Haemostasis
Haemostasis is a complex process involving vasoconstriction, platelet aggregation, blood coagulation and the interactions between them. The descriptions of the processes of platelet aggregation and coagulation pathways in this chapter are restricted to essential knowledge required for understanding the actions of pharmacological agents.
Platelets and platelet aggregation
Platelets are critical components of the blood for initiating thrombus formation, and have a lifespan in the circulation of 8–10 days, with about 10–12% being replaced each day. Platelets aggregate following adhesion to an injured blood vessel and consequent activation. When the integrity of vascular endothelium is breached, subendothelial proteins such as von Willebrand factor (vWF) and collagen come into contact with blood. These proteins interact with a family of platelet-surface glycoprotein (GP) receptors (integrin receptors), particularly GPIb (vWF receptor) and GPVI (collagen receptor), resulting in platelets adhering at the site of injury and to each other (primary reversible aggregation) and the formation of a platelet plug (Fig. 11.1). Platelet adhesion then initiates a process known as platelet activation.
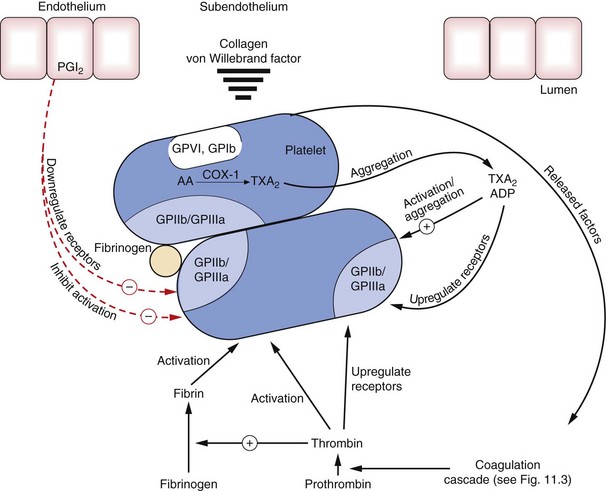
Fig. 11.1 Platelets and platelet aggregation.
Subendothelial macromolecules such as von Willebrand factor and collagen interact with glycoprotein receptors (GPVI and GPIb) on platelets, causing activation of platelets and upregulation of GPIIb/IIIa receptors, which are crosslinked by fibrinogen, resulting in aggregation. During the initial processes of aggregation, stimulation of the synthesis and release of a number of platelet-derived substances, such as thromboxane A2 (TXA2) synthesised from arachidonic acid (AA) by cyclo-oxygenase-1 (COX-1), ADP, and other factors (see text) further promote aggregation by upregulation of GPIIb/IIIa receptors. Conversely, prostacyclin (PGI2) from endothelial cells inhibits activation and upregulation of GPIIb/IIIa receptors. Thrombin is generated by the action of factor Xa on prothrombin (see Fig. 11.3).
Extension of the platelet plug requires activation of platelets and their subsequent aggregation together (homotypic aggregation). Platelets are initially activated by exposure to soluble agonists, such as thrombin generated by local coagulation, ADP released from endothelial cells and collagen. These activators lead to an increase in intracellular Ca2+ and activation of (MLCK). MLCK phosphorylates myosin light chains in the platelet which interact with actin, disrupt the platelet cytoskeleton and change the shape of the platelet.
The increase in platelet intracellular Ca2+ activates phospholipase A2, which liberates arachidonic acid (AA) from membrane phospholipids. AA is then converted by cyclo-oxygenase type 1 (COX-1) in the platelet to thromboxane A2, the most potent naturally occurring pro-aggregating agent, which diffuses from the platelet. Significant disruption of the platelet cytoskeleton as the platelet changes shape also initiates a platelet release reaction, which expels mediators in platelet dense storage granules from the cell. Fusion of dense granules with the platelet cell membrane releases platelet factor 4, adrenaline, ADP and serotonin. Outside the platelet ADP, thrombin and thromboxane A2 interact with specific platelet surface receptors and trigger intracellular pathways that express and activate GPIIb/IIIa collagen receptors on the surface of the platelets (Figs 11.1 and 11.2). Therefore, ADP released from platelets acts as a mediator for initiators of platelet activation, such as collagen and thrombin. Secondary irreversible homotypic platelet–platelet aggregation follows platelet activation when the activated GPIIb/IIIa receptors on the surface of the platelets are crosslinked by fibrinogen in the plasma.
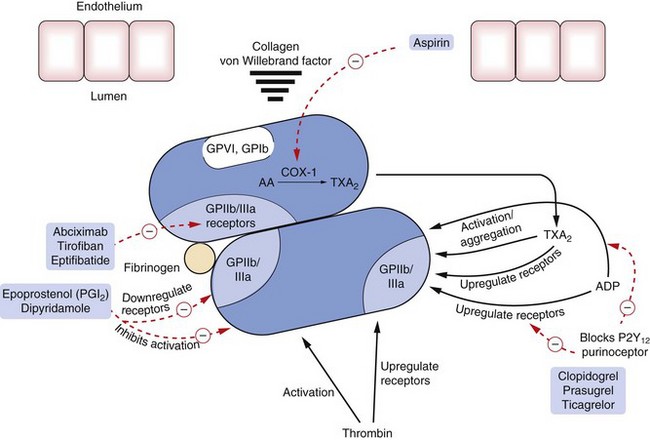
Fig. 11.2 Sites of action of major drugs used in haemostasis.
Drugs act directly or indirectly to inhibit activation of platelets or to block or reduce upregulation of the glycoprotein GPIIb/IIIa receptors (integrin receptor family), which are necessary for aggregation of platelets. Abciximab is an antibody, tirofiban a non-peptide inhibitor and eptifibatide a peptide inhibitor of these glycoprotein receptors. Epoprostenol and dipyridamole inhibit activation of platelets and downregulate the glycoprotein receptors by increasing cAMP. Clopidogrel inhibits ADP receptors and prevents ADP-induced upregulation of the glycoprotein GPIIb/IIIa receptors and platelet aggregation. Aspirin inhibits the generation of thromboxane A2 (TXA2) by cyclo-oxygenase-1 (COX-1), which otherwise causes activation of platelets and upregulation of GPIIb/IIIa receptors. AA, arachidonic acid. For direct and indirect inhibitors of thrombin, see Fig. 11.3.
The substances released from platelet dense granules also facilitate haemostasis by:
 reducing prostacyclin (prostaglandin I2, PGI2) synthesis by vascular endothelium; prostacyclin is a vasodilator and a potent inhibitor of platelet aggregation,
reducing prostacyclin (prostaglandin I2, PGI2) synthesis by vascular endothelium; prostacyclin is a vasodilator and a potent inhibitor of platelet aggregation,
inhibiting the action of heparin sulphate produced by vascular endothelium; this enhances activity of the coagulation cascade.
Expression of platelet GPIIb/IIIa surface receptors can be inhibited by an increase in the concentration of cyclic nucleotides (cAMP, cGMP) in the platelet. This is the mechanism by which prostacyclin (PGI2) inhibits platelet aggregation (Figs 11.1 and 11.2).
