Systemic and pulmonary hypertension
Systemic hypertension
The cause of systemic hypertension in the majority of people is unknown (essential hypertension), with a complex interplay between genetic and environmental influences. Abnormal regulation of the physiological mechanisms that normally control arterial blood pressure may be an important factor. A small number of people with hypertension have an identifiable underlying cause (secondary hypertension).
Circulatory reflexes and the control of systemic blood pressure
Systemic blood pressure (BP) is determined by the cardiac output (CO) and total peripheral resistance (TPR).
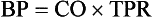
Blood pressure is maintained within fairly narrow limits due to modulation by a series of physiological reflexes that are triggered by both acute and chronic changes in blood pressure. There are both short-term and long-term control mechanisms. Important regulatory systems are:
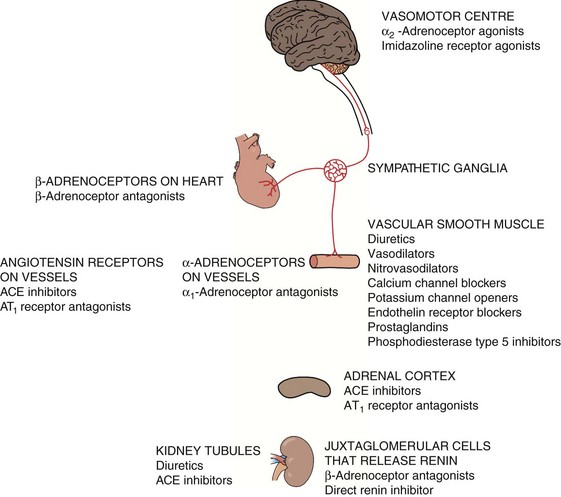
The autonomic nervous system regulates arterial blood pressure in several ways.
 In the heart, the sympathetic nervous system acts mainly through β1-adrenoceptors to increase myocardial contractility and heart rate, generating a greater cardiac output and increasing blood pressure (Ch. 4). An increase in parasympathetic nervous system tone reduces heart rate and therefore cardiac output and blood pressure
In the heart, the sympathetic nervous system acts mainly through β1-adrenoceptors to increase myocardial contractility and heart rate, generating a greater cardiac output and increasing blood pressure (Ch. 4). An increase in parasympathetic nervous system tone reduces heart rate and therefore cardiac output and blood pressure
In arterial resistance vessels, sympathetic nervous stimulation produces arteriolar vasoconstriction through stimulation of postsynaptic α1-adrenoceptors. Arterial vasoconstriction raises blood pressure, but also increases the afterload on the heart. In the healthy heart, cardiac output is maintained by an increase in cardiac contractility via β1-adrenoceptors.
In venous capacitance vessels, sympathetic stimulation of postsynaptic α1-adrenoceptors produces venous constriction. This increases venous return to the heart (preload), raises cardiac output and increases blood pressure (Ch. 7).
The autonomic nervous system is normally responsible for immediate regulation of blood pressure. Change in systemic blood pressure is detected by baroreceptors (stretch receptors) in the aorta and carotid arteries. When blood pressure rises, stretch of the baroreceptors increases afferent nerve impulses to a coordinating area in the medulla which controls the autonomic outflow to the cardiovascular system. The increase in afferent impulses inhibits sympathetic nervous system output and increases parasympathetic nervous system outflow, returning the blood pressure to normal. The opposite occurs when blood pressure falls (Fig. 6.1).
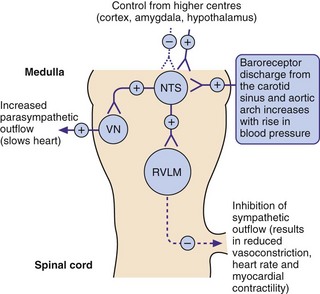
Fig. 6.1 The role of baroreceptors.
Increased blood pressure increases neural discharge from the baroreceptors (stretch receptors) in the carotid sinus and aortic arch, resulting in a compensatory inhibition in sympathetic outflow from the rostral ventrolateral medulla (RVLM) and an increase in the parasympathetic outflow from the cardioinhibitory vagal nucleus (VN). Both effects act to lower blood pressure, which is also influenced by control from higher centres acting at the nucleus of the tractus solitarius (NTS). See Fig. 6.7 for sites of action of centrally acting antihypertensive drugs. +, stimulation; –, inhibition.
A slower compensatory mechanism for a reduction in blood pressure is initiated by the release of renin from the juxtaglomerular apparatus of the kidney (Fig. 6.2). The major stimuli leading to renin release are reduced renal blood flow (often as a result of a decrease in blood pressure), decreased Na+ in the renal distal tubule and direct sympathetic stimulation via β1-adrenoceptors. Renin is a protease that acts on circulating renin substrate (angiotensinogen) to release the decapeptide angiotensin I. This is further cleaved by angiotensin-converting enzyme the octapeptide angiotensin II. There are several additional enzymatic pathways for generating angiotensin II that do not involve ACE (see Fig. 6.8). Angiotensin II acts on various tissues via specific angiotensin AT1 and AT2 receptors (Fig. 6.2). Its action at the AT1 receptor produces potent direct vasoconstriction, and also enhances sympathetic nervous tone by facilitating presynaptic neuronal release of noradrenaline and through stimulation of central sympathetic outflow. Angiotensin II has a number of additional properties which promote salt and water retention (Fig. 6.3), one of the most powerful being the release of aldosterone from the adrenal cortex. Aldosterone acts at the distal renal tubule to conserve salt and water at the expense of K+ loss (Ch. 14). Therefore, angiotensin II and aldosterone raise blood pressure by vasoconstriction and by increasing circulating blood volume. Some of the effects of angiotensin at the AT1 receptor do not raise blood pressure; in the endothelium, AT1 receptor stimulation enhances nitric oxide production, resulting in vasodilation and dampening the pressor effects of angiotensin II. Angiotensin II has additional actions at the AT2 receptor that appear to oppose some of those at the AT1 receptor (Fig. 6.2).
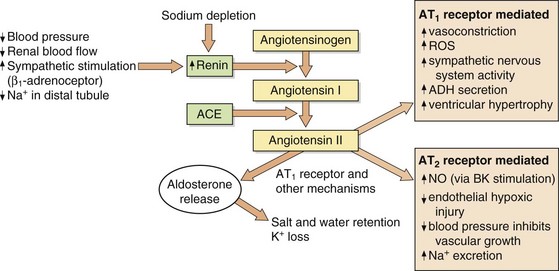
Fig. 6.2 Formation and actions of angiotensin II.
Angiotensin II acts on type 1 (AT1) and type 2 (AT2) receptors. Current therapeutic drugs act predominantly to block AT1 receptors. The number of AT2 receptors is low relative to that of AT1 receptors but increases in pathological conditions. ACE, angiotensin-converting enzyme; ADH, antidiuretic hormone; BK, bradykinin, NO, nitric oxide; ROS, reactive oxygen species.
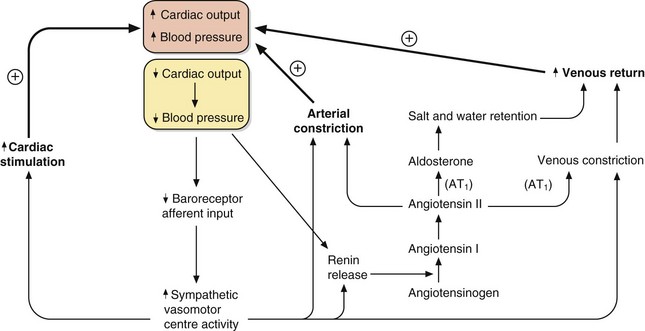
Fig. 6.3 The control of blood pressure via the sympathetic and renin–angiotensin–aldosterone systems.
A fall in cardiac output or blood pressure (yellow box) produces relatively rapid responses mediated by increased sympathetic activity and slower responses mediated by renin–angiotensin–aldosterone mechanisms. The outcomes are increased cardiac stimulation, increased arterial constriction and increased venous return, restoring blood pressure. AT1, angiotensin II type 1 receptor.
The integration of the fast-responding sympathetic nervous system and the slower-responding renin–angiotensin–aldosterone system in response to a fall in blood pressure is shown in Figure 6.3. These mechanisms prevent hypotension due to peripheral pooling of blood on standing and during exercise.
