Extrapyramidal movement disorders and spasticity
The group of nuclei in the area of the brain known as the basal ganglia (Fig. 24.1) are part of an integrative loop motor circuit (the cortico-basal ganglia-thalamo-cortical loop). This loop is intimately involved in the coordination of motor function. Nuclei in the basal ganglia feed neuronal output to the cortex and receive input from the cortex. Degeneration of vital neurons in the basal ganglia produces disordered regulation of neuronal activity and dysfunctional motor activity. Treatment for these disorders is directed at restoring the balance among the neurotransmitters in the basal ganglia.
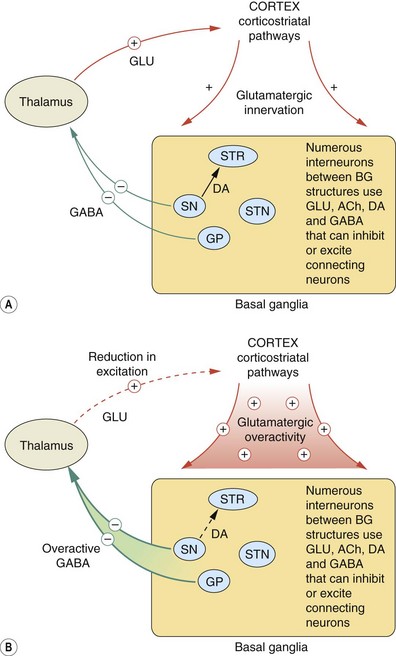
Fig. 24.1 The cortico-basal ganglia-thalamo-cortical loop.
(A) Main pathways connecting the basal ganglia, the thalamus and the cortex involved in movement; (B) indicates how they are disordered in Parkinson's disease relative to (A). In Parkinson's disease the pathological changes in the basal ganglia, principally the loss of dopaminergic activity required to initiate movement, result in increased inhibitory γ-aminobutyric acid (GABA) transmission in pathways from the substantia nigra and the globus pallidus to the thalamus; consequently there is excessive inhibition of thalamic-cortical brainstem motor networks. Hyperactivity in the glutamatergic cortico-basal ganglia pathways and also in cholinergic pathways within the basal ganglia exacerbates the predominant inhibitory influence of the basal ganglia on movement. BG, basal ganglia; DA, dopamine; GLU, glutamate; GP, globus pallidus; SN, substantia nigra; STN, subthalamic nucleus; STR, striatum; −, inhibition; +, stimulation; dashed line, reduced activity compared to normal function in control (A).
Parkinson's disease and parkinsonism
Parkinson's disease and parkinsonism arise from dysfunction in the part of the brain called the basal ganglia, which is involved in the control of movement. The basal ganglia system includes several nuclei such as the substantia nigra, the striatum, the globus pallidus and the subthalamic nucleus (Fig. 24.1). Between these nuclei there are many complex internal neuronal loop circuits that use glutamate, dopamine, acetylcholine or γ-aminobutyric acid (GABA) as neurotransmitters. In addition there are external neuronal loop circuits that integrate neurons outside of the basal ganglia with the internal circuits of the basal ganglia. For example, basal ganglia nuclei feed into and receive information from the cortex (via the cortico-basal ganglia-thalamo-cortical loop). The precise details of the complex interplay between the neuronal circuits are beyond the scope of this book and only general principles are given.
Parkinson's disease is a disorder characterised by a triad of:
There are two clinical subtypes of Parkinson's disease: the akinetic-rigid form and the tremor-dominant type. The trigger for the condition is unknown, but environmental toxins have been implicated in a small and specific gene mutations may be responsible in a small proportion of cases.
The underlying pathology involves loss of neurons in the substantia nigra pars compacta and deposition of intracytoplasmic Lewy bodies. Lewy bodies are complex structures that produce functional changes in dopaminergic neurons of the nigrostriatal pathway, possibly involving impaired handling of free radicals generated during dopamine metabolism. This ultimately leads to progressive neuronal death and degeneration of the nigrostriatal pathway (Fig. 24.1). More than 50% of substantia nigra pars compacta neurons must undergo degeneration before symptoms are apparent.
In Parkinson's disease, as a result of degeneration of the nigrostriatal dopaminergic pathways, there is destabilisation of the motor control networks. The basal ganglia normally provide a persistent inhibitory influence on the initiation of movement through GABAergic inhibition of the thalamus. When movement is required, the nigrostriatal dopaminergic pathways act to release the relevant thalamic and cortical motor systems from this inhibition. Denervation of the substantia nigra results in reduced stimulation of D1 and D2 receptors in the striatum. The consequence of this is excessive GABAergic inhibition of the thalamus and reduced glutamatergic activation of cortical systems (Fig. 24.1B shows changes in Parkinson's disease relative to normal function in Fig. 24.1A). At the same time, hyperactivity in the glutamatergic pathways that connect the cortex to the basal ganglia exacerbates the predominant inhibitory influence of the basal ganglia on movement. Cholinergic transmission in the basal ganglia is also enhanced in Parkinson's disease, contributing particularly to tremor.
In some conditions that have clinical similarities to Parkinson's disease, for example the Steele–Richardson–Olszewski (progressive supranuclear palsy) and Shy–Drager syndromes, the GABA neurons also degenerate, which explains the poor response of these conditions to treatment with dopamine-replacement therapy. Drugs that block striatal dopamine receptors, such as antipsychotic drugs (Ch. 21), can produce a parkinsonian syndrome which also responds poorly to dopamine-replacement therapy.