Polyunsaturated (omega-3) fatty acids in fish oils are precursors for thromboxane A3, which causes less platelet aggregation than thromboxane A2; they also increase production of a modified form of prostacyclin (PGI3) by vascular endothelium which has equal anti-aggregatory activity to PGI2. Therefore, a high intake of fish oils creates a state in which platelets are less able to aggregate.
Heterotypic platelet aggregation can also arise when platelets aggregate with leucocytes (and particularly monocytes) in circulating blood. This process has been detected close to atherosclerotic lesions but also in a variety of inflammatory conditions, and may follow initial activation of platelets by vascular damage. Heterotypic aggregation results from expression of P-selectin on the surface of the platelet. P-selectin is one of several molecules found in platelet alpha-granules, which are released when the platelet cytoskeleton is only minimally disrupted by thrombin- or ADP-mediated activation of the cell. Heterotypic platelet aggregation is not inhibited by some antiplatelet drugs (such as aspirin) to the same extent as homotypic aggregation
Blood coagulation and the coagulation cascade
Both coagulant and anticoagulant factors regulate haemostasis. Activation of the coagulation cascade is divided into extrinsic and intrinsic pathways (Fig. 11.3). The factors involved in these cascades amplify the coagulation response and work together to produce a thrombus. The extrinsic pathway accounts for most of the coagulation in vivo, but both coagulation pathways respond to breaches in endothelial integrity much more slowly than platelet aggregation. The following description of the pathways is simplified to identify the key steps at which drugs can modulate coagulation.
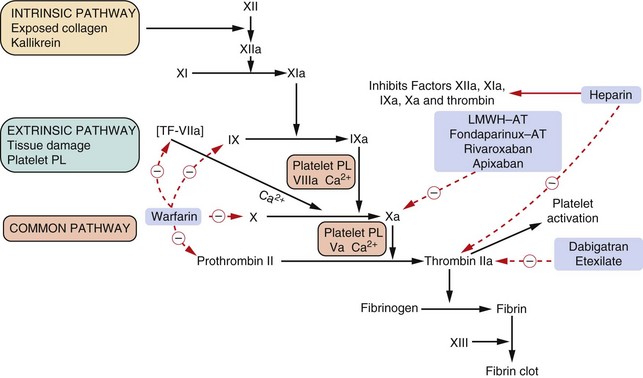
Fig. 11.3 The coagulation cascade and action of anticoagulants.
The complex cascade of clotting factor synthesis is initiated extrinsically by tissue damage. Activation of the clotting factors after damage depends upon platelet factors, tissue factor, phospholipids, Ca2+ and vitamin K. The provision of platelet products is further enhanced by the formation of thrombin, which then activates further platelets as well as causing fibrin formation. Heparin acts at various sites in the cascade by complexing with the anticlotting factor antithrombin III (AT) and inhibiting thrombin (IIa) and the other activated clotting factors shown. Low-molecular-weight heparin (LMWH) complexes with AT but in a different manner to unfractionated heparin and inhibits only factor Xa. Bivalirudin and dabigatran etexilate inhibit thrombin (IIa) action. Warfarin inhibits the synthesis of the vitamin K-dependent clotting factors VII, IX, X and II (prothrombin). Roman numerals indicate the individual clotting factors; PL, phospholipid; TF, tissue factor.
The extrinsic coagulation pathway is initiated by exposure of blood to tissue factor (TF) on the surface of subendothelial cells after vascular injury, and is activated rapidly within minutes of endothelial disruption. Formation of complexes of TF with factor VIIa, and the presence of phospholipids and Ca2+, result in the conversion of inactive factor X to active Xa.
The intrinsic pathway is triggered by contact of blood with a negatively charged surface such as subendothelial collagen, and its activation is delayed by more than 10 min after tissue disruption. The intrinsic coagulation pathway comprises a series of enzyme-mediated reactions involving activation of several clotting factors and eventually activation of factor X.
Activation of factor X, which mediates the hydrolysis of prothrombin to thrombin (factor IIa), is the point at which the two pathways of coagulation converge (Fig. 11.3). The actions of thrombin (factor IIa) and several other activated coagulation factors (Fig 11.3) are inhibited by circulating antithrombin. Antithrombin inhibits coagulation factors after forming complexes with heparin-like molecules that are produced by intact endothelial cells, and with heparin released from mast cells. Once sufficient thrombin has been produced to overcome the effect of circulating antithrombin, the soluble protein fibrinogen is converted to an insoluble fibrin gel. Thrombin also activates factor XIII, which crosslinks the fibrin polymers and forms a fibrin mesh that traps circulating platelets, leucocytes and red blood cells.
Each activated clotting factor is inactivated extremely rapidly so that the coagulation process remains localised at the site of the initiating event. However, in some circumstances, aggregates of platelets combined with fibrin thrombi can embolise and occlude more distal parts of the circulation.
Arterial and venous thrombosis
There are differences in the composition of an arterial or venous thrombus. Arterial thrombosis occurs in the setting of high flow and high shear stress, and platelets play a prominent role in the initiation and growth of the thrombus. In contrast, venous thrombi form in a low-flow, low-shear stress environment. Venous thrombus usually forms initially in the valve pockets of deep veins, and consists mainly of fibrin and red cells with few platelets.
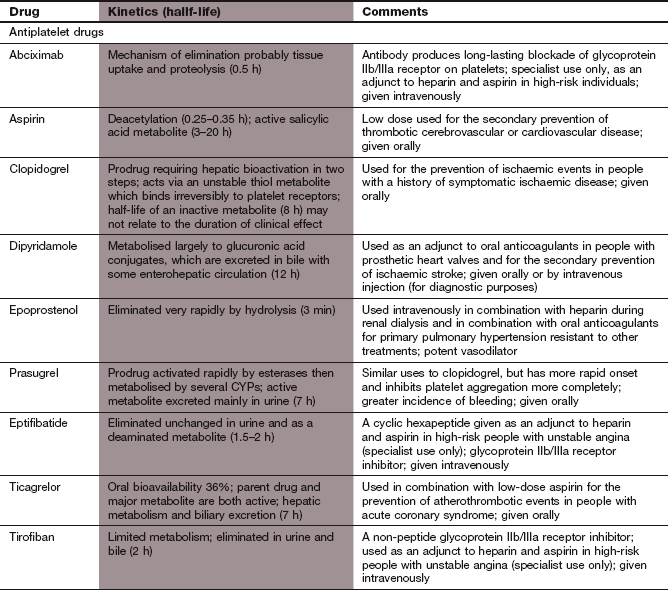
Antiplatelet drugs
Mechanism of action on platelets: The highly potent platelet-aggregating agent thromboxane A2 is formed in platelets from AA by the enzyme COX-1. After release from the platelet, thromboxane A2 acts via TP receptors on the surface of the platelet to generate the intracellular second messengers inositol triphosphate (IP3) and diacylglycerol (DAG). These lead to Ca2+ release in the cell and expression and activation of GPIIb/IIIa receptors.
