Anxiolytics, sedatives and hypnotics
There is considerable overlap in the pharmacology of drugs that have anxiolytic (anxiety-relieving) and hypnotic (sleep-inducing) properties. Compounds with sedative properties (moderating excitement and calming) at low doses often have hypnotic effects at higher doses. In addition, sedative drugs may have anxiolytic properties when used at doses that are too low to produce sedation. Compounds such as buspirone have been developed that have anxiolytic properties but do not sedate.
Anxiety disorders
Biological basis of anxiety disorders
Anxiety disorders are among the most common psychiatric syndromes and affect 15% of the general population at some time during their life. The clinical manifestations of anxiety are both psychological and physical. Anxiety is only pathological when it is inappropriate to the degree of stress to which the individual is exposed. A variety of anxiety disorders is recognised, of which the most common are generalised anxiety disorder, panic disorder, phobic disorder and obsessive compulsive disorder. Mixed anxiety and depressive disorder is the most common clinical presentation. Many anxiety syndromes present early in life, and tend to become chronic if untreated. They are often associated with substance abuse.
The symptoms vary among the anxiety disorders, but usually include apprehension, worry, fear and nervousness. Increased sympathetic nervous system activity frequently accompanies these feelings, causing sweating, tachycardia and epigastric discomfort. Sleep is often disturbed, with difficulty getting to sleep being a common feature. Physical symptoms can be prominent (somatisation) and disabling, and are sometimes difficult to interpret because anxiety can coexist with an underlying chronic physical condition.
Dysfunction of neurotransmission in the limbic region of the brain underlies the genesis of anxiety. The amygdala is a central part of the system that processes a fear stimulus and selects a response based on previous experience. Implementation of the response is through the locus coeruleus (autonomic and neuroendocrine responses) and nucleus paragigantocellularis (autonomic responses) in the brainstem, and through the hypothalamus. There are many neurobiological theories that attempt to explain the origin of anxiety disorders. These try to integrate our understanding of the neurochemical disturbances with genetic predisposition and environmental triggers. It is now thought that generalised anxiety disorder and major depression may share a genetic basis, and that expression of the clinical syndrome is determined by environmental factors. Structural changes in the neural pathways from the amygdala to the cortex have been identified in anxiety disorders, which may underlie hyperactive sensory processing of threat stimuli. In addition, the cognitive control mechanisms that terminate the emotional response to the sensory cues is deficient.
Decreased serotonergic (5-hydroxytryptamine, 5-HT) neurotransmission in the limbic system has been implicated in many anxiety syndromes. Overactivity of noradrenergic systems may also be important. Deficient inhibition of limbic neurotransmission by γ-aminobutyric acid (GABA) interneurons is found in many anxiety disorders, with reduced sensitivity of postsynaptic GABAA receptors. Excessive activity in excitatory glutamatergic neurons at N-methyl-D-aspartate (NMDA) receptors in the amygdala has also been implicated, and may be responsible for fear conditioning. Supersensitivity of receptors for peptide neurotransmitters such as cholecystokinin and neuropeptide Y may also occur. There is increasing evidence for the central role of brain-derived neurotrophic factor (BDNF) in modulating neural plasticity in anxiety states (see Fig. 22.2). BDNF is regulated by most of the neurotransmitters implicated in the genesis of anxiety states, and a decrease in BDNF correlates with anxiety and memory deficits.
In some anxiety syndromes there is excess secretion of corticotropin-releasing hormone (CRH), but a low plasma cortisol concentration and upregulation of glucocorticoid receptors. CRH is a neurotransmitter in the limbic system, and upregulation may occur from early adverse experiences, leading to conditioning of those with a genetic predisposition to anxiety disorder in later life.
Drug therapy for anxiety
Drugs used to treat anxiety are called anxiolytics.
Benzodiazepines
In addition to their anxiolytic effect, benzodiazepines have several other properties that are clinically useful. This section also considers drugs that are not used primarily for treatment of anxiety.
Mechanism of action and effects: Benzodiazepines act by potentiating the actions of GABA, the primary inhibitory neurotransmitter in the central nervous system (CNS). They act at a regulatory site closely linked to the GABAA receptor which mediates fast inhibitory synaptic neurotransmission (Fig. 20.1). GABA increases the influx of Cl− into the neuron, hyperpolarises the cell membrane and decreases cell excitability. Binding of a benzodiazepine to subunits of the receptor induces a conformational change in the GABA receptor that enhances its affinity for the neurotransmitter (Fig. 20.1). Benzodiazepines act only in the presence of GABA to enhance GABA-mediated opening of the ion channel; they have no direct action on the channel (Fig. 20.1).
