4 ALTERED CELLULAR FUNCTION
INTRODUCTION
Knowledge of the structural and functional reactions of cells to injurious agents is key to understanding disease processes. Altered cellular biology can result from adaptation, injury, disease, cancer or ageing. Adaptation occurs in response to both normal (or physiological) conditions and adverse (or pathological) conditions. For example, the uterus adapts to pregnancy — a normal physiological state — by enlarging to accommodate the growing fetus. Enlargement occurs because of an increase in the size and number of uterine cells. In contrast, when an adverse condition such as high blood pressure arises, heart cells are stimulated to enlarge because of the increased pressure. However, unlike most of the body’s adaptive mechanisms, cellular adaptations to adverse conditions are usually successful for only a short period of time. If the stress on the cell is severe or long term it will eventually overwhelm the adaptive processes, and cellular injury or death ensues.
Cellular injury may be reversible or irreversible. Cellular injuries from various causes have different clinical and pathophysiological manifestations. Cellular death is confirmed by structural changes seen when cells are examined under a microscope.
Cellular ageing causes structural and functional changes that eventually may lead to cellular death or a decreased capacity to recover from injury. Mechanisms explaining how and why cells age are currently not fully understood, and distinguishing between pathological changes and physiological changes that occur with ageing is often difficult. Ageing clearly causes alterations in cellular structure and function, yet growing old is both inevitable and normal. Furthermore, many older people are able to maintain a remarkable state of health, with little or no apparent disease.
CAUSES OF CELLULAR INJURY
Most diseases begin with cell injury. Cellular injury occurs if the cell is unable to maintain homeostasis — a normal or adaptive steady state — in the face of injurious stimuli. Injured cells may recover or die. Stimuli that can cause damage to the cell include a lack of adequate oxygen supply (hypoxia), chemical agents, physical agents, infectious agents, genetic factors and nutritional imbalances. All of these noxious stimuli (noxious meaning that they can cause harm) may contribute to cell injury, depending on a number of factors.
If cellular injury does occur, the extent of injury depends on the type, state (that is, if the cell is dividing or fully matured) and adaptive processes of the cell. In addition, it is also influenced by the type, severity and duration of the injurious stimulus. Moreover, two individuals exposed to an identical stimulus may incur different degrees of cellular injury — modifying factors, such as nutritional status, can profoundly influence the extent of injury. While the scientific community has a good knowledge of the types and effects of these stimuli on cells, the precise ‘point of no return’ that leads to cellular death remains a biochemical puzzle and the exact mechanisms responsible for the transition from reversible to irreversible cellular damage are not fully understood.
Some of the different causes of cellular injury are summarised in Table 4-1.
Table 4-1 SOME CAUSES OF CELLULAR INJURY
| CAUSE | CHARACTERISTICS | EXAMPLES |
|---|---|---|
| Hypoxia | Lack of cellular oxygen causes an increase in anaerobic respiration leading to a lack of sodium and potassium transport across the cell membrane. Eventually, the cells swell due to fluid accumulation and die. | |
| Nutritional imbalances | Pathophysiological cellular effects develop when nutrients are not consumed in the diet and so are not transported to the body’s cells, or when excessive amounts of nutrients are consumed and transported to the body’s cells. | |
| Physical agents | ||
| Hypothermic injury | Caused by chilling or freezing of cells, creating high intracellular sodium concentrations; or abrupt drops in temperature leading to vasoconstriction and increased viscosity of the blood, and causing ischaemic injury, infarction and necrosis | |
| Hyperthermic injury | Caused by excessive heat; varies in severity according to the nature, intensity and extent of the heat | |
| Atmospheric pressure | Tissue injury caused by compressive waves of air or fluid impinging on the body, followed by a sudden wave of decreased pressure. Changes may collapse the lungs, rupture internal solid organs and cause widespread haemorrhage (bleeding). Also, carbon dioxide and nitrogen, which are normally dissolved in the blood, come out of solution and form small bubbles (called gas emboli), causing hypoxic injury and pain. | |
| Sunlight | Prolonged exposure to sunlight has been strongly linked to skin cancers. | |
| Trauma | Injury is caused by physical impact or irritation; they may be overt or cumulative. | |
Hypoxia
Hypoxia, or lack of sufficient oxygen, is the single most common cause of cellular injury. Hypoxia can result for many reasons, but these can be broadly classified into problems with oxygen entering the blood and problems with transporting oxygen around the body to the cells (see Figure 4-1).
