Depression, attention deficit hyperactivity disorder and narcolepsy
Depression
Clinical depression is characterised by diverse psychological and physical symptoms. Major depressive disorder is considered to be present if there are at least five of the following symptoms present for at least two months, or producing marked functional impairment for at least two weeks: depressed mood, loss of interest and enjoyment of activities, loss of energy, loss of appetite or weight disturbance (significant weight loss or gain), sleep disturbance, psychomotor change, a sense of guilt and worthlessness, concentration difficulties or indecisiveness or thoughts of death or suicide. Depressed mood or loss of interest must be present, but otherwise depression can present with mainly physical rather than psychological symptoms. The existence of mixed anxiety–depression disorder is now also well accepted.
Major depressive disorder is an episodic illness that has a lifetime prevalence of about 15% of the population, and recurs in almost three-quarters of people who experience an episode. In about 25% of people with depression the condition may become chronic, with duration of symptoms of more than two years, while up to 40% of people report reduced psychosocial functioning even after recovery from a depressive episode. As the number of depressive episodes increases the threshold for precipitation of a further episode by life stresses appears to decrease, a process referred to as ‘kindling’. Both a genetic predisposition and the effects of adverse events in early life may determine whether a person is susceptible to depressive illness in later life.
Biological basis of depression
The cause of depression is unknown and there may not be a single mechanism. The neurotrophic hypothesis postulates that there are changes in the connectivity between key structures in the brain that are involved in regulation of mood and the stress response, including limbic structures (amygdala, hippocampus and nucleus accumbens) and the prefrontal cortex. It has been suggested that disruption to limbic connections with the prefrontal cortex impairs the normal feedback from the cortex that regulates limbic activity. Depression involves a negative emotional bias that may lead the person to preferentially recall negative events. This negative information is processed in the amygdala, which is hyperactive in people with depression.
In people who are genetically predisposed to depression, stress can initiate remodelling and elimination of hippocampal circuits involved in regulation of mood, cognition and behaviour. High plasma concentrations of the stress peptide corticotropin-releasing hormone (CRH; also known as corticotropin-releasing factor) and the stress hormone cortisol lead to impaired neuroplasticity. Atrophy of the hippocampus blunts the negative feedback from this structure on neuroendocrine function and leads to a perpetuation of the stress response.
The monoamine theory, which has underpinned treatment of depression for many years, is complementary to the neurotrophic hypothesis. Depression is associated with reduced central nervous system (CNS) monoaminergic neurotransmission, possibly as a result of high monoamine oxidase A activity. This affects both serotonergic and noradrenergic pathways, which are closely involved in regulation of mood.
Corticotropin-releasing hormone in depression
In depression, there is hypersecretion of the ‘stressor’ peptide CRH (Ch. 43), which has detrimental effects on neural synaptic plasticity and neurogenesis, and promotes neuronal excitotoxicity. CRH depresses serotonergic neurotransmission and is also a neurotransmitter that orchestrates the CNS control of many behavioural, endocrine, autonomic and immunological responses. Many of these circuits involve glutamatergic neurotransmission via the excitatory N-methyl-D-aspartate (NMDA) receptor. Depressed neurotransmission in these pathways provides a potential target for the treatment of depression. CRH also increases cortisol secretion, which contributes to the loss of neuroplasticity.
Serotonergic and noradrenergic pathways in the brain
Most serotonergic neurons are found in the raphe area of the midbrain, from where they project to the hippocampus in the limbic system and to the cerebral cortex. Presynaptic α2-adrenoceptors and 5-hydroxytryptamine 1 (5-HT1) autoreceptors (not shown in Fig. 22.1) inhibit serotonin release. Somatodendritic 5-HT1, α1- and β-adrenoceptor autoreceptors on the neuronal cell bodies of the raphe nuclei also regulate firing in serotonergic neurons. Postsynaptic 5-HT2 receptors mediate the effects of serotonin and are widely distributed in the cerebral cortex, but especially the prefrontal cortex. A schematic of these mechanisms is shown in Figure 22.1.
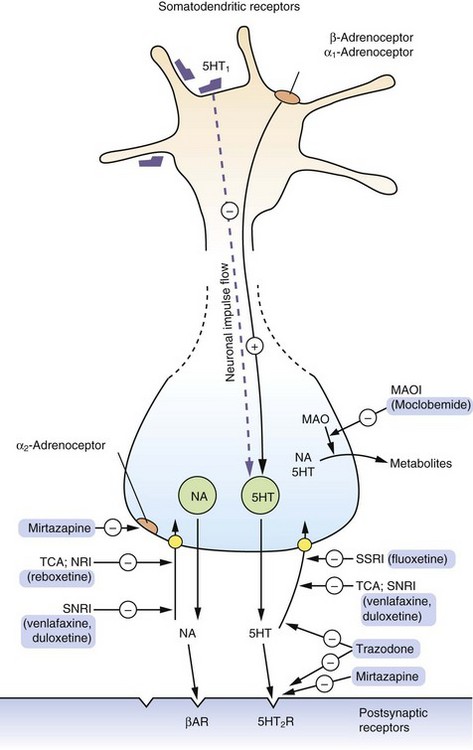
Fig. 22.1 The actions of drugs used in the treatment of depression on CNS serotonergic and adrenergic functioning.
The primary action of many drugs in current clinical use is to enhance serotonin (5-HT, 5-hydroxytryptamine) and noradrenaline (NA) availability. The majority of released serotonin and noradrenaline is rapidly removed from the synapse by reuptake into the neuron (yellow circles). Antidepressants vary in their abilities to inhibit the reuptake of serotonin or noradrenaline, thus enhancing the synaptic concentrations of these transmitters. Stimulation of presynaptic α2-adrenoceptors reduces monoamine release; mirtazapine, by blocking these presynaptic autoreceptors, increases noradrenaline and serotonin release and transmission. Other drugs act by significantly blocking postsynaptic receptors which are upregulated in depression. βAR, β-adrenoceptor; MAO, monoamine oxidase; MAOI, monoamine oxidase inhibitor; NA, noradrenaline; NRI, (selective) noradrenaline reuptake inhibitor; SNRI, serotonin and noradrenaline reuptake inhibitor; SSRI, selective serotonin reuptake inhibitor; TCA, tricyclic antidepressant.
In contrast, most noradrenergic neurons arise in the locus coeruleus and the lateral tegmental areas of the brainstem. The locus coeruleus and the raphe region have many reciprocal neural projections, and therefore the pathways are interdependent. For example, noradrenergic neurotransmission stimulates serotonergic neurons by activating somatodendritic α1-adrenoceptors, but also inhibits serotonin synthesis and release through presynaptic α2-adrenoceptors.
