Neurotransmission and the peripheral autonomic nervous system
Arrangement of the central and peripheral nervous systems
Principles of neurotransmission
The peripheral autonomic nervous system
The sympathetic nervous system and noradrenergic transmission
Synthesis and storage of catecholamines: noradrenaline, adrenaline and dopamine
The parasympathetic nervous system and cholinergic transmission
This chapter deals predominantly with details of the peripheral autonomic nervous system; the general principles relating to the central nervous system and the somatic nervous system are similar, but specific details are dealt with in Section 5 and Chapter 27, respectively.
Arrangement of the central and peripheral nervous systems
There are two principal neuronal control systems in the body. Functionally they are highly integrated and should be considered holistically, but for educational clarity they are introduced separately.
 The central nervous system (CNS) comprises neuronal networks of the brain, brainstem and spinal cord (Section 5).
The central nervous system (CNS) comprises neuronal networks of the brain, brainstem and spinal cord (Section 5).
The peripheral nervous systems, which interconnect the CNS to the organs of the body, include:
the autonomic (automatic or involuntary) nervous system, which comprises sympathetic and parasympathetic nervous systems and also includes the enteric nervous system of the gut,
the somatic (voluntary) nervous system, which innervates skeletal muscle (Ch. 27).
The CNS integrates, processes and responds to sensory messages.
It receives sensory information from all parts of the body, including visceral sensory afferent nerves (e.g. from viscera, smooth muscle and cardiac muscle) and somatic sensory afferents (e.g. from skeletal muscle).
It responds by sending instructions via the autonomic efferent nerves of the sympathetic and parasympathetic systems (e.g. to glands, smooth muscle and cardiac muscle) and via somatic motor efferents to skeletal muscle.
Principles of neurotransmission
Action potentials (APs) passing along axons provide instructions to other neurons or non-neuronal cells (e.g. smooth muscle cells). Instructions are transferred by the release of chemical neurotransmitters from the presynaptic endings of the neuron, which then diffuse through a small physical space, called the synaptic cleft, and stimulate the receiving (postsynaptic) cells via recognition proteins (receptors) (Fig. 4.1). The transmitters may instruct the receiving cells to increase (excite) or reduce (inhibit) their activity.
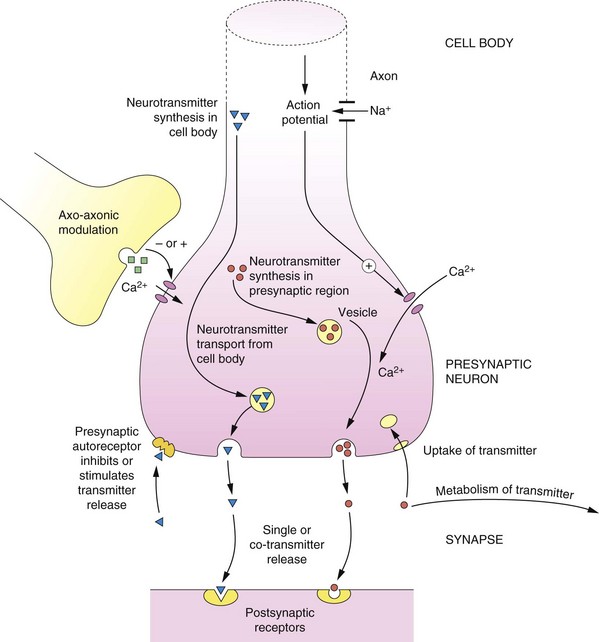
Fig. 4.1 Principles of neurotransmission at a synapse.
Basic pathways of synthesis, storage, release, action and inactivation of a typical neurotransmitter are shown, as described in the text. At many synapses co-transmission of different neurotransmitters occurs.
Neurotransmitters can be either synthesised within the presynaptic region (e.g. noradrenaline) or transported from the cell body to the synaptic region (e.g. peptides). The neurotransmitter is taken up from the cytosol using specific vesicular transporters within the nerve ending and stored within membrane vesicles. The transmitter in the vesicle may form a complex; for example, noradrenaline forms a complex with adenosine triphosphate (ATP), which reduces the free concentration of noradrenaline within the vesicle.
The release of the neurotransmitter can be ‘fine-tuned’ by axo-axonic connections and by presynaptic receptors (which are discussed below). A generalised scheme for neurotransmission is as follows (Fig. 4.1):
a. The cell body (or soma) responds to an appropriate stimulus by generating an AP.
b. The AP is conducted along the axon by the opening of voltage-gated Na+ channels and the influx of Na+; when the AP reaches the presynaptic nerve terminal it results in an influx of Ca2+ through voltage-dependent channels.
c. Ca2+-dependent processes result in fusion of neurotransmitter-containing vesicles with the presynaptic membrane and the release of stored neurotransmitter into the synaptic space.
d. The released neurotransmitter binds to and stimulates the appropriate receptors in the postsynaptic membranes and generates biochemical changes in the recipient cells; these may be functional changes in cells (e.g. smooth muscle contraction) or excitation or inhibition of another neuron (e.g. transmission of the AP to postsynaptic nerve fibres).
e. The released neurotransmitter may also stimulate autoreceptors in the presynaptic membranes, and thereby modulate the further release of the neurotransmitter.
f. The transmitter is degraded by enzymes or taken back into the presynaptic neuron for re-use.
Neurons may release a single transmitter, but often more than one transmitter is released; there are many examples of co-transmission, which are described later in this book.
Presynaptic receptors and modulation of transmitter release
An important characteristic of neurons is the presence of presynaptic receptors (Fig. 4.1 and Table 4.1). Presynaptic receptors may increase or decrease the release of the neurotransmitter and are described as facilitatory and inhibitory, respectively. There are two main sources of ligands for presynaptic receptors:
Table 4.1
The control of transmitter release by presynaptic receptor mechanisms
| Neurotransmitter | Presynaptic receptors inhibiting release | Presynaptic receptors facilitating release |
| Acetylcholine (ACh) | M2, α2, D2/D3, 5-HT3 | N1, NMDA |
| Dopamine | D2/D3, M2 | N1, NMDA |
| γ-Aminobutyric acid (GABA) | GABAB | – |
| Histamine | H3 | – |
| Serotonin (5-hydroxytryptamine, 5-HT) | 5-HT1D, α2 | 5-HT3 |
| Noradrenaline | α2., H3, M2, D2, opioid | β2, N1, angiotensin II |
neurotransmitter released from the vesicles that can act presynaptically (autoreceptors),
neurotransmitter released from other neurons, usually by axo-axonal synapses (Fig. 4.1), involving a different neurotransmitter to that released by the neuron itself (heteroreceptors).
Inhibition of transmitter release is usually achieved by limiting Ca2+ entry through voltage-gated ion channels into the neuron.
The first recognition of a clinically important presynaptic receptor came with the discovery that the antihypertensive agent clonidine lowers blood pressure via stimulation of presynaptic α2-adrenoceptors, with subsequent inhibition of the release of vasoconstricting noradrenaline. Presynaptic receptors (Table 4.1) are increasingly recognised to have important roles in the clinical effects produced by many drugs.
The peripheral autonomic nervous system
The peripheral autonomic nervous system (ANS) is an important site for drug action because:
the ANS either controls or contributes to the control of the functioning of nearly all of the major organ systems of the body,
ANS dysfunction is present in many diseases,
ANS dysfunction can occur as an unwanted effect of drug treatment,
the ANS utilises two major different neurotransmitters and a number of receptor subtypes; these provide a variety of sites for drug action (Box 4.1), which allows modification of particular body functions with some degree of selectivity.
Box 4.1 Targets for drug action within the ANS
Muscarinic receptors at postganglionic nerve endings in the parasympathetic nervous system (muscarinic receptor subtypes)
Adrenergic receptors for noradrenaline and adrenaline in the sympathetic nervous system (α- and β-adrenoceptor subtypes)
Presynaptic receptors in the parasympathetic and sympathetic nervous systems
Modification of synthesis, storage, release and inactivation of ACh
Modification of synthesis, storage, release and inactivation of noradrenaline
The peripheral ANS is subdivided into two main branches (Fig. 4.2, Box 4.2):
Box 4.2 Organisation of the ANS
Parasympathetic and sympathetic efferent nerves from the spinal cord synapse at intermediate ganglia before synapsing with the effector cells at their postganglionic nerve endings
ACh and noradrenaline are the principal neurotransmitters in the ANS but other transmitters also have neurotransmitter roles.
