Endocrine Pancreas
The endocrine pancreas consists of about 1 million microscopic clusters of cells, the islets of Langerhans, which contain four major cell types—beta, alpha, delta, and PP (pancreatic polypeptide) cells. The cells can be differentiated morphologically by their staining properties, by the ultrastructural characteristics of their granules, and by their hormone content. The beta cell produces insulin, which is the most potent anabolic hormone known, with multiple synthetic and growth-promoting effects; the alpha cell secretes glucagon, inducing hyperglycemia by its glycogenolytic activity in the liver; delta cells contain somatostatin, which suppresses both insulin and glucagon release; and PP cells contain a unique pancreatic polypeptide, VIP, that exerts several gastrointestinal effects, such as stimulation of secretion of gastric and intestinal enzymes and inhibition of intestinal motility. The most important disease of the endocrine pancreas is diabetes mellitus, caused by deficient production or action of insulin.
Diabetes Mellitus
Diabetes mellitus is not a single disease entity but rather a group of metabolic disorders sharing the common underlying feature of hyperglycemia. Hyperglycemia in diabetes results from defects in insulin secretion, insulin action, or, most commonly, both. The chronic hyperglycemia and attendant metabolic deregulation of diabetes mellitus may be associated with secondary damage in multiple organ systems, especially the kidneys, eyes, nerves, and blood vessels. According to the American Diabetes Association, diabetes affects over 20 million children and adults, or 7% of the population, in the United States, nearly a third of whom are currently unaware that they have hyperglycemia. Approximately 1.5 million new cases of diabetes are diagnosed each year in the United States, and diabetes is the leading cause of end-stage renal disease, adult-onset blindness, and nontraumatic lower extremity amputations. A staggering 54 million adults in this country have prediabetes, which is defined as elevated blood sugar that does not reach the criterion accepted for an outright diagnosis of diabetes (discussed next); persons with prediabetes have an elevated risk for development of frank diabetes.
Diagnosis
Blood glucose levels normally are maintained in a very narrow range, usually 70 to 120 mg/dL. The diagnosis of diabetes is established by elevation of blood glucose by any one of three criteria:
1 A random blood glucose concentration of 200 mg/dL or higher, with classical signs and symptoms (discussed next)
2 A fasting glucose concentration of 126 mg/dL or higher on more than one occasion
3 An abnormal oral glucose tolerance test (OGTT), in which the glucose concentration is 200 mg/dL or higher 2 hours after a standard carbohydrate load (75 g of glucose).
Derangements in carbohydrate metabolism proceed along a continuum. Persons with serum fasting glucose values less than 110 mg/dL, or less than 140 mg/dL for an OGTT, are considered to be euglycemic. However, those with serum fasting glucose greater than 110 but less than 126 mg/dL, or OGTT values of greater than 140 but less than 200 mg/dL, are considered to have impaired glucose tolerance, also known as prediabetes. Persons with impaired glucose tolerance have a significant risk for progression to overt diabetes over time, with as many as 5% to 10% advancing to full-fledged diabetes mellitus per year. In addition, those with impaired glucose tolerance are at risk for cardiovascular disease, as a consequence of abnormal carbohydrate metabolism and the coexistence of other risk factors (Chapter 9).
Classification
Although all forms of diabetes mellitus share hyperglycemia as a common feature, the underlying causes of hyperglycemia vary widely. The vast majority of cases of diabetes fall into one of two broad classes:
• Type 1 diabetes (T1D) is characterized by an absolute deficiency of insulin secretion caused by pancreatic beta cell destruction, usually resulting from an autoimmune attack. Type 1 diabetes accounts for approximately 10% of all cases.
• Type 2 diabetes (T2D) is caused by a combination of peripheral resistance to insulin action and an inadequate compensatory response of insulin secretion by the pancreatic beta cells (relative insulin deficiency). Approximately 80% to 90% of patients have type 2 diabetes.
A variety of monogenic and secondary causes make up the remaining cases of diabetes (Table 19–5). An important point is that although the major types of diabetes arise by different pathogenic mechanisms, the long-term complications in kidneys, eyes, nerves, and blood vessels are the same and are the principal causes of morbidity and death.
Table 19–5 Classification of Diabetes Mellitus
| 1. Type 1 Diabetes |
| Beta cell destruction, usually leading to absolute insulin deficiency |
| 2. Type 2 Diabetes |
| Combination of insulin resistance and beta cell dysfunction |
| 3. Genetic Defects of Beta Cell Function |
| Maturity-onset diabetes of the young (MODY), caused by mutations in: |
| Hepatocyte nuclear factor 4α gene (HNF4A)—MODY1 |
| Glucokinase gene (GCK)—MODY2 |
| Hepatocyte nuclear factor 1α gene (HNF1A)—MODY3 |
| Pancreatic and duodenal homeobox 1 gene (PDX1)—MODY4 |
| Hepatocyte nuclear factor 1β gene (HNF1B)—MODY5 |
| Neurogenic differentiation factor 1 gene (NEUROD1)—MODY6 |
| Maternally inherited diabetes and deafness (MIDD) due to mitochondrial DNA mutations (3243A→G) |
| Defects in proinsulin conversion |
| Insulin gene mutations |
| 4. Genetic Defects in Insulin Action |
| Insulin receptor mutations |
| 5. Exocrine Pancreatic Defects |
| Chronic pancreatitis |
| Pancreatectomy |
| Neoplasia |
| Cystic fibrosis |
| Hemochromatosis |
| Fibrocalculous pancreatopathy |
| 6. Endocrinopathies |
| Growth hormone excess (acromegaly) |
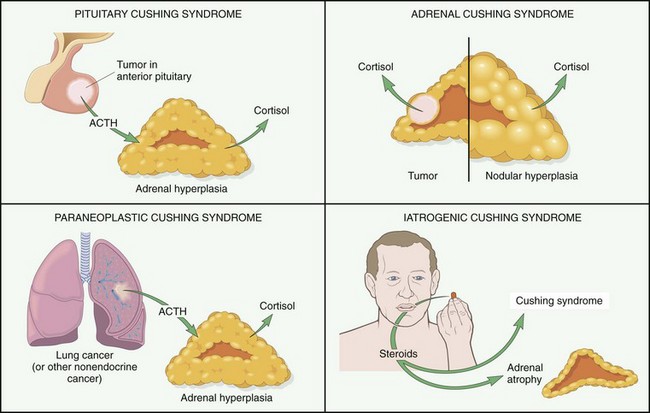
| Cushing syndrome |
| Hyperthyroidism |
| Pheochromocytoma |
| Glucagonoma |
| 7. Infections |
| Cytomegalovirus infection |
| Coxsackievirus B infection |
| Congenital rubella |
| 8. Drugs |
| Glucocorticoids |
| Thyroid hormone |
| β-Adrenergic agonists |
| 9. Genetic Syndromes Associated with Diabetes |
| Down syndrome |
| Klinefelter syndrome |
| Turner syndrome |
| 10. Gestational Diabetes Mellitus |
| Diabetes associated with pregnancy |
Modified from the American Diabetes Association: Position statement from the American Diabetes Association on the diagnosis and classification of diabetes mellitus. Diabetes Care 31(Suppl 1):S55–S60, 2008.
Normal Insulin Physiology and Glucose Homeostasis
Before discussing the pathogenesis of the two major types of diabetes, we briefly review normal insulin physiology and glucose metabolism. Normal glucose homeostasis is tightly regulated by three interrelated processes: (1) glucose production in the liver, (2) glucose uptake and utilization by peripheral tissues, chiefly skeletal muscle, and (3) actions of insulin and counterregulatory hormones (e.g., glucagon).
