Demyelinating Diseases
Demyelinating diseases of the CNS are acquired conditions characterized by preferential damage to myelin, with relative preservation of axons. The clinical deficits are due to the effect of myelin loss on the transmission of electrical impulses along axons. The natural history of demyelinating diseases is determined, in part, by the limited capacity of the CNS to regenerate normal myelin and by the degree of secondary damage to axons that occurs as the disease runs its course.
Several disease processes can cause loss of myelin. These include destruction of myelin by immunological reactions, as in multiple sclerosis, and by infections. In progressive multifocal leukoencephalopathy, JC virus infection of oligodendrocytes results in loss of myelin (described above). In addition, inherited disorders may affect synthesis or turnover of myelin components; these are termed leukodystrophies and are discussed with metabolic disorders.
MULTIPLE SCLEROSIS
Multiple sclerosis (MS) is an autoimmune demyelinating disorder characterized by distinct episodes of neurologic deficits, separated in time, attributable to white matter lesions that are separated in space. It is the most common of the demyelinating disorders, having a prevalence of approximately 1 per 1000 persons in most of the United States and Europe. The disease may become clinically apparent at any age, although onset in childhood or after age 50 years is relatively rare. Women are affected twice as often as are men. In most individuals with MS, the clinical course takes the form of relapsing and remitting episodes of variable duration (weeks to months to years) marked by neurologic defects, followed by gradual, partial recovery of neurologic function. The frequency of relapses tends to decrease during the course of time, but there is a steady neurologic deterioration in most affected individuals.
Pathogenesis.
The lesions of MS are caused by an immune response that is directed against the components of the myelin sheath.27,28 As in other autoimmune disorders, the pathogenesis of this disease involves both genetic and environmental factors (Chapter 6). The incidence of MS is 15-fold higher when the disease is present in a first-degree relative and roughly 150-fold higher with an affected monozygotic twin. Genetic linkage of MS susceptibility to the DR2 extended haplotype of the major histocompatibility complex is also well established. A recent genome-wide screen supported this association and identified additional associations with single-nucleotide polymorphisms in IL-2 and IL-7 receptor genes.29 The current thinking is that these cytokine receptor polymorphisms may influence the balance between pathogenic effector T cells and protective regulatory T cells. These genetic associations point to the importance of the immune system in the susceptibility to MS.
Given the prominence of chronic inflammatory cells within and around MS plaques as well as this genetic validation, immune mechanisms that underlie the destruction of myelin are the focus of much investigation. The available evidence indicates that the disease is initiated by CD4+ TH1 and TH17 T cells that react against self myelin antigens and secrete cytokines. TH1 cells secrete IFNγ, which activates macrophages, and TH17 cells promote the recruitment of leukocytes (Chapter 6). The demyelination is caused by these activated leukocytes and their injurious products. The infiltrate in plaques and surrounding regions of the brain consists of T cells (mainly CD4+, some CD8+) and macrophages. How the autoimmune reaction is initiated is not understood; a role of viral infection (e.g., EBV) in activating self-reactive T cells has been proposed but remains controversial.
Experimental autoimmune encephalomyelitis is an animal model of MS in which demyelination and inflammation occur after immunization of animals with myelin proteins.30 Many of our concepts of MS pathogenesis have been derived from studies in this model. The experimental disorder can be passively transferred to other animals with TH1 and TH17 cells that recognize myelin antigens.