Inhibition of COX-1 by aspirin reduces platelet thromboxane A2 synthesis and inhibits platelet aggregation, but does not eliminate it completely because other pathways for platelet activation still function (Figs 11.1 and 11.2). Aspirin (acetylsalicylic acid) irreversibly inhibits COX-1 by acetylation (Ch. 29) and since platelets lack a nucleus and cannot synthesise new enzyme, their ability to aggregate will be reduced throughout the lifespan of the platelet. The antiplatelet action of aspirin occurs at very low doses that have little analgesic or anti-inflammatory actions. At higher doses, aspirin also inhibits the production of prostacyclin by vascular endothelium which may offset some of the beneficial effects on platelets. Details of the pharmacology of aspirin can be found in Chapter 29.
Phosphodiesterase inhibitors
Mechanism of action: Dipyridamole has multiple mechanisms of action; the most important is probably inhibition of the reuptake of adenosine by cells. The increased plasma concentration of adenosine promotes vasodilation and inhibits platelet aggregation by stimulation of intracellular adenylyl cyclase and production of the intracellular cyclic nucleotides cGMP and cAMP. Dipyridamole also inhibits phosphodiesterase types 3 and 5, which degrade cyclic nucleotides. High intracellular cyclic nucleotide concentrations inhibit activation of cell surface GPIIb/IIIa receptors, leading to reduced platelet activation (Fig. 11.2). Dipyridamole has a number of other actions that are of uncertain significance, including antioxidant properties.
ADP receptor antagonists
Mechanism of action: ADP activates platelets via two purinergic surface receptors, P2Y1 and P2Y12. P2Y1 receptors increase intracellular Ca2+ and initiate platelet shape change. Activation of P2Y12 receptors inhibits adenylyl cyclase, and reduces generation of the intracellular cyclic nucleotides that inhibit activation of GPIIb/IIIa receptors.
ADP receptor antagonists inhibit platelet aggregation by binding selectively to P2Y12 receptors (Fig. 11.2). Inhibition of P2Y12 receptors also reduces the production of thromboxane A2 by the platelet. Clopidogrel and prasugrel are irreversible receptor inhibitors, while ticagrelor binds to a different site on the receptor and produces reversible inhibition. There is considerable inter-individual variability in the degree of platelet inhibition by clopidogrel, and it has a slow onset of action (about 5 days for full effect) without a loading dose. Both prasugrel and ticagrelor are more predictable inhibitors of platelet activation than clopidogrel and have a more rapid onset of action.
Pharmacokinetics: Clopidogrel is a prodrug. It is well absorbed from the gut, and is activated by metabolism in the liver to a derivative that has a half-life of 7 h. Prasugrel is also a prodrug that is well absorbed from the gut and metabolised rapidly in the liver to an active metabolite which has a long half-life of 8 days. Ticagrelor is active as the parent drug, and also has an active metabolite. The offset of action of ticagrelor over 3 days is much slower than would be predicted from the short half-life.
Glycoprotein IIb/IIIa receptor antagonists
Mechanism of action: Abciximab is a murine/human chimaeric monoclonal antibody to the GPIIb/IIIa receptors with the Fc fragment removed to prevent clearance of antibody-bound platelets from the circulation. Abciximab binds irreversibly to the GPIIb/IIIa receptors and blocks the binding of fibrinogen (Fig. 11.2). Abciximab can reduce platelet aggregation by more than 90%.
Eptifibatide is a synthetic peptide that binds reversibly to and blocks the GPIIb/IIIa receptor.
Pharmacokinetics: All GPIIb/IIIa antagonists are given intravenously, usually as an initial bolus to achieve rapid inhibition of platelets followed by continuous infusion. The duration of receptor blockade with abciximab is longer than predicted from its very short half-life of 30 min due to slow dissociation from the receptor over several hours. After stopping abciximab, platelet aggregation largely recovers by 48 h as new platelets are synthesised.
Eptifibatide has a short half-life of about 2.5 h, and is eliminated by the kidney. Platelet aggregation recovers more rapidly after treatment than with abciximab, due to rapid dissociation of the drug from the receptor after a few seconds.
Epoprostenol
Mechanism of action: Epoprostenol (PGI2) increases platelet cAMP, which at low concentrations inhibits platelet aggregation and at higher concentrations reduces platelet adhesion. Epoprostenol is also a peripheral arterial vasodilator.
Clinical uses of antiplatelet drugs
Aspirin has often been used as the sole antiplatelet drug in a variety of clinical settings. However, there are some situations where clopidogrel is more effective, or where combinations of aspirin and an ADP receptor antagonist give better outcomes than aspirin alone. The suppression of thromboxane A2 production by ADP receptor antagonists has called into question whether the concurrent use of aspirin is necessary to achieve optimal clinical outcomes, but this issue is unresolved. A combination of antiplatelet drugs inevitably carries a greater risk of bleeding than a single agent. Main uses of antiplatelet drugs are listed here.
Secondary prevention of embolic stroke and transient cerebral ischaemic attacks (aspirin, clopidogrel, dipyridamole). Clopidogrel alone is more effective than aspirin alone for the secondary prevention of stroke, while the combination has no further advantage despite a higher risk of bleeding. Dipyridamole combined with aspirin is better than aspirin alone for prevention of recurrent transient ischaemic attacks, and is equally effective as clopidogrel after stroke (Ch. 9).
Secondary prevention after acute coronary syndrome (aspirin, clopidogrel, prasugrel, ticagrelor, eptifibitide, tirofiban). The combination of aspirin and clopidogrel is better than aspirin alone for reducing further vascular events after myocardial infarction. Prasugrel and ticagrelor may have advantages over clopidogrel in some situations (Ch. 5).
Reduction of ischaemic complications produced by stent thrombosis following percutaneous coronary intervention (PCI) with stent insertion; these complications include non-fatal myocardial infarction, death and the need for emergency surgical revascularisation. Aspirin with clopidogrel or prasugrel are given for up to a year, often with abciximab or eptifibatide added at the time of the procedure when PCI is carried out following myocardial infarction.
Secondary prevention of myocardial infarction in stable angina or peripheral vascular disease (aspirin, clopidogrel): either aspirin or clopidogrel alone is effective, and there is no evidence to support combination therapy (Chs 5 and 10).
Primary prevention of ischaemic heart disease (aspirin). This is a controversial area, with the potential for serious haemorrhage offsetting much of the potential benefit. Use of aspirin should be confined to people at very high risk of developing cardiovascular disease.
Anticoagulation in extracorporeal circulations; for example, cardiopulmonary bypass and renal haemodialysis (epoprostenol).
Symptom relief in Raynaud's phenomenon (epoprostenol) (Ch. 10).
Dipyridamole is used as a pharmacological stress for the coronary circulation to detect myocardial ischaemia in people who are unable to exercise. This is related to its ability to block the cellular uptake of adenosine. In the heart, adenosine acts on specific receptors in the small resistance coronary arteries to produce vasodilation. Dipyridamole can divert blood away from myocardium supplied by stenosed coronary arteries by preferentially dilating healthy vascular beds (vascular steal).