The principal metabolic function of insulin is to increase the rate of glucose transport into certain cells in the body (Fig. 19–21). These are the striated muscle cells (including myocardial cells) and, to a lesser extent, adipocytes, representing collectively about two thirds of total body weight. Glucose uptake in other peripheral tissues, most notably the brain, is insulin-independent. In muscle cells, glucose is then either stored as glycogen or oxidized to generate adenosine triphosphate (ATP). In adipose tissue, glucose is stored primarily as lipid. Besides promoting lipid synthesis (lipogenesis), insulin also inhibits lipid degradation (lipolysis) in adipocytes. Similarly, insulin promotes amino acid uptake and protein synthesis while inhibiting protein degradation. Thus, the metabolic effects of insulin can be summarized as anabolic, with increased synthesis and reduced degradation of glycogen, lipid, and protein. In addition to these metabolic effects, insulin has several mitogenic functions, including initiation of DNA synthesis in certain cells and stimulation of their growth and differentiation.
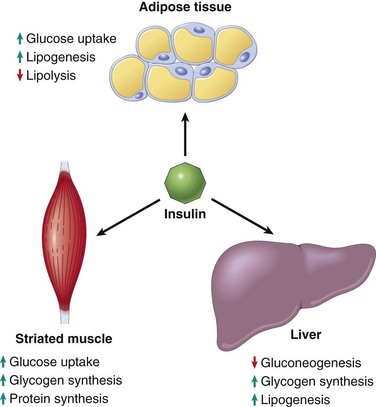
Insulin reduces the production of glucose from the liver. Insulin and glucagon have opposing regulatory effects on glucose homeostasis. During fasting states, low insulin and high glucagon levels facilitate hepatic gluconeogenesis and glycogenolysis (glycogen breakdown) while decreasing glycogen synthesis, thereby preventing hypoglycemia. Thus, fasting plasma glucose levels are determined primarily by hepatic glucose output. After a meal, insulin levels rise and glucagon levels fall in response to the large glucose load. The most important stimulus that triggers insulin release is glucose itself, which initiates insulin synthesis in the pancreatic beta cells. In peripheral tissues (skeletal muscle and adipose tissue), secreted insulin binds to the insulin receptor, triggering a number of intracellular responses that promote glucose uptake and postprandial glucose utilization, thereby maintaining glucose homeostasis. Abnormalities at various points along this complex signaling cascade, from synthesis and release of insulin by beta cells to insulin receptor interactions in peripheral tissues, can result in the diabetic phenotype.
 Pathogenesis
Pathogenesis
Type 1 Diabetes Mellitus
Type 1 diabetes is an autoimmune disease in which islet destruction is caused primarily by immune effector cells reacting against endogenous beta cell antigens. Type 1 diabetes most commonly develops in childhood, becomes manifest at puberty, and progresses with age. Most patients with type 1 diabetes depend on exogenous insulin for survival; without insulin they develop serious metabolic complications such as ketoacidosis and coma.
Although the clinical onset of type 1 diabetes is abrupt, this disease in fact results from a chronic autoimmune attack on beta cells that usually starts many years before the disease becomes evident (Fig. 19–22). The classic manifestations of the disease (hyperglycemia and ketosis) occur late in its course, after more than 90% of the beta cells have been destroyed. The fundamental immune abnormality in type 1 diabetes is a failure of self-tolerance in T cells. This failure of tolerance may be a result of some combination of defective clonal deletion of self-reactive T cells in the thymus, as well as defects in the functions of regulatory T cells or resistance of effector T cells to suppression by regulatory cells. Thus, autoreactive T cells not only survive but are poised to respond to self-antigens. Not surprisingly, autoantibodies against a variety of beta cell antigens, including insulin and the beta cell enzyme glutamic acid decarboxylase, are detected in the blood of 70% to 80% of patients. In the rare cases in which the pancreatic lesions have been examined early in the disease process, the islets show necrosis of beta cells and lymphocytic infiltration (so-called insulitis).
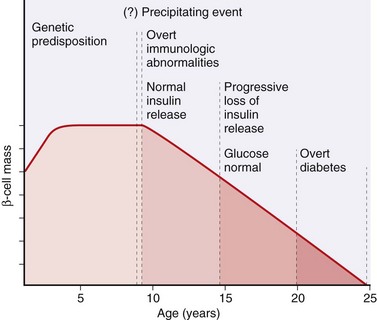
Figure 19–22 Stages in the development of type 1 diabetes mellitus. The stages are listed from left to right, and hypothetical beta cell mass is plotted against age.
(From Eisenbarth GE: Type 1 diabetes—a chronic autoimmune disease. N Engl J Med 314:1360, 1986.)
As with most autoimmune diseases, the pathogenesis of type 1 diabetes involves the interplay of genetic susceptibility and environmental factors. Genome-wide association studies (Chapter 6) have identified over 20 susceptibility loci for type 1 diabetes. Of these, the principal susceptibility locus for type 1 diabetes resides in the chromosomal region that encodes the class II MHC molecules on 6p21 (HLA-D). Between 90% and 95% of white patients with type 1 diabetes have HLA-DR3, or DR4, or both, in contrast with about 40% of normal subjects, and 40% to 50% of patients are DR3/DR4 heterozygotes, in contrast with 5% of normal subjects. Of note, despite the high relative risk in persons with particular class II alleles, most people who inherit these alleles do not develop diabetes. Several non-HLA genes also confer susceptibility to type 1 diabetes, including polymorphisms within the gene encoding insulin itself, as well as CTLA4 and PTPN22. CTLA-4 is an inhibitory receptor of T cells and PTPN-22 is a protein tyrosine phosphatase; both are thought to inhibit T cell responses, so polymorphisms that interfere with their functional activity are expected to set the stage for excessive T cell activation. Polymorphisms in the insulin gene may reduce expression of this protein in the thymus, thus reducing the elimination of T cells reactive with this self protein (Chapter 4). Additional evidence suggests that environmental factors, especially infections, may be involved in type 1 diabetes. It has been proposed that certain viruses (mumps, rubella, and coxsackie B viruses, in particular) may be an initiating trigger, perhaps because some viral antigens are antigenically similar to beta cell antigens (molecular mimicry), leading to bystander damage to the islets, but this idea is not conclusively established.
Type 2 Diabetes Mellitus
Type 2 diabetes is a prototypical complex multifactorial disease. Environmental factors, such as a sedentary life style and dietary habits, unequivocally play a role, as described in the subsequent discussion of the association with obesity. Genetic factors are also involved in the pathogenesis, as evidenced by the disease concordance rate of 35% to 60% in monozygotic twins compared with nearly half that in dizygotic twins. Such concordance is even greater than in type 1 diabetes, suggesting perhaps an even larger genetic component in type 2 diabetes. Additional evidence for a genetic basis has emerged from recent large-scale genome-wide association studies, which have identified more than a dozen susceptibility loci called “diabetogenic” genes. Unlike type 1 diabetes, however, the disease is not linked to genes involved in immune tolerance and regulation (e.g., HLA, CTLA4), and evidence of an autoimmune basis is lacking. The two metabolic defects that characterize type 2 diabetes are (1) a decreased ability of peripheral tissues to respond to insulin (insulin resistance) and (2) beta cell dysfunction that is manifested as inadequate insulin secretion in the face of insulin resistance and hyperglycemia (Fig. 19–23). Insulin resistance predates the development of hyperglycemia and usually is accompanied by compensatory beta cell hyperfunction and hyperinsulinemia in the early stages of the evolution of diabetes.
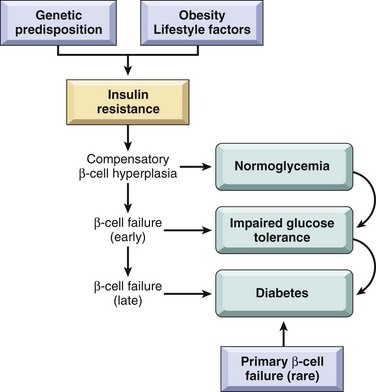
Figure 19–23 Pathogenesis of type 2 diabetes mellitus. Genetic predisposition and environmental influences converge to cause insulin resistance. Compensatory beta cell hyperplasia can maintain normoglycemia, but eventually beta cell secretory dysfunction sets in, leading to impaired glucose tolerance and, ultimately, frank diabetes. Rare instances of primary beta cell failure can lead directly to type 2 diabetes without an intervening state of insulin resistance.