Based on the growing understanding of the pathogenesis of MS, therapies are being developed that modulate or inhibit T cell responses and block the recruitment of T cells into the brain. A potential contribution of humoral immunity has also been suspected for a long time, based on the early observation of oligoclonal bands of immunoglobulin in CSF.31 The demonstration that B-cell depletion can decrease the incidence of demyelinating lesions lends support to this idea.32
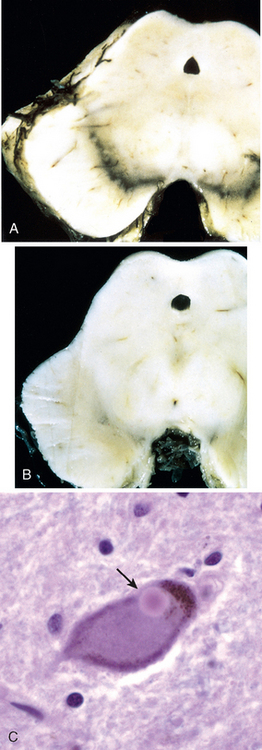
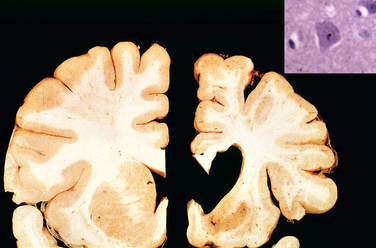
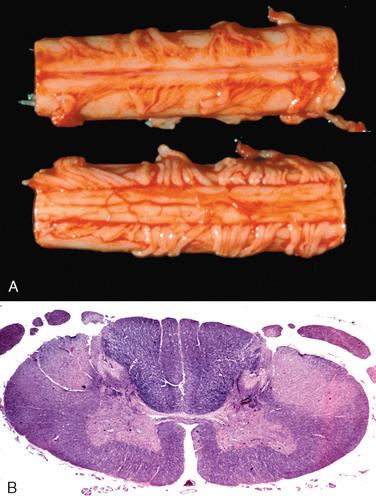
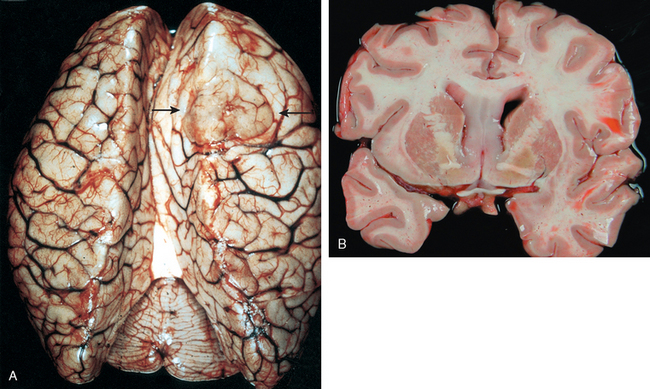
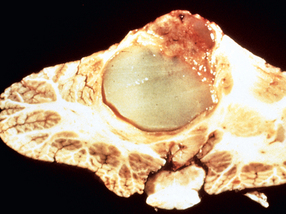
Morphology. MS is a white matter disease that is best appreciated in sections of the brain and spinal cord. Lesions appear as multiple, well-circumscribed, somewhat depressed, glassy, graytan, irregularly shaped plaques (Fig. 28-33). In the fresh state these are firmer than the surrounding white matter (sclerosis). Plaques can be found throughout the white matter and also extend into gray matter, since these have myelinated fibers running through them. The size of lesions varies considerably, from small foci that are only recognizable microscopically to confluent plaques that involve large portions of the centrum semiovale. The lesions often have sharply defined borders (Fig. 28-34). Plaques commonly occur adjacent to the lateral ventricles. They are also frequent in the optic nerves and chiasm, brainstem, ascending and descending fiber tracts, cerebellum, and spinal cord.
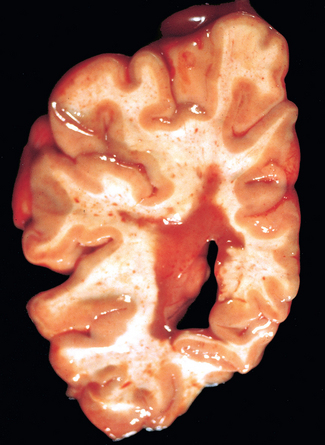
FIGURE 28-33 Multiple sclerosis. Section of fresh brain showing brown plaque around occipital horn of the lateral ventricle.
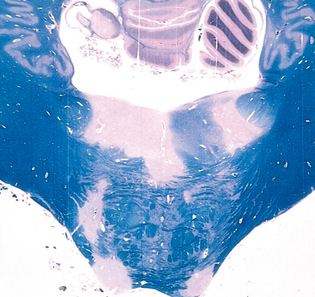
FIGURE 28-34 Multiple sclerosis (MS). Unstained regions of demyelination (MS plaques) around the fourth ventricle (Luxol fast blue PAS stain for myelin).