Additional mechanisms involved in controlling vascular tone and blood volume include circulating or local endothelial hormones and metabolites, such as natriuretic peptides, antidiuretic hoemone, prostaglandins, bradykinin, nitric oxide, endothelin and adenosine. Their relative importance may differ in health and disease states.
Aetiology and pathogenesis of systemic hypertension
There is no absolute cut-off between normal and high blood pressure. Blood pressure in all populations is normally distributed with a slight skew because of a small number of individuals with very high blood pressures. The risk of complications is also a continuous variable, increasing as blood pressure rises. Defining a point at which blood pressure is ‘high’ is therefore somewhat arbitrary but it is usually set at values greater than 140/90 mmHg. Using this definition, hypertension is a common condition found in 20–30% of the population of the developed world, with the prevalence increasing with age.
Hypertension does not usually cause symptoms but produces progressive structural changes in the heart and circulation. These include formation of atheroma, microaneurysms on intracerebral blood vessels and remodelling of the muscle in the left ventricle and arterial resistance vessels. These changes predispose to clinical complications that are often referred to as ‘target organ damage’. The principal complications of hypertension are ischaemic heart disease (especially in middle-aged Europeans and Americans), and cerebrovascular disease (especially in Asians and older people) which usually presents as thromboembolic stroke or, less commonly, as cerebral haemorrhage (Ch. 9). Ischaemic complications of hypertension are more likely to occur if it is accompanied by hypercholesterolaemia, diabetes mellitus and smoking. The underlying vascular lesions that occur in hypertension and their resulting complications are shown in Figure 6.4.
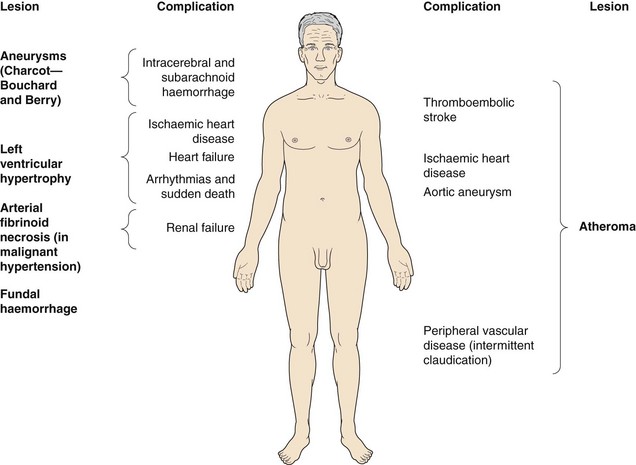
Fig. 6.4 Complications of hypertension.
Hypertension causes vascular lesions and damage throughout the body.
Sustained hypertension predisposes to left ventricular muscle hypertrophy (LVH). LVH is an independent risk factor for the complications of hypertension, particularly ischaemic heart disease (since the muscle outgrows its blood supply), heart failure with preserved ejection fraction (Ch. 7) and arrhythmias leading to sudden death (Ch. 8).
If target organ damage or diabetes is present, treatment reduces complications when introduced at blood pressures above 140/90 mmHg. However, if there is no target organ damage treatment may not alter outcome until the systolic pressure is sustained above 160 mmHg, or the diastolic blood pressure is sustained above 100 mmHg.
Hypertension should ideally be confirmed by ambulatory 24 h blood pressure monitoring, unless target organ damage gives a clear indication treatment is necessary. Sometimes, the blood pressure is raised only when the measurement is taken by a doctor or, to a lesser extent, by a nurse. This phenomenon is termed ‘white coat’ or ‘office’ hypertension and appears to carry little risk of complications over the subsequent few years but can eventually lead to sustained hypertension. It can be detected by ambulatory 24 h blood pressure monitoring. White coat hypertension often persists despite drug treatment, which can then result in quite troublesome hypotension away from the surgery or clinic. There is also a phenomenon of ‘masked’ hypertension, when blood pressure is normal in the clinic but high at home. This appears to carry a similar risk of complications as sustained hypertension.
Malignant or accelerated hypertension is an infrequently encountered condition, produced by very high blood pressure or a rapid rise in blood pressure. It is characterised pathologically by arterial fibrinoid necrosis, and identified clinically by the presence of flame-shaped haemorrhages, hard exudates and ‘cotton wool’ spots in the retina or by papilloedema, which can lead to visual disturbance. If untreated, accelerated hypertension usually leads to death from renal failure, heart failure or stroke within 5 years.
Hypertension is usually characterised by increased peripheral arterial resistance, which arises from arteriolar smooth muscle constriction and hypertrophy leaving a smaller vessel lumen and an increase in the wall-to-lumen ratio (vascular remodelling). Cardiac output is often normal in younger people with hypertension, but is usually reduced in the elderly. The cause of the inappropriately raised peripheral resistance is unknown in the majority of people with hypertension, who are said to have ‘essential hypertension’. Essential hypertension probably has a polygenic inheritance, leading to several clinical subtypes with different underlying pathogenic mechanisms. Environmental influences and factors such as diet, level of exercise, obesity and alcohol intake all interact with the genetic programming to determine the final level of blood pressure. There is evidence that reduced renal Na+ excretion plays a central role in the pathogenesis of essential hypertension, and the kidney requires a higher-than-normal blood pressure to maintain a normal extracellular fluid volume. However, the disturbance in essential hypertension is much more widespread than the kidney, with cell membrane abnormalities found in many organs.
Isolated systolic hypertension, usually found in older people, is the consequence of stiffening of large ‘conductance’ arteries. These vessels normally expand to accommodate the blood expelled from the heart in systole, which slows the pulse wave and increases the time taken for it to reach the peripheral resistance vessels. The pulse wave is normally reflected back from the peripheral vessels in diastole, and supports the diastolic blood pressure and therefore coronary artery perfusion. If the compliance of the large arteries is reduced, then the pulse wave reaches the peripheral vessels early and is reflected back in systole. This increases systolic blood pressure and reduces diastolic pressure. In isolated systolic hypertension, coronary artery perfusion can be impaired, while cardiac work is increased.
A secondary underlying cause of high blood pressure, which often has a renal or endocrine origin, can be identified in about 5% of people with hypertension (Table 6.1).
Table 6.1
Principal causes of secondary hypertension
| Causes | |
| Renal | Renal artery stenosis, glomerulonephritis, interstitial nephritis, arteritis, polycystic disease, chronic pyelonephritis |
| Endocrine | Conn's syndrome (aldosterone excess), Cushing's syndrome (glucocorticoid excess), phaeochromocytoma (catecholamine excess), acromegaly |
| Pregnancy | Pre-eclampsia and eclampsia |
| Drugs | Oestrogen, corticosteroids, non-steroidal anti-inflammatory drugs (NSAIDs), ciclosporin |
Not surprisingly, since the cause of hypertension is unclear, treatment cannot be directed precisely at the underlying mechanism(s). Most antihypertensive drugs are vasodilators, and they often modulate the natural hormonal or neuronal mechanisms responsible for blood pressure regulation. Less commonly, a hypotensive action is partially achieved by reducing cardiac output. The principal classes of antihypertensive drugs and their sites of action are shown in Table 6.2 and Figure 6.5.
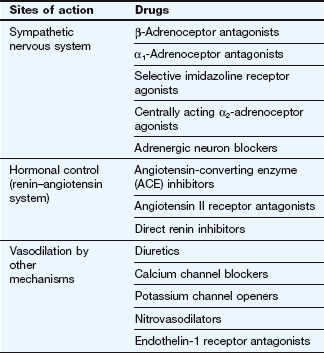
Antihypertensive drugs
Drugs acting on the sympathetic nervous system
Beta-adrenoceptor antagonists (β-blockers):
Mechanism of action in hypertension: Beta-adrenoceptor antagonists reduce blood pressure in several ways (Fig. 6.6). Selective β1-adrenoceptor antagonists are as effective as non-selective drugs, indicating that β2-adrenoceptor blockade makes little contribution. The more important actions are probably:
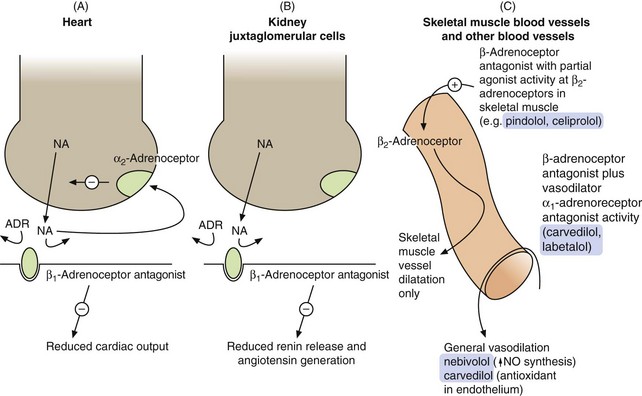
Fig. 6.6 Sites of action of the β-adrenoceptor antagonists relevant to their use as antihypertensive agents.