Insulin Resistance
Insulin resistance is defined as the failure of target tissues to respond normally to insulin. It leads to decreased uptake of glucose in muscle, reduced glycolysis and fatty acid oxidation in the liver, and an inability to suppress hepatic gluconeogenesis. A variety of functional defects have been reported in the insulin signaling pathway in states of insulin resistance (for example, reduced phosphorylation-dependent activation of the insulin receptor and its downstream components), which attenuate signal transduction. Few factors play as important a role in the development of insulin resistance as obesity.
Obesity and Insulin Resistance
The association of obesity with type 2 diabetes has been recognized for decades, with visceral obesity being common in a majority of affected patients. Insulin resistance is present even with simple obesity unaccompanied by hyperglycemia, indicating a fundamental abnormality of insulin signaling in states of fatty excess. In fact, the term metabolic syndrome has been applied to a constellation of findings dominated by visceral obesity, which is accompanied by insulin resistance, glucose intolerance, and cardiovascular risk factors such as hypertension and abnormal lipid profiles (Chapter 7). In the absence of weight loss and lifestyle modifications, persons with metabolic syndrome are at significant risk for the development of frank type 2 diabetes, underscoring the importance of obesity to the pathogenesis of this disease. The risk of diabetes increases as the body mass index (a measure of body fat content) increases, suggesting a dose-response relationship between body fat and insulin resistance. Although many details of the so-called adipo-insulin axis remain to be elucidated, recognition of some of the putative pathways leading to insulin resistance has increased substantially (Fig. 19–24):
• Role of excess free fatty acids (FFAs): Cross-sectional studies have demonstrated an inverse correlation between fasting plasma FFAs and insulin sensitivity. The level of intracellular triglycerides often is markedly increased in muscle and liver tissues in obese persons, presumably because excess circulating FFAs are deposited in these organs. Intracellular triglycerides and products of fatty acid metabolism are potent inhibitors of insulin signaling and result in an acquired insulin resistance state. These lipotoxic effects of FFAs are mediated through a decrease in activity of key insulin-signaling proteins.
• Role of inflammation: Over the past several years, inflammation has emerged as a major player in the pathogenesis of type 2 diabetes. It is now known that a permissive inflammatory milieu (mediated not by an autoimmune process as in type 1 diabetes but rather by pro-inflammatory cytokines that are secreted in response to excess nutrients such as FFAs) results in both peripheral insulin resistance and beta cell dysfunction (see later). Excess FFAs within macrophages and beta cells can engage the inflammasome, a multiprotein cytoplasmic complex that leads to secretion of the cytokine interleukin IL-1β (Chapter 2). IL-1β, in turn, mediates the secretion of additional pro-inflammatory cytokines from macrophages, islets, and other cells that are released into the circulation and act on the major sites of insulin action to promote insulin resistance. Thus, excess FFAs can impede insulin signaling directly within peripheral tissues, as well as indirectly through the release of pro-inflammatory cytokines. Not surprisingly, there are now several ongoing trials of cytokine antagonists (particularly of IL-1β) in patients with type 2 diabetes.
• Role of adipokines: Adipose tissue is not merely a passive storage depot for fat; it can operate as a functional endocrine organ, releasing so-called adipokines in response to extracellular stimuli or changes in metabolic status. Thus, adipocytes also release IL-1β and other pro-inflammatory cytokines into the circulation in response to excess FFAs, which promote peripheral insulin resistance. By contrast, adiponectin is an adipokine with insulin sensitizing activity, which probably acts by dampening the inflammatory response.
• Peroxisome proliferator-activated receptor-γ (PPARγ): PPARγ is a nuclear receptor and transcription factor expressed in adipose tissue and plays a seminal role in adipocyte differentiation. A class of antidiabetic medications known as thiazolidinediones acts as agonist ligands for PPARγ and improves insulin sensitivity. Activation of PPARγ promotes secretion of antihyperglycemic adipokines such as adiponectin, and shifts the deposition of FFAs toward adipose tissue and away from liver and skeletal muscle.
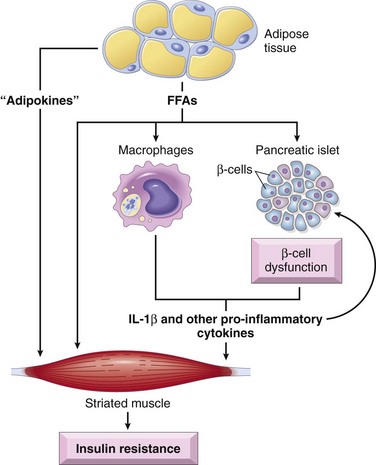
Figure 19–24 Mechanisms of beta cell dysfunction and insulin resistance in type 2 diabetes. Free fatty acids directly cause beta cell dysfunction and induce insulin resistance in target tissues (such as striated muscle, shown here), and also induce the secretion of pro-inflammatory cytokines that cause more beta cell dysfunction and insulin resistance.
Beta Cell Dysfunction
Beta cell dysfunction in type 2 diabetes reflects the inability of these cells to adapt themselves to the long-term demands of peripheral insulin resistance and increased insulin secretion. In states of insulin resistance, insulin secretion initially is higher for each level of glucose than in controls. This hyperinsulinemic state is a compensation for peripheral resistance and often can maintain normal plasma glucose for years. Eventually, however, beta cell compensation becomes inadequate, and there is progression to hyperglycemia, which is accompanied by an absolute loss in beta cell mass. The molecular mechanisms underlying beta cell dysfunction in type 2 diabetes are multifactorial and in many instances overlap with those implicated in insulin resistance. Thus, excess nutrients such as FFAs and glucose can promote the secretion of pro-inflammatory cytokines from beta cells, which leads to recruitment of mononuclear cells (macrophages and T cells) into the islets, resulting in more local cytokine production. The consequences of this abnormal inflammatory microenvironment are beta cell dysfunction and, ultimately, beta cell death. Amyloid replacement of islets is a characteristic finding in persons with long-standing type 2 diabetes and is present in more than 90% of diabetic islets examined (see later). The islet amyloid polypetide (IAPP), also known as amylin, is secreted by the beta cells in conjunction with insulin, and its abnormal aggregation results in amyloid. IAPP also engages the inflammasome and promotes IL-1β secretion, thus sustaining the inflammatory onslaught on surviving beta cells even late in the disease.
Monogenic Forms of Diabetes
Type 1 and type 2 diabetes are genetically complex, and despite the associations with multiple susceptibility loci, no single-gene defect (mutation) can account for predisposition to these entities. By contrast, monogenic forms of diabetes (Table 19–5) are uncommon examples of the diabetic phenotype occurring as a result of loss-of-function mutations within a single gene. Monogenic causes of diabetes include either a primary defect in beta cell function or a defect in insulin receptor signaling. The largest subgroup of patients in this category traditionally was designated as having maturity-onset diabetes of the young (MODY) because of its superficial resemblance to type 2 diabetes and its occurrence in younger patients; MODY can be the result of inactivating mutations in one of six genes. Other uncommon causes include maternally inherited diabetes and bilateral deafness, secondary to mitochondrial DNA mutations, and mutations within the insulin gene itself, which most commonly manifests with diabetes in the neonatal period. Finally, rare instances of insulin receptor mutations that affect receptor synthesis, insulin binding, or downstream signal transduction can cause severe insulin resistance, accompanied by hyperinsulinemia and diabetes.
