Cardiac arrhythmias
Basic cardiac electrophysiology
Control of cell depolarisation in pacemaker and non-pacemaker cells
Control of cell repolarisation in pacemaker and non-pacemaker cells
Mechanisms of arrhythmogenesis
Basic cardiac electrophysiology
Action potentials in myocardial cells and the resultant highly regulated cardiac contractions are a product of transmembrane ion currents generated by the movement of ions through membrane channels (Ch. 1). A variety of specific channels exist for transmembrane Na+, Ca2+ and K+ transport in the myocardium (Figs 8.1 and 8.2). These channels cycle through three states: resting, open or closed (inactive and refractory). Whether the ion channels are open to allow ion flow or are closed is determined by the electrical potential across the cell membrane. Therefore they are called voltage-gated ion channels. The direction in which ions move is dependent upon the type of channel, the concentration gradient of the ions and the transmembrane electrical potential (Figs 8.1 and 8.2). As a result of activity in these ion channels, the resting potential inside a cardiac cell is approximately −70 to −80 mV compared with the extracellular environment, although this varies among different types of cells in the heart.
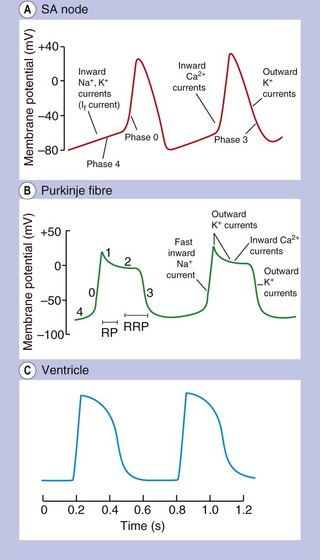
Fig. 8.1 Action potentials show variations in patterns among different populations of myocytes in different regions of the heart. The patterns are determined by the opening and closing of selective gates for Na+, Ca2+ and K+, and this figure should be studied in conjunction with Figure 8.2. The overall stability of the resting transmembrane ionic balance is controlled by active pumps, such as the Na+/K+-ATPase pump, which maintain the Na+ concentration gradient of 140 mM outside the cell versus 10–15 mM inside, and the K+ concentration gradient of 140 mM inside and 4 mM outside the cell. This results in an electrical potential at rest of approximately −70 to −80 mV inside the cell relative to 0 mV outside the cell. Large ion fluxes at rest are prevented by specific pumps and closure of voltage-gated gates. Action potentials in the atrioventricular (AV) node, bundle of His and ventricle are controlled by the sinoatrial (SA) node when the heart is in sinus rhythm. The rate of spontaneous depolarisation of the SA node determines its primacy as a pacemaker in the healthy heart. Phase 0 (B, C) occurs when the membrane potential reaches a defined threshold potential and an ‘all-or-none’ influx of Na+ through voltage-dependent fast Na+ channels occurs; this is transient and the gates close after a few milliseconds. Phase 0 is much slower in the SA and AV nodes than in ventricular cells, and depends mainly upon Ca2+ influx (A). This causes the conduction velocity in the SA node to be considerably less than that in the Purkinje fibres, and the refractory period is longer in proportion to the total duration of the action potential. Phase 1, called the early repolarisation and notch, results from K+ efflux (the transient outward (Ito) current) and reduced Na+ influx (Fig. 8.2). The phase 2 plateau is primarily a result of Ca2+ influx (slow inward, or SI, current) which is balanced by K+ efflux over a slow time course. Phase 3 repolarisation results from inactivation of Ca2+ influx and increasing K+ efflux via a number of currents (see text, Table 8.1 and Fig. 8.2). Part of the overall importance of the K+ currents is to maintain a stable resting membrane potential. Phase 4 (A, B) is termed the diastolic or pacemaker depolarisation generated on hyperpolarisation. The phase 4 inward ‘funny’ current (If) involves Na+ and K+ and is gated both by changes in voltage and by cAMP. If controls the rate of spontaneous beating of the heart and is regulated by the sympathetic and parasympathetic nervous systems. Ca2+ currents may also be involved in pacemaker activity in Phase 4. RP, absolute refractory period; RRP, relative refractory period.
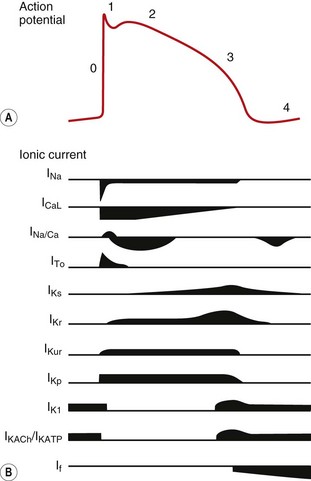
Fig. 8.2 A schematic representation of the influx and efflux of Na+, Ca2+ and K+ in Purkinje fibres. This figure should be examined in conjunction with Table 8.1, with other explanations given in the text. A downward inflection represents influx of the ion, and upward represents efflux. (Modified and reproduced with permission from Tamargo et al. 2004.)
When activated, depolarisation of these cells generates an action potential. The sinoatrial (SA) node, atrioventricular (AV) node, bundle of His and Purkinje system are part of the specialised conducting system of the heart that ensures the depolarizing impulse is conducted rapidly and synchronously through ventricular myocardium. In addition, the AV node slows impulse conduction to allow time for completion of mechanical events in the atrium and for ventricular filling to be completed. Once the impulse is passed to myocytes, depolarisation initiates contraction and also triggers depolarisation in adjacent myocytes. This ensures conduction of the impulse progressively through the myocardium.
The action potentials in cells of the sinoatrial (SA) and atrioventricular (AV) nodes, Purkinje fibres and ventricular muscle vary substantially in their characteristics (Figs 8.1, 8.2 and see Fig. 8.5). Action potentials in cardiac cells can be divided into four phases (Figs 8.1A and 8.2), although phases 1 and 2 are not clearly evident in the SA and AV nodes because of the different ion channels that are activated (Fig. 8.1A).
Cells with pacemaker activity
The myocardial cells of the specialised conducting system (SA node, AV node, bundle of His and Purkinje system) are distinguished electrophysiologically from cardiac myocytes by their intrinsic ability to depolarise spontaneously in phase 4, and independently generate an action potential. These cells are said to possess automaticity and are termed pacemaker cells. Cardiac myocytes have a stable resting potential and do not show phase 4 spontaneous depolarisation (compare Fig. 8.1A and 8.1C).
The primary pacemaker that drives normal repetitive cardiac contractions is the SA node (producing a normal sinus rhythm of 60–100 impulses per minute at rest). These secondary pacemaker, the AV node, depolarises more slowly and can generate 40–60 impulses per minute, while the tertiary pacemakers (the bundle of His, its branches and the Purkinje fibres) can fire 20–40 times per minute. The secondary and tertiary pacemakers will only be utilised if there is a failure of pacemakers that have a faster rate of spontaneous depolarisation.
Control of cell depolarisation in pacemaker and non-pacemaker cells
Slow spontaneous depolarisation in phase 4 in all pacemaker cells results from influx of positive ions (Na+ and K+) into the cell through f-channels that generate the strangely termed cardiac pacemaker ‘funny’ current (If) (Figs 8.1A and 8.2). The If is unusual in being generated by mixed-ion transport through a single channel, in being activated by the negative intracellular potential in diastole and in having slow activation and deactivation rates. Non-pacemaker cells have a minimal or no funny current.
The intrinsic rate of firing of a pacemaker cell depends on four factors:
 the resting intracellular potential,
the resting intracellular potential,
the resting potential at which the If is activated,
Activation of the If in the SA node is modulated by the autonomic nervous system via intracellular cAMP. Stimulation of β1-adrenoceptors generates cAMP and shifts activation of the channel to a less negative intracellular voltage, while vagal stimulation via muscarinic M2 receptors inhibits cAMP production and then activation of the channel requires a more negative intracellular voltage.
Full depolarisation of pacemaker cells occurs when, as a result of the If, the internal membrane potential reaches a threshold potential that opens voltage-gated L-type Ca2+ channels so that Ca2+ influx into the cell occurs (phase 0) (Fig. 8.1A; ICaL in Fig. 8.2). The SA node is the dominant pacemaker because activation of the If current in the SA node occurs at a less negative intracellular potential and has a faster intrinsic activation rate than in the AV node or Purkinje fibres. Therefore, spontaneous diastolic depolarisation in the SA node is initiated earlier and the threshold potential for full depolarisation is reached sooner.
