Chapter 28 The Central Nervous System
The principal functional unit of the central nervous system (CNS) is the neuron. Of all the cells in the body, neurons have a unique ability to receive, store, and transmit information. Neurons of different types and in different locations have distinct properties, including functional roles, distribution of their connections, neurotransmitters used, metabolic requirements, and levels of electrical activity at a given moment. A set of neurons, not necessarily clustered together in a region of the brain, may thus show selective vulnerability to various insults because it shares one or more of these properties. Since most mature neurons are incapable of cell division, destruction of even a small number of neurons essential for a specific function may leave the individual with a neurologic deficit. Stem cell populations in the brain may represent a potential mechanism for repair after injury.1 The CNS is affected by a number of unique neurological disorders, and also responds to common insults (e.g., ischemia, infection) in a manner that is distinct from other tissues.2,3
Cellular Pathology of the Central Nervous System
Reactions of Neurons to Injury.
Neurons vary considerably in structure and size throughout the nervous system and within a given brain region. Structural specializations associated with neuronal function include those related to synaptic transmission as well as axonal and dendritic differentiation. Neurons share pathways for response to injury, including apoptotic mechanisms, with cells in other tissues. During development, neuronal apoptosis has an important role in defining neuronal number; it comes into play in a variety of disease states as well, including certain neurodegenerative diseases. The principal patterns of neuronal injury are the following:
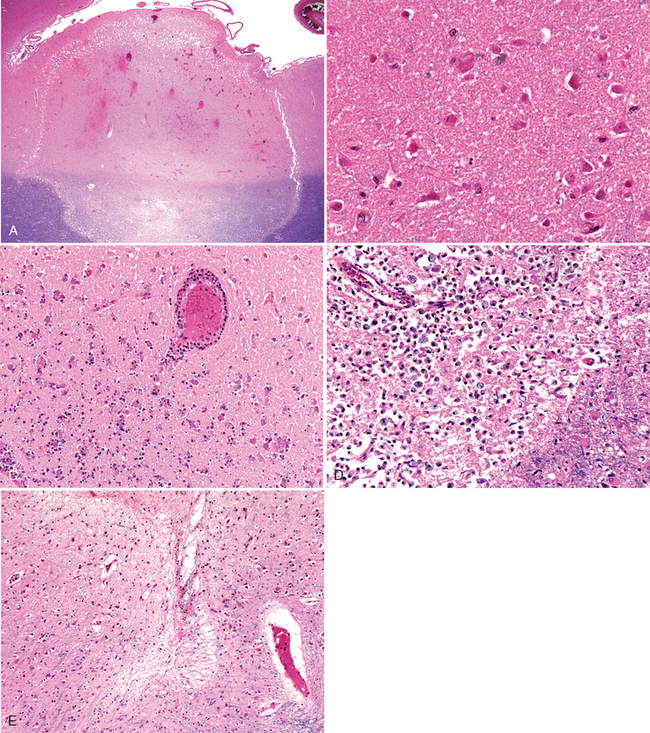
FIGURE 28-13 Cerebral infarction. A, At low magnification it is possible to see the demarcated areas of an acute infarction. In the underlying white matter, the areas of infarction are well shown by the myelin stain. B, Acute ischemic injury causes diffuse eosinophilia of neurons, which are beginning to shrink. C, Infiltration of a cerebral infarct by neutrophils begins at the edges of the lesion where vascular supply has remained intact. D, After about 10 days, an area of infarction is characterized by the presence of macrophages and surrounding reactive gliosis. E, Remote small intracortical infarcts are seen as areas of tissue loss with residual gliosis.
Reactions of Astrocytes to Injury.
The astrocyte derives its name from its star-shaped appearance. These cells have multipolar, branching cytoplasmic processes that emanate from the cell body and contain the glial fibrillary acidic protein (GFAP), a cell type–specific intermediate filament (Fig. 28-1). Astrocytes act as metabolic buffers and detoxifiers within the brain. Additionally, through the foot processes, which surround capillaries or extend to the subpial and subependymal zones, they contribute to barrier functions controlling the flow of macromolecules between the blood, the cerebrospinal fluid (CSF), and the brain. Gliosis (or astrogliosis) is the most important histopathologic indicator of CNS injury, regardless of etiology, and is characterized by both hypertrophy and hyperplasia. In this reaction, the nuclei of astrocytes, which are typically round to oval (10 μm wide) with evenly dispersed, pale chromatin, enlarge, become vesicular, and develop prominent nucleoli. The previously scant cytoplasm expands to a bright pink, somewhat irregular swath around an eccentric nucleus, from which emerge numerous stout, ramifying processes; these cells are called gemistocytic astrocytes.
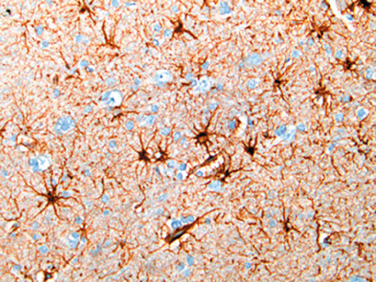
FIGURE 28-1 Astrocytes and their processes. Immunohistochemical staining for GFAP reveals astrocytic perinuclear cytoplasm and well-developed processes (brown).
When directly injured, astrocytes can react with cytoplasmic swelling. This is seen in acute insults that cause the cell’s ATP-dependent ion channels to fail, as occurs in hypoxia, hypoglycemia, and toxic injuries. The Alzheimer type II astrocyte is a gray-matter cell with a large (two to three times normal) nucleus, pale-staining central chromatin, an intranuclear glycogen droplet, and a prominent nuclear membrane and nucleolus. Its name is a misnomer, as it is mainly seen not in Alzheimer disease but in individuals with long-standing hyperammonemia due to chronic liver disease, Wilson disease, or hereditary metabolic disorders of the urea cycle.
Astrocytes are not spared from processes that cause the formation of cytoplasmic inclusion bodies. Rosenthal fibers are thick, elongated, brightly eosinophilic, somewhat irregular structures that occur within astrocytic processes, and contain two heat-shock proteins (αB-crystallin and hsp27) as well as ubiquitin. Rosenthal fibers are typically found in regions of long-standing gliosis; they are also characteristic of one type of glial tumor, pilocytic astrocytoma. In Alexander disease, a leukodystrophy associated with a mutations in the gene encoding GFAP, abundant Rosenthal fibers are found in periventricular, perivascular, and subpial locations. More commonly seen are corpora amylacea, or polyglucosan bodies. These are round, faintly basophilic, periodic acid–Schiff (PAS)–positive, concentrically lamellated structures of 5 to 50 μm in diameter that are located wherever there are astrocytic end processes, especially in the subpial and perivascular zones. Though consisting primarily of glycosaminoglycan polymers, they also contain heat-shock proteins and ubiquitin. They occur in increasing numbers with advancing age and are thought to represent a degenerative change in the astrocyte. The Lafora bodies that are seen in the cytoplasm of neurons (as well as hepatocytes, myocytes, and other cells) in myoclonic epilepsy (Lafora body myoclonus with epilepsy) have a similar structure and biochemical composition.
Reactions of Other Glial Cells to Injury.
In contrast to astrocytes, oligodendrocytes and ependyma do not participate in the active response to CNS injury and show a more limited repertoire of reactions. Oligodendroglial cytoplasmic processes wrap around exons and form myelin. Each oligodendrocyte myelinates numerous internodes on multiple axons. Injury or apoptosis of oligodendroglial cells is a feature of acquired demyelinating disorders and leukodystrophies. Oligodendroglial nuclei may harbor viral inclusions in progressive multifocal leukoencephalopathy. Glial cytoplasmic inclusions, primarily composed of α-synuclein, are found in oligodendrocytes in multiple system atrophy (MSA).
Ependymal cells, the ciliated columnar epithelial cells lining the ventricles, do not have specific patterns of reaction. When there is inflammation or marked dilation of the ventricular system, disruption of the ependymal lining is paired with proliferation of subependymal astrocytes to produce small irregularities on the ventricular surfaces (ependymal granulations). Certain infectious agents, particularly CMV, may produce extensive ependymal injury, with viral inclusions in ependymal cells.
Reactions of Microglia to Injury.
Microglia are mesoderm-derived cells whose primary function is to serve as a fixed macrophage system in the CNS. They share many surface markers with peripheral monocytes/macrophages (such as CR3 and CD68). They respond to injury by (1) proliferating; (2) developing elongated nuclei (rod cells), as in neurosyphilis; (3) forming aggregates about small foci of tissue necrosis (microglial nodules); or (4) congregating around cell bodies of dying neurons (neuronophagia). In addition to resident microglia, blood-derived macrophages are the principal phagocytic cells present in inflammatory foci.
Cerebral Edema, Hydrocephalus, and Raised Intracranial Pressure and Herniation
The brain and spinal cord are protected by the rigid compartment defined by the skull, vertebral bodies, and dura mater. Generalized brain edema, increased CSF volume (hydrocephalus), and focally expanding mass lesions may increase intracranial pressure. Depending on the degree and rapidity of this increase and the nature of the underlying lesion, the consequences range from subtle neurologic deficits to death.
CEREBRAL EDEMA
Cerebral edema or, more precisely, brain parenchymal edema, is of two principal types:
In practice, conditions associated with generalized edema often have elements of both vasogenic and cytotoxic edema.
Interstitial edema (hydrocephalic edema) occurs especially around the lateral ventricles when an increase in intravascular pressure causes an abnormal flow of fluid from the intraventricular CSF across the ependymal lining to the periventricular white matter. In generalized edema, the gyri are flattened, the intervening sulci are narrowed, and the ventricular cavities are compressed. As the brain expands, herniation may occur.
HYDROCEPHALUS
The choroid plexus within the ventricular system produces CSF, which normally circulates through the ventricular system and enters the cisterna magna at the base of the brain stem through the foramina of Luschka and Magendie. Subarachnoid CSF bathes the superior cerebral convexities and is absorbed by the arachnoid granulations. Hydrocephalus refers to the accumulation of excessive CSF within the ventricular system (Fig. 28-2). Most cases occur as a consequence of impaired flow and resorption of CSF; only rarely is overproduction the cause of hydrocephalus (e.g., with tumors of the choroid plexus). An increased volume of CSF within the ventricles expands them and can elevate the intracranial pressure.
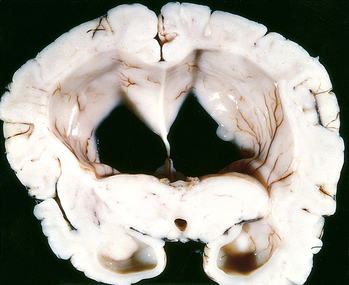
FIGURE 28-2 Hydrocephalus. Dilated lateral ventricles seen in a coronal section through the midthalamus.
When hydrocephalus develops in infancy before closure of the cranial sutures, there is enlargement of the head, manifested by an increase in head circumference. Hydrocephalus developing after this period, in contrast, is associated with expansion of the ventricles and increased intracranial pressure, without a change in head circumference. If only a portion of the ventricular system is enlarged because of excess CSF, as may occur because of a mass in the third ventricle, the pattern is called noncommunicating hydrocephalus. In communicating hydrocephalus there is enlargement of the entire ventricular system. The term hydrocephalus ex vacuo refers to dilation of the ventricular system with a compensatory increase in CSF volume secondary to a loss of brain parenchyma.
RAISED INTRACRANIAL PRESSURE AND HERNIATION
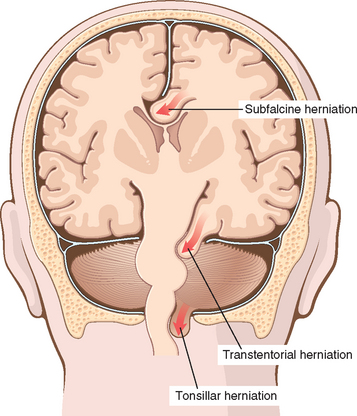
When the volume of the brain increases beyond the limit permitted by compression of veins and displacement of CSF, the pressure within the skull will increase. Most cases are associated with a mass effect, either diffuse, as in generalized brain edema, or focal, as with tumors, abscesses, or hemorrhages. Elevated intracranial pressure may also reduce perfusion of the brain, further exacerbating cerebral edema. Because the cranial vault is divided by rigid dural folds (the falx and tentorium), localized expansion of the brain may cause it to be displaced in relation to these partitions. If the expansion is sufficiently severe, a herniation syndrome may occur (Fig. 28-3).
Malformations and Developmental Diseases
Although the pathogenesis and etiology of CNS malformations are largely unknown, both genetic and environmental influences appear to be involved. Aberrations of signaling molecules and mutations of homeotic genes that control body patterning are being increasingly identified as causes of developmental disorders of the CNS. Many toxic compounds and infectious agents are also known to have teratogenic effects.5
NEURAL TUBE DEFECTS
Failure of a portion of the neural tube to close, or reopening of a region of the tube after successful closure, may lead to one of several malformations.6 All are characterized by abnormalities involving some combination of neural tissue, meninges, and overlying bone or soft tissues. An encephalocele is a diverticulum of malformed CNS tissue extending through a defect in the cranium. It most often occurs in the occipital region or in the posterior fossa. Collectively, neural tube defects account for most CNS malformations.
The most common neural tube defects involve the spinal cord and are caused by a failure of closure or by reopening of the caudal portions of the neural tube. Spinal dysraphism or spina bifida may be an asymptomatic bony defect (spina bifida occulta) or a severe malformation with a flattened, disorganized segment of spinal cord, associated with an overlying meningeal outpouching. Myelomeningocele (or meningomyelocele) refers to extension of CNS tissue through a defect in the vertebral column; the term meningocele applies when there is only a meningeal extrusion. Myelomeningoceles occur most commonly in the lumbosacral region, and affected individuals manifest clinical deficits referable to motor and sensory function in the lower extremities as well as disturbances of bowel and bladder control from both the structural abnormality of the cord itself and superimposed infection that extends from the thin, overlying skin.
The frequency of neural tube defects varies widely among different ethnic groups. Both genetic and environmental factors are involved. The concordance rate is high among monozygotic twins, and the overall recurrence rate for a neural tube defect in subsequent pregnancies has been estimated at 4% to 5%. Folate deficiency during the initial weeks of gestation has been implicated as a risk factor; differences in rates of neural tube defects between populations can be attributed in part to polymorphisms in enzymes of folic acid metabolism. Folate deficiency may affect cell division during critical periods that coincide with closure of the neural tube. Antenatal diagnosis is based on imaging and the screening of maternal blood samples for elevation of α-fetoprotein.
Anencephaly is a malformation of the anterior end of the neural tube, with absence of the brain and calvarium. Forebrain development is disrupted at approximately 28 days of gestation, and all that remains in its place is the area cerebrovasculosa, a flattened remnant of disorganized brain tissue with admixed ependyma, choroid plexus, and meningothelial cells. The posterior fossa structures may be spared, depending on the extent of the skull deficit; descending tracts associated with disrupted structures are, as expected, absent.