Stimulation of the sympathetic nervous system has a widespread effect in the body because of interconnections between efferent fibres, whereas the parasympathetic nervous system is more organ-specific (Fig. 4.2).
The neurotransmitters are synthesised in the presynaptic neuron, stored and released into the synapse in response to depolarization and Ca2+ influx caused by an AP.
At all ganglia, the neurotransmitter is ACh, acting on nicotinic N1 receptors, which then elicits an AP in the postganglionic nerve.
Parasympathetic efferents have muscarinic receptors at neuroeffector synapses.
Most sympathetic efferents have noradrenergic receptors at neuroeffector synapses.
Adrenaline and noradrenaline are synthesised and released in the adrenal medulla in response to sympathetic stimulation and enhance the effects of local noradrenaline release.
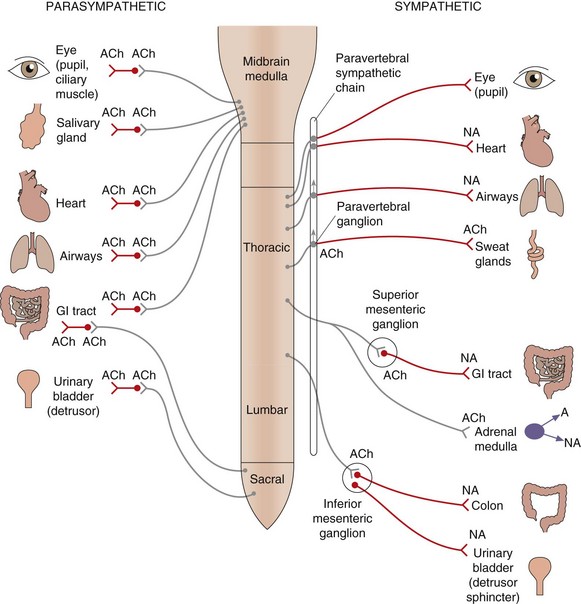
Fig. 4.2 Organisation of the parasympathetic and sympathetic autonomic nervous systems.
Activation of the sympathetic nervous system leads to widespread neuronal release of noradrenaline supplemented by release into the circulation of adrenaline (and noradrenaline) from the adrenal medulla; these act at α- and β-adrenoceptors (see Table 4.2). Stimulation of the parasympathetic nervous system is more localised to particular organs, mediated by acetylcholine acting at muscarinic receptors (see Table 4.3). Some tissues, such as airways, have sparse sympathetic innervation and sympathetic effects are mainly a result of circulating adrenaline. Sympathetic innervation of sweat glands is mediated, unusually, by acetylcholine. The ganglia innervating some organs are not part of the paravertebral chain but grouped together to form the coeliac, superior mesenteric or inferior mesenteric ganglia; the transmitter at all ganglia is acetylcholine acting at nicotinic type 1 receptors. ACh, acetylcholine; NA, noradrenaline; A, adrenaline; GI, gastrointestinal.
parasympathetic nervous system, which utilises acetylcholine (ACh) as the final transmitter at muscarinic receptors on the cells that are being stimulated (called the innervated or effector cells or organs),
sympathetic nervous system, which utilises noradrenaline as the transmitter at adrenoceptors on most, but not all, effector organs; the release of adrenaline and noradrenaline from the adrenal medulla during sympathetic nervous system stimulation is also an important and integral part of the sympathetic nervous system response.
Anatomically, in both branches of the ANS, the efferent neurons innervating effector organs are linked to neurons in the CNS via ganglia. The distribution and neuronal interconnections differ between the two branches (Fig. 4.2).
The parasympathetic efferents give more discrete innervation of organs; the ganglia are close to the innervated organs and they therefore have long preganglionic fibres; there are few or no interconnections between ganglia, so that innervated organs can be affected independently.
The sympathetic efferents are classically described as being involved in the flight-or-fight response and affect many body systems simultaneously. Many of the ganglia are close to the spinal column in the paravertebral sympathetic ganglion chain that lies along each side of the spinal column, so these nerves have long postganglionic fibres to effector organs. All neurons in the sympathetic system can be activated simultaneously because of numerous neuronal interconnections within the paravertebral chain; also, axons passing through the chain without synapsing can interconnect with ganglia such as the inferior mesenteric ganglion and can then diversify to innervate several organs (Fig. 4.2).
Many organs are innervated by both the parasympathetic and sympathetic nervous systems, which act in concert and may have opposite effects on the organ function. Physiological functions therefore often require inverse coordination of sympathetic and parasympathetic activities; for example, urination is brought about by a decreased sympathetic activity on the sphincter muscle and increased parasympathetic activity on the detrusor muscle (see urinary bladder, Tables 4.2 and 4.3, and Ch. 15).
Table 4.2
Effects of sympathetic nervous system activity via adrenoceptor subtypes in major tissues
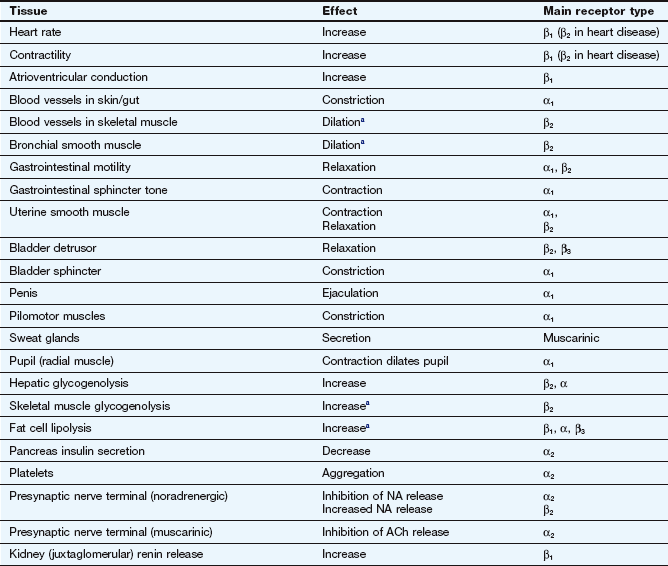
aRespond to circulating adrenaline; little noradrenergic innervation.
Table 4.3
Effect of stimulation of parasympathetic nerves (via muscarinic M receptor subtypes) in major tissues
| Tissue | Effect | Main receptor type |
| Heart rate | Decrease | M2 |
| Contractility of atria | Decrease | M2 |
| Atrioventricular conduction velocity | Decrease | M2 |
| Vascular endothelium | Dilates blood vessel: NO release | M1, M3 |
| Bronchial smooth muscle | Constriction | M2, M3 |
| Gut motility | Contraction, relaxation | M2, M3 |
| Gut sphincter tone | Increased | M3 |
| Gut secretions | Increased | M3 |
| Bladder detrusor | Contraction | M3 |
| Bladder sphincter | Relaxation | M3 |
| Penis | Erection | M3 |
| Eye pupil circular muscle | Contraction (miosis) | M3 |
| Ciliary muscle | Contraction (accommodates for near vision) | M3 |
| Pancreatic insulin secretion | Increased | M1, M3 |
| Salivary glands | Secretion | M1, M3 |
| Emesis | Increased | M3 |
The concept of opposing actions, although imperfect, can be useful in remembering the effects that each part of the nervous system has on tissue function. Tables 4.2 and 4.3 show the effects that stimulation of the sympathetic or parasympathetic nervous systems have on major tissues and the primary receptors that are involved. Under resting conditions, the predominant drive to many organs is from the parasympathetic nervous system.
The sympathetic nervous system and noradrenergic transmission
Noradrenaline and adrenaline are members of a group of amine transmitters called catecholamines (a catechol is a benzene ring with two adjacent hydroxyl groups; Fig. 4.3A). Both the catechol and amino groups are important for receptor binding. The receptors that noradrenaline and adrenaline stimulate are described as adrenoceptors and the effects of noradrenaline and adrenaline at these receptors are described as noradrenergic and adrenergic effects.