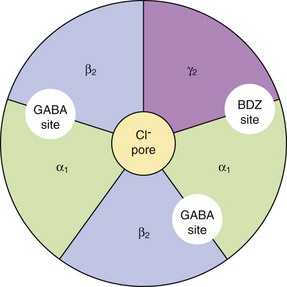
Fig. 20.1 The GABAA receptor.
The GABAA receptor consists of five transmembrane subunits configured from the 19 possible subunits that have been identified; thus many configurations of the GABA receptor exist, which vary in their sensitivity to benzodiazepines. A common configuration comprises two α1, two β2 and one γ2 subunit. Binding of GABA to the receptor at the interfaces of the α1 and β2 subunits mediates opening of the Cl− channel and an influx of Cl− ions, resulting in hyperpolarisation of the cell. This action is enhanced by drugs stimulating allosteric regulatory sites on the GABA receptor, distinct from the GABA-binding site. Diazepam, lorazepam and other ‘classic’ benzodiazepines (BDZs) bind at the interface of the α1 and γ2 subunits. Compounds such as zolpidem bind with high affinity for the α1-subunits and also enhance Cl− ion influx. The intravenous anaesthetics propofol and etomidate bind to β2- and β3-subunits (Ch. 17).
The GABAA receptor has α, β and γ subunits, arranged in a group of five (usually two α, two β and one γ, although only α and β are essential) around a central pore (see Fig. 20.1 and legend). There are many subtypes of each subunit, and therefore many receptor configurations that show differences in their regional distributions in the brain. Benzodiazepines bind between an α and γ subunit. The presence of an α1- or α5-subunit confers the sedative and amnesic properties of benzodiazepines, while both α2 and α3 appear to be involved in the anxiolytic and muscle relaxant effects. Anticonvulsant activity is conferred by several α subunits. The minority of GABA receptors with only α4 or α6 subunits do not bind benzodiazepines.
The increase in inhibitory neurotransmission produced by benzodiazepines has the following potentially useful effects:
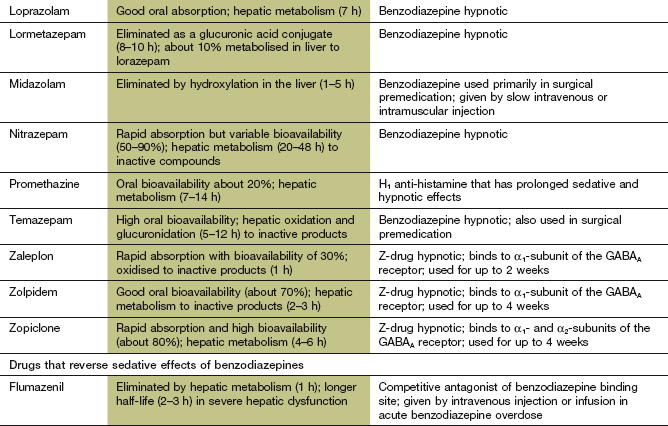
Pharmacokinetics: Benzodiazepines are well absorbed from the gut and their lipid solubility ensures ready penetration into the brain. The pharmacokinetics of individual benzodiazepines determines their major clinical uses. Benzodiazepines that are useful for inducing sleep (e.g. temazepam) are rapidly absorbed from the gut. This produces a fast onset of sedation, then sleep. Metabolism of short-acting benzodiazepines produces inactive derivatives. A brief duration of action is desirable for hypnotics, to avoid hangover sedation in the morning
Long-acting benzodiazepines, such as diazepam, are metabolised in the liver to active compounds (see Fig. 2.12) that contribute to their duration of action through relatively slow elimination from the body. Repeated dosing with long-acting compounds, such as diazepam, increases the risk of accumulation and a prolonged sedative effect. The anxiolytic properties of benzodiazepines are best exploited by using a compound with a long duration of action. Smaller doses can then be used to minimise sedation, and the rebound in anxiety symptoms that can occur between doses of a short-acting drug is avoided.