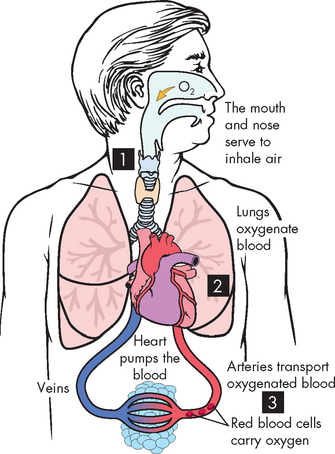
FIGURE 4-1 The different causes of hypoxia.
1 Interruption of oxygen supply to the lungs. 2 Inadequate oxygen entering the blood in the lungs. 3 Inadequate transport of oxygen in the circulation.
Source: Based on Damjanov I. Pathology for the health professions. 3rd edn. St Louis: Saunders; 2006.
Conditions that cause inadequate oxygen delivery to the blood include:
Hypoxia due to insufficient transport of oxygen through the body can occur because of:
 a decrease in haemoglobin (which is the molecule inside red blood cells that carries oxygen around the blood)
a decrease in haemoglobin (which is the molecule inside red blood cells that carries oxygen around the blood)But by far the most common cause of hypoxia in the body is a reduction in blood supply to the cells. This is termed ischaemia, which literally means a restriction in blood flow. This may be observed in many hospital patients and contributes significantly to mortality. For instance, patients with ischaemia of the heart and brain often die because these organs need a constant supply of oxygen.
Ischaemic injury is often caused by the gradual narrowing of arteries (termed arteriosclerosis; see Chapter 23) and complete blockage by blood clots (thrombosis). Progressive hypoxia caused by gradual arterial obstruction is better tolerated than the sudden acute anoxia (total lack of oxygen) caused by a sudden obstruction, as with a blood clot or other plug in the circulation. An acute obstruction in a coronary artery can cause cell death within minutes if the blood supply is not restored.
Chemical agents
Chemical injury to the cell begins with a biochemical interaction between a toxic substance and the cell membrane, which is ultimately damaged, leading to increased permeability. This means that more substances, both harmful and non-harmful, can pass through the cell membrane. There are two general mechanisms:
Many chemical agents cause cellular injury. Highly toxic substances are known as poisons.
Minute amounts of some agents, such as cyanide, can rapidly destroy enough cells to cause the death of the individual in a very short period of time after exposure. Chronic exposure to air pollutants, insecticides and herbicides can also cause cellular injury. Carbon monoxide (in the exhaust of car fumes) and social drugs such as alcohol can significantly alter cellular function and injure cellular structures. Recreational, over-the-counter and prescribed drugs also may cause cellular injury, sometimes leading to death. Accidental or suicidal poisonings by chemical agents cause numerous deaths.
Physical agents
Physical agents that lead to cell injury include blunt trauma to the body resulting in damage to the tissues and penetrating wounds that pass the skin and enter the body, such as surgical incisions. Other forms of physical agents include environmental stressors such as high and low temperatures and sunlight.
Infectious agents
Infectious microorganisms can enter the body and multiply rapidly to cause disease. They may enter the cell, like viruses, or release toxins that are harmful to the cell. Infections in hospital patients are one of the most common clinical presentations and, accordingly, Chapter 14 examines the effects of infection.