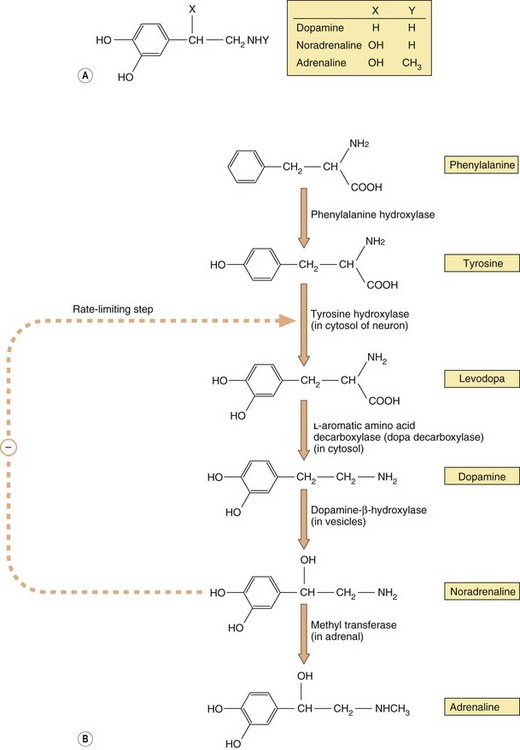
Fig. 4.3 The structure of the main physiological catecholamines (A) and their synthesis from amino acid precursors (B).
The catecholamine synthetic pathway is described in the text.
The approved European names of noradrenaline and adrenaline when they are formulated and used therapeutically as medicines are norepinephrine and epinephrine, respectively; however, when their physiological actions are being described, the terms noradrenaline and adrenaline are retained. Most preparations of adrenaline and noradrenaline in Europe are dual-labelled with both terms. In the USA, the terms epinephrine and norepinephrine are used for both therapeutic and physiological descriptions.
Synthesis and storage of catecholamines: noradrenaline, adrenaline and dopamine
Catecholamine neurotransmitters are synthesised from inactive precursors (Fig. 4.3B). The basic carbon skeleton of catecholamines is derived from phenylalanine or tyrosine, which are aromatic amino acids used predominantly in protein synthesis. The sequence of synthesis of adrenaline (via dopamine and noradrenaline) is shown in Figure 4.3B. The oxidation of tyrosine to levodopa by tyrosine hydroxylase, which occurs within the neuron, commits the molecule to become a neurotransmitter. This step is subject to negative feedback by the catecholamines that are subsequently produced, thereby regulating supply. Conversion of levodopa to dopamine is catalysed by a cytosolic enzyme, aromatic L-amino acid decarboxylase (also known as dopa decarboxylase). The amine product, dopamine, is taken up by vesicles via a specific transporter termed the vesicular monoamine transporter 2 (VMAT2). In neurons that use dopamine as their primary transmitter this is the end of the synthetic pathway. Dopamine is a vital neurotransmitter in some parts of the peripheral nervous system and also widely in the CNS (Chs 7, 21 and 24).
The vesicles present in noradrenergic neurons contain dopamine-β-hydroxylase. This enzyme is largely present in the membranes of the vesicles, but on exocytosis some is lost into the synapse, after which it diffuses into the bloodstream and is slowly cleared; its levels in the blood can therefore be used as a marker of peripheral noradrenaline release. In noradrenergic neurons this is the end of the synthetic pathway. Noradrenaline and its precursor dopamine are stored in the vesicles complexed with ATP and proteoglycans.
The chromaffin cells of the adrenal medulla contain an additional cytosolic enzyme (phenylethanolamine-N-methyl transferase), which converts noradrenaline to adrenaline by the addition of a methyl group (Fig. 4.3B). Adrenaline is then transferred into chromaffin cell granules by vesicular monoamine transporter 1 (VMAT1), where it is stored ready for release.
Noradrenaline release
Release in response to a nerve impulse occurs following Ca2+ influx and Ca2+-mediated fusion of the noradrenaline-containing vesicle with the cytoplasmic membrane.
Noradrenaline in the presynaptic neuron may also be released by so-called indirectly acting sympathomimetic amines, which are low-molecular-weight basic compounds; for example, food constituents (such as tyramine), therapeutic drugs (such as ephedrine) and some drugs of abuse (such as amfetamine and metamfetamine). These compounds are taken into the presynaptic cytosol and into the vesicles, from which they displace noradrenaline. Increased noradrenaline release into the synapse is responsible for the biological effects produced by ingestion of compounds like tyramine, ephedrine and amfetamine.
Uptake and metabolism of released noradrenaline
The principal mechanism for the removal of noradrenaline from the synapse is uptake (approximately 70–90%) into the presynaptic neuron via a specific high-affinity carrier called uptake 1 (or the norepinephrine transporter, NET); it also takes up dopamine but not adrenaline (Fig. 4.4). Both noradrenaline and dopamine can then be transferred from the cytosol into the vesicles by VMAT2. Some of the noradrenaline remaining in the synapse is taken up into non-neuronal tissues by a low-affinity carrier called uptake 2, which can also transport adrenaline; some of the remaining noradrenaline and the majority of any adrenaline released into the circulation as a co-transmitter is metabolised. Separate transporters exist for reuptake of serotonin (5-hydroxytryptamine, 5-HT) and dopamine from their respective neurons, termed the serotonin transporter (SERT) and dopamine transporter (DAT), respectively. Therapeutic agents that block NET or SERT increase the amount of neurotransmitter in the synaptic cleft and are used in treating depression (Ch. 22). The uptake of noradrenaline by NET is also blocked by cocaine and amfetamine.
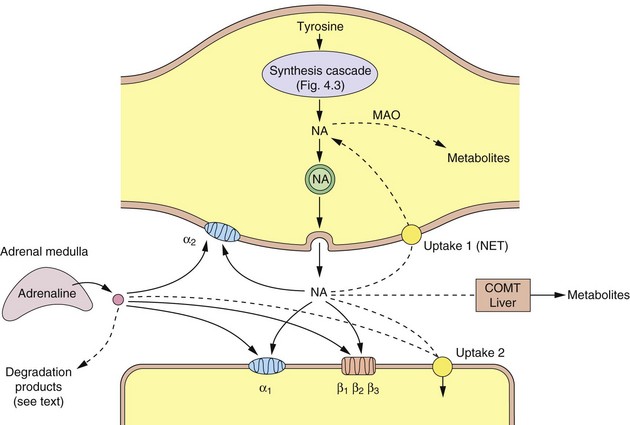
Fig. 4.4 A noradrenergic nerve terminal (varicosity) showing the synthesis and sites of action of noradrenaline and adrenaline.
Dashed lines show the ways that the actions of the catecholamines are curtailed. COMT, catechol-O-methyltransferase; MAO, monoamine oxidase; NA, noradrenaline; NET, norepinephrine transporter (uptake 1).
There are two main enzymes involved in the initial steps in the catabolism of noradrenaline: monoamine oxidase (MAO) and catechol-O-methyltransferase (COMT).
Monoamine oxidase
MAO is present on the surface of the mitochondria of the presynaptic neuron, where it oxidises free cytoplasmic noradrenaline. It is also present in many other sites such as the gastrointestinal epithelium and liver. Oxidative removal of the amino group on noradrenaline via MAO is the major pathway of catabolism of noradrenaline and other aminergic neurotransmitters. Loss of the amino group prevents binding to the postsynaptic receptor, and therefore is an inactivation process. In the periphery, metabolism results in the formation of vanillylmandelic acid, which is the main urinary metabolite. In the CNS, the metabolites are conjugated with sulphate before being excreted in the urine. There are two main MAO isoenzymes, MAO-A and MAO-B, which differ in their organ distribution and substrate affinities (Table 4.4). The use of inhibitors of MAO-A and/or MAO-B isoenzymes in depression and Parkinson's disease is discussed in Chapters 22 and 24.
