Epilepsy
Pathological basis of epilepsy
Epilepsy affects up to 0.6% of the population and is characterised by recurrent epileptic seizures without any immediate provoking cause. Epileptic seizures are sudden, transient and usually unpredictable episodes of motor, sensory, autonomic or psychic disturbance triggered by abnormal neuronal discharges in the brain. The clinical manifestations depend on the site of the discharge.
 In partial or focal seizures the discharge starts in a localised area of the brain and may remain localised or may secondarily spread to affect the whole brain.
In partial or focal seizures the discharge starts in a localised area of the brain and may remain localised or may secondarily spread to affect the whole brain.
In generalised seizures the abnormal discharge affects the whole of the brain (Table 23.1).
Table 23.1
Simplified classification of epileptic seizures
| Seizure type | Characteristics |
| Partial (focal) seizures | |
| Simple partial seizures | Motor, somatosensory or psychic symptoms; consciousness is not impaired |
| Complex partial seizures | Temporal lobe, psychomotor; consciousness is impaired |
| Secondary generalised seizures | These begin as partial seizures |
| Generalised seizures | Affect whole brain with loss of consciousness |
| Clonic, tonic or tonic–clonic | Initial rigid extensor spasm, respiration stops, defaecation, micturition and salivation occur (tonic phase, ≈1 min); violent synchronous jerks (clonic phase, 2–4 min) |
| Myoclonic | Seizures of a muscle or group of muscles |
| Absence | Abrupt loss of awareness of surroundings, little motor disturbance (occur in children) |
| Atonic | Loss of muscle tone/strength |
| Unclassified seizures |
Identification of the type of seizure is important in the selection of the most appropriate therapy.
The origin of epilepsy is complex. For most people with epilepsy the initial focus of abnormal neuronal activity is structural damage in the brain such as that resulting from trauma, tumours, cerebrovascular disease or haemorrhage. Seizures can also be caused by metabolic disturbance, such as hypoglycaemia or alcohol abuse. In about 30% of cases of epilepsy where there is no identifiable structural or metabolic disorder (idiopathic epilepsy) there is an important genetic component.
Neurotransmitters and epilepsy
Coordinated activity among neurons depends on a controlled balance between excitatory and inhibitory influences on the electrical activity across neuronal cell membranes. Neuronal networks cooperate by oscillatory electrical activity between different parts of the brain. Generalised epilepsy involves a change from these oscillations to abnormally synchronised activity across large-scale neuronal networks, in particular involving both the cortex and subcortical structures such as the thalamus. Structural changes in neuronal networks in the brain are often found in people with epilepsy and this may provide the basis for generation of the abnormal discharges. These arise from focal lesions in the neocortex and limbic structures (especially hippocampus and amygdala) that promote formation of abnormal regional hyperexcitable circuits.
In healthy neuronal circuits, depolarising inward Na+ and Ca2+ ionic currents are mainly activated by excitatory glutamate N-methyl-D-aspartate (NMDA) receptors. Depolarisation is followed by repolarising outward K+ currents activated by GABAB receptors which act as a feedback inhibitory circuit in response to excitation. Influx of Cl− ions into the neuron produced by GABAA receptor activation hyperpolarises the cell and inhibits impulse generation. Neuropeptide Y is co-released with γ-aminobutyric acid (GABA) and potentiates the inhibition. Membrane-bound ATPase pumps contribute to maintenance of the correct resting membrane potential by active transport of ions across the cell membrane in the resting phase (Ch. 8).
An epileptic seizure probably arises from a localised imbalance between excitatory neurotransmission, principally mediated by glutamate, and inhibitory neurotransmission, mediated by GABA, which leads to a focus of neuronal instability. In some forms of epilepsy there may be a defect in the neuronal currents that results in incomplete repolarisation of the cell. This will leave the neuron closer to its threshold potential for firing and create a hyperexcitable state. Such instability could initiate the burst of firing that produces epileptiform activity. Once an electrical discharge is triggered, spontaneous repetitive firing of the focus is maintained by a feedback mechanism known as post-tetanic potentiation. The synchronisation of the electrical charge that is necessary to generate a seizure may also be enhanced by neural plasticity. Remodelling of the neural connections of an individual GABA neuron can lead to simultaneous hyperpolarisation of a large group of glutamatergic neurons. Neurogenesis can be triggered by seizures and potentiate the development of this type of circuitry, creating a group of hyperpolarised cells. Repolarisation activates currents that generate rebound excitation, producing a synchronised discharge. Generalisation of this discharge probably relies on increased synaptic connections from the excitatory cells.
Several inherited epilepsy syndromes have now been characterised at a cellular level, and arise from mutations of proteins involved in ion channel function. Reduction in the activity of membrane-bound ATPases linked to neuronal transmembrane ion pumps has also been found in the brains of people with primary generalised epilepsy. Ion channel dysfunction may therefore provide the basis for the genesis of many types of generalised seizures, but defective GABA-mediated inhibitory neurotransmission also appears to be a key factor. The genesis of partial seizures is less well understood. These circuits may be generated by disruption of glial cell function and changes in the neuronal microenvironment.
Antiepileptic drugs
Most antiepileptic drugs produce their main antiepileptic effects either by blockade of depolarising ion channels, or by enhancing the inhibitory actions of GABA. Many drugs have multiple sites of action, so the drugs below are grouped by their principal mode of action.