Diazepam, lorazepam and midazolam can also be given by intravenous injection to provide rapid sedation pre-operatively or before procedures such as endoscopy. Intravenous lorazepam and diazepam are also given for emergency treatment of generalised seizures and status epilepticus (Ch. 23). Long-acting benzodiazepines, such as clobazam, clonazepam, diazepam and lorazepam, are used in the prophylaxis of epilepsy (see Ch. 23).
 Drowsiness, which may cause problems with driving or operating machinery.
Drowsiness, which may cause problems with driving or operating machinery.
Confusion, especially in the elderly.
Paradoxical increase in aggression.
Potentiation of the sedative effects of other CNS-depressant drugs, such as alcohol. In overdose, such combinations can lead to severe respiratory depression. Flumazenil is a competitive antagonist of benzodiazepines and can be used in acute overdose to reverse respiratory depression (Ch. 53)
Tolerance to the therapeutic effects of benzodiazepines is common. There is widespread concern that their hypnotic effects are lost quite early, although there is little evidence to support this. However, rebound insomnia on withdrawal can perpetuate benzodiazepine use.
Dependence with physical and psychological withdrawal symptoms occurs during long-term treatment. The risk is highest in people with personality disorders, or a previous history of dependence on alcohol or drugs, and is more likely to occur if high doses of benzodiazepines are used. Restricting use to a maximum of 4 weeks will minimise the risk of dependence. With long-acting drugs, withdrawal symptoms may be delayed by up to 3 weeks after stopping. Anxiety is the most frequent symptom, while insomnia, depression and abnormalities of perception, such as altered sensitivity to noise, light or touch, also occur. More severe reactions such as psychosis or convulsions arise occasionally. Some withdrawal symptoms may resemble those for which the drug was originally prescribed, encouraging continued use. Gradual withdrawal of a benzodiazepine over 4–8 weeks is desirable after long-term use, although complete withdrawal may take up to a year. Lorazepam is a potent benzodiazepine with a relatively short duration of action that proves particularly difficult to stop because of the intensity of withdrawal symptoms that begin a few hours after cessation of treatment. Substitution with the longer-acting drug diazepam may be helpful before withdrawal is attempted. There are no proven treatments for reducing symptoms associated with withdrawal. Beta-adrenoceptor antagonists (Ch. 5) are sometimes helpful, or an antidepressant (Ch. 22) if there are depressive symptoms or panic attacks.
Azapirones
Mechanism of action and effects: Buspirone is a partial agonist at presynaptic 5-HT1A receptors, producing negative feedback to inhibit serotonin release. It has no effect on GABA receptors. Initial exacerbation of anxiety may occur, possibly caused by postsynaptic 5-HT1A receptor stimulation. The onset of the anxiolytic action of buspirone is slow, beginning after 2 weeks and reaching a maximum effect at approximately 4 weeks. The mechanism of action may involve gradual changes in neural plasticity (enhancement of neural performance or changes in neural connections; Ch. 22). Buspirone has no sedative action, and is ineffective for panic attacks.
Management of anxiety
If substance misuse is identified it should be treated first and may improve symptoms, while comorbid depression may require an antidepressant. Symptoms of anxiety, if mild, often respond to counselling or psychotherapy, using relaxation training or cognitive behavioural therapy without drug therapy.
Generalised anxiety disorder often requires long-term treatment, and there is now considerable evidence that antidepressants (Ch. 22) are useful in this situation. Selective serotonin reuptake inhibitors (SSRIs) such as sertraline are the treatment of choice, or a serotonin and noradrenaline reuptake inhibitor (SNRI) such as venlafaxine if an SSRI is ineffective. Antidepressants can initially exacerbate anxiety, and a benzodiazepine may be necessary for the first 2–3 weeks of treatment to prevent this. The optimal duration of antidepressant treatment in generalised anxiety disorder is uncertain, but similar treatment periods as for depression (Ch. 22) are usually recommended. Pregabalin, which increases inhibitory neurotransmission, is an effective alternative to antidepressants (Ch. 23).
