Rejection of Transplants
The major barrier to transplantation of organs from one individual to another of the same species (called allografts) is immunologic rejection of the transplanted tissue. Rejection is a complex phenomenon involving both cell- and antibody-mediated reactions that destroy the graft. The key to successful transplantation has been the development of therapies that prevent or minimize rejection. Discussed next is how grafts are recognized as foreign and how they are rejected.
Immune Recognition of Allografts
Rejection of allografts is a response mainly to MHC molecules, which are so polymorphic that most individuals in an outbred population differ in at least some of the MHC molecules they express (except, of course, for identical twins). There are two main mechanisms by which the host immune system recognizes and responds to the MHC molecules on the graft (Fig. 4–23):
• Direct recognition. Host T cells directly recognize the allogeneic (foreign) MHC molecules that are expressed on graft cells. Direct recognition of foreign MHC seems to violate the rule of MHC restriction, which states that in every individual, all of the T cells are educated to recognize foreign antigens displayed by only that individual’s MHC molecules. It is postulated that allogeneic MHC molecules (with any bound peptides) structurally mimic self MHC and foreign peptide, and so direct recognition of the allogeneic MHC is essentially an immunologic cross-reaction. Because DCs in the graft express high levels of MHC as well as costimulatory molecules, they are believed to be the major culprits contributing to direct recognition. The most important consequence of direct recognition is the activation of host CD8+ T cells that recognize class I MHC (HLA-A, -B) molecules in the graft. These T cells differentiate into CTLs, which kill the cells in the graft. Host CD4+ helper T cells may be triggered into proliferation and cytokine production by recognition of donor class II MHC (HLA-D) molecules and drive an inflammatory response.
• Indirect recognition. In this pathway, host CD4+ T cells recognize donor MHC molecules after these molecules are picked up, processed, and presented by the host’s own APCs. This sequence is similar to the physiologic processing and presentation of other foreign (e.g., microbial) antigens. The activated CD4+ T cells then recognize APCs displaying graft antigens and secrete cytokines that induce inflammation and damage the graft. The indirect pathway is also involved in the production of antibodies against graft alloantigens; if these antigens are proteins, they are picked up by host B cells, and peptides are presented to helper T cells, which then stimulate antibody responses.
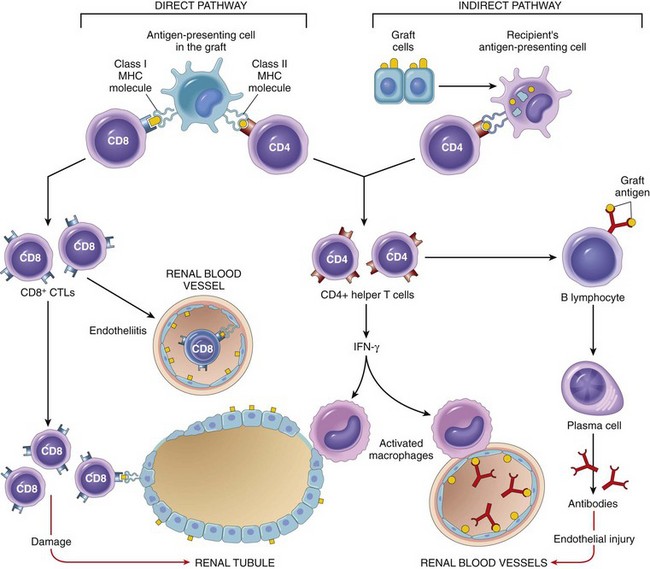
Figure 4–23 Recognition and rejection of allografts. In the direct pathway, donor class I and class II MHC antigens on antigen-presenting cells (APCs) in the graft (along with costimulators, not shown) are recognized by host CD8+ cytotoxic T cells and CD4+ helper T cells, respectively. CD4+ cells proliferate and produce cytokines (e.g., IFN-γ), which induce tissue damage by a local delayed-type hypersensitivity reaction. CD8+ T cells responding to graft antigens differentiate into CTLs that kill graft cells. In the indirect pathway, graft antigens are displayed by host APCs and activate CD4+ T cells, which damage the graft by a local delayed-type hypersensitivity reaction and stimulate B lymphocytes to produce antibodies. IFN-γ, interferon-γ; MHC, major histocompatibility complex.