FOREBRAIN ANOMALIES
The volume of brain may be abnormally large (megalencephaly) or small (microencephaly). Microencephaly, by far the more common of the two, can occur in a wide range of settings, including chromosome abnormalities, fetal alcohol syndrome, and human immunodeficiency virus 1 (HIV-1) infection acquired in utero. It is postulated that the underlying anomaly is a reduction in the number of neurons that reach the neocortex and this leads to a simplification of the gyral folding—a model supported by experimental results in mouse models. The pool of proliferating precursor cells in the developing brain lies adjacent to the ventricular system. Neuronal number is determined by the fraction of proliferating cells that undergo transition into migrating cells with each cell cycle. Early on, most cell divisions yield two more progenitor cells, while as development progresses there are more asymmetric divisions yielding both a progenitor cell and a cell headed for the developing cortex. If excess cells exit the proliferating pool too early, then the overall generation of neurons is reduced; if too few exit during early rounds of division, then the geometric expansion of the proliferating population results in an eventual overproduction of neurons.7-9
Among the recognizable malformations10 are conditions that can range from a noticeable decrease in the number of gyri to total absence, leaving a smooth-surfaced brain, lissencephaly (agyria) (Fig. 28-5). A variety of forms of lissencephaly with distinct genetic causes have been described. One of the best understood causes is mutations in the gene encoding the microtubule-associated protein LIS-1, which complexes with dynein and affects the function of the centrosome in nuclear movement. Lissencephaly can also occur from a series of mutations in the genes encoding enzymes responsible for the glycosylation of α-dystroglycan; when this receptor for extracellular matrix components does not have appropriate post-translational modifications, its stability is diminished.
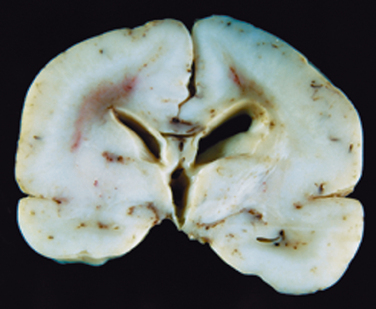
FIGURE 28-5 Lissencephaly. The absence of cortical gyri defines this abnormality, seen here in the brain from a full-term infant.
Polymicrogyria is characterized by small, unusually numerous, and irregularly formed cerebral convolutions. The gray matter is composed of four layers (or fewer), with entrapment of apparent meningeal tissue at points of fusion that would otherwise be the cortical surface. Polymicrogyria can be induced by localized tissue injury toward the end of neuronal migration, although genetically determined forms, which are typically bilateral and symmetric, are also recognized.11
Neuronal heterotopias are a group of migrational disorders that are commonly associated with epilepsy.12 They consist of collections of neurons in inappropriate locations along the migrational pathways. As might be expected, one location in which heterotopias can be found is along the ventricular surface—as though the cells never managed to leave their place of birth. Periventricular heterotopias can be caused by mutations in the gene encoding filamin A, an actin-binding protein responsible for assembly of complex meshworks of filaments. This gene is on the X chromosome, and the mutant allele causes male lethality; in females the process of X inactivation separates neurons into those with a normal allele (in the correct location) and those with the mutant allele (in the heterotopia). Another microtubule-associated protein, doublecortin (DCX), is also encoded by a gene on the X chromosome; mutations in this gene result in lissencephaly in males and in subcortical band heterotopias in females. These heterotopias may consist of discrete nodules of neurons sitting in the subcortical white matter or complete ribbons that parody the overlying cortex.
Holoprosencephaly is a spectrum of malformations characterized by incomplete separation of the cerebral hemispheres across the midline. Severe forms manifest midline facial abnormalities, including cyclopia; less severe variants (arrhinencephaly) show absence of the olfactory cranial nerves and related structures. Intrauterine diagnosis of severe forms by ultrasound examination is now possible. Holoprosencephaly is associated with trisomy 13 as well as other genetic syndromes.13 Sonic hedgehog is a member of a family of secreted proteins synthesized by the notochord and neural plate during neural development. Mutations affecting sonic hedgehog or its signaling pathway may result in holoprosencephaly.
In agenesis of the corpus callosum, a relatively common malformation, there is an absence of the white matter bundles that carry cortical projections from one hemisphere to the other (Fig. 28-6). Radiologic imaging studies show misshapen lateral ventricles (“bat-wing” deformity); on coronal whole-mount sections of the brain, bundles of anteroposteriorly oriented white matter can be demonstrated. Agenesis of the corpus callosum can be associated with mental retardation or may occur in clinically normal individuals. It can be present in isolation or can be associated with a wide range of other malformations. Unlike patients with surgical callosal section, who show clinical evidence of hemispheric disconnection, individuals with this malformation can have minimal deficits.
POSTERIOR FOSSA ANOMALIES
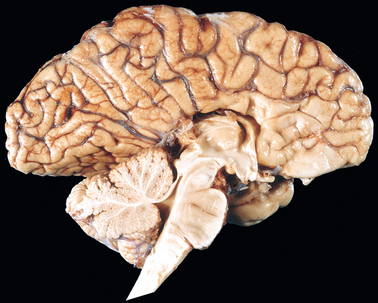
The Dandy-Walker malformation is characterized by an enlarged posterior fossa. The cerebellar vermis is absent or present only in rudimentary form in its anterior portion. In its place is a large midline cyst that is lined by ependyma and is contiguous with leptomeninges on its outer surface. This cyst represents the expanded, roofless fourth ventricle in the absence of a normally formed vermis. Dysplasias of brainstem nuclei are commonly found in association with Dandy-Walker malformation.
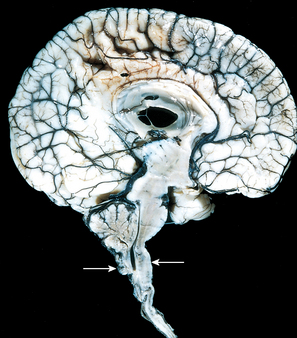
The Arnold-Chiari malformation (Chiari type II malformation) consists of a small posterior fossa, a misshapen midline cerebellum with downward extension of vermis through the foramen magnum (Fig. 28-7), and, almost invariably, hydrocephalus and a lumbar myelomeningocele. Other associated changes may include caudal displacement of the medulla, malformation of the tectum, aqueductal stenosis, cerebral heterotopias, and hydromyelia (see later). In the Chiari I malformation, low-lying cerebellar tonsils extend down into the vertebral canal. In contrast to the significant clinical consequences of the preceding two malformations, this may be a silent abnormality or may cause symptoms referable to obstruction of CSF flow and medullary compression; if present, these symptoms can usually be corrected by neurosurgical intervention.
SYRINGOMYELIA AND HYDROMYELIA
These are disorders characterized by a discontinuous multisegmental or confluent expansion of the ependyma-lined central canal of the cord (hydromyelia) or by the formation of a fluid-filled cleftlike cavity in the inner portion of the cord (syringomyelia, syrinx) that may extend into the brainstem (syringobulbia).
Syringomyelia may be associated with the Chiari I malformation; it may also occur in association with intraspinal tumors or following traumatic injury. In general, the histologic appearance is similar in all these conditions, with destruction of the adjacent gray and white matter, surrounded by a dense feltwork of reactive gliosis. The disease generally becomes manifest in the second or third decade of life. The distinctive symptoms and signs of a syrinx are the isolated loss of pain and temperature sensation in the upper extremities because of the predilection for early involvement of the crossing anterior spinal commissural fibers of the spinal cord.
Perinatal Brain Injury
Brain injury occurring in the perinatal period is an important cause of childhood neurologic disability. Injuries that occur early in gestation may destroy brain tissue without evoking the usual “reactive” changes in the parenchyma and may be difficult to distinguish from malformations.
The broad clinical term cerebral palsy refers to a nonprogressive neurologic motor deficit characterized by combinations of spasticity, dystonia, ataxia/athetosis, and paresis attributable to insults occurring during the prenatal and perinatal periods. Signs and symptoms may not be apparent at birth and only later declare themselves, as development proceeds. Postmortem examinations of children with this syndrome have shown a wide range of neuropathologic findings, including destructive lesions traced to remote events that may have caused hemorrhage and infarction.
In premature infants there is an increased risk of intraparenchymal hemorrhage within the germinal matrix, often near the junction between the thalamus and the caudate nucleus. Hemorrhages may remain localized or extend into the ventricular system and thence to the subarachnoid space, sometimes leading to hydrocephalus.
Infarcts may occur in the supratentorial periventricular white matter (periventricular leukomalacia), especially in premature infants. These are chalky yellow plaques consisting of discrete regions of white matter necrosis and calcification. When both gray and white matter are involved by extensive ischemic damage, large destructive cystic lesions develop throughout the hemispheres; this condition is termed multicystic encephalopathy (Fig. 28-8).
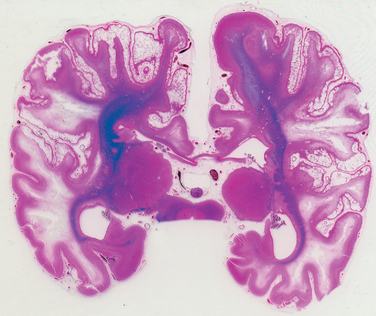
FIGURE 28-8 Multicystic leukoencephalopathy. Numerous cystic spaces representing the consequences of widespread ischemic injury are present.
In perinatal ischemic lesions of the cerebral cortex, the depths of sulci bear the brunt of injury and result in thinned-out, gliotic gyri (ulegyria). The basal ganglia and thalamus may also suffer ischemic injury, with patchy neuronal loss and reactive gliosis. Later, aberrant and irregular myelinization gives rise to a marble-like appearance of the deep nuclei: status marmoratus. Because the lesions are in the caudate, putamen, and thalamus, choreoathetosis and related movement disorders are common clinical sequelae.
Trauma
The anatomic location of the lesion and the limited capacity of the brain for functional repair are major determinants of the consequences of CNS trauma. Injury of several cubic centimeters of brain parenchyma may be clinically silent (e.g., in the frontal lobe), severely disabling (in the spinal cord), or fatal (in the brainstem).
The physical forces associated with head injury may result in skull fractures, parenchymal injury, and vascular injury; all three can coexist. The magnitude and distribution of a traumatic brain lesion depend on the shape of the object causing the trauma, the force of impact, and whether the head is in motion at the time of injury. A blow to the head may be penetrating or blunt; it may cause either an open or a closed injury.
SKULL FRACTURES
A fracture in which bone is displaced into the cranial cavity by a distance greater than the thickness of the bone is called a displaced skull fracture. The thickness of the cranial bones varies; therefore, their resistance to fracture differs greatly. Also, the relative incidence of fractures among skull bones is related to the pattern of falls. When an individual falls while awake, such as might occur when stepping off a ladder, the site of impact is often in the occipital portion of the skull; in contrast, a fall that follows loss of consciousness, as might follow a syncopal attack, commonly results in a frontal impact. Symptoms referable to the lower cranial nerves or the cervicomedullary region, and the presence of orbital or mastoid hematomas distant from the point of impact, raise the suspicion of a basal skull fracture, which typically follows impact to the occiput or sides of the head. CSF discharge from the nose or ear and infection (meningitis) may follow. The kinetic energy that causes a fracture is dissipated at a fused suture; fractures that cross sutures are termed diastatic. With multiple points of impact or repeated blows to the head, the fracture lines of subsequent injuries do not extend across fracture lines of prior injury.
PARENCHYMAL INJURIES
Concussion
Concussion is a clinical syndrome of altered consciousness secondary to head injury typically brought about by a change in the momentum of the head (when a moving head is suddenly arrested by impact on a rigid surface). The characteristic neurologic picture includes instantaneous onset of transient neurologic dysfunction, including loss of consciousness, temporary respiratory arrest, and loss of reflexes. Although neurologic recovery is complete, amnesia for the event persists. The pathogenesis of the sudden disruption of neurologic function is unknown; it probably involves dysregulation of the reticular activating system in the brainstem. Postconcussive neuropsychiatric syndromes, typically associated with repetitive injuries, are well recognized.
Direct Parenchymal Injury
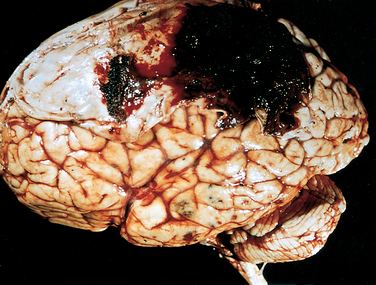
Contusion and laceration are lesions associated with direct parenchymal injury of the brain, either through transmission of kinetic energy to the brain and bruising analogous to what is seen in soft tissues (contusion) or by penetration of an object and tearing of tissue (laceration). As with any other organ, a blow to the surface of the brain, transmitted through the skull, leads to rapid tissue displacement, disruption of vascular channels, and subsequent hemorrhage, tissue injury, and edema (Fig. 28-9). The crests of gyri are most susceptible, since this is where the direct force is greatest. The most common locations for contusions correspond to the most frequent sites of direct impact and to regions of the brain that overlie a rough and irregular inner skull surface, such as the frontal lobes along the orbital ridges and the temporal lobes. Contusions are less frequent over the occipital lobes, brainstem, and cerebellum unless these sites are adjacent to a skull fracture (fracture contusions).
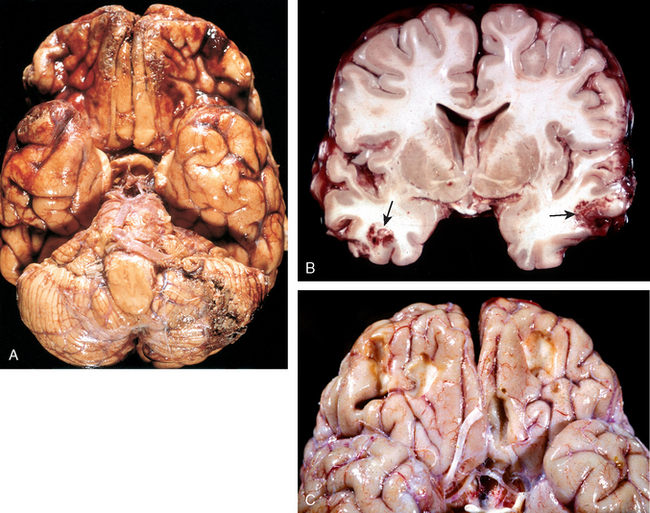
FIGURE 28-9 A, Multiple contusions involving the inferior surfaces of frontal lobes, anterior temporal lobes, and cerebellum. B, Acute contusions are present in both temporal lobes, with areas of hemorrhage and tissue disruption (arrows). C, Remote contusions are present on the inferior frontal surface of this brain, with a yellow color (associated with the term plaque jaune).
