Asthma and chronic obstructive pulmonary disease
Asthma and chronic obstructive pulmonary disease (COPD) show several similarities in their clinical features but have some distinct pathophysiological – including immunological – differences. Both are inflammatory disorders of the bronchi. Clinically, they are characterised by airflow obstruction (a forced expiratory volume in 1 second [FEV1] below 80% of predicted and a ratio of FEV1 to forced vital capacity of less than 70%).
Asthma
The characteristic feature of asthma is reversible airflow obstruction. Asthma is often associated with an atopic disposition, and exposure to allergens or other environmental air pollutants may then result in expression of the condition. However, more severe and adult-onset asthma is often non-allergic and accounts for 10–30% of cases.
The most common symptoms of asthma are chest tightness, wheeze, breathlessness and cough, although cough may be the only symptom in younger people, especially at night. Airflow obstruction in asthma typically shows marked variability over time and greater than 15% improvement in response to any inhaled bronchodilator (see below). The symptoms are due to a combination of smooth muscle constriction in the airway and bronchial inflammation.
The pathogenesis of asthma is complex, and our knowledge is incomplete (Figs 12.1 and 12.2). Immune dysfunction leads to airway inflammation in asthma and may result from impaired regulation and imbalance between different T-lymphocyte subsets and also epithelial and airway dendritic cells. Chronic inflammation of the bronchial mucosa is prominent, with infiltration of activated T-helper lymphocytes, eosinophils, mast cells, basophils and sometimes neutrophils. These inflammatory cells release several powerful chemical mediators (Figs 12.1 and 12.2). The result is hyperplasia of bronchial smooth muscle with abnormal reactivity, oedema of the bronchial wall, deposition of subepithelial collagen and increased airway secretions. Dysregulation of numerous inflammatory mediators appears to be involved in asthma, including cysteinyl-leukotrienes, histamine, proteases and a variety of cytokines and chemokines but, unlike COPD, there is relatively little evidence of an increase in reactive oxygen species.
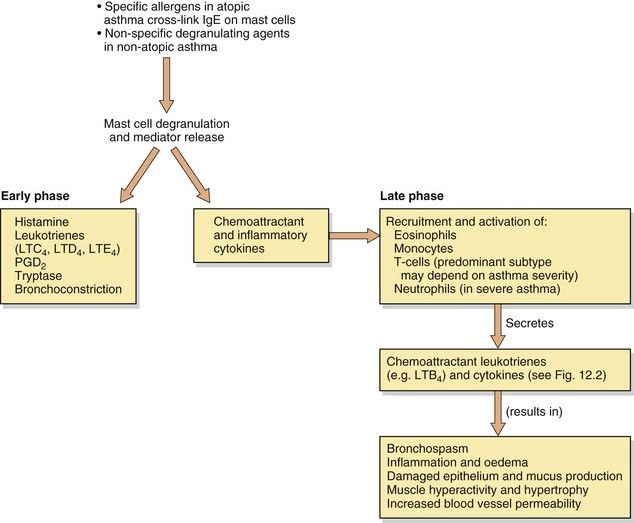
Fig. 12.1 Some aspects of the early and late-phase responses in asthma.
Crosslinking of the over-expressed IgE on mast cells of atopic individuals, and non-immunogenic stimuli in more severe non-atopic asthma, can degranulate mast cells, resulting in secretion of mediators that contribute to the pathogenesis of asthma. These mediators directly produce bronchoconstriction, initiate the acute inflammatory response, and attract and activate cells responsible for further inflammatory mediator production and persistent chronic inflammation. LT, leukotriene; PG, prostaglandin.
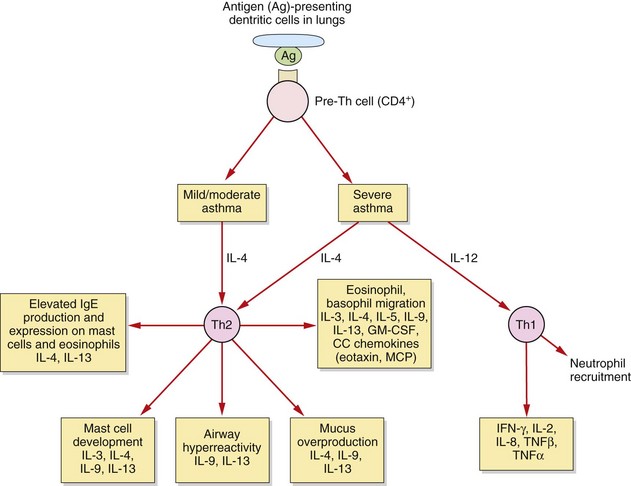
Fig. 12.2 T-cells and asthma.
In allergic asthma there are complex and still poorly understood imbalances in the immune system. These include alterations in the functioning of several T-cell subsets and additional dysregulation in epithelial cells, fibroblasts and airway dendritic cells. In mild to moderate allergic asthma, the T-helper cell type 2 (Th2) response is amplified, and Th2 cytokines contribute to many of the pathophysiological features of asthma. In severe asthma there is an additional pathological role for T-helper type 1 (Th1) cytokines and neutrophils. IL, interleukin; GM-CSF, granulocyte–macrophage colony-stimulating factor; IFN, interferon; MCP, monocyte chemoattractant proteins, TNF, tumour necrosis factor.
Figure 12.1 shows how exposure of atopic individuals to a relevant allergen (such as pollen or the faeces of house-dust mite) crosslinks IgE bound to mast cell membrane receptors and causes mast cell degranulation. Degranulation and the subsequent pathological processes can also occur in hypersensitive non-atopic asthmatics with normal levels of IgE, triggered by other factors such as upper respiratory tract infections, particularly with human rhinovirus. Degranulation of mast cells produces immediate bronchoconstriction (early phase) due to the release of a number of spasmogens, of which the most potent are cysteinyl-leukotrienes (Fig. 12.1). Chemotactic mediators are also released, promoting an influx of inflammatory cells which 4–6 h later results in a delayed bronchoconstrictor response (late-phase) and the commencement of a cascade of other pathological events in the airways. The persistent release of spasmogens and inflammatory mediators by these infiltrating cells can leave the bronchi hyperreactive to various irritants for several weeks. The inflammatory mediators produce mucosal oedema, which narrows the airways, and stimulate smooth muscle contraction leading to bronchoconstriction. Excessive production of mucus can cause further airways obstruction by plugging the bronchiolar lumen.
In mild to moderate asthma there is an increase in the number and activation of eosinophils (accompanied by some neutrophils and macrophages) in the airway and hyperresponsiveness of the airways to irritants and spasmogens. There is a persistent and excessive T-helper cell type 2 (Th2) immune response (Fig. 12.2). All airways are involved in the inflammatory process in mild to moderate asthma, but the degree of submucosal fibrosis and mucus secretion is modest, with no parenchymal destruction.
In severe asthma there is evidence of additional, greater infiltration of neutrophils, tissue destruction and airways remodelling, with progressive thickening and loss of elastic recoil, especially in the peripheral airways. In addition to the changes seen in mild to moderate asthma, in severe disease there is increased expression of T-helper cell type 1 (Th1)-derived cytokines.
Chronic obstructive pulmonary disease
About 95% of people with COPD are, or have been, cigarette smokers. There is wide variability in the rate of decline in pulmonary function in persistent smokers, with about 10–20% showing an accelerated decline that may reflect a genetic susceptibility. Less common causes of COPD are exposure to air pollution (including biomass fuels in the developing world) and inherited α1-antiprotease deficiency.
COPD is a symptom complex that is characterised by persistent airflow obstruction, with most people showing limited reversibility in response to a bronchodilator; however, about 10% of people with COPD do show considerable bronchodilator-induced reversibility of the airflow obstruction, and have a mixed inflammatory pattern in the airways, which probably represents an overlap between asthma and COPD (wheezy bronchitis). The airflow obstruction in COPD is usually slowly progressive and results from a combination of decreased bronchial luminal diameter (produced by wall thickening, intraluminal mucus and changes in the fluid lining the small airways) and dynamic airways collapse due to emphysema (see below). It is often accompanied by chronic bronchitis (production of mucoid sputum for all or part of the year).
The most frequent symptoms of COPD are gradually progressive breathlessness and cough. The cough is often productive and usually worse in the morning, but its severity is unrelated to the degree of airflow obstruction. Repeated respiratory infections are common, and are often associated with exacerbations of the airflow obstruction and symptomatic deterioration.