Slow influx of Ca2+ in phase 0 is also the reason for slow conduction of the impulse through the AV node. In contrast, phase 0 of the action potential in Purkinje fibres and in atrial and ventricular myocytes is initiated by a rapid influx of Na+ through voltage-gated Na+ ion channels (fast Na+ channels) (Fig. 8.1B; INa in Fig. 8.2).
At the end of phase 0, the intracellular voltage potential briefly becomes positive, at which point a voltage-triggered ‘gate’ closes and inactivates the Na+ or Ca2+ channels and prevents further depolarisation (Fig. 8.1a,b).
Control of cell repolarisation in pacemaker and non-pacemaker cells
Once depolarised, cardiac cells then undergo a process of repolarisation to return the membrane potential to its resting level. This creates the conditions for the next action potential to be initiated (Figs 8.1 and 8.2). In both pacemaker and non-pacemaker cells, repolarisation (phase 3) is achieved by the opening of cell membrane K+ channels known as rectifiers (see Table 8.1 for explanation). In the Purkinje system and non-pacemaker cardiac cells, repolarisation is delayed by influx of Ca2+ through L-type channels (phase 2, the plateau phase, ICaL in Fig. 8.2), which balances K+ efflux (Fig. 8.1B, phase 2). Eventually in these cells the K+ current dominates and the cell returns to a negative intracellular resting potential.
Table 8.1
Selected examples of K+ channels and associated currents
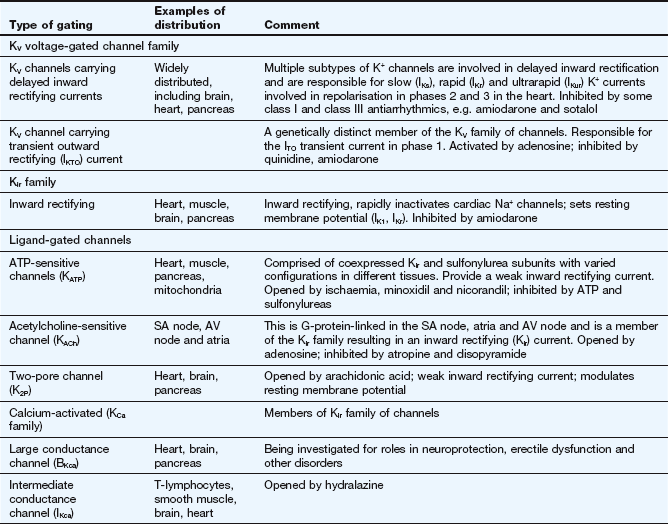
This table should be studied in conjunction with Fig. 8.2.
Potassium channels are diverse in structure and behaviour. Each channel consists of four membrane-spanning subunits, with each subunit consisting of two to six linked membrane segments which make up the water-filled pore. This allows many different configurations of K+ channels, many of which have particular physiological roles. Channels with subunit variations are associated with different types of current which are involved in repolarisation at different phases of the cardiac action potential. The channels can be open or closed depending upon the voltage across the cell or the presence of a selective ligand.
Rectifying current: an inward rectifying current means that under conditions of equivalent but opposing electrochemical potentials these channels pass more current inwards than outwards. An outward rectifying current is similar but in an outward direction.
In the resting phase between action potentials, Na+ and K+ transmembrane concentration gradients are restored by a separate exchange pump (Na+/K+-ATPase; see Fig. 7.4), and intracellular Ca2+ concentration is restored by a Na+/Ca2+ exchange pump. The negative internal resting membrane potential is maintained by high K+ permeability of resting cell membranes through inward rectifying voltage- and ligand-gated K+ channels which close when the cell depolarises (see also Ch. 1).
During the period between phase 0 and the end of phase 2 of the action potential the myocardial cell is refractory to further depolarisation (the absolute refractory period, RP). This is because the depolarising channels are inactivated until a sufficiently negative potential is restored inside the cell. During phase 3, a large depolarising stimulus can open sufficient Na+ or Ca2+ channels (many of which will have recovered to the resting state) to overcome the K+ efflux and initiate a further action potential. This part of the action potential is the relative refractory period (RRP; Fig. 8.1B).
The sum of the individual electrical currents that pass from one cell to another through the heart can be recorded as the surface electrocardiogram (ECG) (Fig. 8.3).
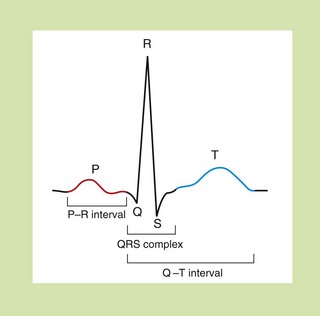
Fig. 8.3 The waveform for cardiac events seen on a surface electrocardiogram. The P wave represents the spread of depolarisation through the atria, and the QRS complex is the spread through the ventricles. The T wave represents repolarisation of the ventricle. The P–R interval is the time of conductance from atrium to ventricles, and the QRS time is the time the ventricles are activated. The duration of the ventricle action potential is given by the Q–T interval.
Mechanisms of arrhythmogenesis
Arrhythmias are disorders of rate and rhythm of the heart, which can arise as the result of either abnormal impulse generation or abnormal impulse conduction. There are three principal mechanisms of arrhythmogenesis.
Increased automaticity. Dominant ectopic pacemakers (pacemakers other than the SA node) can arise when pacemaker cells in the specialised conducting tissue develop a more rapid phase 4 depolarisation than the SA node. Ectopic pacemakers can also arise when rapid spontaneous phase 4 depolarisation develops in myocytes that usually have a stable phase 4. Ischaemia, or other changes in the microcellular environment, can create conditions that allow a non-specialised myocardial cell to express f-channels and become a pacemaker.
Re-entry. This is the cause of most clinically important arrhythmias. It is initiated when a depolarising impulse (often a premature ectopic beat from elsewhere in the heart) arrives at a part of the myocardium that is still in its refractory period. This is usually a fast conducting pathway with slow recovery from depolarisation. The impulse will bypass the refractory tissue. If this impulse is conducted through an adjacent part of the myocardium with slower conduction but fast recovery, then the impulse may subsequently arrive at the distal part of the refractory tissue when this tissue has had sufficient time to repolarise. The impulse will then be conducted retrogradely through this previously refractory tissue (Fig. 8.4C). If there has been sufficient time for the fast recovery myocardium proximal to the block to repolarise, a self-perpetuating circuit of electrical activity will be initiated (a re-entry circuit). The re-entry circuit acts as a pacemaker that initiates impulses that are then propagated through the heart. Such functional re-entry circuits can be localised within a small area of myocardium that has been damaged by fibrosis or ischaemia (micro re-entry). Micro re-entry circuits may arise in the atria, within the AV node or in the ventricles. The myocardium can also support large anatomical re-entry circuits (macro re-entry). These circuits arise when there is a congenital accessory pathway that bypasses the AV node, and conducts electrical activity between the atria and ventricles. The re-entry circuit between the atria and ventricles includes both the accessory pathway and the AV node (such as occurs in the Wolff–Parkinson–White syndrome).
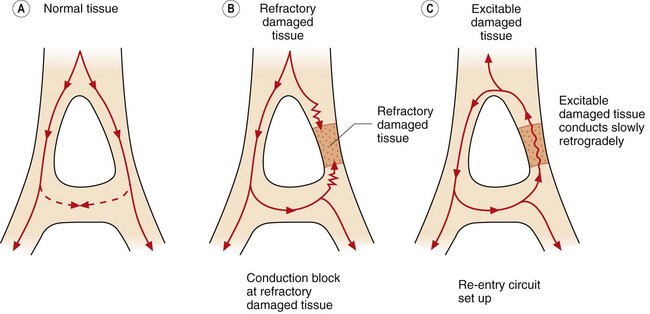
Fig. 8.4 Conduction in normal and damaged cardiac tissue. (A) In normal tissue, conduction is carefully ordered. When an action potential has been generated, the cells cannot be immediately reactivated because of refractoriness of the myocardial cells. If conducted impulses meet, they die out. (B) If an area of damage or dysfunction is present, impulses are conducted abnormally. If an action potential arrives at the area of damaged tissue and it is fully refractory, the impulse is blocked and an arrhythmia will not develop. (C) If an action potential arrives at a damaged area and it is capable of being excited and conducting in a retrograde direction, a perpetuating abnormal re-entry circuit may be set up.