Benzodiazepines can be considered as a short-term measure for anxiety to treat crises, since they have a rapid onset of action over 15–60 min. However, the potential for dependence should limit their use to a maximum of 4 weeks, and the dose should be gradually reduced after the first 2 weeks. Buspirone has similar efficacy to benzodiazepines, but the slow onset of action (3 days) makes it less versatile for managing short-term anxiety. In addition, anxiety that responds well to benzodiazepines often responds less well to buspirone, possibly due to a relative lack of effect of buspirone on somatic symptoms. Somatic symptoms of anxiety (e.g. tremor, palpitations) that are produced by overactivity in the sympathetic nervous system are often helped by a non-selective β-adrenoceptor antagonist such as propranolol (Ch. 5).
Social anxiety disorder responds to monoamine oxidase inhibitors (MAOIs; Ch. 22) better than to most other agents. Moclobemide is the treatment of choice, but phenelzine is also used. Phobic disorders usually need a different approach, and cognitive behavioural therapy is often most effective. Panic disorder is usually treated with tricyclic antidepressants or SSRIs, with MAOIs reserved for people who do not respond.
Insomnia
Defining insomnia is complicated by the considerable variability in the normal pattern of sleep. Most healthy adults sleep between 7 and 9 h per night, but much shorter or even longer periods can be normal. Insomnia is considered to be present if there is repeated inability to initiate or maintain sleep, despite adequate opportunity and time for sleep. There are three major categories of insomnia, defined by duration of symptoms (Table 20.1). Symptoms include sleep-onset insomnia (difficulty falling asleep, more common in younger people), frequent nocturnal awakening (difficulty maintaining sleep, more common in older people), early morning awakening (with difficulty getting back to sleep) and difficulty functioning in the daytime due to perceived poor sleep. Obstructive sleep apnoea is a common cause of sleep disturbance, affecting up to 10% of people who report insomnia.
Table 20.1
| Type of insomnia | Duration | Likely causes |
| Transient | 2–3 days | Acute situational or environmental stress (e.g. jet lag, shift work) |
| Short-term | <3 weeks | Ongoing personal stress |
| Long-term | >3 weeks | Psychiatric illness, behavioural reasons, medical reasons |
The reticular formation in the midbrain, medulla and pons is responsible for maintaining wakefulness. Activity in the reticular formation is dependent on sensory input via collateral connections from the main sensory pathways. Neurotransmitter systems involved in the regulation of sleep are complex. Cortical arousal is regulated by noradrenergic pathways from the locus coeruleus, cholinergic ascending tracts from brainstem nuclei, histaminergic neurons from the tuberomammillary nucleus and serotonergic neurons from the raphe nuclei. Hypocretins are important neuropeptide transmitters found in the lateral hypothalamus which, through connections with other hypothalamic and brainstem nuclei, promote wakefulness (see also narcolepsy, Ch. 22). Sleep is induced by neurotransmission by GABA, melatonin and galanin (a predominantly inhibitory neuropeptide) from the anterior hypothalamus, which inhibits the arousal neurons.
Sleep patterns
The two main types of sleep pattern are non-rapid-eye-movement (non-REM) sleep and rapid-eye-movement (REM) sleep. These sleep patterns occur in cycles (Fig. 20.2), with non-REM sleep varying between light sleep (stages 1 and 2) and slow-wave sleep (stages 3 and 4). Two-thirds of sleep is usually spent in stages 2–4, characterised by continuous or intermittent delta waves (slow waves) on the electroencephalogram. These deeper stages of sleep are the recuperative phase, while most dreaming occurs during the REM sleep periods. Increasing age is associated with more nocturnal awakening and longer periods of REM sleep.
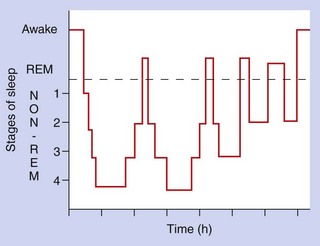
Drugs for treating insomnia (hypnotics)
Non-benzodiazepine hypnotics that modulate the GABAA/chloride channel
Mechanism of action and effects: Zaleplon, zolpidem and zopiclone (the so-called Z drugs) belong to different chemical classes but interact in a similar manner with the postsynaptic GABAA receptor on neuronal membranes. They bind to regulatory binding sites on the receptor that are close to, but distinct from, the benzodiazepine-binding site (Fig. 20.1). Like the benzodiazepines, they increase GABA-mediated Cl− influx into the cell, which inhibits neurotransmission. Zolpidem and zaleplon are selective for the α1-subunit in the GABA receptor. Zopiclone also acts on the α2-subunit of the GABA receptor. Although zopiclone also possesses anxiolytic and anticonvulsant activity, its short duration of action makes it unsuitable for these indications.