Pathways mediated by glutamate, γ-aminobutyric acid (GABA) and substance P also modulate monoaminergic neurotransmission.
Monoamine neurotransmitters and depression
Serotonergic pathways in the CNS are believed to be mainly involved in mood, while noradrenergic pathways are involved in stress systems, drive and energy state. These monoaminergic circuits in the brain are closely integrated. Simplistically, it has been hypothesised that the following biological changes in the monoamine system are important in depression:
 low levels of monoamine neurotransmitters,
low levels of monoamine neurotransmitters,
upregulation of postsynaptic monoamine receptors,
upregulation of the inhibitory presynaptic and somatodendritic autoreceptors that control monoamine release.
There are increased 5-HT2 receptor numbers in the frontal cortex of depressed suicide victims, whereas other studies have indicated that serotonin and noradrenaline concentrations in the brain are reduced in depression. Overall, evidence for the ‘monoamine’ theory as a molecular basis for depression is limited, but the response to drugs that increase monoamine neurotransmission supports the concept.
Regulation of brain-derived neurotrophic factor in depression
Most antidepressant drugs increase the CNS monoamine concentrations rapidly, but the clinical benefit of antidepressant therapy is delayed. This suggests that more gradual adaptive changes occur as a result of increased monoaminergic neurotransmission. These pharmacologically induced changes are incompletely understood, but they may help to normalise the fundamental dysfunction in intracellular signalling pathways and transduction mechanisms that have been described in depression.
There is evidence for the central role of brain-derived neurotrophic factor (BDNF) in depression. Regulation of BDNF by monoamines is shown in Figure 22.2. BDNF expression is reduced when monoamine neurotransmission is impaired, but also in conditions of stress with elevated serum cortisol. Decreased expression of BDNF has adverse effects on neuronal plasticity can reduce neuronal networks, and may be a major factor in loss of neuronal circuitry and hippocampal atrophy. There is some evidence that successful antidepressant treatment is associated with increased BDNF expression and a restoration of hippocampal function and neuroendocrine regulation.
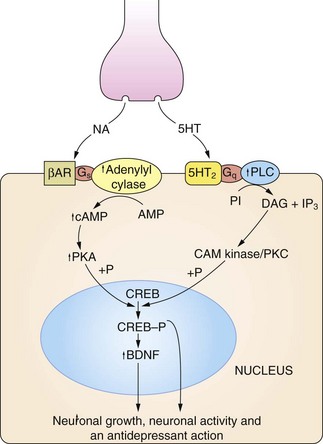
Fig 22.2 The regulation of neuronal growth and plasticity by monoamines and brain-derived neurotrophic factor (BDNF).
Adequate levels of monoamines, cAMP response element-binding protein (CREB-P) and BDNF are considered necessary for neuronal growth and plasticity. An increase in cAMP can result from noradrenaline (NA) acting on β-adrenoceptor (βAR) subtypes, and an increase in diacylglycerol (DAG) signalling can result from serotonin (5-HT) acting on 5-HT2 type receptors. There is also evidence that cAMP can be increased by serotonin acting on 5-HT4 and 5-HT7 receptors, and DAG by noradrenaline acting on α1-adrenoceptors (not shown). Critical points in this cascade may be dysfunctional in depressed individuals, including reduced synthesis of the monamine transmitters, genetic polymorphism affecting the function or expression of monoamine receptors, anomalies in coupling of Gs to adenylyl cyclase, reduced protein kinase A (PKA) activity and reduced phosphorylation of CREB. These may result in reduced BDNF activity, leading to neuronal atrophy and cell death in the hippocampus and cortex. CAM kinase, calmodulin-dependent protein kinase; IP3, inositol triphosphate; PI, phosphatidylinositol; PKC, protein kinase C; PLC, phospholipase C.
Antidepressant drug action
Most of the antidepressant drugs currently used clinically target the mechanisms involved in the control of monoamine neurotransmitter turnover or monoamine receptor function. There seems to be little difference in efficacy between drugs that act predominantly on serotonergic or on noradrenergic pathways, although they differ in their side-effect profiles. The ways that major antidepressants work to modify monoamine turnover and function are shown in Figure 22.1.
Long-term treatment with antidepressants promotes both the structural and functional integrity of the neural circuits that regulate mood. The mechanisms by which they achieve this are complex.
Enhanced CNS monoamine levels. The initial action of most drugs used in the treatment of depression is to enhance neurotransmission by CNS monoamines, particularly serotonin but also noradrenaline and dopamine. Increased noradrenergic activity further enhances serotonergic neurotransmission by stimulating somatodendritic α1-adrenoceptors on serotonergic neurons. However, although antidepressants rapidly increase synaptic monoamine levels, clinical improvement is delayed. In part, this may be due to slow reduction in the number of upregulated somatodendritic and presynaptic 5-HT1 inhibitory autoreceptors (see above), which is necessary before activity increases in serotonergic pathways.
Effects on postsynaptic monoamine receptor expression and intracellular signal transduction. During treatment with antidepressants there is a gradual increase in responsiveness to serotonin in the prefrontal cortex. There is considerable evidence that antidepressants reverse the changes in intracellular signalling that are found in depression (Fig. 22.2). They enhance the response to monoamine receptor stimulation, which increases expression of BDNF and its receptor. As a result there is enhanced differentiation of progenitor cells into neurons and increased neuronal survival. Of note, electroconvulsive therapy can also increase the expression and activity of BDNF.
Regulation of CRH production. During long-term treatment with antidepressants there is normalisation of overexpressed CRH secretion. This may be related to upregulation of CNS glucocorticoid receptors, with feedback inhibition of CRH.
Antagonism of NMDA receptor action. Antidepressant drugs bind to a site in NMDA receptor-associated ion channels in the hippocampus and cerebral cortex, and may protect cells against stress-induced ‘glutamate excitotoxicity’.
Antidepressant drugs
Tricyclic antidepressant drugs
Mechanism of action: Tricyclic antidepressants (TCAs) inhibit the reuptake of monoamine neurotransmitters into the presynaptic neuron by competitive inhibition of monoamine transporter (MAT) proteins, particularly the noradrenaline transporter NET and the serotonin transporter SERT (Fig. 22.1). Some drugs show little monoamine selectivity, while other compounds are more selective for one monoamine (Table 22.1). However, the degree of monoamine selectivity has not been shown to influence efficacy. The subsequent effects on the CNS are described above.