(A) In the heart, the β1-adrenoceptor antagonist drugs reduce stimulation of the β1-adrenoceptors by noradrenaline (NA) and circulating adrenaline (ADR). Presynaptic stimulation of α2-adrenoceptors, which inhibits noradrenaline release, still functions normally. (B) In the kidney, β1-adrenoceptor blockade reduces the activity of the renin–angiotensin system. (C) Some selective β1-adrenoceptor antagonists have hybrid activity: pindolol and celiprolol have intrinsic sympathomimetic activity, acting as partial agonists at β2-adrenoceptors in skeletal muscle blood vessels, leading to vasodilation and reduced peripheral resistance. As partial agonists these drugs reduce heart rate and cardiac output less than full antagonists. Nebivolol may dilate blood vessels more generally by releasing nitric oxide (NO). Carvedilol and labetalol also have α1-adrenoceptor antagonist activity, reducing peripheral resistance.
reduction of heart rate and myocardial contractility, which decreases cardiac output,
blockade of renal juxtaglomerular β1-adrenoceptors, which reduces renin secretion and therefore generation of angiotensin II and aldosterone. This produces vasodilation and reduces plasma volume,
peripheral arterial vasodilation is not a direct effect of pure β-adrenoceptor antagonists, but is an additional property of some compounds that have a hybrid action (such as nebivolol). These produce vasodilation by mechanisms other than β1-adrenoceptor antagonism (Fig. 6.6C and see especially Ch. 5),
blockade of presynaptic β-adrenoceptors in sympathetic neurons supplying arteriolar resistance vessels. This may reduce the release of noradrenaline and thus attenuate reflex arterial vasoconstriction, but the clinical importance of this effect is uncertain.
For further details about β-adrenoceptor antagonists and their many uses, see Chapters 5 and 8, and Box 6.1.
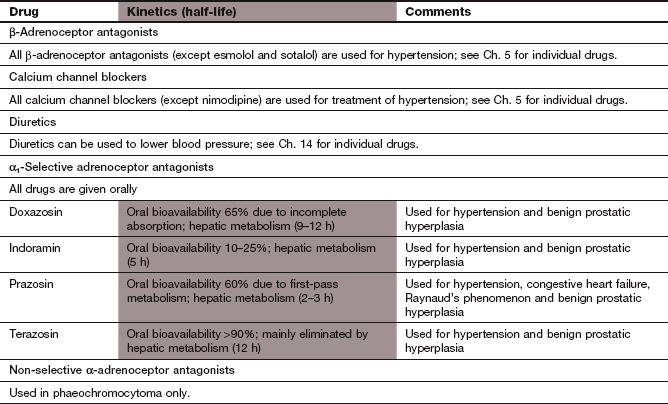
Alpha-adrenoceptor antagonists (α-blockers):
Mechanisms of action: Alpha-adrenoceptor antagonists (often referred to as α-blockers) lower blood pressure by blockade of postsynaptic α1-adrenoceptors, leading to:
dilation of arteriolar resistance vessels, which lowers peripheral resistance,
dilation of venous capacitance vessels, which reduces venous return and therefore cardiac output.
When blood pressure falls as a result of using an α-adrenoceptor antagonist, this is detected by arterial baroreceptors. The baroreceptors initiate an increase in sympathetic discharge from the medulla, causing a reflex tachycardia (Figs 6.1 and 6.3). However, because noradrenaline released from cardiac sympathetic nerve terminals also stimulates inhibitory α2-adrenoceptors on the presynaptic sympathetic neuron, the degree of sympathetic stimulation and reflex tachycardia is attenuated (Fig. 6.6A). Selective α1-adrenoceptor antagonists do not block the presynaptic α2-adrenoceptors on sympathetic nerve terminals, and therefore reflex tachycardia is unusual with these compounds.
By contrast, non-selective α-adrenoceptor antagonists block both postsynaptic α1-adrenoceptors and presynaptic α2-adrenoceptors, and their use is accompanied by a marked reflex tachycardia. Non-selective agents now have little place in clinical practice except for the perioperative management of phaeochromocytoma.
Alpha-adrenoceptor antagonists produce a potentially beneficial effect on plasma lipids by increasing high-density lipoprotein (HDL) cholesterol and reducing triglycerides (Ch. 48). Whether this has any relevance for the prevention of atheroma in individuals with hypertension is uncertain. Selective α1-adrenoceptor antagonists are also used to treat the symptoms of bladder outlet obstruction (Ch. 15).
Pharmacokinetics: Selective α1-adrenoceptor antagonists are well absorbed from the gut and undergo extensive first-pass metabolism and subsequent elimination by the liver. The compounds differ principally in their half-lives and, therefore, duration of action; for example, prazosin has a half-life of 3 h whereas doxazosin has a half-life of 9–12 h.
Centrally acting antihypertensive drugs
Selective imidazoline receptor agonists:
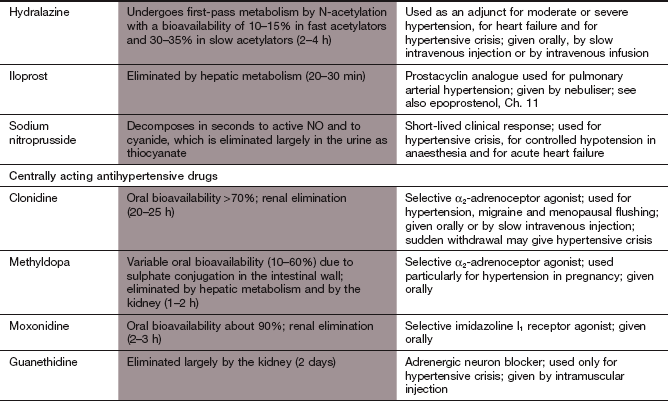
Mechanism of action: Imidazoline I1 receptors are important for the regulation of sympathetic drive (Figs 6.1 and 6.7). They are concentrated in the rostral ventrolateral medulla, a part of the brainstem involved in control of blood pressure. Increased neuronal activity in this area, either through baroreceptor stimulation or by direct stimulation of I1 receptors by moxonidine, will decrease sympathetic outflow, which results in a fall in blood pressure with no reflex tachycardia. Unlike other centrally acting drugs (clonidine and methyldopa), moxonidine has a low affinity for presynaptic α2-adrenoceptors.
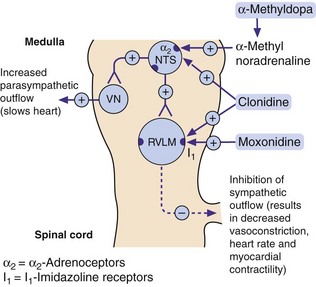
Fig. 6.7 Mechanisms of centrally acting antihypertensive drugs.
These drugs act on the same medullary centres that respond to raised blood pressure (Fig. 6.1). Methylnoradrenaline and clonidine stimulate α2-adrenoceptors in the nucleus of the tractus solitarius (NTS). Moxonidine, and possibly also clonidine, act on imidazoline I1 receptors in the rostral ventrolateral medulla (RVLM). VN, vagal nucleus (cardioinhibitory centre).
Centrally acting α2-adrenoceptor agonists:
Unwanted effects limit the use of the centrally acting α2-adrenoceptor agonists, although methyldopa is a drug of choice in the treatment of hypertension in pregnancy (see below).
Mechanisms of action: The α2-adrenoceptor agonists act at presynaptic autoreceptors in the central nervous system (CNS) to reduce sympathetic nervous outflow and increase vagal outflow from the medulla (Fig. 6.7). This reduces both peripheral arterial and venous tone.
Methyldopa is a prodrug that is metabolised in the nerve terminal as a ‘false substrate’ in the biosynthetic pathway for noradrenaline, to produce α-methylnoradrenaline, a potent α2-adrenoceptor agonist. Clonidine is a direct-acting α2-adrenoceptor agonist that is also an agonist at imidazoline I1 receptors (see above). Clonidine has some peripheral postsynaptic α1-adrenoceptor agonist activity, which produces direct peripheral vasoconstriction; this initially offsets some of the central blood pressure-lowering effect.