Triggered activity. A cell can develop transient depolarisations during or following repolarisation (‘afterdepolarisations’), which will initiate an action potential if they reach the threshold potential of the cell. Afterdepolarisations are said to be ‘early’ if they occur during repolarisation (relative refractory period in Fig. 8.1B), or ‘delayed’ if they occur in phase 4. Early afterdepolarisations are due to opening of L-type Ca2+ channels or fast Na+ channels. Delayed afterdepolarisations are due to intracellular Ca2+ overload and follow spontaneous Ca2+ release from the sarcoplasmic reticulum during prolonged repolarisation. This activates the 3Na+/Ca2+ exchange transporter and produces a net depolarising current. Triggered activity is an uncommon mechanism of arrhythmogenesis but may be responsible for the proarrhythmic activity of class Ia and III antiarrhythmic agents (early afterdepolarisations) and digitalis glycosides (delayed afterdepolarisations) (see below).
Classification of antiarrhythmic drugs
A widely used classification of antiarrhythmic drugs (the Vaughan Williams classification) is based on their effects on the action potential (Fig. 8.5). This classification has many flaws and does not take account of the multiple actions possessed by some drugs, or the fact that the effects of drugs on diseased myocardium may be different from that on healthy myocardium. However, there is no widely accepted alternative classification.
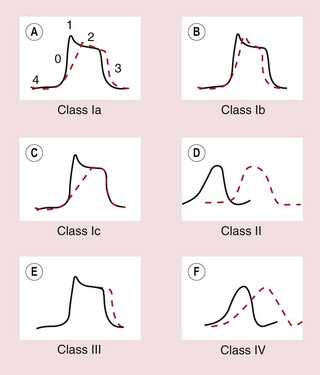
Fig. 8.5 Effects of different classes of antiarrhythmic drug on the cardiac action potential. (A, B, C, E) Drug effects on ventricular cells; (D, F) effects on the AV node. (A) Class Ia drugs. Block fast Na+ channels in phase 0 with moderate potency, and some K+ channels. Repolarisation is prolonged. (B) Class Ib drugs. Weakly block fast Na+ channels in phase 0 only in abnormal tissue; little effect on K+ channels. (C) Class Ic drugs. Potently block fast Na+ channels and weakly block Ca2+ channels and some K+ channels. (D) Class II drugs. Reduce phase 4 and phase 0 depolarisation in AV and SA nodes. Repolarisation in the AV node is prolonged. (E) Class III drugs. Block some K+ channels, inhibiting repolarisation and prolonging the action potential. (F) Class IV drugs. Block L-type Ca2+ channels, slowing phase 0 and phase 4 depolarisation, particularly in the AV node, with less effect in the SA node. Repolarisation is prolonged.
Vaughan Williams classification
The Vaughan Williams classification recognises four classes of drug (Table 8.2).

Class I
All class I drugs inhibit fast Na+ channels and slow the rate of rise of phase 0, and therefore reduce the excitability of the myocardial cell. They are often called membrane stabilisers. They readily penetrate the phospholipid bilayers of the cell membrane, where they concentrate in the hydrophobic core and bind to hydrophobic amino acids in the Na+ channel. Class I drugs are subdivided according to their effects on the duration of the action potential.
Class Ia drugs, such as disopyramide, slow impulse conduction by producing moderate Na+ channel blockade. In addition, they block some K+ channels (Table 8.1, Figs 8.1 and 8.5A), which prolongs repolarisation and therefore extends the duration of the action potential. They are effective for the treatment of both supraventricular and ventricular arrhythmias (Table 8.2).
Class Ib drugs, such as lidocaine, slow impulse conduction by producing weak Na+ channel blockade in abnormal tissue (such as ischaemic myocardium) with no effect in healthy tissue. They do not block K+ channels and have either no effect on repolarisation or may shorten it (Fig. 8.5B). They are only effective for the treatment of ventricular arrhythmias.
Class Ic drugs, such as flecainide, slow impulse conduction by producing marked Na+ channel blockade. They produce weak blockade of some K+ channels (Table 8.1), and also block inward Ca2+ channels. There is minimal effect on repolarisation (Fig. 8.5C). They are effective for the treatment of both atrial and ventricular arrhythmias.
The different effects of the class I subgroups result from their diverse ion-channel binding characteristics. During the time course of the action potential, the access of the drug to its binding site is intermittent and dependent on the state of the channel. Class Ib drugs (such as lidocaine) show marked use-dependency; that is, the action increases with the frequency of opening of the channel. They associate more rapidly with Na+ channels during depolarisation and they rapidly dissociate from the channel when it returns to the resting state. Therefore they are more effective when there are repetitive depolarisations, and they will effectively block premature impulses. Cells in ischaemic myocardium are more likely to be depolarised, which explains the selectivity of class Ib drugs for ventricular arrhythmias in ischaemic heart disease (ischaemia mainly affects the ventricles). Class Ic drugs (such as flecainide) also show use-dependent binding, but dissociate slowly from their binding sites in the Na+ channel and therefore produce prolonged blockade. This results in a widespread reduction in cellular excitability. Class Ia drugs (such as disopyramide) have binding characteristics between those of the other two subgroups.
Class II
The class II drugs are the β-adrenoceptor antagonists (β-blockers) which block the actions of catecholamines on the heart. They reduce the rate of spontaneous depolarisation of SA and AV nodal tissue and reduce conduction through the AV node through their action on adrenoceptor-sensitive f-channels (Fig. 8.5D). They can also inhibit ectopic pacemakers that have developed automaticity. Beta-adrenoceptor antagonists are effective for the treatment of both supraventricular and ventricular arrhythmias, particularly if these are catecholamine-dependent.
Class III
Class III drugs such as amiodarone prolong the duration of the action potential by inhibiting some K+ channels involved in repolarisation, thus increasing the absolute refractory period (Table 8.1, Fig. 8.5E). They are effective for the treatment of both supraventricular and ventricular arrhythmias.
Class IV
There are two types of Ca2+ channel in the heart but the class IV drugs are calcium channel blockers that selectively block the L-type Ca2+ channel. They slow conduction of the action potential particularly in the AV node. They have a lesser effect on the rate of depolarisation at the SA node (Fig. 8.5F). They are only effective for the treatment of supraventricular arrhythmias.
Proarrhythmic activity of antiarrhythmic drugs
Many antiarrhythmic drugs have the potential to precipitate serious arrhythmias, such as incessant ventricular tachycardia. Several of them (particularly class Ia agents and sotalol) prolong the Q–T interval on the ECG (Fig. 8.3). This predisposes to a polymorphic ventricular tachycardia known as torsade de pointes, which has a characteristic twisting QRS axis on the ECG and can degenerate into ventricular fibrillation. Drug-induced ventricular rhythm disturbances are particularly refractory to treatment.
There are probably multiple mechanisms of drug-induced arrhythmogenesis. Several risk factors have been identified, including the following.
Excessive slowing of cardiac impulse conduction, such as occurs with marked blockade of Na+ channels.
Excessive prolongation of the action potential, especially if this is due to blockade of the IKr repolarising current (Table 8.1, Fig. 8.2). Prolonged repolarisation may cause early afterdepolarisations (see above), or a selective effect of the drugs on some cells in the myocardium can lead to differential rates of repolarisation that predisposes to re-entry circuits.
Mutations in genes coding for channels that regulate Na+, K+ and Ca2+ transmembrane ion flows exist in 5–10% of people, and are probably subclinical variants of the congenital long QT syndrome. These individuals are more susceptible to torsade de pointes when exposed to a drug that prolongs the Q–T interval.
Structural heart disease, especially ischaemic heart disease, with greater slowing of conduction in diseased myocardium.
Female sex: about 70% of those who develop torsade de pointes are women.
Class Ia drugs
Disopyramide is no longer widely used in the UK because of its unwanted effects.
Pharmacokinetics: Oral absorption of disopyramide is almost complete. Metabolism in the liver generates a compound with less antiarrhythmic activity but with greater antimuscarinic activity.