Pharmacokinetics: The Z drugs are rapidly absorbed from the gut, and are metabolised in the liver. They have short half-lives (1–6 h), which makes them well suited to their use as hypnotics.
Management of insomnia
Drugs play only a small part in the treatment of insomnia. Explanation of the normal variations in sleep patterns and avoidance of diuretics, drinks containing caffeine, cigarettes or alcohol in the hours before retiring can help. Eliminating excessive noise or heat in the bedroom, encouraging regular exercise in the day and minimising daytime napping may also be useful.
Hypnotic drugs are reserved for times when abnormal sleep markedly affects quality of life. The ideal hypnotic would induce good-quality prolonged sleep without disturbance of the normal sleep pattern. It should have a rapid onset of action, with no ‘hangover’ sedation in the morning, and should not produce tolerance or dependence. Few drugs come close to this ideal profile. Benzodiazepines reduce sleep latency (the time between settling down and falling asleep) and prolong sleep duration. However, they reduce the time spent in REM sleep, with more time spent in stage 2 sleep. Short-acting drugs (such as zolpidem) are preferred if there is delayed onset of sleep, and medium-acting drugs (such as temazepam or zopiclone) for those who wake in the middle of the night. Long-acting drugs are used to suppress daytime anxiety, but carry the risk of hangover sedation the following day. The Z drugs produce less disturbance of sleep ‘architecture’ than other drugs, having less effect on the amount of REM sleep while increasing the duration of deeper (slow-wave) sleep.
Hypnotic drugs should be used only for short periods and intermittently if possible, since tolerance to hypnotics frequently occurs after 2 weeks. If a benzodiazepine is used continuously for 4–6 weeks then rebound insomnia, caused by mild dependence, is common when the drug is stopped. Nevertheless, benzodiazepines are still widely used. The Z drugs carry a similar risk of dependence.
Of the other hypnotics, chloral derivatives and clomethiazole (see compendium) should usually be avoided. Compounds with sedative actions as a part of their therapeutic profile are sometimes used as hypnotics. For example, a sedative anti-histamine such as promazine (Ch. 39) can be helpful for children with somnambulism (sleep walking) or night terrors, but daytime sedation and weight gain can be a problem. Sedative tricyclic antidepressants (Ch. 22), such as amitriptyline, should be considered if there is an underlying depressive illness. If less-sedating antidepressants are used then short-term concurrent use of a benzodiazepine may be necessary while awaiting the onset of the antidepressant effect.
Melatonin is effective for insomnia caused by jet lag or shift work, but is ineffective for primary insomnia.
True/false questions
1. Benzodiazepines with a medium to long duration of action are useful for treating anxiety states.
2. Long-term use of benzodiazepines is recommended in anxiety states.
3. Potentiation of the CNS effects of benzodiazepines occurs with concurrent use of alcohol.
4. The CNS-depressant effects of benzodiazepines can be reversed with flumazenil.
5. Lower doses of benzodiazepines should be used in the elderly.
6. Buspirone is more sedative than temazepam.
7. Benzodiazepines used to treat anxiety should be administered for as short a time as possible.
8. Benzodiazepines have no effect on sleep patterns as measured by the duration of REM sleep.
One-best-answer (OBA) question
Choose the most correct statement from the following.
A Benzodiazepines act to potentiate the inhibitory actions of γ-aminobutyric acid (GABA) at its receptor.
B Withdrawal symptoms abate within 3 weeks of abruptly stopping diazepam.
C Barbiturates are the drugs of choice in patients with insomnia and anxiety.
D Buspirone decreases anxiety by acting at the GABA receptor site.
E If there is no response to one hypnotic, it is advisable to switch to another.
Case-based questions
Mrs FL is a 46-year-old mother of three who is finding it very hard to cope following the sudden death of her husband 3 months ago. She has returned to work but does not sleep properly, experiences occasional periods of anxiety during the day and feels that she is at risk of losing her job because tiredness and anxiety about her financial difficulties prevent her concentrating on her work.
A What drug might you prescribe to help Mrs FL's insomnia, and what factors determine your choice of this drug?
B How does your chosen drug work to reduce insomnia and anxiety?
C What potential unwanted effects and drug interactions should you warn Mrs FL about?