Drugs for Parkinson's disease
Targets for drug therapy in Parkinson's disease are:
Glutamatergic dysregulation has not yet provided suitable targets for drug therapy.
Dopaminergic drugs
Mechanism of action: Dopamine cannot be given to replace the underlying deficiency in the basal ganglia because it does not cross the blood–brain barrier. However, levodopa (L-DOPA) is the immediate precursor of dopamine that is carried by the large neutral amino acid transporter into the brain. It is taken up into dopaminergic neurons and converted to dopamine by L-aromatic amino acid decarboxylase, also known as DOPA decarboxylase (Ch. 2).
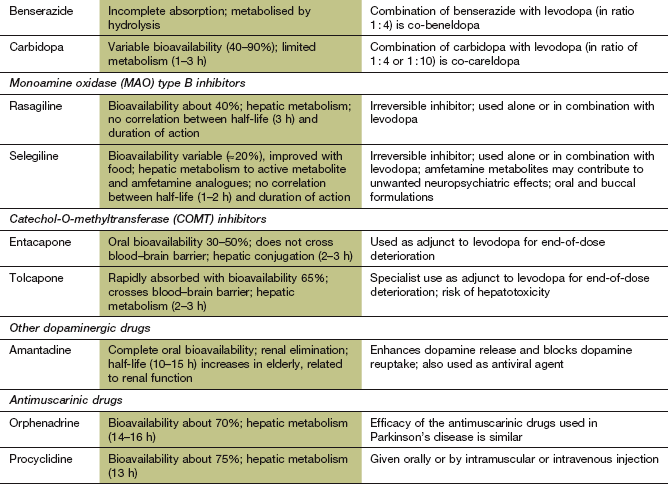
Pharmacokinetics: Levodopa is absorbed from the small intestine by an active transport mechanism for large neutral amino acids. A similar transport system transfers levodopa across the blood–brain barrier. When it is given alone, levodopa is extensively decarboxylated to dopamine in peripheral tissues such as the gut wall, liver and kidney. This reduces the amount of levodopa that reaches the brain to about 1% of an oral dose, while the peripheral dopamine that is generated produces unwanted effects. Therefore, levodopa is given in combination with a peripheral dopa decarboxylase inhibitor (with carbidopa as co-careldopa or with benserazide as co-beneldopa). Inhibition of the peripheral metabolism of levodopa increases the amount that crosses the blood–brain barrier to 5–10% of the oral dose. The dopa decarboxylase inhibitor does not cross the blood–brain barrier and therefore does not inhibit the required conversion of levodopa to dopamine by DOPA decarboxylase within the central nervous system (CNS).
The half-life of levodopa is short (about 1 h). In the early stages of Parkinson's disease synthesis and storage of dopamine in striatal neurons is sufficient to ensure a stable response despite infrequent doses of levodopa. This becomes less reliable as the disease progresses and more neurons are lost. Modified-release formulations of levodopa provide a more continuous supply of drug to the neurons and can be useful to treat ‘end-of-dose’ deterioration in symptoms. Transition from conventional levodopa to a modified-release formulation requires care, because the latter has a lower bioavailability which makes it difficult to estimate the equivalent dose.
 Peripheral formation of dopamine produces nausea and vomiting due to stimulation of the chemoreceptor trigger zone (CTZ) of the medullary vomiting centre which lies outside the blood–brain barrier. Nausea and vomiting are rarely dose-limiting but can be reduced by domperidone, a peripheral dopamine antagonist (Ch. 32). Peripheral dopamine may also cause arrhythmias, and vasodilation may cause postural hypotension and flushing.
Peripheral formation of dopamine produces nausea and vomiting due to stimulation of the chemoreceptor trigger zone (CTZ) of the medullary vomiting centre which lies outside the blood–brain barrier. Nausea and vomiting are rarely dose-limiting but can be reduced by domperidone, a peripheral dopamine antagonist (Ch. 32). Peripheral dopamine may also cause arrhythmias, and vasodilation may cause postural hypotension and flushing.
Excessive dopamine generation within the CNS can produce dyskinetic involuntary movements, especially of the face and neck, or akathisia (restlessness). Psychological disturbance can also occur, including hallucinations, anxiety, confusion, pathological gambling, increased libido and psychosis.
Dopamine receptor agonists
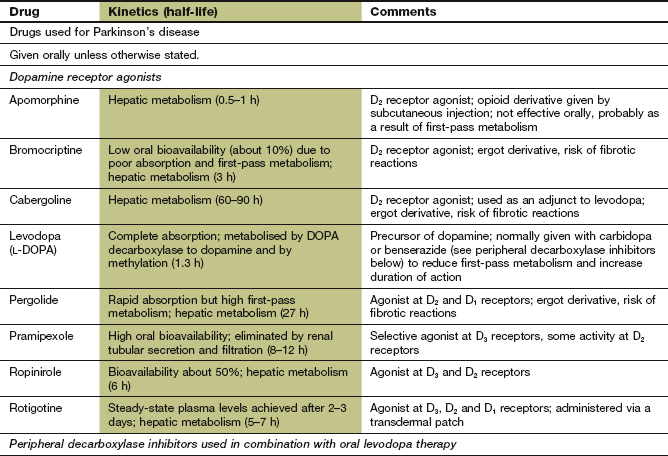
Mechanism of action: In contrast to levodopa, these drugs are direct agonists at central dopaminergic receptors (Ch. 4). They have a longer duration of action than levodopa. The orally active drugs act with varying patterns of selectivity on dopamine receptors of the D1-like family (D1, D5) and the D2-like family (D2, D3, D4); activity at receptors of the D2-like family is thought to underlie their therapeutic effect in Parkinson's disease (Fig. 24.2). Dopamine agonists that are structurally related to ergot alkaloids (Ch. 26), such as bromocriptine and pergolide, have been associated with fibrotic reactions (see below) and these drugs are now rarely used. The non-ergot drugs ropinirole, pramipexole and rotigotine are also used to treat restless legs syndrome.