Table 4.4
Monoamine oxidase (MAO) and its inhibitors
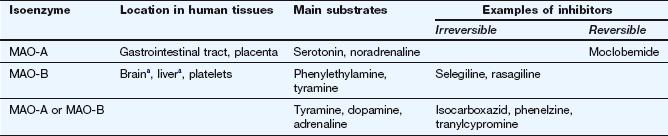
aBoth isoenzymes are present, but in humans the amount of MAO-B exceeds that of MAO-A.
Catechol-O-methyltransferase
COMT occurs only at low levels in noradrenergic neurons, but is present in many other tissues, including the adrenal gland and liver. The enzyme catalyses the transfer of a methyl group onto the aromatic ring; this removes the catechol centre and prevents binding to the postsynaptic receptor. COMT is a minor route of inactivation of both dopamine and noradrenaline. Inhibitors of COMT are used as an adjunct to levodopa therapy for Parkinson's disease (Ch. 24).
Sympathetic nervous system receptors
All ganglia utilise ACh as a neurotransmitter, acting predominantly on nicotinic type 1 (N1) receptors to elicit an AP in the postganglionic axon. The receptor type at most postganglionic nerve endings in the sympathetic nervous system are adrenergic receptors (adrenoceptors).
Based on the effects of a number of agonists and antagonists, the adrenoceptors are divided into two types, α and β, and further into α-subtypes (α1 and α2) and β-subtypes (β1, β2 and β3) (see Table 4.2 and the drug receptor table at the end of Ch. 1). It is now understood that there are multiple forms of some of these subtypes (i.e. α1A, α1B and α1D, and α2A, α2B and α2C) and these are discussed in later chapters where clinically relevant. The receptor subtypes show different affinities for the endogenous catecholamines, noradrenaline and adrenaline:
α1: noradrenaline ≥ adrenaline
α2: adrenaline > noradrenaline
β1: noradrenaline ≥ adrenaline
Selective stimulation or blockade of individual adrenoceptor subtypes forms the basis of significant areas of pharmacology and therapeutics and is dealt with in relevant chapters.
The parasympathetic nervous system and cholinergic transmission
ACh is synthesised within the cytosol of the cholinergic neuron from choline and acetyl-CoA (Fig. 4.5). Choline is a highly polar, quaternary amino compound that is also present in phosphatidylcholine; it is obtained largely from the diet. Because of its fixed positive charge it does not readily cross cell membranes and there are specific transporters to allow uptake into the presynaptic neuron (Fig. 4.5) and from the gastrointestinal tract and across the blood–brain barrier (Ch. 2). Acetylation of choline to form ACh is catalysed by choline acetyltransferase. The rate of synthesis of ACh is closely controlled and related to ACh turnover, so that rapid release of ACh stores is associated with enhanced synthesis.
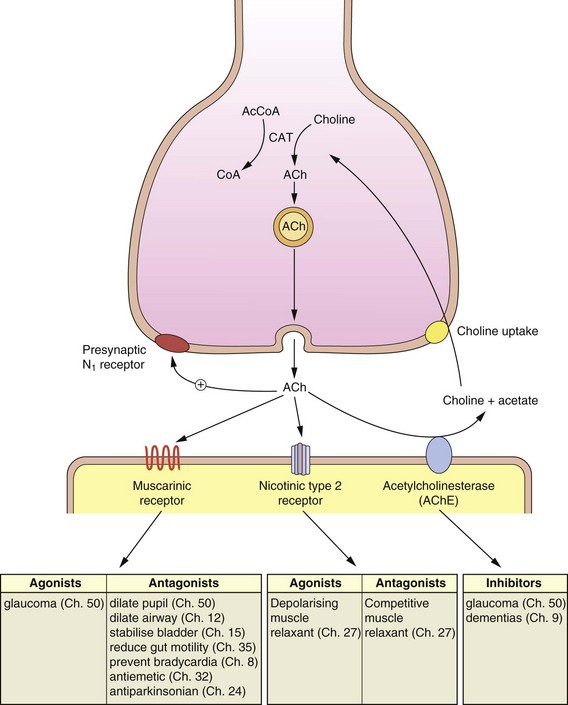
Fig. 4.5 The mechanisms involved in the synthesis, release and inactivation of acetylcholine.
The actions of agonists and antagonists of muscarinic (M) and nicotinic receptors and acetylcholinesterase are shown with the relevant chapters dealing with their pharmacology. ACh, acetylcholine; AcCoA, acetyl-CoA; CAT, choline acetyltransferase.
Storage of acetylcholine
Cytosolic ACh is taken up into membrane vesicles by a specific transmembrane transporter (the vesicular ACh transporter, VAChT) and is stored in the vesicles in association with ATP and acidic proteoglycans (which are also released on exocytosis of the vesicles). Each vesicle contains 1000–50 000 ACh molecules, and neuromuscular junctions (Ch. 27) contain about 300 000 vesicles.
Release of acetylcholine
Release occurs by Ca2+-mediated fusion of the vesicle membrane with the cytoplasmic membrane and exocytosis (Fig. 4.5). This process can be inhibited by botulinum toxin from Clostridium botulinum bacteria and stimulated by latrotoxin from the black widow spider (Latrodectus spp.). The number of vesicles released depends on the site of the synapse, with between 30 and 300 vesicles undergoing exocytosis, releasing from 30 000 to over 3 million ACh molecules into the synaptic cleft. Neurons within the CNS are more sensitive to ACh release and require fewer ACh molecules to be released to stimulate a recipient axon compared with the neuromuscular junction, which requires millions of ACh molecules for skeletal muscle contractility to occur.
Metabolism and inactivation of released acetylcholine
Both presynaptic and postsynaptic membranes are rich in acetylcholinesterase (AChE); hence the released ACh is hydrolysed very rapidly (in usually <1 ms) to give choline and acetate. This rapid hydrolysis, and the rapid equilibration between ACh bound to the receptor and free in the synapse, mean that the ‘receptor phase’ of the transmission process only lasts for 1–2 ms (the postsynaptic changes may be more prolonged; see below).
AChE is an important target for drug action and for the toxic effects of some chemicals; the active site of the esterase has two critical features involved in the metabolism of ACh (Fig. 4.6):
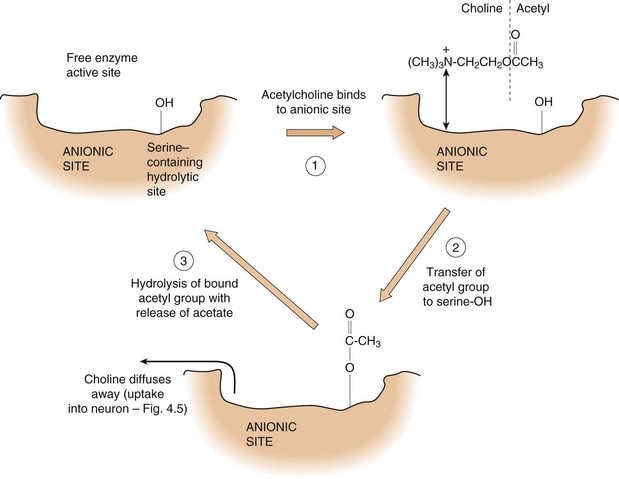
Fig. 4.6 The mechanism of hydrolysis of acetylcholine by acetylcholinesterase.
The choline moiety of acetylcholine binds to the anionic site of the acetylcholinesterase active site, allowing hydrolysis of the acetyl group, with subsequent release of choline and acetate.
an anionic site, which forms an ionic bond with the quaternary nitrogen of the choline part of ACh,
a hydrolytic site, which contains a serine moiety; the hydroxyl group of the serine accepts the acetyl group from ACh and very rapidly transfers it to water to complete the hydrolysis reaction.
Inhibition of AChE will prevent the breakdown of ACh and lead to prolonged receptor occupancy, the consequences of which depend on the nature of the receptor and the innervated cell/tissue.
AChE inhibitors can be divided into three types.
AChE inhibitors that bind to the anionic site. The enzyme can be inhibited by an agent binding reversibly to the anionic site, for example edrophonium (Ch. 28).