Pharmacokinetics: Methyldopa is incompletely absorbed from the gut and undergoes dose-dependent first-pass metabolism, with an elimination half-life of 1–2 h. Clonidine is completely absorbed from the gut and has a half-life of about 24 h.
Sympathetic blockade: failure of ejaculation, and postural or exertional hypotension (unusual with clonidine, owing to its direct peripheral action).
Unopposed parasympathetic action: diarrhoea.
CNS effects: sedation and drowsiness occur in up to 50% of people who take methyldopa; depression is occasionally seen.
Fluid retention with peripheral oedema.
Methyldopa induces a reversible positive Coombs' test in 20% of people, resulting from production of IgG to red cell membrane constituents; however, haemolytic anaemia is rare.
Sudden withdrawal of clonidine can produce severe rebound hypertension with tachycardia, sweating and anxiety.
Drugs affecting the renin–angiotensin system
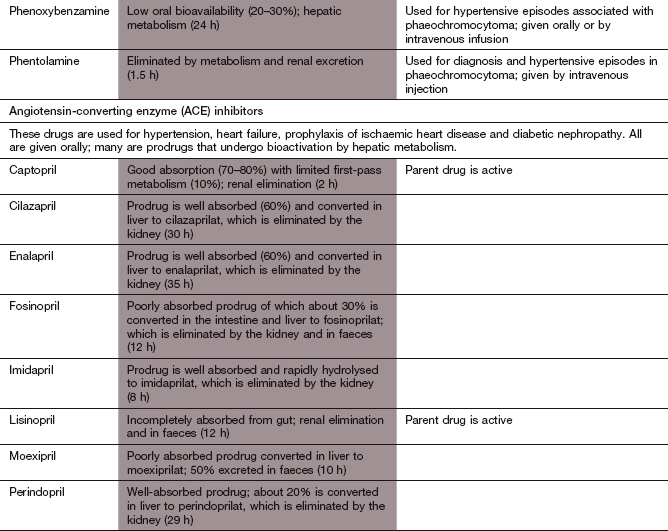
Angiotensin-converting enzyme inhibitors:
Mechanisms of action: The ACE inhibitors lower blood pressure by several mechanisms (see Figs 6.2, 6.5 and 6.8).
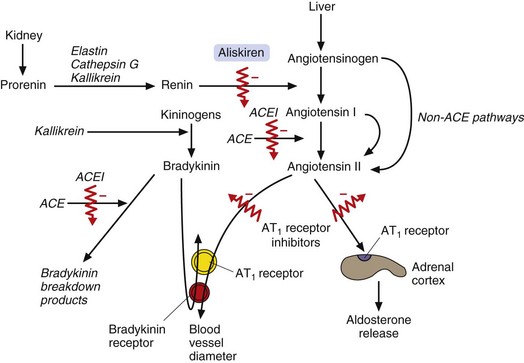
Fig. 6.8 The biological actions of angiotensin II and bradykinin and drugs that modify these actions.
Angiotensin II causes vasoconstriction by stimulating AT1 receptors in the blood vessels and causes Na+ retention by stimulating AT1 receptors in the adrenal cortex, which results in aldosterone release. Bradykinin causes vasodilation by acting on vascular smooth muscle cells and on endothelial cells. Angiotensin-converting enzyme inhibitors (ACEI) block angiotensin II formation from angiotensin I (although alternative, non-ACE-dependent protease pathways remain that can result in some angiotensin II formation from angiotensinogen or angiotensin I). ACE inhibitors also reduce the breakdown of bradykinin, contributing to their vasodilator effects. Angiotensin II receptor antagonists block AT1 receptors in blood vessels and the adrenal cortex. Aliskiren directly inhibits the actions of renin on angiotensinogen.
Competitive inhibition of plasma ACE reduces generation of circulating angiotensin II and consequently reduces the release of aldosterone (Fig. 6.8).
Inhibition of tissue ACE in the vascular wall is central to the hypotensive effect of these drugs (Fig. 6.8). Reduced tissue concentrations of angiotensin II lead to arterial dilation and, to a lesser extent, venous dilation. Angiotensin II production is not completely inhibited owing to alternative pathways for its generation by proteases including chymase and chymotrypsin-like angiotensin-II-generating enzyme (CAGE). The activity in these pathways is enhanced due to stimulation of renin release by the fall in blood pressure after ACE inhibition.
Competitive inhibition of plasma ACE reduces generation of circulating angiotensin II and consequently reduces the release of aldosterone (Fig. 6.8).
Reduction in angiotensin II-mediated potentiation of the sympathetic nervous system (Figs 6.2 and 6.3) prevents reflex tachycardia.
Angiotensin II is implicated in the development of arterial remodelling and LVH in hypertension. ACE inhibitors are more effective in regressing LVH than diuretics or β-adrenoceptor antagonists.
ACE also degrades other peptides including bradykinin (Fig. 6.8). Increased bradykinin in the vascular wall may contribute to the hypotensive actions of ACE inhibitors.
There are many clinical uses of ACE inhibitors other than hypertension; these are listed in Box 6.2.
Pharmacokinetics: Many ACE inhibitors are given orally as prodrugs, because the active forms are polar and poorly absorbed from the gut. The prodrugs are converted in the liver to the active agent; for example, ramipril is converted to the active compound ramiprilat. In contrast, captopril and lisinopril are absorbed adequately as an active molecule. Most ACE inhibitors are excreted in the active form by the kidney. The half-lives range from about 2 h (for example, captopril and ramiprilat) to about 30–35 h (for example, enalaprilat).
Persistent dry cough that is not dose-related and may be caused by accumulation of kinins in the lung. This occurs in 10–30% of people who take ACE inhibitors, is more common in women and can develop after many months of treatment.
Postural hypotension, which is rare unless there is salt and water depletion, for example as a result of therapy with diuretics. Profound hypotension can occur in such individuals, particularly after the first dose. This is rarely a problem in the treatment of hypertension, but can be in the treatment of severe heart failure (Ch. 7).
Renal impairment, especially in those with severe bilateral renal artery stenosis who rely on angiotensin-mediated efferent glomerular arterial vasoconstriction to maintain glomerular perfusion pressure.
Disturbance of taste (which may be permanent), nausea, vomiting, dyspepsia or bowel disturbance.
Angioedema, which is more frequent in people of Afro-Caribbean origin.
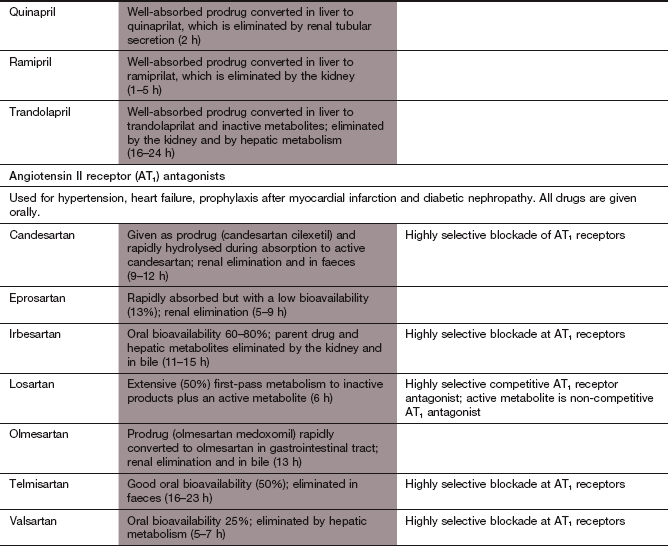
Angiotensin II receptor antagonists:
Mechanism of action: The angiotensin II receptor antagonists are selective for the AT1 receptor subtype, which is found in the heart, blood vessels, kidney, adrenal cortex, lung and brain, and have less effect at the AT2 receptor subtype (Fig. 6.2). Actions of angiotensin II via the AT1 receptor include vasoconstriction, cell growth and proliferation, aldosterone release, sympathetic stimulation, salt and water retention, inhibition of renin release and an increase in reactive oxygen species. In contrast to the use of ACE inhibitors, kinin degradation is unaffected by angiotensin II receptor antagonists and inhibition of the effects of angiotensin II is more complete (Fig. 6.8). There are many clinical uses of angiotensin II receptor antagonists other than hypertension; these are listed in Box 6.2.
Pharmacokinetics: Losartan is well absorbed from the gut and partially converted to an active metabolite, which is responsible for most of the pharmacological effects and has a longer half-life (6 h) than the parent drug (2 h). Candesartan and valsartan are given as prodrugs. Most angiotensin II receptor antagonists are given once daily.