In COPD there is an inflammatory process that particularly affects the peripheral airways. The predominant infiltrating cells are neutrophils and macrophages, but ongoing damage to the lung even after the trigger is removed is probably due to T-lymphocyte-mediated inflammation (Fig. 12.3). There is increased oxidative stress due to reactive oxygen species derived from cigarette smoke and other pollutants and released from neutrophils and inflammatory macrophages. The inflammation produces a marked fibrotic reaction with parenchymal destruction and excessive bronchial mucus secretion. Corresponding histological changes include an increase in goblet cells in the bronchial mucosa and an increase in muscle mass in the bronchial wall, accompanied by interstitial fibrosis. Apoptosis of endothelial and alveolar cells reduces the ability of the lung to repair itself in response to sustained injury from the inhaled pollutants.
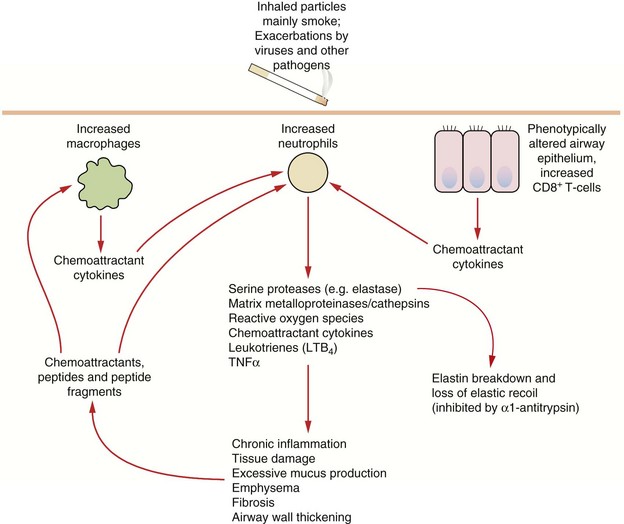
Fig. 12.3 Some pathophysiological factors in COPD.
A small percentage of people who smoke are particularly susceptible to the development of COPD; susceptibility may be determined by variability in inflammatory or protective genes. Chronic alterations in the recruitment, activation and control in function of neutrophils, macrophages and subsets of T-cells results in chronic parenchymal damage, loss of elastic recoil and episodes of infection. TNF, tumour necrosis factor.
Emphysema is a pathological description, and is defined as enlargement of airways distal to the terminal bronchioles owing to destructive changes that may involve the entire acinus (panacinar) or the central part of the acinus (centriacinar). Lung parenchymal destruction is largely mediated by tissue proteases and cathepsins that are released by neutrophils and macrophages. Generation of excessive amounts of reactive oxygen species inhibits the antiproteases that normally protect the lung against such attack. This explains the susceptibility of people with inherited α1-antiprotease deficiency to emphysema. Tissue destruction leads to a loss of lung recoil on expiration, which is a major factor in generating expiratory flow. Emphysema is probably the dominant factor in severe COPD.
Drugs for asthma and chronic obstructive pulmonary disease
For the treatment of airways disease, direct delivery of drug to the lung by inhalation allows the use of smaller doses and therefore reduces the incidence of unwanted systemic effects (see Table 12.1). It also allows rapid onset of action of ‘rescue’ medication. The drug is usually delivered to the airways in an aerosol. The size of the aerosol particle that is inhaled is an important factor that determines whether or not it will reach the airways and where in the airways it will be deposited. The optimal particle size for treatment is 2–5 µm. Particles larger than 10 µm impact on the upper airways and will be swallowed. Particles smaller than 1 µm will not be deposited in the lower respiratory tract, and will either reach the alveoli and are absorbed into the blood or are exhaled. Other factors that influence aerosol deposition include the pattern of inhalation, the properties of the carrier and the type and severity of the lung disease. There are several methods for delivery of inhaled drug.
Table 12.1
Comparison of aerosol and oral therapy for asthma
| Aerosol | Oral | |
| Ideal pharmacokinetics | Slow absorption from the lung surface and rapid systemic clearance | Good oral absorption and slow systemic clearance |
| Dose | Low dose delivered rapidly to target | High systemic dose necessary to achieve an appropriate concentration in the lung |
| Systemic drug concentration | Low | High |
| Incidence of unwanted effects | Low | High (but depends on drug) |
| Distribution in the lung | Reduced in severe disease | Unaffected by disease |
| Compliance | Good with bronchodilators, poor with anti-inflammatory drugs | Good |
| Ease of administration | Difficult for small children and infirm peoplea | Good |
| Effectiveness | Good in mild to moderate disease | Good even in severe disease |
aMay be improved by breath-actuated inhalers or spacing devices. Nebulisers can be used for severe exacerbations.
Pressurised metered-dose inhaler
This is the most common device for delivery of bronchodilator and anti-inflammatory drugs used in the treatment of asthma and COPD. The propellant in the device is a pressurised hydrofluoroalkane (HFA) (having replaced chloroflurocarbons, or CFCs) and activation delivers a measured dose of aerosol via an atomization nozzle. Manually activated inhalers are widely used since they are convenient and inexpensive, but they require coordination of device activation and inhalation. The delivery and uptake of the drug are suboptimal if inspiratory flow is low, if inspiration is not full and is not preceded by full expiration, or if inspiration is followed by a breath hold of less than 6 s. About one-third of users find coordination difficult and, even if it is optimal, up to 70–90% of the aerosol may be deposited in the oropharynx and swallowed. The inhaler should be shaken before use.
Pressurised metered-dose inhaler with a spacer
A spacer device (a plastic reservoir) can be attached to the pressurised metered-dose inhaler to act as a chamber from which the suspended aerosol particles can be inhaled. The use of a spacer removes the need to coordinate aerosol activation and inspiration. A spacer can be large volume which retains more of the aerosol, or small volume which is more convenient but in which the aerosol impacts to a greater extent on the wall of the spacer. The spacer can be designed as a holding chamber by incorporating a one-way valve that retains the aerosol in the chamber for longer. A spacer is essential for young children, and for very young children a holding chamber can be attached to a facemask. The inhaler is activated into the spacer, and the person breathes normally through the mouthpiece. Inhalation of the contents should be completed within 10 s. The spacer allows evaporation of propellant and may create more droplets of the correct size to deposit in the airways. It also reduces drug deposition in the oropharynx, due to reduced particle velocity. Electrostatic charge on the plastic wall can attract particles and reduce drug delivery, so non-electrostatic materials are preferred. The device should be washed in mild detergent and air-dried to minimise the electrostatic charge. Addition of a spacer makes a metered-dose inhaler system less portable and may reduce adherence with treatment.
Breath-actuated metered-dose inhaler
There are several types of breath-actuated metered-dose inhaler, delivering either an aerosol or dry powder. The aerosol type is a modified metered-dose inhaler that is activated by inspiration. Actuation requires air to be drawn through the mouthpiece at a flow rate of at least 30 L⋅min−1. People who have severe airflow obstruction cannot achieve this. Breath-actuated metered-dose inhaler devices cannot be used with a spacer.
Dry-powder inhaler
Dry-powder inhalers contain particles of drug of optimal size for deposition. Inspiration through the device generates turbulence, which disperses the particles in the inspired air. Some devices use a single dose capsule, while in others the source is a bulk powder with the device metering the dose. The delivered dose is dependent on the inspiratory effort, unless the device is power-assisted by a battery or vibrating piezoelectric crystals.
Multi-dose liquid inhaler
This novel delivery device uses a spring to force a metered dose of liquid through a narrow nozzle. It creates more fine particles and gives high drug delivery to the lungs.
Nebulisers
Nebulisers are devices that are used with a facemask or mouthpiece to deliver drug from a reservoir solution. There are two types.
 Jet nebulisers use compressed air or oxygen passing through a narrow orifice at 6–8 L⋅min−1 to suck drug solution from a reservoir into a feed tube. There are fine ligaments in this tube, and the impact of the solution on these ligaments generates droplets (Venturi principle). Baffles trap the larger droplets.
Jet nebulisers use compressed air or oxygen passing through a narrow orifice at 6–8 L⋅min−1 to suck drug solution from a reservoir into a feed tube. There are fine ligaments in this tube, and the impact of the solution on these ligaments generates droplets (Venturi principle). Baffles trap the larger droplets.