Sodium channel blockers
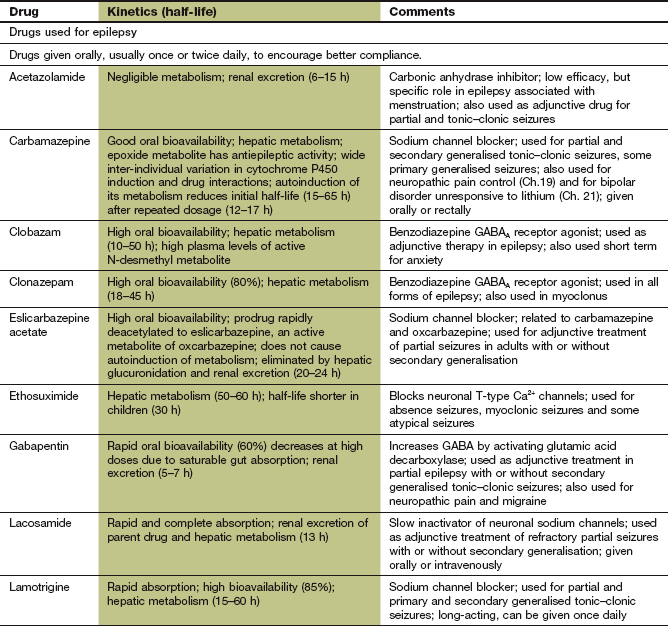
Carbamazepine, oxcarbazepine, eslicarbazepine
Mechanism of action and uses: Carbamazepine and oxcarbazepine are effective in most types of epilepsy, except for myoclonic epilepsy and absences, which they can exacerbate. Eslicarbazepine is the major active metabolite of oxcarbazepine, given as an acetate prodrug. Their mechanisms of action are incompletely understood but include:
use-dependent blockade of Na+ channels by stabilising them in the inactive state, which inhibits repetitive neuronal firing. This is the main mechanism of action,
attenuation of the action of glutamate at NMDA receptors, and reduced glutamate release.
Carbamazepine and oxcarbazepine are also used in the management of trigeminal neuralgia (Ch. 19), and carbamazepine in the management of diabetic neuropathy (Ch. 19) and bipolar disorder (Ch. 21).
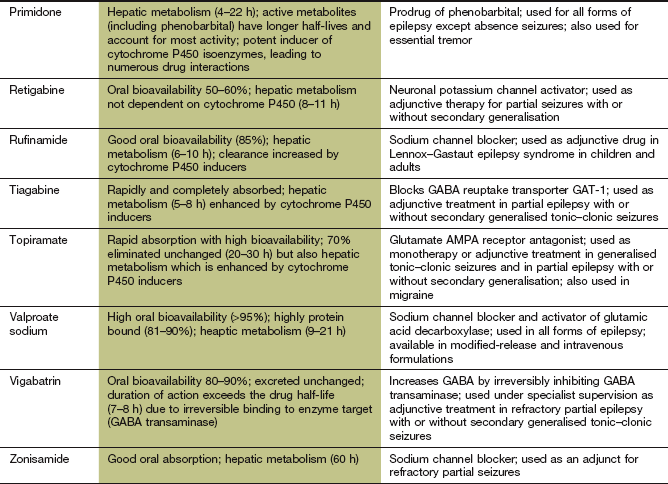
Pharmacokinetics: Carbamazepine is metabolised in the liver to an active epoxide metabolite. The half-life of carbamazepine is initially very long, at about 1.5 days, but decreases by about a half over the first 2–3 weeks of treatment because of ‘autoinduction’ of its own metabolism in the liver. Seizure control may then require an increase in dose. Transient unwanted neurological effects, which may occur in association with the peak plasma drug concentration when using the conventional formulation of carbamazepine, can be minimised by the use of a modified-release formulation.
Oxcarbazepine is well absorbed orally and is rapidly and extensively converted in the liver to active metabolites, including S-licarbazepine. Eslicarbazepine acetate is a prodrug converted rapidly to S-licarbazepine after oral administration, and is metabolised by glucuronidation and renal excretion.
Unwanted effects: The unwanted effects of oxcarbazepine and eslicarbazepine are less severe than those of carbamazepine.
Nausea and vomiting (dose-related, and especially early in treatment), constipation, diarrhoea, anorexia.
Rashes, especially transient generalised erythema but more severe reactions also occur. If a rash is produced by carbamazepine, oxcarbazepine can often be given without recurrence. Stevens–Johnson syndrome occasionally occurs with carbamazepine, and is more frequent in people with HLA-B*1502, who are most often of Han Chinese or Thai origin. Testing for this allele is recommended in such people before using carbamazepine.
Central nervous system toxicity leads to headache, double vision, dizziness, drowsiness, confusion or ataxia. These are most common early in treatment and are dose-related.
Transient leucopenia is common, especially early in treatment, but severe bone marrow depression is rare.
Hyponatraemia, caused by potentiation of the action of antidiuretic hormone on the kidney, can lead to confusion and decreased control of seizures. This may be more pronounced with oxcarbazepine.
Teratogenicity in the form of neural tube defects is common (see below).
Induction of hepatic CYP3A4 (Table 2.7) by carbamazepine can lead to drug interactions. The most common interaction is with the oral combined contraceptive pill (Ch. 45) and the dose of oestrogen should be increased to avoid failure of contraception. The metabolism of warfarin (Ch. 11) and ciclosporin (Ch. 38) are also accelerated. Inhibition of CYP3A4 by erythromycin, clarithromycin or diltiazem can increase the plasma concentration of carbamazepine. Interactions of carbamazepine with other antiepileptic drugs are discussed below. Oxcarbazepine and eslicarbazepine have little effect on cytochrome P450 and therefore have few drug interactions.
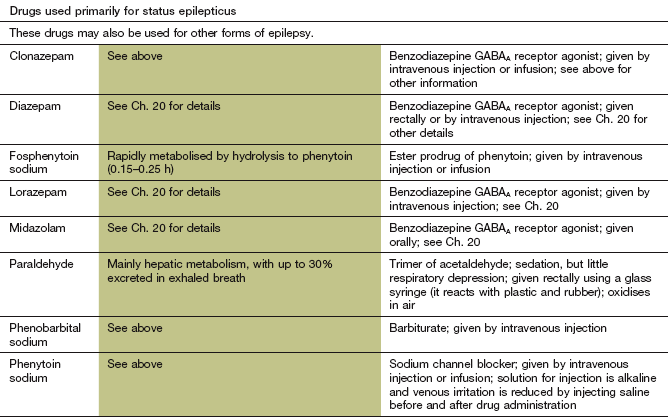
Phenytoin and fosphenytoin
Mechanism of action and uses: Phenytoin and its prodrug fosphenytoin have a broad spectrum of activity and are effective against all forms of epilepsy, except absences. They have several actions that may contribute to the antiepileptic activity:
use-dependent blockade of Na+ channels, which reduces cell excitability, is the main mechanism of action,
Phenytoin is sometimes used in the management of trigeminal neuralgia (Ch. 19).