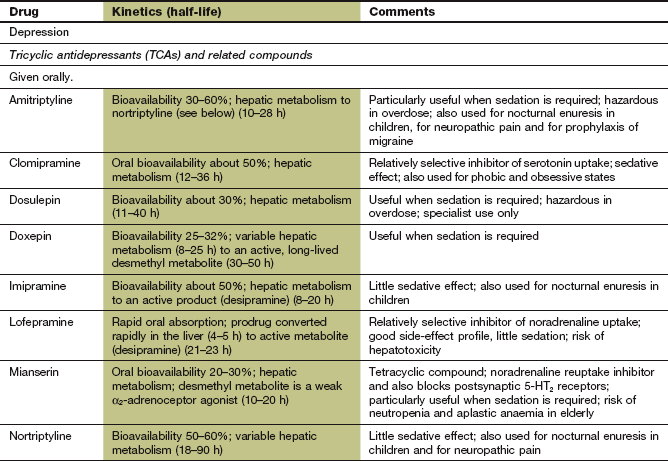
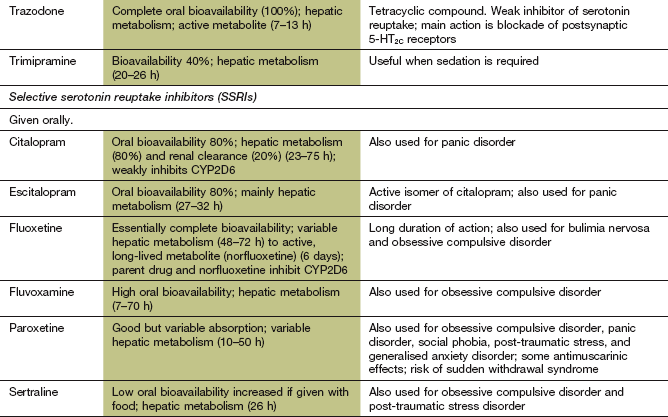
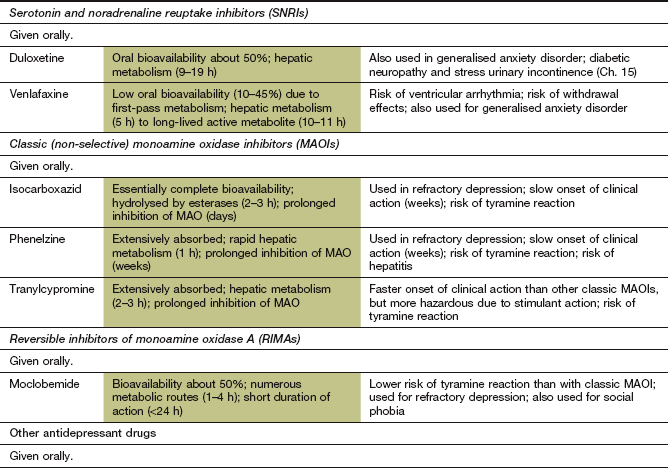
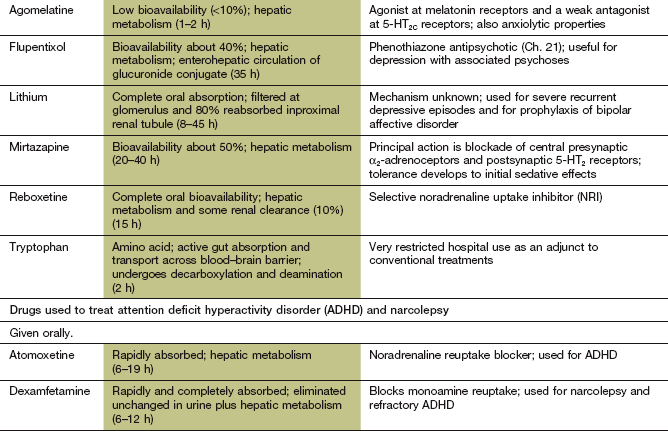
Table 22.1
Comparative properties of some commonly used antidepressant drugs
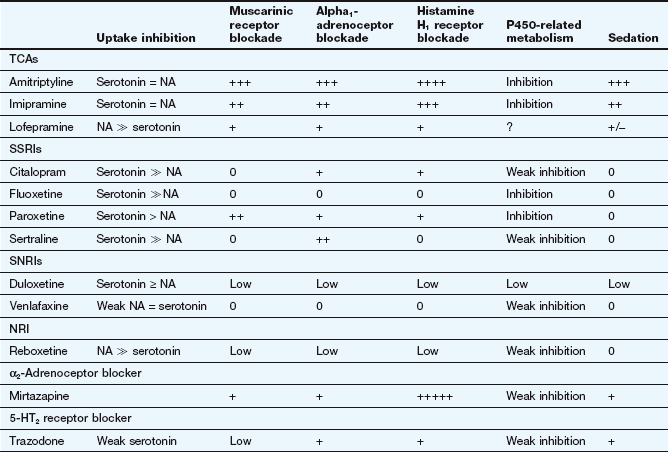
Drugs are listed under their conventional groupings but many have mixed or uncertain mechanisms of action. Differential blockade of muscarinic receptors, α1-adrenoceptors and histamine H1 receptors contributes to the side-effect profiles of antidepressant drugs. Other antidepressant drugs, including monoamine oxidase inhibitors (MAOIs), are listed in the Compendium at the end of the chapter. NA, noradrenaline; SSRI, selective serotonin reuptake inhibitor; SNRI, serotonin and noradrenaline reuptake inhibitor; NRI, noradrenaline reuptake inhibitor; TCA, tricyclic antidepressant.
The table is constructed from data in Richelson E (2002) The clinical relevance of antidepressant interaction with neurotransmitter transporters and receptors. Psychopharmacol Bull 36(4), 133–150 and other sources for approximate comparison only.
Many of the unwanted effects of these drugs are a consequence of blockade of other postsynaptic receptors (e.g. muscarinic and histamine H1 receptors and α1-adrenoceptors) (Table 22.1), which do not influence their antidepressant action.
Pharmacokinetics: All TCAs are well absorbed from the gut and highly protein bound in plasma. They undergo extensive first-pass metabolism in the liver, and active metabolites are formed which are partially responsible for the variable effective half-lives of these drugs (8–90 h; see Compendium at the end of this chapter). The combination of high first-pass metabolism and high clearance but a long elimination half-life is explained by high apparent volumes of distribution (10–50 L⋅kg−1 body weight). There is considerable inter-individual variability in the first-pass metabolism of most TCAs, leading to up to 40-fold differences in the plasma concentrations of the parent drug. There is no clear dose relationship for the therapeutic effects, although unwanted effects are dose-related. Dose titration is usually necessary to optimise the therapeutic response; this should be gradual over 1–2 weeks to minimise unwanted effects.