Ultrasonic nebulisers use a piezoelectric crystal vibrating at high frequency to create the aerosol, and do not require gas flow. The vibrations are transmitted through a buffer to the drug solution and form a fountain of liquid in the nebulisation chamber. Ultrasonic nebulisers produce a more uniform particle size than jet nebulisers, but are less widely used due to cost.
Up to 10 times the amount of drug is required in a nebuliser to produce the same degree of bronchodilation achieved by a metered-dose inhaler. Drug delivery is more efficient via a mouthpiece than via a mask from which drug can be deposited in the nasal passages.
Symptom-relieving drugs for airflow obstruction (bronchodilators; ‘relievers’)
Mechanism of action and effects: Beta2-adrenoceptors are widely distributed in the lung, and the receptor density is higher in bronchial smooth muscle than in other cell types such as epithelial and endothelial cells and mast cells. Stimulation of these receptors by an agonist stabilises the receptor in its active rather than inactive configuration. This results in increased generation of cAMP by adenylyl cyclase, and activation of protein kinase A (PKA), which phosphorylates proteins that are central to the regulation of smooth muscle tone. Major beneficial actions of a β2-adrenoceptor agonist are:
bronchodilation due to reduced Ca2+ release from intracellular stores and reduced Ca2+ entry into smooth muscle cells,
inhibition of mediator release from mast cells and monocytes,
However, in addition to their beneficial effects on the airway, use of a β2-adrenoceptor agonist in asthma can enhance Th2 inflammatory pathways and also downregulate β2-adrenoceptors. Therefore, regular use of a β2-adrenoceptor agonist without an inhaled corticosteroid is not advised. There is evidence of synergy between inhaled corticosteroids and inhaled β2-adrenoceptor agonists, with the latter enhancing the gene-transcription effects of corticosteroids and corticosteroids enhancing β2-adrenoceptor gene transcription.
Some β2-adrenoceptor agonists, such as salbutamol, terbutaline and salmeterol, have about 60% partial agonist activity at the receptor (low-efficacy agonists) compared with formoterol and indacaterol which have full agonist activity (high-efficacy agonists). The relevance of these differences to treatment outcomes and unwanted effects is unclear.
Pharmacokinetics: The selectivity of β2-adrenoceptor agonists for the β2-adrenoceptor subtype is dose-dependent. Inhalation of drug aids selectivity since it delivers small but effective doses to the airways and minimises systemic exposure and stimulation of β-adrenoceptors outside the lungs (Table 12.1). The dose–response relationship for bronchodilation is log-linear and a 10-fold increase in dose is required to double the effect.
Short-acting β2-adrenoceptor agonists, such as salbutamol, have a rapid onset of action, often within 5 min, and produce bronchodilation for up to about 6 h. Their duration of action is far longer than the natural adrenoceptor agonists such as adrenaline, because they are not substrates for the uptake transporter on the presynaptic neuron or for catechol-O-methyltransferase, the enzyme that metabolises catecholamines outside adrenergic neurons (Ch. 4).
Salmeterol and formoterol have a longer duration of action (up to 12 h) because they are more lipophilic than short-acting agents and bind to the lipid of the cell membrane. Salmeterol has a slower onset of action than short-acting agents, but the onset with formoterol is rapid. Indacaterol is an ultra-long acting (up to 24 h) lipophilic, rapid-onset β2-adrenoceptor agonist.
Salbutamol and terbutaline can also be given orally (as conventional or modified-release formulations), by subcutaneous or intramuscular injection or by intravenous infusion. Much larger doses are required to deliver an adequate amount of drug to the lungs by any of these routes compared to inhaled doses. This reduces the selectivity for β2-adrenoceptors, and systemic unwanted effects can be troublesome.
Fine skeletal muscle tremor from stimulation of β2-adrenoceptors.
Tachycardia and arrhythmias result from both β1- and β2-adrenoceptor stimulation in the heart when high doses of inhaled drug are used, or after oral or parenteral administration.
Hypokalaemia with high doses, due to promotion of cellular uptake of K+ by a cAMP-dependent action of β2-adrenoceptor agonists on the Na+/K+ pump. Nebilised salbutamol is sometimes used as a treatment for hyperkalaemia. Hypomagnesaemia and hyperglycaemia can also occur. These effects do not persist during long-term use.
Paradoxical bronchospasm has been reported with inhalation, usually when given for the first time or with a new canister.
Tolerance to the bronchodilator effects with prolonged use of β2-adrenoceptor agonists is modest, but desensitisation and downregulation of the β2-adrenoceptor does occur. The process of receptor desensitisation appears to be more rapid for mast cells than for bronchial smooth muscle, and the prevention of exercise-induced bronchoconstriction is more affected than the symptom relief that these drugs produce. Corticosteroids reduce desensitisation by increasing β2-adrenoceptor gene transcription and enhancing coupling of the receptor to adenylyl cyclase.
Regular use of high doses of short-acting or inhaled long-acting β2-adrenoceptor agonists has been linked with asthma deaths. One possibility is that they precipitate serious cardiac arrhythmias during severe asthma exacerbations. It is also possible that their use might allow people to tolerate initial exposure to larger doses of allergens or irritants, which then produce an enhanced late asthmatic response. The excess mortality, although of concern, is extremely low. Recent investigation has raised the possibility that β2-adrenoceptor polymorphism may modify the response to β2-adrenoceptor agonists in some individuals, but it is not known whether this explains the risk of adverse events.
Antimuscarinic agents
Mechanism of action and effects: Many cell types in the respiratory system, including both neuronal and non-neuronal cells, have nicotinic and muscarinic surface receptors. These mediate a multitude of actions in response to parasympathetic nervous system stimulation. There are two main types of muscarinic receptors in the airways, as follows.
M3 receptors mediate direct bronchoconstriction and glandular mucus secretion and also enhance mucociliary clearance from the bronchi. M3 receptor stimulation activates phospholipase C with subsequent formation of inositol triphosphate (IP3) and diacylglycerol (DAG), which are key events in the signalling pathway that increases intracellular Ca2+ (Ch. 1, Fig. 1.5).
M2 receptors are more numerous and inhibit ciliary activity as well as limiting β2-adrenoceptor-mediated bronchodilation by inhibition of adenylyl cyclase.
Therefore, blocking both M2 and M3 receptors could be beneficial in bronchoconstriction. However, M2 autoreceptors are also present on presynaptic parasympathetic nerves supplying the lungs. Stimulation of these autoreceptors inhibits acetylcholine release and attenuates vagally mediated bronchoconstriction. Blocking these M2 autoreceptors may blunt the beneficial effect of non-selective muscarinic antagonists.
Ipratropium is a non-selective muscarinic receptor antagonist, and binds to all muscarinic receptors in the lung including the presynaptic M2 autoreceptor. Ipratropium therefore has the potential to augment vagally mediated bronchoconstriction. The recommended dose is determined by unwanted effects and is well below the dose that produces maximal bronchodilation. By contrast, tiotropium is functionally selective for the M3 receptor. Although it has a high affinity for all muscarinic receptors, it dissociates rapidly from M2 receptors.
The main benefit of muscarinic antagonists is in COPD; they are of less value for bronchodilation in acute mild to moderate asthma, but ipratropium has a place when added to a β2-adrenoceptor agonist in severe exacerbations of asthma.
Pharmacokinetics: The antimuscarinic drugs used for bronchodilation are N-quaternary congeners of the tertiary-structured atropine, and are poorly absorbed orally and do not cross the blood–brain barrier. They are given exclusively by inhalation as a powder or aerosol or via a nebuliser. They have a slower onset of action (30–60 min) than salbutamol (5–10 min), probably due to slow absorption from the surface of the airways. The duration of action is related to the rate of removal locally from the airways, and not the half-life of elimination from the circulation.
Unwanted effects: Similarly to inhaled β2-adrenoceptor agonists, direct delivery of antimuscarinic drugs to the lung is the main reason for the relative lack of unwanted systemic effects.
Dry mouth is the most common unwanted effect.
Tiotropium can cause urinary retention in men with prostatism.
Exacerbation of angle-closure glaucoma (Ch. 50).