Pharmacokinetics: Phenytoin is well absorbed from the gut, but this is a slow process. Slow intravenous injection can be used if a rapid onset of action is needed. Intramuscular injection of phenytoin should be avoided since absorption by this route is erratic and muscle damage can occur. Phenytoin is eliminated by hepatic metabolism, but metabolism is readily saturated so its elimination changes from first-order (linear) kinetics to zero-order (non-linear) kinetics (Ch. 2; Fig. 23.1). This occurs in some individuals at plasma drug concentrations below or near the lower end of the therapeutic range. A small change in dose then produces a large change in the plasma concentration (Ch. 2) and the elimination half-life is increased from about 12 h to almost 2 days. Plasma phenytoin concentrations are closely correlated to the clinical effect, and their measurement is useful as a guide to dosing. When the plasma concentration is close to or within the therapeutic range then any increase in dose should be small. Phenytoin is highly protein-bound (about 90%) and can be displaced from its binding sites by sodium valproate and salicylates, which briefly enhance the clinical effect of phenytoin to an unpredictable extent. The concentration of phenytoin in saliva reflects the free drug concentration in plasma, and measurement of the salivary concentration can be useful to guide dose adjustment in pregnancy or renal failure, or to avoid blood sampling in children.
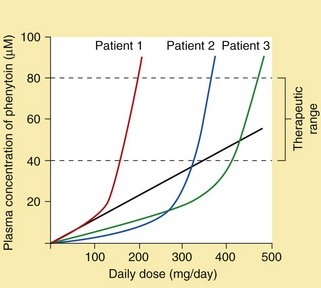
Fig. 23.1 Inter-individual variation in the plasma concentration of phenytoin at steady state in relation to the daily dose.
The figure illustrates the relationship between daily dose and plasma concentrations in three different individuals. The straight line illustrates the increase in plasma concentrations of phenytoin in Patient 1 that would occur if the metabolism were not saturated (i.e. first-order kinetics). Non-linearity within the desired therapeutic range due to saturation of metabolism can lead to difficulties in dosage adjustment.
Fosphenytoin is a prodrug of phenytoin that is only available for parenteral use. It can be given by intramuscular injection (absorption from this route is good, unlike that of phenytoin) or by intravenous infusion, and is completely metabolised to phenytoin.
Unwanted effects: Most unwanted effects of phenytoin and fosphenytoin are dose-related.
Nausea, vomiting, constipation, anorexia.
CNS effects: impaired brainstem and cerebellar function, producing confusion, dizziness, tremor, nervousness or insomnia. Nystagmus, blurred vision, ataxia and dysarthria are signs of overdosage.
Chronic connective tissue effects: gum hypertrophy, coarsening of facial features, hirsutism and acne. It is therefore usual to avoid phenytoin in young women or adolescents.
Rashes. Stevens–Johnson syndrome occasionally occurs with phenytoin, and is more frequent in people with HLA-B*1502, who are most often of Han Chinese or Thai origin. Testing for this allele is recommended in such people before using phenytoin.
Folic acid metabolism is increased by phenytoin, producing megaloblastic haemopoiesis, although anaemia with a macrocytic blood picture is rare.
Increased vitamin D metabolism can produce vitamin D deficiency; in rare cases this results in osteomalacia.
Teratogenicity with carbamazepine, including facial and digital malformations, occurs in up to 10% of pregnancies.
Induction of hepatic cytochrome P450 enzymes (Ch. 2) predisposes to several drug interactions. In particular, the metabolism of warfarin and ciclosporin are increased; interactions with other antiepileptic drugs are discussed below.
Lacosamide
Mechanism of action and uses: Lacosamide is used for adjunctive treatment of refractory partial seizures with or without secondary generalisation. Its major mechanism of action is enhancing the slow inactivation of neuronal Na+ channels, which stabilises cell membranes. It also binds to collapsing response mediator protein-2 (CRMP-2), which is involved in neuronal differentiation and axonal outgrowth, but it is not known whether this is important for the antiepileptic action.
Lamotrigine
Mechanism of action and uses: Lamotrigine has a wide spectrum of efficacy for partial and generalised seizures. It produces use-dependent inhibition of neuronal voltage-gated Na+ channels. Unlike carbamazepine and phenytoin, it selectively targets dendrites of pyramidal neurons that synthesise glutamate and aspartate and lamotrigine reduces glutamate release.
Lamotrigine is also used for prophylaxis of depression in bipolar disorder (Ch. 21).
Pharmacokinetics: Lamotrigine is well absorbed orally and is metabolised in the liver. The half-life is long (15–60 h).
Nausea, vomiting, diarrhoea, dry mouth.
CNS effects: drowsiness, headache, fatigue, dizziness, double vision and ataxia; tremor can be troublesome at high dosages.
Hypersensitivity syndrome with fever, rash, lymphadenopathy and hepatic dysfunction. This is more common if lamotrigine is used together with sodium valproate.
Rashes: some disappear despite continued treatment, but severe skin reactions, including Stevens–Johnson syndrome and toxic epidermal necrolysis, occasionally occur, particularly in children, following rapid dose escalation or with concurrent use of sodium valproate.
Zonisamide
Mechanism of action and uses: Zonisamide is used for adjunctive treatment of refractory partial seizures with or without secondary generalisation. Its mechanisms of action include:
GABA receptor agonists
Mechanism of action and uses: These drugs enhance the action of the inhibitory neurotransmitter GABA (Ch. 20). Clonazepam and clobazam are used orally for prophylaxis, usually as an adjunct to other drugs. Lorazepam, diazepam or clonazepam can be used intravenously to treat individual seizures, or if intravenous access is not available then rectal diazepam or buccal or intranasal midazolam can be used. Intravenous diazepam is formulated as an emulsion to reduce the incidence of thrombophlebitis.