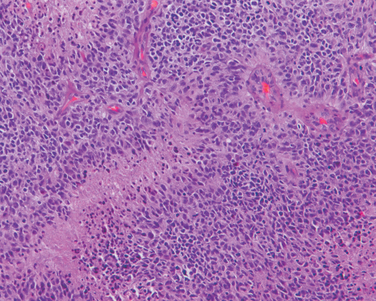
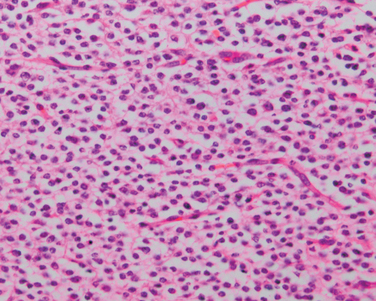
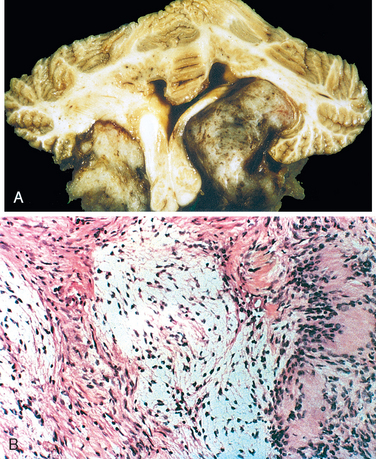
Microscopically, in an active plaque there is evidence of ongoing myelin breakdown with abundantmacrophages containing lipid-rich, PAS-positive debris. Inflammatory cells, including both lymphocytes and monocytes, are present, mostly as perivascular cuffs, especially at the outer edge of the lesion (Fig. 28-35A). Active lesions are often centered on small veins. Within a plaque there is relative preservation of axons (Fig. 28-35B) and depletion of oligodendrocytes. In time, astrocytes undergo reactive changes. As lesions become quiescent, the inflammatory cells slowly disappear. Within inactive plaques, little to no myelin is found, and there is a reduction in the number of oligodendrocyte nuclei; instead, astrocytic proliferation and gliosis are prominent. Axons in old gliotic plaques show severe depletion of myelin and are also greatly diminished in number.
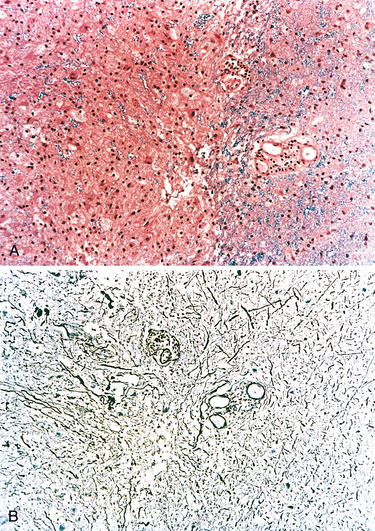
FIGURE 28-35 Multiple sclerosis. A, Myelin-stained section shows the sharp edge of a demyelinated plaque and perivascular lymphocytic cuffs. B, The same lesion stained for axons shows relative preservation.
Active plaques can also be grouped into four basic patterns: those that are sharply demarcated and centered on blood vessels, either with (pattern I) or without (pattern II) deposition of immunoglobulin and complement, and those that are less well demarcated and are not centered on vessels (patterns III and IV). These latter two are distinguished by the distribution of oligodendrocyte apoptosis (III, widespread; IV, central only). It has been observed that only one pair of patterns (I/II or III/IV) may be present in a given individual, suggesting that these may reflect distinct mechanisms rather than different stages of lesion.
In some MS plaques (shadow plaques) the border between normal and affected white matter is not sharply circumscribed. In this type of lesion some abnormally thinned-out myelin sheaths can be demonstrated, especially at the outer edges. This phenomenon is most commonly interpreted as evidence of partial and incomplete remyelination by surviving oligodendrocytes. Abnormally myelinated fibers have also been observed at the edges of typical plaques. Although these histologic findings suggest a limited potential for remyelination in the CNS, the remaining axons within most MS plaques remain unmyelinated; studies aimed at promoting remyelination are an important focus of research.33
Clinical Features.
Although MS lesions can occur anywhere in the CNS and consequently may induce a wide range of clinical manifestations, certain patterns of neurologic symptoms and signs are commonly observed. Unilateral visual impairment, due to involvement of the optic nerve (optic neuritis, retrobulbar neuritis), is a frequent initial manifestation of MS. However, only some affected individuals (10% to 50%, depending on the population studied) with optic neuritis go on to develop MS. Involvement of the brainstem produces cranial nerve signs, ataxia, nystagmus, and internuclear ophthalmoplegia from interruption of the fibers of the medial longitudinal fasciculus. Spinal cord lesions give rise to motor and sensory impairment of trunk and limbs, spasticity, and difficulties with the voluntary control of bladder function. Examination of the CSF in individuals with MS shows a mildly elevated protein level, and in one third of cases, there is moderate pleocytosis. IgG levels in the CSF are increased and oligoclonal IgG bands are usually observed on immunoelectrophoresis; these are indicative of the presence of a small number of activated B cell clones, postulated to be self-reactive, in the CNS. Radiologic studies using magnetic resonance imaging, typically based on identifying gadolinium-enhancing lesions, have taken on a prominent role in assessing disease progression; these studies, when correlated with autopsy studies as well as clinical findings, have indicated that some plaques may be clinically silent even in otherwise symptomatic patients.