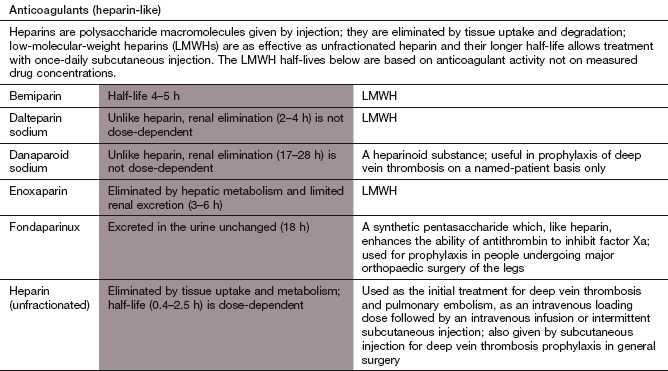
Anticoagulant drugs
Anticoagulation can be achieved with either injectable or oral drug therapy. Increasingly, oral anticoagulant therapy with newer agents is likely to supersede the long-established use of heparin followed by warfarin to initiate anticoagulation.
Injectable anticoagulants
Heparins are a family of highly sulphated acidic mucopolysaccharides (glycosaminoglycans) that are found in mast cells, basophils and endothelium. Heparins have a variable molecular weight of between 3000 and 30 000 Da according to the numbers of polysaccharide subunits.
Mechanism of action and effects: Heparin is available as an unfractionated preparation, or as low-molecular-weight heparins (LMWHs), which consist of the heparin subfractions that have molecular weights of less than 7000 Da.
Unfractionated heparin forms a complex with and alters the conformation of antithrombin III; this complex can then inactivate thrombin and factors IXa, Xa, XIa and XIIa (Fig. 11.3). LMWH interacts with antithrombin III in a different manner to unfractionated heparin, and the LMWH–antithrombin complexes have a more selective anticoagulant action, mainly inhibiting factor Xa (Fig. 11.3).
Additional actions of the heparins are as follows.
Promotion of tissue factor pathway inhibitor (TFPI) release from the vascular wall contributes to the antithrombotic effects of heparin. TFPI inhibits formation of factor Xa.
Inhibition of platelet aggregation through binding to platelet factor 4 (mainly unfractionated heparin).
Activation of lipoprotein lipase, which in addition to promoting lipolysis also reduces platelet adhesiveness.
Pharmacokinetics: Heparins are inactive orally and are given intravenously or by subcutaneous injection. They have a rapid onset of action. Heparins do not cross the placenta or enter breast milk. The two principal forms of heparin have different pharmacokinetic properties.
Unfractionated heparin: This is extracted from porcine intestinal mucosa or bovine lung, and consists of a mean of 45 polysaccharide units. It has dose-dependent pharmacokinetics: the half-life is very short (about 30 min) at low doses, increasing some five-fold at higher doses. Variable binding to plasma proteins contributes to inter-individual variation in the dose required to achieve target levels of anticoagulation. Most heparin is metabolised in endothelial cells after binding to cell surface receptors. Unfractionated heparin is usually given by continuous intravenous infusion for full anticoagulation. Low-dose subcutaneous injections are used for prophylaxis against venous thrombosis, although bioavailability by this route is only about 30%.
Low-molecular-weight heparins: LMWHs have a mean of 15 polysaccharide units. They are almost completely absorbed after subcutaneous administration and only need to be given once or twice daily by subcutaneous injection for full anticoagulation. LMWHs have a low affinity for plasma protein binding sites and for endothelial cell heparin receptors. They have two routes of elimination: a rapid, saturable liver uptake and slower renal excretion. The different LMWHs have half-lives in the range 2–6 h. When the dose of a LMWH is based on body weight they produce a more predictable anticoagulant effect compared with unfractionated heparin.
Control of heparin therapy: The therapeutic index for heparin is low. The degree of anticoagulation with unfractionated heparin is usually monitored with the activated partial thromboplastin time (APTT; a global test of the intrinsic coagulation pathway), which should be prolonged by 1.5–2.0 times the control value for full anticoagulation. Monitoring is not required when unfractionated heparin is used subcutaneously for prophylaxis. The anticoagulant effect of LMWHs can be monitored by the degree of factor Xa inhibition, but this is not carried out routinely since their effect is much more predictable than that of unfractionated heparin.
Haemorrhage is the most common problem. The risk is greater in the elderly, especially if there is a history of heavy alcohol intake. The effect of unfractionated heparin can be rapidly reversed by intravenous injection of protamine sulphate, a basic peptide which binds strongly to the acidic heparin components. Protamine binds poorly to LMWHs and only partially reverses their action.
Osteoporosis is a rare complication which can occur when heparin is given for several weeks; heparin binds to osteoblasts, and inhibits their activity. The risk is less with LMWH.
Heparin-induced thrombocytopenia (HIT) usually occurs 5–15 days after starting intravenous heparin in about 2% of people, and arises from the development of antibodies to the heparin–platelet factor 4 complex. This causes platelet activation, aggregation and thrombosis. LMWHs are much less likely to cause HIT, since they have much less affinity for platelet factor 4 and their lower binding to endothelium also reduces interference with platelet–vessel wall interaction. Danaparoid (see the Compendium at the end of this chapter) is often used if continued parenteral anticoagulation is necessary.
Hyperkalaemia by inhibition of aldosterone secretion. This is most likely to occur after at least 7 days of treatment.
Fondaparinux
Mechanism of action: Fondaparinux is a synthetic pentasaccharide almost identical to the natural pentasaccharide sequence of heparin that binds to antithrombin. Like LMWH, it enhances the innate ability of antithrombin to inhibit factor Xa.
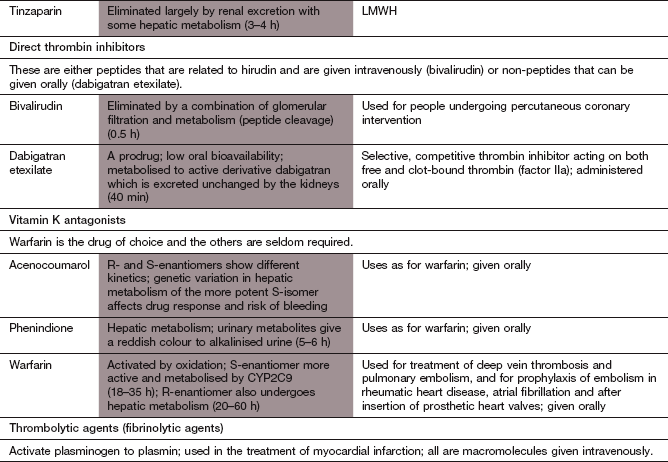
Oral anticoagulants
Mechanism of action: These drugs inhibit hepatic vitamin K epoxide reductase, which is the enzyme that converts vitamin K to its active (hydroquinone) form. As a result, the hepatic synthesis of the vitamin K-dependent clotting factors II (prothrombin), VII, IX and X is impaired (Fig. 11.3). There is a delay in the onset of the anticoagulant effect, due to the presence of previously synthesised clotting factors, which must be cleared from the circulation.