Pharmacokinetics: These are long-acting benzodiazepines, discussed in detail in Chapter 20.
Unwanted effects: These are discussed in Chapter 20. Partial or complete tolerance to the antiepileptic action of benzodiazepines often occurs after about 4–6 months of continuous treatment.
Phenobarbital and primidone
Mechanism of action and effects: These drugs have a wide spectrum of activity and are effective in most forms of epilepsy, but unwanted effects limit their use. Phenobarbital is a barbiturate, and its major mechanism of action is activation of postsynaptic neuronal GABAA receptors (see also Ch. 20). This increases the duration of opening of the transmembrane Cl− channel associated with the receptor, and the neuronal membrane is therefore hyperpolarised and less likely to fire. In contrast to benzodiazepines, the GABA receptors are activated by phenobarbital independently of the presence of the inhibitory amino acid neurotransmitter, but phenobarbital will also potentiate the effect of GABA (Ch. 20). The action of primidone is due in part to its conversion to phenobarbital. Primidone has no advantage over phenobarbital and is generally less well tolerated. People with epilepsy who do not respond to phenobarbital or who tolerate it poorly are unlikely to benefit from primidone.
Pharmacokinetics: Oral absorption of phenobarbital is almost complete. Elimination is by hepatic metabolism and renal excretion. The half-life is very long, at about 4 days, but with considerable inter-individual variation. Primidone is well absorbed orally and is converted in the liver to two active metabolites, one of which is phenobarbital.
The plasma concentrations of phenobarbital and primidone relate poorly to the control of seizures; they are only useful as a guide to adherence to treatment. Control of seizures or unwanted effects should be used to determine dosages.
CNS effects: sedation, fatigue and memory impairment are common in adults; paradoxical excitement, confusion and restlessness can occur in the elderly, and hyperactivity can occur in children.
Folic acid metabolism is increased by phenobarbital, producing megaloblastic haemopoiesis, although anaemia with a macrocytic blood picture is rare.
Increased vitamin D metabolism can produce vitamin D deficiency; in rare cases this results in osteomalacia.
Tolerance to both unwanted and therapeutic effects occurs during long-term treatment.
Dependence with a physical withdrawal reaction is seen after long-term treatment.
Induction of hepatic cytochrome P450 enzymes (Ch. 2) leads to increased metabolism of phenobarbital itself and warfarin, ciclosporin and oestrogen (reducing the effectiveness of oral contraception). Interactions with other antiepileptic drugs are considered below.
GABA reuptake inhibitor
Mechanism of action and uses: Tiagabine is used as an adjunctive therapy for partial seizures with or without secondary generalisation. It is a potent inhibitor of GABA transporter 1 (GAT-1) and decreases glial and presynaptic neuronal uptake of the inhibitory amino acid GABA. Uptake by GAT-1 is the mechanism that normally limits the duration of action of GABA at its receptor. The action of tiagabine is relatively selective for the hippocampus and thalamus.
GABA transaminase inhibitor
Mechanism of action and uses: Vigabatrin is only used in combination with other drugs to treat epilepsy that is resistant to other drug combinations, or when they are poorly tolerated. It is effective in partial epilepsy with or without secondary generalisation, but its use is now restricted because of the unacceptably high risk of visual field defects (see below). It is, however, still useful for infantile spasms. Vigabatrin is a structural analogue of GABA and produces irreversible inhibition of GABA transaminase (GABA-T), the enzyme that inactivates GABA. The generalised increase in CNS concentrations of GABA inhibits the spread of epileptic discharges.
Pharmacokinetics: Vigabatrin is rapidly absorbed from the gut and is excreted unchanged by the kidney. Irreversible drug binding to its target enzyme GABA-T means that its duration of action is determined by the time required for GABA-T synthesis, rather than the half-life of elimination of the drug. GABA-T activity recovers to about 60% of baseline after 5 days. The efficacy of vigabatrin, therefore, is unrelated to the plasma drug concentration, and blood concentration monitoring is of no value.
CNS effects: sedation and fatigue, dizziness, nervousness, irritability, depression, impaired concentration, headache, blurred vision, diplopia, nystagmus and tremor.
Psychotic reactions, especially if there is a history of psychiatric disorder.
Severe peripheral visual field defects during prolonged use; they can arise from 1 month to several years after starting treatment, and are usually irreversible; regular monitoring of visual fields at 6-month intervals is recommended.
Drugs with a potential GABA-related mechanism of action
Mechanism of action and uses: Sodium valproate has a wide spectrum of antiepileptic activity, and suppresses the initial seizure discharge as well as the spread of seizure activity. It is effective for all forms of epilepsy. The mechanisms of action of sodium valproate are uncertain, but include:
potentiation of the effect of the inhibitory amino acid GABA, by enhancing the activity of glutamic acid decarboxylase (GAD), which converts glutamate to GABA.
use-dependent blockade of transmembrane Na+ channels, thus stabilising neuronal membranes.
The immediate antiepileptic effects may be due to extracellular actions on neuronal ion channels, but slow diffusion into neurons produces delayed intracellular effects. Inhibition of histone deacetylase (HDAC) by valproate modulates the transcription of multiple genes encoding signalling proteins and ion channels. The full benefit of treatment may not be apparent for several weeks. Valproate is also used for the management of neuropathic pain (Ch. 19) and bipolar disorder (Ch. 21) and the prophylaxis of migraine (Ch. 26).
Pharmacokinetics: Sodium valproate is well absorbed from the gut. Conventional-formulation tablets should be taken with food to reduce gastric upset. Valproate is highly protein-bound in plasma (90–95%) at low to moderate drug concentrations, but the proportion of free drug rises with increasing drug concentration. Valproate is highly ionised at physiological pH but is rapidly transported across the blood–brain barrier via an anion exchange transporter. Subsequent diffusion into and out of neurons is slow, partly explaining why the drug concentration in plasma does not correlate well with its therapeutic effect. The monitoring of blood concentrations is only useful to assess compliance.