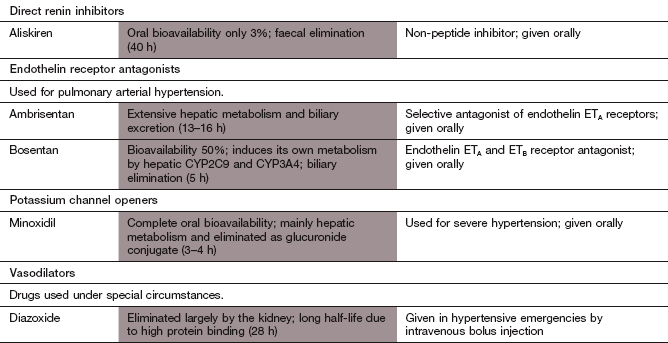
Mechanism of action: Aliskiren is a selective renin inhibitor with low affinity for other proteases. It binds competitively to the active site of the enzyme and inhibits the generation of angiotensin I (Fig. 6.8). Vasodilation is achieved by reduced angiotensin II synthesis, without the compensatory increase in plasma renin activity that occurs with an ACE inhibitor or angiotensin II receptor antagonist. The place of aliskiren in the treatment of hypertension remains to be established.
Vasodilators
Thiazide or thiazide-type diuretics are most frequently used to lower blood pressure, but loop and potassium-sparing diuretics are used in some situations.
Mechanism of action in hypertension: Full details of the sites and mechanisms of action of diuretics on the kidney and their unwanted effects are considered in Chapter 14. Actions involved in lowering blood pressure include the following.
An initial hypotensive effect is produced by intravascular salt and water depletion. However, compensatory mechanisms such as activation of the renin–angiotensin–aldosterone system largely restore plasma and extracellular fluid volumes (see Fig. 6.3) (unless salt and water retention was a major component of the initial hypertension, e.g. in advanced renal failure or as a consequence of other antihypertensive treatment).
Direct arterial dilation is responsible for the longer-term reduction in blood pressure. The mechanism of vasodilation is not well understood, but may result from reduced Ca2+ entry into the smooth muscle of the arteriolar resistance vessel walls (perhaps as a consequence of intracellular Na+ depletion).
Thiazide and thiazide-type diuretics: These produce their maximum blood pressure-lowering effect at doses lower than those required for significant diuretic activity. This is an advantage, since most unwanted effects are dose-related.
Loop diuretics: Loop diuretics are usually less effective than thiazides in the treatment of essential hypertension. Despite having a more powerful diuretic action, their duration of action is too short. However, hypertension with advanced renal impairment or hypertension resistant to multiple drug treatment is more likely to be associated with fluid retention and often responds better to a loop diuretic than to a thiazide.
Potassium-sparing diuretics: Spironolactone, a specific aldosterone antagonist, is particularly effective for hypertension caused by primary hyperaldosteronism (Conn's syndrome). However, it can also be useful to treat resistant hypertension. Amiloride and triamterene (Ch. 14) are less effective than thiazides in essential hypertension.
The calcium channel blockers lower blood pressure principally by arterial vasodilation. For clinical uses, see Box 6.3. For further details, see Chapter 5.
Mechanism of action: Vascular smooth muscle possesses ATP-sensitive K+ channels (KATP) that are responsible for repolarisation of the cell (see also Ch. 8). Minoxidil opens KATP channels, causing an efflux of K+ which hyperpolarises the cell and leads to closure of voltage-gated Ca2+ channels and muscle relaxation (see also potassium channel openers, Ch. 5). Minoxidil is one of the most powerful peripheral arterial dilators.
Pharmacokinetics: Minoxidil is well absorbed from the gut and mainly metabolised in the liver. It has a short half-life (3–4 h).
Arterial vasodilation produces flushing and headache.
The reflex sympathetic nervous system response to vasodilation causes tachycardia and palpitation (which can be blunted by concurrent use of a β-adrenoceptor antagonist, ACE inhibitor or angiotensin II receptor antagonist).
Salt and water retention occur through stimulation of the renin–angiotensin–aldosterone system (Fig. 6.2). This, along with increased transcapillary pressure from vasodilation, can produce peripheral oedema, which can be reduced by the concurrent use of diuretics.
Hydralazine: Hydralazine is rarely used for long-term treatment of hypertension, but is an important treatment for hypertension in late pregnacy (pre-eclampsia), since it maintains uterine blood flow.
Mechanism of action: The mechanism of action of hydralazine is uncertain, but it may activate guanylyl cyclase, leading to the intracellular production of cGMP, and act as a nitric oxide donor. This will produce smooth muscle relaxation by mechanisms similar to those of organic nitrates (see Fig. 5.3).
Pharmacokinetics: Hydralazine undergoes extensive first-pass metabolism in the gut wall and liver, principally by N-acetylation. Some individuals, who are genetically determined slow acetylators (Ch. 2), require lower doses of hydralazine to achieve a clinical effect but are more susceptible to some of the unwanted effects.
Arterial vasodilation with reflex sympathetic activation produces tachycardia, flushing, hypotension and fluid retention.
A systemic lupus erythematosus (SLE)-like syndrome, which usually occurs after several months of treatment, is dose-related and is more common in slow acetylators. It resembles the naturally occurring disease but does not produce renal or cerebral damage and is slowly reversed if treatment is stopped. A positive antinuclear antibody is found in many individuals who do not develop the syndrome.
Because it must be given intravenously, the use of nitroprusside is limited to the emergency management of hypertensive crises.
Mechanism of action: Nitroprusside is a nitrovasodilator with a mechanism of action similar to that of organic nitrates (Ch. 5). It produces dilation of arterioles and veins, reducing both peripheral resistance and venous return.
Pharmacokinetics: Nitroprusside is given by intravenous infusion and its duration of action is less than 5 min. Metabolism to cyanide within red blood cells (by electron transfer from haemoglobin iron) terminates its effect. The cyanide is partly bound in the erythrocyte and partly liberated, when it is taken up by cells and inhibits intracellular cytochrome oxidase. Free cyanide is converted in the liver to less toxic thiocyanate but thiocyanate accumulates with prolonged infusion. Therefore, treatment with nitroprusside is usually limited to a maximum of 3 days.
Treatment of systemic hypertension
Morbidity and premature deaths associated with untreated hypertension are considerable (Fig. 6.4) and increase with advancing age. Therefore, treatment of older people with hypertension prevents more events in the short term than treating a similar number of younger people. However, early treatment will prevent vascular damage occurring in the younger individuals with hypertension – an important consideration since the vascular changes are not completely reversible once established.
The optimal target blood pressure in uncomplicated hypertension is a systolic blood pressure below 140 mmHg and a diastolic pressure (phase V Korotkoff sound) below 90 mmHg. When there is target organ damage or diabetes, a lower target of 130/80 mmHg is recommended, to minimise the risk of progressive vascular disease. There is no lower limit for blood pressure reduction, except in people with significant coronary artery disease. In this situation, lowering the diastolic blood pressure much below 70 mmHg may reduce coronary artery perfusion and increase the risk of myocardial infarction. Even if the target pressures cannot be achieved, any blood pressure reduction in severe hypertension will reduce the risk of complications. Treating isolated systolic hypertension (systolic >160 mmHg, diastolic <90 mmHg) in the elderly gives similar benefits to the treatment of diastolic hypertension in this age group.
It is rarely possible to correct the underlying cause of hypertension. Lifestyle modifications such as weight loss, restriction of alcohol and salt intake, and increasing exercise may be enough to lower the blood pressure satisfactorily in some individuals with mild hypertension. In people with more severe hypertension these measures can produce a substantial reduction in blood pressure but rarely restore it to normal values. It is important to advise all those with hypertension not to smoke, because smoking doubles the risk of cardiac and cerebrovascular events at any level of blood pressure.
The decision to treat hypertension with drugs should be determined largely by an assessment of the overall risk of complications in that individual. Drug treatment is usually started if blood pressure remains higher than the levels discussed above despite non-pharmacological approaches.