Pharmacokinetics: Warfarin is the most widely used vitamin K antagonist. It is almost completely absorbed from the gut and is highly bound to plasma albumin. It is eliminated by cytochrome P450-mediated hepatic metabolism (CYP2C9) and has a very long half-life of 1–2 days. Functional CYP2C9 polymorphisms contribute to considerable inter-individual variability in warfarin sensitivity. The plasma concentration of warfarin does not correlate directly with the clinical effect of the drug, which is determined by the balance between the rates of synthesis and degradation of clotting factors. The maximum effect of an individual dose of warfarin is reflected in the blood coagulation time some 24–36 h later. On stopping treatment, the duration of anticoagulant action is determined largely by the time required to synthesise new clotting factors.
Control of oral anticoagulant therapy: Factor VII is the clotting factor that is most sensitive to vitamin K deficiency, since it has the shortest half-life of the vitamin K-sensitive clotting factors. Therefore, a test of the extrinsic coagulation pathway – the prothrombin time – is used as a measure of effectiveness. The degree of prolongation of the prothrombin time is standardised by comparison with control plasma from a single source, and referred to as the international normalised ratio (INR). Therapeutic INR ranges differ according to the condition being treated:
INR 2–2.5 for prophylaxis of deep vein thrombosis (thromboprophylaxis),
INR 2–3 for thromboprophylaxis in hip surgery and fractured femur operations, for treatment of deep vein thrombosis and pulmonary embolism, and for prevention of thromboembolism in atrial fibrillation,
INR 3–4.5 for prevention of recurrent deep vein thrombosis and for preventing thrombosis on mechanical prosthetic heart valves.
Unwanted effects: Warfarin is an important example of a drug that has a narrow therapeutic index.
Haemorrhage. The most effective antidote to warfarin is phytomenadione (vitamin K1). For major bleeding, this is given intravenously and controls bleeding within 6 h. An immediate coagulant effect is achieved by also giving an intravenous injection of prothrombin complex concentrate (vitamin K-dependent clotting factors) or an infusion of fresh frozen plasma. After giving a large dose of phytomenadione, it can be difficult to restore therapeutic anticoagulation with warfarin for up to 3 weeks. If the INR is >8.0 but there is no bleeding or only minor bleeding, then a smaller dose of phytomenadione can be given intravenously or orally with less disturbance to subsequent anticoagulation.
Alopecia, skin necrosis and hypersensitivity reactions occur rarely.
Warfarin crosses the placenta and can have undesirable effects on the fetus. It is teratogenic and should be avoided in the first trimester of pregnancy, except when essential; furthermore, it should not be used in the last trimester, as it increases the risk of intracranial haemorrhage in the baby during delivery.
Drug interactions are particularly important. The anticoagulant effect of warfarin can be increased by broad-spectrum antibacterial agents that suppress the production of vitamin K by gut bacteria. Drugs such as amiodarone (Ch. 8) and the histamine H2 receptor antagonist cimetidine (Ch. 33), which inhibit CYP2C9-mediated metabolism of warfarin, enhance its effects. Drugs that induce CYP2C9 – for example, phenytoin (Ch. 23) and alcohol (Ch. 54) – reduce the effect of warfarin by increasing its elimination.
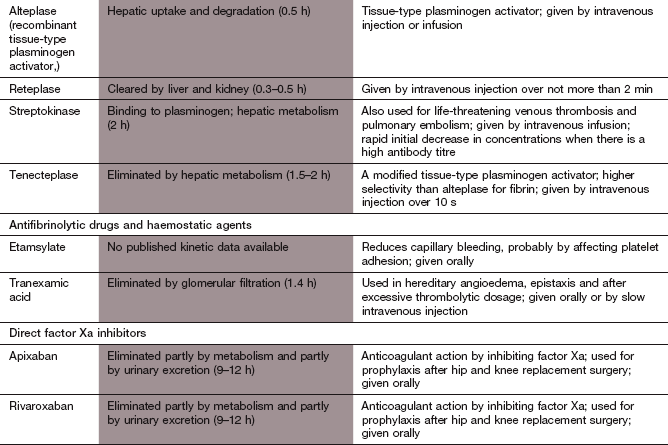
Direct factor Xa inhibitors
Mechanism of action: Apixaban and rivaroxaban are orally active factor Xa inhibitors that bind reversibly to the active site of factor Xa. They inhibit both free factor Xa and that bound to the prothrombinase complex, and unlike warfarin produce a rapid onset of predictable anticoagulation.
Pharmacokinetics: Apixaban and rivaroxaban are well absorbed from the gut. They are partially metabolised in the liver and partially excreted by the kidneys. Their half-lives are around 10 h.
Nausea, and less often other gastrointestinal upset.
Haemorrhage. If bleeding occurs, the short half-life means that stopping treatment may be all that is required. There is no direct antidote, but serious bleeding can be reduced with intravenous prothrombin complex concentrates or activated factor X.
Drug interactions: rivaroxaban is a substrate for P-glycoprotein (P-gp), and its excretion is reduced by drugs that inhibit P-gp, such as ketoconazole.
Direct thrombin inhibitors
Mechanism of action: Dabigatran is a selective, direct competitive thrombin inhibitor that binds to and inhibits both free circulating and thrombus-bound thrombin (factor IIa). It produces a rapid onset of predictable anticoagulation.
Clinical uses of anticoagulants
Until recently, rapid anticoagulation was usually achieved with LMWH or unfractionated heparin, and warfarin was started simultaneously for long-term anticoagulation. The heparin is stopped when the INR reaches the desired therapeutic range. The newer oral anticoagulants have a rapid onset of action (so heparin is not needed) and compared to warfarin they have fewer drug interactions and do not need monitoring of their anticoagulant effect. In most situations where warfarin is used, they have similar or greater efficacy compared to warfarin with either similar or lower risk of bleeding. Therefore, increasing use of drugs such as rivaroxaban and dabigatran in place of warfarin is likely.
Venous thromboembolism
Acute pulmonary embolism can present with a wide variety of symptoms. Massive emboli produce shock or sustained hypotension, while smaller emboli can present with chest pain, dyspnoea or haemoptysis. Pulmonary embolism is a major cause of morbidity and death. Most serious pulmonary emboli arise from deep vein thrombosis in the lower limb, particularly if the thrombus extends to the larger veins above the calf. Massive pulmonary emboli causing haemodynamic instability are fatal in about 60% of cases if untreated. Mortality is much lower in stable patients. Many episodes of deep vein thrombosis occur in hospital, particularly in people over 40 years of age following major illness, trauma or surgery. Pulmonary embolism has been estimated to be responsible for 10% of all deaths in hospital. Chronic pulmonary embolic disease can lead to pulmonary arterial hypertension with progressive dyspnoea (Ch. 6).
Factors predisposing to venous thromboembolism in hospital (Table 11.1) include prolonged immobility and a variety of coexisting medical conditions such as cancer. Spontaneous thromboembolism can occur after long journeys, such as by road or air, and in various inherited or acquired disorders of the coagulation system. Use of the combined oral contraceptive pill by older women who smoke (see Ch. 45) is also a factor.