Gastrointestinal upset: nausea, vomiting, anorexia, abdominal pain and bowel disturbance. These can be minimised by gradual dosage titration. Valproate may rarely cause pancreatitis, and serum amylase should be measured if symptoms such as abdominal pain or nausea and vomiting arise.
Weight gain caused by appetite stimulation.
Transient hair loss, with hair regrowth being curly.
Ataxia, tremor, confusion and, rarely, encephalopathy and coma. These can be minimised by slow dosage titration.
Thrombocytopenia or impaired platelet activity.
Severe hepatotoxicity can develop but is rare, and usually occurs in the first six months of therapy. This is most frequent in children under three years of age or in people with organic brain disorders who are receiving multiple drug therapy for seizures. Transiently raised liver enzymes are common but usually do not progress to more serious liver dysfunction.
Teratogenicity in the form of neural tube defects (see below).
Inhibition of hepatic cytochrome P450 enzymes, leading to interactions with other antiepileptic drugs (see below).
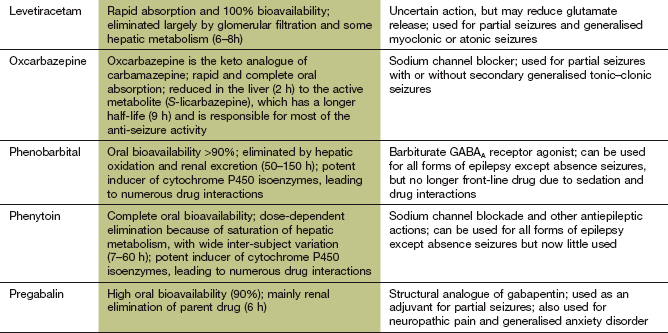
Gabapentin and pregabalin
Mechanism of action and uses: The major use of gabapentin and pregabalin is in partial seizures with or without secondary generalisation. Although designed as a structural analogue of GABA, gabapentin does not mimic GABA in the brain. The mechanisms of action of gabapentin and pregabalin are unclear, but probably include:
potentiation of the inhibitory effect of GABA by enhancing activity of GAD, which converts glutamate to GABA,
reduced synthesis of the excitatory neurotransmitter glutamate,
inhibition of P/Q-type voltage-gated Ca2+ channels in the neocortex and hippocampus. The drugs reduce Ca2+ entry into neurons, which may inhibit release of excitatory neurotransmitters such as glutamate.
Gabapentin and pregabalin are also used in the management of neuropathic pain (Ch. 19), and pregabalin for generalised anxiety disorder (Ch. 20).
Pharmacokinetics: Gabapentin is incompletely absorbed from the gut via a saturable transport mechanism, while pregabalin is better absorbed. Both drugs are excreted largely unchanged by the kidney with half-lives of about 6 h.
Glutamate receptor antagonist
Mechanism of action and uses: Topiramate is used alone or as an add-on treatment for drug-resistant partial or generalised seizures. Various mechanisms of action have been proposed:
antagonist activity at the α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA)/kainite subtype of receptor for the excitatory amino acid glutamate,
use-dependent blockade of neuronal Na+ channels,
enhancement of the action of GABA at GABAA receptors, although the mechanism of this interaction is unknown,
inhibition of carbonic anhydrase isoenzymes, producing multiple effects on transmembrane ionic fluxes.
Topiramate is also used for prophylaxis of migraine (Ch. 26).
Pharmacokinetics: Topiramate is rapidly absorbed orally and up to 70% is eliminated unchanged by the kidney, while the rest is metabolised in the liver. It has a long half-life (20–30 h).
Neuronal calcium channel blocker
Mechanism of action and uses: Ethosuximide is a drug of choice in absence seizures, and may be effective for myoclonic seizures, and tonic or atonic seizures. It is ineffective in other types of epilepsy. In absence seizures, T-type Ca2+ channels are believed to be responsible for generating excessive activity in thalamocortical relay neurons. Ethosuximide blocks these channels and prevents synchronised neuronal firing.
Pharmacokinetics: Absorption of ethosuximide from the gut is almost complete. Metabolism in the liver is extensive and the half-life is very long, at 2–3 days, although it is shorter in children. Plasma and salivary drug concentrations correlate well with control of seizures and can be used to monitor treatment.
Neuronal potassium channel opener
Mechanism of action and uses: Retigabine is used as an adjunctive (add-on) therapy for partial seizures with or without secondary generalisation. It is an activator of neuronal voltage-receptor K+ channels, which hyperpolarises neurons. It may also enhance GABA-mediated neurotransmission, and reduce the release of the excitatory neurotransmitter glutamate.
Drugs with other mechanisms of action
Mechanism of action and uses: Levetiracetam is used for adjunctive treatment of partial seizures with or without secondary generalisation. Its mechanisms of action remain uncertain, although levetiracetam binds to a protein (synaptic vesicle protein 2A) on the presynaptic neuronal plasma membrane and modulates release of excitatory neurotransmitters, such as glutamate. It produces selective inhibition of synchronised epileptiform burst firing and propagation of seizure activity in the hippocampus, without affecting neuronal excitability.
Interactions among antiepileptic drugs
Many antiepileptics affect hepatic drug-metabolising enzymes, especially cytochrome P450 isoenzymes (see Ch. 2, Table 2.7); therefore, drug interactions are frequent. Interactions when two or more antiepileptics are used together can have major clinical implications for seizure control and/or toxicity. However, the extent of the interaction is variable and unpredictable. Common interactions are listed below.
Carbamazepine is an enzyme inducer that often lowers the plasma concentrations of clobazam, clonazepam, lamotrigine, tiagabine, topiramate, sodium valproate, zonisamide and an active metabolite of oxcarbazepine.