Sedation as a result of histamine H1 receptor and α1-adrenoceptor blockade (Ch. 39). Some compounds are highly sedative, for example amitriptyline, and others less so. Sedation can be useful to help restore sleep patterns in depression (using a larger dose of a sedative drug at night) but can be troublesome or even dangerous during the day.
Antimuscarinic effects (see Ch. 4): dry mouth is a frequent occurrence, and less commonly constipation, urinary retention, impotence and visual disturbance. Tolerance to these effects can occur and gradual increases in dose may reduce their incidence.
Excessive sweating and tremor.
Postural hypotension produced by peripheral α1-adrenoceptor blockade (Ch. 4) can be particularly troublesome in the elderly, although tolerance can occur.
Epileptogenic effects: TCAs lower the convulsive threshold, and seizures can be provoked, even when there is no previous clinical history.
Cardiotoxicity in overdose: most tricyclic drugs depress myocardial contractility. They can produce tachycardia and severe arrhythmias when taken in overdose, due to both antimuscarinic effects and excessive noradrenergic stimulation. Lofepramine is less cardiotoxic than other drugs in this class.
Weight gain: appetite stimulation is common, probably due to histamine H1 receptor blockade.
Hyponatraemia from inappropriate antidiuretic hormone (ADH; vasopressin) secretion, leading to drowsiness, confusion and convulsions.
Sexual dysfunction with reduced interest in sex, erectile dysfunction in men and diminished arousal in women, and difficulty attaining orgasm.
Sudden withdrawal syndrome: during long-term treatment doses should be gradually reduced over four weeks to avoid agitation, headache, malaise, sweating and gastrointestinal upset, which can accompany sudden withdrawal. These may result from excessive cholinergic activity following prolonged muscarinic receptor blockade.
Drug interactions: Several important drug interactions are recognised. TCAs potentiate the central depressant activity of many drugs, including alcohol. A dangerous interaction can result from giving a monoamine oxidase (MAO) inhibitor (MAOI) (see below) and a TCA together due to prolonged action of the increased serotonin released from the neuron. The interaction can lead to hyperpyrexia, convulsions and coma, and can occur up to two weeks after stopping an MAOI due to the long duration of MAO inhibition.
The risk of serious arrhythmias is increased when TCAs are taken with drugs that prolong the Q–T interval on the electrocardiogram (Ch. 8). Such drugs include the class III antiarrhythmic sotalol, and all class I antiarrhythmics.
Selective serotonin reuptake inhibitors and related antidepressants
Mechanism of action: Unlike the TCAs, the selective serotonin reuptake inhibitors (SSRIs) reduce the neuronal reuptake of serotonin by its presynaptic transporter protein (SERT), but have little or no effect on noradrenaline reuptake (Table 22.1). They have a more favourable profile of unwanted effects than TCAs because of their low affinity for muscarinic and histamine receptors and α1-adrenoceptors. Paroxetine is unusual among SSRIs in having affinity for muscarinic M3 receptors, found in the brain, salivary glands and smooth muscle.
The proposed mechanism of action of SSRIs is as follows.
The increase in synaptic serotonin concentration as a result of reduced neuronal uptake leads to downregulation of the 5-HT1 somatodendritic and axon terminal presynaptic inhibitory autoreceptors on serotonergic neurons.
Reduced inhibitory autoreceptor activity increases serotonin release at the axon terminal. The prolonged increase in synaptic serotonin concentration (as a result of both increased release and reduced neuronal uptake) downregulates postsynaptic 5-HT2 receptors.
Subsequent changes in intracellular function are described above.
Pharmacokinetics: SSRIs are well absorbed from the gut and metabolised in the liver. Paroxetine has a long half-life (10–20 h), which is greatest in poor metabolisers of CYP2D6 substrates (30–50 h). Citalopram, fluoxetine and sertraline have very long half-lives (23–75 h). The active metabolite of fluoxetine has a half-life of 6 days, and the resulting very long duration of action can be a disadvantage if an MAOI is used subsequently (see below).
Unwanted effects: In contrast to the TCAs, SSRIs have few antimuscarinic effects (apart from paroxetine), cause little sedation or weight gain and are not cardiotoxic in overdose. However, they may cause:
nausea (frequent), dyspepsia, abdominal pain or diarrhoea (less frequent),
insomnia, anxiety and agitation,
hyponatraemia, due to inappropriate secretion of ADH and leading to drowsiness, confusion and convulsions, is more frequent than with TCAs,
dry mouth and constipation with paroxetine,
sexual dysfunction with reduced interest in sex, erectile dysfunction in men and diminished arousal in women, and difficulty attaining orgasm. This affects up to three-quarters of people taking SSRIs,
increased risk of bleeding due to platelet dysfunction,
SSRIs lower the convulsive threshold, and seizures can be provoked, even when there is no previous clinical history,
sudden withdrawal syndrome after long-term use, which may be most troublesome with paroxetine; it presents with gastrointestinal symptoms, headache, anxiety, dizziness, paraesthesia, electric shock sensations in the head, neck and spine, sleep disturbance and sweating and usually begins 24–72 h after stopping treatment. The dose should be gradually reduced over at least 4 weeks,
increase in suicidal thoughts and self harm in people aged under 25 years.
Drug interactions: The most serious interaction is with MAOIs (see TCAs above). An interval of five weeks is recommended after stopping fluoxetine, or two weeks after paroxetine or sertraline, before an MAOI (including selegiline, Ch. 24) is taken. Fluoxetine and other SSRIs inhibit hepatic CYP2D6 (Table 2.7), and this can increase the plasma concentration of drugs metabolised by this enzyme.
Serotonin and noradrenaline reuptake inhibitors
Mechanism of action and uses: Venlafaxine and duloxetine are classified as serotonin and noradrenaline reuptake inhibitors (SNRIs) although at lower doses they have a greater effect on serotonin reuptake (Table 22.1). Like the TCAs, they inhibit neuronal reuptake of both serotonin and noradrenaline, but share with SSRIs a low affinity for muscarinic and histamine receptors and α1-adrenoceptors. Their unwanted effect profiles are therefore closer to those of the SSRIs than those of the TCAs. There is some evidence that clinical improvement with venlafaxine may begin earlier than with other antidepressant drugs.
Duloxetine is also used as an adjunctive treatment for smoking cessation (Ch. 54), and in urinary stress incontinence (Ch. 15).