Gastrointestinal disturbances.
Powerful negative inotropic effect; disopyramide should be avoided in people with left ventricular dysfunction.
Proarrhythmic effects (see above).
Antimuscarinic effects (see Ch. 4), especially urinary retention, dry mouth and blurred vision.
Class Ib drugs
Pharmacokinetics: Extensive first-pass metabolism to a potentially toxic metabolite means that oral administration of lidocaine is not practicable. It is given intravenously, initially as a loading dose by bolus injection followed by an infusion. Lidocaine is extensively metabolised in the liver to compounds with little antiarrhythmic activity, although one can cause seizures. The half-life of lidocaine is 2 h.
Class Ic drugs
Propafenone
Propafenone has weak β-adrenoceptor antagonist activity in addition to its class Ic action.
Pharmacokinetics: Oral absorption of propafenone is almost complete, but extensive first-pass metabolism by cytochrome P450-mediated oxidation is saturable so the half-life is dose-dependent. Elimination is much slower in subjects with CYP2D6 genetic polymorphism.
Class II drugs
Beta-adrenoceptor antagonists (β-blockers)
The antagonist activity of β1-adrenoceptor is responsible for the therapeutic effects of this class. The most widely used agents for treatment of rhythm disturbances are atenolol, bisoprolol and propranolol, but all drugs in this class have antiarrhythmic activity. Beta-adrenoceptor antagonists are discussed in more detail in Chapter 5.
Esmolol is an ultra-short-acting β1-adrenoceptor-selective (cardioselective) agent that is used by bolus intravenous injection exclusively for the treatment of arrhythmias. It is most often used when arrhythmias arise during anaesthesia.
Class III drugs
Amiodarone is a drug with multiple antiarrhythmic actions. It has class III actions by blocking several K+ channels and shows use-dependence (see above). However, amiodarone also has a class Ib-like action on Na+ channels, as well as non-competitive β-adrenoceptor antagonist (class II) activity and calcium channel-blocking (class IV) actions. The antiarrhythmic effects produced early after intravenous infusion are believed to be due to β-adrenoceptor antagonist activity, since the class III effect is delayed.
Pharmacokinetics: Amiodarone is incompletely absorbed orally and has a large volume of distribution as a result of extensive uptake into adipose tissue. An intravenous formulation is available. Both amiodarone and its major hepatic metabolite have extremely long half-lives (50–60 days), so a prolonged loading dose regimen is used for both routes of administration.
Gastrointestinal disturbances, for example constipation and nausea, most often occur during the loading period.
Reversible corneal microdeposits develop in almost all people, and can cause dazzling by lights when driving at night.
Amiodarone has a high iodine content and a structural relationship to thyroid hormone. In iodine-sufficient areas (such as the UK), inhibition of intracellular thyroxine (T4) transport and 5′-deiodinase by amiodarone reduces the conversion of T4 to active triiodothyronine (T3) (Ch. 41). This produces hypothyroidism in about 10% of those treated. Hypothyroidism can be treated by thyroxine replacement without stopping amiodarone (Ch. 41). Amiodarone can also exacerbate underlying asymptomatic autoimmune thyroid disease. By contrast, in people who are iodine-deficient amiodarone can produce a destructive thyroiditis with release of preformed thyroid hormone leading to thyrotoxicosis in up to 10% of those taking it. Amiodarone-induced thyrotoxicosis is often resistant to treatment. Thyroid function should be checked every 6 months during treatment.
Photosensitive skin rashes are common, and use of wide-spectrum sunscreen is recommended. Slate-grey skin discoloration can also occur.
Peripheral neuropathy or myopathy.
Hepatitis and cirrhosis occur rarely.
Progressive pneumonitis and lung fibrosis are rare but serious effects of long-term treatment.
Drug interactions: the plasma concentrations of warfarin (Ch. 11) and digoxin (Ch. 7) are increased by amiodarone, with consequent potentiation of their effects. Amiodarone inhibits the metabolism of warfarin. It displaces digoxin from tissue stores and inhibits its renal excretion, both actions which increase the risk of digoxin toxicity.
Unlike most antiarrhythmic drugs, amiodarone does not have negative inotropic effects.
Sotalol
Sotalol is a non-selective β-adrenoceptor antagonist (Ch. 5) with additional class III properties. It selectively blocks the IKr K+ current (which is particularly involved in phase 2 and 3 repolarisation), and shows reverse use-dependency (higher receptor binding when the channel is closed) so that sotalol is most effective at slow rates of cell depolarisation (bradycardia). Sotalol is a racemic mixture; the L-isomer has both β-adrenoceptor antagonist and class III activity, whereas the D-isomer has only class III activity. The class III activity gives sotalol a greater proarrhythmic potential than other β-adrenoceptor antagonists (see above). Sotalol is reserved for treatment of significant cardiac rhythm disturbances and is not used for the other indications for β-adrenoceptor antagonists.
Pharmacokinetics: Sotalol is almost completely absorbed from the gut and excreted unchanged in the urine. Its half-life varies between 7 and 18 h.
Unwanted effects: These are discussed in Chapter 5. Sotalol also has proarrhythmic activity (see above).
Class IV drugs
Verapamil and diltiazem (Ch. 5), but not the dihydropyridine derivatives such as amlodipine, have antiarrhythmic activity. Verapamil can be given intravenously for a rapid effect, but should not be given together with a β-adrenoceptor antagonist, because of summation of myocardial depression and AV nodal conduction block. Details of calcium channel blockers can be found in Chapter 5.
Other drugs for cardiac rhythm disturbances
Those drugs used for the management of rhythm disturbances that do not fit into the Vaughan Williams classification are considered here.
Digitalis glycosides
Digitalis glycosides (such as digoxin) are not strictly antiarrhythmic, but they are useful for controlling ventricular rate in atrial flutter and atrial fibrillation by reducing conduction through the AV node. Digitalis glycosides are discussed in Chapter 7.
Adenosine
Mechanism of action and effects: Adenosine is a purine nucleoside with electrophysiological actions mediated by the A1 subtype of specific G-protein-coupled adenosine receptors. These receptors activate inward rectifier KACh channels which enhances the flow of K+ out of myocardial cells and hyperpolarises the resting cell (see Table 8.1). In addition, adenosine antagonises the stimulatory effects of noradrenaline on Ca2+ currents. These actions combine to stabilise the myocardial cell transmembrane ion fluxes. Adenosine has potent effects on the SA node, producing sinus bradycardia. It also slows impulse conduction through the AV node, but has no effect on conduction in the ventricles. Consequently, it is useful only for the management of supraventricular arrhythmias, particularly those caused by AV nodal re-entry mechanisms.
The action of adenosine at the A2 adenosine receptor reduces Ca2+ uptake in vascular smooth muscle, and produces vasodilation. In the coronary circulation, preferential dilation of healthy arteries produces coronary blood flow ‘steal’ that reduces flow in stenosed arteries. This receptor action enables adenosine to be used as a pharmacological stress to induce ischaemia in people with coronary artery disease, which can then be assessed by radionuclide scanning, magnetic resonance imaging or echocardiography.
Pharmacokinetics: Adenosine is given by rapid bolus intravenous injection. The effect is terminated by uptake into erythrocytes and endothelial cells, followed by metabolism to inosine and hypoxanthine. Adenosine has a half-life of less than 10 s and its duration of action is less than 1 min.
Unwanted effects: Unwanted effects are common and occur in about 25% of those given adenosine, but last less than 1 min:
Atropine
Atropine (see Ch. 4) is an antimuscarinic drug that is given by intravenous bolus injection and reduces the inhibitory effect of the vagus nerve on the heart. Blockade of muscarinic M2 receptors increases the rate of firing of the SA node and increases conduction through the AV node. This is due to inhibition of inward rectifying KACh channels which prevents hyperpolarisation of the cell membrane (Table 8.1) and enhanced activation of pacemaker f-channels. Atropine is used specifically for the treatment of sinus bradycardia and AV block. It is metabolised in the liver and has a half-life of 2–5 h.