Phenobarbital and primidone are enzyme inducers that often lower the plasma concentrations of clonazepam, lamotrigine, phenytoin, sodium valproate, tiagabine, zonisamide and an active metabolite of oxcarbazepine.
Phenytoin is an enzyme inducer and it often lowers the plasma concentrations of carbamazepine, clonazepam, lamotrigine, tiagabine, topiramate, sodium valproate, zonisamide and an active metabolite of oxcarbazepine.
Valproate inhibits hepatic drug metabolism which often increases the plasma concentrations of phenobarbital and lamotrigine, as well as those of an active metabolite of carbamazepine. Sodium valproate can displace phenytoin from plasma protein binding sites but also inhibits the metabolism of phenytoin, and the net result is an increase in the active free component.
Vigabatrin often reduces plasma phenytoin concentration by an unknown mechanism.
Management of epilepsy
Treatment of individual seizures
The initial management of a seizure involves positioning the person to avoid injury. Particular attention must also be given to maintaining the airways and ensuring adequate oxygenation. A correctable cause such as hypoglycaemia should be sought and treated, and intravenous thiamine given if alcohol abuse is suspected.
Prolonged or repetitive seizures (status epilepticus) usually require urgent parenteral drug treatment. Intravenous lorazepam is the drug of choice, with clonazepam as an alternative. Diazepam can be used, but it can cause thrombophlebitis and has a shorter duration of action owing to more rapid tissue distribution. A second dose of benzodiazepine can be given if necessary after 10 min. If intravenous access is not available, then midazolam can be given by the buccal or intranasal routes. Diazepam is available as a rectal solution, which may be particularly useful for children or for initial treatment out of hospital. Close observation for signs of drug-induced respiratory depression should be maintained after giving a benzodiazepine.
If there is no response after 25 min, or seizures recur, then a slow intravenous injection of phenytoin, or a more rapid injection of fosphenytoin or phenobarbital, should be given. If seizures are still not controlled with these measures, then full anaesthesia using thiopental or propofol (Ch. 17) with assisted respiration in an intensive care unit will be necessary.
Prophylaxis for seizures
A diagnosis of epilepsy requires two or more spontaneous seizures. After a single event, up to 80% of people will have a second fit within three years. If a predisposing cause cannot be identified and avoided (e.g. alcohol withdrawal, photosensitive epilepsy precipitated by viewing a television from too close a distance), drug treatment will usually be recommended after a second seizure, unless the seizures were separated by very long intervals or were mild. Occasionally, treatment will be recommended after a first fit, such as when there is structural brain damage.
Treatment should begin with a single drug, the choice depending on the type of epilepsy and relative toxicity of the drugs (Table 23.2). For generalised seizures or unclassifiable seizures, in the absence of factors that would lead to an alternative choice, sodium valproate is often recommended since it has the broadest spectrum of activity. Lamotrigine is usually considered the drug of first choice for partial seizures. The initial drug dose should be low, with gradual titration to minimise unwanted effects. If seizures continue, then the maximum tolerated dose should be taken. If seizures are not controlled with the first-choice drug it becomes more important to accurately identify the type of seizure. A second single drug should then be introduced and titrated to an adequate dose before the first drug is gradually withdrawn (Table 23.2). A single drug will usually control seizures in up to 90% of people with epilepsy, although this may not be achieved with the first drug chosen. However, if the first drug does not control the seizures, then the chance of a second single drug being successful is 13%, and with a third only 4%.
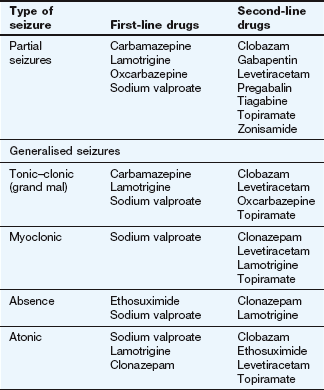
Refractory epilepsy can indicate poor adherence to treatment, inappropriate drug choice or dosage, or that the seizures are ‘pseudoseizures’ (non-epileptic attack disorder) rather than true epilepsy. Multiple drug treatment (initially with two first-line drugs, or a first- and a second-line drug) should be reserved for seizures that have not been controlled by treatment with two or three first- or second-line drugs given alone. Drugs like tiagabine, vigabatrin and zonisamide are usually used in combination with other agents.
Combination therapy at maximally tolerated doses does not control the seizures in some people with epilepsy, even when there is good adherence to treatment recommendations. Lack of seizure control is more frequent if the onset was at an early age, if there are generalised, atonic or absence seizures, or if there is underlying structural brain damage. Some data suggest that resistance can arise from overexpression of proteins that transport drugs out of the CNS, such as P-glycoprotein, but the evidence for this is conflicting. Alternatively, resistance may arise from genetic variation affecting targets for drug action. For temporal lobe epilepsy there is now good evidence that surgical treatment should be considered if more than two consecutive antiepileptic drugs fail to control the seizures. Surgery for other forms of epilepsy may provide some amelioration of seizure frequency.
It is not usually necessary to monitor plasma drug concentrations unless seizure control is poor, or if poor adherence or drug toxicity is suspected. Good seizure control will often be achieved at plasma drug concentrations that are below the accepted therapeutic range, and under such circumstances an increase in dosage would not be necessary. Conversely, people who continue to have seizures may need plasma drug concentrations above the standard therapeutic range to achieve seizure control, provided the drug is well tolerated. The only drug for which monitoring is of proven benefit for dosage adjustment is phenytoin, primarily because metabolism may be saturated at therapeutic doses and the kinetics become non-linear (Fig. 23.1). Adjustment of the dosages of carbamazepine or ethosuximide may be easier if the plasma concentration is known; however, for other drugs monitoring is only of value to confirm that the drug is being taken.
In the UK, a driving licence is revoked until the individual has been seizure-free for one year, or has suffered only nocturnal seizures for three years. Driving is not advisable during withdrawal of antiepileptic drugs, or for six months afterwards.