A person who suffers a blow to the head may develop a contusion at the point of contact (a coup injury) or a contusion on the brain surface diametrically opposite to it (a contrecoup injury). Since their macroscopic and microscopic appearance is indistinguishable, the distinction between them is based on forensic identification of the point of impact and the circumstances attending the incident. In general, if the head is immobile at the time of trauma, only a coup injury is found. If the head is mobile, both coup and contrecoup lesions may be found. Whereas the coup lesion is caused by the contact between brain and skull at the site of impact, the contrecoup contusion is thought to develop when the brain strikes the opposite inner surface of the skull after sudden deceleration.
Sudden impacts that result in violent posterior or lateral hyperextension of the neck (as occurs when a pedestrian is struck from the rear by a vehicle) may avulse the pons from the medulla or the medulla from the cervical cord, causing instantaneous death.
Morphology. When seen on cross-section, contusions are wedge shaped, with the broad base lying along the surface, deep to the point of impact (Fig. 28-9B). The histologic appearance of contusions is independent of the type of trauma. In the earliest stages, there is edema and hemorrhage, which is often pericapillary. During the next few hours, the extravasation of blood extends throughout the involved tissue, across the width of the cerebral cortex, and into the white matter and subarachnoid space. Morphologic evidence of neuronal injury (pyknosis of the nucleus, eosinophilia of the cytoplasm, and disintegration of the cell) takes about 24 hours to appear, although functional deficits may occur earlier. Axonal swellings develop in the vicinity of damaged neurons or at great distances away. The inflammatory response to the injured tissue follows its usual course, with the appearance of neutrophils followed by macrophages. Old traumatic lesions on the surface of the brain have a characteristic macroscopic appearance. They are depressed, retracted, yellowish brown patches involving the crests of gyri most commonly located at the sites of contrecoup lesions (inferior frontal cortex, temporal and occipital poles). The term plaque jaune is applied to these lesions (Fig. 28-9C); they can become epileptic foci. More extensive hemorrhagic regions of brain trauma give rise to larger cavitated lesions, which can resemble remote infarcts. In sites of old contusions, gliosis and residual hemosiderin-laden macrophages predominate.
Diffuse Axonal Injury
Although it is most often affected, the surface of the brain is not the only region of damage in traumatic injury. Also affected may be the deep white matter regions (the corpus callosum, paraventricular, and hippocampal areas in the supratentorial compartment), cerebral peduncles, brachium conjunctivum, superior colliculi, and deep reticular formation in the brainstem. The microscopic findings include axonal swelling, indicative of diffuse axonal injury, and focal hemorrhagic lesions. Angular acceleration alone, in the absence of impact, can cause diffuse axonal injury as well as hemorrhage. As many as 50% of individuals who develop coma shortly after trauma, even without cerebral contusions, are believed to have diffuse axonal injury. The mechanical forces associated with trauma are believed to damage the integrity of the axon at the node of Ranvier, with subsequent alterations in axoplasmic flow.
Morphology. Diffuse axonal injury is characterized by the widespread but often asymmetric axonal swellings that appear within hours of the injury and may persist for much longer. These are best demonstrated with silver impregnation techniques or with immunoperoxidase stains for axonally transported proteins, including amyloid precursor protein and α-synuclein. Later, there are increased numbers of microglia in related areas of the cerebral cortex and, subsequently, degeneration of the involved fiber tracts.
TRAUMATIC VASCULAR INJURY
Vascular injury is a frequent component of CNS trauma. It results from direct trauma and disruption of the vessel wall, and leads to hemorrhage. Depending on the anatomic posi tion of the ruptured vessel, hemorrhage may occur in the epidural, subdural, subarachnoid, and intraparenchymal compartments, sometimes in combination (Fig. 28-10). Subarachnoid and intraparenchymal hemorrhages most often occur concomitantly in the setting of brain trauma that also results in superficial contusions and lacerations. A traumatic tear of the carotid artery where it traverses the carotid sinus may lead to the formation of an arteriovenous fistula.
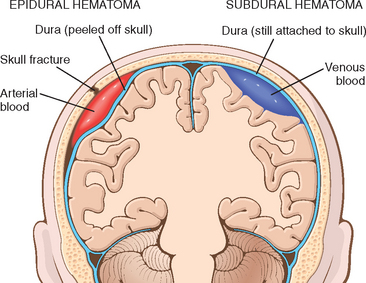
FIGURE 28-10 Epidural hematoma (left) in which rupture of a meningeal artery, usually associated with a skull fracture, leads to accumulation of arterial blood between the dura and the skull. In a subdural hematoma (right), damage to bridging veins between the brain and the superior sagittal sinus leads to the accumulation of blood between the dura and the arachnoid.
Epidural Hematoma
Normally the dura is fused with the periosteum on the internal surface of the skull. Dural arteries, most importantly the middle meningeal artery, are vulnerable to injury, particularly with temporal skull fractures in which the fracture lines cross the course of the vessel. In children, in whom the skull is deformable, a temporary displacement of the skull bones leading to laceration of a vessel can occur in the absence of a skull fracture.
Once a vessel has been torn, the extravasation of blood under arterial pressure can cause the dura to separate from the inner surface of the skull (Fig. 28-11). The expanding hematoma has a smooth inner contour that compresses the brain surface. When blood accumulates slowly patients may be lucid for several hours before the onset of neurologic signs. An epidural hematoma may expand rapidly and is a neurosurgical emergency requiring prompt drainage.
Subdural Hematoma
Between the inner surface of the dura mater and the outer arachnoid layer of the leptomeninges lies the subdural space. Bridging veins travel from the convexities of the cerebral hemispheres through the subarachnoid space and the subdural space to empty into the superior sagittal sinus. Similar anatomic relationships exist with other dural sinuses. These vessels are particularly prone to tearing along their course through the subdural space and are the source of bleeding in most cases of subdural hematoma. It is thought that the brain, floating freely bathed in CSF, can move within the skull, but the venous sinuses are fixed. The displacement of the brain that occurs in trauma can tear the veins at the point where they penetrate the dura. In elderly individuals with brain atrophy, the bridging veins are stretched out and the brain has additional space for movement, hence the increased rate of subdural hematomas in these patients, even after relatively minor head trauma. Infants are also particularly susceptible to subdural hematomas because their bridging veins are thin-walled.
Morphology. On macroscopic examination, the acute subdural hematoma appears as a collection of freshly clotted blood along the brain surface, without extension into the depths of sulci (Fig. 28-12). The underlying brain is flattened and the subarachnoid space is often clear. Typically, venous bleeding is self-limited; breakdown and organization of the hematoma take place over time. This usually occurs in the following sequence:
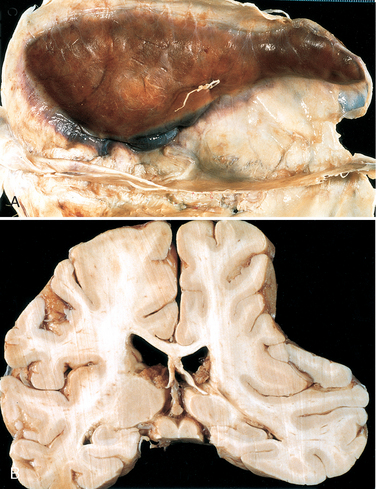
FIGURE 28-12 A, Large organizing subdural hematoma attached to the dura. B, Coronal section of the brain showing compression of the hemisphere underlying the subdural hematoma shown in A.
Typically, the organized hematoma is firmly attached by fibrous tissue only to the inner surface of the dura and is not adherent to the underlying smooth arachnoid, which does not contribute to its formation. The lesion can eventually retract as the granulation tissue matures, until there is only a thin layer of reactive connective tissue (“subdural membranes”). A common finding in subdural hematomas, however, is the occurrence of multiple episodes of repeat bleeding (chronic subdural hematomas), presumably from the thin-walled vessels of the granulation tissue. The risk of repeat bleeding is greatest in the first few months after the initial hemorrhage.
Clinical Features.
Subdural hematomas most often manifest within 48 hours of injury. They are most common over the lateral aspects of the cerebral hemispheres and are bilateral in about 10% of cases. Neurologic signs commonly observed are attributable to the pressure exerted on the adjacent brain. There may be focal signs, but often the clinical manifestations are nonlocalizing and include headache and confusion. Slowly progressive neurologic deterioration is typical, but acute decompensation may occur. The treatment of subdural hematomas is to remove the blood and associated organizing tissue.
SEQUELAE OF BRAIN TRAUMA
A broad range of neurologic syndromes may become manifest months or years after brain trauma of any cause. These have gained increasing notice in the context of legal medicine and litigation involving issues of compensation for those in the civilian work force and the military services. Post-traumatic hydrocephalus is largely due to obstruction of CSF resorption from hemorrhage into the subarachnoid spaces. Post-traumatic dementia and the punch-drunk syndrome (dementia pugilistica) follow repeated head trauma during a protracted period; the neuropathologic findings include hydrocephalus, thinning of the corpus callosum, diffuse axonal injury, neurofibrillary tangles (mainly in the medial temporal areas), and diffuse amyloid β (Aβ)-positive plaques (see “Alzheimer Disease”). Other important sequelae of brain trauma include post-traumatic epilepsy, tumors (meningioma), infectious diseases, and psychiatric disorders.3
SPINAL CORD TRAUMA
The spinal cord, normally protected within the bony vertebral canal, is vulnerable to trauma from its skeletal encasement. Most injuries that damage the cord are associated with displacement of the vertebral column, either rapid and temporary or persistent. The level of cord injury determines the extent of neurologic manifestations: lesions involving the thoracic vertebrae or below can lead to paraplegia; cervical lesions result in quadriplegia; those above C4 can, in addition, lead to respiratory compromise from paralysis of the diaphragm. Segmental damage to the descending and ascending white matter tracts isolates the distal spinal cord from its cortical connections with the cerebrum and brainstem; this interruption, rather than the segmental gray matter damage that may occur at the level of the impact, is the principal cause of neurological deficits.
Morphology. The histologic changes of traumatic injury of the spinal cord are similar to those found at other sites in the CNS. At the level of injury the acute phase consists of hemorrhage, necrosis, and axonal swelling in the surrounding white matter. The lesion tapers above and below the level of injury. In time the central necrotic lesion becomes cystic and gliotic; cord sections above and below the lesion show secondary ascending and descending wallerian degeneration, respectively, involving the long white-matter tracts affected at the site of trauma.
Cerebrovascular Diseases
Cerebrovascular disease is the third leading cause of death (after heart disease and cancer) in the United States; it is also the most prevalent neurologic disorder in terms of both morbidity and mortality. Cerebrovascular diseases include the expected three major categories, thrombosis, embolism, and hemorrhage, with patient management differing between groups. “Stroke” is the clinical designation that applies to all these conditions, particularly when symptoms begin acutely. From the standpoint of pathophysiology and pathologic anatomy, it is convenient to consider cerebrovascular disease as two processes:
The most common cerebrovascular disorders are global ischemia, embolism, hypertensive intraparenchymal hemorrhage, and ruptured aneurysm.
HYPOXIA, ISCHEMIA, AND INFARCTION
The brain requires a constant supply of glucose and oxygen, which is delivered by the blood. Although the brain accounts for only 1% to 2% of body weight, it receives 15% of the resting cardiac output and accounts for 20% of the total body oxygen consumption. Cerebral blood flow remains relatively constant over a wide range of blood pressure and intracranial pressure because of autoregulation of vascular resistance. The brain is a highly aerobic tissue, in which oxygen rather than metabolic substrate is limiting. The brain may be deprived of oxygen by several mechanisms: hypoxia caused by a low partial pressure of oxygen (Po2), impairment of the blood’s oxygen-carrying capacity, or inhibition of oxygen use in the tissue; or ischemia, either transient or permanent, after interruption of the normal circulatory flow. Cessation of blood flow can result from a reduction in perfusion pressure (as in hypotension), small- or large-vessel obstruction, or both.
When blood flow to a portion of the brain is reduced, the survival of the tissue at risk depends on the presence of collateral circulation, the duration of ischemia, and the magnitude and rapidity of the reduction of flow. These factors determine, in turn, the precise anatomic site and size of the lesion and, consequently, the clinical deficit. Two principal types of acute ischemic injury are recognized:
The general biochemical changes that attend cellular ischemia are discussed in Chapter 1. Here we describe several special responses to ischemia in the CNS.14-16 The metabolic depletion of energy associated with ischemia can result in inappropriate release of excitatory amino acid neurotransmitters such as glutamate, initiating cell damage by allowing excessive influx of calcium ions through NMDA-type glutamate receptors. This elevation of cellular calcium ions can, in turn, trigger a wide range of processes including inappropriate activation of signaling cascades, free radical generation, and mitochondrial injury. All told, these together result in cell death, mostly through necrosis. In the region of transition between necrotic tissue and the normal brain, there is an area of “at-risk” tissue, referred to as the penumbra. This region can be rescued from injury in many animal models with a variety of anti-apoptotic interventions, implying that it may undergo damage by apoptosis as well.
Hypotension, Hypoperfusion, and Low-Flow States (Global Cerebral Ischemia)
The clinical outcome of a severe hypotensive episode that produces global cerebral ischemia (diffuse hypoxic/ischemic encephalopathy) varies with the severity of the insult. In mild cases there may be only a transient post-ischemic confusional state followed by complete recovery and no irreversible tissue damage. However, irreversible damage to CNS tissue may occur in some individuals who suffer mild or transient global ischemic insults. There is a hierarchy of sensitivity among CNS cells: neurons are the most sensitive, although glial cells (oligodendrocytes and astrocytes) are also vulnerable. There is also variability in the susceptibility of populations of neurons in different regions of the CNS (selective vulnerability), based in part on differences in regional cerebral blood flow and cellular metabolic requirements. With severe global cerebral ischemia, widespread neuronal death occurs, irrespective of regional vulnerability. Patients who survive this injury often remain in a persistent vegetative state. Other patients meet the current clinical criteria for “brain death,” including evidence of irreversible diffuse cortical injury (isoelectric, or “flat,” electroencephalogram) and brainstem damage, such as absent reflexes and respiratory drive, and absent cerebral perfusion. When individuals with this pervasive form of injury are maintained on mechanical ventilation, the brain gradually undergoes an autolytic process—so-called “respirator brain.”
Border zone (“watershed”) infarcts occur in the regions of the brain or spinal cord that lie at the most distal reaches of the arterial blood supply, the border zones between arterial territories. In the cerebral hemispheres, the border zone between the anterior and the middle cerebral artery distributions is at greatest risk. Damage to this region produces a sickle-shaped band of necrosis over the cerebral convexity a few centimeters lateral to the interhemispheric fissure. Border zone infarcts are usually seen after hypotensive episodes.