Genetic causes
Genetic abnormalities can produce defects in cellular metabolism, alter the structure and function of cells and make cells more susceptible to injury. Genetic abnormalities and environmental influences on genes are discussed in Chapter 37.
MECHANISMS OF CELLULAR INJURY
There are four common biochemical themes that are important in understanding cell injury and cell death regardless of the injurious agent. These are ATP depletion, oxygen and oxygen-derived free radicals, increased intracellular calcium and loss of calcium steady state, and defects in cell membrane permeability (see Table 4-2). However, the mechanisms that drive cellular damage can be grouped under three main areas:
Table 4-2 MECHANISMS OF CELL INJURY
| MECHANISM | EFFECTS ON THE CELL |
|---|---|
| ATP depletion | Loss of mitochondrial ATP and decreased ATP production. Results include cellular swelling, decreased protein synthesis and decreased membrane transport — all changes that contribute to loss of integrity of the cell membrane. |
| Oxygen and oxygen-derived free radicals | Lack of oxygen is key in the progression of cell injury in ischaemia (reduced blood supply); reactive oxygen species cause destruction of the cell membrane and cell structure. |
| Intracellular calcium increased | Normally, intracellular calcium concentrations in the cytoplasm are very low. Ischaemia causes an increase in cytoplasmic calcium concentrations; sustained levels of calcium continue to increase with damage to the cell membrane. |
| Defects in cell membrane permeability | Early loss of selective membrane permeability is found in all forms of cell injury. |
We start with hypoxic injury, which can be relatively common in patients in hospital settings.
Hypoxic injury
Cellular responses to hypoxic injury caused by ischaemia have been demonstrated in studies of the heart muscle. Within 1 minute of blood supply to the heart muscle (myocardium) being interrupted, the heart becomes pale and has difficulty contracting normally. Within 3–5 minutes, the ischaemic portion of the myocardium ceases to contract because of a rapid decrease in the ability of the mitochondria to produce ATP. Lack of ATP leads to increased anaerobic metabolism, which can only generate ATP from glycogen (a stored form of glucose) when there is insufficient oxygen. When glycogen stores are depleted, even anaerobic metabolism ceases and no ATP production occurs.
A reduction in ATP levels causes the cell membrane’s sodium–potassium (Na+–K+) pump and sodium–calcium exchange to fail, which leads to an intracellular accumulation of sodium and calcium and diffusion of potassium out of the cell. The sodium–potassium pump uses ATP to actively pump sodium out of the cell and potassium into the cell, which is vital to cell homeostasis. Sodium and water then can enter the cell freely causing cellular swelling; as well, early dilation of the endoplasmic reticulum results. Dilation causes the ribosomes to detach from the rough endoplasmic reticulum, reducing protein synthesis. With continued hypoxia, the entire cell becomes markedly swollen, with increased concentrations of sodium, water and chloride, and decreased concentrations of potassium.
It should be noted that these disruptions will be reversed if oxygen is made available and restored. However, if oxygen is not restored, swelling of lysosomes and marked mitochondrial swelling occurs. Continued hypoxic injury with accumulation of calcium subsequently activates multiple enzyme systems, resulting in membrane damage, cytoskeleton disruption, activation of inflammation, DNA degradation and eventual cell death (see Figure 4-2). Structurally, with plasma membrane damage, calcium from outside the cell readily moves into the cell and intracellular calcium stores are released. Intracellular calcium activates enzymes that can further damage cell membrane proteins, ATP and nucleic acids. Irreversible damage is characterised by two events:
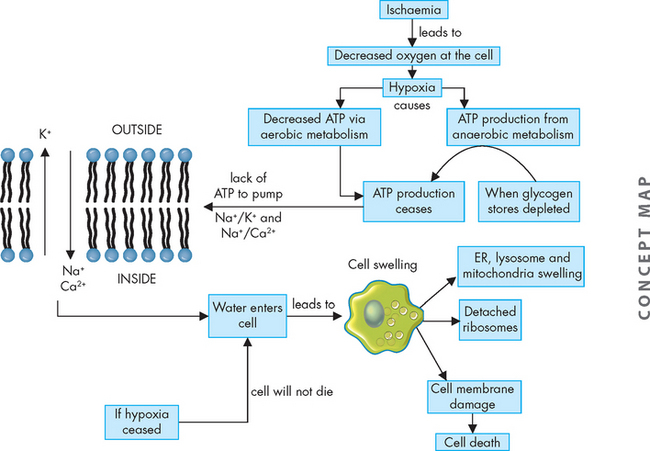
Ischaemia causing hypoxia which leads to reduction in ATP production. If the hypoxic insult continues, a series of intracellular changes occur leading to cell death.