Drug regimens in hypertension
Lowering blood pressure by a very modest amount with drugs (even if the target levels described above are not achieved) produces a substantial (≈40%) reduction in the risk of stroke, as well as reducing the risk of heart failure by 50% and reducing the incidence of renal failure. Drug treatment also reduces the risk of coronary artery disease in the elderly by about 25%; evidence for a similar reduction in the young is less convincing, but this may reflect the short duration of the trials (up to 5 years). In people with evidence of LVH, regression of left ventricular mass during treatment of hypertension will reduce cardiovascular events by 60% compared to those in whom left ventricular mass is unchanged. ACE inhibitors, angiotensin II receptor antagonists and calcium channel blockers may be most effective in this regard.
Treatment regimens that are based on diuretics, calcium channel blockers, ACE inhibitors or angiotensin II receptor antagonists have generally shown equal efficacy in reducing vascular events. In contrast, β-adrenoceptor antagonists are less effective at preventing the complications of hypertension and are no longer recommended as first-line therapy. Treatment of hypertension should follow a ‘stepped care’ approach (Fig. 6.9). A single drug will achieve good blood pressure control in about one-third of people with hypertension. If the initial choice of drug fails to produce a sufficient reduction in blood pressure, then the first drug should be continued and a second drug should be added.
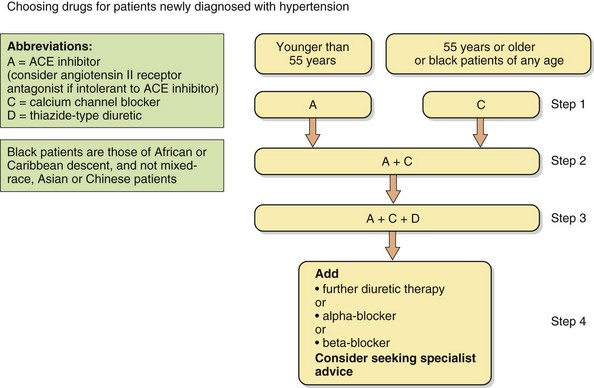
Fig. 6.9 The British Hypertension Society recommendations for combining blood pressure-lowering drugs.
Steps 1–4 reflect hypertension severity and risk, and initial treatment is influenced by patient age and ethnic origin.
The British Hypertension Society has endorsed a protocol for combining blood pressure-lowering drugs, which is based on their mode of action (Fig. 6.9). The underlying principle is that younger people with hypertension are more likely to have high plasma renin concentrations, and therefore a drug that suppresses the renin–angiotensin–aldosterone system is most likely to be effective. An ACE inhibitor is usually the drug of choice, or an angiotensin II receptor antagonist if an ACE inhibitor is not tolerated. Conversely, elderly people with hypertension are more likely to have ‘low renin’ hypertension and a calcium channel blocker or thiazide diuretic is more likely to produce a substantial reduction in blood pressure. Use of a drug that suppresses the renin–angiotensin system creates the equivalent of a low-renin state, whereas calcium channel blockers and diuretics increase plasma renin. This provides the rationale for combination therapy with drugs from complementary classes. The recommendations are based on the probability of achieving optimal blood pressure control and the evidence that the achieved blood pressure, rather than the means by which it was achieved, is important for improving outcome. However, the combination of an ACE inhibitor with a calcium channel blocker may be more effective at preventing cardiovascular disease than an ACE inhibitor with a thiazide diuretic. Consequently, a calcium channel blocker is preferred as the first-line treatment of older people with hypertension.
Optimal third-step therapy is an ACE inhibitor, calcium channel blocker and a thiazide or thiazide-like diuretic. Beta-adrenoceptor antagonists are only recommended as fourth-line treatment for uncomplicated hypertension. Several studies have shown that atenolol is less effective at preventing complications of hypertension than are other classes of drugs, and there is little information on outcome with other β-adrenoceptor antagonists.
Both thiazide diuretics and β-adrenoceptor antagonists increase the risk of developing new-onset diabetes, particularly when used together. This combination is not recommended for those who are at increased risk of glucose intolerance, such as people who are obese, those with a strong family history of type 2 diabetes or people of South Asian or Afro-Caribbean origin who have a higher risk of developing diabetes. In contrast, both ACE inhibitors and angiotensin II receptor antagonists reduce the risk of developing diabetes.
Resistant hypertension: If three drugs with complementary actions, taken in adequate dosage, are insufficient to control the blood pressure, then the person is said to have ‘resistant’ hypertension. There are several possible causes of apparently resistant hypertension. These include:
poor adherence to prescribed therapy (see Ch. 55),
‘white coat’ hypertension, which responds poorly to drug treatment,
secondary hypertension, most often caused by renal artery stenosis or Conn's syndrome,
concurrent drug use, such as a non-steroidal anti-inflammatory drug or a glucocorticoid, or excessive alcohol consumption,
intravascular volume expansion, due to antihypertensive drug therapy or renal failure.
Some people with resistant hypertension benefit from treatment with a loop diuretic rather than a thiazide, which will help if expansion of the plasma volume is contributing to drug resistance. Spironolactone can be effective when added to a thiazide diuretic, especially if there is evidence of increased production of aldosterone. An α-adrenoceptor antagonist or β-adrenoceptor antagonist are further options as the fourth drug for resistant hypertension. In men, minoxidil is a particularly powerful hypotensive agent, but excess hair growth limits its use for women. In a few individuals, treatment with five or more drugs may be necessary.
Additional treatment to reduce risk of vascular complications: Hypertension should be treated as part of a strategy to tackle all factors that increase the risk of cardiovascular disease. Advice on stopping smoking is important. A statin is recommended for primary prevention of cardiovascular disease in people with hypertension and a predicted risk of cardiovascular disease greater than 20% in the subsequent 10 years, or in those with diabetes mellitus (Ch. 48).
Hypertension in special groups
There may be reasons for selecting particular classes of drugs, particularly if there are other comorbid conditions that need treatment (Table 6.3). Ethnic differences may also be important: for example, people of Afro-Caribbean origin respond less well to ACE inhibitors or angiotensin II receptor antagonists as first-line therapy than do Caucasians, since at all ages they are more likely to have low-renin hypertension.
Table 6.3
Selection of antihypertensive drugs for coexisting conditions
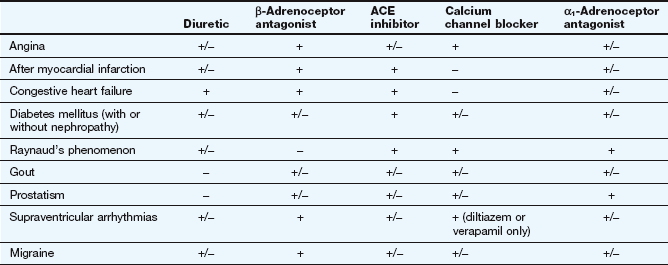
+, Treatment of choice; +/−, no obvious advantage/not preferred; −, usually contraindicated.
Malignant or accelerated hypertension: Immediate treatment is important for people with hypertension who have retinal haemorrhages and exudates or papilloedema. Rapid blood pressure reduction is potentially dangerous, since autoregulation of cerebral blood flow is reset at a higher level than normal. A sudden fall in perfusion pressure can lead to a profound drop in cerebral blood flow and ischaemic cerebral damage. Intravenous drugs should usually be avoided, and oral amlodipine is the most widely recommended treatment, which gradually reduces the blood pressure over 24 h or more.
Renal artery stenosis: ACE inhibitors or angiotensin II receptor antagonists usually produce an excellent reduction in blood pressure if hypertension is caused by renal artery stenosis, but they can lead to deterioration in renal function, especially if there are bilateral stenoses. Renal artery angioplasty with insertion of a stent is not currently recommended for either preservation of renal function or blood pressure control unless the stenosis is caused by fibromuscular dysplasia. In atherosclerotic disease, embolisation of small distal arteries in the kidney during angioplasty may cause worsening of renal function.
Diabetic nephropathy: ACE inhibitors and angiotensin II receptor antagonists appear to protect the kidney more than other classes of antihypertensive drug in diabetic nephropathy. In particular, they reduce progression from microalbuminuria to overt nephropathy, and can be more effective when combined than using either drug alone. The benefit from these classes of antihypertensive drugs is probably not simply due to blood pressure reduction. They produce afferent glomerular artery vasodilation by inhibiting the generation or action of angiotensin II, and therefore reduce glomerular perfusion pressure. Other complications of hypertension in people with diabetes are prevented equally well by other antihypertensive drugs.