Morphology. In the setting of global ischemia, the brain is swollen, the gyri are widened, and the sulci are narrowed. The cut surface shows poor demarcation between gray and white matter. The microscopic changes of irreversible ischemic injury (infarction) are grouped into three categories. Early changes, occurring 12 to 24 hours after the insult, include acute neuronal changes (red neurons; Figs. 28-13A and Figs. 28-13B) characterized at first by microvacuolization, then eosinophilia of the neuronal cytoplasm, and later nuclear pyknosis and karyorrhexis. Similar acute changes occur somewhat later in astrocytes and oligodendroglia. Pyramidal cells in CA1 of the hippocampus (Sommer sector), Purkinje cells of the cerebellum, and cortical pyramidal neurons are the most susceptible to global ischemia of short duration. After the acute injury, the reaction to tissue damage begins with infiltration by neutrophils (Fig. 28-13C). Subacute changes, occurring at 24 hours to 2 weeks, include necrosis of tissue, influx of macrophages, vascular proliferation, and reactive gliosis (Fig. 28-13D). Repair, robust after approximately 2 weeks, is characterized by eventual removal of all necrotic tissue, loss of normally organized CNS structure, and gliosis (Fig. 28-13E). In the cerebral cortex the neuronal loss and gliosis produce an uneven destruction of the neocortex, with preservation of some layers and involvement of others, a pattern termed pseudolaminar necrosis.
Infarction from Obstruction of Local Blood Supply (Focal Cerebral Ischemia)
Cerebral arterial occlusion may lead to focal ischemia and, if sustained, to infarction of a specific region within the territory of distribution of the compromised vessel. The size, location, and shape of the infarct and the extent of tissue damage that results are determined by modifying factors mentioned earlier, the most important being the adequacy of collateral flow. The major source of collateral flow is the circle of Willis (supplemented by the external carotid-ophthalmic pathway). Partial and inconstant reinforcement is available over the surface of the brain for the distal branches of the anterior, middle, and posterior cerebral arteries through cortical-leptomeningeal anastomoses. In contrast, there is little if any collateral flow for the deep penetrating vessels supplying structures such as the thalamus, basal ganglia, and deep white matter.
Occlusive vascular disease of severity sufficient to lead to cerebral infarction may be due to in situ thrombosis, embolization from a distant source, or various forms of vasculitides; the basic pathology of these conditions is discussed in Chapters 4 and 11.
The majority of thrombotic occlusions are due to atherosclerosis. The most common sites of primary thrombosis causing cerebral infarction are the carotid bifurcation, the origin of the middle cerebral artery, and either end of the basilar artery. The evolution of arterial stenosis varies from progressive narrowing of the lumen and thrombosis, which may be accompanied by anterograde extension, to fragmentation and distal embolization. Another important aspect of occlusive cerebrovascular disease is its frequent association with systemic diseases such as hypertension and diabetes.
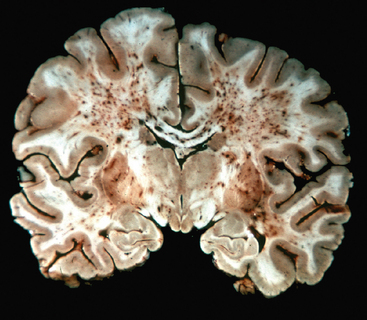
Embolism to the brain occurs from a wide range of origins. Cardiac mural thrombi are among the most common sources; myocardial infarct, valvular disease, and atrial fibrillation are important predisposing factors. Next in importance are thromboemboli arising in arteries, most often originating over atheromatous plaques within the carotid arteries. Other sources of emboli include paradoxical emboli, particularly in children with cardiac anomalies; emboli associated with cardiac surgery; and emboli of other material (tumor, fat, or air). The territory of distribution of the middle cerebral artery—the direct extension of the internal carotid artery—is most frequently affected by embolic infarction; the incidence is about equal in the two hemispheres. Emboli tend to lodge where blood vessels branch or in areas of preexisting luminal stenosis. “Shower embolization,” as in fat embolism, may occur after fractures; affected individuals manifest generalized cerebral dysfunction with disturbances of higher cortical function and consciousness, often without localizing signs. Widespread hemorrhagic lesions involving the white matter are characteristic of embolization of bone marrow after trauma (Fig. 28-14).
A variety of inflammatory processes that involve blood vessels may also lead to luminal narrowing and cerebral infarcts. While infectious vasculitis of small and large vessels was once most commonly associated with syphilis and tuberculosis, it is now more common in the setting of immunosuppression and opportunistic infection (such as aspergillosis or CMV encephalitis). Polyarteritis nodosa and other non-infections vasculitides may involve cerebral vessels and cause single or multiple infarcts throughout the brain. Primary angiitis of the CNS is an inflammatory disorder that involves multiple small- to medium-sized parenchymal and subarachnoid vessels and is characterized by chronic inflammation, multinucleated giant cells, and destruction of the vessel wall. Granulomas may be found in association with the giant cells, leading to the alternative name of granulomatous angiitis of the nervous system. Affected individuals manifest a diffuse encephalopathic or multifocal clinical picture, often with cognitive dysfunction; patients improve with steroid and immunosuppressive treatment. Other conditions that may cause thrombosis and infarction (and intracranial hemorrhage) include hypercoagulable states, dissecting aneurysm of extracranial arteries in the neck supplying the brain, and drug abuse (amphetamines, heroin, cocaine).
Infarcts are subdivided into two broad groups based on the presence of hemorrhage. Hemorrhagic (red) infarction, characterized by multiple, sometimes confluent, petechial hemorrhages, is typically associated with embolic events (Fig. 28-15A). The hemorrhage is presumed to be secondary to reperfusion of damaged vessels and tissue, either through collaterals or directly after dissolution of intravascular occlusive material. In contrast, nonhemorrhagic (pale, bland, anemic) infarcts are usually associated with thrombosis (Fig. 28-15B). The clinical management of patients with these two types of infarcts differs greatly as thrombolytic therapy may be used in cases of thrombosis but is contraindicated in hemorrhagic infarcts. Thrombolytic therapy is beneficial only during a narrow time window after onset of symptoms; therefore, rapid medical attention is essential.
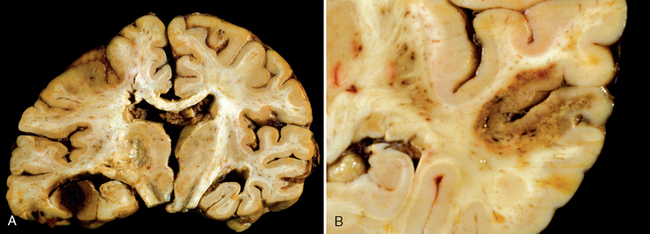
FIGURE 28-15 A, A hemorrhagic infarction is present in the inferior temporal lobe of the left side of this brain. B, A bland infarct with punctate hemorrhages, consistent with ischemia-reperfusion injury, is present in the temporal lobe.
Morphology. The macroscopic appearance of a nonhemorrhagic infarct varies with the time after loss of blood supply. During the first 6 hours of irreversible injury, little can be observed. By 48 hours the tissue becomes pale, soft, and swollen, and the corticomedullary junction becomes indistinct. From 2 to 10 days, the brain becomes gelatinous and friable, and the previously ill-defined boundary between normal and abnormal tissue becomes more distinct as edema resolves in the adjacent tissue that has survived. From 10 days to 3 weeks, the tissue liquefies, eventually leaving a fluid-filled cavity lined by dark gray tissue, which gradually expands as dead tissue is removed (Fig. 28-16).
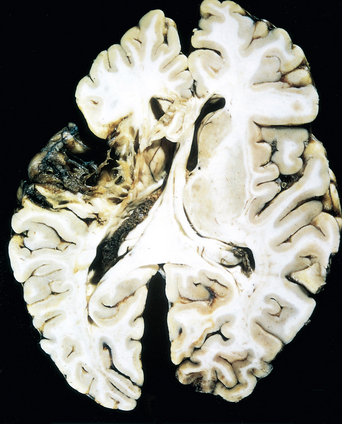
On microscopic examination the tissue reaction evolves along the following sequence: After the first 12 hours, ischemic neuronal change (red neurons; see earlier) and both cytotoxic and vasogenic edema predominate. There is loss of the usual tinctorial characteristics of white- and gray-matter structures. Endothelial and glial cells, mainly astrocytes, swell, and myelinated fibers begin to disintegrate. Up to 48 hours, neutrophilic emigration progressively increases and then falls off. Phagocytic cells, derived from circulating monocytes and activated microglia, are evident at 48 hours and become the predominant celltype in the ensuing 2 to 3 weeks. The macrophages become stuffed with the products of myelin breakdown or blood and may persist in the lesion for months to years. As the process of liquefaction and phagocytosis proceeds, astrocytes at the edges of the lesion progressively enlarge, divide, and develop a prominent network of cytoplasmic extensions. Reactive astrocytes can be seen as early as 1 week after the insult.
After several months, the astrocytic response recedes, leaving behind a dense meshwork of glial fibers admixed with new capillaries and some perivascular connective tissue. In the cerebral cortex, the cavity is separated from the meninges and subarachnoid space by a gliotic layer of tissue, derived from the molecular layer of the cortex. The pia and arachnoid are not affected and do not contribute to the healing process. Infarcts undergo these reactive and reparative stages from the edges inward; thus, different areas of a lesion may look different, particularly during the early stages, revealing the natural progression of the response.
The microscopic picture and evolution of hemorrhagic infarction parallel ischemic infarction, with the addition of blood extravasation and resorption. In individuals receiving anticoagulant treatment, hemorrhagic infarcts may be associated with extensive intracerebral hematomas. Venous infarcts are often hemorrhagic and may occur after thrombotic occlusion of the superior sagittal sinus or other sinuses or occlusion of the deep cerebral veins. Carcinoma, localized infections, and other conditions leading to a hypercoagulable state increase the risk for venous thrombosis.
Spinal cord infarction may be seen in the setting of hypoperfusion or as a consequence of interruption of the feeding tributaries derived from the aorta. Occlusion of the anterior spinal artery is rarer and may occur as a result of embolism or vasculitis.
Clinical Features.
Deficits associated with infarction are determined by the brain region involved rather than the underlying pathologic process. Neurologic symptoms referable to the area of injury often develop rapidly, over minutes, and may continue to evolve over hours. There can be improvement in severity of symptoms associated with reversal of injury in the ischemic penumbra as well as with resolution of associated local edema. In general, there is often a degree of slow improvement during a period of months. Because strokes are frequently associated with atherosclerosis, many of the genetic and lifestyle risk factors are the same as those for atherosclerotic disease.
HYPERTENSIVE CEREBROVASCULAR DISEASE
The most important effects of hypertension on the brain include lacunar infarcts, slit hemorrhages, and hypertensive encephalopathy, as well as massive hypertensive intracerebral hemorrhage. The incidence of these disorders is likely to decline with increased screening for hypertension and aggressive management of blood pressure.
Lacunar Infarcts
Hypertension affects the deep penetrating arteries and arterioles that supply the basal ganglia and hemispheric white matter as well as the brainstem. These cerebral vessels develop arteriolar sclerosis and may become occluded; the structural changes are similar to those described in the systemic vessels of individuals with hypertension (Chapter 11). An important clinical and pathologic consequence of CNS arterial lesions is the development of single or multiple, small, cavitary infarcts known as lacunae (Fig. 28-17). These are lake-like spaces, less than 15 mm wide, which occur in the lenticular nucleus, thalamus, internal capsule, deep white matter, caudate nucleus, and pons, in descending order of frequency. On microscopic examination they consist of areas of tissue loss with scattered lipid-laden macrophages and surrounding gliosis. Depending on their location in the CNS, lacunae can either be clinically silent or cause severe neurologic impairment. Affected vesselsmay also be associated with widening of the perivascular spaces but without tissue infarction (état criblé).

Slit Hemorrhages
Hypertension also gives rise to rupture of the small-caliber penetrating vessels and the development of small hemorrhages. In time these hemorrhages resorb, leaving behind a slitlike cavity (slit hemorrhage) surrounded by brownish discoloration; on microscopic examination, slit hemorrhages show focal tissue destruction, pigment-laden macrophages, and gliosis.
Hypertensive Encephalopathy
Acute hypertensive encephalopathy is a clinicopathologic syndrome arising in an individual with malignant hypertension, and is characterized by diffuse cerebral dysfunction, including headaches, confusion, vomiting, and convulsions, sometimes leading to coma. Rapid therapeutic intervention to reduce the accompanying increased intracranial pressure is required, since the syndrome often does not remit spontaneously. At postmortem examination such individuals may show an edematous brain with or without transtentorial or tonsillar herniation. Petechiae and fibrinoid necrosis of arterioles in the gray and white matter may be seen microscopically.
Individuals who, over the course of many months and years, suffer multiple, bilateral, gray matter (cortex, thalamus, basal ganglia) and white matter (centrum semiovale) infarcts may develop a distinctive clinical syndrome characterized by dementia, gait abnormalities, and pseudobulbar signs, often with superimposed focal neurologic deficits. The syndrome, generally referred to as vascular (multi-infarct) dementia, is caused by multifocal vascular disease of several types, including (1) cerebral atherosclerosis, (2) vessel thrombosis or embolization from carotid vessels or from the heart, and (3) cerebral arteriolar sclerosis from chronic hypertension. When the pattern of injury preferentially involves large areas of the subcortical white matter with myelin and axon loss, the disorder is referred to as Binswanger disease; this distribution of vascular white-matter injury must be distinguished clinically and radiologically from other diseases that affect the hemispheral white matter.
INTRACRANIAL HEMORRHAGE
Hemorrhages may occur at any site within the CNS. In some instances they may be a secondary phenomenon occurring, for example, within infarcts in arterial border zones or in infarcts caused by only partial or transient vascular obstruction. Primary hemorrhages within the epidural or subdural space are typically related to trauma and were discussed earlier with traumatic lesions. Hemorrhages within the brain parenchyma and subarachnoid space, in contrast, are more often a manifestation of underlying cerebrovascular disease, although trauma may also cause hemorrhage in these sites.
Intracerebral (Intraparenchymal) Hemorrhage
Spontaneous (nontraumatic) intraparenchymal hemorrhages occur most commonly in middle to late adult life, with a peak incidence at about age 60 years. Most are caused by rupture of a small intraparenchymal vessel. When the hemorrhages occur in the basal ganglia and thalamus, they are designated ganglionic hemorrhages to distinguish them from those that occur in the lobes of the cerebral hemispheres, which are called lobar hemorrhages. The two major underlying etiologies of this form of cerebrovascular disease are hypertension and cerebral amyloid angiopathy (CAA). In addition, other local and systemic factors may cause or contribute to nontraumatic hemorrhage, including systemic coagulation disorders, neoplasms, vasculitis, aneurysms, and vascular malformations.