Drug treatment of arrhythmias
Arrhythmias can be asymptomatic, or they can produce a variety of consequences that range from mild symptoms to life-threatening effects on cardiac output. The probability of developing symptoms depends on several factors, the most important of which are the rate of an abnormal rhythm and the presence of underlying heart disease. The range of consequences of rhythm disturbances includes:
Treatment may not be necessary for benign or self-terminating arrhythmias and reassurance may be all that is required. In some cases it may be possible to remove or treat an underlying cause.
The choice of treatment depends on the situation. With most tachyarrhythmias, sinus rhythm should be restored if possible. Direct current (DC) cardioversion is used to achieve this in severe, life-threatening or drug-resistant arrhythmias. Drug therapy is used if there is less need for an immediate effect, or to control the ventricular rate if the abnormal rhythm cannot be terminated. Radiofrequency ablation of an arrhythmogenic focus or pathway is increasingly used to prevent arrhythmia. This is carried out after intracardiac electrophysiological studies, using a cardiac catheter. Long-term drug treatment for bradyarrhythmias is not possible and an implanted pacemaker may be necessary.
Supraventricular tachyarrhythmias
Atrial premature beats are very common and usually benign, but sometimes they are a consequence of digoxin toxicity, and frequent multifocal atrial ectopics can result from organic heart disease. Other than treatment of an underlying cause, specific drug therapy is rarely needed. Some people are disturbed by a post-ectopic pause followed by a more forceful beat when sinus rhythm recommences. If treatment is required, then a β-adrenoceptor antagonist, or a calcium channel blocker such as verapamil or diltiazem, can be used to suppress the ectopics.
Atrial tachycardia
Atrial tachycardia is an infrequent rhythm disturbance usually arising from an automatic focus that produces an atrial rate of 150–250 beats⋅min−1. There is usually AV conduction block that results in a slower ventricular rate. Atrial tachycardia is not usually associated with significant cardiac disease, but can be a manifestation of digitalis toxicity. If drug therapy is necessary an AV nodal blocking agent such as a β-adrenoceptor antagonist or a calcium channel blocker (verapamil or diltiazem) will control the ventricular rate but rarely restores sinus rhythm. Sinus rhythm can be achieved with a class Ic antiarrhythmic agent such as flecainide, given with an AV nodal blocking drug (flecainide alone increases the risk of 1 : 1 AV nodal conduction if the atrial rate slows sufficiently but sinus rhythm is not restored). Sotalol or amiodarone can also be used to maintain sinus rhythm. Ablation of the initiating focus may also be considered.
A less common form of atrial tachycardia is multifocal atrial tachycardia arising from several ectopic foci, usually in people with severe pulmonary disease. Calcium channel blockers are usually used for ventricular rate control if treatment is needed.
Atrial flutter
In atrial flutter, the atrial rate is usually 250–350 beats⋅min−1 and the impulses are conducted to the ventricles with 2 : 1 or greater degrees of AV block. Flutter waves may be obvious on the ECG, or appear if the ventricular rate is slowed by transiently increasing the degree of AV block using vagal stimulation (such as carotid sinus massage) or the administration of adenosine. Atrial flutter usually arises from a macro re-entrant circuit in the right atrium. Underlying causes include recent cardiac surgery, cor pulmonale and congenital heart disease, but it can arise for no obvious reason. It may be paroxysmal, and it can degenerate into atrial fibrillation.
Drug therapy is relatively unsuccessful for restoring sinus rhythm, and DC cardioversion (synchronised to discharge on the R wave of the ECG) or rapid ‘overdrive’ electrical pacing to capture the ventricle followed by a gradual reduction in the paced rate may be required. Class Ic and III antiarrhythmic agents can prevent recurrence of paroxysmal atrial flutter. If a class Ic agent such as flecainide is used, then an AV nodal blocking drug should be given concurrently, since the atrial rate could slow with 1 : 1 AV conduction, causing an unacceptably high ventricular rate.
Control of the ventricular rate in atrial flutter can be achieved in a similar manner to that in atrial fibrillation (see below), but treatment is often less successful. For this reason, radiofrequency ablation of the re-entrant pathway via a cardiac catheter is becoming increasingly popular. Prophylaxis against thromboembolism should be given, similar to that for atrial fibrillation.
Atrial fibrillation
Atrial fibrillation is the most common rhythm disturbance in clinical practice (apart from ectopic beats). It has a variety of underlying causes (Box 8.1), some of which may be treatable. In younger people, atrial fibrillation often occurs without any obvious underlying cause, when it is called ‘lone’ atrial fibrillation. The arrhythmia usually arises from multiple re-entry circuits in the atria, although a rapid ectopic focus in a pulmonary vein may be responsible for triggering paroxysmal atrial fibrillation. The ventricular rate will depend on AV nodal function, so that when the AV node conducts well, atrial fibrillation produces a rapid ventricular rate. Atrial fibrillation predisposes to left atrial thrombus formation and subsequent systemic emboli, which most commonly cause stroke. Clinically, three forms of atrial fibrillation are recognised: paroxysmal (intermittent self-limiting episodes), persistent (present for more than 7 days but less than 1 year) and permanent (present for more than 1 year after unsuccessful attempts to maintain sinus rhythm, or if a decision has been made not to attempt this). Management has four underlying aims.
To identify and treat the underlying cause.
To restore or maintain sinus rhythm in paroxysmal or persistent atrial fibrillation (Box 8.2). It is desirable to attempt to restore sinus rhythm in younger people or those who tolerate the rhythm disturbance poorly. In these individuals, symptoms and exercise tolerance are usually improved by restoring sinus rhythm, but the risk of stroke is not removed (see below). However, the case for restoring sinus rhythm is less clear-cut for older people who tolerate the rhythm well, because there is no reduction in the risk of thromboembolic events (see below), and their quality of life may not improve. Restoration of sinus rhythm is usually possible in lone atrial fibrillation or when a precipitating factor has been treated. It can be achieved with drugs (pharmacological or chemical cardioversion), especially if the rhythm disturbance is of recent onset (40–80% success rate if the arrhythmia is of less than 7 days' duration), but often requires QRS-synchronised DC cardioversion. Pharmacological cardioversion is most rapidly achieved by using a single oral dose of a class Ic drug such as flecainide or propafenone. Intravenous amiodarone is also effective, but takes longer to restore sinus rhythm. Recurrence of atrial fibrillation is most frequent during the first 3–6 months after restoration of sinus rhythm. Drugs are not always recommended for prophylaxis to maintain sinus rhythm after a first cardioversion, because of their proarrhythmic effects. However, if there is a high risk of recurrence, then sinus rhythm can be maintained with a class Ic drug, sotalol, amiodarone or dronedarone (see Compendium at end of chapter). Amiodarone is the most successful single drug for long-term prevention of recurrence; although it maintains sinus rhythm in only about 75% of people at 1 year, this is superior to the 40% maintenance of sinus rhythm with the other drugs. Digitalis glycosides are ineffective for restoring or maintaining sinus rhythm in paroxysmal atrial fibrillation and should be avoided. Radiofrequency isolation of a pulmonary vein trigger area via a cardiac catheter is becoming increasingly used for younger people with paroxysmal atrial fibrillation. Other curative procedures are infrequently used.
To control a rapid ventricular response in persistent or permanent atrial fibrillation. For ventricular rate control both at rest and on exercise a drug that slows AV nodal conduction, such as a β-adrenoceptor antagonist, verapamil or diltiazem, should be used. Rate control at rest can be achieved with digoxin, but a rapid heart rate often still occurs during exercise (see Ch. 7), so it is only used alone for sedentary people. A β-adrenoceptor antagonist, verapamil, diltiazem or amiodarone can be used together with digoxin if rate control is difficult to achieve. Sotalol has no additional benefit in sustained atrial fibrillation and should be avoided because of its greater proarrhythmic activity compared with that of other β-adrenoceptor antagonists. Dronedarone is not used for rate control due to evidence of increased mortality. If drug combinations do not provide satisfactory rate control, then AV nodal ablation with insertion of a pacemaker can be considered.