AChE inhibitors that carbamylate the serine group. Some inhibitors bind to the anionic site and transfer a carbamoyl group instead of an acetyl group from the drug to the serine hydroxyl group. The carbamoyl group is hydrolysed more slowly from the serine than is an acetyl group and, as a result, prolonged and profound (but reversible) inhibition of the enzyme occurs; examples include neostigmine and pyridostigmine, which are used for reversing neuromuscular block produced by competitive neuromuscular junction blockers (Ch. 27) and in treatment of myasthenia gravis (Ch. 28). Reversible AChE inhibitors such as donepezil and rivastigmine are used in treatment of Alzheimer's disease (Ch. 9).
AChE inhibitors that phosphorylate the serine group. Some inhibitors such as the organophosphates react with the serine hydroxyl group (with or without binding to the anionic site) to produce a phosphorylated enzyme. The phosphorylated enzyme is resistant to hydrolysis and, therefore, these inhibitors cause inhibition which is irreversible (or very slowly and partially reversible). Such permanent changes in enzyme activity are of limited clinical use. The drug ecothiopate acts via phosphorylation of AChE and has a limited clinical use in ophthalmology. Compounds in this group may also be encountered clinically in people suffering accidental or intentional poisoning with organophosphate pesticides, and there has been concern in recent years over low-level exposure of agricultural workers to such compounds, for example in sheep dips. Organophosphates have also been developed as nerve gases for chemical warfare (e.g. sarin). The active serine hydroxyl group may be regenerated soon after exposure by administration of the drug pralidoxime (2-PAM), which is an antidote to organophosphate poisoning, although its efficacy is contentious. A few hours after exposure the phosphorylated enzyme undergoes changes, known as ageing, and pralidoxime cannot reactivate the enzyme after ageing has occurred.
Unlike many other neurotransmitters, ACh is not inactivated by a specific reuptake process, but because choline is a limited resource there is a specific reuptake mechanism to allow choline to re-enter the presynaptic neuron for re-use, rather than simply diffuse away. No such process occurs for acetate because it is readily available from intermediary metabolism. Presynaptic uptake of choline can be inhibited by structural analogues, such as hemicholinium, but such drugs are not useful clinically because of the widespread and non-specific consequences of impairment of ACh uptake, synthesis and release.
Cholinergic receptors
The cholinergic receptors can be divided into nicotinic and muscarinic types. Two nicotinic and five muscarinic subtypes have been characterised (see drug receptor table at the end of Ch. 1). The receptors were originally named after nitrogen-containing basic compounds (alkaloids) present in plants (nicotine) or fungi (muscarine). Figure 4.5 gives information about the general effects of stimulants and inhibitors of muscarinic and nicotinic type 1 receptors and AChE inhibitors and identifies the chapter(s) describing the clinical relevance of these actions.
Nicotinic (N1) receptors
These occur within the CNS and on the postsynaptic membranes of all ganglia of both the sympathetic and parasympathetic branches of the ANS.
Nicotinic (N2) receptors
These occur at the junction between the somatic motor nerves and skeletal muscles (the neuromuscular junction; see Ch. 27).
The nicotinic receptor is a ligand-gated ion channel of five subunits (see Fig. 1.1), with disulphide cross-linking between adjacent subunits; there are different types of subunit (α, β, γ and δ), and different combinations give rise to neuronal N1 receptors and neuromuscular junction N2 receptors. The differences between N1 and N2 receptors in their agonist/antagonist binding characteristics are clinically very important, because they allow neuromuscular blockade (paralysis) without major effects on the ANS.
Muscarinic (M) receptors
These are G-protein-coupled receptors (GPCRs) widely distributed in the CNS and in pre- and postganglionic fibre/effector organ junctions of the parasympathetic branch of the ANS. They are also present on most sweat glands (other than the palms of the hands), which are, however, innervated by the sympathetic branch of the ANS. Table 4.3 shows the effect of stimulation of the muscarinic receptors in major tissues and the principal muscarinic receptor subtype that is involved. Application of molecular biology has identified five subtypes of muscarinic receptor (M1–M5; see drug receptor table at the end of Ch. 1). The distribution and functions of M1, M2 and M3 receptors have been well characterised (Table 4.3).
In addition to occurring on postsynaptic sites, N1 and M receptors are also found presynaptically; recent data suggest that the main role of N1 receptors in the CNS may be as a presynaptic neuromodulator.
It should be appreciated that AChE inhibitors increase the concentrations of ACh at all nicotinic and muscarinic receptor sites, and therefore produce a diverse array of effects. For example, when an AChE inhibitor is used to overcome reversible neuromuscular blockade (see Ch. 27) it increases ACh-mediated effects produced via the parasympathetic nervous system, such as on the gastrointestinal tract and heart. These unwanted effects of ACh can be blocked by co-administration of an antimuscarinic agent (see drug receptor table at the end of Ch. 1).
The distribution of key neurotransmitters and receptors of the somatic and autonomic nervous systems is summarised in Figure 4.7. Students should familiarise themselves with Figure 4.7 and with the possible sites of drug action (Tables 4.2 and 4.3). Such knowledge is fundamental to understanding both the principal mechanisms of action for many drugs and the source of unwanted effects for others.
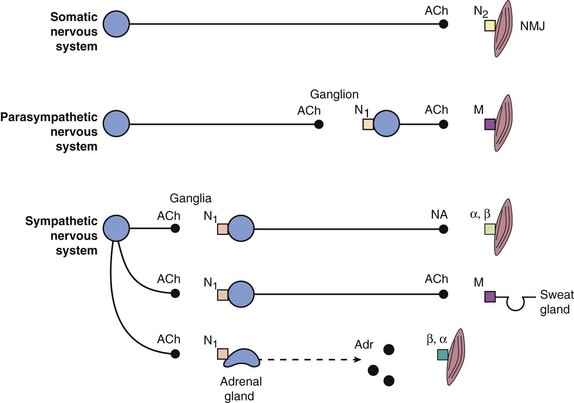
Fig. 4.7 Schematic diagram of the distribution of the main neurotransmitters and receptors of the somatic, parasympathetic and sympathetic nervous systems.
α, β, adrenoceptors; ACh, acetylcholine; Adr, adrenaline; M, muscarinic receptor; N1, N2, nicotinic receptors; NA, noradrenaline; NMJ, neuromuscular junction.
Other transmitters in the peripheral nervous system
In addition to ACh and noradrenaline, there are other transmitters with roles in neurotransmission and function in the peripheral nervous system. Many of these are also of considerable importance in the CNS. The different transmitters are dealt with in the chapters that describe their clinical importance, and include:
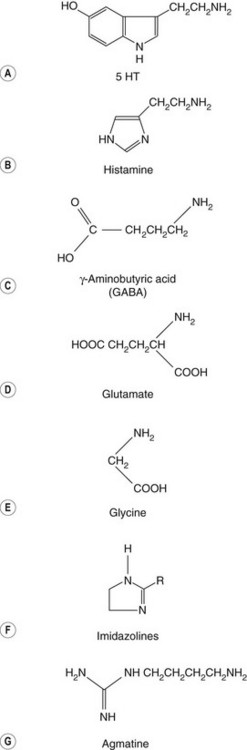
amines, e.g. dopamine, histamine, serotonin,
amino acids, e.g. glutamate, glycine, γ-aminobutyric acid (GABA), agmatine,
Nitric oxide, calcitonin gene-related peptide, vasoactive intestinal peptide (VIP), neuropeptide Y, ghrelin and others are described later in the book.
Amines
Dopamine is an important neurotransmitter in both the CNS and the periphery, and subsequent chapters cover its actions (Chs 7, 21, 24 and 32).
Synthesis and storage of dopamine: Synthesis and storage of dopamine have been described above under noradrenaline.
Release of dopamine: Nerve stimulation causes release of dopamine present in vesicles (see noradrenaline). Dopaminergic neurons are not important in the clinical responses to indirectly acting sympathomimetics, although certain behavioural responses to amfetamines are linked to dopamine D2 receptor activity. The antiviral drug amantadine, which is of some value in Parkinson's disease, causes release of dopamine (Ch. 24).