Effector Mechanisms of Graft Rejection
Both T cells and antibodies reactive with the graft are involved in the rejection of most solid-organ allografts (Fig. 4–23).
T Cell–Mediated Rejection
CTLs kill cells in the grafted tissue, causing parenchymal and endothelial cell death (the latter resulting in thrombosis and graft ischemia). Cytokine-secreting CD4+ T cells trigger inflammatory reactions resembling DTH in the tissues and blood vessels, with local accumulation of mononuclear cells (lymphocytes and macrophages). Activated microphages can injure graft cells and vasculature. The microvascular injury also results in tissue ischemia, which contributes to graft destruction.
Antibody-Mediated Rejection
Although T cells are of paramount importance in allograft rejection, antibodies also mediate some forms of rejection. Alloantibodies directed against graft MHC molecules and other alloantigens bind to the graft endothelium and cause vascular injury through complement activation and recruitment of leukocytes. Superimposed on the resulting endothelial damage and dysfunction is thrombosis, adding further ischemic insult to the injury.
Hyperacute rejection is a special form of rejection occurring if pre-formed anti-donor antibodies are present in the circulation of the host before transplantation. This may happen in multiparous women who have anti-HLA antibodies against paternal antigens encountered during pregnancy, or in individuals exposed to foreign HLA (on platelets or leukocytes) from previous blood transfusions. Obviously, such antibodies also may be present in a patient who has previously rejected an organ transplant. Subsequent transplantation in such patients will result in immediate rejection (within minutes to hours) because the circulating antibodies rapidly bind to the endothelium of the grafted organ, with resultant complement activation and vascular thrombosis. With the current practice of screening potential recipients for pre-formed anti-HLA antibodies and cross-matching (testing recipients for the presence of antibodies directed against the donor’s lymphocytes), hyperacute rejection occurs in less than 0.4% of transplant recipients.
 Morphology
Morphology
On the basis of the time course and morphology of rejection reactions, they have been classified as hyperacute, acute, and chronic (Fig. 4–24). This classification is helpful for understanding the mechanism of rejection, because each pattern is caused by a different type of dominant immunologic reaction. The morphology of these patterns is described in the context of renal transplants; however, similar changes are encountered in other vascularized organ transplants.
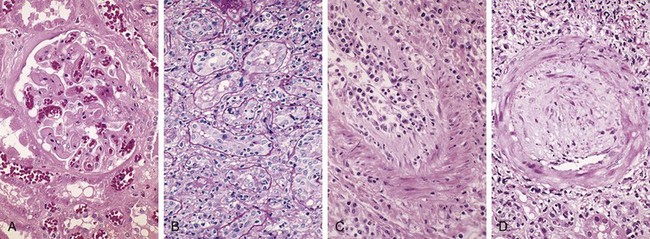
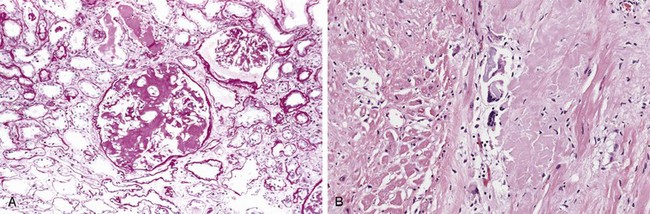
Figure 4–24 Morphologic patterns of graft rejection. A, Hyperacute rejection of a kidney allograft associated with endothelial damage and thrombi in a glomerulus. B, Acute cellular rejection of a kidney allograft with inflammatory cells in the interstitium and between epithelial cells of the tubules. C, Acute humoral rejection of a kidney allograft (rejection vasculitis) with inflammatory cells and proliferating smooth muscle cells in the intima. D, Chronic rejection in a kidney allograft with graft arteriosclerosis. The arterial lumen is replaced by an accumulation of smooth muscle cells and connective tissue in the intima.