To reduce thromboembolism by long-term anticoagulation (Ch. 11). Anticoagulation, usually with warfarin, is the prophylactic of choice in atrial fibrillation associated with rheumatic heart disease or thyrotoxicosis, and for 1 month before and at least 1 month after DC cardioversion. In non-rheumatic atrial fibrillation, the risk of emboli is related to the number of associated risk factors (Table 8.3). Most people with atrial fibrillation, whether sustained or paroxysmal, should take thromboprophylaxis. Warfarin (maintaining the international normalised ratio [INR] between 2 and 3; see Ch. 11) reduces the risk of thromboembolism by about two-thirds, and low-dose aspirin reduces the risk of thromboembolism by one-quarter. There are also newer oral anticoagulants with similar efficacy to warfarin, but with a lower risk of serious bleeding and no need for monitoring, such as apixaban, dabigatran and rivaroxaban (Ch. 11). Oral anticoagulation is preferred for people at moderate or high risk of embolism, but has little advantage in those at low risk, when the increased risk of bleeding outweighs the benefit. Even after restoration of sinus rhythm in paroxysmal or persistent atrial fibrillation, people at high risk of thromboembolic events should continue to take thromboprophylaxis, since the risk of stroke does not decrease. This may reflect the high risk of recurrence (often asymptomatic) of atrial fibrillation.
Table 8.3
Risk of stroke in non-rheumatic atrial fibrillation
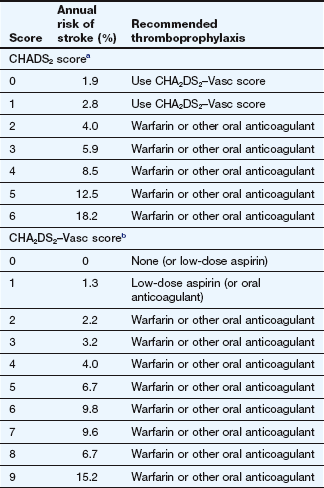
Based on guidelines for the management of atrial fibrillation of the European Society of Cardiology (2010).
aThe CHADS2 score is a summation of the following component scores: congestive heart failure, 1; hypertension, 1; age >75 years, 1; diabetes mellitus, 1; prior stroke or transient ischaemic attack, 2.
bThe CHA2DS2–Vasc score is a summation of the following component scores: congestive heart failure, 1; hypertension, 1; age >75 years, 2; diabetes mellitus, 1; prior stroke or transient ischaemic attack, 2; age 64–74 years, 1; vascular disease (prior myocardial infarction, peripheral arterial disease, aortic plaque), 1; Age 64–74, 1; Female sex, 1.
Junctional (nodal) tachycardias
Junctional tachycardias usually arise from a re-entry circuit, and are often initiated by an ectopic beat. A micro re-entry circuit can form within the AV node if there are two functional intranodal pathways with different recovery times (AV nodal re-entry tachycardia; AVNRT). Such circuits account for 60% of supraventricular tachycardias other than atrial fibrillation/flutter, and are not usually associated with structural cardiac disease. Alternatively, a macro re-entry circuit may involve an accessory AV pathway connecting the atria and ventricles such as in Wolff–Parkinson–White syndrome (AV re-entrant tachycardia; AVRT), which accounts for 30% of supraventricular tachycardias. Termination of an acute attack of nodal tachycardia can often be achieved with vagotonic manoeuvres such as carotid sinus massage, or by adenosine. For AVNRT, β-adrenoceptor antagonists, diltiazem or verapamil can be used to treat acute episodes or for prophylaxis. However, if there is an accessory AV pathway diltiazem, verapamil and digoxin should be avoided because selective blockade of the AV node by these drugs can predispose to rapid conduction of atrial arrhythmias through the accessory pathway. Junctional tachycardias involving an accessory pathway often respond well to flecainide, sotalol or amiodarone. Radiofrequency ablation of the re-entry circuit, via a cardiac catheter, is being employed increasingly for troublesome junctional tachycardias.
Immediate management of narrow-complex tachycardia of uncertain origin
If the rhythm is regular it is often not possible to determine from the ECG whether a narrow-complex tachycardia has an atrial or nodal origin. If vagotonic manoeuvres are unsuccessful, and the person is haemodynamically stable, intravenous adenosine should be given. This often converts a junctional tachycardia to sinus rhythm or can slow the ventricular rate sufficiently to identify the origin of the rhythm on an ECG. If there is a history of severe asthma, intravenous verapamil may be preferred. DC cardioversion should be considered if there is haemodynamic instability.
Ventricular tachyarrhythmias
Ventricular ectopic beats can occur in healthy individuals or in association with a variety of cardiac disorders such as ischaemic heart disease and heart failure. Frequent ventricular ectopic beats after myocardial infarction predict a poorer long-term outcome; however, suppressing such ectopics with class I antiarrhythmic drugs increases mortality and should be avoided. In contrast, β-adrenoceptor antagonists after myocardial infarction reduce the risk of sudden death (Ch. 5). A β-adrenoceptor antagonist can also suppress ventricular ectopic beats induced by stress or anxiety. In other situations, symptomatic ventricular ectopic beats can be suppressed by a class I drug such as flecainide.
Ventricular tachycardia
Ventricular tachycardia presents with broad QRS complexes on the ECG (broad-complex tachycardia). Although broad complexes can arise with supraventricular tachycardias (when there is bundle branch block), broad-complex tachycardia is usually treated on the assumption that it is ventricular tachycardia. Ventricular tachycardia is often associated with serious underlying heart disease, such as ischaemic heart disease or heart failure, and is more common following myocardial infarction. It can be either sustained or non-sustained. Sustained ventricular tachycardia can be associated with a minimal or absent cardiac output (‘pulseless’ ventricular tachycardia), when it is treated in the same way as ventricular fibrillation (see below). Polymorphic or incessant ventricular tachycardias can arise as a complication of antiarrhythmic drug therapy (see above) and with other drugs that prolong the Q–T interval on the ECG.
For sustained ventricular tachycardias, drug options include class Ib antiarrhythmic agents such as lidocaine (especially after myocardial infarction), and amiodarone. Sustained ventricular tachycardia is often associated with a poor long-term outlook in ischaemic heart disease, and coronary revascularisation or an automatic implantable cardiac defibrillator (ICD) may be beneficial. During and after the acute phase of myocardial infarction, a β-adrenoceptor antagonist is the treatment of choice to suppress non-sustained ventricular tachycardias.
Polymorphic or incessant ventricular tachycardias do not respond well to conventional treatments. Withdrawal of a precipitant drug, correction of electrolyte imbalance and intravenous magnesium sulphate are the therapies of choice. Temporary transvenous overpacing at a rate of 90–110 beats⋅min−1 may prevent recurrence. In the congenital form of long QT syndrome a β-adrenoceptor antagonist is the mainstay of treatment.
Ventricular fibrillation
Ventricular fibrillation is a potentially lethal arrhythmia that constitutes one form of ‘cardiac arrest’. An algorithm for the management of cardiac arrest is regularly updated by the European Resuscitation Council and is shown in Figure 8.6. The important principles of prolonged resuscitation are the maintenance of adequate cardiac output by external chest compression, and oxygenation by artificial inflation of the lungs, while attempting to restore sinus rhythm. Ventricular fibrillation is the commonest arrhythmia in acute cardiac arrest and it should be assumed to be present unless an ECG is available to show otherwise. It should be treated with immediate DC cardioversion. Adrenaline (epinephrine; Ch. 4) may be given to vasoconstrict the peripheries and thus maintain pressure in the central arteries perfusing the heart and brain. For recurrent ventricular fibrillation, suppression can be achieved by long-term use of antiarrhythmic drugs such as sotalol or amiodarone (often combined with a β-adrenoceptor antagonist), but frequently requires an automatic ICD.
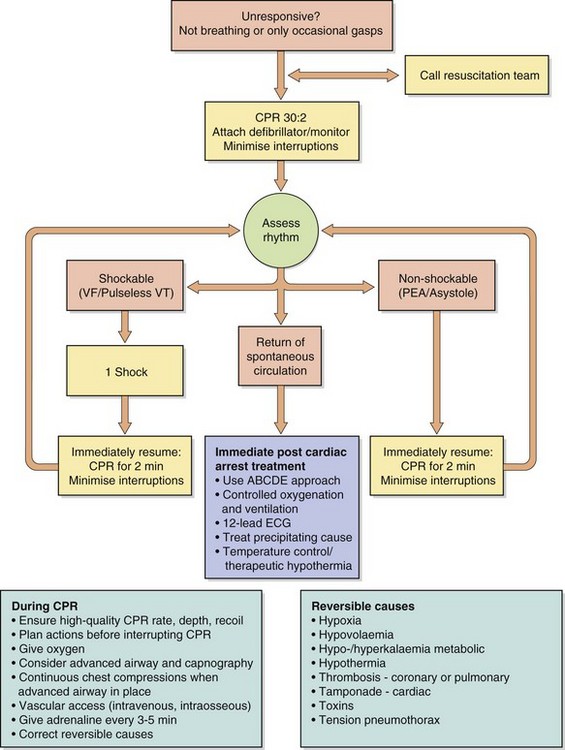
Fig. 8.6 An algorithm for the management of cardiac arrest. CPR, cardiopulmonary resuscitation; EMS, emergency medical services; PEA, pulseless electrical activity; VF, ventricular fibrillation; VT, ventricular tachycardia. Adapted from the 2010 European Resuscitation Council (ERC) Guidelines for Resuscitation (Nolan et al. 2010).