Pharmacokinetics: Venlafaxine and duloxetine are well absorbed from the gut and undergo extensive first-pass metabolism in the liver. The main active metabolite of venlaxafine has a long half-life (11 h) and the half-life of duloxetine is 9–19 h.
Nausea, vomiting, anorexia, dyspepsia, constipation.
Drowsiness, insomnia, dizziness, confusion, headache, tremor.
QT segment prolongation on the ECG with venlafaxine, which predisposes to ventricular arrhythmias (see Ch. 8); it should be avoided in people at high risk of arrhythmias.
Sudden withdrawal symptoms are more frequent than with other antidepressants, with gastrointestinal symptoms, headache, anxiety, dizziness, paraesthesia, tremor, sleep disturbance and sweating.
Selective noradrenaline reuptake inhibitors
Mechanism of action: Reboxetine is related to fluoxetine but is a selective noradrenaline reuptake inhibitor (NRI). Increased noradrenergic activity at somatodendritic α1-adrenoceptors enhances serotonergic neurotransmission. Reboxetine, in common with the SSRIs, has little activity at muscarinic and histamine receptors and α1-adrenoceptors. It therefore has fewer unwanted effects than do TCAs.
Presynaptic α2-adrenoceptor blockers
Mechanism of action: Mirtazapine is a tetracyclic drug unrelated structurally to the TCAs. In addition to potent postsynaptic 5-HT2C receptor-blocking activity in the cortex (see serotonin receptor blockers below), mirtazapine blocks presynaptic α2-adrenoceptors (Fig. 22.1). This reduces negative feedback inhibition of serotonin release from raphe nucleus neurons in their terminal projections to regions such as the cortex and hippocampus. Mirtazapine blocks histamine H1 receptors but has a low affinity for muscarinic receptors and postsynaptic α1-adrenoceptors. It has minimal effects on monoamine reuptake.
Serotonin receptor blockers
Mechanism of action: Trazodone is a tetracyclic compound. Its most significant antidepressant action is blockade of postsynaptic 5-HT2C receptors, which increases activity of dopamine and noradrenaline in the frontal cortex. It also produces weak inhibition of presynaptic serotonin reuptake, but does not inhibit noradrenaline reuptake. Trazodone blocks α1-adrenoceptors and weakly blocks muscarinic and histamine H1 receptors.
Classic (non-selective) monoamine oxidase inhibitors
Mechanism of action: The mechanism of action of classic (non-selective) monoamine oxidase inhibitors (MAOIs) is complex, but their primary action is irreversible inhibition of intracellular MAO, which is the enzyme responsible for degrading free monoamines in the presynaptic nerve terminal. This leads to accumulation of monoamine neurotransmitters in the presynaptic neuron and increased release when the nerve is stimulated (Fig. 22.1). There are two MAO isoenzymes (Fig. 22.3). MAO-B is the predominant enzyme in many parts of the brain, but MAO-A is present in noradrenergic and serotonergic neurons, especially in the locus coeruleus and other cells of the brainstem, as well as being the main enzyme in peripheral tissues. Inhibition of MAO-A in the brain produces the therapeutic effects of these drugs, but classic inhibitors (MAOIs) are not selective for this isoenzyme. Inhibition of both MAO-A and MAO-B in the gut wall and liver has important consequences (see below). MAOIs also inhibit various drug-metabolising enzymes in the liver, which predisposes to drug interactions but does not contribute to clinical efficacy.
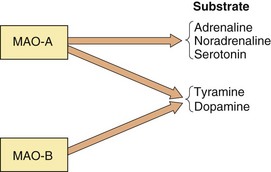
Fig. 22.3 Actions of monoamine oxidase (MAO) and MAO inhibitors.
The relative selectivity of the substrates for MAO is shown. MAO-A is the target for drugs useful in treating depression. Non-selective inhibition of both MAO-A and MAO-B blocks the metabolism of tyramine, which is responsible for the adverse food reaction that occurs with these drugs. Reversible inhibitors of MAO-A (RIMAs) block only subtype A. Therefore, tyramine is still metabolised by subtype B and the food reaction is reduced. Selective inhibitors of MAO-B enhance CNS dopamine levels and are used in Parkinson's disease (Ch. 24).
Pharmacokinetics: All drugs in this class are well absorbed from the gut. They are irreversible enzyme inhibitors and therefore their prolonged duration of action is unrelated to the half-life of the drug. Drug withdrawal is followed by gradual restoration of normal MAO activity over about 2 weeks as new enzyme is synthesised.
Compared with the TCAs, antimuscarinic effects are unusual and there is no predisposition to seizures (except in overdose; see below).
Dose-related postural hypotension can occur. Unlike with the TCAs, tolerance does not develop. The mechanism may involve conversion of tyramine (normally degraded by MAO) to octopamine, a false neurotransmitter which competes with noradrenaline at sympathetic nerve terminals.
Due to its structural similarity to amfetamine (Ch. 54), tranylcypromine causes CNS stimulation, leading to irritability and insomnia. Doses should be given early in the day to avoid disturbing sleep.
Hepatitis is a rare idiosyncratic reaction to the hydrazine derivative phenelzine.
Acute overdose produces delayed toxic effects after some 12 h. Excessive adrenergic stimulation leads to chest pain, headache and hyperactivity, progressing to confusion and severe hypertension, with eventually profound hypotension and seizures.
Food interactions can occur because MAO in the gut wall and liver usually prevents the absorption of natural amines, particularly tyramine, which is an indirect-acting sympathomimetic (Ch. 4). If food containing tyramine, for example cheese, yeast extracts (such as Bovril, Oxo or Marmite), pickled herrings, chianti and caviar, or broad bean pods (which contain L-dopa) is eaten, the increased absorption of tyramine and consequently greater release of noradrenaline result in vasoconstriction and severe hypertension. The first indication of this is a throbbing headache. A warning card should be supplied to people who take MAOIs.
Drug interactions: A number of drug interactions can occur. Indirect-acting sympathomimetics (Ch. 4) in cold remedies (e.g. ephedrine, phenylpropanolamine), and levodopa (treatment of Parkinson's disease, Ch. 24) will be more active, with a risk of hypertensive crisis. The toxicity of the triptans (5-HT1 receptor agonists used for treatment of migraine, Ch. 26) will be potentiated. All these drugs should be avoided for two weeks after stopping an MAOI. The combination of MAOIs with TCAs or SSRIs (see above) can be dangerous. Other important interactions occur because MAOIs can impair the hepatic metabolism of certain drugs, especially opioid analgesics.