Removal of activity of released dopamine: Dopamine is removed by similar mechanisms to those described above for noradrenaline, with reuptake by the dopamine transporter (DAT) representing the major pathway.
Dopamine receptors: There are a number of types of dopamine receptors, and relatively selective therapeutic agents are available for some of these (see drug receptor table at the end of Ch. 1). Dopamine receptors are classified into those that increase cAMP (D1 and D5) and those that decrease cAMP (D2, D3 and D4). The D4 receptor shows polymorphic expression; subtypes D2 and D4 are associated with schizophrenia and relatively selective antagonists of each are valuable antipsychotic drugs and have some different biological properties (Ch. 21).
Serotonin (5-hydroxytryptamine)
Serotonin (or 5-HT; Fig. 4.8A) is a neurotransmitter in the CNS and periphery that has properties similar to the catecholamines.
Synthesis of serotonin: Serotonin is synthesised from the amino acid tryptophan by two reactions that are similar to those involved in the conversion of tyrosine to dopamine. The first reaction is oxidation of the benzene ring to form 5-hydroxytryptophan, catalysed by tryptophan hydroxylase, which is the rate-limiting step and only found in serotonin-producing cells. Conversion to serotonin is catalysed by aromatic L-amino acid decarboxylase (see noradrenaline synthesis).
Serotonin is present in the diet but undergoes essentially complete first-pass metabolism by MAO-A in the gut wall and liver. Serotonin is not synthesised by blood platelets, but they accumulate high concentrations of serotonin from the circulation which can be released when platelets aggregate and during migraine episodes.
Storage of serotonin: The major site of serotonin storage in the body (>90%) is the enterochromaffin cells of the gastrointestinal tract. Platelets accumulate serotonin and neurons utilizing serotonin are widely distributed in the brain. In presynaptic neurons serotonin is stored in vesicles as a complex with ATP, and there is an active uptake process which transfers cytoplasmic serotonin into the storage vesicle.
Release of serotonin: The release of serotonin vesicles is by Ca2+-mediated exocytosis. A rise in intraluminal pressure in the gastrointestinal tract stimulates the release of serotonin from the chromaffin cells. Release of serotonin from chromaffin cells contributes to nausea following cancer chemotherapy with cytotoxic drugs by stimulation of the chemoreceptor trigger zone (Ch. 32) and of sensory receptors within the gastrointestinal tract. There is a significant release of platelet serotonin in migraine (Ch. 26).
Metabolism and removal of serotonin activity: The principal mechanism of inactivation of released serotonin is via its reuptake into the presynaptic nerve by the serotonin transporter (SERT), which shows a high affinity for serotonin and is distinct from the noradrenaline transporter (NET). Dual inhibitors of serotonin and noradrenaline reuptake (SNRIs) and selective serotonin reuptake inhibitors (SSRIs) are useful antidepressants (Ch. 22). Serotonin reuptake is also carried out by a low-affinity plasma membrane monoamine transporter (PMAT), which is insensitive to SSRIs.
Metabolism within the neuron is by MAO, which generates the excretory product 5-hydroxyindoleacetic acid (5-HIAA). There is a considerable turnover of serotonin in the chromaffin and nerve cells, and 5-HIAA is a normal constituent of human urine.
Serotonin receptors: There is a family of serotonin receptors, which has allowed the development of selective drugs (see drug receptor table at the end of Ch. 1). Thus far, the different serotonin receptors comprise 13 different G-protein-coupled 7TM receptors and one ligand-gated ion channel, which are divided into seven classes (5-HT1 to 5-HT7) on the basis of their structural and functional characteristics. Not all of the subtypes of receptors have recognised physiological roles. Receptors in the 5-HT1 group are mostly presynaptic and inhibit adenylyl cyclase, whereas those in the 5-HT2 group are mostly postsynaptic in the periphery and activate phospholipase C. Identification of receptor subtype functions and selective inhibitors or stimulants has facilitated progress in the treatment of diseases including depression (Ch. 22) and migraine (Ch. 26).
Histamine
Histamine (Fig. 4.8B) is an important transmitter both in the CNS and in the periphery, as well as being an allergic mediator released from mast cells and basophils.
Synthesis of histamine: The amino acid histidine is decarboxylated to histamine by histidine decarboxylase. In addition to the synthesis and storage of histamine by mast cells and basophils there is continual synthesis, release and metabolic inactivation by growing tissues and in wound healing.
Storage of histamine: Most attention has focused on the storage of histamine in mediator-releasing cells such as mast cells and basophils (Ch. 12). In these cells it is present in granules associated with heparin. The presence of histidine decarboxylase and the synthesis of histamine in neurons in the CNS, although less well explored, appear to be associated mainly with the hypothalamus, from where projections run to many parts of the brain. Histamine plays a role in wakefulness, memory, appetite and many other functions.
Release of histamine: The release of histamine from mast cells and basophils has been studied extensively in relation to allergic reactions (Chs 12 and 39). In chromaffin cells in the gut and enterochromaffin-like (ECL) cells in the gastric mucosa, histamine is synthesised rapidly when required (Ch. 33). The release of histamine from neurons may be similar to the release of other amine neurotransmitters, but this has not been demonstrated unequivocally.
Removal of histamine activity: Histamine is rapidly inactivated by oxidation to imidazole acetic acid. Histamine is not a substrate for MAO and the oxidation is catalysed by diamine oxidase (or histaminase). A second, minor route of metabolism is methylation by histamine-N-methyltransferase, and the product is then a substrate for MAO. Histamine is also eliminated as an N-acetyl conjugate.
Histamine receptors: There are four receptors for histamine (see drug receptor table at the end of Ch. 1). H1 receptors have been studied extensively in relation to inflammation and allergy (Chs 12 and 39). Histamine-containing neurons are found in the brain, particularly in the brainstem, with pathways projecting into the cerebral cortex. H1 receptors are probably important in these pathways, because sedation is a serious problem with H1 receptor antagonists that are able to cross the blood–brain barrier (Ch. 2). The second generation of H1 antihistamines produce less sedation. H1 receptors are also involved in emesis (Ch. 32).
The discovery of H2 receptors affecting the release of gastric acid led to the development of important H2-selective antihistamines that reduce acid secretion and contribute to the treatment of dyspepsia and to ulcer healing (Ch. 33). H2 receptors are also present in the brain and are probably responsible for the confusional state associated with the use of the H2 receptor antagonist cimetidine. H3 receptors are found in the CNS and other sites and H4 receptors are localised mainly to leucocytes, but their functions are poorly understood.
Amino acids
GABA is an important inhibitory neurotransmitter responsible for about 40% of all inhibitory activity in the CNS (Fig. 4.8C).
Synthesis and storage of GABA: GABA is formed by the decarboxylation of glutamate by glutamate decarboxylase in GABAergic neurons. GABA is stored in membrane vesicles in the brain and in interneurons in the spinal cord (particularly laminae II and III).
Release of GABA: GABA is released by Ca2+-mediated exocytosis. Co-transmitters such as glycine, metenkephalin and neuropeptide Y are stored in GABA vesicles and released with GABA.
Removal of GABA activity: Uptake by the GAT family of transporters is the principal mechanism for the removal of GABA from the synaptic cleft. The antiepileptic drug tiagabine may act as an inhibitor of GABA uptake by GAT-1 (Ch. 23).
GABA is metabolised by transamination with α-ketoglutarate, which forms the corresponding aldehyde (succinic semialdehyde) and amino acid (glutamic acid). The antiepileptic drug vigabatrin inhibits GABA transamination.
GABA receptors: There are two main types of GABA receptor, with different mechanisms of action (see drug receptor table at the end of Ch. 1). Stimulation of both receptors produces hyperpolarisation of the cell membrane, with GABAA causing rapid inhibition and GABAB producing a slower and more prolonged response. The GABAA receptor comprises a number of subunits. There are multiple forms of each subunit and numerous possible combinations (see Fig. 20.1); consequently, the GABAA receptor should be regarded as a family of receptors. Hyperpolarisation following GABAA receptor stimulation results from the opening of Cl− channels and influx of Cl−. GABAB receptors are G-protein-linked receptors that hyperpolarise the cell indirectly by closing Ca2+ channels and opening K+ channels. A subtype of GABAA receptor (GABAA-ρ) is found in the retina, where its significance remains unclear. Both GABAA and GABAB receptors are found presynaptically and inhibit neurotransmitter release by hyperpolarising the cell (by opening Cl− or K+ channels) and reducing release of the vesicles of the innervating cell (by closing Ca2+ channels). Many important drugs act by altering GABA breakdown or by enhancing GABA activity at its receptor (Chs 20 and 23).