Table 11.1
Risk of thromboembolism in people admitted to hospital
| Risk | Procedure |
| Low | Minor surgery, no other risk factor Major surgery, age <40 years, no other risk factors Minor trauma or illness |
| Moderate | Major surgery, age ≥40 years or other risk factor Heart failure, recent myocardial infarction, malignancy, inflammatory bowel disease Major trauma or burns Minor surgery, trauma or illness in patient with previous deep vein thrombosis or pulmonary embolism |
| High | Fracture or major orthopaedic surgery of pelvis, hips or lower limb Major pelvic or abdominal surgery for cancer Major surgery, trauma or illness in patient with previous deep vein thrombosis or pulmonary embolism Lower limb paralysis Major lower limb amputation |
After an initial spontaneous deep vein thrombosis, the risk of recurrence is about 25% after 4 years, but is much lower after postoperative thrombosis. Following a deep vein thrombosis, chronic post-phlebitic syndrome can develop, with pain, swelling and ulceration of the affected leg.
Prevention of deep vein thrombosis
In hospitalised people, the most appropriate method to prevent deep vein thrombosis will depend on the degree of risk.
Mechanical methods: These are used for people in hospital who are at moderate risk of thromboembolism and include graduated elastic compression stockings and intermittent pneumatic compression devices to improve venous flow and limit stasis in venous valve pockets. They can also be used to supplement pharmacological prophylaxis in people at high risk.
Low-dose subcutaneous heparin: This is the treatment of choice for many people in hospital who are at high or moderate risk of thromboembolism. Heparin reduces both initiation and extension of fibrin-rich thrombi at doses that have little effect on other measurements of blood coagulation; therefore, laboratory monitoring is unnecessary. Low-dose unfractionated heparin reduces deep venous thrombosis and fatal pulmonary emboli by about two-thirds, with minimal risk of serious bleeding, although minor bleeding is increased. LMWHs or fondaparinux are more effective than unfractionated heparin for those at highest risk.
Oral anticoagulants: Low-dose dabigatran, apixaban and rivaroxaban are at least as effective as LMWHs for thromboprophylaxis in people undergoing hip and knee orthopaedic surgery. Bleeding rates with dabigatran, apixaban and LMWH are similar, but may be higher with rivaroxaban. Prophylaxis should be started before surgery. Warfarin may be more effective than heparin for prophylaxis in people at highest risk of thromboembolism, but is not widely used. Although a meta-analysis of several studies suggests that low-dose aspirin reduces deep venous thrombosis, it is less effective than heparin.
Treatment of established venous thromboembolism
The goals of treatment for deep vein thrombosis are to prevent pulmonary emboli and to restore patency of the occluded vessel, with preservation of the function of venous valves. In about 50% of people with deep venous thrombosis, the vessel will recanalise within 3 months if appropriately treated. Use of compression stockings in the first few weeks after a deep venous thrombosis of the leg reduces the incidence of post-phlebitic syndrome.
Therapeutic anticoagulation: This is the treatment of choice for deep vein thrombosis and for most pulmonary emboli since anticoagulation substantially reduces mortality. Heparin is still the most widely used initial treatment for its rapid onset of effect. LMWH or fondaparinux given subcutaneously are preferred to unfractionated heparin, except in people with significant renal impairment. Heparin is usually given for 3–5 days, with concurrent initiation of treatment with warfarin. Heparin can be stopped once warfarin has produced adequate anticoagulation (i.e. the INR is within the therapeutic range; see above). When deep vein thrombosis occurs in someone with cancer there is a high risk of both bleeding and recurrence during treatment with warfarin. In this situation, prolonged treatment with LMWH (6 months, or lifelong if remission is not achieved) is usually advocated. The optimal duration of anticoagulant therapy is not well defined, but suggested periods are shown in Table 11.2.
Table 11.2
Suggested duration of anticoagulant therapy for venous thromboembolism
| Risk of recurrence | Clinical setting | Duration |
| Low | Temporary risk factors for thromboembolism | 3 months |
| Intermediate | Continuing medical risk factors for thromboembolism | 3–6 months |
| High | Recurrent thromboembolism; inherited thrombophilic tendency | Indefinite |
Oral anticoagulants such as rivaroxaban can be used instead of sequential heparin and warfarin, and have equal efficacy. At present there is only evidence for the efficacy of dabigatran after initial treatment with LMWH.
Surgical venous thrombectomy: This may be required for massive iliofemoral thrombosis if it threatens the viability of the limb. Pulmonary embolectomy is occasionally carried out for large pulmonary emboli.
Thrombolysis and percutaneous thrombectomy: Pharmacological thrombolysis (see below) has no advantage over warfarin in uncomplicated deep venous thrombosis. However, it is used to disintegrate massive pulmonary emboli, and reduces mortality in haemodynamically unstable patients. Percutaneous mechanical thrombectomy (fragmentation and removal of the thrombus) and surgical embolectomy are occasionally used if there are contraindications to thrombolysis.
Arterial thromboembolism
Warfarin is used long term for the prevention of thrombosis on prosthetic heart valves. Atrial fibrillation and mural thrombus in the left ventricle following a myocardial infarction predispose to arterial embolism and are indications for anticoagulation with warfarin. Dabigatran, apixaban and rivaroxaban are at least as effective as warfarin for prevention of thromboembolism in atrial fibrillation and may have a lower risk of haemorrhage (Ch. 8). When combined with antiplatelet therapy, apixaban reduces the composite endpoint of mortality, reinfarction and ischaemic stroke after an acute coronary syndrome (Ch. 5).
The fibrinolytic system
Fibrinolysis is the physiological mechanism for dissolving the fibrin meshwork in a thrombus. The process is initiated by activation of plasminogen, a circulating α2-globulin (Fig. 11.4). Tissue plasminogen activator (t-PA) is released from damaged vessels and cleaves plasminogen to the active enzyme plasmin. In the circulation, plasminogen activator inhibitors 1 and 2 rapidly inactivate t-PA. However, t-PA binds to fibrin locally at the site of release, and converts fibrin-bound plasminogen to plasmin. Plasmin splits both fibrinogen and fibrin into degradation products, and if this occurs at the site of a thrombus it produces lysis of the clot matrix. Fibrinolytic therapy (also called thrombolytic therapy) is achieved by using a plasminogen activator in such large quantities that the inhibitory controls are overwhelmed.
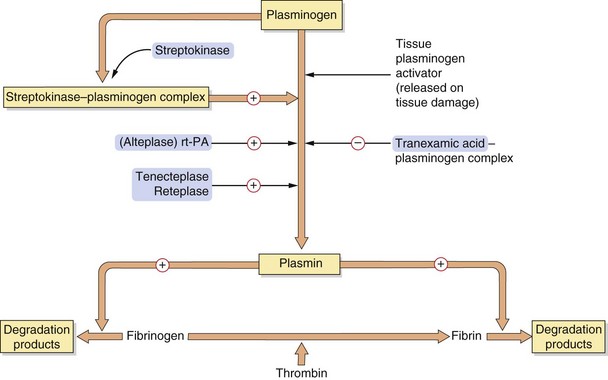
Fig. 11.4 The fibrinolytic system.