Reversible inhibitors of monoamine oxidase A (RIMAs)
Mechanism of action and effects: Moclobemide is a reversible inhibitor of MAO-A, the same isoenzyme target for the antidepressant action of classic MAOIs. However, moclobemide does not inhibit MAO-B, so tyramine absorbed from the gut can still be degraded and the food reaction described above for classic MAOIs is very unlikely to occur. Also, since the action of moclobemide on MAO-A is reversible, high concentrations of tyramine will displace the drug from the enzyme, further facilitating degradation of tyramine. If moclobemide is taken after meals, then inhibition of MAO-A in the gut during absorption of tyramine will be minimised, providing further protection. MAO-A inhibition by moclobemide lasts less than 24 h after a single dose.
Pharmacokinetics: Oral absorption of moclobemide is good but there is substantial first-pass metabolism, partially to an active metabolite. Moclobemide undergoes hepatic metabolism and has a short half-life (1–2 h).
Drug interactions: Inhibition of cytochrome P450 activity in the liver by cimetidine (Ch. 33) substantially reduces the metabolism of moclobemide, and smaller starting doses are recommended in this situation. Moclobemide should not be given with other antidepressants, and the recommendations for stopping these drugs before prescribing moclobemide are the same as for classic MAOIs.
Melatonin receptor agonist and serotonin receptor antagonist
Mechanism of action and effects: Agomelatine is a synthetic agonist of the naturally occurring substance melatonin, which is secreted by the pineal gland and is involved in regulation of circadian rhythms and therefore sleep pattern. Agomelatine is an agonist at both melatonin MT1 receptors (attenuating alerting signals to the cortex) and MT2 receptors (producing a phase-shifting action on the circadian rhythm of sleep). Agomelatine significantly improves sleep quality when taken at night to mimic the natural rhythm of melatonin release. It is a weak antagonist at 5-HT2C receptors (see serotonin receptor blockers above), and increases noradrenaline and dopamine release in the frontal cortex. The antidepressant efficacy of agomelatine is similar to that of SSRIs.
Management of depression
Drugs form only part of the management of depression but are usually necessary for moderate, severe or protracted symptoms. However, in mild to moderate depression, cognitive therapy is as effective as drug treatment and should be tried first. The herbal remedy Hypericum perforatum (St John's wort) is comparable to antidepressant drugs for treatment of mild depression, but neither St John's wort nor antidepressant drugs show convincing efficacy compared to placebo in mild disease. St John's wort does not often cause unwanted effects, but concentrations of the active constituents vary among different preparations. St John's wort can induce cytochrome P450, and should not be taken with a prescribed antidepressant. All antidepressant drugs have a delayed onset of action, and people who are severely ill should be considered for electroconvulsive therapy (ECT), which gives a more rapid response.
The TCAs are now less frequently prescribed for depression. They have serious cardiotoxic effects when taken in overdose and should usually be avoided when treating people who are at high risk of attempting suicide. Encouraging adherence to treatment with TCAs may initially be difficult, since antimuscarinic unwanted effects can be troublesome before any benefit is perceived. Starting with a small dose of TCA followed by gradual dose titration can minimise unwanted effects. There is now evidence that large doses of a TCA do not necessarily enhance the treatment response but do increase unwanted effects, so the use of low dosages may therefore be preferred.
The newer drugs, for example SSRIs and SNRIs, are no more effective than TCAs and do not work any faster, but they are better tolerated. They are certainly safer than TCAs when there is a high risk of suicide. The use of SSRIs or SNRIs as the first-line treatment for depression is now well established.
Up to 70% of depressed people will respond to drug therapy if the dosage is adequate, compared with about 30% taking placebo. However, only 50% will respond to an individual drug, and up to a further 20% of people with depression will respond if the drug is changed after failure of the initial treatment. Responders show an initial improvement in sleep pattern within a few days. Psychomotor retardation responds more gradually over several days, leading to greater involvement with everyday activities and enjoyment of life. Improvement in the depressed mood is delayed, beginning up to two or more weeks after commencing treatment with an adequate drug dosage. The response of most symptoms tends to be erratic, with good and bad days.
Initial treatment with an antidepressant should be for 4–6 weeks. If there is no response after this time, and if it is believed that adherence to treatment is not a problem, then either the dose should be increased, if unwanted effects permit, or an alternative drug can be substituted. If there is a good response then the dosage can usually be reduced, but maintenance treatment should be continued for at least 4–6 months after the first episode of depression to minimise the risk of relapse. A longer period of maintenance treatment to prevent recurrence (at least one year and often longer) is often recommended for the elderly, for others who are at high risk of recurrence, and for people who have experienced two or more depressive episodes. About half of all people who experience depression only have a single episode. In individuals with recurrent illness, relapse occurs within a year in up to 65% of those who stop treatment, but only in 15% of people who continue treatment. Risk factors for relapse in major depression include:
Classic MAOIs are usually reserved for atypical depression with hypochondriacal and phobic symptoms, or when SSRIs have failed. The therapeutic place of the newer antidepressants such as SNRIs and RIMAs in those who fail to respond to an SSRI has yet to be established. Small doses of the phenothiazine antipsychotic drug flupentixol (Ch. 21) are sometimes used for treatment of depressed elderly people. The evidence for a true antidepressant effect of flupentixol is slight, but some symptoms undoubtedly do improve.
Treatment is most difficult in severe depression, especially if there are psychotic features, or where depression forms part of a bipolar affective disorder (Ch. 21). ECT is used for treatment-resistant depression and in the elderly, who are particularly likely to show a response. Overall, ECT is probably more effective than drug therapy but does produce some lasting cognitive impairment, especially if given bilaterally rather than unilaterally, or given frequently or with high currents. ECT should be combined with prolonged antidepressant drug treatment. Lithium (Ch. 21) is used for people with severe recurrent depressive episodes and for prophylaxis of bipolar affective disorder. The effect of lithium can take several months to become fully established. The treatment of depression in bipolar disorder is discussed further in Chapter 21.
Attention deficit hyperactivity disorder and narcolepsy
Several drugs with similar mechanisms of action to antidepressants, as well as central stimulant drugs, have limited uses in the management of attention deficit hyperactivity disorder (ADHD) and narcolepsy.
Attention deficit hyperactivity disorder
ADHD is the most common behavioural and cognitive disorder in children of school age, but often remains a problem in adult life. The mechanism is poorly understood, but may involve dopamine deficiency in the prefrontal cortex. There are three subtypes:
predominantly inattentive subtype: failing to pay attention to details, difficulty with sustained attention, disorganisation and forgetfulness,
predominantly hyperactive-impulsive subtype: excessive fidgeting and squirming, restlessness, frequently interrupting and intruding on others,
predominantly inattentive/hyperactive-impulsive subtype: features of both subtypes in two areas of life and causing dysfunction in at least one area.