Methylxanthines
Mechanism of action and effects: Methylxanthines are a group of naturally occurring substances found in coffee, tea, chocolate and related foodstuffs. Naturally occurring theophylline (1,3-dimethylxanthine), and its ester derivative aminophylline, are the only compounds in clinical use. They are chemically similar to caffeine. Methylxanthines have vasodilatory, anti-inflammatory and immunomodulatory actions. The mechanisms of action of methylxanthines are multiple, controversial and of uncertain importance.
Inhibition of the enzyme phosphodiesterase (PDE), which degrades cyclic nucleotide second messengers, may partly explain the actions of methylxanthines. Theophylline preferentially inhibits the isoenzymes PDE3 (which degrades cAMP and cGMP) and PDE4 (which degrades cAMP). PDE3 is found in bronchial smooth muscle and PDE4 in several inflammatory cell types, including mast cells. The rise in intracellular cAMP in bronchial smooth muscle stimulates large-conductance voltage-gated Ca2+-activated K+channels (BKCa) in the cell membrane, leading to cell hyperpolarisation and muscle relaxation. However, theophylline only produces bronchodilation at relatively high plasma concentrations (10–20 mg⋅L−1) and drugs that are more effective PDE inhibitors (such as dipyridamole) do not bronchodilate. Prolonging the duration of action of cyclic nucleotides may potentiate the action of β2-adrenoceptor agonists and produce a synergistic dilator effect on bronchial smooth muscle. PDE inhibition also stimulates ciliary beat frequency in the airways and enhances water transport across the airway epithelium, which increase mucociliary clearance. In contrast, theophylline increases the force and rate of contraction of cardiac muscle through its effect on cAMP (Ch. 7), but also causes arterial vasodilation by inhibiting the breakdown of cGMP.
Increased diaphragmatic contractility and reduced fatigue have been reported at lower plasma theophylline concentrations than those required for bronchodilation. This may improve lung ventilation.
Adenosine receptor antagonism may be relevant to some of the clinical effects of methylxanthines (see also adenosine; Ch. 8). Adenosine releases histamine and leukotrienes from mast cells, which results in the constriction of hyperresponsive airways in individuals with asthma. Theophylline is a potent antagonist at adenosine A1 and A2 receptors (Ch. 1) and may reduce bronchoconstriction by this mechanism. Adenosine receptor antagonism is responsible for central nervous system (CNS) stimulation, which improves mental performance and alertness, and in the kidney reduces tubular Na+ reabsorption and leads to natriuresis and diuresis.
Activation of histone deacetylases (HDACs; see corticosteroids, below): acetylation of core histones, which form part of the structure of chromatin, activates gene transcription, while their deacetylation suppresses gene transcription, including transcription of pro-inflammatory genes. Theophylline at low concentrations activates HDAC in nuclear extracts, indicating an action independent of adenosine and other surface receptors, and also increases HDAC activity in bronchial biopsies of asthmatic patients. Anti-inflammatory effects of theophylline occur at drug plasma concentrations of 5–10 mg⋅L−1, similar to those that produce clinical benefit. The action of theophylline on HDAC may potentiate the anti-inflammatory effects of corticosteroids (see Ch. 44).
Pharmacokinetics: The extent of absorption of theophylline from the gut is unpredictable, with considerable inter-individual variation. This, and the short but highly variable plasma half-life, has resulted in the widespread use of modified-release formulations. Theophylline has a narrow therapeutic index, and since different formulations vary in their release characteristics they are not readily interchangeable. Theophylline is metabolised in the liver by cytochrome P450 (CYP1A2 and, to a lesser extent, by CYP3A4), giving the potential for drug interactions. Aminophylline is a more water-soluble ester prodrug, which is hydrolysed rapidly after absorption from the gut to theophylline and ethylenediamine. Aminophylline can also be given by intravenous infusion. Measurement of blood theophylline concentrations is valuable as a guide to effective dosing.
Unwanted effects: Most are dose-related and can arise within the accepted therapeutic plasma concentration range.
Gastrointestinal upset, including nausea, vomiting (from PDE4 inhibition in the vomiting centre) and diarrhoea.
CNS stimulation, including insomnia, irritability, headache (from PDE3 inhibition) and occasionally seizures at high plasma concentrations (from adenosine receptor antagonism).
Hypotension from peripheral vasodilation (from PDE3 inhibition in the smooth muscle cells of many blood vessels). In contrast, cerebral arteries are constricted by methylxanthines (adenosine is a vasodilator of cranial blood vessels and methylxanthines may act as adenosine receptor antagonists in this vascular bed).
Cardiac stimulation produces various arrhythmias.
Hypokalaemia can occur acutely, especially after intravenous injection, which also promotes cardiac arrhythmias.
Tolerance to the beneficial effects of methylxanthines can occur.
Drug interactions can be troublesome, due to the narrow therapeutic index of theophylline. Hepatic CYP1A2 enzyme inhibitors such as ciprofloxacin, erythromycin, clarithromycin, fluconazole and ketoconazole (Ch. 51, Table 2.7) can precipitate theophylline toxicity.
Anti-inflammatory drugs for airways obstruction (‘preventers’)
Mechanism of action and effects: Corticosteroids with powerful glucocorticoid activity (but without significant mineralocorticoid activity) are the most effective class of drug in the treatment of chronic asthma, but are relatively ineffective in COPD. They suppress inflammation and the immune response but are not bronchodilators and are therefore of no benefit in the initial stages of an acute attack of asthma.
Intracellular events involved in the anti-inflammatory action of corticosteroids are described in Chapters 38 and 44. Inhibition of transcription of genes coding for the cytokines involved in inflammation is particularly important in asthma. Higher glucocorticoid concentrations also activate anti-inflammatory genes and genes linked to glucocorticoid unwanted effects. Following a delay of 6–12 h, corticosteroids reduce airway responsiveness to several bronchoconstrictor mediators and, with chronic therapy, inhibit both the early and late reactions to allergen.
Anti-inflammatory effects of corticosteroids in asthma include:
reduced airway oedema and leucocyte recruitment by induction of tight junctions in vascular endothelial cells,
reduced inflammatory cell activation (including macrophages, T-lymphocytes, eosinophils and airway epithelial cells) with reduced inflammatory cytokine, chemokine, adhesion molecule and inflammatory enzyme expression (Ch. 38). In allergic disease, suppression of Th2 cells and their cytokines is particularly important,
reduced inflammatory cell recruitment to the airways (eosinophils, T-lymphocytes, mast cells and dendritic cells) through reduction in chemotactic mediators and adhesion molecules, and reduced survival (enhanced apoptosis) of airway inflammatory cells,
decreased local generation of inflammatory prostaglandins and leukotrienes by inhibition of phospholipase A2 which reduces mucosal oedema (see also Ch. 29),
beta2-adrenoceptor upregulation and better coupling to adenylyl cyclase, which restores responsiveness to β2-adrenoceptor agonists,
enhanced activity of the M2 autoreceptors on acetylcholine nerve endings inhibits acetylcholine release and relieves vagally mediated bronchoconstriction,
suppression of the excess epithelial cell shedding and goblet cell hyperplasia found in the bronchial epithelium in asthma.
Inhaled corticosteroids produce some improvement in asthmatic symptoms after 24 h and a maximum response after 1–2 weeks. Reduction in airway responsiveness to allergens and irritants occurs gradually over several months, but many of the chronic structural changes in the airways in asthma are not affected by corticosteroids.
Pharmacokinetics: Whenever possible, corticosteroids are given by inhalation of an aerosol or dry powder in order to minimise systemic unwanted effects, but they can be used intravenously or orally in severe asthma. Desirable properties of an inhaled corticosteroid include a low rate of absorption across mucosal surfaces (such as the lung, but also the gut for swallowed drug) and rapid inactivation once absorbed. Beclometasone dipropionate fulfils the former criterion, but it is only slowly inactivated once it reaches the systemic circulation. Inhaled budesonide (which is inactivated by extensive first-pass metabolism in the liver following oral absorption) or fluticasone (which is very poorly absorbed from the gut) may be preferred if high doses of inhaled drug are needed, or for the treatment of children, in whom the systemic effects can be more problematic.
Unwanted effects: The unwanted effects of oral and parenteral corticosteroids are described in Chapter 44. Inhaled corticosteroids only have systemic actions when given in high doses. The amount of swallowed drug can be minimised by using a large-volume spacer (see above); large aerosol particles, which would otherwise be deposited on the oropharyngeal mucosa, are trapped in the spacer and only the smaller particles are inhaled.