Restoration of oxygen, however, can cause additional injury called reperfusion injury (see Figure 4-3). Briefly, this occurs during the process of ATP production in the mitochondria, where oxygen is transformed to water and small amounts of partially reduced oxygen are formed, called free radicals. Reperfusion injury results from the generation of free radicals, which are chemically highly unstable and can cause damage to the cell membrane by undergoing destructive chemical reactions. This process is sometimes referred to as oxidative stress and is discussed in the next section.
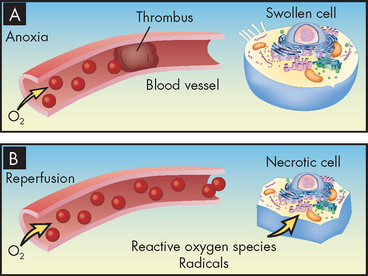
FIGURE 4-3 Reperfusion injury.
A Without oxygen, or anoxia, the cells display hypoxic injury and become swollen. B With reoxygenation, reperfusion injury increases because of the formation of reactive oxygen radicals that can cause cell necrosis.
Source: Based on Damjanov I. Pathology for the health professions. 3rd edn. St Louis: Saunders; 2006.
The impact of oxygen and oxygen-derived free radicals
Although we require oxygen to survive, oxygen can actually be particularly harmful to cells — this is extremely surprising! To understand how this occurs, we need to delve into some chemistry. Free radicals are formed during ATP production from partially reduced oxygen molecules called reactive oxygen species. A free radical is an atom that is electrically uncharged yet is unstable; this means that it is prone to stealing or donating a charge from another molecule. When a molecule has its charge stolen, it becomes a free radical. Therefore, it is capable of injurious chemical bonding with proteins, lipids and carbohydrates — key molecules in membranes and nucleic acids (see Chapter 2).
Free radicals are highly reactive and can react with most molecules close by. Normally, this is not an issue, as the body produces only a small amount of free radicals. Furthermore, very efficient scavengers, called antioxidants, clean up these molecules before they can undergo chemical reactions. However, during periods of stress, more reactive oxygen species are produced and free radicals become more difficult to control. They accumulate when cells are injured and initiate many chain reactions, causing damage. The large numbers of reactive oxygen species overwhelm the balance by antioxidants. This inefficiency of antioxidants is even more serious in mitochondria because mitochondria do not have an enzyme to degrade the free radicals.1 Consequently, excessive production of reactive oxygen species in mitochondria will damage lipids, proteins and mitochondrial DNA (mDNA), leading to irreversible cell death (see Figure 4-4).2–5 3 4 5 Mitochondrial oxidative stress has been implicated in heart disease, Alzheimer’s disease and Parkinson’s disease, as well as ageing itself.5–8 6 7 8
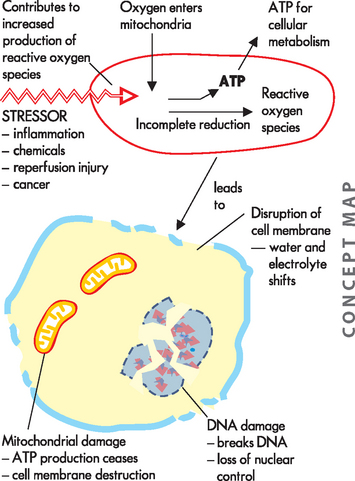
Natural antioxidants