Hypertension is the most common underlying cause of primary brain parenchymal hemorrhage, accounting for more than 50% of clinically significant hemorrhages and for roughly 15% of deaths among individuals with chronic hypertension. Hypertension causes a number of abnormalities in vessel walls, including accelerated atherosclerosis in larger arteries; hyaline arteriolosclerosis in smaller vessels; and, in severe cases, proliferative changes and frank necrosis of arterioles. Arteriolar walls affected by hyaline change are presumably weaker than are normal vessels and are therefore more vulnerable to rupture. In some instances chronic hypertension is associated with the development of minute aneurysms, termed Charcot-Bouchard microaneurysms, which may be the site of rupture. Charcot-Bouchard aneurysms, not to be confused with saccular aneurysms of larger intracranial vessels, occur in vessels that are less than 300 μm in diameter, most commonly within the basal ganglia.
Morphology. Hypertensive intraparenchymal hemorrhage may originate in the putamen (50% to 60% of cases), thalamus, pons, cerebellar hemispheres (rarely), and other regions of the brain (Fig. 28-18A). Acute hemorrhages, independent of etiology, are characterized by extravasation of blood with compression of the adjacent parenchyma. Old hemorrhages show an area of cavitary destruction of brain with a rim of brownish discoloration. On microscopic examination the early lesion consists of a central core of clotted blood surrounded by a rim of brain tissue showing anoxic neuronal and glial changes as well as edema. Eventually the edema resolves, pigment- and lipid-laden macrophages appear, and proliferation of reactive astrocytes is seen at the periphery of the lesion. The cellular events then follow the same time course that is observed after cerebral infarction.
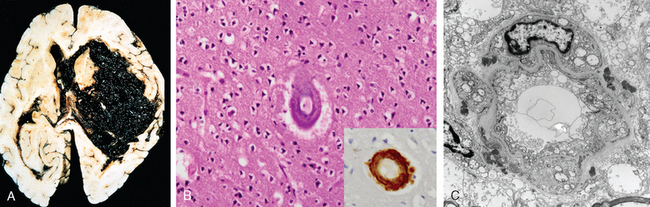
FIGURE 28-18 A, Massive hypertensive hemorrhage rupturing into a lateral ventricle. B, Amyloid deposition in a cortical arteriole in cerebral amyloid angiopathy; inset, Immunohistochemical staining for Aβ shows the deposited material in the vessel wall. C, Electron micrograph shows granular osmophilic material in a case of CADASIL.
CAA is a condition in which amyloidogenic peptides, nearly always the same one found in Alzheimer disease (Aβ40; see the discussion below), deposit in the walls of medium- and small-caliber meningeal and cortical vessels. This deposition can result in weakening of the vessel wall and risk of hemorrhage. As with Alzheimer disease, in which there is a relationship between a polymorphism in the gene that encodes apolipoprotein E (ApoE) and risk of disease, there is an effect of the ApoE genotype on the risk of recurrence of hemorrhage from sporadic CAA. The presence of either an ε2 or ε4 allele increases the risk of repeat bleeding. While some mutations in the precursor protein for the Aβ peptide (amyloid precursor protein, APP) cause familial Alzheimer disease, others result in autosomal dominant forms of CAA.
Morphology. The underlying vascular abnormality of CAA is typically restricted to the leptomeningeal and cerebral cortical arterioles and capillaries, although involvement of the molecular layer of the cerebellum can be observed as well. Involved vessels appear “stiff” on microscopic sections, remaining open with round lumens through tissue processing. Unlike with arteriolar sclerosis, there is no fibrosis; rather, dense and uniform deposits of amyloid are present (Fig. 28-18B).
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) is a rare hereditary form of stroke caused by mutations in the gene encoding the Notch3 receptor.17 The disease is characterized clinically by recurrent strokes (usually infarcts, less often hemorrhages) and dementia. Histopathologic study has shown abnormalities of white matter and leptomeningeal arteries (also involving non-CNS vessels) consisting of concentric thickening of the media and adventitia. Basophilic, PAS-positive deposits, which appear as osmiophilic compact granular material by electron microscopy, have been consistently detected in the walls of affected vessels, as has loss of smooth muscle cells (Fig. 28-18C). The diagnosis can be made through the identification of these deposits in other tissues, such as skin or muscle biopsies, or through genetic approaches. Many of the causative mutations disrupt the normal folding of the extracellular domain of Notch3, and the characteristic deposits appear to be comprised of Notch3 ectodomains. How these deposits relate to the disease is not understood; a toxic gain-of-function mechanism affecting vascular smooth muscle has been proposed.17
Clinical Features.
Intracerebral hemorrhage, independent of cause, can be clinically devastating if it affects large portions of the brain and extends into the ventricular system, or it can affect small regions and either be clinically silent or evolve like an infarct. Over weeks or months there is a gradual resolution of the hematoma, sometimes with considerable clinical improvement. Again, the location of the hemorrhage determines the clinical manifestations.
Subarachnoid Hemorrhage and Ruptured Saccular Aneurysms
The most frequent cause of clinically significant subarachnoid hemorrhage is rupture of a saccular (berry) aneurysm. Subarachnoid hemorrhage may also result from extension of a traumatic hematoma, rupture of a hypertensive intracerebral hemorrhage into the ventricular system, vascular malformation, hematologic disturbances, and tumors.
Saccular aneurysm is the most common type of intracranial aneurysm. Other aneurysm types include atherosclerotic (fusiform; mostly of the basilar artery), mycotic, traumatic, and dissecting. These latter three, like saccular aneurysms, are most often found in the anterior circulation, but differ in that they more often cause cerebral infarction rather than subarachnoid hemorrhage.
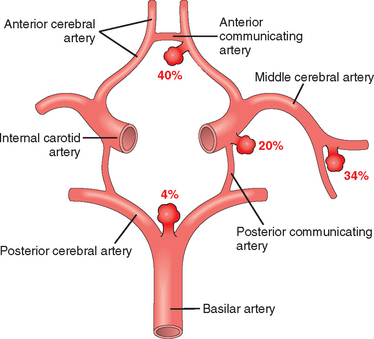
Saccular aneurysms are found in about 2% of the population according to recent data from community-based radiologic studies. About 90% of saccular aneurysms are found near major arterial branch points in the anterior circulation (Fig. 28-19); multiple aneurysms exist in 20% to 30% of cases in autopsy series.
Pathogenesis of Saccular Aneurysms.
The etiology of saccular aneurysms is unknown. Although the majority occur sporadically, genetic factors may be important in their pathogenesis, since there is an increased incidence of aneurysms in first-degree relatives of those affected. There is also an increased incidence in individuals with certain mendelian disorders (such as autosomal dominant polycystic kidney disease, Ehlers-Danlos syndrome type IV, neurofibromatosis type 1 [NF1], and Marfan syndrome), fibromuscular dysplasia of extracranial arteries, and coarctation of the aorta. The predisposing factors include cigarette smoking and hypertension (estimated to be present in about half of these patients). Although they are sometimes referred to as congenital, theaneurysms are not present at birth but develop over time because of an underlying defect in the media of the vessel.
Morphology. An unruptured saccular aneurysm is a thin-walled outpouching, usually at an arterial branch point along the circle of Willis or a major vessel just beyond. Saccular aneurysms measure from a few millimeters to 2 or 3 cm in diameter and have a bright red, shiny surface and a thin, translucent wall (Fig. 28-20). Atheromatous plaques, calcification, or thrombotic occlusion of the sac may be found in the wall or lumen of the aneurysm. Brownish discoloration of the adjacent brain and meninges is evidence of prior hemorrhage. The neck of the aneurysm may be either wide or narrow. Rupture usually occurs at the apex of the sac with extravasation of blood into the subarachnoid space, the substance of the brain, or both. The arterial wall adjacent to the neck of the aneurysm often shows some intimal thickening and gradual attenuation of the media as it approaches the neck. At the neck of the aneurysm, the muscular wall and intimal elastic lamina stop short and are absent from the aneurysm sac itself. The sac is made up of thickened hyalinized intima. The adventitia covering the sac is continuous with that of the parent artery.
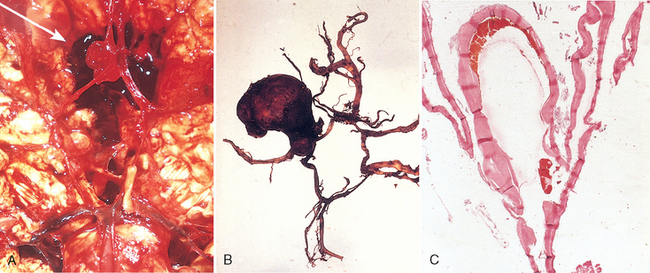
Clinical Features.
Rupture of an aneurysm with clinically significant subarachnoid hemorrhage is most frequent in the fifth decade and is slightly more frequent in females. Overall, the rate of bleeding is roughly 1.3% per year, with the probability of rupture increasing with the size of the lesion. Aneurysms greater than 10 mm in diameter have a roughly 50% risk of bleeding per year. Rupture may occur at any time, but in about one third of cases it is associated with acute increases in intracranial pressure, such as with straining at stool or sexual orgasm. Blood under arterial pressure is forced into the subarachnoid space and affected individuals are stricken with a sudden, excruciating headache (“the worst headache I’ve ever had”), rapidly losing consciousness. Between 25% and 50% of patients die with the first rupture, but patients who survive often improve and recover consciousness in minutes. Repeat bleeding is common in survivors, and it is currently not possible to predict in which patients repeat bleeding will occur. With each episode of bleeding, the prognosis is worse.
The clinical consequences of blood in the subarachnoid space can be separated into acute events, occurring within hours to days after the hemorrhage, and late sequelae associated with the healing process. In the first few days after a subarachnoid hemorrhage, regardless of the etiology, there is an increased risk of additional ischemic injury from vasospasm affecting vessels bathed in the extravasated blood. This problem is of greatest significance in cases of basal subarachnoid hemorrhage, in which vasospasm can involve major vessels of the circle of Willis. Various mediators have been proposed to have a role in this reactive process, including endothelins, nitric oxide, and arachidonic acid metabolites. In the healing phase of subarachnoid hemorrhage, meningeal fibrosis and scarring occur, sometimes leading to obstruction of CSF flow as well as interruption of the normal pathways of CSF resorption.
Vascular Malformations
Vascular malformations of the brain are classified into four principal groups: arteriovenous malformations, cavernous malformations, capillary telangiectasias, and venous angiomas. Of these, the first two are the types associated with risk of hemorrhage and development of neurologic symptoms.
Morphology. Arteriovenous malformations (AVM) involve vessels in the subarachnoid space extending into brain parenchyma or may occur exclusively within the brain. This tangled network of wormlike vascular channels has prominent, pulsatile arteriovenous shunting with high blood flow. They are composed of greatly enlarged blood vessels separated by gliotic tissue, often with evidence of prior hemorrhage. Some vessels can be recognized as arteries with duplication and fragmentation of the internal elastic lamina, while others show marked thickening or partial replacement of the media by hyalinized connective tissue.
Cavernous malformations consist of greatly distended, loosely organized vascular channels with thin, collagenized walls and are devoid of intervening nervous tissue (thus distinguishing them from capillary telangiectasias). They occur most often in the cerebellum, pons, and subcortical regions, in decreasing order of frequency, and have a low flow without arteriovenous shunting. Foci of old hemorrhage, infarction, and calcification frequently surround the abnormal vessels. Capillary telangiectasias are microscopic foci of dilated, thin-walled vascular channels separated by relatively normal brain parenchyma and occurring most frequently in the pons. Venous angiomas (varices) consist of aggregates of ectatic venous channels. Foix-Alajouanine disease (angiodysgenetic necrotizing myelopathy) is a venous angiomatous malformation of the spinal cord and overlying meninges, most often in the lumbosacral region, associated with ischemic myelomalacia and slowly progressive neurologic symptoms.
Clinical Features.
Arteriovenous malformations are the most common type of clinically significant vascular malfor mation. Males are affected twice as frequently as females, and the lesion is often recognized clinically between the ages of 10 and 30 years, presenting as a seizure disorder, an intracerebral hemorrhage, or a subarachnoid hemorrhage. The most common site is the territory of the middle cerebral artery, particularly its posterior branches. Large arteriovenous malformations occurring in the newborn period can lead to congestive heart failure because of shunt effects, especially if the malformation involves the vein of Galen. Cavernous malformations are unique among this class of lesion in that familial forms are relatively common, with a variety of identified genetic loci.18 Multiplicity of lesions is an additional hallmark of these autosomal dominant disorders with high penetrance.
Infections
With infection, damage to nervous tissue may be the consequence of direct injury of neurons or glia by the infectious agent or may occur indirectly through the elaboration of microbial toxins, destructive effects of the inflammatory response, or the result of immune-mediated mechanisms. There are four principal routes by which infectious microbes enter the nervous system. Hematogenous spread is the most common means of entry; infectious agents ordinarily enter through the arterial circulation, but retrograde venous spread can occur through anastomoses with veins of the face. Direct implantation of microorganisms is almost invariably traumatic or is associated with congenital malformations (such as meningomyelocele). Local extension can come from any of several adjacent structures (air sinuses, an infected tooth, cranial or spinal osteomyelitis). Transport along the peripheral nervous system occurs with certain viruses, such as rabies and herpes zoster. General aspects of the pathology of infectious agents are discussed in Chapter 8; here we focus on some of the distinctive forms of CNS infections.
ACUTE MENINGITIS
Meningitis refers to an inflammatory process of the leptomeninges and CSF within the subarachnoid space, while meningoencephalitis combines this with inflammation of the brain parenchyma. Meningitis is usually caused by an infection, but may also occur in response to a nonbacterial irritant introduced into the subarachnoid space (chemical meningitis). Infectious meningitis is broadly classified into acute pyogenic (usually bacterial meningitis), aseptic (usually acute viral meningitis), and chronic (usually tuberculous, spirochetal, or cryptococcal) on the basis of the characteristics of inflammatory exudate on CSF examination and the clinical evolution of the illness.
Acute Pyogenic (Bacterial) Meningitis
The microorganisms that cause acute pyogenic meningitis vary with the age of the affected individual. In neonates, they include Escherichia coli and the group B streptococci; at the other extreme of life, Streptococcus pneumoniae and Listeria monocytogenes are more common. Among adolescents and in young adults, Neisseria meningitidis is the most common pathogen, with clusters of cases causing frequent public health concerns. The introduction of immunization against Haemophilus influenzae has markedly reduced the incidence of meningitis associated with this organism in the developed world; the population that was previously at highest risk (infants) now has a much lower overall risk of meningitis, with S. pneumoniae being the most prevalent organism.