Complications of Diabetes
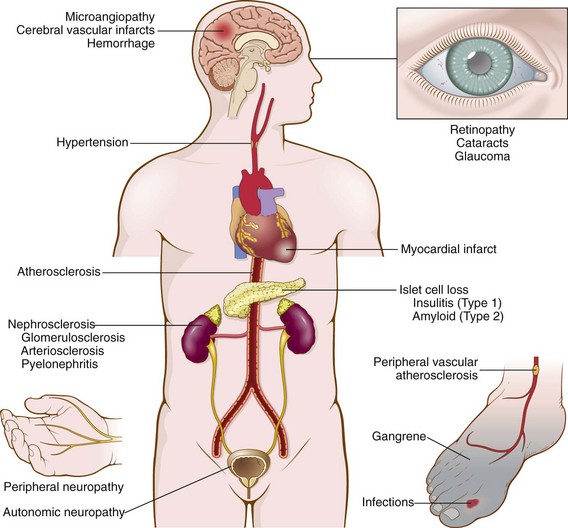
Diabetes can be a devastating disease because the abnormal glucose metabolism and other metabolic derangements have serious pathologic effects on virtually all the systems of the body. The most significant complications of diabetes are vascular abnormalities, renal damage, and lesions affecting the peripheral nerves and eyes (Fig. 19–25). The pathologic findings in these tissues and their clinical consequences are described below. There is extreme variability among patients in the time of onset of these complications, their severity, and the particular organ or organs involved. In persons with tight control of their diabetes, the onset may be delayed.
The pathogenesis of the long-term complications of diabetes is multifactorial, although persistent hyperglycemia (glucotoxicity) seems to be a key mediator. At least three distinct metabolic pathways seem to be involved in the pathogenesis of long-term complications; it is likely that all of them play a role in a tissue-specific manner.
1 Formation of advanced glycation end products (AGEs). AGEs are formed as a result of nonenzymatic reactions between intracellular glucose-derived precursors (glyoxal, methylglyoxal, and 3-deoxyglucosone) with the amino groups of both intracellular and extracellular proteins. The natural rate of AGE formation is greatly accelerated in the presence of hyperglycemia. AGEs bind to a specific receptor (RAGE), which is expressed on inflammatory cells (macrophages and T cells) and in endothelium and vascular smooth muscle. The detrimental effects of the AGE-RAGE signaling axis within the vascular compartment include
In addition to receptor-mediated effects, AGEs can directly cross-link extracellular matrix proteins, which decreases protein removal while enhancing protein deposition. AGEs cross-linked proteins can trap other plasma or interstitial proteins; for example, low-density lipoprotein (LDL) gets trapped within AGE-modified large vessel walls, accelerating atherosclerosis (Chapter 9), while albumin can get trapped within capillaries, accounting in part for the basement membrane thickening that is characteristic of diabetic microangiopathy (see later).
2 Activation of protein kinase C. Activation of intracellular protein kinase C (PKC) by calcium ions and the second messenger diacylglycerol (DAG) is an important signal transduction pathway in many cellular systems. Intracellular hyperglycemia can stimulate the de novo synthesis of DAG from glycolytic intermediates and hence cause activation of PKC. The downstream effects of PKC activation are numerous and include production of proangiogenic molecules such as vascular endothelial growth factor (VEGF), implicated in the neovascularization seen in diabetic retinopathy, and profibrogenic molecules such as transforming growth factor-β, leading to increased deposition of extracellular matrix and basement membrane material.
3 Disturbances in polyol pathways. In some tissues that do not require insulin for glucose transport (e.g., nerves, lens, kidneys, blood vessels), hyperglycemia leads to an increase in intracellular glucose that is then metabolized by the enzyme aldose reductase to sorbitol, a polyol, and eventually to fructose, in a reaction that uses NADPH (the reduced form of nicotinamide dinucleotide phosphate) as a cofactor. NADPH is also required by the enzyme glutathione reductase in a reaction that regenerates reduced glutathione (GSH). As described in Chapter 1, GSH is one of the important antioxidant mechanisms in the cell, and any reduction in GSH increases cellular susceptibility to oxidative stress. In neurons, persistent hyperglycemia appears to be the major underlying cause of diabetic neuropathy (glucose neurotoxicity).
 Morphology
Morphology
Diabetes and Its Late Complications
Pathologic findings in the diabetic pancreas are variable and not necessarily dramatic. The important morphologic changes are related to the many late systemic complications of diabetes. In most patients, morphologic changes are likely to be found in arteries (macrovascular disease), basement membranes of small vessels (microangiopathy), kidneys (diabetic nephropathy), retina (retinopathy), nerves (neuropathy), and other tissues. These changes are seen in both type 1 and type 2 diabetes (Fig. 19–25).
Pancreas
Lesions in the pancreas are inconstant and rarely of diagnostic value. One or more of the following alterations may be present:
• Reduction in the number and size of islets. This change most often is seen in type 1 diabetes, particularly with rapidly advancing disease. Most of the islets are small, inconspicuous, and not easily detected.
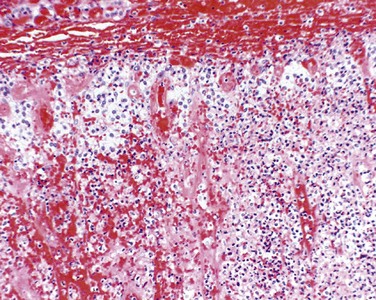
• Leukocytic infiltration of the islets, which are principally composed of mononuclear cells (lymphocytes and macrophages) (Fig. 19–26, A). Of note, both type 1 and type 2 diabetes may demonstrate islet inflammation early in the disease, although it is typically more severe in T1D. In both types inflammation is often absent by the time the disease is clinically evident.
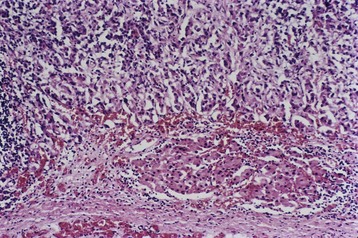
• Amyloid replacement of islets in long-standing type 2 diabetes, appearing as deposition of pink, amorphous material beginning in and around capillaries and between cells. At advanced stages the islets may be virtually obliterated (Fig. 19–26, B); fibrosis also may be observed. While inflammation is observed early in the natural history of type 2 diabetes, amyloid deposition occurs in long-standing cases.
• An increase in the number and size of islets, especially characteristic of nondiabetic newborns of diabetic mothers. Presumably, fetal islets undergo hyperplasia in response to the maternal hyperglycemia.
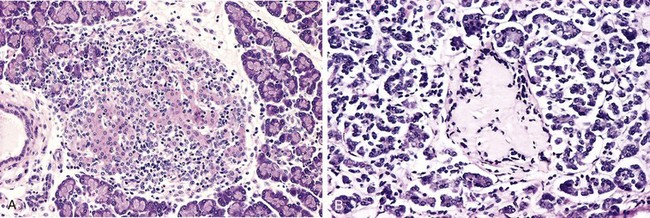
Figure 19–26 A, Autoimmune insulitis in a rat (BB) model of autoimmune diabetes. This disorder also is seen in type 1 human diabetes. B, Amyloidosis of a pancreatic islet in type 2 diabetes. Amyloidosis typically is observed late in the natural history of this form of diabetes, with islet inflammation noted at earlier observations.
(A, Courtesy of Dr. Arthur Like, University of Massachusetts, Worcester, Massachusetts.)