NEUROMYELITIS OPTICA
The development of synchronous (or near synchronous) bilateral optic neuritis and spinal cord demyelination is referred to as neuromyelitis optica or Devic disease. White cells are common in the CSF, often including neutrophils. Within the damaged areas of white matter, there is typically necrosis, an inflammatory infiltrate including neutrophils, and vascular deposition of immunoglobulin and complement. These lesions have been suggested to be mediated by humoral immune mechanisms.34 Many affected individuals show antibodies to aquaporins, which are in part responsible for maintenance of astrocytic foot process and thus the integrity of the blood-brain barrier.35,36
ACUTE DISSEMINATED ENCEPHALOMYELITIS AND ACUTE NECROTIZING HEMORRHAGIC ENCEPHALOMYELITIS
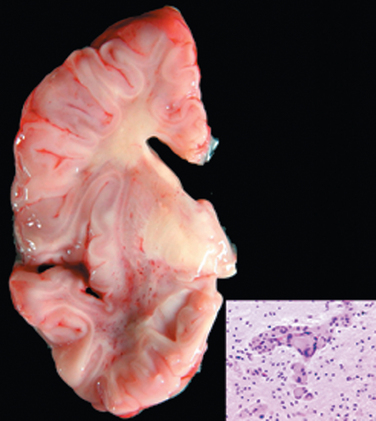
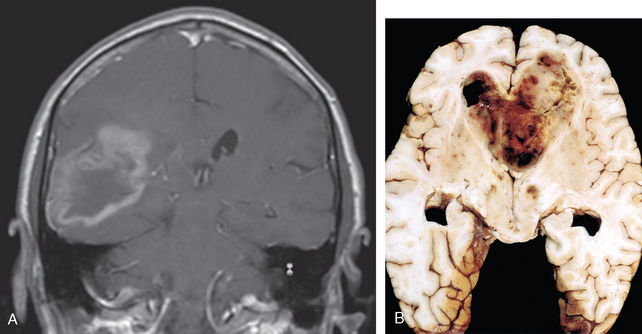
Acute disseminated encephalomyelitis (ADEM, perivenous encephalomyelitis) is a diffuse, monophasic demyelinating disease that follows either a viral infection or, rarely, a viral immunization. Symptoms typically develop a week or two after the antecedent infection and include headache, lethargy, and coma rather than focal findings, as seen in MS. The clinical course is rapid, and as many as 20% of those affected die; the remaining patients recover completely.
Acute necrotizing hemorrhagic encephalomyelitis (ANHE, acute hemorrhagic leukoencephalitis of Weston Hurst) is a fulminant syndrome of CNS demyelination, typically affecting young adults and children. The illness is almost invariably preceded by a recent episode of upper respiratory infection, most often of unknown cause. The disease is fatal in many patients, with significant deficits present in most survivors.
Morphology. In ADEM, macroscopic examination of the brain shows only grayish discoloration around white-matter vessels. On microscopic examination, myelin loss with relative preservation of axons can be found throughout the white matter. In the early stages, polymorphonuclear leukocytes can be found within the lesions; later, mononuclear infiltrates predominate. The breakdown of myelin is associated with the accumulation of lipid-laden macrophages. In contrast with MS, all lesions appear similar, consistent with the clinically monophasic nature of the disorder.
ANHE shows histologic similarities with ADEM, including a perivenular distribution of demyelination and widespread dissemination throughout the CNS (sometimes with extensive confluence of lesions). However, the lesions are much more severe than those of ADEM and include destruction of small blood vessels, disseminated necrosis of white and gray matter with acute hemorrhage, fibrin deposition, and abundant neutrophils. Scattered lymphocytes are seen in foci of demyelination.
The lesions of ADEM are similar to those induced by immunization of animals with myelin components or with early rabies vaccines that had been prepared from brains of infected animals. This has suggested that ADEM may represent an acute autoimmune reaction to myelin and that ANHE may represent a hyperacute variant, although no inciting antigens have been identified.
OTHER DISEASES WITH DEMYELINATION
Central pontine myelinolysis is characterized by loss of myelin (with relative preservation of axons and neuronal cell bodies) in a roughly symmetric pattern involving the basis pontis and portions of the pontine tegmentum but sparing the periventricular and subpial regions.37 Lesions may be found more rostrally; it is extremely rare for the process to extend below the pontomedullary junction. Extra-pontine lesions occur in the supratentorial compartment, with similar appearance and apparent etiology. The condition is most commonly associated with rapid correction of hyponatremia, although it can be associated with other severe electrolyte or osmolar imbalance, as well as orthotopic liver transplantation. The clinical presentation of central pontine myelinolysis is that of a rapidly evolving quadriplegia; radiologic imaging studies localize the lesion to the basis pontis. Morphologically there is myelin loss without evidence of inflammation; neurons and axons are well preserved. Again, because of the monophasic nature of the disease all lesions appear to be at the same stage of myelin loss and reaction.