Once started, treatment should usually be continued for at least 2–3 years after the last seizure. Treatment should probably be lifelong if there is a continuing predisposing condition or the person wishes to continue to drive. If a decision is made to withdraw treatment then it should be gradual over at least 2–3 months to minimise the risk of rebound seizures. When several drugs are used, one should be withdrawn at a time.
Prophylaxis for seizures is often given for up to three months following neurosurgical procedures or head injury, particularly if there was a depressed skull fracture or an associated intracranial haematoma. Evidence that such routine use is beneficial is not secure.
Febrile convulsions occur commonly in infancy and usually do not lead to epilepsy or produce CNS damage. About 4% of children have them and they recur in about one-third. It is important to reduce pyrexia during subsequent febrile episodes, such as by removal of clothes and use of paracetamol (Ch. 29). Routine prophylaxis with antiepileptic drugs is not recommended, but rectal diazepam is sometimes given when a child who has previously had a febrile convulsion becomes pyrexial.
Antiepileptic drugs in pregnancy
No antiepileptic drug has a proven safety record in pregnancy and their use may carry a high risk of teratogenesis if the fetus is exposed in the first trimester (see Table 56.1). Fetal abnormalities are most frequent if more than one drug is used. Neural tube defects and other problems are particularly common with sodium valproate (malformations in about 10% of pregnancies), and to a lesser extent with carbamazepine and oxcarbazepine (2–4%). Developmental abnormalities also occur with phenobarbital, phenytoin, lamotrigine and topiramate, but there is too little information about many of the other drugs. There is also increasing evidence of language and neurocognitive defects in children born to mothers who are taking antiepileptic drugs.
Women of child-bearing age who are taking antiepileptic drugs should be given contraceptive advice. If they wish to become pregnant they should be counselled about the risk and offered antenatal screening during pregnancy, with α-fetoprotein measurement (to detect neural tube defects) and second-trimester ultrasound scanning. Folic acid supplements may reduce the risk of neural tube defects and should be recommended before and during pregnancy. It is important to advise a potential mother with epilepsy that the risks of uncontrolled seizures during pregnancy, to both her and the fetus, may be greater than the risk associated with drug therapy.
When the mother is taking carbamazepine, phenobarbital or phenytoin there is an increased risk of neonatal bleeding. Prophylactic vitamin K1 should be given to the mother from 36 weeks of pregnancy, and to the newborn immediately after birth.
True/false questions
1. Generalised seizures include tonic–clonic and absence seizures.
2. Absence seizures occur mainly in adults.
3. Partial seizures cause motor, sensory or psychic symptoms without loss of consciousness.
4. Generalised muscle contractions do not occur in partial seizures.
5. The excitatory amino acid glutamate is increased in some seizures.
6. There are currently no antiepileptic drugs that act by reducing excessive glutamatergic activity.
7. Antiepileptic drugs that stimulate γ-aminobutyric acid (GABA) receptors or enhance GABA stability act by inhibiting Na+ influx in neurons.
8. A major mechanism of antiepileptic drug action is the inhibition of Na+ channels.
9. The metabolism of carbamazepine diminishes with regular use.
10. Activation of K+ channels by retigabine hyperpolarises neurons.
11. Ethosuximide blocks neuronal Ca2+ channels.
12. The plasma concentrations of phenytoin increase in a linear manner with increasing dosage of the drug.
13. Vigabatrin is a first-line drug for the treatment of all types of epilepsy.
14. Tiagabine enhances GABA levels in synapses by reducing its reuptake.
15. The abrupt withdrawal of antiepileptics should be avoided.
16. The antiepileptic drug gabapentin is also used for neuropathic pain.
One-best-answer (OBA) question
Which of the following is the most accurate statement about antiepileptic drugs?
B The use of diazepam in epilepsy is confined to long-term prophylaxis in tonic–clonic seizures.
C Valproate induces drug-metabolising enzymes in the liver.
D The effectiveness of phenobarbital diminishes with time.
E The risk of teratogenicity can be reduced in pregnancy by combining antiepileptic drugs.
Case-based questions
Case 1: a 7-year-old boy was described as ‘dreamy’ by his mother. He was making slow progress at school and his mother and teachers commented that he could not concentrate and had frequent episodes of staring vacantly for a few seconds, then carrying on as normal. Following an electroencephalogram (EEG) a synchronised electrical discharge characteristic of an absence form of epilepsy was demonstrated.
A Which of the following would be suitable as a drug of first choice: phenytoin, phenobarbital, sodium valproate, ethosuximide?
B What are the major unwanted effects of the drug(s) you have chosen?
C If control of absence seizures is inadequate with your chosen antiepileptic, can combination therapy be given?
Case 2: a 19-year-old woman had a long-term history of epilepsy of the complex partial seizure type, which often gravitated to generalised seizures. For several years her epilepsy had been well controlled with a stable drug regimen. She now sought advice on contraception.
A What antiepileptic drugs might be effective in the type of epilepsy this woman has?
B What suitable options are available for contraception?
C What potential problems can arise if the woman takes the combined oral hormonal contraceptive?
D Would an injected progestogen contraceptive be worth considering?
E If the combined oral hormonal contraceptive were the chosen method, what strategies should be adopted to ensure its efficacy?
F Would the oral progestogen-only contraceptive be a suitable method of contraception?
1. True. Tonic–clonic seizures (formerly termed grand mal) and absence seizures (formerly petit mal) are two types of generalised seizure affecting the whole brain.
2. False. Absences, manifested by transient unawareness of surroundings and generally without motor disturbance, occur in children.
3. False. Simple partial (or focal) seizures do not cause loss of conciousness, but consciousness can be impaired in complex partial seizures and in secondary generalised seizures that arise from partial seizures.
4. False. In ‘Jacksonian epilepsy’ an epileptic focus in the primary motor cortex causes jerking localised to a specific group of muscles, which gradually spreads to involve many other muscle groups.
5. True. Glutamatergic over-activity is implicated in some types of epilepsy and may cause neuronal excitotoxicity.
6. False. Reducing glutamatergic activity may account in part for the action of antiepileptic drugs such as topiramate, which can block glutamate AMPA receptors, and others such as lamotrigine, levetiracetam and retigabine, which decrease glutamate release.