Affected individuals typically show systemic signs of infection superimposed on clinical evidence of meningeal irritation and neurologic impairment, including headache, photophobia, irritability, clouding of consciousness, and neck stiffness. A spinal tap yields cloudy or frankly purulent CSF, under increased pressure, with as many as 90,000 neutrophils per cubic millimeter, an increased protein concentration, and a markedly reduced glucose content. Bacteria may be seen on a smear or can be cultured, sometimes a few hours before the neutrophils appear. Untreated pyogenic meningitis can be fatal, while effective antimicrobial agents markedly reduce mortality. The Waterhouse-Friderichsen syndrome results from meningitis-associated septicemia with hemorrhagic infarction of the adrenal glands and cutaneous petechiae (Chapter 24). It occurs most often with meningococcal and pneumococcal meningitis. In the immunosuppressed individual, purulent meningitis may be caused by other agents, such as Klebsiella or an anaerobic organism, and may have an atypical course and uncharacteristic CSF findings, all of which make the diagnosis more difficult.
Morphology. The normally clear CSF is cloudy and sometimes frankly purulent. In acute meningitis, an exudate is evident within the leptomeninges over the surface of the brain (Fig. 28-21). The meningeal vessels are engorged and stand out prominently. The location of the exudate varies; in H. influenzae meningitis, for example, it is usually basal, whereas in pneumococcal meningitis it is often densest over the cerebral convexities near the sagittal sinus. From the areas of greatest accumulation, tracts of pus can be followed along blood vessels on the surface of the brain. When the meningitis is fulminant, the inflammation may extend to the ventricles, producing ventriculitis.
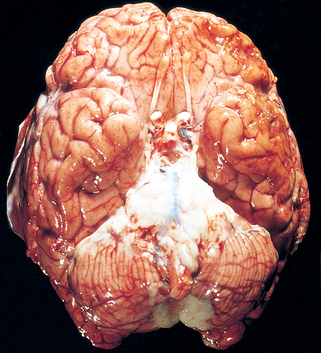
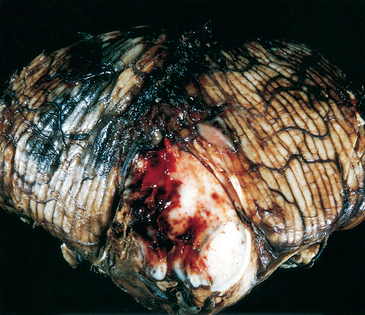
FIGURE 28-21 Pyogenic meningitis. A thick layer of suppurative exudate covers the brainstem and cerebellum and thickens the leptomeninges.
(From Golden JA, Louis DN: Images in clinical medicine: acute bacterial meningitis. N Engl J Med 333:364, 1994.)
On microscopic examination, neutrophils fill the subarachnoid space in severely affected areas and are found predominantly around the leptomeningeal blood vessels in less severe cases. In untreated meningitis, Gram stain reveals varying numbers of the causative organism, although they are frequently not demonstrable in treated cases. In fulminant meningitis, the inflammatory cells infiltrate the walls of the leptomeningeal veins and may extend into the substance of the brain (focal cerebritis). Phlebitis may also lead to venous thrombosis and hemorrhagic infarction of the underlying brain.
Leptomeningeal fibrosis may follow pyogenic meningitis and cause hydrocephalus. In some infections, particularly in pneumococcal meningitis, large quantities of the capsular polysaccharide of the organism produce a particularly gelatinous exudate that encourages arachnoid fibrosis, called chronic adhesive arachnoiditis.
Acute Aseptic (Viral) Meningitis
Aseptic meningitis is a clinical term referring to the absence of recognizable organisms in a patient with meningeal irritation, fever, and alterations of consciousness of relatively acute onset. The name is a misnomer, as the disease is generally of viral, and rarely of bacterial or other, etiology. The clinical course is less fulminant than that of pyogenic meningitis, and the CSF findings also differ; in aseptic meningitis there is a lymphocytic pleocytosis, the protein elevation is only moderate, and the glucose content is nearly always normal. The viral aseptic meningitides are usually self-limiting and are treated symptomatically. Remarkably, even with molecular methods for detection of pathogens, the etiologic agent is identified in only a minority of cases.19 The spectrum of pathogens varies seasonally and geographically. An aseptic meningitis–like picture may also develop subsequent to rupture of an epidermoid cyst into the subarachnoid space or the introduction of a chemical irritant (“chemical” meningitis). In these cases the CSF is sterile, there is pleocytosis with neutrophils and an increased protein concentration, but the sugar content is usually normal.
ACUTE FOCAL SUPPURATIVE INFECTIONS
Brain Abscess
Brain abscesses may arise by direct implantation of organisms, local extension from adjacent foci (mastoiditis, paranasal sinusitis), or hematogenous spread (usually from a primary site in the heart, lungs, or distal bones or after tooth extraction). Predisposing conditions include acute bacterial endocarditis, which tends to produce multiple abscesses; congenital heart disease with right-to-left shunting and loss of pulmonary filtration of organisms; chronic pulmonary sepsis, as can be seen with bronchiectasis; and immunosuppression. Streptococci and staphylococci are the most common offending organisms identified in nonimmunosuppressed populations.
Morphology. Grossly, abscesses are discrete lesions with central liquefactive necrosis surrounded by fibrosis and swelling (Fig. 28-22). On microscopic examination there is exuberant granulation tissue with neovascularization around the necrosis that is responsible for marked vasogenic edema. A collagenous capsule is produced by fibroblasts derived from the walls of blood vessels. Outside the fibrous capsule is a zone of reactive gliosis with numerous gemistocytic astrocytes.
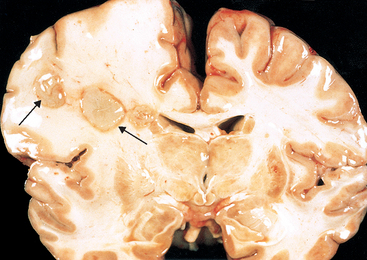
Cerebral abscesses are destructive lesions and patients almost invariably present clinically with progressive focal deficits in addition to the general signs of raised intracranial pressure. The CSF is under increased pressure, the white cell count is raised, and protein concentration is increased, but the glucose content is normal. The source of infection may be apparent, or it may be a small systemic focus that is not symptomatic. The increased intracranial pressure and progressive herniation can be fatal, and abscess rupture can lead to ventriculitis, meningitis, and venous sinus thrombosis. With surgery and antibiotics, the otherwise high mortality rate can be reduced to less than 10%.
Subdural Empyema
Bacterial or occasionally fungal infection of the skull bones or air sinuses can spread to the subdural space, producing a subdural empyema. The underlying arachnoid and subarachnoid spaces are usually unaffected, but a large subdural empyema may produce a mass effect. Further, a thrombophlebitis may develop in the bridging veins that cross the subdural space, resulting in venous occlusion and infarction of the brain. With treatment, including surgical drainage, resolution of the empyema occurs from the dural side, and, if it is comp lete, a thickened dura may be the only residual finding. Symptoms include those referable to the source of the infection. In addition, most patients are febrile, with headache and neck stiffness, and, if untreated, may develop focal neurologic signs, lethargy, and coma. The CSF profile is similar to that seen in brain abscesses, because both are parameningeal infectious processes. If diagnosis and treatment are prompt, complete recovery is usual.
Extradural Abscess
Extradural abscess, commonly associated with osteomyelitis, often arises from an adjacent focus of infection, such as sinusitis or a surgical procedure. When the process occurs in the spinal epidural space, it may cause spinal cord compression and constitute a neurosurgical emergency.
CHRONIC BACTERIAL MENINGOENCEPHALITIS
Chronic bacterial infection of the meninges and the brain may be caused by M. tuberculosis, T. pallidum, and Borrelia species. Each of these is briefly described next.
Tuberculosis
Tuberculosis of the brain may be part of systemic disease or apparently isolated, the brain having been seeded from a silent, usually pulmonary, lesion. It may involve the meninges or the brain, often together.
Morphology. On macroscopic examination, the subarachnoid space contains a gelatinous or fibrinous exudate, most often at the base of the brain, obliterating the cisterns and encasing cranial nerves. There may be discrete, white granules scattered over the leptomeninges. The most common pattern of involvement is a diffuse meningoencephalitis. On microscopic examination, there are mixtures of lymphocytes, plasma cells, and macrophages. Florid cases show well-formed granulomas, often with caseous necrosis and giant cells. Arteries running through the subarachnoid space may show obliterative endarteritis with inflammatory infiltrates in their walls and marked intimal thickening. Organisms can often be seen with acid-fast stains. The infectious process may spread to the choroid plexus and ependymal surface, traveling through the CSF. In cases of long-standing duration, a dense, fibrous adhesive arachnoiditis may develop, most conspicuous around the base of the brain. Hydrocephalus may result.
Another manifestation of the disease is the development of a single (or often multiple) wellcircumscribed intraparenchymal mass (tuberculoma), which may be associated with meningitis. A tuberculoma may be as large as several centimeters in diameter, causing significant mass effect. On microscopic examination, there is usually a central core of caseous necrosis surrounded by a typical tuberculous granulomatous reaction; calcification may occur in inactive lesions.
Clinical Features.
Patients with tuberculous meningitis usually have symptoms of headache, malaise, mental confusion, and vomiting. There is only a moderate CSF pleocytosis made up of mononuclear cells or a mixture of polymorphonuclear and mononuclear cells; the protein concentration is elevated, often strikingly so; and the glucose content typically is moderately reduced or normal. The most serious complications of chronic tuberculous meningitis are arachnoid fibrosis producing hydrocephalus, and obliterative endarteritis producing arterial occlusion and infarction of underlying brain. When the process involves the spinal cord subarachnoid space, nerve roots may also be affected. With tuberculomas, the symptoms are typical of space-occupying lesions of the brain, and have to be distinguished from CNS tumors.
Infection by Mycobacterium tuberculosis in individuals with acquired immunodeficiency syndrome (AIDS) is often similar to that in individuals not suffering from AIDS, but there may be less host reaction. HIV-positive individuals are also at risk for infection by M. avium-intracellulare, usually in the setting of disseminated infection. These lesions typically contain confluent sheets of macrophages filled with organisms, with little or no associated granulomatous reaction.
Neurosyphilis
Neurosyphilis is a manifestation of the tertiary stage of syphilis and occurs in only about 10% of individuals with untreated infection. The major patterns of CNS involvement are meningovascular neurosyphilis, paretic neurosyphilis, and tabes dorsalis; affected individuals often show incomplete or mixed pictures, most commonly the combination of tabes dorsalis and paretic disease (taboparesis). Individuals infected with HIV are at increased risk for neurosyphilis, particularly as an acute syphilitic meningitis or meningovascular disease, because of impaired cell-mediated immunity. The rate of progression and severity of the disease seem to be accelerated, possibly for the same reason.
Morphology. Meningovascular neurosyphilis is a chronic meningitis involving the base of the brain and more variably the cerebral convexities and the spinal leptomeninges. In addition, there may be an associated obliterative endarteritis (Heubner arteritis) accompanied by a distinctive perivascular inflammatory reaction rich in plasma cells and lymphocytes. Cerebral gummas (plasma cell–rich mass lesions) may also occur in the meninges and extend into the parenchyma.
Paretic neurosyphilis is caused by invasion of the brain by Treponema pallidum and is clinically manifested as insidious but progressive mental deficits associated with mood alterations (including delusions of grandeur) that terminate in severe dementia (general paresis of the insane). On microscopic examination, inflammatory lesions are associated with parenchymal damage in the cerebral cortex (particularly the frontal lobe but also affecting other areas of the isocortex) characterized by loss of neurons, proliferations of microglia (rod cells), gliosis, and iron deposits. The latter are demonstrable with the Prussian blue stain perivascularly and in the neuropil, and are presumably the sequelae of small bleeds stemming from damage to the microcirculation. The spirochetes can, at times, be demonstrated in tissue sections. There is often an associated hydrocephalus with damage to the ependymal lining and proliferation of subependymal glia, called granular ependymitis.
Tabes dorsalis is the result of damage by the spirochetes to the sensory nerves in the dorsal roots, which produces impaired joint position sense and resultant ataxia (locomotor ataxia); loss of pain sensation, leading to skin and joint damage (Charcot joints); other sensory disturbances, particularly the characteristic “lightning pains”; and absence of deep tendon reflexes. On microscopic examination there is loss of both axons and myelin in the dorsal roots, with corresponding pallor and atrophy in the dorsal columns of the spinal cord. Organisms are not demonstrable in the cord lesions.
Neuroborreliosis (Lyme Disease)
Lyme disease is caused by the spirochete Borrelia burgdorferi, transmitted by various species of Ixodes tick; involvement of the nervous system is referred to as neuroborreliosis. Neurologic symptoms are highly variable and include aseptic meningitis, facial nerve palsies and other polyneuropathies, as well as encephalopathy. The rare cases that have come to autopsy have shown a focal proliferation of microglial cells in the brain as well as scattered extracellular organisms (identified by Dieterle stain).
VIRAL MENINGOENCEPHALITIS
Viral encephalitis is a parenchymal infection of the brain almost invariably associated with meningeal inflammation (meningoencephalitis) and sometimes with simultaneous involvement of the spinal cord (encephalomyelitis).
Some viruses tend to infect the nervous system. Such neural tropism takes several forms: some viruses infect specific cell types (such as oligodendrocytes), while others preferentially involve particular areas of the brain (such as medial temporal lobes or the limbic system). Latency is an important facet of several viral infections of the CNS (e.g., herpes zoster, progressive multifocal leukoencephalopathy). Systemic viral infections in the absence of direct evidence of viral penetration into the CNS may be followed by an immune-mediated disease, such as perivenous demyelination (see “Acute Disseminated Encephalomyelitis and Acute Necrotizing Hemorrhagic Encephalomyelitis”). Intrauterine viral infection may cause congenital malformations, as occurs with rubella. A slowly progressive degenerative disease syndrome may follow many years after a viral illness; an example is post-encephalitic parkinsonism after the 1918 viral influenza pandemic.
Arthropod-Borne Viral Encephalitis
Arboviruses are an important cause of epidemic encephalitis, especially in tropical regions of the world, and they are capable of causing serious morbidity and high mortality. In the Western hemisphere the most important types are Eastern and Western equine, West Nile, Venezuelan, St. Louis, and La Crosse; elsewhere in the world, pathogenic arboviruses include Japanese B (Far East), Murray Valley (Australia and New Guinea), and tick-borne (Russia and Eastern Europe). All have animal hosts and mosquito vectors, except for the tick-borne type. Clinically, affected individuals develop generalized neurologic deficits, such as seizures, confusion, delirium, and stupor or coma, and often focal signs, such as reflex asymmetry and ocular palsies. Involvement of the spinal cord in West Nile encephalitis can lead to a polio-like syndrome with paralysis. The CSF is usually colorless but with a slightly elevated pressure and, initially, a neutrophilic pleocytosis that rapidly converts to lymphocytes; the protein concentration is elevated, but glucose content is normal.