Nutrient antioxidants — vitamin C, vitamin E and beta-carotene, a precursor to vitamin A — work by inactivating free radicals. Especially important is the prevention of oxidative damage to mitochondrial DNA. Vitamin C, found in citrus fruits, broccoli and potatoes, is probably the most notable of the antioxidant nutrients. A water-soluble vitamin, it is the first line of defence, scavenging free radicals before they enter cell membranes. Vitamin C promotes wound healing, growth and tissue repair. It also enhances the effect of vitamin E. It is known to lower the risk of heart disease and cataracts. Vitamin E, which is fat-soluble and available in unprocessed oils, wheat germ, hazelnuts, almonds, egg yolk and butter, does much of its protective antioxidant work within the lipid-rich cell membrane. It is an anticoagulant and important in the formation of blood cells. It also helps to utilise vitamin K, and it reduces the risk of cataracts. Beta-carotene, found in carrots, dark green and yellow-orange vegetables and fruits, leafy vegetables, tomatoes, spinach, squash and broccoli, is converted to vitamin A in the small intestine and may be associated with reduced risk of cancer, cataracts and heart disease.
Free radicals cause several damaging effects by (1) lipid peroxidation, which is the destruction of lipids leading to membrane damage and increased permeability; (2) attacking critical proteins that affect ion pumps and transport mechanisms; (3) fragmenting DNA, causing decreased protein synthesis; and (4) damaging mitochondria, causing the liberation of calcium into the cytoplasm. Because of the increased understanding of free radicals, a growing number of diseases and disorders have been linked either directly or indirectly to these oxygen reactive species (see Box 4-1).
Alteration to calcium homeostasis
The concentration of calcium inside the cell is quite low compared with the concentration in the extracellular fluid. Also, it tends to be stored in the mitochondria and endoplasmic reticulum and is released only when needed, so the amount of calcium actually in the cytoplasm is low. The role of calcium is to activate enzymes in the cell and it is intricately involved in muscle contraction. Therefore, like the sodium–potassium pump, calcium too is pumped out of the cell to keep the concentration low. However, if the cell membrane is damaged, such as occurs when the cell is hypoxic, calcium enters the cell and the concentration in the cytoplasm increases. Stored calcium is also released, further increasing the cytoplasmic concentration. Unfortunately, this increased intracellular calcium causes cell injury by:
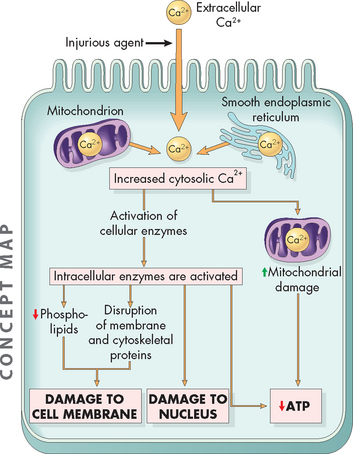
FIGURE 4-5 The mechanism by which increases in intracellular calcium cause cellular damage.
Calcium enters the cell and is released from the stores in the mitochondria and endoplasmic reticulum. This activates enzymes that cause a decrease in ATP production and damage the cell membrane and nucleus.
Source: Based on Kumar V. Robbins & Cotran pathological basis of disease. 8th edn. Philadelphia: Saunders; 2010.
CELLULAR ADAPTATION
We have looked at the causes of cellular injury and how these agents affect cell structure and function. Now we need to explore what cells do in response to these insults. A number of factors determine cellular function and structure — namely genetics, the type of surrounding cells and whether the cell can readily obtain nutrients and oxygen — but if cells are placed under continual stress, they have the ability to adapt. Cells adapt to their environment to escape and protect themselves from injury. An adapted cell is neither normal nor injured — its condition lies somewhere between these two states.