The fibrinolytic system is linked intimately with the coagulation cascade and platelet function. When a clot is formed via the prothrombotic system, activation of plasminogen to the fibrinolytically active plasmin is initiated by several tissue plasminogen activators, thus lysing the clot. The drugs promoting this act as plasminogen activators (alteplase (recombinant tissue-type plasminogen activator, rt-PA) and derivatives) or bind to plasminogen (streptokinase), promoting plasmin activity. The antifibrinolytic drug tranexamic acid inhibits plasminogen activation.
Fibrinolytic (thrombolytic) agents
Mechanism of action: Fibrinolytic drugs enhance fibrinolysis by substituting for the naturally occurring t-PA. They bind to and activate plasminogen, which degrades fibrin thrombi. Alteplase is a genetically engineered copy of the naturally occurring t-PA that binds directly to fibrinogen and fibrin. It has a wide range of clinical uses. Tenecteplase is a genetically engineered modified form of t-PA with increased fibrin specificity, less sensitivity to plasminogen activator inhibitors and a longer duration of action than alteplase. Tenecteplase is only licensed for treatment of myocardial infarction
Streptokinase is obtained from haemolytic streptococci, and is inactive until it forms a complex with circulating plasminogen; the resultant streptokinase–plasminogen activator complex substitutes for t-PA in the fibrinolytic cascade, causing plasminogen activation. Streptokinase is now used less frequently than other fibrinolytic drugs.
The effectiveness of any fibrinolytic agent is greatest with fresh thrombus and if a large surface area of thrombus is exposed to the drug.
Pharmacokinetics: All fibrinolytic agents are given intravenously or intra-arterially. Alteplase and related compounds are metabolised in the liver. The streptokinase–plasminogen activator complex is degraded enzymatically in the circulation. Some streptokinase is cleared from the plasma before it forms an active complex, by combining with circulating neutralising antibody formed during previous exposure to streptokinase. After the use of streptokinase, or following a streptococcal infection, neutralising antibodies can persist in high titre for several years and substantially reduce the effectiveness of subsequent therapy with streptokinase. For this reason, repeat use of streptokinase is not recommended.
Alteplase and its derivatives have a more rapid onset of action than streptokinase and consequently the reperfusion of occluded vessels is faster. Infusions of alteplase are given over 1–3 h, depending on the condition being treated. Tenecteplase is given as a single bolus. Because of a short duration of action, when alteplase or its derivatives have been used to lyse coronary artery thrombus, subsequent anticoagulation with heparin for 48 h is necessary to reduce the risk of reocclusion. Streptokinase is usually given as a short (1 h) infusion for the treatment of coronary artery occlusion, although longer infusions are usual for peripheral arterial occlusions or pulmonary embolism. The long duration of action means that heparin is not necessary after streptokinase has been given.
Haemorrhage is usually minor, but serious bleeding, for example intracerebral haemorrhage, occurs in about 1% of those treated. Bleeding can be stopped by antifibrinolytic drugs (see below) or by transfusion of fresh frozen plasma.
Hypotension: this is dose-related and more common with streptokinase. It may be caused by enzymatic release of the vasodilator bradykinin from its circulating precursor. If the infusion of the fibrinolytic is stopped for a brief period, the blood pressure usually recovers rapidly and treatment can be continued.
Allergic reactions: these are rare but can occur with streptokinase, as a consequence of its bacterial origin.
Clinical uses of fibrinolytic agents
Fibrinolytic agents are used to treat the following:
acute myocardial infarction (although this use is rapidly declining with the greater availability of primary coronary angioplasty) (Ch. 5),
ischaemic stroke (alteplase only) (Ch. 9),
pulmonary embolism or deep venous thrombosis, in a minority of cases (see above),
peripheral arterial thromboembolism (Ch. 10),
central venous catheters occluded by clot (alteplase): this is particularly useful to restore patency of ‘long lines’ inserted for intravenous nutrition or administration of cytotoxic drugs.
Antifibrinolytic and haemostatic agents
Mechanisms of action: Tranexamic acid competitively inhibits the activation of plasminogen, so fibrinolysis is inhibited. The theoretical risk of creating a thrombotic tendency does not appear to be a clinical problem.
Desmopressin
Desmopressin (Ch. 43) briefly increases the plasma concentrations of clotting factor VIII and von Willebrand factor, an adhesion protein in blood vessel walls. Factor VIII accelerates the process of fibrin formation and von Willebrand factor enhances platelet adhesion to subendothelial tissue.
Clinical uses of antifibrinolytic and haemostatic agents
The use of haemostatic agents is limited, but includes the following.
Tranexamic acid is used to prevent bleeding after surgery, for example prostatectomy or bladder surgery, or after dental extraction in individuals with haemophilia.
Desmopressin is used in mild congenital bleeding disorders such as haemophilia A or von Willebrand's disease; it is given to reduce spontaneous or traumatic bleeding, or as a prophylactic before surgery.
Tranexamic acid is used for the treatment of menorrhagia and epistaxis, or for bleeding following overdose of a fibrinolytic drug.
Tranexamic acid is used for treatment of hereditary angioedema.
True/false questions
1. Unfractionated heparin and low-molecular-weight heparin (LMWH) directly inhibit thrombin.
2. Heparin can be used to prevent clotting of blood collected in laboratory test tubes.
3. Once administered, the action of heparin cannot be reversed.
4. Fondaparinux is a pentapeptide activator of antithrombin III.
5. Warfarin readily crosses the placenta.
6. Warfarin prevents the activation of clotting factors which depend upon vitamin K for their synthesis.
7. Anticoagulant activity of warfarin is inhibited by broad-spectrum antibacterial agents.
8. Clopidogrel has its antithrombotic action by enhancing the action of ADP on platelets.
9. Abciximab is an antibody directed against the glycoprotein GPIIb/IIIa receptor on platelets.
10. Aspirin is a reversible inhibitor of cyclo-oxygenase type 1 (COX-1).
11. Aspirin inhibits platelet aggregation at doses below those needed for an anti-inflammatory effect.
12. Fibrinolytic infusions of recombinant tissue-type plasminogen activator (rt-PA; alteplase) for myocardial infarction are usually given over 1 h whereas streptokinase is given for 3–24 h.
13. Tenecteplase is a modified form of t-PA with a longer half-life than alteplase.
14. Apixaban is a direct inhibitor of factor Xa.
15. Dabigatran inhibits thrombin in the plasma and within the thrombus.
One-best-answer (OBA) question
Choose the one correct statement about anticoagulant drugs.
A INR would need to be regularly monitored in people treated with heparin.
B If an oral anticoagulant is required for rapid anticoagulant activity before surgery, heparin is the drug of choice.
C Dosage adjustment of warfarin would be required if a person was prescribed concomitant treatment with the H2 receptor antagonist cimetidine.
D In overdose, the effects of warfarin cannot be reversed.
E During treatment with a broad-spectrum antibacterial the anticoagulant effects of enoxaparin may be inhibited.
Case-based questions
A 51-year-old obese female was treated with oestrogen replacement therapy for 18 months because of perimenopausal symptoms. She was scheduled for a hip replacement.