D Mrs FL returns 2 weeks later, saying that she regularly wakes at 4 a.m. and cannot get back to sleep; consider the advantages and disadvantages of changing her to a longer-acting drug or to another, ‘newer’ hypnotic.
E What are the problems associated with long-term use of benzodiazepines?
F What other options should be considered to help to manage Mrs FL's problems in the long term?
Extended-matching-item questions
Keeping in mind the equally important roles of psychological and psychiatric help, choose the most appropriate statement A–E for the next pharmacological course of action in the case scenarios 1 and 2 described below.
A Gradual tapering of the medication over many months.
B Gradual tapering of the medication over several days.
C Prescribing another course of the same benzodiazepine.
D Considering giving paroxetine.
E Immediate withdrawal of all medications.
Scenario 1. A 54-year-old woman has a history of anxiety. Seven years earlier she had taken regular lorazepam and for the last 3 years her doctor had been refilling prescription requests without reassessing the clinical need. There is no indication of depressive illness. The woman now wishes to stop her medication.
Scenario 2. You have been treating a woman aged 25 for a year. She has been having up to 10 intense panic attacks a month. At any time of day, she suddenly develops a peculiar and very strong feeling of being lightheaded, jumpy and being smothered. Her heart rate increases dramatically and the episodes come on so quickly and severely that she feels she might be dying. She then feels shaky, sweaty and unsteady. Each attack quickly reaches such intensity that she is often unable to continue work and needs to go home. She has been treated with intermittent courses of diazepam for a year without improvement.
1. True. Benzodiazepines used in anxiety, such as diazepam, have long-lived metabolites that contribute to the duration of action.
2. False. Dependence, tolerance and withdrawal symptoms occur with long-term continuous use of benzodiazepines.
3. True. Alcohol, older anti-histamines and barbiturates can potentiate CNS depression by benzodiazepines.
4. True. Flumazenil is a competitive benzodiazepine antagonist used in acute overdose.
5. True. Hepatic metabolism of benzodiazepines is reduced in the elderly, who are also more sensitive to their effects.
6. False. Buspirone has less sedative action than benzodiazepines such as temazepam.
7. True. Using the lowest possible dose of benzodiazepine for the shortest duration reduces the risk of tolerance and withdrawal effects.
8. False. Benzodiazepines affect sleep structure, with loss of REM sleep. Zolpidem has less effect than benzodiazepines.
OBA answer
A Correct. Benzodiazepines potentiate the entry of Cl− through the receptor-operated channel which is part of the GABAA receptor.
B Incorrect. With long-acting benzodiazepines withdrawal symptoms may take more than 3 weeks to appear.
C Incorrect. Barbiturates have been superseded as hypnotics because of unwanted effects, tolerance and dependence liability.
D Incorrect. Buspirone acts at presynaptic 5-HT1A receptors to reduce serotonin (5-HT) release.
E Incorrect. An alternative hypnotic is unlikely to work and switching between hypnotics is not good practice.
Case-based answers
A Mrs FL's insomnia and anxiety are a response to bereavement and might present fewer long-term problems than chronic ‘endogenous’ anxiety. Benzodiazepines and the newer hypnotics are safer than barbiturates. Nevertheless, the central concept in benzodiazepine therapy is to use the minimal effective dose for the shortest possible period. A short-acting benzodiazepine (e.g. temazepam) taken at night should help restore her sleep pattern and may improve her daytime tiredness. The relatively short half-life of temazepam (5–12 h) should minimise risk of sedation during the working day. However, if the daytime anxiety also warrants treatment, a long-acting benzodiazepine (e.g. diazepam) given at night may be the drug to choose. Only short or intermittent courses of treatment should be given. The anxiolytic buspirone is not sedative, but it is ineffective against panic attacks.
B Benzodiazepines are GABAA agonists that enhance GABAA-mediated inhibition of neuronal activity in the brain and spinal cord. Benzodiazepines bind to GABAA receptors at a site separate from GABA itself and increase the frequency of GABA-induced channel opening, enhancing Cl− entry into the cell and neuronal hyperpolarisation.
C Benzodiazepines are relatively free of serious unwanted effects if used correctly and are safe in overdose, but Mrs FL should be advised that they cause sedation and may interfere markedly with driving and operating machinery (worsened by interaction with alcohol, barbiturates and sedative anti-histamines). Other unwanted effects include headache, dry mouth, hypotension, anterograde amnesia, skin rashes and blood dyscrasias.