Glutamate
Glutamate (Fig. 4.8D) is an important excitatory amino acid neurotransmitter with wide-reaching actions in physiological and pathological conditions. The functions of glutamate are described in later chapters. Aspartate (which is similar to glutamate but has only one CH2 group) acts at the same receptors. Administration of glutamate or aspartate causes CNS excitation, tachycardia, nausea and headache, and convulsions at very high doses. Hyperactivity at glutamate receptors has been proposed as a factor in the generation of epilepsy (Ch. 23).
Synthesis and storage of glutamate: Glutamate (glutamic acid) is an amino acid that is found in most cells and is widely distributed within the CNS. Glutamate is stored in presynaptic vesicles in the neurons.
Release of glutamate: Exocytosis of vesicles is mediated via the influx of Ca2+ into the presynaptic nerve terminal, as occurs for other neurotransmitters. Some antiepileptic drugs, for example lamotrigine and valproate (Ch. 23), inhibit glutamate release.
Removal of glutamate activity: The action of glutamate in the synapse is terminated by excitatory amino acid transporters (EAATs), which take up glutamate (and aspartate) into the neuron and surrounding glial cells.
Glutamate receptors: There are two major types of glutamate receptor, the ionotropic family (AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid)/kainate/NMDA (N-methyl-D-aspartate)) and the metabotropic family (metabotropic glutamate receptors, or mGluRs), which have a range of biological actions (see drug receptor table at the end of Ch. 1). The NMDA antagonist memantine is used in treating Alzheimer's disease (Ch. 9).
Glycine
Glycine (Fig. 4.8E) is a widely available amino acid that acts as an inhibitory neurotransmitter. It is released in response to nerve stimulation and acts in the spine, lower brainstem and retina.
Synthesis and storage of glycine: Glycine is present in all cells and is accumulated by neurons. It is stored within neurons in vesicles.
Release of glycine: Vesicle release accompanies an AP, as described above for other neurotransmitters. Tetanus toxin prevents glycine release, and the decrease in glycine-mediated inhibition results in reflex hyperexcitability.
Removal of glycine activity: Released glycine undergoes cellular uptake via the high-affinity transporters GLYT-1 and GLYT-2 in glial and neuronal cells.
Glycine receptors: Glycine receptors (GlyRs) are ligand-gated Cl− channels similar in structure to GABAA channels: they are present mainly on interneurons in the spinal cord. Strychnine produces convulsions through the blockade of glycine receptors. Glycine is important for the activity of NMDA receptors (see drug receptor table at the end of Ch. 1).
Imidazoline receptor ligands
Studies of the centrally acting α2-adrenoceptor agonists clonidine, moxonidine and rilmenidine showed their antihypertensive effects could be not be interpreted wholly by actions on the α2-adrenoceptor. These imidazoline compounds (Fig. 4.8F) are thought to act at least partly at imidazoline (I)-binding sites, of which there are three main types. The I1 receptor mediates the sympatho-inhibitory actions on blood pressure in the brainstem, the I2 receptor is an allosteric binding site on monoamine oxidase and the I3 receptor regulates insulin secretion from pancreatic β cells. The putative natural ligand for I receptors, agmatine (Fig. 4.8G), is a decarboxylated derivative of the amino acid arginine; it also binds to α2-adrenoceptors and activates nitric oxide synthase, but its role in disease is unclear.
Peptides
The importance of peptides as neurotransmitters has been appreciated in recent years, largely because of the development of highly specific and sensitive probes, combined with histochemical techniques, which has allowed their detection and measurement. Unlike other classes of neurotransmitter, peptides are synthesised in the cell body as precursors, which are transported down the axon to the site of storage. There are specific receptors for different peptides (see drug receptor table at the end of Ch. 1). An AP causes the release of the peptide from its precursor; inactivation is probably via hydrolysis by a local peptidase.
Peptide neurotransmitters are often found stored in the same nerve endings as other transmitters (described above) and undergo simultaneous release (co-transmission).
Peptides do not cross the blood–brain barrier readily. A major problem for exploiting our increasing knowledge of the importance of peptides is devising ways to deliver the novel products derived from molecular biology to the sites within the brain where they can have an effect.
Substance P is released from C-fibres (Ch. 19) by a Ca2+-linked mechanism and is an important neurotransmitter for sensory afferents in the dorsal horn. It is also present in the substantia nigra, associated with dopaminergic neurons, and may be involved in the control of movement.
Opioid peptides are a range of endogenous peptides that are the natural ligands for opioid receptors; opioid receptors were recognised in the brain and gastrointestinal tract for many years before the natural ligands were identified. These are discussed in Chapter 19.
A number of other peptides are detectable in the CNS particularly in the hypothalamus and/or pituitary gland (e.g. neurotensin, oxytocin, somatostatin, vasopressin; see Chs 43 and 45) or in the gastrointestinal tract (e.g. cholecystokinin and vasoactive intestinal peptide).
Purines
Adenosine and guanosine are endogenous purines and exist in the body in the free form, attached to ribose or deoxyribose (as nucleosides), and as mono-, bi- or triphosphorylated nucleotides. Purines within cells are usually incorporated into nucleotides, which are involved in the energetics of biochemical processes (e.g. ATP), act as intracellular signals (e.g. cAMP and cGMP; see Ch. 1) and are involved in the synthesis of RNA and DNA. ATP is present in the presynaptic vesicles of some other neurotransmitters and is released along with the primary neurotransmitter, following which it may act on postsynaptic receptors (co-transmission). Extracellular ATP is rapidly hydrolysed via adenosine diphosphate (ADP) to adenosine. Adenosine itself is very rapidly metabolised and inactivated.
There is a family of purine receptors that show individual selectivity for different purines and give different responses (see drug receptor table at the end of Ch. 1). G-protein-coupled purinergic receptors (P2Y) are specific for the adenosine and uridine phosphates, and ADP causes platelet aggregation via P2Y12-type receptors. This effect of ADP can be inhibited with clopidogrel and ticagrelor, which have important anti-aggregatory actions (Ch. 11). Ligand-gated P2X receptors for ATP are widely distributed in the brain. The adenosine receptors A1–A3, formerly called P1 receptors, show very high selectivity for adenosine itself. Adenosine is used therapeutically to terminate supraventricular tachycardia.
True/false questions
1. The sympathetic division of the ANS utilises adrenaline as its primary transmitter substance.
2. The parasympathetic and sympathetic nervous systems have opposite effects in every organ.
3. Sympathetic nervous stimulation to the gut inhibits gut motility and sphincter tone.
4. Acetylcholine is metabolised by plasma cholinesterase in the synaptic cleft.
5. Dopamine and noradrenaline are synthesised from levodopa.
6. Dopamine is a transmitter in the peripheral autonomic nervous system.
7. Tyramine is metabolised by both isoenzymes of monoamine oxidase (MAO-A and MAO-B).
8. Both α1- and α2-adrenoceptor antagonists can be used to lower blood pressure.
9. Botulism is caused by poisoning with a bacterial toxin.
10. Botulinum toxin enhances acetylcholine release from all cholinergic neurons.
11. There are two subtypes of β-adrenoceptor.
12. Blockade of presynaptic adrenoceptors by propranolol increases noradrenaline release.
13. The actions of synaptic serotonin and noradrenaline are curtailed mainly by metabolism by MAO and COMT.
14. The synaptic uptake of noradrenaline and serotonin can be inhibited selectively.
15. The vagal cranial nerve to the eye decreases pupil size.
16. Blockade of H1 histamine receptors reduces gastric acid secretion.
17. Glutamate and glycine are inhibitory amino acid transmitters.
18. Substance P is a transmitter in the dorsal horn of the spinal cord
One-best-answer (OBA) question
Which of the following is the most accurate statement about neurotransmission?