(A–D, Courtesy of Dr. Helmut Rennke, Department of Pathology, Brigham and Women’s Hospital and Harvard Medical School, Boston, Massachusetts.)
Hyperacute Rejection
Hyperacute rejection occurs within minutes to a few hours after transplantation in a presensitized host and typically is recognized by the surgeon just after the vascular anastomosis is completed. In contrast with a nonrejecting kidney graft, which regains a normal pink color and tissue turgor and promptly excretes urine, a hyperacutely rejecting kidney rapidly becomes cyanotic, mottled, and flaccid and may excrete only a few drops of bloody fluid. The histologic picture is characterized by widespread acute arteritis and arteriolitis, vessel thrombosis, and ischemic necrosis, all resulting from the binding of preformed antibodies to graft endothelium. Virtually all arterioles and arteries exhibit characteristic acute fibrinoid necrosis of their walls, with narrowing or complete occlusion of the lumens by precipitated fibrin and cellular debris (Fig. 4–24, A).
Acute Rejection
Acute rejection may occur within days to weeks of transplantation in a nonimmunosuppressed host or may appear months or even years later, even in the presence of adequate immunosuppression. Acute rejection is caused by both cellular and humoral immune mechanisms, and in any one patient, one or the other may predominate, or both may be present. On histologic examination, cellular rejection is marked by an interstitial mononuclear cell infiltrate with associated edema and parenchymal injury, whereas humoral rejection is associated with vasculitis.
Acute cellular rejection most commonly is seen within the first months after transplantation and typically is accompanied by clinical signs of renal failure. Histologic examination usually shows extensive interstitial CD4+ and CD8+ T cell infiltration with edema and mild interstitial hemorrhage (Fig. 4–24, B). Glomerular and peritubular capillaries contain large numbers of mononuclear cells, which also may invade the tubules, leading to focal tubular necrosis. In addition to tubular injury, CD8+ T cells also may injure the endothelium, causing an endothelitis. Cyclosporine (a widely used immunosuppressive agent) is also nephrotoxic and induces so-called arteriolar hyaline deposits. Renal biopsy is used to distinguish rejection from drug toxicity. Accurate recognition of cellular rejection is important, because patients typically respond promptly to increased immunosuppressive therapy.
Acute humoral rejection (rejection vasculitis) caused by antidonor antibodies also may participate in acute graft rejection. The histologic lesions may take the form of necrotizing vasculitis with endothelial cell necrosis; neutrophilic infiltration; deposition of antibody, complement, and fibrin; and thrombosis. Such lesions may be associated with ischemic necrosis of the renal parenchyma. Somewhat older subacute lesions are characterized by marked thickening of the intima by proliferating fibroblasts, myocytes, and foamy macrophages (Fig. 4–24, C). The resultant narrowing of the arterioles may cause infarction or renal cortical atrophy. The proliferative vascular lesions mimic arteriosclerotic thickening and are believed to be caused by cytokines that stimulate proliferation of vascular smooth muscle cells. Local deposition of complement breakdown products (specifically C4d) is used to detect antibody-mediated rejection of kidney allografts.
Chronic Rejection
Patients present with chronic rejection late after transplantation (months to years) with a progressive rise in serum creatinine levels (an index of renal function) over a period of 4 to 6 months. Chronic rejection is dominated by vascular changes, interstitial fibrosis, and loss of renal parenchyma; there are typically only mild or no ongoing cellular parenchymal infiltrates. The vascular changes occur predominantly in the arteries and arterioles, which exhibit intimal smooth muscle cell proliferation and extracellular matrix synthesis (Fig. 4–24, D). These lesions ultimately compromise vascular perfusion and result in renal ischemia manifested by loss or hyalinization of glomeruli, interstitial fibrosis, and tubular atrophy. The vascular lesion may be caused by cytokines released by activated T cells that act on the cells of the vascular wall, and it may be the end stage of the proliferative arteritis described earlier.