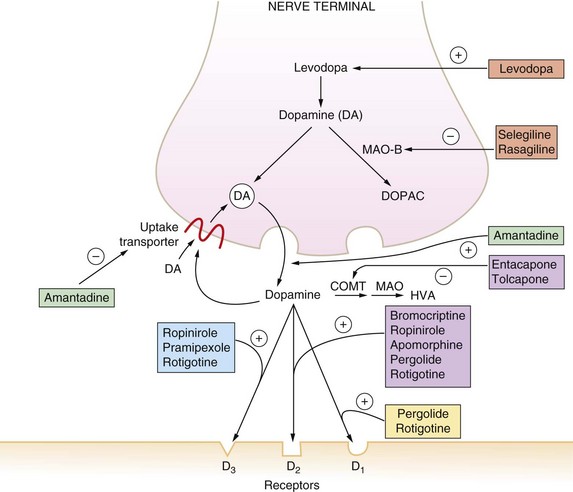
Fig. 24.2 The major effects of drugs on the dopaminergic nerve terminal in the CNS.
Drugs act at a number of different sites to amplify dopaminergic signalling. COMT, catechol-O-methyltransferase; DA, dopamine; DOPAC, 3,4-dihydroxyphenylacetic acid; HVA, homovanillic acid; MAO-B, monoamine oxidase B; +, stimulation; −, inhibition.
Pharmacokinetics: Pramipexole is well absorbed from the gut and is eliminated by hepatic metabolism. It has a half-life of 8–12 h. Ropinirole has a lower bioavailability and is eliminated by the kidneys, with a half-life of 6 h. Rotigotine is only formulated for delivery via a transdermal delivery patch to provide a more continuous supply of the drug.
Apomorphine is given parenterally by subcutaneous injection or continuous infusion, giving a very rapid onset of action. It has a short duration of action because of rapid hepatic metabolism.
Unwanted effects: Gradual dosage titration over several months may limit unwanted effects, which may include:
nausea, vomiting, dyspepsia, abdominal pain,
dizziness, nervousness, fatigue,
neuropsychiatric effects with hallucinations and confusion which are more frequent than with levodopa,
sedation, sudden onset of sleep,
skin reactions (with transdermal patches),
postural hypotension, peripheral oedema,
ergot-derived drugs such as bromocriptine and pergolide (now rarely used) may cause peripheral vasospasm, especially in people with Raynaud's phenomenon,
ergot-derived drugs may also cause pulmonary, pericardial and retroperitoneal fibrosis, and cardiac valve lesions,
respiratory depression with high dosages of apomorphine (an opioid derivative), which is antagonised by naloxone (Ch. 19).
Amantadine
Mechanism of action and uses: Amantadine is believed to act in Parkinson's disease by stimulating release of dopamine stored in nerve terminals and by reducing reuptake of released dopamine by the presynaptic neuron (Fig. 24.2). It is also a weak glutamate N-methyl-D-aspartate (NMDA) receptor antagonist. Its effectiveness tends to be short-lived because of the development of tolerance, but it can be beneficial for treatment of levodopa-induced dyskinesias. Amantidine is also used as an antiviral drug (Ch. 51).
Selective monoamine oxidase type B inhibitors
Mechanism of action and effects: These drugs are irreversible inhibitors of the enzyme monoamine oxidase (MAO), which is responsible for the intraneuronal degradation of monoamine neurotransmitters (Ch. 4 and Fig. 22.3). They are relatively selective at low doses for the isoenzyme (MAO-B) found in the striatum. This isoenzyme is distinct from MAO-A, which is also present in the gut wall and other peripheral tissues. Interactions with drugs and foods containing tyramine, which is a problem with conventional non-selective MAO inhibitor (MAOI) antidepressants (Ch. 22), do not occur with these MAO-B-selective drugs. Selective MAO-B inhibitors prolong the duration of action of dopamine and reduce the levodopa dosage requirement by about one-third, but only produce modest clinical benefit when used alone.
Pharmacokinetics: Selegiline and rasagiline have relatively low oral bioavailability (20–40%) and both drugs have short half-lives (1–3 h) due to rapid hepatic metabolism, but their duration of action is longer due to irreversible inhibition of their enzyme target. Selegiline undergoes metabolism in part to the L-isomers of amfetamine and metamfetamine, which have long half-lives and may contribute to unwanted neuropsychiatric effects.