Bradycardias
Treatment with atropine may be necessary if sinus bradycardia is causing symptoms (e.g. after myocardial infarction or an overdose with a β-adrenoceptor antagonist). Hypotension precipitated by drugs such as streptokinase (Ch. 11) or the first dose of an angiotensin-converting enzyme (ACE) inhibitor (Ch. 6) is often associated with vagally mediated bradycardia, which will respond to atropine.
Atrioventricular block (‘heart block’)
AV block can be congenital or may accompany a variety of heart diseases. When occuring after myocardial infarction it is usually temporary if the infarct is inferior but is often permanent after anterior infarction. First-degree heart block (prolongation of the P–R interval on the ECG but with all P waves conducted to the ventricles) or Wenckebach (Mobitz type 1) second-degree heart block (progressive P–R prolongation until there is a non-conducted P wave) rarely require treatment, but higher degrees of block (second-degree, Mobitz type 2) and third-degree heart or complete heart block (with non-conducted P waves) should be treated. If complete AV block arises suddenly, then loss of consciousness (Stokes–Adams attack) or death can occur. If the onset is acute, atropine should be given intravenously to increase AV conduction, or an intravenous infusion of the non-selective β-adrenoceptor agonist isoprenaline can be used (Ch. 7). However, external or temporary transvenous electrical cardiac pacing is usually required in an emergency. If the AV block is permanent, the implantation of a permanent electrical cardiac pacemaker is usually necessary.
True/false questions
1. The sinoatrial (SA) node and the atrioventricular (AV) node have pacemaker activity.
2. Pacemaker cells in the SA node discharge at a higher frequency than those in other parts of the heart.
3. Spontaneous or pacemaker depolarisation during diastole results solely from the influx of Na+.
4. The influx of Na+ during phase 0 lasts only for milliseconds.
5. Cells are unable to generate further action potentials during phases 0, 1 and 2 of the action potential.
6. Reducing the gradient of the slope of phase 4 will slow the normal pacemaker rate.
7. Sympathetic and vagal stimulation reduce the slope of phase 4 depolarisation and reduce pacemaker rate.
8. Healthy non-pacemaker cells remain quiescent if not excited by an impulse arising from other regions in the heart.
9. Flecainide blocks Na+ channels.
10. Beta-adrenoceptor antagonist drugs are useful in stress-induced tachycardias.
11. The antiarrhythmic action of amiodarone depends only on blockade of K+ channels.
12. Amiodarone reaches steady-state concentrations after several months of treatment.
13. Adenosine is effective in the treatment of ventricular arrhythmias.
14. Verapamil affects both the plateau phase 2 and phase 4 of the action potential cycle.
15. Combining verapamil and a β-adrenoceptor antagonist may cause AV nodal conduction block.
One-best-answer (OBA) question
Considering the flow of ions into cardiac myocytes (inward flow) and out of myocytes (outward flow), identify which one statement most correctly describes a situation that would prevent arrhythmias.
A Increased inward flow of Na+ during phase 0 of the action potential
B Increased inflow of Ca2+ during phase 4 of the action potential
C Decreased inflow of Na+ during phase 0 of the action potential
D Increased inflow of Ca2+ during phase 2 of the action potential
E Decreased outflow of K+ in phase 3 of the action potential
Case-based questions
Mr GH, aged 48 years, consulted his GP complaining of palpitations and was found to have an irregular pulse with a rate of 120 beats⋅min−1. He had been suffering from shortness of breath and faintness for the previous 6 h. The symptoms had started after a drinking binge 36 h previously. Examination, blood tests (including thyroid function tests), ECG and chest radiograph revealed no coexisting heart disease, diabetes or hypertension. The ECG confirmed atrial fibrillation.
A What were the options available for treating Mr GH?
Before any treatment could be instituted, Mr GH spontaneously reverted to sinus rhythm. He was well for a year but then returned to his GP with a 3-day history of palpitations, breathlessness, chest pain and dizziness. Examination and an ECG again revealed atrial fibrillation. He was referred to a cardiologist and echocardiography showed no evidence of structural cardiac disease. Electrical DC cardioversion was carried out and the rhythm reverted to sinus rhythm.
Over the next 5 years, episodes of atrial fibrillation occurred with increasing frequency, and, eventually, sinus rhythm could not be restored with a variety of antiarrhythmic drugs or by DC conversion.
B What prophylactic treatment should be considered at the time of DC cardioversion? What drug treatments may be useful after DC cardioversion?
1. True. The SA and AV nodes, the bundle of His and the Purkinje system are pacemaker cells and form the specialised conducting system of the heart.
2. True. Pacemaker cells in the SA node therefore initiate cardiac rhythm.
3. False. Slow spontaneous depolarisation in pacemaker cells results from an inward flow of Na+ and K+ ions (funny current or If).
4. True. The fast Na+ channels for influx close at the end of phase 1.
5. True. However, during phase 3, the cells are only relatively refractory to further depolarising stimuli and a sufficient stimulus could fire an action potential during this phase.
6. True. The slope in phase 4 controls the normal pacemaker rate as it determines the time taken to reach the threshold potential.
7. False. Vagal stimulation reduces the slope of phase 4 and slows the rate of firing, but sympathetic stimulation increases the slope and hence the firing rate.
8. True. However, if the intracellular Ca2+ concentration rises abnormally (e.g. under the influence of cardiac glycosides or catecholamines), this can exchange with Na+ passing inwards, causing membrane depolarisations, called afterdepolarisations or ‘triggered activity’.
9. True. All class I antiarrhythmics such as flecainide (class Ic) block fast Na+ channels and slow the rate of rise of phase 0, therefore reducing myocardial excitability.
10. True. Beta-adrenoceptor antagonists reduce pacemaker depolarisation rate by inhibiting the sympathetic stimulation of the cAMP-dependent funny current (If) in the SA and AV nodes.
11. False. Like other class III agents, amiodarone blocks several types of K+ channel, but also has a class Ib-like action on Na+ channels, class II activity (non-competitive β-adrenoceptor antagonism) and class IV activity (calcium channel blockade).
12. True. Accumulation of amiodarone to steady-state after about 6 months is due to its lipophilicity, resulting in a high apparent volume of distribution and very long half-life (50–60 days).
13. False. Adenosine has no beneficial effect on ventricular arrhythmias. Its main effect involves enhancing K+ conductance and inhibiting Ca2+ influx, resulting in reduced AV nodal conduction and an increase in the AV nodal refractory period. Adenosine is useful in supraventricular arrhythmias, particularly when caused by AV nodal re-entry mechanisms; it has high efficacy and a very short duration of action.
14. True. L-type Ca2+ channels are involved in the phase 2 plateau while both T- and L-type channels contribute to depolarisation in phase 0 and the funny current in phase 4. Verapamil acts on both to slow the rate of the pacemaker depolarisation and to reduce the plateau phase, thus shortening the action potential. These effects make verapamil useful in supraventricular tachycardias but not in ventricular arrhythmias.
15. True. Verapamil is a highly negatively inotropic calcium channel blocker, reduces cardiac output, slows the heart rate, and impairs atrioventricular conduction, so it may cause AV nodal block or heart failure when used with β-adrenoceptor antagonists.
OBA answer
A, B Incorrect. Each of these would increase depolarisation rate in phase 4 and the rate of firing of the SA and AV nodes.
C Correct. This would slow the rate of depolarisation in phase 0 and is one of the mechanisms by which class I antiarrhythmics exert their therapeutic actions.