Cellular adaptations are a common and central part of many disease states. In the early stages of a successful adaptive response, cells may have enhanced function; thus, it is hard to know whether the response is pathological or an extreme adaptation to an excessive functional demand. The most significant adaptive changes in cells are:
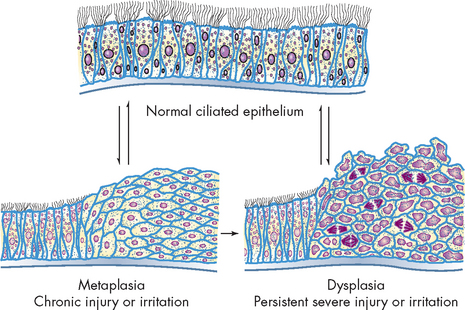
Another change, which is not truly adaptive, is dysplasia. This involves abnormal change in the size, shape and arrangement of mature cells. These changes are shown in Figure 4-6.
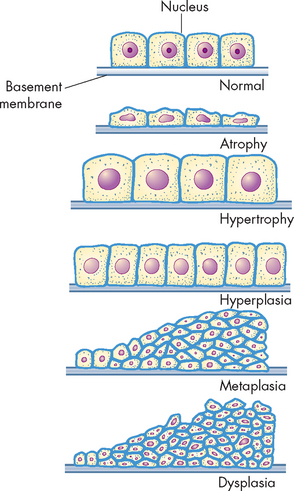
Atrophy
Atrophy is a decrease or shrinkage in cellular size. If atrophy occurs in a sufficient number of an organ’s cells, the entire organ shrinks and becomes smaller. Atrophy can affect any organ, but it is most common in skeletal muscle, the heart, secondary sex organs and the brain, and is especially related to ageing (see Figure 4-7). Atrophy can be classified as physiological or pathological. Physiological atrophy occurs with early development. For example, the thymus gland, a gland involved in immune system function, undergoes the normal process of physiological atrophy during childhood. The reasons for this are unknown. Pathological atrophy occurs as a result of decreases in workload, pressure, use, blood supply, nutrition and hormonal and nervous system stimulation. Individuals immobilised in bed for a prolonged time exhibit a type of skeletal muscle atrophy called disuse atrophy. This is prevalent in hospitalised patients who cannot be mobilised; for instance, patients with multiple bone fractures or significant obesity. Ageing causes brain cells to become atrophic and endocrine-dependent organs, such as the gonads (testes or ovaries), to shrink as hormonal stimulation decreases. Whether atrophy is caused by normal physiological conditions or by pathological conditions, atrophic cells exhibit the same basic changes.
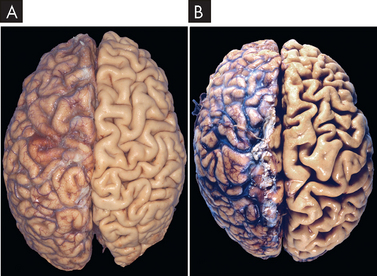
FIGURE 4-7 Atrophy of the brain.
A Normal brain of a young adult. B Atrophy of the brain of an 82-year-old male with cerebrovascular disease, resulting in reduced blood supply. Note: the meninges (a vascular layer of the brain) have been stripped from the right half of each specimen to reveal the surface of the brain. Viewed from the top of the brain.
Source: Kumar V. Robbins & Cotran pathological basis of disease. 8th edn. Philadelphia: Saunders; 2010.