D Rebound wakefulness may indicate a need for a longer-acting benzodiazepine such as nitrazepam or diazepam, which may also help to reduce Mrs FL's daytime anxiety and panic attacks. Conversely, daytime sedation may interfere with driving and work, exacerbated by long-acting metabolites of these drugs. An alternative may be to prescribe buspirone; however, this requires 1–2 weeks for a response. Switching to a newer hypnotic such as zolpidem is unlikely to make a difference.
E Long-term use of a benzodiazepine is associated with dependence, manifested mainly as a withdrawal reaction, which may include rebound anxiety, tremor, nausea, irritability, anorexia and dysphoria. Together with rapid development of tolerance (especially to hypnotic action), these contraindicate benzodiazepine treatment for more than 3 weeks.
F In the longer term a course of antidepressants may be indicated. Mrs FL's recovery from bereavement may be aided by psychological counselling and support from family and employer.
Extended-matching-item answers
Scenario 1: Answer A. Withdrawal from long courses of benzodiazepines is difficult. She is liable to show withdrawal symptoms or return of the original complaints that determined the original prescription. Psychological and other forms of counselling may be advisable. Withdrawal should include gradual dosage reduction and anxiety management. Long-term psychological support is equally important for successful outcome, particularly for reducing the incidence and severity of post-withdrawal syndromes.
Scenario 2: Answer D. Continuing with a benzodiazepine is unlikely to improve matters after a year of treatment. The use of anxiolytics may be masking depression. An option might be to assess for depression and use a selective serotonin reuptake inhibitor (SSRI), such as paroxetine, licensed for the treatment of anxiety and panic disorders. General assessment is also recommended to rule out other disorders and non-pharmacological treatments should be considered.
Abramowitz, JS, Taylor, S, McKay, D. Obsessive compulsive disorder. Lancet. 2009;374:491–499.
Baldwin, D, Woods, R, Lawson, R, et al. Efficacy of drug treatment for generalized anxiety disorder: systematic review and meta-analysis. BMJ. 2011;342:d1199.
Ebert, B, Wafford, KA, Deacon, S. Treating insomnia: current and investigational pharmacological approaches. Pharmacol Ther. 2006;112:612–629.
Falloon, K, Arroll, B, Elley, CR, et al. The assessment and management of insomnia in primary care. BMJ. 2011;342:d2899.
Fricehione, G. Generalized anxiety disorder. N Engl J Med. 2004;351:675–682.
Gale, C, Davidson, O. Generalized anxiety disorder. BMJ. 2007;334:579–581.
Gottesmann, C. GABA mechanisms and sleep. Neuroscience. 2002;111:231–239.
Lader, MH. Limitations on the use of benzodiazepines in anxiety and insomnia: are they justified? Eur Neuropsychopharmacol. 1999;9(Suppl 6):S399–S405.
Lerch, C, Park, GR. Sedation and analgesia. Br Med Bull. 1999;55:76–95.
Michels, G, Moss, SJ. GABAA receptors: properties and trafficking. Crit Rev Biochem Mol Biol. 2007;42:3–14.
Morin, CM, Benca, R. Chronic insomnia. Lancet. 2012;379:1129–1141.
Roy-Byrne, PP, Craske, MG, Stein, MB. Panic disorder. Lancet. 2006;368:1023–1032.
Schenk, CH, Mahowald, MW, Sack, RL. Assessment and management of insomnia. JAMA. 2003;289:2475–2479.
Schneier, FR. Social anxiety disorder. N Engl J Med. 2006;355:1029–1036.
Szabadi, E. Drugs for sleep disorders: mechanisms and therapeutic prospects. Br J Clin Pharmacol. 2006;61:761–766.
Tyrer, P, Baldwin, D. Generalised anxiety disorder. Lancet. 2006;368:2156–2166.
Wilson, SJ, Nutt, DJ, Alford, C, et al. British Association for Psychopharmacology consensus statement on evidence-based treatment of insomnia, parasomnias and circadian rhythm disorders. J Psychopharmacol. 2010;11:1577–1601.
Young, C, Knudsen, N, Hilton, A, Reves, JG. Sedation in the intensive care unit. Crit Care Med. 2000;28:853–866.