D, E Incorrect. Each of these would shorten action potential duration, increasing the likelihood of arrhythmias.
Case-based answers
A The aim at this stage is to restore and maintain sinus rhythm in Mr GH, who appears to have no structural heart disease. Since the arrhythmia is of short duration, pharmacological cardioversion may be successful. This could be achieved by flecainide, propafenone or amiodarone. Amiodarone is usually reserved for people with significant cardiac dysfunction or those refractory to other agents. Flecainide and propafenone should be avoided in people with significant cardiac dysfunction or concomitant ischaemic heart disease. However, they are probably suitable for this man. Digoxin, calcium channel blockers and β-adrenoceptor antagonists are ineffective for terminating atrial fibrillation. Synchronised DC cardioversion is successful in up to 90% of people with atrial fibrillation who have no structural heart disease or heart failure, who are aged less than 50 years and whose duration of atrial fibrillation is less than 1 year. It could be considered if drugs are unsuccessful. About 50% of the time, recent-onset atrial fibrillation (less than 48 h duration) spontaneously converts to sinus rhythm. In Mr GH, the atrial fibrillation could have been brought on by excess alcohol (so-called ‘holiday heart’). If he moderates his alcohol intake then prophylaxis would not be necessary after a single attack.
B Anticoagulation with warfarin is essential for at least 3–4 weeks before and 4 weeks after a DC cardioversion to minimise the risk of a systemic embolus. For prophylaxis against recurrence, antifibrillatory drugs are usually given for at least 3–6 months following DC cardioversion, since this is the period of highest risk of recurrence. Digoxin, verapamil and β-adrenoceptor antagonists are not effective for prophylaxis. After 5 years of recurrence of atrial fibrillation, sinus rhythm could not be restored. Therefore, the aim in Mr GH is to control ventricular rate. Digoxin suppresses AV nodal conduction and can reduce the ventricular response rate. This is mediated through potentiation of vagal effects on the heart and is less effective during exercise; therefore, a β-adrenoceptor antagonist or calcium channel blocker (such as verapamil) is preferred. However, β-adrenoceptor antagonists (in high doses) and verapamil are negatively inotropic, and if there is significant cardiac dysfunction or heart failure they are contraindicated. The positive inotropic action of digoxin might be helpful if there is coexisting left ventricular impairment. The major long-term consequence of atrial fibrillation is the risk of thromboembolism and this is greatest in those over 75 years of age. For Mr GH, aspirin or no thromboprophylaxis is appropriate as he is at a relatively low risk of stroke because of his age and lack of any coexisting hypertension, diabetes or significant left ventricular impairment.
Compendium: Drugs used to treat cardiac arrhythmias
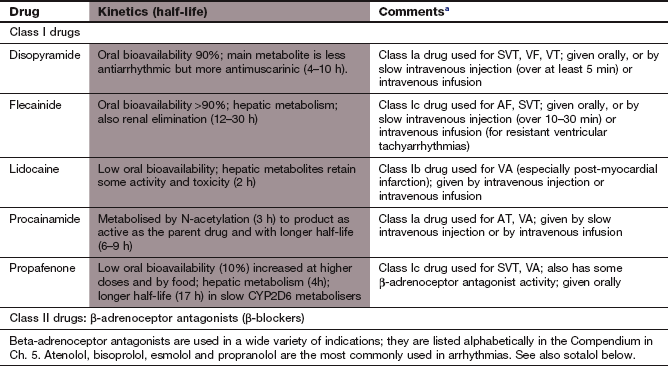
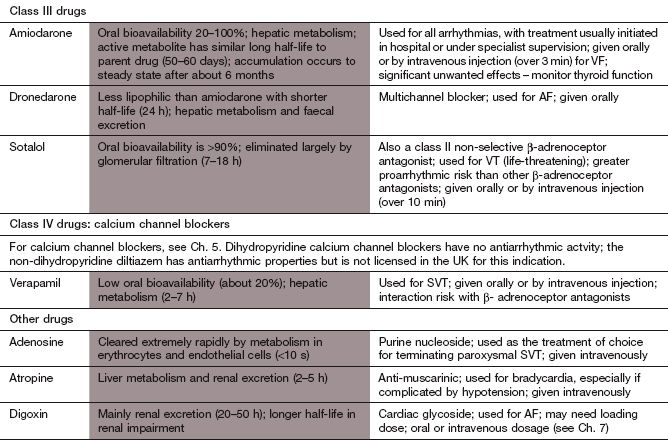
aThe types of arrhythmias commonly treated with different drugs are: AF, atrial fibrillation; AT, atrial tachycardia; SVT, supraventricular tachycardia; VA, ventricular arrhythmias; VF, ventricular fibrillation; VT, ventricular tachycardia.
CalòL, Sciarra, L, Lamberti, F, et al. Electropharmacological effects of antiarrhythmic drugs on atrial fibrillation termination. Part 1: molecular and ionic fundamentals of antiarrhythmic drug actions. Ital Heart J. 2003;4:430–441.
Delacrétaz, E. Supraventricular tachycardias. N Engl J Med. 2006;354:1039–1051.
Grant, AO. Molecular biology of sodium channels and their role in cardiac arrhythmias. Am J Med. 2001;110:296–305.
Gupta, A, Lawrence, AT, Krishnan, K, et al. Current concepts in the mechanisms and management of drug-induced QT prolongation and torsade de pointes. Am Heart J. 2007;153:891–899.
Hart, RG, Pearce, LA, Aguilar, MI. Meta-analysis: antithrombotic therapy to prevent stroke in patients who have nonvalvular atrial fibrillation. Ann Intern Med. 2007;146:857–867.
International Liaison Committee on Resuscitation. 2005 International Consensus on Cardiopulmonary Resuscitation and Emergency Cardiovascular Care Science with Treatment Recommendations, Part 1: Introduction. Resuscitation. 2005;67:181–186.
Iqbal, MB, Taneja, AK, Lip, GYH, et al. Recent developments in atrial fibrillation. BMJ. 2005;330:238–243.
Katz, AM. Selectivity and toxicity of antiarrhythmic drugs: molecular interactions with ion channels. Am J Med. 1998;104:179–195.
Lafuente-Lafuente, C, Mouly, S, Longás-Tejero, MA, et al. Antiarrhythmic drugs for maintaining sinus rhythm after cardioversion of atrial fibrillation. Arch Intern Med. 2006;166:719–728.
Lau, W, Newman, D, Dorian, P. Can antiarrhythmic agents be selected based on mechanism of action? Drugs. 2000;60:1315–1328.
Lip, GYH, Tse, H-F. Management of atrial fibrillation. Lancet. 2007;370:604–618.
Nattel, S, Opie, LH. Controversies in atrial fibrillation. Lancet. 2006;367:262–272.
Nolan, JP, Soar, J, Zideman, DA, Biarent, D, Bossaert, LL, Deakin, C, Koster, RW, Wyllie, J, Böttiger, B, on behalf of ERC Guidelines Writing Group. European Resuscitation Council Guidelines for Resuscitation 2010 Section 1: Executive Summary. Resuscitation. 2010;81:1219–1276.
Page, RL. Newly diagnosed atrial fibrillation. N Engl J Med. 2004;351:2408–2416.
Reiffel, JA, Reiter, MJ, Blitzer, M. Antiarrhythmic drugs and devices for the management of ventricular tachyarrhythmias in ischemic heart disease. Am J Cardiol. 1998;82:31I–40I.
Reiter, MJ, Reiffel, JA. Importance of beta blockade in the therapy of serious ventricular arrhythmias. Am J Cardiol. 1998;82:9I–19I.
Roden, DM, Balser, JR, George, AL, Jr., Anderson, ME. Cardiac ion channels. Annu Rev Physiol. 2002;64:431–475.
Shorofsky, SR, Balke, CW. Calcium currents and arrhythmias: insights from molecular biology. Am J Med. 2001;110:127–140.
Tamargo, J, Caballero, R, Gomez, R, et al. Pharmacology of cardiac potassium channels. Cardiovasc Res. 2004;62:9–33.
Task Force on the Management of Atrial Fibrillation of the European Society of Cardiology. Guidelines for the management of atrial fibrillation. Eur Heart J. 2010;31:2369–2429.