There are some specific problems with inhaled corticosteroids:
dysphonia (hoarseness), caused by deposition on vocal cords and myopathy of laryngeal muscles, occurs in up to one-third of those using inhaled corticosteroids. This may be less troublesome with breath-actuated delivery, since the method of inspiration leads to protection of the vocal cords by the false cords,
oral candidiasis can occur but can be prevented by using a spacer device or by gargling with water after use of the inhaler,
prolonged use of high doses of inhaled corticosteroid has been associated with systemic unwanted effects. These include adrenal suppression, osteoporosis and reduced growth velocity in children. In older people with COPD there is an increased risk of pneumonia.
Cromones
The cromones are used to prevent asthma attacks, but they are usually less effective than inhaled corticosteroids and only about one-third of people benefit from treatment. Cromones have no bronchodilator activity and are of no use in acute attacks of asthma. The major use of cromones is as prophylactic agents in the treatment of mild to moderate antigen-, pollutant- and exercise-induced asthma. They are also used as nasal inhalants to treat seasonal allergic rhinitis (Ch. 39) and in ophthalmic solutions to treat allergic conjunctivitis (Ch. 50).
Mechanisms of action and effects:
Mast cell stabilisation. Sodium cromoglicate was originally introduced as a mast cell stabiliser. It enhances phosphorylation of a protein that normally forms a substrate for intracellular protein kinase C, and interferes with the signal transduction for inflammatory mediator release. This action may protect against immediate bronchoconstriction induced by allergens, exercise or cold air.
Inhibition of sensory C-fibre neurons by antagonism of the effects of the tachykinins, substance P and neurokinin B, which are involved in generation of sensory stimuli. This is probably responsible for protection against bronchoconstriction produced by irritants such as sulphur dioxide.
Inhibition of accumulation of eosinophils in the lungs and reduced activation of eosinophils, neutrophils and macrophages in inflamed lung tissue. These actions may be important in preventing the ‘late-phase’ response to allergen and the development of bronchial hyperreactivity.
Inhibition of B-cell switching to IgE production probably also contributes to the long-term effects.
A single dose of either nedocromil sodium or sodium cromoglicate will prevent the early-phase bronchoconstrictor response to allergen, but treatment for 1–2 months may be necessary to block the late-phase reaction.
Pharmacokinetics: Both sodium cromoglicate and nedocromil sodium are highly ionised and poorly absorbed across biological membranes. They are therefore largely retained at the site of action on bronchial mucosa after inhalation as a powder or from a metered-dose aerosol inhaler. Swallowed drug is unabsorbed and voided in the faeces.
Leukotriene receptor antagonists
Mechanisms of action and effects: The leukotriene receptor antagonists are given orally and inhibit the bronchoconstriction induced by the cysteinyl-leukotrienes (LTC4, LTD4 and LTE4) (Ch. 29) by blocking the CysLT1 receptor on bronchial smooth muscle. Cysteinyl-leukotrienes are released from various cells, including activated mast cells and eosinophils, in response to several airway insults, and their synthesis is increased by many mediators, such as cytokines. Cysteinyl-leukotrienes can also contribute to airway oedema, enhanced secretion of mucus and airway eosinophilia (Fig. 12.1).
Leukotriene receptor antagonists reduce both the early and late bronchoconstrictor responses to inhaled allergen, and may be most useful in mild and moderate asthma, exercise-induced bronchoconstriction and asthma provoked by non-steroidal anti-inflammatory drugs (NSAIDs; Ch. 29). The effects are additive to those of inhaled corticosteroid.
Phosphodiesterase type 4 inhibitors
Mechanism of action and effects: Phosphodiesterase type 4 (PDE4) is the main isoenzyme present in cells involved in the inflammatory process in COPD. PDE4 degrades the intracellular second messenger cAMP, and inhibition of this enzyme with roflumilast has several anti-inflammatory actions:
decreased cytokine and chemokine release from neutrophils, eosinophils, macrophages and T-lymphocytes,
decreased expression of adhesion molecules on T-lymphocytes and other inflammatory cells. Along with the reduced chemokine release, this results in less accumulation of these cells in the airway,
decreased apoptosis of airway cells, which may assist in sputum clearance.
Roflumilast is highly selective for PDE4, and therefore has little action in tissues that express different PDE isoenzymes. It is effective in COPD patients with chronic bronchitis (prominent cough and sputum production) who have frequent exacerbations. In these individuals, roflumilast improves lung function and reduces exacerbation frequency.
Magnesium sulphate
Mechanism of action and effects: Intravenous magnesium sulphate can be given for the treatment of severe asthma in adults if life-threatening features are present. Magnesium bronchodilates by blocking Ca2+ channels in smooth muscle cell membranes, therefore reducing Ca2+ influx into the cell.
Antibodies to immunoglobulin E (IgE)
Mechanism of action: Omalizumab is a recombinant humanised IgG1k monoclonal antibody that binds selectively to IgE, to form complexes which are removed from the circulation. This leads to a reduction in IgE receptor expression and mediator release from mast cells, basophils and dendritic cells. Treatment with omalizumab gradually reduces airway inflammation in asthma, with a peak response after 12–16 weeks. Omalizumab is used for the treatment of persistent severe allergic asthma in adults and children over 12 years that cannot be controlled with high-dose inhaled corticosteroids with a long-acting β2-adrenoceptor agonist and other standard therapies for asthma.
Pharmacokinetics: Omalizumab is given subcutaneously every 2–4 weeks in a dose that is determined by the recipient's plasma IgE concentration and body weight. It forms complexes with IgE that are removed by the reticuloendothelial system and endothelial cells. Omalizumab has a very long half-life of about 26 days.
Management of asthma
Treatment of asthma has two aims:
The acute attack
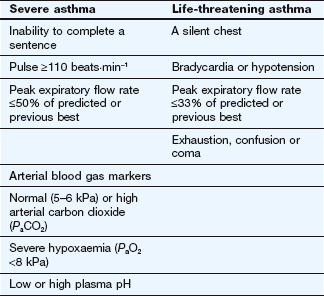
Mild infrequent attacks of asthma can often be controlled by occasional use of a short-acting inhaled β2-adrenoceptor agonist. Antimuscarinic agents are less effective unless asthma coexists with chronic obstructive airways disease. More severe attacks of asthma require intensive treatment with bronchodilators and systemic corticosteroids. The signs of severe and life-threatening asthma are shown in Table 12.2.
Treatment of acute severe asthma should include:
high-flow oxygen via a facemask to achieve an arterial oxygen saturation of above 90%,
inhaled short-acting β2-adrenoceptor agonist such as salbutamol, preferably via an oxygen-driven nebuliser, or a via a metered-dose inhaler with a large-volume spacer if a nebuliser is not available,
high-dose oral prednisolone or initial intravenous hydrocortisone followed by oral prednisolone.
If response to treatment is poor after 15–30 min, or if there are life-threatening features, additional treatment should be given:
If response is poor after a further 15–30 min, then consider:
After recovery from a severe asthma attack, prednisolone should be continued for at least 5 days or until there are no residual symptoms, especially at night, and the peak expiratory flow rate is at least 80% of the person's previous best. High doses of prednisolone can be stopped abruptly if used for 3 weeks or less, but should be reduced gradually if they have been used for a longer period (Ch. 44).
Prophylaxis of chronic asthma
An initial attempt should be made to identify and exclude precipitating factors; for example, allergens, occupational precipitants, NSAIDs (see below) and β-adrenoceptor antagonists (including eye drops) (Ch. 5). Long-term treatment is guided by a stepwise treatment plan.
Step 1. Mild intermittent asthma. Inhaled short-acting β2-adrenoceptor agonist, such as salbutamol, taken as required. For those who are intolerant to this treatment, inhaled ipratropium and oral theophylline are alternative options but there is a higher risk of unwanted effects with the latter. Step 2 treatment should be considered if more than two doses of short-acting β2-adrenoceptor agonist are required in a week, or if there has been an exacerbation of asthma in the previous 2 years.
Step 2. Regular inhaled preventer therapy. For adults, a regular standard-dose inhaled corticosteroid such as beclometasone is used in addition to step 1 therapy. In children under 5 years, a leukotriene receptor antagonist such as montelukast could be tried, but is generally less effective than inhaled corticosteroid.