Adults with ADHD often present with poor occupational performance, marital instability, poor self-discipline or self-organisation, and restlessness. Sleep disturbances may also be prominent.
Narcolepsy
Narcolepsy usually begins in adolescence and is characterised by overwhelming daytime sleepiness, even if night-time sleep has been adequate. Sudden daytime naps are frequent and there may be prolonged periods of drowsiness. Hallucinations may be troublesome on falling asleep or awakening. The condition may coexist with cataplexy, characterised by sudden loss of muscle function ranging from weakness to collapse that is often precipitated by laughter. People with narcolepsy have an abnormal sleep pattern, with rapid-eye-movement (REM) sleep at the onset of sleep rather than after a period of non-REM sleep (Ch. 20). Narcolepsy has a genetic predisposition and there may be an autoimmune component. There is loss of hypocretin-secreting neurons in the hypothalamus, possibly as a result of autoimmune-mediated cell destruction. Hypocretins are neurotransmitters that regulate wakefulness by releasing dopamine and noradrenaline in the hypothalamus, which excite histaminergic tuberomammillary neurons. The tuberomammillary nucleus is involved in control of arousal, sleep and circadian rhythms.
Drugs for attention deficit hyperactivity disorder and narcolepsy
Mechanism of action: Atomoxetine is a selective inhibitor of presynaptic neuronal uptake of noradrenaline. There is a secondary increase in dopaminergic activity in the prefrontal cortex. It has antidepressant activity, but the mechanism of action in ADHD is not known.
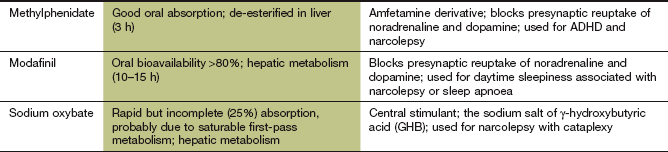
Methylphenidate
Mechanism of action: Methylphenidate is an amfetamine derivative that activates the brainstem arousal system. Its mechanism is not established, but there is evidence that the drug blocks the presynaptic reuptake of noradrenaline and dopamine, and increases dopaminergic neurotransmission in the prefrontal cortex.
Dexamfetamine
Dexamfetamine selectively releases monoamines such as noradrenaline, serotonin and dopamine from CNS neurons in the mesolimbic pathway and also the reticular formation that regulates alertness and sleep (see Ch.54).
Modafinil
Mechanism of action: Modafinil blocks the presynaptic reuptake of noradrenaline and dopamine, increasing dopaminergic neurotransmission in the prefrontal cortex. It may also reduce inhibitory GABA-mediated neurotransmission. There is also evidence that modafinil activates hypocretin-releasing neurons in the hypothalamus.
Management of attention deficit hyperactivity disorder
Behaviour management and psychotherapy have a role, but are more effective when combined with drug treatment. However, there is some concern that the benefits of the currently available compounds may be short-lived, with loss of efficacy after about three years. Methylphenidate is usually the first choice, while dexamfetamine can be tried if there is no improvement with methylphenidate. Atomoxetine is used when stimulant drugs are ineffective.
True/false questions
1. The processes underlying depression are all explained by the monoamine hypothesis.
2. Downregulation of serotonin (5-HT2) receptors occurs during antidepressant treatment.
3. Most tricyclic antidepressants (TCAs) inhibit the reuptake of noradrenaline and serotonin equally.
4. TCAs have a less satisfactory therapeutic ratio than do selective serotonin reuptake inhibitors (SSRIs).
5. Selective inhibitors of noradrenaline or serotonin reuptake are equally effective as antidepressants.
6. TCAs potentiate the central depressant effects of alcohol.
7. Lofepramine is more cardiotoxic than amitriptyline.
8. Co-administration of an SSRI and a monoamine oxidase inhibitor (MAOI) can cause cardiovascular collapse.
9. The antidepressant activity of mirtazepine is related to blockade of serotonin receptors.
10. Trazodone is a potent inhibitor of monoamine reuptake.
11. Venlafaxine has marked sedative and antimuscarinic actions.
12. Venlafaxine has a low incidence of cardiovascular toxicity.
13. Only 30–40% of people with depression improve as a direct result of antidepressant drug treatment.
14. Lithium is only used for treatment of bipolar affective disorder.
15. Methylphenidate is an amfetamine derivative used in the management of ADHD.
16. Narcolepsy is associated with loss of neurons that secrete hypocretins.
One-best-answer (OBA) question
Which statement concerning antidepressant drugs is the most accurate?
A A TCA would be more suitable than an SSRI for a patient with urinary outflow problems due to benign prostate hypertrophy.
B The antidepressant action of trazodone is due mainly to muscarinic receptor blockade.
C People taking moclobemide are likely to get an adverse reaction if they eat cheese.
D An SSRI would be more suitable than a TCA to treat a person with serious depression and suicidal tendencies.
E Increases in levels of brain monoamine levels occur only after 2–3 weeks of treatment with a TCA.
Case-based questions
JA, a 34-year-old female financier, is a former international athlete now working in the City of London. In 2006 she was appointed manager of the emerging countries fund of a large unit trust company. In early 2009 the company was taken over and JA had a new boss and was demoted to assistant manager of the fund. From 2009 to 2010 she had put on 10 kg in weight, and started a strict diet and worked out at a gym four times a week. She visited her GP with a 6-month history of increasing insomnia, lack of concentration, irritability and anxiety. She had begun to withdraw from a busy social calendar and was becoming indecisive. This was now affecting her work. For the previous four weeks she had had recurrent thoughts of suicide. She had also lost 4 kg in weight during that period. The GP diagnosed that she was depressed, arranged a psychiatric consultation for her and started her on a TCA.
1. False. The monoamine hypothesis cannot alone explain all features of depression, including the slow onset of antidepressant action of drugs that can rapidly increase CNS levels of monamines.
2. True. Downregulation of 5-HT2 receptors parallels the time course of improvement of clinical condition, whereas the time course of the increase in amine transmitters does not.
3. False. TCAs vary in their ability to affect noradrenaline and serotonin reuptake.
4. True. TCAs have a greater potential to produce serious unwanted effects, e.g. causing cardiac arrhythmias in acute overdose.
5. True. Antidepressant drugs differ more in their profiles of unwanted effects profile than in their clinical efficacy.
6. True. Alcohol should not be consumed by people taking TCAs.
7. False. Lofepramine is among the least cardiotoxic of the tricyclic and related antidepressants; it also causes little sedation.