 Summary
Summary
Recognition and Rejection of Organ Transplants (Allografts)
• The graft rejection response is initiated mainly by host T cells that recognize the foreign HLA antigens of the graft, either directly (on APCs in the graft) or indirectly (after uptake and presentation by host APCs).
• Types and mechanisms of rejection comprise the following:
 Hyperacute rejection: Pre-formed antidonor antibodies bind to graft endothelium immediately after transplantation, leading to thrombosis, ischemic damage, and rapid graft failure.
Hyperacute rejection: Pre-formed antidonor antibodies bind to graft endothelium immediately after transplantation, leading to thrombosis, ischemic damage, and rapid graft failure.Methods of Improving Graft Survival
Because HLA molecules are the major targets in transplant rejection, better matching of the donor and the recipient improves graft survival. The benefits of HLA matching are most dramatic for living related donor kidney transplants, and survival improves with increasing number of loci matched. However, as drugs for immunosuppression have improved, HLA matching is not even attempted in some situations, such as heart, lung, liver, and islet transplantation; in such instances, the recipient often needs a transplant urgently and other considerations, such as anatomic (size) compatibility, are of greater practical importance.
Immunosuppression of the recipient is a practical necessity in all organ transplantation except in the case of identical twins. At present, drugs such as cyclosporine, the related FK506, mofetil mycophenolate (MMF), rapamycin, azathioprine, corticosteroids, antilymphocyte globulin, and monoclonal antibodies (e.g., monoclonal anti-CD3) are used. Cyclosporine and FK506 suppress T cell–mediated immunity by inhibiting transcription of cytokine genes, in particular, the gene for IL-2. Although immunosuppression has made transplantation of many organs feasible, there is still a price to be paid. Global immunosuppression results in increased susceptibility to opportunistic fungal, viral, and other infections. These patients are also at increased risk for developing Epstein-Barr virus (EBV)–induced lymphomas, human papillomavirus–induced squamous cell carcinomas, and Kaposi sarcoma. To circumvent the untoward effects of immunosuppression, much effort is devoted to trying to induce donor-specific tolerance in host T cells. One strategy being pursued is to prevent host T cells from receiving costimulatory signals from donor DCs during the initial phase of sensitization. This can be accomplished by administration of agents to interrupt the interaction between the B7 molecules on the DCs of the graft with the CD28 receptors on host T cells. This will interrupt the second signal for T cell activation and either induce apoptosis or render the T cells functionally unresponsive. The improvement in immunosuppressive therapy has led to improving survival of grafts and acute rejection is becoming less of a concern, especially for kidney and heart grafts. Chronic rejection remains a serious problem, however, especially because it responds much less effectively than does acute rejection to available immunosuppressive agents.
Transplantation of Hematopoietic Stem Cells
Hematopoietic stem cell (HSC) transplantation is used as therapy for hematopoietic and some nonhematopoietic malignancies, aplastic anemias, and certain inherited disorders, particularly immune deficiency states and severe forms of thalassemia. HSCs historically were obtained solely from donor bone marrow, but are now increasingly harvested from the peripheral blood after mobilization by administration of hematopoietic growth factors, or from the umbilical cord blood of newborns, a readily available rich source of HSCs. The recipient receives chemotherapy and/or irradiation to destroy malignant cells (e.g., in leukemia) and to create a graft bed; then, HSCs are infused into the peripheral blood, from which they home to their bone marrow niches. Rejection of allogeneic HSC transplants seems to be mediated by some combination of host T cells and NK cells that are resistant to radiation therapy and chemotherapy. Two major problems complicate this form of transplantation: graft-versus-host disease and immune deficiency.
Graft-Versus-Host Disease (GVHD)
This occurs when immunologically competent T cells (or their precursors) are transplanted into recipients who are immunologically compromised. Although GVHD happens most commonly in the setting of allogeneic HSC transplantation (usually involving minor histocompatibility mismatches between donor and recipient), it also may occur after transplantation of solid organs rich in lymphoid cells (e.g., the liver) or after transfusion of nonirradiated blood. On receiving allogeneic HSCs, an immunologically compromised host cannot reject the graft, but T cells present in the donor graft perceive the recipient’s tissue as “foreign” and react against it. This results in the activation of both CD4+ and CD8+ T cells, ultimately causing inflammation and killing host cells.