Morphology. The encephalitides caused by various arboviruses differ in epidemiology and prognosis, but the histopathologic picture is similar, except for variations in the severity and extent of the lesions within the CNS. Characteristically, there is a lymphocytic meningoencephalitis (sometimes with neutrophils), and a tendency for inflammatory cells to accumulate perivascularly (Fig. 28-23A). Multiple foci of necrosis of gray and white matter are found; in particular, there is evidence of single-cell neuronal necrosis with phagocytosis of the debris (neuronophagia). Microglial cells form small aggregates around foci of necrosis, called microglial nodules (Fig. 28-23B). In severe cases there may be a necrotizing vasculitis with associated focal hemorrhages. While some viruses reveal their presence by inclusion bodies, the identification of viral pathogens is most often by a combination of ultrastructural, immunohistochemical, and molecular methods.19
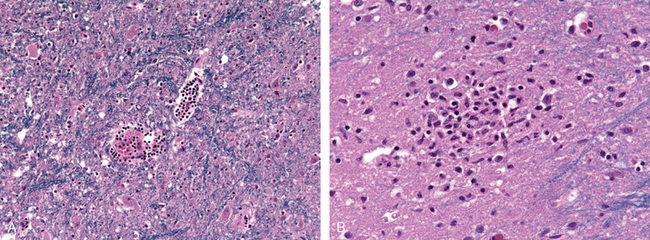
Herpes Simplex Virus Type 1
Herpes simplex virus type 1 (HSV-1) encephalitis is most common in children and young adults. Only about 10% of the affected individuals have a history of prior herpes. The most commonly observed clinical presenting symptoms in herpes encephalitis are alterations in mood, memory, and behavior. Polymerase chain reaction (PCR)–based methods for virus detection in CSF samples have increased the ease of diagnosis and the recognition of a subset of patients with less severe disease. Antiviral agents now provide effective treatment in many cases, with a significant reduction in the mortality rate. In some individuals, HSV-1 encephalitis follows a subacute course with clinical manifestations (weakness, lethargy, ataxia, seizures) that evolve during a more protracted period (4 to 6 weeks).
Morphology. This encephalitis starts in, and most severely involves, the inferior and medial regions of the temporal lobes and the orbital gyri of the frontal lobes (Fig. 28-24). The infection is necrotizing and often hemorrhagic in the most severely affected regions. Perivascular inflammatory infiltrates are usually present, and Cowdry type A intranuclear viral inclusion bodies may be found in both neurons and glia. In individuals with slowly evolving HSV-1 encephalitis, there is more diffuse involvement of the brain.
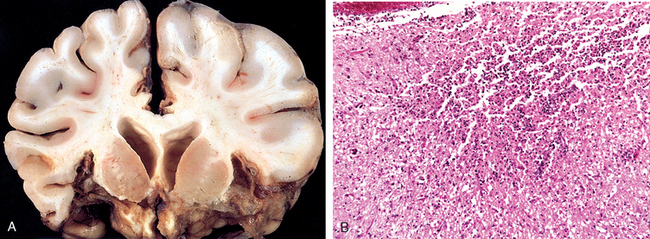
Herpes Simplex Virus Type 2
Herpes simplex virus type 2 (HSV-2) also infects the nervous system; in adults it causes meningitis, but as many as 50% of neonates born by vaginal delivery to women with active primary HSV genital infections acquire the infection during passage through the birth canal and develop severe encephalitis. In the face of active HIV infection, HSV-2 may cause an acute, hemorrhagic, necrotizing encephalitis.
Varicella-Zoster Virus (Herpes Zoster)
Primary varicella infection presents as one of the childhood exanthems (chickenpox), ordinarily without any evidence of neurologic involvement. Following the cutaneous infection, the virus enters a latent phase within sensory neurons of the dorsal root or trigeminal ganglia. Reactivation of infection in adults (“shingles”) usually manifests as a painful, vesicular skin eruption in a single or limited dermatomal distribution. Herpes zoster reactivation is usually a self-limited process, but there may be a persistent postherpetic neuralgia syndrome particularly after age 60, including both persistent pain as well as painful sensation following nonpainful stimuli.
Overt CNS involvement with herpes zoster is much rarer but can be severe. Herpes zoster has been associated with a granulomatous arteritis; immunocytochemical and electron microscopic evidence of viral involvement has been obtained in a few of these cases. In immunosuppressed individuals, herpes zoster may cause acute encephalitis with numerous sharply circumscribed lesions characterized by demyelination followed by necrosis.
Cytomegalovirus
This infection of the nervous system occurs in fetuses and immunosuppressed individuals. The outcome of infection in utero is periventricular necrosis that produces severe brain destruction followed later by microcephaly and periventricular calcification. CMV is a common opportunistic viral pathogen in individuals with AIDS, with CNS involvement being common in this setting.
Morphology. In the immunosuppressed individual, the most common pattern of involvement is that of a subacute encephalitis, which may be associated with CMV inclusion-bearing cells (see Fig. 8-15). Although any type of cell within the CNS (neurons, glia, ependyma, endothelium) can be infected by CMV, there is a tendency for the virus to localize in the paraventricular subependymal regions of the brain. This results in a severe hemorrhagic necrotizing ventriculoencephalitis and a choroid plexitis. The virus can also attack the lower spinal cord and roots, producing a painful radiculoneuritis. Prominent cytomegalic cells with intranuclear and intracytoplasmic inclusions can be readily identified by conventional light microscopy and confirmed as CMV by immunohistochemistry.
Poliomyelitis
While paralytic poliomyelitis has been effectively eradicated by vaccination in many parts of the world, there are still regions where it remains a problem. In nonimmunized individuals poliovirus infection causes a subclinical or mild gastroenteritis, similar to that caused by other members of the picorna group of enteroviruses. In a small fraction of the vulnerable population, however, it secondarily invades the nervous system.
Morphology. Acute cases show mononuclear cell perivascular cuffs and neuronophagia of the anterior-horn motor neurons of the spinal cord. The inflammatory reaction is usually confined to the anterior horns but may extend into the posterior horns, and the damage is occasionally severe enough to produce cavitation. In situ reverse transcriptase–PCR has shown poliovirus RNA in anterior-horn cell motor neurons. The cranial motor nuclei are sometimes involved. Postmortem examination in long-term survivors of symptomatic poliomyelitis shows loss of neurons and gliosis in the affected anterior horns of the spinal cord, some residual inflammation, atrophy of the anterior (motor) spinal roots, and neurogenic atrophy of denervated muscle.
Clinical Features.
CNS infection manifests initially with meningeal irritation and a CSF picture of aseptic meningitis. The disease may progress no further or advance to involve the spinal cord. When the disease affects the motor neurons of the spinal cord, it produces a flaccid paralysis with muscle wasting and hyporeflexia in the corresponding region of the body—the permanent neurologic residue of poliomyelitis. In the acute disease, death can occur from paralysis of the respiratory muscles, and a myocarditis sometimes complicates the clinical course. Because of the destruction of motor neurons, paresis or paralysis follows; when it involves the innervation of the diaphragm and intercostal muscles, severe respiratory compromise may occur and cause long-term morbidity. Post-polio syndrome can develop in patients 25 to 35 years after the resolution of the initial illness. It is characterized by progressive weakness associated with decreased muscle mass and pain, and has an unclear pathogenesis.
Rabies
Rabies is a severe encephalitis transmitted to humans by the bite of a rabid animal, usually a dog or various wild mammals that form natural reservoirs. Exposure to certain species of bats, even without a known bite, can also lead to rabies.
Morphology. On macroscopic examination the brain shows intense edema and vascular congestion. On microscopic examination there is widespread neuronal degeneration and an inflammatory reaction that is most severe in the brainstem. The basal ganglia, spinal cord, and dorsal root ganglia may also be involved. Negri bodies, the pathognomonic microscopic finding, are cytoplasmic, round to oval, eosinophilic inclusions that can be found in pyramidal neurons of the hippocampus and Purkinje cells of the cerebellum, sites usually devoid of inflammation (Fig. 28-25). The presence of rabies virus can be detected within Negri bodies by ultrastructural and immunohistochemical examination.
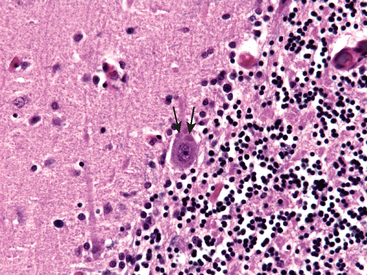
Clinical Features.
Since the virus enters the CNS by ascending along the peripheral nerves from the wound site, the incuba tion period (commonly between 1 and 3 months) depends on the distance between the wound and the brain. The disease begins with nonspecific symptoms of malaise, headache, and fever, but the conjunction of these symptoms with local paresthesias around the wound is diagnostic. As the infection advances, the affected individual exhibits extraordinary CNS excitability; the slightest touch is painful, with violent motor responses progressing to convulsions. Contracture of the pharyngeal musculature on swallowing produces foaming at the mouth, which may create an aversion to swallowing even water (hydrophobia). There is meningismus and, as the disease progresses, flaccid paralysis. Periods of alternating mania and stupor progress to coma and death from respiratory center failure.
Human Immunodeficiency Virus
In the period before the availability of effective anti-retroviral therapy, neuropathologic changes were demonstrated at postmortem examination in as many as 80% to 90% of cases of AIDS. These included direct effects of virus on the nervous system, opportunistic infections, and primary CNS lymphoma. Since the early days, there has been a decrease in the frequency of these secondary effects of HIV infection, in those who receive intensive multidrug anti-retroviral therapy.20,21
HIV aseptic meningitis occurs within 1 to 2 weeks of seroconversion in about 10% of patients; antibodies to HIV can be demonstrated and the virus can be isolated from the CSF. The few neuropathologic studies of the early and acute phases of symptomatic or asymptomatic HIV invasion of the nervous system have shown a mild lymphocytic meningitis, perivascular inflammation, and some myelin loss in the hemispheres. Among the cell types of the CNS, only microglia have the appropriate combination of CD4 and a chemokine receptor (CCR5 or CXCR4) to allow for efficient infection by HIV.22 During the chronic phase, an HIV encephalitis is commonly found when symptomatic individuals come to autopsy.
Morphology. HIV encephalitis is best characterized microscopically as a chronic inflammatory reaction with widely distributed infiltrates of microglial nodules, sometimes with associated foci of tissue necrosis and reactive gliosis (Fig. 28-26). The microglial nodules are also found in the vicinity of small blood vessels, which show abnormally prominent endothelial cells and perivascular foamy or pigment-laden macrophages. These changes occur especially in the subcortical white matter, diencephalon, and brainstem. An important component of the microglial nodule is the macrophage-derived multinucleated giant cell. In some cases there is also a disorder of white matter characterized by multifocal or diffuse areas of myelin pallor, axonal swelling and gliosis. HIV can be detected in CD4+ mononuclear and multinucleated macrophages and microglia by immunoperoxidase and molecular methods.
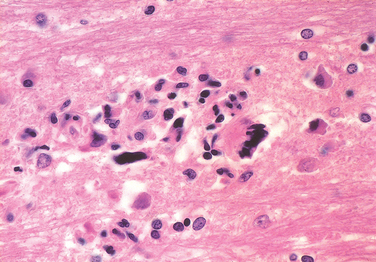
Cognitive changes, both mild and florid enough to be termed HIV-associated dementia, appear to have persisted into the era of effective anti-HIV treatment regimens. Rather than having a specific pathologic lesion as its correlate, this disorder is most tightly related to the extent of activated microglia in the brain, not all of which are necessarily HIV-infected. A wide range of possible mechanisms for neuronal dysfunction and injury in this setting have been proposed, including actions of cytokines and activation of an inflammatory cascade as well as a cavalcade of toxic effects of HIV-derived proteins; in all probability, each of these pathways has a contributory role in the pathogenesis of neural injury (Chapter 6).
Progressive Multifocal Leukoencephalopathy
Progressive multifocal leukoencephalopathy (PML) is a viral encephalitis caused by the JC polyomavirus; because the virus preferentially infects oligodendrocytes, demyelination is its principal pathologic effect. The disease occurs almost exclusively in immunosuppressed individuals in various clinical settings, including chronic lymphoproliferative or myeloproliferative illnesses, immunosuppressive chemotherapy including monoclonal antibody therapy targeting integrins, granulomatous diseases, and HIV/AIDS.
Although most people have serologic evidence of exposure to JC virus by the age of 14 years, no clinical disease has been associated with primary infection by the virus. It is thought that PML results from the reactivation of virus in the setting of immunosuppression. Clinically, affected individuals develop focal and relentlessly progressive neurologic symptoms and signs, and imaging studies show extensive, often multifocal, lesions in the hemispheric or cerebellar white matter.
Morphology. The lesions consist of patches of irregular, ill-defined destruction of the white matter ranging in size from millimeters to extensive involvement of an entire lobe of the brain (Fig. 28-27). On microscopic examination the typical lesion consists of a patch of demyelination, most often in a subcortical location, in the center of which are scattered lipid-laden macrophages and a reduced number of axons. At the edge of the lesion are greatly enlarged oligodendrocyte nuclei with glassy amphophilic viral inclusions (Fig. 28-27, inset), which contain viral antigens by immunohistochemistry. Within the lesions, there may be bizarre giant astrocytes with one to several irregular, hyperchromatic nuclei mixed with more typical reactive astrocytes.
Subacute Sclerosing Panencephalitis
Subacute sclerosing panencephalitis (SSPE) is a rare progressive clinical syndrome characterized by cognitive decline, spasticity of limbs, and seizures. It occurs in children or young adults, months or years after an initial, early-age acute infection with measles. The disease represents persistent, but nonproductive, infection of the CNS by an altered measles virus; changes in several viral genes have been associated with the disease. On microscopic examination, there are widespread gliosis and myelin degeneration; viral inclusions, largely within the nuclei, of oligodendrocytes and neurons; variable inflammation of white and gray matter; and neurofibrillary tangles. Ultrastructural study shows that the inclusions contain nucleocapsids characteristic of measles; immunohistochemistry for measles virus antigen is positive. The disease has largely disappeared with the spread of vaccination programs; however, there are still cases being reported from nonimmunized populations.
FUNGAL MENINGOENCEPHALITIS
Fungal disease of the CNS is encountered primarily in immunocompromised individuals. The brain is usually involved when there is widespread hematogenous dissemination of the fungus, most often Candida albicans, Mucor species, Aspergillus fumigatus, and Cryptococcus neoformans. In endemic areas, pathogens such as Histoplasma capsulatum, Coccidioides immitis, and Blastomyces dermatitidis may involve the CNS after a primary pulmonary or cutaneous infection; again, this often follows immunosuppression.