Catechol-O-methyltransferase inhibitors
Mechanism of action and effects: Catechol-O-methyltransferase (COMT) is responsible for breakdown of between 10 and 30% of levodopa, both peripherally and in the CNS (Ch. 4), but, in the presence of a peripheral dopa decarboxylase inhibitor, COMT is responsible for most of the peripheral metabolism of levodopa. Inhibition of COMT doubles the half-life of levodopa (when it is used with a dopa decarboxylase inhibitor) and produces a 50% increase in the motor response to any given dose, and reduces end-of-dose deterioration. The dose of levodopa may therefore need to be reduced when a COMT inhibitor is started.
Pharmacokinetics: Entacapone and tolcapone are rapidly but variably absorbed from the gut. They are metabolised in the liver and have short half-lives of 2–3 h. Entacapone does not cross the blood–brain barrier, and therefore only inhibits peripheral COMT. Tolcapone crosses the blood–brain barrier and has greater efficacy but causes more unwanted effects.
Antimuscarinic drugs
Mechanism of action and effects
Drugs that block central muscarinic receptors (Ch. 4) help to restore the balance between CNS cholinergic and dopaminergic activity. They have little effect on bradykinesia, and are less effective than levodopa for treating tremor and rigidity.
Pharmacokinetics
Antimuscarinic drugs used for parkinsonian symptoms are well absorbed from the gut and undergo hepatic metabolism. They have half-lives in the range of 3–16 h.
Unwanted effects
These result from blockade of peripheral muscarinic receptors (Ch. 4) causing constipation, dry mouth, urinary retention and blurred vision. Reduced saliva production can be helpful in some people with Parkinson's disease, in whom sialorrhoea is a problem. Blockade of CNS muscarinic receptors can produce confusion, memory impairment and restlesness in the elderly. Tolerability of the antimuscarinic drugs varies, and changing to an alternative may be helpful if there are unwanted effects.
Management of Parkinson's disease and parkinsonian syndromes
Treatment is usually delayed in Parkinson's disease until symptoms affect quality of life. Levodopa (with a peripheral decarboxylase inhibitor) is still widely used for the initial treatment of Parkinson's disease, and a useful clinical response is achieved in about 70% of those treated. Levodopa is particularly useful for reducing bradykinesia. Poor responses to individual doses of levodopa may be due to interference with its absorption by a high-protein meal or by delayed gastric emptying, and the response may be improved by taking the drug before meals. There is increasing reluctance to use levodopa in the early stages of Parkinson's disease, since it is possible that pulsatile dopaminergic stimulation produced by oral doses of levodopa, with its short half-life, may increase the risk of dyskinesias and response fluctuations developing later in treatment. Various strategies to reduce this problem are under investigation.
If a dopamine receptor agonist, such as pramipexole, is used as initial treatment of Parkinson's disease there is significantly less risk of response fluctuation or dyskinesias with levodopa in advanced disease. Therefore a dopamine receptor agonist is often preferred for treatment of younger people. Levodopa is still preferred to a dopamine receptor agonist for older people (over 65 years) or those with cognitive impairment, because of its lower propensity to cause confusion.
Levodopa can produce disabling motor complications immediately on starting treatment, but they become progressively more likely with prolonged use. These complications are due to a change from a long-duration response to levodopa to a short-duration response as the population of dopaminergic neurons reduces and their dopamine storage capacity is limited. The duration of symptomatic benefit after each dose may be reduced (‘wearing off’), the dose may take longer to work (‘delayed on’) or it may sometimes fail to produce any improvement (‘no on’). The ‘on–off’ phenomenon with rapid swings between severe bradykinesia and toxic dyskinesias should be treated by a reduction in total levodopa dosage and by adding another drug with the aim of maintaining more stable delivery of levodopa to the neurons. Successful combinations include levodopa and a peripheral decarboxylase inhibitor with either an MAO-B inhibitor such as selegiline or a COMT inhibitor such as entacapone. Alternatively, a dopaminergic receptor agonist could be added. The rapid action of subcutaneous apomorphine can be invaluable to abort the ‘off’ state. Apomorphine is highly emetogenic and may need to be given with domperidone, an anti-emetic dopamine receptor blocker that does not cross the blood–brain barrier (Ch. 32). Domperidone should be taken 30 min before apomorphine, but it is often necessary to ‘load’ with domperidone for 24 h before starting apomorphine. Infusion of levodopa gel into the jejunum via a gastrostomy tube can improve motor function in late-stage disease. The addition of amantadine may reduce levodopa-associated dyskinesias.
High-frequency bilateral electrical stimulation of the subthalamic nuclei via implanted electrodes is used to switch off their activity. This strategy is effective for people who respond to levodopa but continue to have marked motor complications despite optimising therapy. It is used as an alternative to surgical ablation of the nuclei since it allows the clinician to vary the site and area of the stimulation with time. Depression may be a problem with this treatment. Surgical treatment is sometimes advocated in advanced Parkinson's disease. Severe tremor may respond to stereotactic thalamotomy or pallidotomy. Pallidotomy can also be helpful for severe dyskinesias.
Antimuscarinic agents are rarely used for idiopathic Parkinson's disease, but may be given for tremor that responds inadequately to levodopa. They can also be helpful in reducing excessive salivation.
Symptomatic treatment for a variety of associated symptoms may be necessary in Parkinson's disease. These include treatment of autonomic symptoms such as postural hypotension, vomiting, constipation, urinary frequency and impotence. Parkinsonian psychosis should be treated with an atypical antipsychotic drug, such as clozapine or quetiapine (Ch. 21).