1. Was anticoagulant therapy necessary for this woman?
2. Should thromboprophylaxis have been started before surgery?
3. Should heparin or warfarin have been chosen for prophylaxis and what routes of administration were appropriate?
The hip replacement was carried out successfully and the woman was discharged from hospital after 5 days, although heparin therapy was continued for a further 5 days.
4. Why was therapy continued for this extended period and what out-of-hospital therapeutic prophylaxis could be considered?
1. False. Heparins first form a complex with antithrombin III; the complex then inactivates thrombin and other clotting factors including factors IXa, Xa and XIa. Complexes of LMWH with antithrombin have a more selective action on factor Xa.
2. True. The complexing of heparin with antithrombin in plasma means it can anticoagulate blood in vitro.
3. False. The action of unfractionated heparin (but not LMWH) can be reversed by the strongly basic protein protamine, which rapidly binds to it, forming an inactive complex.
4. False. All heparins are mucopolysaccharides. Fondaparinux is similar in structure to the pentasaccharide sequence within heparin that binds to antithrombin
5. True. Warfarin can cause fetal abnormalities and, unless essential, should not be given in early or late pregnancy.
6. True. Warfarin is a vitamin K antagonist that impairs vitamin K-dependent hepatic synthesis of factors II (prothrombin), VII, IX and X.
7. False. Vitamin K is produced by gut bacteria. Alteration of gut flora by broad-spectrum antibacterials will reduce vitamin K formation and hence reduce vitamin K-dependent clotting factor synthesis, enhancing the activity of warfarin.
8. False. Clopidogrel prevents the platelet aggregatory action of ADP by blocking purinergic (P2Y12) receptors.
9. True. The increased expression of GPIIb/IIIa receptors on platelets is essential for aggregation as fibrinogen links adjacent platelets by binding to GPIIb/IIIa receptors, thereby initiating aggregation.
10. False. Aspirin (acetylsalicylic acid) irreversibly inhibits COX-1 by acetylating its active site.
11. True. Thromboxane A2 (TXA2) required for platelet aggregation is synthesised by COX-1, whereas prostaglandins are synthesised during inflammation predominantly by induced cyclo-oxygenase type 2 (COX-2). Aspirin is 160 times more active at inhibiting COX-1 than COX-2, so it has no anti-inflammatory effect at the low doses required to inhibit TXA2 synthesis.
12. False. Streptokinase is usually infused for 1 h and alteplase for 3 h. Streptokinase has a longer half-life (1 h, alteplase 0.5 h), permitting a shorter infusion time.
13. True. Alteplase is identical to the naturally occurring t-PA, while tenecteplase has been modified for greater fibrin specificity and a longer duration of action.
14. True. The ‘xabans’ (apixaban, rivaroxaban) are orally active direct inhibitors of factor Xa.
15. True. Unlike heparin, which only inhibits plasma thrombin (via anti-thrombin III), dabigatran directly inhibits both free thrombin and thrombus-bound thrombin.
16. False. Tranexamic acid is an antifibrinolytic agent that inhibits plasminogen activation, reducing fibrin degradation and the risk of bleeding.
OBA answer
A Incorrect. Regular INR monitoring is required in people taking warfarin but not heparin, when the activated partial thromboplastin time (APTT) is used. Monitoring is not required when LMWH is used subcutaneously.
B Incorrect. Heparin is inactive orally and must be given by intravenous or subcutaneous routes.
C Correct. Warfarin is metabolised by the liver cytochrome P450 CYP2C9 and cimetidine inhibits this isoenzyme. A reduction in warfarin dose may be required or the replacement of cimetidine with another H2 antihistamine or a proton pump inhibitor without an interaction with warfarin.
D Incorrect. The effects of warfarin can be reversed with vitamin K1.
E Incorrect. Broad-spectrum antibacterials may suppress the production of vitamin K by gut bacteria and increase the activity of warfarin, but would not affect the actions of heparin.
Case-based answers
1. Anticoagulant therapy is necessary. Postoperative venous thromboembolism occurs in 40–50% of people who undergo hip replacement, and fatal pulmonary embolism in 1–5%, if prophylactic anticoagulant therapy is not given. This woman is also at increased risk because of obesity.
2. This is controversial. Initiating prophylaxis postoperatively allows more effective haemostatic control during and immediately after surgery and does not reduce the effectiveness of treatment.
3. Heparins are active given intravenously or subcutaneously and their onset of action is rapid, whereas warfarin takes several days for full effectiveness but can be given orally. Heparin would therefore be chosen if started pre- or postoperatively.
4. The woman was obese, a risk factor for postoperative venous thrombosis. Daily self-administered subcutaneous prophylaxis with LMWH could be used. LMWH has a better bioavailability, a longer half-life and a lower risk of producing thrombocytopenia. Unlike unfractionated heparin, its effect is predictable.
Antiplatelet Trialist's Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction and stroke in high risk patients. BMJ. 2002;324:71–86.
Cryer, B. Reducing the risks of gastrointestinal bleeding with antiplatelet therapies. N Engl J Med. 2005;352:287–289.
Gladding, P, Webster, M, Ormiston, J, et al. Antiplatelet drug unresponsiveness. Am Heart J. 2008;155:591–599.
Gresele, P, Momi, S, Falcinelli, E. Antiplatelet therapy: phosphodiesterase inhibitors. Br J Clin Pharmacol. 2011;72:634–646.
Sambu, N, Curzen, N. Monitoring the effectiveness of antiplatelet therapy: opportunities and limitations. Br J Clin Pharmacol. 2011;72:683–696.
Schneider, DJ. Antiplatelet therapy: glycoprotein IIb-IIIa antagonists. Br J Clin Pharmacol. 2011;72:672–682.
Wijeyeratne, YD, Heppinstall, S. Antiplatelet therapy: ADP receptor antagonists. Br J Clin Pharmacol. 2011;72:647–657.
Agnelli, G, Becattini, C. Acute pulmonary embolism. N Engl J Med. 2010;363:266–274.
Blann, AD, Lip, YH. Venous thromboembolism. BMJ. 2006;332:215–219.
Cayley, WE. Preventing deep vein thrombosis in hospital inpatients. BMJ. 2007;335:147–151.
DeZee, KJ, Shimeall, WT, Douglas, et al. Treatment of excessive anticoagulation with phytonadione (vitamin K). A meta-analysis. Arch Intern Med. 2006;166:391–397.
Ginsberg, JS, Greer, I, Hirsch, J. Use of antithrombotic agents during pregnancy. Chest. 2001;119:s122–s131.
Kazmi, RS, Lwaleed, BA. New anticoagulants: how to deal with treatment failure and bleeding complications. Br J Clin Pharmacol. 2011;72:593–603.
Kyrle, PA, Eichinger, S. Deep vein thrombosis. Lancet. 2005;365:1163–1174.
Lee, CJ, Ansell, JE. Direct thrombin inhibitors. Br J Clin Pharmacol. 2011;72:581–592.
Toschi, V, Lettino, M. Inhibitors of propagation of coagulation: factors V and X. Br J Clin Pharmacol. 2011;72:563–580.