Step 3. Inhaled corticosteroid plus long-acting inhaled β2-adrenoceptor agonist. If symptoms in an adult are not controlled by standard doses of inhaled corticosteroid, a long-acting β2-adrenoceptor agonist such as salmeterol is usually more effective than increasing the dose of corticosteroid. An inhaled short-acting β2-adrenoceptor agonist can still be used as required. If there is no response to the long-acting β2-adrenoceptor agonist, it should be stopped and the corticosteroid dose increased. The corticosteroid dose can also be increased if there was some response to a long-acting β2-adrenoceptor agonist but the symptoms are still not controlled. For persistent poor control, sequential add-on therapy with a leukotriene receptor antagonist, a modified-release theophylline formulation or a modified-release oral β2-adrenoceptor agonist should be tried.
For children under 5 years, a leukotriene receptor antagonist is added to regular inhaled corticosteroid with an inhaled short-acting β2-adrenoceptor agonist as required.
Step 4. High-dose inhaled corticosteroid plus regular bronchodilators. High-dose inhaled corticosteroid with an inhaled long-acting β2-adrenoceptor agonist plus a short-acting β2-adrenoceptor agonist as required, and a sequential trial of one of the following:
Step 5. Regular oral corticosteroid. Oral prednisolone is taken in addition to high-dose inhaled corticosteroid with an inhaled long-acting β2-adrenoceptor agonist plus a short-acting β2-adrenoceptor agonist as required.
Aspirin-intolerant asthma
About 5–20% of people with asthma experience acute exacerbations when they take aspirin or other NSAIDs in a laboratory setting (Ch. 29). Individuals with the clinical syndrome of aspirin-intolerant asthma (or aspirin-exacerbated respiratory disease, AERD) have an eosinophilic rhinosinusitis and nasal polyposis in addition to asthma. The condition may be initiated by priming of the respiratory mucosa by an immune reaction to a viral infection or other insult which chronically upregulates the cysteinyl-leukotriene biosynthetic pathway or CysLT1 receptor expression. Production of bronchoconstrictor leukotrienes nevertheless remains under partial inhibitory control of prostaglandin E2 (PGE2).
Aspirin is an irreversible cyclo-oxygenase type 1 (COX-1) and COX-2 inhibitor with greater effect on COX-1 inhibition. COX inhibition reduces PGE2 synthesis, which removes its inhibition of leukotriene synthesis, provoking acute bronchospasm (see Fig. 29.1). Other NSAIDs that inhibit COX-1 also induce bronchoconstriction, but the selective COX-2 inhibitors do not provoke asthma. In sensitive individuals, asthma symptoms begin within 3 h of ingesting aspirin, accompanied by profuse rhinorrhoea, conjunctival injection and, sometimes, flushing or urticaria. Airways inflammation can persist for many weeks after an aspirin challenge.
Leukotriene receptor antagonists produce symptom relief in some people with aspirin-induced asthma. Treatment of acute aspirin-intolerant asthma is the same as for any other episode. Sometimes, long-term use of an oral corticosteroid is the only way to control persistent symptoms; in such cases, desensitisation to aspirin should be attempted. Nasal polypectomy may be necessary to control rhinosinusitis.
Management of chronic obstructive pulmonary disease
There are two goals in the treatment of COPD: to minimise symptoms (including a reduction in acute exacerbations) and to preserve lung function.
Cessation of smoking. Stopping smoking (see Ch. 54) is the only effective way to alter the natural history of COPD. Smoking cessation slows the rate of decline in lung function to that naturally seen with ageing, although any loss of lung function due to smoking cannot be restored. Occupational exposure to inhaled pollutants should also be minimised.
Pneumococcal and influenza vaccination. These can reduce infective exacerbations in people with COPD.
Inhaled bronchodilators. The principles are similar to those for asthma, although the limited reversibility of the airway obstruction means that the benefit is less marked, except during an acute exacerbation of symptoms. Some improvement in symptoms and functional capacity can occur without changes in standard lung function tests and the main benefit is improved lung emptying during expiration, with reduced hyperinflation at rest. Inhaled bronchodilators reduce the frequency of exacerbations of COPD. Short-acting β2-adrenoceptor agonists and antimuscarinic agents are equally effective for mild symptoms. For continuing breathlessness either a long-acting β2-adrenoceptor agonist (with a short-acting bronchodilator as required) or the long-acting antimuscarinic drug tiotropium (with a short-acting β2-adrenoceptor agonist as required) may give better symptom relief and reduce the risk of exacerbations. Theophylline or roflumilast are usually reserved for advanced COPD when symptoms persist or there are frequent exacerbations despite use of inhaled long-acting bronchodilators and corticosteroid. Nebulised bronchodilators can be useful for severe exacerbations.
Corticosteroids. Many of the chronic inflammatory changes in COPD do not respond to corticosteroids; nevertheless, an oral corticosteroid should be used for 7–14 days when treating an acute exacerbation of symptoms. Long-term use of an inhaled corticosteroid can reduce the number and severity of exacerbations, and should be considered if there are two or more exacerbations in a 12-month period. An inhaled corticosteroid can also produce some symptomatic benefit for people who remain breathless despite the use of a long-acting bronchodilator, or those with a forced expiratory volume of less than 50% of predicted. About 10% of people with COPD will have an improvement in their forced expiratory volume with an inhaled corticosteroid. However, there is an increased risk of pneumonia with inhaled corticosteroid, especially in the elderly with COPD.
Antibacterial drugs. One-third of infective exacerbations are due to viral infection, but antibacterial drugs (Ch. 51) produce earlier symptomatic improvement if there is moderate-to-severe acute exacerbation of symptoms with purulent sputum.
Mucolytic agents. Mecysteine hydrochloride, erdosteine or carbocisteine (Ch. 13) may reduce the frequency of exacerbations of COPD. An initial 1-month trial should be considered if COPD is accompanied by a chronic productive cough or if there are prolonged severe exacerbations. Treatment should only be continued if there is a perceived benefit.
Oxygen therapy. This is extremely important to treat hypoxaemia during acute exacerbations. Care must be taken to raise the arterial oxygen saturation (if possible to ≥90%) without increasing the arterial carbon dioxide tension. To avoid suppressing hypoxic drive in type 2 respiratory failure (hypoxaemia with a raised arterial carbon dioxide concentration), low-dose supplementary oxygen may be necessary (e.g. 24% via Venturi mask or 1–2 L⋅min−1 via nasal cannulae). Long-term domiciliary oxygen treatment, usually from an oxygen concentrator which removes nitrogen from air and delivers via nasal cannulae, improves symptoms and survival in COPD with respiratory failure (with an arterial oxygen tension less than 7.3 kPa). It should only be considered if respiratory failure persists for 3–4 weeks despite optimal drug therapy and without a clinical exacerbation. Those in the household must be warned of the fire risk if people smoke when receiving oxygen therapy. To improve survival in COPD with respiratory failure, oxygen must be used for at least 15 h per day.
Ventilatory support. This may be required during exacerbations. Intubation and mechanical ventilation may be necessary, but non-invasive assisted ventilation is preferable. Nasal intermittent positive-pressure ventilation (NIPPV) is being increasingly used during exacerbations for people who fail to respond to maximal medical therapy, especially if there is carbon dioxide retention.
Pulmonary rehabilitation. This improves exercise capacity and reduces the sensation of breathlessness, and can substantially improve morale.
True/false questions
1. Asthma is defined as irreversible airflow obstruction resulting from chronic airway inflammation.
2. An influx of Th2 lymphocytes occurs in the late airway response to inhaled allergen challenge of people with asthma and atopy.
3. The β2-adrenoceptor agonists are effective in preventing exercise-induced asthma.
4. The β2-adrenoceptor agonists have no effect on mucus clearance.
5. Tolerance to β2-adrenoceptor agonists can occur.
6. The mechanisms of action of theophylline are unclear.
7. The plasma concentration of theophylline is increased by simultaneous administration of erythromycin or ciprofloxacin.
