocardia asteroides causes respiratory infections, while other species, mainly Nocardia brasiliensis, infect the skin. A fifth of N. asteroides infections involve the CNS, presumably after dissemination from the lungs. Most patients with N. asteroides have defects in T cell–mediated immunity, often due to prolonged steroid use or HIV infection, or diabetes mellitus. Respiratory infection with N. asteroides causes an indolent illness with fever, weight loss, and cough, which may be mistaken for tuberculosis or malignancy. CNS infections with N. asteroides are also indolent and cause varying neurologic deficits depending on the site of the infection. Skin infections have a range of manifestations, from rapidly progressive infections resembling those of Staphylococcus or Streptococcus to slowly progressive lesions.
Morphology. Nocardia appear in tissue as slender gram-positive organisms arranged in branching filaments (Fig. 8-24). Irregular staining gives the filaments a beaded appearance. Nocardia stain with modified acid-fast stains (Fite-Faraco stain), unlike Actinomyces, which may appear similar on Gram stain of tissue. At any site of infection, Nocardia elicit a suppurative response with central liquefaction and surrounding granulation and fibrosis. Granulomas do not form.