A Neurotransmitters are synthesised in the presynaptic nerve terminal.
B Neurotransmitters are taken up into the presynaptic neuron by passive diffusion.
C Acetylcholine release is modified by receptors on the presynaptic membrane.
D Each postganglionic sympathetic neuron releases a single neurotransmitter.
E Fusion of vesicles with the presynaptic membrane is facilitated by K+ influx triggered by the AP.
1. False. Noradrenaline is the main transmitter substance at the sympathetic postganglionic nerve endings. Adrenaline is released only from the adrenal medulla and acetylcholine is the transmitter only in sweat glands and hair follicles.
2. False. While the two autonomic systems have broadly opposing actions on many organs, other organs may be controlled by only one system (e.g. the lens of the eye).
3. False. Sympathetic nervous stimulation releases noradrenaline and inhibits motility but increases the tone of the sphincters.
4. False. Within the synaptic cleft acetylcholine is broken down rapidly by acetylcholinesterase.
5. True. Levodopa is converted into dopamine by DOPA decarboxylase and then to noradrenaline by dopamine-β-hydroxylase.
6. True. Dopamine is predominantly an important transmitter in the CNS but also in some peripheral sites, for example the renal vascular smooth muscle.
7. True. This is important, as selective inhibitors of MAO-A used in the treatment of depression leave MAO-B unaffected so this is available to metabolise tyramine in food, thereby avoiding the ‘cheese reaction’ (see Ch. 22).
8. False. Antagonism of α1-adrenoceptors on peripheral resistance vessels causes relaxation and lowers blood pressure, but presynaptic α2-adrenoceptors reduce noradrenaline release so blockade of these receptors would increase noradrenaline release and raise blood pressure.
9. True. Botulinum toxin from the anaerobic bacterium Clostridium botulinum can cause fatal poisoning.
10. False. Botulinum toxin inhibits acetylcholine release and causes skeletal muscle paralysis; it can be used locally where there is muscle spasm or excessive sweating.
11. False. A third type, the β3-adrenoceptor, is found in adipocytes, the heart, colon, bladder and some other tissues, but is less widespread than the β1- and β2-adrenoceptors.
12. True. Propranolol is a non-selective antagonist of β-adrenoceptors, and the role of the presynaptic β2-adrenoceptor is to increase noradrenaline release. (Noradrenaline release is decreased by presynaptic α2-adrenoceptors.)
13. False. The actions of serotonin and noradrenaline are curtailed mainly by reuptake into the presynaptic neuron by their respective transporters, SERT and NET.
14. True. The SERT and NET uptake transporters can be inhibited by SSRIs and other selective antidepressant drugs.
15. True. Vagal (parasympathetic) stimulation causes miosis of the pupil and also accommodates the lens for near vision.
16. False. Gastric acid secretion is promoted by histamine released from enterochromaffin-like (ECL) cells acting at H2 receptors, and is reduced by H2 antihistamines such as ranitidine.
17. False. Glycine is an inhibitory transmitter but glutamate is excitatory.
18. True. Substance P in the dorsal horn is an important transmitter in sensory afferents.
OBA answer
A Incorrect. Peptides are synthesised in the cell body and transported to the postganglionic nerve ending.
B Incorrect. Active transporters transfer neurotransmitters back into the presynaptic neuron.
C Correct. On parasympathetic nerve endings, stimulation of presynaptic N1 receptors increases acetylcholine release whereas stimulation of presynaptic M2 receptors decreases acetylcholine release.
D Incorrect. Co-transmission is common, such as noradrenaline and vasoactive intestinal polypeptide released from sympathetic nerve endings to the gut.
E Incorrect. An influx of Ca2+ is associated with transmitter release.
Abrams, P, Andersson, K-E, Buccafusco, J, et al. Muscarinic receptors: their distribution and function in body systems, and the implications for treating overactive bladder. Br J Pharmacol. 2006;148:565–578.
Berger, M, Gray, J, Roth, BL. The expanded biology of serotonin. Annu Rev Med. 2009;60:355–366.
Bowery, NG, Bettler, B, Froestl, W, et al. International Union of Pharmacology. XXXIII. Mammalian gamma-aminobutyric acid (B) receptors: structure and function. Pharmacol Rev. 2002;54:247–264.
Burnstock, G. Purinergic signalling. Br J Pharmacol. 2006;147(Suppl. 1):S172–S181.
Dajas-Bailador, F, Wonnacott, S. Nicotinic acetylcholine receptors and the regulation of neuronal signalling. Trends Pharmacol Sci. 2004;25:317–324.
Dani, JA, Bertrand, D. Nicotinic acetylcholine receptors and nicotinic cholinergic mechanisms of the central nervous system. Ann Rev Pharmacol Toxicol. 2007;47:699–729.
Eglen, RM, Choppin, A, Dillon, MP, Hegde, S. Muscarinic receptor ligands and their therapeutic potential. Curr Opin Chem Biol. 1999;3:426–432.
Filip, M, Bader, M. Overview on 5-HT receptors and their role in physiology and pathology of the central nervous system. Pharmacol Rep. 2009;61:761–777.
Foster, AC, Kemp, JA. Glutamate- and GABA-based CNS therapeutics. Curr Opin Pharmacol. 2006;6:7–17.
Frishman, WH, Kotob, F. Alpha-adrenergic blocking drugs in clinical medicine. J Clin Pharmacol. 1999;39:7–16.
Gether, U, Andersen, PH, Larsson, OM, Schousboe, A. Neurotransmitter transporters: molecular function of important drug targets. Trends Pharmacol Sci. 2006;27:375–383.
Grace, AA, Gerfen, CR, Aston-Jones, G. Catecholamines in the central nervous system. Overview. Adv Pharmacol. 1998;42:655–670.
Haas, HL, Sergeeva, OA, Selbach, O. Histamine in the nervous system. Physiol Rev. 2008;88:1183–1241.
Head, GA, Mayorov, DN. Imidazoline receptors, novel agents and therapeutic potential. Cardiovasc Hematol Agents Med Chem. 2006;4:17–32.
Hieble, JP. Adrenoceptor subclassification: an approach to improved cardiovascular therapeutics. Pharm Acta Helv. 2000;74:163–171.
Insel, PA. Adrenoceptors – evolving concepts and clinical implications. N Engl J Med. 1996;334:580–585.
Kirstein, SL, Insel, PA. Autonomic nervous system pharmacogenomics: a progress report. Pharmacol Rev. 2004;56:31–52.
Ogden, KK, Traynelis, SF. New advances in NMDA receptor pharmacology. Trends Pharmacol Sci. 2011;32:726–733.
Olsen, RW, Sieghart, W. GABAA receptors: subtypes provide diversity of function and pharmacology. Neuropharmacology. 2008;56:141–148.
Piascik, MT, Perez, DM. Alpha-1 adrenergic receptors: new insights and directions. J Pharmacol Exp Ther. 2001;298:403–410.
Romanelli, MN, Gualtieri, F. Cholinergic nicotinic receptors: competitive ligands, allosteric modulators, and their potential applications. Med Res Rev. 2003;23:393–426.
Salio, C, Lossi, L, Ferrini, F, Merighi, A. Neuropeptides as synaptic transmitters. Cell Tissue Res. 2006;326:583–598.
Simons, FE. Advances in antihistamines. N Engl J Med. 2004;351:2203–2217.
Small, KM, McGraw, DW, Liggett, SB. Pharmacology and physiology of human adrenergic receptor polymorphisms. Annu Rev Pharmacol Toxicol. 2003;43:381–411.
Thurmond, RL, Gelfand, EW, Dunford, PJ. The role of histamine H1 and H4 receptors in allergic inflammation: the search for new antihistamines. Nat Rev Drug Discov. 2008;7:41–53.
Wallukat, G. The beta-adrenergic receptors. Herz. 2002;27:683–690.
Youdim, MBH, Edmondson, D, Tipton, K. The therapeutic potential of monoamine oxidase inhibitors. Nat Rev Neurosci. 2006;7:295–309.