8. Methylxanthines cause drowsiness.
9. An unwanted effect of theophylline is stimulation of the heart.
10. Ipratropium is more effective than salbutamol for preventing bronchospasm following challenge with an allergen.
11. Tiotropium is a selective antagonist of muscarinic M2 receptors.
12. Ipratropium causes bradycardia.
13. Ipratropium is poorly absorbed from the bronchi into the systemic circulation.
14. Leukotriene C4 is an important bronchodilator released from eosinophils.
15. Cysteinyl-leukotrienes are important in the precipitation of asthma in people who are intolerant to NSAIDs.
16. Montelukast inhibits 5-lipoxygenase that converts arachidonic acid to leukotrienes.
17. The leukotriene receptor antagonists (LTRA) are only given prophylactically.
18. Glucocorticoids reduce eosinophil recruitment to the bronchial mucosa.
19. Roflumilast is a selective inhibitor of phosphodiesterase (PDE) type 4.
20. Omalizumab binds to IgE receptors on mast cell membranes, preventing degranulation.
One-best-answer (OBA) questions
1. Which one of the following is the most likely unwanted effect of high-dose salbutamol?
2. Which of the following is the most accurate statement about the mechanism of action of glucocorticoids in asthma therapy?
A They reduce airway inflammation by inhibiting eosinophil apoptosis
B They reduce airway narrowing by relaxing bronchial smooth muscle
C They reduce release of mast cell mediators by blocking allergen–IgE interaction
D They downregulate β2-adrenoceptors on bronchial smooth muscle
E They reduce oedema by inducing endothelial tight junctions
Extended-matching-item questions
1. Match each statement below to the most appropriate option A–G.
2. Which option A–H is the most appropriate for add-on treatment to the current medication prescribed in each case scenario below?
E Modified-release theophylline
F Intravenous magnesium sulphate
H Modified-release theophylline.
2.1. A 25-year-old woman was admitted to the accident and emergency department with an acute exacerbation of her asthma. Her peak expiratory flow was 150 L⋅min−1. Her pulse rate was 145 beats⋅min−1, her respiratory rate was 30 min−1, respiration was shallow and she was confused. She was treated with 60% oxygen, nebulised salbutamol, nebulised ipratropium, intravenous aminophylline and intravenous hydrocortisone. Arterial blood gases on admission, breathing air, showed a PO2 of 8.4 kPa, PCO2 7.2 kPa and pH 7.29. There was little clinical improvement and she was transferred to the intensive care unit.
2.2. A 64-year-old man had mild asthma that was well controlled taking salbutamol two to three times a week and inhaled beclometasone twice daily. He complained of soreness of the mouth and hoarseness and was advised about oral hygiene.
2.3. A 67-year-old man had COPD with a chronic cough producing clear sputum. The cough and sputum production had not recently changed. He had stopped smoking 3 months previously because of his dyspnoea. Prior to that time, he had smoked 20 cigarettes a day for 50 years. He denied alcohol use. He had no other significant medical illnesses. His FEV1 was 1.34 L (about 45% of that predicted). He was taking salbutamol four times daily. A trial of inhaled beclometasone 3 months previously had provided no benefit and had been stopped.
2.4. A 60-year-old woman attended the accident and emergency department with increasing shortness of breath, increased production of green–yellow sputum and fever over the previous 4 days. She was known to have COPD. She was taking daily salbutamol and ipratropium by breath-actuated inhalers.
2.5. A 30-year-old man had mild asthma and allergic rhinitis. He was taking inhaled salbutamol and beclometasone, both twice daily. Recently he had been waking most nights with a persistent cough. He was a non-smoker and had no other medical history.
1. False. Airflow obstruction in asthma is mostly reversible, either spontaneously or as a result of treatment.
2. True. Th2 lymphocytes generate cytokines that promote activation, recruitment and survival of eosinophils and other leucocytes and increase the expression of IgE receptors on mast cells and eosinophils.
3. True. Salbutamol is effective taken before exercise but the longer-acting β2-adrenoceptor agonists are slower in onset. Cromoglicate taken prophylactically may also be effective.
4. False. Beta2-adrenoceptor agonists increase ciliary action and mucus clearance.
5. True. Tolerance to β2-adrenoceptor agonists may occur due to downregulation of their target receptor, an effect counteracted by administration of corticosteroids.
6. True. Methylxanthines may bronchodilate by inhibiting phosphodiesterases (PDEs) or blocking adenosine receptors on airway smooth muscle, while inhibition of histone deacetylase may account for reported anti-inflammatory effects at low doses.
7. True. Erythromycin and ciprofloxacin inhibit liver cytochrome P450 enzymes, resulting in slower metabolism of theophylline.
8. False. Methylxanthines present in coffee increase alertness and can cause irritability and headache.
9. True. All methylxanthines have positive inotropic and chronotropic activity and a narrow therapeutic index.
10. False. Ipratropium is less effective against allergen challenge but can be useful as an adjunct and in the management of COPD.
11. False. Tiotropium is a selective antagonist of M3 receptors on airway smooth muscle, with less antagonism of inhibitory M2 autoreceptors on parasympathetic nerves than ipratropium.
12. False. Ipratropium can cause a modest tachycardia owing to blockade of muscarinic receptors in the heart.
13. True. Ipratropium and tiotropium have quaternary structures and are therefore only poorly absorbed, minimising unwanted systemic effects.
14. False. Leukotriene C4 (and its extracellular metabolite LTD4) are potent bronchoconstrictors, and also increase mucus secretion, oedema and eosinophilia in the airway.
15. True. In susceptible individuals, NSAIDs that inhibit cyclo-oxygenase (COX)-1 may cause acute bronchospasm either by shunting arachidonic acid from the COX pathway to the leukotriene pathway or, more probably, by liberating the leukotriene pathway from partial inhibition by COX-derived prostaglandin E2.
16. False. Montelukast and other lukast drugs are antagonists of the cysteinyl-leukotriene receptor type 1 (CysLT1), not inhibitors of 5-lipoxygenase.
17. True. The LTRA are oral drugs taken once or twice daily for prophylaxis of asthma; they may also benefit comorbid conditions such as allergic rhinitis.
18. True. Glucocorticoids act at the transcriptional level to inhibit numerous steps in the inflammatory pathways involved in the pathogenesis of asthma, including the proliferation, recruitment, activation and survival of eosinophils.
19. True. Roflumilast is a highly selective inhibitor of PDE4, which is found in inflammatory cells.
20. False. Omalizumab binds to circulating and tissue IgE, leading to its clearance by endothelial cells. This results in a reduction in the numbers of IgE receptors on mast cells and other cells.
OBA answers
1. Answer B is correct. Salbutamol is a β2-adrenoceptor agonist that can produce hypokalaemia by increasing cellular uptake of K+. At higher doses it may cause tachycardia, hyperglycaemia and hypomagnesaemia, while mydriasis is not likely with a β-adrenoceptor agonist.
2. Answer E is correct. Glucocorticoids induce endothelial tight junctions, which reduces vascular permeability and leucocyte migration into tissue.
Extended-matching-item answers
1.1. Answer D. Salbutamol acts selectively on the β2-adrenoceptors in airways, activating adenylyl cyclase and increasing cAMP.
1.2. Answer A. Theophylline inhibits the breakdown of cAMP by phosphodiesterases.
1.3. Answer F. Indometacin (and other NSAIDs that inhibit COX-1) can induce bronchoconstriction in aspirin-sensitive people with asthma. Celecoxib, a selective COX-2 inhibitor, is unlikely to cause this reaction, but should still be used with care.
1.4. Answer E. Montelukast is a selective antagonist of CysLT1 receptors for the bronchoconstrictor cysteinyl-leukotrienes (LTC4/D4/E4), synthesis of which is triggered by non-selective NSAIDs.
1.5. Answer C. Prostaglandin D2 is a bronchoconstrictor released by mast cells.
2.1. Answer F. This woman is being treated according to the British Thoracic Society guidelines; an appropriate add-on medication for use in this life-threatening situation is intravenous magnesium sulphate.
2.2. Answer D. This man should additionally be advised to use a spacer with all inhaled drugs. This improves the effectiveness of the medication and will reduce deposition of corticosteroid in the mouth and oropharynx, reducing the occurrence of fungal growth and hoarseness.
2.3. Answer A. The antimuscarinic drug ipratropium provides equal or greater benefit to β2-adrenoceptor agonists in COPD and will reduce the volume of sputum produced. Corticosteroids are of modest benefit in only a small percentage of people with COPD.
2.4. Answer B. This woman has an infection-related exacerbation of her COPD and should be treated with an appropriate antibiotic. Nebulised salbutamol and ipratropium should also be started.