• Acute GVHD (occurring days to weeks after transplantation) causes epithelial cell necrosis in three principal target organs: liver, skin, and gut. Destruction of small bile ducts gives rise to jaundice, and mucosal ulceration of the gut results in bloody diarrhea. Cutaneous involvement is manifested by a generalized rash.
• Chronic GVHD may follow the acute syndrome or may occur insidiously. The patients develop skin lesions resembling those of SS (discussed earlier) and manifestations mimicking other autoimmune disorders.
GVHD is a potentially lethal complication that can be minimized but not eliminated by HLA matching. As another potential solution, donor T cells can be depleted before marrow transplant. This protocol has proved to be a mixed blessing: The risk of GVHD is reduced, but the incidence of graft failure and (in those with the disease) the recurrence of leukemia increase. It seems that the multifunctional T cells not only mediate GVHD but also are required for the efficient engraftment of the transplanted HSCs and elimination of leukemia cells (so-called graft-versus-leukemia effect).
Immune Deficiencies
These are often of prolonged duration in recipients of HSC transplants. Among the many reasons for this impairment is the slow reconstitution of the recipient’s adaptive immune system, which is destroyed or suppressed to allow the graft to take and requires many months to recover. During this vulnerable period, recipients are susceptible to a variety of infections, mostly viral, such as cytomegalovirus (CMV) and EBV infections.
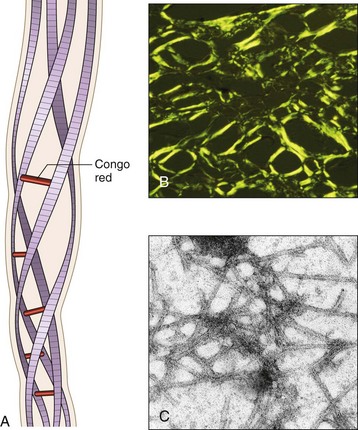
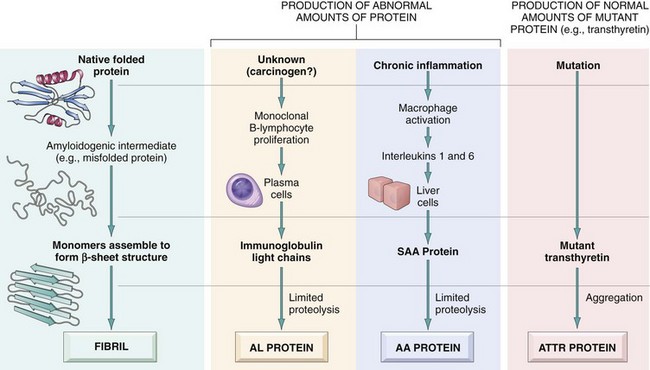
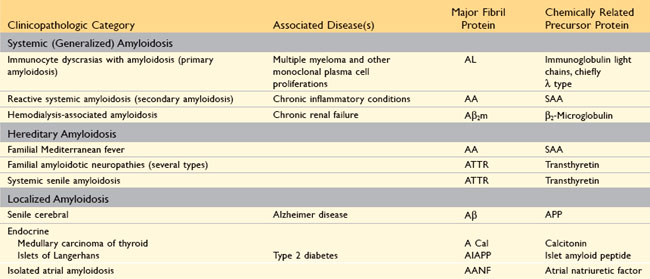
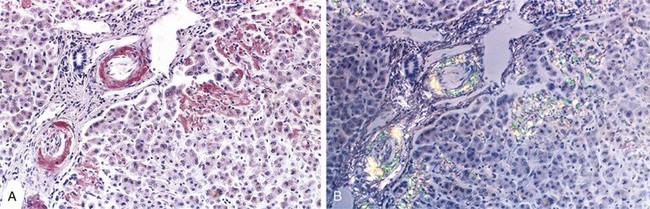
 Pathogenesis of Amyloid Deposition
Pathogenesis of Amyloid Deposition