Phaeochromocytoma: Phaeochromocytoma is a catecholamine-secreting tumour, often arising from the adrenal gland. Noradrenaline-secreting tumours most often lead to sustained hypertension, through vasoconstriction mediated by α1-adrenoceptor stimulation. Treatment should be started with an α-adrenoceptor antagonist (usually phenoxybenzamine) to prevent excessive vasoconstriction, followed by a β-adrenoceptor antagonist to block the arrhythmogenic effects of the catecholamines on the heart. Definitive treatment, whenever possible, is by surgical removal of the tumour.
Primary hyperaldosteronism: This can be caused by bilateral adrenal hyperplasia or, less commonly, by an adrenal adenoma (Conn's syndrome). The drug treatment of choice is spironolactone, to directly block the effects of aldosterone at its renal tubular receptor. If there is a tumour, surgical excision should be considered.
Pregnancy: There are two issues peculiar to pregnancy.
Pre-existing chronic hypertension. The risk of hypertension to mother and fetus is probably not great until the systolic blood pressure reaches 150 mmHg, or the diastolic blood pressure reaches 95 mmHg. Treatment of blood pressure at lower levels carries a risk of impairment of fetal growth. Many antihypertensive drugs should be avoided if possible in early pregnancy because they are teratogenic, or their potential for teratogenicity is not known (Ch. 56). The drugs with the best safety record for pre-existing hypertension in women who wish to become pregnant are methyldopa, nifedipine and labetalol. In the second trimester, the risk of fetal malformations is lower, but thiazide diuretics and pure β-adrenoceptor antagonists are usually avoided because they may retard fetal growth by reducing placental blood flow. ACE inhibitors or angiotensin II receptor antagonists may cause oligohydramnios (reduced amniotic fluid production), renal failure and hypotension in the fetus, or intra-uterine death, and should be avoided at all stages of pregnancy.
Pre-eclampsia. This usually occurs after 20 weeks of gestation. It presents as hypertension with oedema and proteinuria or hyperuricaemia in women whose blood pressure had previously been normal. If this condition is untreated there is a risk to the mother of convulsions, cerebral haemorrhage, abruptio placentae, pulmonary oedema and renal failure, and a risk to the fetus of severe growth retardation or even death. Once the diagnosis is established, bed rest is supplemented by antihypertensive drugs as described above for pre-existing hypertension in pregnancy. Labetalol given by intravenous infusion is favoured in severe pre-eclampsia.
Pulmonary hypertension
Pulmonary hypertension is most commonly found in people with chronic obstructive lung disease and some other lung disorders, where it arises as a result of destructive changes affecting the structure of the vascular bed. It also occurs with multiple small pulmonary emboli which silt up the peripheral pulmonary arteries and increase vascular resistance. However, some people develop increased pulmonary vascular resistance for unknown reasons (primary pulmonary hypertension, PPH), which has distinctive pathological findings of either the formation of plexiform vascular lesions or thrombotic arteriopathy. The most common presenting complaint in PPH is shortness of breath, although fatigue, chest pain, syncope, peripheral oedema and palpitation also frequently occur. The sustained increase in pulmonary vascular resistance leads to progressive right heart failure.
Drugs for treating pulmonary hypertension
Endothelin receptor antagonists
Mechanism of action: In PPH the expression of endothelin is increased in the pulmonary vasculature. Endothelin-1 is a powerful vasoconstrictor and smooth muscle mitogen which exerts its effects via two receptors, ETA and ETB. ETA receptors on vascular smooth muscle cells primarily mediate vasoconstriction and cell proliferation, while a smaller population of ETB receptors on endothelial cells mediate vasodilation via nitric oxide release; they are also responsible for clearance of endothelin from the circulation. Bosentan is an antagonist at both endothelin ETA and ETB receptors, while ambrisentan is selective for ETA receptors.
Prostaglandins
Epoprostenol is naturally occurring prostacyclin (prostaglandin I2, PGI2) and iloprost is a synthetic PGI2 analogue. Prostacyclin is a vasodilator that also inhibits platelet aggregation (Ch. 11), and both effects may be useful in the management of PPH. Iloprost is given by inhalation, but its short duration of action requires use every 2–3 h and it has a high incidence of flushing, headache, jaw pain and cough. Epoprostenol must be given by continuous intravenous infusion, so is only used when other treatments are ineffective.
Phosphodiesterase inhibitors
cGMP production in the pulmonary vasculature may be a protective mechanism against PPH. Oral phosphodiesterase type 5 inhibitors that inhibit breakdown of cGMP, such as sildenafil and tadalafil (Ch. 16), reduce pulmonary artery pressure, and the effects are additive to those of inhaled iloprost.
Management of pulmonary hypertension
Secondary pulmonary hypertension in chronic lung disease is most effectively managed by alleviating hypoxaemia when possible, using bronchodilators or long-term domiciliary oxygen therapy. There is no specific drug therapy. Chronic pulmonary embolic disease is treated by life-long anticoagulation, usually with warfarin.
PPH can be treated with drugs that reduce pulmonary vascular resistance. About 25% of people with PPH maintain a vasoactive pulmonary vascular bed (defined as a 20% decrease in pulmonary vascular resistance on acute challenge with a vasodilator). In this situation, treatment with a calcium channel blocker such as nifedipine will improve both symptoms and survival. However, most people with PPH show little evidence of vascular reactivity, and conventional vasodilators tend to produce excessive systemic hypotension before useful pulmonary vasodilation is achieved. For such individuals, treatment is considered with an endothelin antagonist, inhaled prostacyclin or a phosphodiesterase V inhibitor. All these drugs can improve symptoms and quality of life but have not been shown to improve survival. The role of combination therapy is under investigation.
True/false questions
1. Stretch of baroreceptors increases the afferent impulses to the vasomotor centre, resulting in a rise in blood pressure.
2. Nifedipine lowers blood pressure principally by arterial vasodilation.
3. Moxonidine stimulates imidazoline receptors in the medulla.
4. Propranolol lowers blood pressure by peripheral vasodilation.
5. Thiazide diuretics reduce sodium and water reabsorption in the distal convoluted tubule.
6. Thiazide diuretics reduce blood pressure in the long term at doses that do not cause diuresis.
7. Thiazide diuretics are the drugs of choice for treating pregnancy-related hypertension.
8. The potassium-sparing diuretics amiloride and spironolactone have the same mechanism of action in the distal tubule and early collecting duct.
9. Selective blockade of α1-adrenoceptors by prazosin increases noradrenaline release.
10. ACE inhibitors prevent the conversion of angiotensinogen to angiotensin I.
11. ACE inhibitors prevent the breakdown of bradykinin.
12. Minoxidil blocks K+ channels in smooth muscle cell membranes.
13. Nitroprusside can be given for up to 3 months.
14. Phosphodiesterase type 5 inhibitors are used in primary pulmonary hypertension (PPH).
15. Ambrisentan selectively blocks pulmonary vasoconstriction mediated by endothelin ETA receptors.
One-best-answer (OBA) question
Which of the following statements about antihypertensive drugs is the least accurate?
A Thiazide diuretics increase the risk of diabetes mellitus.
B β-Adrenoceptor antagonists are first-line therapy for hypertension.
C Blood pressure is not always lowered adequately by a single drug.
D Angiotensin II receptor antagonists selectively block angiotensin AT1 receptors.
E Nifedipine is more likely to cause reflex tachycardia than verapamil.
Extended-matching-item question
1. Choose which of the following drug classes A–D would be good choices for initial treatment of the people newly diagnosed with hypertension in the scenarios 1.1–1.3 below. Each drug may be selected once, more than once or never.
A An ACE inhibitor or angiotensin II receptor antagonist
B A non-selective β-adrenoceptor antagonist
1.1. A black, clinically obese male aged 75 years with a blood pressure of 150/100 mmHg and no other pathology
1.2. A white female aged 40 years with type 1 diabetes mellitus and a blood pressure of 150/100 mmHg
1.3. A woman 24 weeks pregnant with pre-existing chronic hypertension and a blood pressure of 150/100 mmHg
Case-based question
Mr AT, a 60-year-old man with type 2 diabetes mellitus, smoked 20 cigarettes a day. His plasma lipid levels were normal and there was no proteinuria. His ECG was normal. His height was 1.70 m and his weight 95.5 kg. He had no evidence of fluid retention or heart failure. Following treatment with a calcium channel blocker, his blood pressure was reduced from 175/110 mmHg but he remained hypertensive (155/95 mmHg) and he then had a small myocardial infarction. What changes in his therapy would you consider?
1. False. Baroreceptor impulses to the vasomotor centre reduce sympathetic outflow, enhance vagal outflow and lower blood pressure.