Drugs improve symptoms and quality of life in idiopathic Parkinson's disease, but there is little evidence that they alter the underlying rate of neuronal degeneration. Levodopa therapy increases life expectancy, probably by reducing complications such as aspiration pneumonia. Several studies are underway to look at a potential neuroprotective effect of dopamine receptor agonists. However, neuroprotective strategies have so far proved disappointing.
Drug-induced parkinsonism (e.g. with antipsychotics; Ch. 21) responds poorly to levodopa because the causative drug occupies the D2 receptors. Parkinsonism resulting from antipsychotic drug therapy responds best to withdrawal of the drug. If this is not possible, then an atypical antipsychotic drug should be used and an antimuscarinic drug used to treat residual symptoms.
Other involuntary movement disorders (dyskinesias)
Dyskinesias are abnormal involuntary movement disorders that can present in several ways.
Tremor is a rhythmic sinusoidal movement caused by repetitive muscle contractions. It may be an exaggeration of the normal physiological tremor, or an abnormal movement such as is seen in Parkinson's disease.
Akathisia is a compulsive need to move, often in stereotyped patterns.
Chorea is irregular, unpredictable, jerky and non-stereotyped movement that involves several different parts of the body.
Myoclonus is rapid shock-like movements that are often repetitive.
Tics are rapid repetitive movements that can sometimes be controlled voluntarily, but with difficulty, for short periods.
Dystonias are sustained spasms of muscle contraction that distort a part of the body into a dystonic posture. The dystonia is often exaggerated by voluntary movement. Examples include spasmodic torticollis (twisted neck) and oculogyric crisis.
Movement disorders have numerous causes, and can be precipitated by drug therapy. For example, a tremor can be caused by lithium, sodium valproate, tricyclic antidepressants and sympathomimetics. Antipsychotic drugs (Ch. 21) are associated with a wide variety of movement disorders, ranging from acute dystonia to akathisia, and tardive dyskinesias (involving choreodystonic movements often of the face and mouth). The dopamine receptor antagonist metoclopramide (Ch. 32) can produce acute dystonias, especially in children and young adults, and occasionally tardive dyskinesias.
Some movement disorders have a genetic origin. An example is Huntington's disease, an autosomal dominant hereditary condition, which presents in adult life with progressive impairment of motor coordination, bizarre limb movements and dementia. The pathology is a loss of GABA inhibitory neurons within the neostriatum, which connect with the substantia nigra. There is a consequent reduction of inhibitory activity on dopaminergic cells in the substantia nigra and cells in the globus pallidus. Therefore, these cells generate uncoordinated discharges that produce bursts of excess motor activity.
Drug treatment
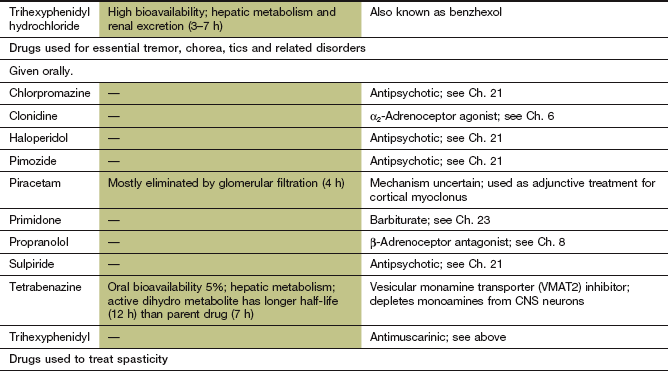
Mechanism of action: Tetrabenazine inhibits the vesicular monamine transporter 2 protein (VMAT2) in CNS neurons that transports newly synthesised monoamines from the cytosol into synaptic vesicles for storage and later release. The monamines, including dopamine, are instead degraded prematurely by MAO. Tetrabenazine is mainly used for Huntington's disease and related disorders.
Management of dyskinesias and dystonias
Treatment options depend on the cause. Some common strategies are listed below.
Withdrawal of a provoking drug. Symptoms may initially be exacerbated but then usually settle. Withdrawal dyskinesias usually respond to gradual drug discontinuation. Tardive dyskinesias associated with antipsychotic treatment may become worse on drug withdrawal, but then slowly improve over many months.
Exaggerated physiological tremor (e.g. anxiety tremor or tremor of thyrotoxicosis) may respond to a non-selective β-adrenoceptor antagonist such as propranolol (Ch. 5). Benign essential tremor is an action tremor that may be suppressed by a β-adrenoceptor antagonist or by primidone (Ch. 23). Gabapentin is an alternative second-line treatment (Ch. 23).
Tetrabenazine is sometimes effective for treatment of choreiform movements.
Many acute dystonias will respond to an antimuscarinic drug such as trihexyphenidyl given orally, or procyclidine given by intramuscular or intravenous injection for more severe symptoms.
Enhancing inhibitory GABA neurotransmitter activity with baclofen (see below), sodium valproate or clonazepam (Ch. 23) may help some dystonias.