2.5. Answer C. Approximately 80% of severe asthmatic attacks occur between midnight and 8 am. Salbutamol is a short-acting β2-adrenoceptor agonist, providing relief for 2–6 h. A trial of salmeterol or formoterol, which provide bronchodilation for 12 h or longer, should be considered. The long-acting drugs should not be used for relief of acute asthma episodes.
Compendium: drugs used to treat asthma or chronic obstructive pulmonary disease
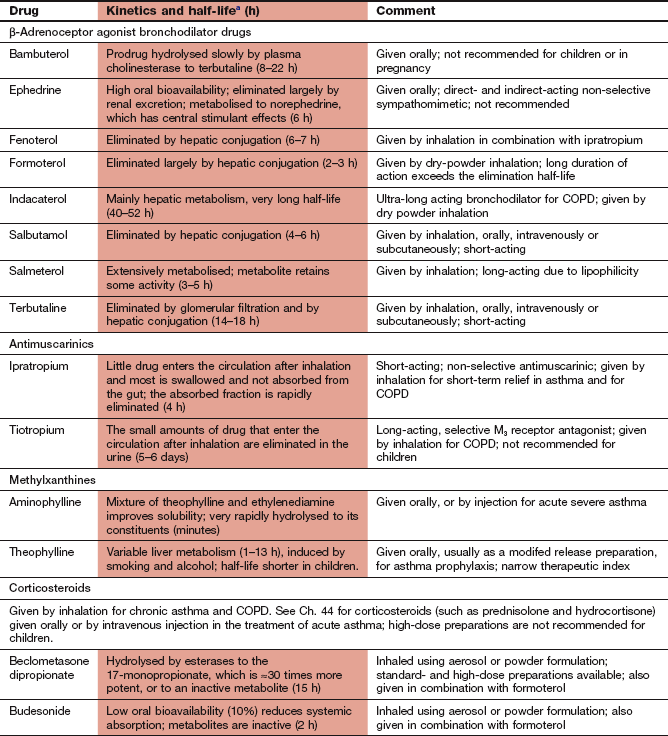
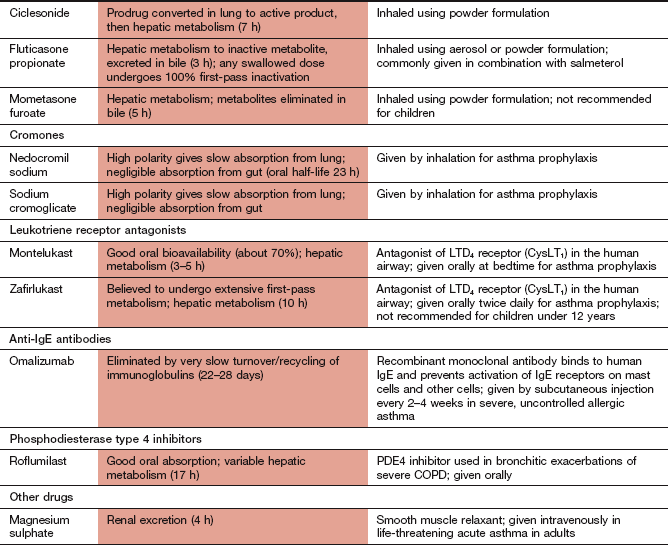
COPD, chronic obstructive pulmonary disease.
aThe half-life refers to the systemic elimination half-life from the general circulation, which may not correlate with the duration of action following inhalation when this is dependent on very slow uptake from the airways.
Barnes, PJ. Glucocorticoids: current and future direction. Br J Pharmacol. 2011;163:29–43.
Cazzola, M, Calzetta, L, Matera, MG. β2-Adrenoceptor agonists: current and future direction. Br J Pharmacol. 2011;163:4–17.
Dolovich, MB, Dhand, R. Aerosol drug delivery: developments in device design and clinical use. Lancet. 2011;377:1032–1045.
Moulton, BC, Fryer, AD. Muscarinic receptor antagonists, from folklore to pharmacology; finding drugs that actually work in asthma and COPD. Br J Pharmacol. 2011;163:44–52.
Adcock, IM, Caramori, G, Chung, KF. New targets for drug development in asthma. Lancet. 2008;372:1073–1087.
Bush, A, Saglani, S. Management of severe asthma in children. Lancet. 2010;376:814–825.
Fanta, CH. Drug therapy: Asthma. N Engl J Med. 2009;360:1002–1014.
Farooque, SP, Lee, TH. Aspirin-sensitive respiratory disease. Annu Rev Physiol. 2009;71:465–487.
Holgate, ST, Polosa, R. The mechanisms, diagnosis and management of severe asthma in adults. Lancet. 2006;368:780–793.
Johnson, M. Molecular mechanisms of β2 adrenergic receptor function, response and regulation. J Allergy Clin Immunol. 2006;117:18–24.
Lazarus, SC. Emergency treatment of asthma. N Engl J Med. 2010;363:755–764.
O'Byrne, PM, Parameswaran, K. Pharmacological management of mild or moderate persistent asthma. Lancet. 2006;368:794–803.
Ormiston, TM, Salpeter, EE. Respiratory tolerance to regular β2-agonist use in patients with asthma. Ann Intern Med. 2004;140:802–814.
Rees, J. Asthma control in adults. BMJ. 2006;332:767–771.
Rowe, BH, Bretzlaff, JA, Bourdon, C, Bota, GW, Camargo, CA, Jr. Intravenous magnesium sulfate treatment for acute asthma in the emergency department: a systematic review of the literature. Ann Emerg Med. 2000;3:6181–6190.
Salpeter, SR, Buckley, NS, Ormiston, TM, Salpeter, EE. Meta-analysis: effect of long-acting β-agonists on severe exacerbations and asthma-related deaths. Ann Intern Med. 2006;144:904–912.
Stevenson, DD. Aspirin sensitivity and desensitization for asthma and sinusitis. Curr Allergy Asthma Rep. 2009;9:155–163.
Chronic obstructive pulmonary disease
Anzueto, A. Clinical course of chronic obstructive pulmonary disease: review of therapeutic interventions. Am J Med. 2006;119:S46–S53.
Barnes, PJ. Theophylline. New perspectives for an old drug. Am J Respir Crit Care Med. 2002;167:813–818.
Barnes, PJ, Ito, K, Adcock, IM. Corticosteroid resistance in chronic obstructive pulmonary disease: inactivation of histone deacetylase. Lancet. 2004;363:731–733.
Brusselle, GG, Joos, GF, Bracke, KR. New insights into the immunology of chronic obstructive pulmonary disease. Lancet. 2012;378:1015–1026.
Criner, GJ. Optimal treatment of chronic obstructive pulmonary disease: the search for the magic combination of inhaled bronchodilators and corticosteroids. Ann Intern Med. 2007;146:606–608.
Decramer, M, Janssens, W, Miravitelles, M. Chronic obstructive pulmonary disease. Lancet. 2012;379:1341–1351.
Hansel, T, Barnes, P. New drugs for exacerbations of chronic obstructive pulmonary disease. Lancet. 2009;374:744–755.
Lipworth, BJ. Phosphodiesterase-4 inhibitors for asthma and chronic obstructive pulmonary disease. Lancet. 2005;365:167–175.
Martinez, FJ, Donohue, JF, Rennard, S. The future of chronic obstructive pulmonary disease treatment – difficulties of and barriers to drug development. Lancet. 2011;378:1027–1037.
Niewoehner, DE. Outpatient management of severe COPD. N Engl J Med. 2010;362:1407–1416.
Plant, PK, Elliot, MW. Chronic obstructive pulmonary disease 9: management of ventilatory failure in COPD. Thorax. 2003;58:537–542.
Rabe, KF, Wedzicha, JA. Controversies in treatment of chronic obstructive pulmonary disease. Lancet. 2011;378:1038–1047.
Wilt, J, Niewoehner, D, MacDonald, R, et al. Management of stable chronic obstructive pulmonary disease: a systematic review for a clinical practice guideline. Ann Intern Med. 2007;147:639–653.
Wilt, J, Weinberger, S, Shekelle, P, et al. Diagnosis and management of stable chronic obstructive pulmonary disease: a clinical practice guideline from the American College of Physicians. Ann Intern Med. 2007;147:633–638.