Diabetic Macrovascular Disease
Diabetes exacts a heavy toll on the vascular system. The hallmark of diabetic macrovascular disease is accelerated atherosclerosis affecting the aorta and large and medium-sized arteries. Except for its greater severity and earlier age at onset, atherosclerosis in diabetics is indistinguishable from that in nondiabetics (Chapter 9). Myocardial infarction, caused by atherosclerosis of the coronary arteries, is the most common cause of death in diabetics. Significantly, it is almost as common in diabetic women as in diabetic men. By contrast, myocardial infarction is uncommon in nondiabetic women of reproductive age. Gangrene of the lower extremities, as a result of advanced vascular disease, is about 100 times more common in persons with diabetes than in the general population. The larger renal arteries also are subject to severe atherosclerosis, but the most damaging effect of diabetes on the kidneys is exerted at the level of the glomeruli and the microcirculation, as discussed later on.
Hyaline arteriolosclerosis, the vascular lesion associated with hypertension (Chapters 9 and 13), is both more prevalent and more severe in diabetics than in nondiabetics, but it is not specific for diabetes and may be seen in elderly persons who do not suffer from either diabetes or hypertension. It takes the form of an amorphous, hyaline thickening of the wall of the arterioles, which causes narrowing of the lumen (Fig. 19–27). Not surprisingly, in diabetic patients, its severity is related not only to the duration of the disease but also to the presence or absence of hypertension.
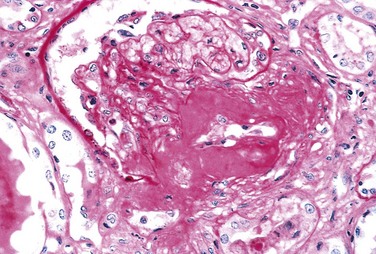
Figure 19–27 Severe renal hyaline arteriolosclerosis in a periodic acid–Schiff stained specimen. Note the markedly thickened, tortuous afferent arteriole. The amorphous nature of the thickened vascular wall is evident.
(Courtesy of Dr. M.A. Venkatachalam, Department of Pathology, University of Texas Health Science Center, San Antonio, Texas.)
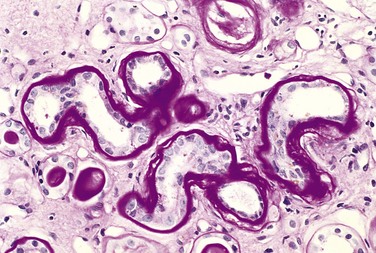
Diabetic Microangiopathy
One of the most consistent morphologic features of diabetes is diffuse thickening of basement membranes. The thickening is most evident in the capillaries of the skin, skeletal muscle, retina, renal glomeruli, and renal medulla. However, it also may be seen in such nonvascular structures as renal tubules, the Bowman capsule, peripheral nerves, and placenta. By both light and electron microscopy, the basal lamina separating parenchymal or endothelial cells from the surrounding tissue is markedly thickened by concentric layers of hyaline material composed predominantly of type IV collagen (Fig. 19–28). Of note, despite the increase in the thickness of basement membranes, diabetic capillaries are more leaky than normal to plasma proteins. The microangiopathy underlies the development of diabetic nephropathy, retinopathy, and some forms of neuropathy. An indistinguishable microangiopathy can be found in aged nondiabetic patients, but rarely to the extent seen in persons with long-standing diabetes.
Diabetic Nephropathy
The kidneys are prime targets of diabetes (see also Chapter 13). Renal failure is second only to myocardial infarction as a cause of death from this disease. Three lesions are encountered: (1) glomerular lesions; (2) renal vascular lesions, principally arteriolosclerosis; and (3) pyelonephritis, including necrotizing papillitis.
The most important glomerular lesions are capillary basement membrane thickening, diffuse mesangial sclerosis, and nodular glomerulosclerosis. The glomerular capillary basement membranes are thickened along their entire length. This change can be detected by electron microscopy within a few years of the onset of diabetes, sometimes without any associated change in renal function (Fig. 19–29).
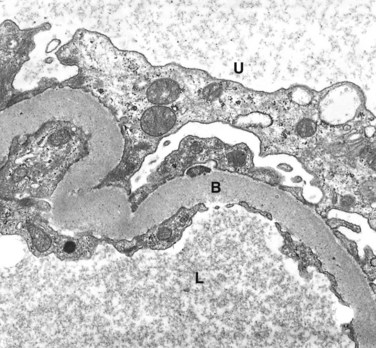
Figure 19–29 Renal glomerulus showing markedly thickened glomerular basement membrane (B) in a diabetic. L, glomerular capillary lumen; U, urinary space.
(Courtesy of Dr. Michael Kashgarian, Department of Pathology, Yale University School of Medicine, New Haven, Connecticut.)
Diffuse mesangial sclerosis consists of a diffuse increase in mesangial matrix along with mesangial cell proliferation and is always associated with basement membrane thickening. It is found in most individuals with disease of more than 10 years’ duration. When glomerulosclerosis becomes marked, patients manifest the nephrotic syndrome, characterized by proteinuria, hypoalbuminemia, and edema (Chapter 13).
Nodular glomerulosclerosis describes a glomerular lesion made distinctive by ball-like deposits of a laminated matrix situated in the periphery of the glomerulus (Fig. 19–30). These nodules are PAS-positive and usually contain trapped mesangial cells. This distinctive change has been called the Kimmelstiel-Wilson lesion, after the two pathologists who first described it. Nodular glomerulosclerosis is encountered in approximately 15% to 30% of persons with long-term diabetes and is a major contributor to morbidity and mortality. Diffuse mesangial sclerosis also may be seen in association with old age and hypertension; by contrast, the nodular form of glomerulosclerosis, once certain unusual forms of nephropathies have been excluded (Chapter 13), is essentially pathognomonic of diabetes. Both the diffuse and the nodular forms of glomerulosclerosis induce sufficient ischemia to cause scarring of the kidneys, manifested by a finely granular-appearing cortical surface (Fig. 19–31).
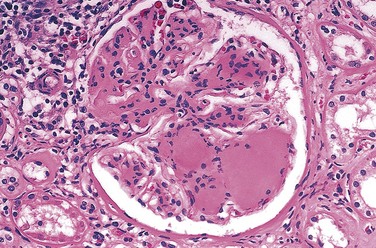
Figure 19–30 Nodular glomerulosclerosis in a renal specimen from a patient with long-standing diabetes.
(Courtesy of Dr. Lisa Yerian, Department of Pathology, University of Chicago, Chicago, Illinois.)
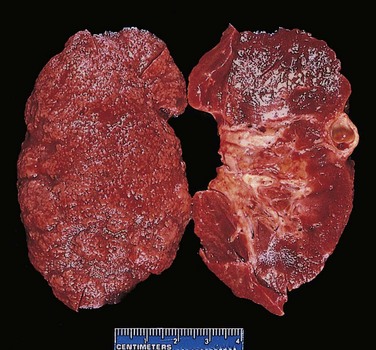
Figure 19–31 Nephrosclerosis in a patient with long-standing diabetes. The kidney has been bisected to demonstrate both diffuse granular transformation of the surface (left) and marked thinning of the cortical tissue (right). Additional features include some irregular depressions, the result of pyelonephritis, and an incidental cortical cyst (far right).
Renal atherosclerosis and arteriolosclerosis constitute part of the macrovascular disease seen in diabetics. The kidney is one of the most frequently and severely affected organs; however, the changes in the arteries and arterioles are similar to those found throughout the body. Hyaline arteriolosclerosis affects not only the afferent but also the efferent arterioles. Such efferent arteriolosclerosis is rarely if ever encountered in persons who do not have diabetes.
Pyelonephritis is an acute or chronic inflammation of the kidneys that usually begins in the interstitial tissue and then spreads to involve the tubules. Both the acute and chronic forms of this disease occur in nondiabetics as well as in diabetics but are more common in persons with diabetes than in the general population; once affected, diabetics tend to have more severe involvement. One special pattern of acute pyelonephritis, necrotizing papillitis (or papillary necrosis), is much more prevalent in diabetics than in nondiabetics.