7. False. GABA hyperpolarises neurons by increasing the influx of Cl− ions.
8. True. Blockade of Na+ channels reduces repetitive neuronal firing and is a major mechanism of action of many antiepileptic drugs.
9. False. The sodium channel blocker carbamazepine induces its own metabolism (autoinduction), so its elimination accelerates with regular use.
10. True. The K+ channel activator retigabine hyperolarises neurons and modulates GABA and glutamate release.
11. True. The action of ethosuximide in absence seizures rests on its blockade of T-type Ca2+ channels in thalamocortical relay neurons.
12. False. Phenytoin exhibits first-order kinetics at low doses, but at higher doses it has zero-order kinetics because the liver drug-metabolising enzymes become saturated.
13. False. Vigabatrin is effective in all types of epilepsy, inhibiting the breakdown of GABA by GABA transaminase, but due to the risk of irreversible narrowing of the visual field it is reserved for people who are resistant to other drugs.
14. True. Tiagabine reduces GABA reuptake by glial cells and presynaptic neurons by inhibiting the GABA transporter GAT-1.
15. True. Withdrawal of an antiepileptic drug should be gradual over 2–3 months to prevent rebound seizures.
16. True. Gabapentin and pregabalin are inhibitors of glutamic acid decarboxylase used for epilepsy and neuropathic pain.
OBA answer
A Incorrect. Phenytoin causes hair growth (hirsutism); other unwanted effects include gingival hyperplasia, acne and facial coarsening.
B Incorrect. Diazepam is used as an adjunct to other treatments for prophylaxis but also alone in status epilepticus.
C Incorrect. Valproate inhibits drug-metabolising enzymes in the liver, increasing plasma concentrations of many other drugs.
D Correct. Tolerance to the therapeutic effects and unwanted effects of phenobarbital develops with time.
E Incorrect. The risk of teratogenesis is increased if more than one drug is given.
Case-based answers
A The absence seizures experienced by this boy should respond well to sodium valproate or ethosuximide; phenytoin and phenobarbital are ineffective in absence seizures.
B Sodium valproate causes nausea, reversible hair loss and weight gain. Uncommonly, liver damage can occur. Ethosuximide causes nausea, anorexia and headache.
C Monotherapy with ethosuximide or sodium valproate should be tried, and adherence with monotherapy checked, before combining drugs. Sodium valproate reduces the clearance of ethosuximide and may cause toxicity.
A A variety of antiepileptic drugs could be used by this woman with complex partial seizures. First-line drugs usually include carbamazepine, oxcarbazepine, lamotrigine or sodium valproate.
B Non-hormonal contraceptives such as barrier methods or intra-uterine devices, are effective and do not carry the risk of drug interactions. However, many women will want to use a hormonal method.
C Carbamazepine, phenytoin, phenobarbital and topiramate all induce liver enzymes that increase the metabolism of sex steroids and reduce the efficacy of oral contraceptives.
D The metabolism of injected medroxyprogesterone acetate (MPA) is affected less than that of sex steroids taken orally. The interval between MPA injections should nevertheless be reduced to 10 weeks. MPA may also reduce the incidence of seizures.
E Because oestrogen metabolism is enhanced by several antiepileptic drugs, it is recommended that, if one of these drugs needs to be prescribed, formulations containing a high concentration of oestrogen (at least 50 µg) should be used. Sometimes more than 100 µg oestrogen daily in split doses may be required to prevent breakthrough bleeding. The pill-free period can also be reduced. If any change in medication for her epilepsy is made, additional barrier methods of contraception should be used until medication is stabilised.
F The progestogen-only oral contraceptive would be unsafe, as its metabolism is increased.
Duncan, JS, Sander, JW, Sisodiya, SM, et al. Adult epilepsy. Lancet. 2006;367:1087–1100.
French, JA, Pedley, TA. Initial management of epilepsy. N Engl J Med. 2008;359:166–176.
Guerrini, R. Epilepsy in children. Lancet. 2006;367:499–524.
Kalviainen, R, Tomson, T. Optimising treatment of epilepsy during pregnancy. Neurology. 2006;67:S59–S63.
Lason, W, Dudra-Jastzebska, M, Rejdak, K, et al. Basic mechanisms of antiepileptic drugs and their pharmacokinetic/pharmacodynamic interactions: an update. Pharmacol Rep. 2011;63:271–292.
Marson, AG, Al-Kharusi, AM, Alwaidh, M, et al. The SANAD study of effectiveness of carbamazepine, gabapentin, lamotrigine, oxcarbazepine, or topiramate for treatment of partial epilepsy: an unblinded randomised controlled trial. Lancet. 2007;369:1000–1015.
Marson, AG, Al-Kharusi, AM, Alwaidh, M, et al. The SANAD study of effectiveness of valproate, lamotrigine, or topiramate for generalised and unclassified epilepsy: an unblinded randomised controlled trial. Lancet. 2007;369:1016–1026.
Perruca, E. Birth defects after prenatal exposure to antiepileptic drugs. Lancet Neurol. 2005;4:781–786.
Perruca, E, Tomson, T. The pharmacological treatment of epilepsy in adults. Lancet Neurol. 2011;10:446–456.
Pohlmann-Eden, B, Beghi, E, Camfield, C, et al. The first seizure and its management in adults and children. BMJ. 2006;332:339–342.
Rugg-Gunn, FJ, Sander, JW. Management of chronic epilepsy. BMJ. 2012;345:e4576.
Shorvon, S. The treatment of status epilepticus. Curr Opin Neurol. 2011;4:165–170.
Tatum, WO, IV., Liporace, J, Benbadia, SR, et al. Updates on the treatment of epilepsy in women. Arch Intern Med. 2004;164:137–146.
Torbjorn, T, Hiilesmaa, V. Epilepsy in pregnancy. BMJ. 2007;335:769–773.