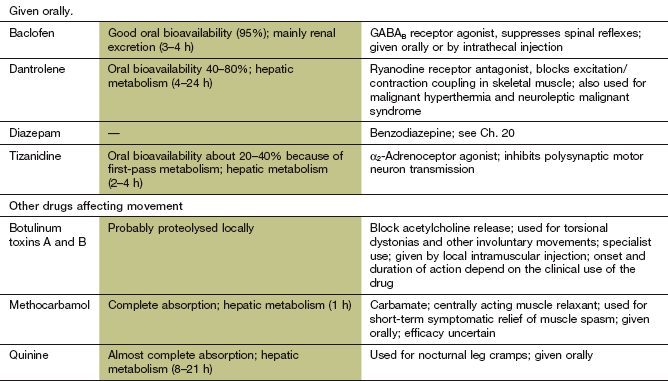
Botulinum toxin (Ch. 27), which impairs acetylcholine release from cholinergic nerve endings, can be injected into dystonic muscles to provide temporary relief by blocking transmission at the neuromuscular junction. Spread of the paralytic effect to adjacent muscles can cause problems; for example, dysphagia after injection of neck muscles for torticollis. There are two serologically distinct types of botulinum toxin used therapeutically; some people who are refractory to botulinum toxin A may respond to botulinum toxin B.
Spasticity
Spasticity is a state of sustained muscle tone or tension which is often associated with an increase in stretch reflexes. The increase in muscle tone can arise from continued spinal reflex activity in the absence of inhibitory input from the motor cortex, such as can result from a stroke or spinal cord injury or in multiple sclerosis. Spasticity in skeletal muscles is often associated with partial or complete loss of voluntary movement and can produce painful and deforming shortening of the muscle (contractures).
Drugs for spasticity
Diazepam (and other benzodiazepines) enhances spinal inhibitory pathways by facilitating GABA-mediated opening of Cl− channels (Ch. 20). The main disadvantage is sedation, a result of inhibitory activity in higher centres at the doses necessary for a spasmolytic action.
Baclofen
Mechanism of action: Baclofen is an analogue of GABA that inhibits excitatory activity at mono- and polysynaptic reflexes at the spinal level. Its precise mechanism of action is uncertain. However, it binds stereoselectively to GABAB receptors, and has an agonist action which hyperpolarises neurons by increasing K+ conductance. This inhibits reflex pathways by blocking presynaptic Ca2+ influx and thus reducing excitatory neurotransmitter release. Baclofen also has an analgesic effect, probably by inhibition of the release of substance P in pain pathways (Ch. 19).
Tizanidine
Mechanisms of action: Tizanidine is an α2-adrenoceptor agonist that increases presynaptic inhibition of motor neurons in the spinal cord via descending noradrenergic pathways. Inhibition is greatest in polysynaptic rather than monosynaptic pathways. Tizanidine has only 10% of the antihypertensive activity of the α2-adrenoceptor agonist clonidine.
Dantrolene
Mechanism of action and uses: Dantrolene is an antagonist at the ryanodine receptor (RyR1) that inhibits the release of Ca2+ from the sarcoplasmic reticulum of skeletal muscles (Ch. 5) and uncouples muscle excitation from activation of the contractile apparatus.
Dantrolene is also used for the treatment of malignant hyperthermia (Ch. 17) and as an adjunctive treatment in neuroleptic malignant syndrome (Ch. 21).
Management of spasticity
Mild spasticity can be useful, since the increased tone may provide support for a weak limb and should not be treated with drugs. Excessive spasticity following a stroke or in multiple sclerosis is most effectively prevented by adequate physiotherapy, and can be helped with orthoses that support the limb and correct deformities.
Drugs are most useful for deforming or painful spasticity, particularly if the person is not ambulant, since muscle hypotonia is a common problem in the drug therapy of spasticity. The primary sites of action of these agents are the spinal reflexes or the release of Ca2+ in the muscle fibre, rather than the neuromuscular junction. Drugs that block the neuromuscular junction (Ch. 27) are not used to treat spasticity, because their main effect would be a further loss of voluntary movement. Baclofen is usually the drug of first choice, and is most often required for spasticity associated with multiple sclerosis or spinal cord injury. Cannabis extract (Ch. 54) is used for spasticity in multiple sclerosis that does not respond to standard treatments. Intramuscular injection of botulinum toxin (see above, and Ch. 27) produces ‘chemodenervation’ and is used for severe focal spasticity. It produces an effect after 24–72 h, which is maximal after 2 weeks and lasts for 2–3 months. In severe spasticity that fails to respond to standard treatments, intrathecal baclofen infusion can provide relief.
True/false questions
1. In Parkinson's disease, there is abnormally low dopaminergic activity and increased GABAergic and cholinergic activity.
2. Symptoms of Parkinson's disease become apparent when approximately 25% of dopaminergic neurons have been lost.
3. Glutamate receptor antagonists are being investigated for use in Parkinson's disease.
4. Levodopa has a half-life of 24 h.
5. Treatment with levodopa may lead to dyskinesias and on–off fluctuations.
6. In early Parkinson's disease the dopamine receptor agonist ropinirole is as effective as levodopa.
7. Antimuscarinic drugs such as trihexyphenidyl have a low incidence of unwanted effects.
8. Pramipexole is a potent agonist at dopamine D3 receptors.
9. Some of the movement disorder in Parkinson's disease arises from abnormalities in non-dopaminergic innervated areas of the brain.
10. The chemoreceptor trigger zone (CTZ) is stimulated by peripheral dopamine generated from levodopa.
11. The monoamine oxidase inhibitor (MAOI) selegiline causes the ‘cheese’ reaction with ingestion of tyramine-containing foods.