Ocular Complications of Diabetes
Visual impairment, sometimes even total blindness, is one of the more feared consequences of long-standing diabetes. The ocular involvement may take the form of retinopathy, cataract formation, or glaucoma. Retinopathy, the most common pattern, consists of a constellation of changes that together are considered by many ophthalmologists to be virtually diagnostic of the disease. The lesion in the retina takes two forms: nonproliferative (background) retinopathy and proliferative retinopathy.
Nonproliferative retinopathy includes intraretinal or preretinal hemorrhages, retinal exudates, microaneurysms, venous dilations, edema, and, most importantly, thickening of the retinal capillaries (microangiopathy). The retinal exudates can be either “soft” (microinfarcts) or “hard” (deposits of plasma proteins and lipids) (Fig. 19–32). The microaneurysms are discrete saccular dilations of retinal choroidal capillaries that appear through the ophthalmoscope as small red dots. Dilations tend to occur at focal points of weakening, resulting from loss of pericytes. Retinal edema presumably results from excessive capillary permeability. Underlying all of these changes is the microangiopathy, which is thought to lead to loss of capillary pericytes and hence to focal weakening of capillary structure.
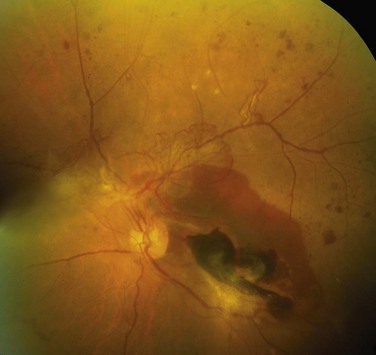
Figure 19–32 Characteristic morphologic changes of diabetic retinopathy. Features include advanced proliferative retinopathy with retinal hemorrhages, exudates, neovascularization, and tractional retinal detachment (lower right corner).
(Courtesy of Dr. Rajendra Apte, Washington University School of Medicine, St. Louis, Missouri.)
The so-called proliferative retinopathy is a process of neovascularization and fibrosis. This lesion leads to serious consequences, including blindness, especially if it involves the macula. Vitreous hemorrhages can result from rupture of newly formed capillaries; the subsequent organization of the hemorrhage can pull the retina off its substratum (retinal detachment).
Diabetic Neuropathy
The central and peripheral nervous systems are not spared by diabetes. The most frequent pattern of involvement is that of a peripheral, symmetric neuropathy of the lower extremities affecting both motor and sensory function, particularly the latter. Other forms include autonomic neuropathy, which produces disturbances in bowel and bladder function and sometimes sexual impotence, and diabetic mononeuropathy, which may manifest as sudden footdrop or wristdrop or isolated cranial nerve palsies. The neurologic changes may be the result of microangiopathy and increased permeability of the capillaries that supply the nerves, as well as direct axonal damage.
Clinical Features
It is difficult to discuss with brevity the diverse clinical presentations of diabetes mellitus. Only a few characteristic patterns are presented here. In the initial 1 or 2 years after manifestation of overt type 1 diabetes (referred to as the “honeymoon period”), exogenous insulin requirements may be minimal to none because of residual ongoing endogenous insulin secretion, but thereafter the beta cell reserve is exhausted and insulin requirements increase dramatically. Although beta cell destruction is a gradual process, the transition from impaired glucose tolerance to overt diabetes may be abrupt, heralded by an event associated with increased insulin requirements such as infection. The onset is marked by polyuria, polydipsia, polyphagia, and in severe cases, ketoacidosis, all resulting from metabolic derangements (Fig. 19–33).
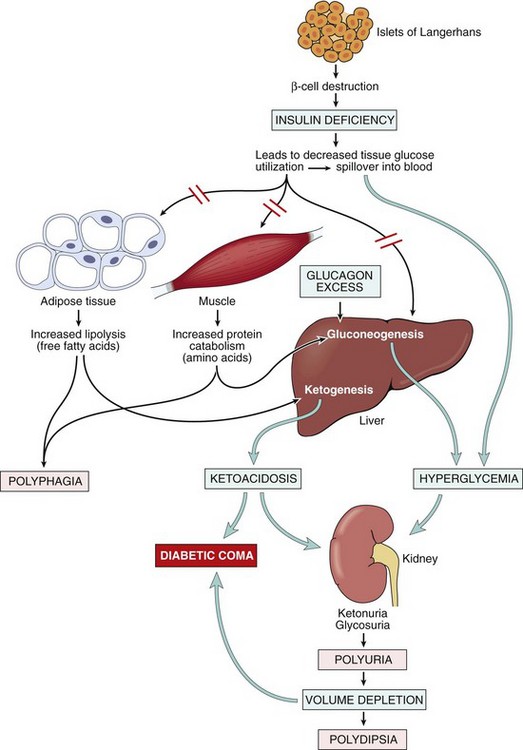
Figure 19–33 Sequence of metabolic derangements leading to diabetic coma in type 1 diabetes mellitus. An absolute insulin deficiency leads to a catabolic state, eventuating in ketoacidosis and severe volume depletion. These derangements bring about sufficient central nervous system compromise to cause coma and, eventually, death if left untreated.
Since insulin is a major anabolic hormone in the body, deficiency of insulin results in a catabolic state that affects not only glucose metabolism but also fat and protein metabolism. The assimilation of glucose into muscle and adipose tissue is sharply diminished or abolished. Not only does storage of glycogen in liver and muscle cease, but also reserves are depleted by glycogenolysis. The resultant hyperglycemia exceeds the renal threshold for reabsorption, and glycosuria ensues. The glycosuria induces an osmotic diuresis and, consequently, polyuria, causing a profound loss of water and electrolytes. The obligatory renal water loss combined with the hyperosmolarity resulting from the increased levels of glucose in the blood tends to deplete intracellular water, triggering the osmoreceptors of the thirst centers of the brain. This sequence of events generates intense thirst (polydipsia). With a deficiency of insulin, the scales swing from insulin-promoted anabolism to catabolism of proteins and fats. Proteolysis follows, and the gluconeogenic amino acids are removed by the liver and used as building blocks for glucose. The catabolism of proteins and fats tends to induce a negative energy balance, which in turn leads to increasing appetite (polyphagia), thus completing the classic triad of diabetes: polyuria, polydipsia, and polyphagia. Despite the increased appetite, catabolic effects prevail, resulting in weight loss and muscle weakness. The combination of polyphagia and weight loss is paradoxical and should always point to the diagnostic possibility of diabetes.
In patients with type 1 diabetes, deviations from normal dietary intake, unusual physical activity, infection, or any other forms of stress may rapidly influence the treacherously fragile metabolic balance, predisposing the affected person to diabetic ketoacidosis. The plasma glucose usually is in the range of 500 to 700 mg/dL as a result of absolute insulin deficiency and unopposed effects of counterregulatory hormones (epinephrine, glucagon). The marked hyperglycemia causes an osmotic diuresis and dehydration characteristic of the ketoacidotic state. The second major effect is activation of the ketogenic machinery. Insulin deficiency leads to activation of lipoprotein lipase, with resultant excessive breakdown of adipose stores, giving rise to increased FFAs, which are oxidized by the liver to produce ketones. Ketogenesis is an adaptive phenomenon in times of starvation, generating ketones as a source of energy for consumption by vital organs (e.g., brain). The rate at which ketones are formed may exceed the rate at which they can be used by peripheral tissues, leading to ketonemia and ketonuria. If the urinary excretion of ketones is compromised by dehydration, the accumulating ketones decrease blood pH, resulting in metabolic ketoacidosis.
Type 2 diabetes mellitus also may manifest with polyuria and polydipsia, but unlike in type 1 diabetes, patients often are older than 40 years and frequently are obese. Unfortunately, with the increase in obesity and sedentary life style in Western society, type 2 diabetes is now seen in children and adolescents with increasing frequency. In some cases, medical attention is sought because of unexplained weakness or weight loss. Most frequently, however, the diagnosis is made after routine blood or urine testing in asymptomatic persons.