There are three main patterns of fungal infection in the CNS: chronic meningitis, vasculitis, and parenchymal invasion. Vasculitis is most frequently seen with mucormycosis and aspergillosis, both of which are characterized by direct fungal invasion of blood vessel walls, but it occasionally occurs with other infections such as candidiasis. The resultant vascular thrombosis produces infarction that is often strikingly hemorrhagic and that subsequently becomes septic from ingrowth of the causative fungus.
Parenchymal invasion, usually in the form of granulomas or abscesses, can occur with most of the fungi and often coexists with meningitis. The most commonly encountered fungi invading the brain are Candida and Cryptococcus. Candidiasis usually produces multiple microabscesses, with or without granuloma formation. Although most fungi invade the brain by hematogenous dissemination, direct extension may also occur, particularly in mucormycosis, most commonly in diabetics with ketoacidosis.
Cryptococcal meningitis, a common opportunistic infection in the setting of HIV/AIDS, may be fulminant and fatal in as little as 2 weeks or indolent, evolving over months or years. The CSF may contain few cells but usually has a high concentration of protein. The mucoid-encapsulated yeasts can be visualized in the CSF by India ink preparations and in tissue sections by PAS, mucicarmine, and silver stains.
Morphology. With cryptococcal infection, the brain shows a chronic meningitis affecting the basal leptomeninges, which are opaque and thickened by reactive connective tissue that may obstruct the outflow of CSF from the foramina of Luschka and Magendie, giving rise to hydrocephalus. Sections of the brain disclose a gelatinous material within the subarachnoid space and small cysts within the parenchyma (“soap bubbles”), which are especially prominent in the basal ganglia in the distribution of the lenticulostriate arteries (Fig. 28-28A). Parenchymal lesions consist of aggregates of organisms within expanded perivascular (Virchow-Robin) spaces associated with minimal or absent inflammation or gliosis (Fig. 28-28B). The meningeal infiltrates consist of chronic inflammatory cells and fibroblasts admixed with cryptococci.
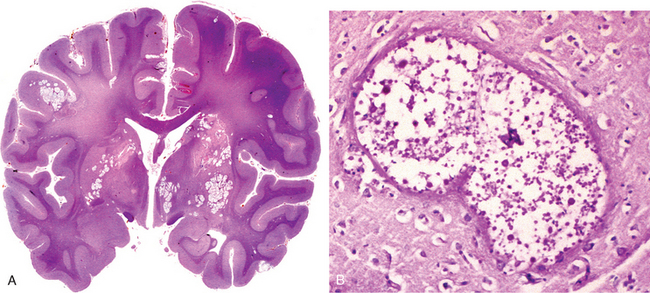
OTHER INFECTIOUS DISEASES OF THE NERVOUS SYSTEM
Protozoal diseases (including malaria, toxoplasmosis, amebiasis, and trypanosomiasis), rickettsial infections (such as typhus and Rocky Mountain spotted fever), and metazoal diseases (especially cysticercosis and echinococcosis) may also involve the CNS and are discussed in Chapter 8.
Cerebral toxoplasmosis is another of the opportunistic infections commonly found in the setting of HIV-associated immunosuppression. The clinical symptoms of infection of the brain by Toxoplasma gondii are subacute, evolving during a 1- or 2-week period, and may be both focal and diffuse. Computed tomography and magnetic resonance imaging studies may show multiple ring-enhancing lesions; however, this radiographic appearance is not pathognomonic, since similar findings may be associated with CNS lymphoma, tuberculosis, and fungal infections. In nonimmunosuppressed hosts, the impact of toxoplasmosis on the brain is most often seen when primary maternal infection occurs early in the pregnancy. It may be followed by a cerebritis in the fetus, with the production of multifocal cerebral necrotizing lesions that may calcify, producing severe damage to the developing brain.
Morphology. Toxoplasmosis of the CNS produces brain abscesses, which are found most often in the cerebral cortex (near the gray-white junction) and deep gray nuclei, less often in the cerebellum and brainstem, and rarely in the spinal cord (Fig. 28-29). Acute lesions exhibit central foci of necrosis, petechial hemorrhages surrounded by acute and chronic inflammation, macrophage infiltration, and vascular proliferation. Both free tachyzoites and encysted bradyzoites (Fig. 28-29B) may be found at the periphery of the necrotic foci. The organisms are usually seen on routine H&E or Giemsa stains, but are more easily recognized by immunocytochemical methods. The blood vessels in the vicinity of these lesions may show marked intimal proliferation or even frank vasculitis with fibrinoid necrosis and thrombosis. After treatment, the lesions consist of large, well-demarcated areas of coagulation necrosis surrounded by lipid-laden macrophages. Cysts and free tachyzoites can also be found adjacent to these lesions but may be considerably reduced in number or absent if therapy has been effective. Chronic lesions consist of small cystic spaces containing scattered lipid- and hemosiderin-laden macrophages that are surrounded by gliotic brain. Organisms are difficult to detect in these older lesions.
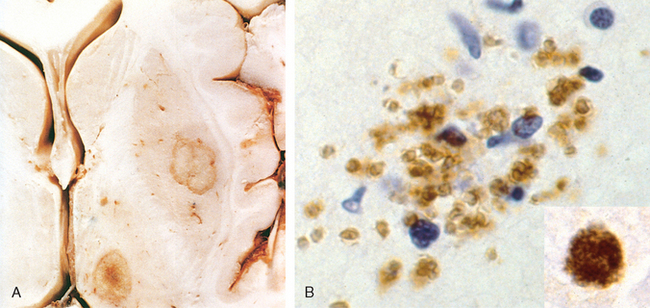
Cerebrial amebiasis.
A rapidly fatal necrotizing encephalitis occurs with infection with Naegleria species, and a chronic granulomatous meningoencephalitis has been associated with infection with Acanthamoeba. The amebas may sometimes be difficult to distinguish from activated macrophages (Fig. 28-30). Methenamine silver or PAS stains are helpful in visualizing the organisms, although definitive identification ultimately depends on combined immunofluorescence studies, morphology, culture, and molecular methods.
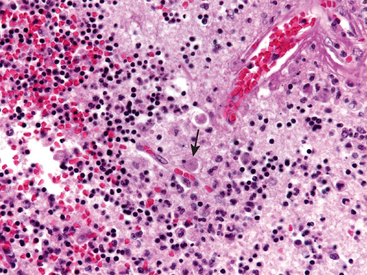
Transmissible Spongiform Encephalopathies (Prion Diseases)
Prions are abnormal forms of a cellular protein that cause transmissible neurodegenerative disorders.23,24 This group of diseases—which includes Creutzfeldt-Jakob disease (CJD), Gerstmann-Sträussler-Scheinker syndrome (GSS), fatal familial insomnia, and kuru in humans; scrapie in sheep and goats; mink-transmissible encephalopathy; chronic wasting disease of deer and elk; and bovine spongiform encephalopathy—share this etiologic basis that distinguishes them from other neurodegenerative and infectious diseases. While differences exist among these disorders, they are all associated with abnormal forms of a specific protein, termed prion protein (PrP), that is both infectious and transmissible. As the name implies, they are predominantly characterized by “spongiform change” caused by intracellular vacuoles in neurons and glia. Clinically, most of the affected patients develop progressive dementia. The most common disorder is CJD. The sporadic form of CJD has an annual incidence of approximately 1 case per 1,000,000 population and accounts for about 90% of cases; familial and transmitted forms make up the rest.
Pathogenesis and Molecular Genetics.
Normal PrP is a 30-kD cellular protein present in neurons. Disease occurs when the PrP undergoes a conformational change from its normal α-helix-containing isoform (PrPc) to an abnormal β-pleated sheet isoform, usually termed PrPsc (for scrapie) (Fig. 28-31). Associated with the conformational change, PrP acquires resistance to digestion with proteases, such as proteinase K. Accumulation of PrPsc in neural tissue seems to be the cause of the pathology in these diseases, but how this material induces the development of cytoplasmic vacuoles and eventual neuronal death is still unknown. Western blotting of tissue extracts after partial digestion with proteinase K allows detection of PrPsc, which is diagnostic.
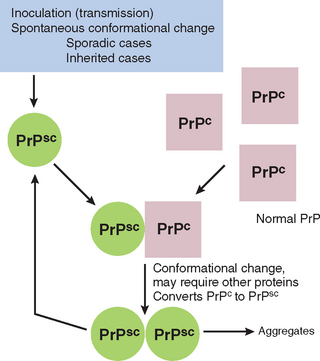
FIGURE 28-31 Proposed mechanism for the conversion of PrPc through protein–protein interactions. The initiating molecules of PrPsc may arise through inoculation (as in directly transmitted cases) or through an extremely low-rate spontaneous conformational change. The effect of the mutations in PrPc is to increase the rate of the conformational change once PrPsc is able to recruit and convert other molecules of PrPc into the abnormal form of the protein. Although the model is drawn with no other proteins involved, it is possible that other proteins play critical roles in the conversion of PrPc to PrPsc.
The conformational change resulting in PrPsc may occur spontaneously at an extremely low rate (resulting in sporadic cases) or at a higher rate if various mutations are present in PrPc, such as occurs in familial forms of CJD and in GSS and fatal familial insomnia. PrPsc, independent of the means by which it originates, then facilitates, in a cooperative fashion, the conversion of other PrPc molecules to PrPsc molecules. It is this activity of PrPsc that accounts for the infectious nature of prion diseases.
The gene encoding PrP, termed PRNP, shows a high degree of conservation across species. A variety of mutations in PRNP have been found to underlie familial forms of prion diseases. In addition, a polymorphism at codon 129 that encodes either methionine (Met) or valine (Val) has been found to influence the disease: individuals who are homozygous for either Met or Val are over-represented among cases of CJD compared with the general population, implying that heterozygosity at codon 129 is protective against development of the disease. Interestingly, this protection also applies against iatrogenic CJD. It has been suggested that the amino-acid at this polymorphic site influences disease by altering the kinetics of aggregation and the conformations of PrP molecules.25
Creutzfeldt-Jakob Disease
CJD, the most common prion disease, is a rare disorder that manifests clinically as a rapidly progressive dementia. It is primarily sporadic (about 85% of cases) in its occurrence, and has a worldwide annual incidence of about 1 per million; familial forms also exist that are caused by mutations in PRNP. The disease has a peak incidence in the seventh decade. There are well-established cases of iatrogenic transmission, notably by corneal transplantation, deep implantation electrodes, and contaminated preparations of human growth hormone. The clinical onset is marked by subtle changes in memory and behavior followed by a rapidly progressive dementia, often with pronounced involuntary jerking muscle contractions on sudden stimulation (startle myoclonus). Signs of cerebellar dysfunction, usually manifested as ataxia, are present in a minority of affected individuals. The disease is uniformly fatal, with an average survival of only 7 months from the onset of symptoms, although a few patients have lived for several years. These long-surviving cases show extensive atrophy of involved gray matter.
Variant Creutzfeldt-Jakob Disease
Starting in 1995, a series of cases of a CJD-like illness came to medical attention in the United Kingdom. These new cases were different from typical CJD in several important respects: the disease affected young adults, behavioral disorders figured prominently in the early stages of the disease, and the neurologic syndrome progressed more slowly than in individuals with other forms of CJD. The neuropathologic findings and molecular features of these new cases were similar to those of CJD, suggesting a close relationship between the two illnesses. Pathologically, variant CJD (vCJD) is characterized by the presence of extensive cortical plaques with a surrounding halo of spongiform change. No alterations in the PRNP gene are present; nearly all affected patients are Met/Met homozygotes at codon 129. With the recent report of a case of vCJD arising in a Val/Val homozygote, the possibility of an influence of codon 129 on incubation period rather than susceptibility has emerged.26 Several lines of evidence have linked vCJD with bovine spongiform encephalopathy, raising complex public health issues. There has also been documented transmission of vCJD by blood products.
Morphology. The progression of the dementia in CJD is usually so rapid that there is little if any grossly evident brain atrophy. On microscopic examination, the pathognomonic finding is a spongiform transformation of the cerebral cortex and, often, deep gray-matter structures (caudate, putamen); this multifocal process results in the uneven formation of small, apparently empty, microscopic vacuoles of varying sizes within the neuropil and sometimes in the perikaryon of neurons (Fig. 28-32A). In advanced cases there is severe neuronal loss, reactive gliosis, and sometimes expansion of the vacuolated areas into cystlike spaces (“status spongiosus”). No inflammatory infiltrate is present. Electron microscopy shows the vacuoles to be membrane-bound and located within the cytoplasm of neuronal processes. Kuru plaques are extracellular deposits of aggregated abnormal protein; they are Congo red- and PASpositive and usually occur in the cerebellum (Fig. 28-32B) although they are present in abundance in the cerebral cortex in cases of vCJD, surrounded by the spongiform changes (Fig. 28-32C). In all forms of prion disease immunohistochemical staining demonstrates the presence of proteinase K–resistant PrPsc in tissue.
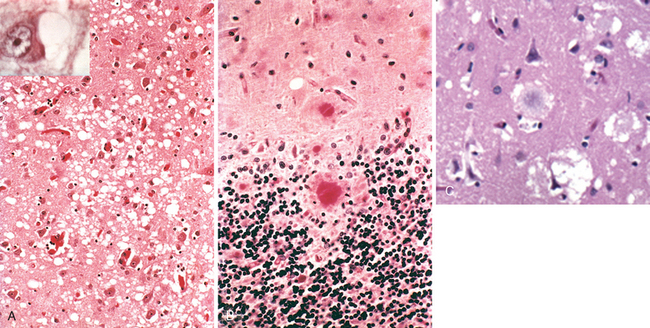
Fatal Familial Insomnia
Fatal familial insomnia (FFI), named in part for the sleep disturbances that characterize its initial stages, is also caused by a specific mutation in the PRNP gene. The mutation, which leads to an aspartate substitution for asparagine at residue 178 of PrPc, results in FFI when it occurs in a PRNP allele encoding methionine at codon 129, but causes CJD when present in tandem with a valine at this position. How these amino acids influence disease phenotype is not understood. In the course of the illness, which typically lasts fewer than 3 years, affected individuals develop other neurologic signs, such as ataxia, autonomic disturbances, stupor, and finally coma. A noninherited form of the disorder (fatal sporadic insomnia) has also been described.
Morphology. Unlike other prion diseases, FFI does not show spongiform pathology. Instead, the most striking alteration is neuronal loss and reactive gliosis in the anterior ventral and dorsomedial nuclei of the thalamus; neuronal loss is also prominent in the inferior olivary nuclei. Proteinase K–resistant PrPsc can be detected by immunostaining or western blotting.