12. Entacapone is a direct dopamine receptor agonist.
13. Tetrabenazine may cause depression.
14. Dantrolene is useful in spasticity as it reduces the Ca2+ release that contributes to contraction of skeletal muscle.
15. Baclofen enhances muscle tone.
16. Botulinum toxin has a duration of action of up to three months.
One-best-answer (OBA) questions
1. Which is the most accurate statement about drugs used for Parkinson's disease?
A Levodopa does not slow the progress of Parkinson's disease.
B Selegiline is a selective MAO-A inhibitor used for Parkinson's disease.
C More than 50% of oral levodopa enters the brain unaltered.
D Currently used drugs for Parkinson's disease selectively stimulate D1-type dopamine receptors.
2. Which one of the following adjunctive therapies will not increase dopamine levels in the brain when given together with levodopa?
3. Which one of the following unwanted effects is least likely to occur following levodopa administration?
Case-based questions
Case 1: a 75-year-old woman had been suffering from progressive symptoms of Parkinson's disease for 5 years. From the outset she had been treated continuously with levodopa, but problems had developed in controlling the symptoms with this drug.
A What is the cause of Parkinson's disease?
B What symptoms is this woman likely to have?
C Levodopa was given as co-beneldopa; what are the benefits of this formulation compared with levodopa alone?
D What difficulties can arise in controlling symptoms with co-beneldopa in the later stages of treatment?
Case 2: a married man aged 40 years was newly diagnosed as suffering from Parkinson's disease. His symptoms were tremor, bradykinesia, hypokinesia and rigidity, which were sufficiently mild that he was still able to carry out his work and pursue his hobbies. He was a security guard and had previously fought as a relatively unsuccessful professional boxer for 10 years, before retiring from the ring at the age of 35.
1. True. Loss of dopaminergic neurons creates abnormal activity within GABAergic and cholinergic pathways.
2. False. Symptoms usually develop when more than 50% of neurons have been lost.
3. True. There is over-activity of some glutamatergic neurons in parkinsonism.
4. False. The short half-life of levodopa (1–2 h) may contribute to end-of-dose deterioration; modified-release formulations provide a more continuous supply of the drug.
5. True. About 50% of people will experience these complications after 5 years treatment with levodopa.
6. True. In very early Parkinson's disease clinical trials up to six months' duration indicate ropinirole is as effective as levodopa.
7. False. Trihexyphenidyl (benzhexol) can cause minor unwanted effects but also severe confusion, particularly in the elderly.
8. True. Pramipexole is a potent D3 agonist, but also a full agonist at D2 receptors.
9. True. Tremor and rigidity in particular may occur because of transmitters other than dopamine.
10. True. The CTZ in the area postrema of the medulla is outside the blood–brain barrier and can be activated by peripheral dopamine.
11. False. Selegiline only inhibits MAO-B, leaving MAO-A intact to metabolise tyramine in cheese and other foods (see Ch. 22).
12. False. Entacapone is a peripheral inhibitor of catechol-O-methyltransferase (COMT), so it preserves levodopa for access to the brain and subsequent conversion to dopamine.
13. True. The risk of depression with tetrabenazene is consistent with its depletion of CNS monamines (see Ch. 22).
14. True. Dantrolene blocks Ca2+ release from sarcoplasmic reticulum so it reduces excitation/contraction coupling in skeletal muscle.
15. False. Baclofen improves spasticity by inhibiting excitatory synapses, probably acting at GABAB receptors.
16. True. Botulinum toxin is used in spasticity by intramuscular injection and inhibits local acetylcholine release for up to three months.
OBA answers
B Incorrect. Selegiline and rasagiline are selective inhibitors for MAO-B, not MAO-A.
C Incorrect. Only 1–2% of an oral dose of levodopa enters the brain in the absence of a peripheral decarboxylase inhibitor.
D Incorrect. Drugs for Parkinson's disease are thought to act mainly by agonism of the D2-like receptor family (D2, D3, D4).
E Incorrect. Carbidopa is a peripheral dopa decarboxylase inhibitor; it does not cross the blood–brain barrier.
A Entacapone inhibits peripheral catechol-O-methyltransferase (COMT), increasing the amount of levodopa crossing into the brain and its subsequent conversion to dopamine.
B Benserazide is a peripheral decarboxylase inhibitor that increases the amount of levodopa crossing into the brain and its subsequent conversion to dopamine.
C Carbidopa is a peripheral decarboxylase inhibitor that increases the amount of levodopa crossing into the brain and its subsequent conversion to dopamine.
D Procyclidine is a muscarinic receptor antagonist and will not affect dopamine levels.
E Rasagiline is an irreversible selective MAO-B inhibitor that will reduce the breakdown of dopamine within the brain.
A Dopamine is a neurotransmitter in the chemoreceptor trigger zone and stimulates nausea and vomiting.
B Dopamine can stimulate β-adrenoceptors in the heart, increasing the likelihood of arrhythmias.
C Orthostatic hypotension is common, particularly in the elderly.
D Dopamine may increase myocardial contractility, but without affecting the heart rate.
E Excessive CNS dopamine concentrations are associated with involuntary movements.
Case-based answers
A Parkinson's disease results from degeneration of more than 50% of dopaminergic neurons in the substantia nigra. The cause of this degeneration is unknown but hypotheses include the actions of reactive oxygen species, neurotoxins, immune disturbances and specific gene mutations. The consequent neurochemical disturbances include over-activity of cholinergic, GABAergic and glutamatergic pathways. Current therapies aim to supplement dopaminergic activity or decrease cholinergic activity.