In the decompensated state, patients with type 2 diabetes may develop hyperosmolar nonketotic coma. This syndrome is engendered by severe dehydration resulting from sustained osmotic diuresis and urinary fluid loss due to chronic hyperglycemia. Typically, the affected person is an elderly diabetic who is disabled by a stroke or an infection and is unable to maintain adequate water intake. The absence of ketoacidosis and its symptoms (nausea, vomiting, respiratory difficulties) delays recognition of the seriousness of the situation until the onset of severe dehydration and coma. Table 19–6 summarizes some of the pertinent clinical, genetic, and histopathologic features that distinguish between type 1 and type 2 diabetes.
Table 19–6 Type 1 Versus Type 2 Diabetes Mellitus
| Type 1 Diabetes Mellitus | Type 2 Diabetes Mellitus |
|---|---|
| Clinical | |
| Onset usually in childhood and adolescence | Onset usually in adulthood; increasing incidence in childhood and adolescence |
| Normal weight or weight loss preceding diagnosis | Vast majority of patients are obese (80%) |
| Progressive decrease in insulin levels | Increased blood insulin (early); normal or moderate decrease in insulin (late) |
| Circulating islet autoantibodies | No islet autoantibodies |
| Diabetic ketoacidosis in absence of insulin therapy | Nonketotic hyperosmolar coma |
| Genetics | |
| Major linkage to MHC class I and II genes; also linked to polymorphisms in CTLA4 and PTPN22 | No HLA linkage; linkage to candidate diabetogenic and obesity-related genes |
| Pathogenesis | |
| Dysfunction in regulatory T cells (Tregs) leading to breakdown in self-tolerance to islet autoantigens | Insulin resistance in peripheral tissues, failure of compensation by beta cells |
| Multiple obesity-associated factors (circulating nonesterified fatty acids, inflammatory mediators, adipocytokines) linked to pathogenesis of insulin resistance | |
| Pathology | |
| Autoimmune “insulitis” | Early: inflammation; late: amyloid deposition in islets |
| Beta cell depletion, islet atrophy | Mild beta cell depletion |
HLA, human leukocyte antigen; MHC, major histocompatibility complex.
As previously discussed, it is the long-term effects of diabetes, more than the acute metabolic complications, which are responsible for the overwhelming preponderance of morbidity and mortality attributable to this disease. In most instances, these complications appear approximately 15 to 20 years after the onset of hyperglycemia.
• In both forms of long-standing diabetes, cardiovascular events such as myocardial infarction, renal vascular insufficiency, and stroke (cerebrovascular accident) are the most common contributors to mortality. The impact of cardiovascular disease can be gauged by its involvement in as many as 80% of deaths among persons with type 2 diabetes; in fact, diabetics have a 3 to 7.5 times greater incidence of death from cardiovascular causes than nondiabetic populations. The hallmark of cardiovascular disease is accelerated atherosclerosis of the large and medium-sized arteries (i.e., macrovascular disease). The importance of obesity in the pathogenesis of insulin resistance has already been discussed, but it also is an independent risk factor for development of atherosclerosis.
• Diabetic nephropathy is a leading cause of end-stage renal disease in the United States. The earliest manifestation of diabetic nephropathy is the appearance of small amounts of albumin in the urine (greater than 30 but less than 300 mg/day—i.e., microalbuminuria). Without specific interventions, approximately 80% of patients with type 1 diabetes and 20% to 40% of those with type 2 diabetes will develop overt nephropathy with macroalbuminuria (excretion of more than 300 mg/day) over the succeeding 10 to 15 years, usually accompanied by the appearance of hypertension. The progression from overt nephropathy to end-stage renal disease can be highly variable and is evidenced by a progressive drop in glomerular filtration rate. By 20 years after diagnosis, more than 75% of persons with type 1 diabetes and about 20% of those with type 2 diabetes with overt nephropathy will develop end-stage renal disease, necessitating dialysis or renal transplantation.
• Visual impairment, sometimes even total blindness, is one of the more feared consequences of long-standing diabetes. This disease currently is the fourth leading cause of acquired blindness in the United States. Approximately 60% to 80% of patients develop some form of diabetic retinopathy approximately 15 to 20 years after diagnosis. In addition to retinopathy, diabetic patients also have an increased propensity for glaucoma and cataract formation, both of which contribute to visual impairment in diabetes.
• Diabetic neuropathy can elicit a variety of clinical syndromes, afflicting the central nervous system, peripheral sensorimotor nerves, and autonomic nervous system. The most frequent pattern of involvement is a distal symmetric polyneuropathy of the lower extremities that affects both motor and sensory function, particularly the latter (Chapter 21). Over time, the upper extremities may be involved as well, thus approximating a “glove and stocking” pattern of polyneuropathy. Other forms include autonomic neuropathy, which produces disturbances in bowel and bladder function and sometimes sexual impotence, and diabetic mononeuropathy, which may manifest as sudden footdrop, wristdrop, or isolated cranial nerve palsies.
• Diabetic patients are plagued by an enhanced susceptibility to infections of the skin, as well as to tuberculosis, pneumonia, and pyelonephritis. Such infections cause about 5% of diabetes-related deaths. In a person with diabetic neuropathy, a trivial infection in a toe may be the first event in a long succession of complications (gangrene, bacteremia, pneumonia) that may ultimately lead to death.
Several large-scale prospective studies have convincingly demonstrated that the long-term complications, and the associated morbidity and mortality, from diabetes are attenuated by strict glycemic control. For patients with type 1 diabetes, insulin replacement therapy is the mainstay of treatment, while non-pharmacologic approaches such as dietary restrictions and exercise (which improves insulin sensitivity) are often the “first line of defense” for type 2 diabetes. Most patients with type 2 diabetes will eventually require therapeutic intervention to reduce hyperglycemia, which can be achieved by administration of a number of agents that lower glucose levels through several distinct mechanisms of action. Glycemic control is assessed clinically by measuring the percentage of glycosylated hemoglobin, also known as HbA1C, which is formed by non-enzymatic addition of glucose moieties to hemoglobin in red cells. Unlike blood glucose levels, HbA1C is a measure of glycemic control over long periods of time (2 to 3 months) and is relatively unaffected by day-to-day variations. An HbA1C below 7% is taken as evidence of tight glycemic control, but patients with HbA1C levels in this range also have an increased risk of potentially life-threatening episodes of therapy-related hypoglycemia, and “optimal” control of glucose levels in diabetic patients remains an unsettled area of clinical investigation.
 Summary
Summary
Diabetes Mellitus: Pathogenesis and Long-Term Complications
• Type 1 diabetes is an autoimmune disease characterized by progressive destruction of islet beta cells, leading to absolute insulin deficiency. Both autoreactive T cells and autoantibodies are involved.
• Type 2 diabetes is caused by insulin resistance and beta cell dysfunction, resulting in relative insulin deficiency. Autoimmunity is not involved.
• Obesity has an important relationship with insulin resistance (and hence type 2 diabetes), probably mediated by cytokines released from adipose tissues (adipocytokines). Other players in the adipo-insulin axis include FFAs (which may cause lipotoxicity) and the PPARγ receptor, which modulates adipocytokine levels.
• Monogenic forms of diabetes are uncommon and are caused by single-gene defects that result in primary beta cell dysfunction (e.g., glucokinase mutation) or lead to abnormalities of insulin–insulin receptor signaling (e.g., insulin receptor gene mutations).
• The long-term complications of diabetes are similar in both types and affect mainly blood vessels, and the kidneys, nerves and eyes. The development of these complications is attributed to three underlying mechanisms: formation of AGEs, activation of PKC, and disturbances in polyol pathways leading to oxidative stress.