8. True. An MAOI and an SSRI should not be combined. The combination can cause CNS excitation, tremor and hyperthermia. An MAOI should not be started until 2–3 weeks after stopping the SSRI, depending upon which SSRI has been taken. Conversely, other antidepressants should not be started until 2–3 weeks after stopping an MAOI.
9. True. The main action of mirtazepine is blockade of postsynaptic 5-HT2 receptors and central presynaptic α2-adrenoceptors.
10. False. Trazodone is only a weak inhibitor of serotonin reuptake, and does not block noradrenaline reuptake; its main action is blockade of postsynaptic 5-HT2 receptors.
11. False. Like the TCAs, the SNRI drug venlafaxine inhibits both noradrenaline and serotonin reuptake, but it lacks the sedative and antimuscarinic effects of TCAs.
12. False. Venlafaxine causes QT segment prolongation and predisposes to ventricular arrhythmia in high-risk depressed people, such as those with uncontrolled blood pressure.
13. True. Depression improves in up to 70% of people taking antidepressants, compared with 30% on placebo.
14. False. Although most commonly used in bipolar affective disorder, lithium is also used in those with severe recurrent depressive episodes who do not respond to other treatment.
15. True. The amfetamine derivative methylphenidate is a first-line drug treatment in ADHD; dexamfetamine may be required in refractory ADHD.
16. True. Hypocretins promote wakefulness by releasing noradrenaline and dopamine.
OBA answer
A Incorrect. Many TCAs have antimuscarinic activity, which could exacerbate urinary retention.
B Incorrect. Trazodone acts mainly by blockade of postsynaptic 5-HT2C receptors.
C Incorrect. Moclobemide is a reversible inhibitor of MAO-A and has relatively little effect on MAO-B, which remains available to metabolise dietary tyramine. The ‘cheese reaction’ is therefore less likely than with classic (non-selective) MAO inhibitors.
D Correct. An SSRI would be more suitable than most TCAs in severe depression with suicidal ideation because of their generally lower toxicity in overdose. The effectiveness of SSRIs and TCAs is similar.
E Incorrect. The increase in brain monoamine levels following treatment with a TCA occurs within hours to days. However, the antidepressant effects on behaviour and mood may take 4–6 weeks or longer to become apparent.
Case-based answers
A Risk factors include sex (more frequent in women), age (peak age 20–40 years), family history of depression, marital status (higher rates in separated and divorced people) and stress. It is possible, but we cannot be certain, that any of the circumstances in the clinical history were contributory to her depression.
B There is a biological association of depression with reduced CNS monoaminergic neurotransmission, notably noradrenaline and serotonin, but it is still unclear whether this is cause or effect. Drugs that deplete monoamines can induce depression, and when monoamines are repleted, symptoms decrease. Probably because of the depletion of monoamines, there is an upregulation of postsynaptic monoamine receptors, including 5-HT2 and β1-adrenoceptors. There are also dysfunctions in other CNS signalling systems (see text). Drugs used to treat depression increase CNS serotonin and/or noradrenaline in the synaptic cleft, which eventually results in postsynaptic receptor downregulation.
C The onset of antidepressant activity is delayed for at least 2–4 weeks. Two-thirds of depressed people improve, one-third do not. One-third of people with depression would improve spontaneously without drug therapy. Whether TCAs prevent recurrence is unknown. Unwanted effects of TCAs include muscarinic receptor blockade (dry mouth, blurred vision, constipation), cardiotoxicity, sedation (variable) and postural hypotension.
D TCAs may not be the most appropriate choice as JA was showing suicidal tendencies; the safety of TCAs in overdose is low. Another antidepressant drug such as an SSRI may have been a better choice as they are safer in overdose and have a generally better profile of unwanted effects, although nausea and sexual dysfunction are relatively common.
Alexopoulos, GS. Depression in the elderly. Lancet. 2005;365:1961–1970.
Belmaker, RH, Agam, G. Major depressive disorder. N Engl J Med. 2008;358:55–68.
Donati, RJ, Rasenick, MM. G-protein signaling and the molecular basis of antidepressant action. Life Sci. 2003;73:1–17.
Ebmeier, KB, Donaghey, C, Steele, JD. Recent developments and current controversies in depression. Lancet. 2006;367:153–167.
Furukawa, TA, McGuire, H, Barbui, C. Meta-analysis of effects and side effects of low dosage tricyclic antidepressants in depression: systematic review. BMJ. 2002;325:991–995.
Geddes, JR, Carney, SM, Davies, C, et al. Relapse prevention with antidepressant drug treatment in depressive disorders: a systematic review. Lancet. 2003;361:653–661.
Hatcher, S, Arroll, B. Newer antidepressants for the treatment of depression in adults. BMJ. 2012;344:d8300.
Krishnan, V, Nestler, EJ. Linking molecules to mood: new insights into the biology of depression. Am J Psychiatry. 2010;167:1305–1320.
Mann, JJ. The medical management of depression. N Engl J Med. 2005;353:1819–1834.
Millan, MJ. Multi-target strategies for the improved treatment of depressive states: conceptual foundations and neuronal substrates, drug discovery and therapeutic application. Pharmacol Ther. 2006;110:135–370.
Nair, A, Vaidya, VA. Cyclic AMP response element binding protein and brain derived neurotrophic factor: molecules that modulate our mood. J Biosci. 2006;31:423–443.
Reid, S, Barbui, C. Long term treatment of depression with selective serotonin reuptake inhibitors and newer antidepressants. BMJ. 2010;340:752–756.
Tanti, A, Belzung, C. Open questions in current models of antidepressant action. Br J Pharmacol. 2010;159:1187–1200.
Timonen, M, LiuKonen, T. Management of depression in adults. BMJ. 2008;336:435–439.
Cao, M. Advances in narcolepsy. Med Clin N Am. 2011;94:541–555.
Jamdar, S, Sathyamoorthy, BT. Management of attention deficit/hyperactivity disorder. Br J Hosp Med. 2007;68:360–366.
Kaplan, G, Newcorn, JH. Pharmacotherapy for child and adolescent attention–deficit hyperactivity disorder. Paed Clin N Am. 2011;58:99–120.
Keam, S, Walker, MC. Therapies for narcolepsy with or without cataplexy: evidence-based review. Curr Opin Neurol. 2007;20:699–703.
Pliszka, SR. Pharmacologic treatment of attention-deficit/hyperactivity disorder: efficacy, safety and mechanisms of action. Neuropsychol Rev. 2007;17:61–72.
Young, TJ, Silber, MH. Hypersomnias of central origin. Chest. 2006;130:913–920.