2. True. Calcium channel blockers act by opening L-type voltage-gated Ca2+ channels, and nifedipine, a dihydropyridine, is relatively selective for these channels in arterial smooth muscle. The non-dihydropyridines verapamil and diltiazem have additional cardiodepressant properties.
3. True. Moxonidine selectively stimulates imidazoline I1 receptors in the ventrolateral medulla; this decreases sympathetic outflow and increases vagal outflow, hence reducing blood pressure.
4. False. Propranolol lowers blood pressure by reducing cardiac output and by reducing renin production, but only β1-adrenoceptor antagonists with partial agonist activity at β2-adrenoceptors (such as pindolol) or those drugs with other hybrid properties (such as nebivolol) produce peripheral vasodilation.
5. True. All diuretics reduce sodium and water reabsorption in the renal tubule; thiazides act at the Na+/Cl− co-transporter (NCC) in the distal convoluted tubule.
6. True. The initial blood-lowering effect of thiazides is due to diuresis, but in the longer term they cause vasodilation by an unknown mechanism even at low doses.
7. False. Thiazide diuretics cause fetal growth retardation by reducing plasma volume and placental blood flow; methyldopa, nifedipine and labetalol are most often used in pregnancy.
8. False. Spironolactone competes with aldosterone for the mineralocorticoid receptor (MR), blocking its stimulation of the Na+/K+-ATPase pump and increased expression of the epithelial Na+ channel (ENaC); this reduces uptake of Na+ and loss of K+ from the tubule. Amiloride, however, directly blocks ENaC (see Ch. 14).
9. False. Prazosin dilates blood vessels by its selective blockade of α1-adrenoceptors; it does not block the presynaptic α2-adrenoceptors, and stimulation of these receptors to limit further noradrenaline release can still take place.
10. False. Angiotensinogen is converted to angiotensin I by renin. ACE effects the subsequent conversion of angiotensin I to angiotensin II.
11. True. ACE inhibitors reduce the breakdown of bradykinin, a potent vasodilator, and this action may contribute to their antihypertensive effects.
12. False. Minoxidil is a potassium channel (KATP) opener; it causes an efflux of K+ ions resulting in hyperpolarisation of arterial smooth muscle cells and vasodilation.
13. False. Nitroprusside is converted to cyanide and then to thiocyanate. The toxicity of these limits its use to 3 days for emergency management of some hypertensive states.
14. True. PPH is often associated with poor vascular reactivity, but vasodilation can be achieved by endothelin antagonists, phosphodiesterase type 5 inhibitors or prostacyclin.
15. True. Ambrisentan selectively blocks vasoconstriction mediated by ETA receptors; endothelial ETB receptors which mediate vasodilation via nitric oxide are unaffected.
OBA answer
Answer B is the least accurate.
A True. Thiazides increase the risk of new-onset diabetes, particularly when combined with β-adrenoceptor antagonists.
B False. β-Adrenoceptor antagonists are now fourth-line therapy in the British Hypertension Society guidelines, as they are less effective in reducing the risk of myocardial infarction and stroke than the other antihypertensive drugs.
C True. Satisfactory lowering of blood pressure is achieved with a single drug in only 30–40% of people with hypertension.
D True. Angiotensin II receptor antagonists block the vasoconstrictor and aldosterone secretory actions of angiotensin II at AT1 receptors; AT2 receptors are involved in vascular growth and are less affected by these drugs.
E True. Nifedipine selective dilates arterioles and may cause reflex tachycardia; this is unlikely with verapamil, which has negative chronotropic activity.
Extended-matching-item answers
1.1. Answer C (a calcium channel blocker) would be a good choice in this 75-year-old black male. The elderly and black people usually have low plasma renin concentrations, so an ACE inhibitor or angiotensin II receptor antagonist would be less effective. A thiazide diuretic or a β-adrenoceptor antagonist might increase the risk of diabetes in this obese man.
1.2. Answer A (an ACE inhibitor or, if poorly tolerated, an angiotensin II receptor antagonist) would be a good choice in a 40-year-old white female. A thiazide diuretic or a β-adrenoceptor antagonist may exacerbate the diabetes.
1.3. Answer C (a calcium channel blocker) would be an appropriate first choice in a pregnant woman with pre-existing hypertension. All the other drugs can cause unwanted effects on the fetus.
Case-based answer
Mr AT's blood pressure has not reached the target level (140/90 mmHg) and he has had a myocardial infarction. His blood pressure may be reduced further by introducing an ACE inhibitor. ACE inhibitors improve survival after a myocardial infarction, especially when there is left ventricular impairment. ACE inhibitors also protect the kidney in diabetic nephropathy and could be considered in this situation. The addition of a β-adrenoceptor antagonist could also be considered for additional long-term prognostic benefit, particularly if there is left ventricular impairment.
August, P. Initial treatment of hypertension. N Engl J Med. 2003;348:610–617.
Blood Pressure Lowering Treatment Trialists’ Collaboration. Effects of different blood pressure lowering regimens on major cardiovascular events: second cycle of prospectively designed overviews. Lancet. 2003;362:1527–1535.
Blood Pressure Lowering Treatment Trialists’ Collaboration. Effects of different regimens to lower blood pressure on major cardiovascular events in older and younger people: meta-analysis of randomised trials. BMJ. 2008;336:1121–1123.
Chiong, JR, Aronow, WS, Khan, IA, et al. Secondary hypertension: current diagnosis and treatment. Int J Cardiol. 2008;124:6–21.
Ernst, ME, Moser, M. Use of diuretics in patients with hypertension. N Engl J Med. 2009;361:2153–2164.
Ferrario, C, Levy, P. Sexual dysfunction in patients with hypertension: implications for therapy. J Clin Hypertens. 2002;4:424–432.
Grossman, E, Messerli, FH. Drug-induced hypertension: an uanappreciated cause of secondary hypertension. Am J Med. 2011;125:14–22.
Kaplan, N, Opie, LH. Controversies in hypertension. Lancet. 2006;367:168–176.
Moser, M, Setaro, JF. Resistant or difficult-to-control hypertension. N Engl J Med. 2006;355:385–392.
Moser, M, Feig, PU. Fifty years of thiazide diuretic therapy for hypertension. Arch Intern Med. 2009;169:1851–1856.
Ong, HT. β blockers in hypertension and cardiovascular disease. BMJ. 2007;334:946–949.
Oparil, S, Zaman, A, Calhoun, DA. Pathogenesis of hypertension. Ann Intern Med. 2003;139:761–776.
Shah, SJ. Pulmonary hypertension. JAMA. 2012;308:1366–1374.
Snow, V, Weiss, KB, Mottur-Pilson, C, et al. The evidence base for tight blood pressure control in the management of type 2 diabetic mellitus. Ann Intern Med. 2004;138:587–592.
Sowers, JR. Treatment of hypertension in patients with diabetes. Arch Intern Med. 2004;164:1850–1857.
Staessen, JA, Wang, J, Bianchi, G, et al. Essential hypertension. Lancet. 2003;361:1629–1641.
Vijan, S, Hayward, RA. Treatment of hypertension in type 2 diabetes mellitus: blood pressure goals, choice of agents, and setting priorities in diabetes. Ann Intern Med. 2003;138:593–602.
Williams, B. Recent hypertension trials: implications and controversies. J Am Coll Cardiol. 2005;45:813–827.
Yoder, SR, Thomberg, LL, Bisognano, JD. Hypertension in pregnancy and women of childbearing age. Am J Med. 2009;122:890–895.
Agarwal, R, Gomberg-Maitland, M. Current therapeutics and practical management strategies for pulmonary arterial hypertension. Am J Med. 2011;162:201–213.
Archer, SL, Michelakis, ED. Phosphodiesterase type 5 inhibitors for pulmonary arterial hypertension. N Engl J Med. 2009;361:1864–1871.
Badesh, DB, Abman, SH, Simmonneau, G, et al. Medical therapy for pulmonary arterial hypertension. Chest. 2007;131:1917–1928.
Macchia, A, Marchioli, R, Marfisi, RM, et al. A meta-analysis of pulmonary hypertension: a clinical condition looking for drugs and research methodology. Am Heart J. 2007;153:1037–1047.
McLaughlin, VV, McGoon, MD. Pulmonary arterial hypertension. Circulation. 2006;114:1417–1431.
Rich, S. The current treatment of pulmonary arterial hypertension: time to redefine success. Chest. 2006;130:1198–1202.