Pancreatic Neuroendocrine Tumors
Pancreatic neuroendocrine tumors (PanNETs), also known as islet cell tumors, are rare in comparison with tumors of the exocrine pancreas, accounting for only 2% of all pancreatic neoplasms. PanNETs are most common in adults and may be single or multifocal; when they are malignant, the liver is the most common site of organ metastases. These tumors have a propensity to elaborate pancreatic hormones, but some are nonfunctional. The latter typically are larger lesions at diagnosis, since they come to clinical attention later in their natural history than functional PanNETs, which often present with symptoms related to excessive hormone production. All PanNETs, with the exception of insulinomas (see later), are regarded as having malignant potential, and in fact, 65% to 80% of PanNETs manifest with overtly malignant features of biologic aggressiveness, such as invasion into local tissues or distant metastases. The proliferative rate of PanNETs (measured using either mitotic counts or nuclear labeling with the proliferation marker Ki-67) is one of the best correlates of outcome. Genomic sequencing of sporadic PanNETs has identified recurrent somatic alterations in three major genes or pathways:
• MEN1, which causes familial MEN syndrome, type 1 (see later), is also mutated in many sporadic neuroendocrine tumors
• Loss-of-function mutations in tumor suppressor genes such as PTEN and TSC2, which are negative regulators of the oncogenic mammalian TOR (mTOR) signaling pathway
• Inactivating mutations in two genes, ATRX and DAXX, which have multiple cellular functions. Of note, nearly half of PanNETs have a somatic mutation in either ATRX or DAXX, but not both, suggesting that the encoded proteins function in a critical but redundant pathway.
Insulinomas
Beta cell tumors (insulinomas) are the most common type of PanNET and may be responsible for the elaboration of sufficient insulin to induce clinically significant hypoglycemia. The characteristic clinical picture is dominated by attacks of hypoglycemia, which occur when plasma blood glucose levels fall below 50 mg/dL. The attacks consist principally of such central nervous system manifestations as confusion, stupor, and loss of consciousness. They are precipitated by fasting or exercise and are promptly relieved by feeding or parenteral administration of glucose. Most insulinomas are cured by surgical resection.
Morphology
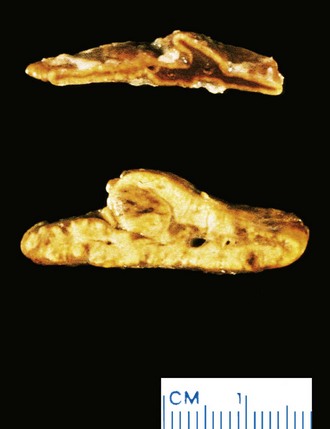
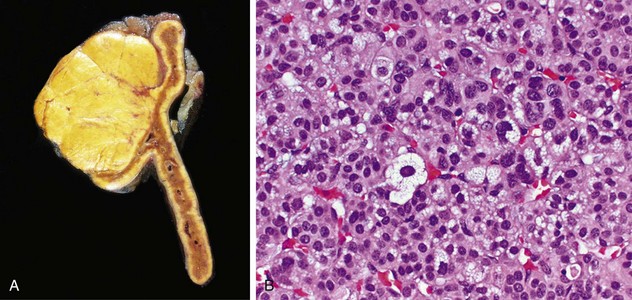
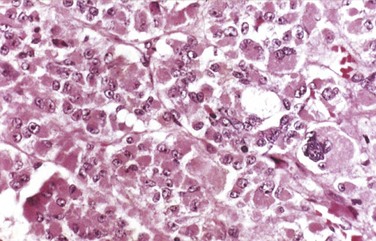
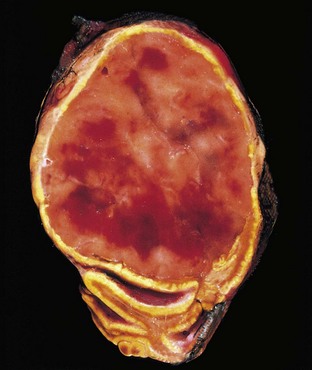
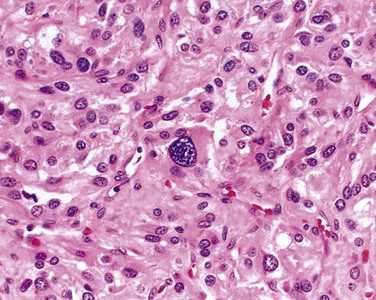
Insulinomas exhibit favorable biologic behavior, possibly because the vast majority are identified while they are small (less than 2 cm in diameter) and localized to the pancreas. Most are solitary lesions, although multifocal tumors or tumors ectopic to the pancreas may be encountered. Malignancy in insulinomas, constituting less than 10% of cases, is diagnosed on the basis of local invasion or metastases. On histologic examination, these benign tumors look remarkably like giant islets, with preservation of the regular cords of monotonous cells and their orientation to the vasculature. Not even malignant lesions present much evidence of anaplasia, and they may be deceptively encapsulated. Deposition of amyloid in the extracellular tissue is a characteristic feature of many insulinomas (Fig. 19–34, A). Under the electron microscope, neoplastic beta cells, like their normal counterparts, display distinctive round granules (Fig. 19–34, B).
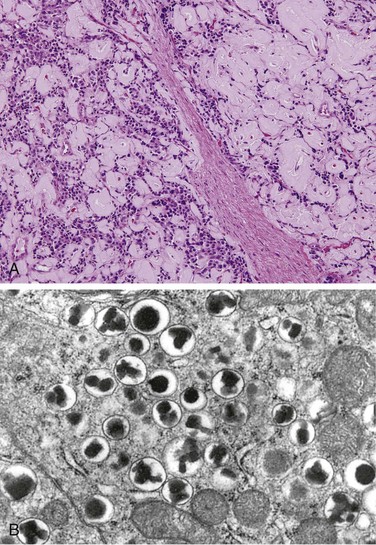
Figure 19–34 Pancreatic neuroendocrine tumor (PanNET), also called islet cell tumor. A, The neoplastic cells are monotonous in appearance and demonstrate minimal pleomorphism or mitotic activity. There is abundant amyloid deposition, characteristic of an insulinoma. On clinical evaluation, the patient had episodic hypoglycemia. B, Electron micrograph of a normal beta cell shows the characteristic membrane-bound granules, each containing a dense, often rectangular core and distinct halo. Insulinomas contain comparable granules.
Gastrinomas
Marked hypersecretion of gastrin usually has its origin in gastrin-producing tumors (gastrinomas), which are just as likely to arise in the duodenum and peripancreatic soft tissues as in the pancreas (the so-called gastrinoma triangle). Zollinger and Ellison first called attention to the association of pancreatic islet cell lesions with hypersecretion of gastric acid and severe peptic ulceration, which are present in 90% to 95% of patients with gastrinomas—the clinical hallmark of Zollinger-Ellison syndrome. In this condition, hypergastrinemia from a pancreatic or duodenal tumor stimulates extreme gastric acid secretion, which in turn causes peptic ulceration. The duodenal and gastric ulcers often are multiple; although they are identical to those found in the general population, they often are unresponsive to usual therapy. In addition, ulcers may occur in unusual locations such as the jejunum; when intractable jejunal ulcers are found, Zollinger-Ellison syndrome should be considered. More than half of the affected patients have diarrhea; in 30%, it is the presenting manifestation.
Morphology
Gastrinomas may arise in the pancreas, the peripancreatic region, or the wall of the duodenum. Over half of gastrin-producing tumors are locally invasive or have already metastasized at the time of diagnosis. In approximately 25% of patients, gastrinomas arise in conjunction with other endocrine tumors, thus conforming to the MEN-1 syndrome (see further on); MEN-1–associated gastrinomas frequently are multifocal, while sporadic gastrinomas usually are single. As with insulin-secreting tumors of the pancreas, gastrin-producing tumors are histologically bland and rarely exhibit marked anaplasia.