B People with Parkinson's disease have akinesia, rigidity and tremor, possibly from inhibition of the motor cortical system, whereas the descending inhibition of the brainstem locomotor areas may contribute to abnormalities of gait and posture.
C Levodopa is the immediate precursor of dopamine and is transported into the CNS by the large neutral amino acid transporter and converted into dopamine, while dopamine itself cannot cross the blood–brain barrier. Levodopa causes nausea and vomiting because it is also metabolised to dopamine in the periphery, which activates the chemoreceptor trigger zone (CTZ). Co-beneldopa is a combination of levodopa with benserazide, a peripheral decarboxylase inhibitor, which prevents the breakdown of levodopa to dopamine in the periphery. The peripheral dopamine receptor antagonist domperidone can also be used to protect against nausea and vomiting.
D Levodopa remains the most effective treatment for Parkinson's disease but there considerable debate about when to start levodopa therapy. There is no convincing evidence that levodopa accelerates neurodegeneration, and survival is reduced if treatment is delayed until greater disability is present. In time, and despite long-term treatment with levodopa, there is an increasing incidence of dyskinesias and on–off fluctuations of effect, although most people continue to derive benefit throughout the duration of their illness. At the end of five years of treatment approximately 50% of those treated will be experiencing reduced effectiveness with levodopa. These motor fluctuations can be a result of unpredictable pharmacokinetic changes, such as unpredictable absorption across the blood–brain barrier or delayed gastric emptying, or because further loss of dopaminergic neurons during disease progression further reduces neuronal storage capacity for dopamine.
E Resolving these problems is highly individual, but dyskinesia and on–off effects may be helped by dosage adjustment (up or down), by shortening the dose interval or by using modified-release formulations. An antimuscarinic drug can be given with levodopa and may be useful in the treatment of tremor, but could cause confusion and hallucinations, particularly in this elderly person. Other drugs which inhibit dopamine metabolism can also be introduced. The selective MAO-B inhibitors selegiline or rasagiline are far more effective as adjuncts to levodopa than as monotherapy. The COMT inhibitor entacapone reduces the peripheral breakdown of levodopa and dopamine and prolongs the benefits of levodopa therapy. A direct dopamine receptor agonist could also be added to levodopa treatment for this woman; non-ergot derivatives including ropinirole, rotigotine (as a transdermal patch) or pramipexole are preferred. Apomorphine, a D2 receptor agonist, given subcutaneously can also be used to counteract the off periods in advanced disease, with domperidone given to reduce its emetic effects.
A There is considerable debate about when to commence levodopa therapy in early-onset Parkinson's disease. The goal should be to improve quality of life and limit long-term unwanted effects. If the degree of disability is not severe there may be no immediate need for therapy, but this is controversial as survival is reduced if treatment with levodopa is delayed until disability develops. If treatment is required in this man, dopamine receptor agonists could be started. These are less likely to produce dyskinesias and could delay the need for levodopa until progressive disabilities start to occur. The possibility of brain damage caused by boxing injury should also be considered; this responds poorly to standard treatments for Parkinson's disease.
Bhidayasiri, R, Truong, DD. Motor complications in Parkinson's disease: clinical manifestations and management. J Neurol Sci. 2008;266:204–215.
Biglan, KM, Ravina, B. Neuroprotection in Parkinson's disease: an elusive goal. Semin Neurol. 2007;27:106–112.
Blanchet, PJ. Antipsychotic drug-induced movement disorders. Can J Neurol Sci. 2003;30(suppl 1):S101–S107.
Clarke, CE. Parkinson's disease. BMJ. 2007;335:441–445.
Gerfen, CR. Molecular effects of dopamine on striatal-projection pathways. Trends Neurosci. 2000;23(suppl):S64–S70.
Hauser, RA, Zesiewicz, TA. Advances in the pharmacological management of early Parkinson disease. Neurologist. 2007;13:126–132.
Hermanowicz, N. Drug therapy for Parkinson's disease. Semin Neurol. 2007;27:97–105.
Hirose, G. Drug induced parkinsonism: a review. J Neurol. 2006;253(suppl 3):iii22–iii24.
Lewitt, PA. Levodopa for the treatment of Parkinson's disease. N Engl J Med. 2008;359:2468–2476.
Obeso, JA, Rodriguez-Oroz, MC, Rodriguez, M, et al. The basal ganglia and disorders of movement: pathophysiological mechanisms. News Physiol Sci. 2002;17:51–55.
Schapira, AH. Treatment options in the modern management of Parkinson's disease. Arch Neurol. 2007;64:1083–1088.
Dyskinesias, dystonias and spasticity
Jancovic, J. Treatment of dystonia. Lancet Neurol. 2006;5:864–872.
Louis, ED. Essential tremor. N Engl J Med. 2001;345:887–891.
Meleger, AL. Muscle relaxants and antispasticity agents. Phys Med Rehab Clin North Am. 2006;17:401–413.
Papapetropoulos, S, Singer, C. Botulinum toxin in movement disorders. Semin Neurol. 2007;27:183–194.
Rekand, T. Clinical assessment and management of spasticity: a review. Acta Neurol Scand Suppl. 2010;190:62–66.
Tarsey, D, Simon, DK. Dystonia. N Engl J Med. 2006;355:818–829.