Cells that have atrophied contain less endoplasmic reticulum and fewer mitochondria than normal cells. These cells also reduce their oxygen consumption and amino acid uptake is reduced. The biochemical changes of atrophy are just beginning to be understood. The mechanisms probably include decreased protein synthesis, increased protein catabolism (breakdown), or both.9
Hypertrophy
Hypertrophy is an increase in the size of cells and consequently in the size of the affected organ. The cells of the heart and kidneys are particularly prone to enlargement. The increased cellular size is associated with an increased accumulation of protein in the cellular components (the cell membrane, endoplasmic reticulum and mitochondria) and not with an increase in cellular fluid. Hypertrophy, like atrophy, can be physiological or pathological and is caused by specific hormone stimulation or by increased functional demand. The triggers for hypertrophy include two types of signals: (1) mechanical signals, such as stretch; and (2) chemical signals, such as growth factors, which stimulate growth. For example, in skeletal muscles (the ones you contract to move bones), physiological hypertrophy occurs in response to heavy work. Hypertrophy is the mechanism that occurs in body builders who lift heavy weights repeatedly — the size of their muscles increases. Muscular hypertrophy tends to diminish if the excessive workload diminishes. Another example of normal or physiological hypertrophy is the increased growth of the uterus and mammary glands in response to pregnancy. A pathological example is pathophysiological hypertrophy of the heart, secondary to mechanical problems, such as faulty heart valves (see Figure 4-8).
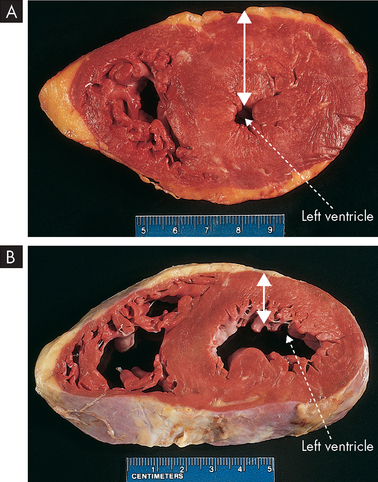
A Hypertrophy of heart muscle in the left ventricle (the major chamber of the heart). B Normal left ventricle size. Arrows indicate thickness of the muscle wall and demonstrate the large increase that occurs with hypertrophy. Images show a transverse slice through the heart.
Source: Kumar V et al. Robbins basic pathology. 8th edn. Philadelphia: Saunders; 2007.
Hyperplasia
Hyperplasia is an increase in the number of cells resulting from an increased rate of cellular division. Hyperplasia, as a response to injury, occurs when the injury has been severe and prolonged enough to have caused cell death. Loss of epithelial cells and cells of the liver and kidneys triggers DNA synthesis and mitotic division (discussed in Chapter 5). Increased cell growth is a multistep process involving the production of growth factors, which stimulate the remaining cells to produce new cell components and, ultimately, to divide. Hyperplasia and hypertrophy often occur together and both take place if the cells can produce DNA; however, in non-dividing cells only hypertrophy occurs.
Two types of normal, or physiological, hyperplasia are (1) compensatory and (2) hormonal. Compensatory hyperplasia is an adaptive mechanism that enables certain organs to regenerate. For example, removal of part of the liver leads to hyperplasia of the remaining liver cells (hepatocytes) to compensate for the loss. Even with removal of 70% of the liver, regeneration of the liver occurs over time! Hormonal hyperplasia occurs in response to release of a hormone. The most common example of this is enlargement of the uterus in the female when pregnancy occurs.
Pathological hyperplasia is the abnormal proliferation of normal cells, usually in response to excessive hormonal stimulation or growth factors on target cells. One of the most common examples of pathological hyperplasia in males is enlargement of the prostate, which occurs in a significant proportion of males over the age of 60 years.10
Metaplasia
Metaplasia is the reversible replacement of one mature cell type by another, sometimes less differentiated, cell type. It is thought to develop from a reprogramming of stem cells (cells that replicate and can turn into one of many different types of cells) in epithelial or connective tissue. These precursor cells mature along a new pathway because of chemical signals in the cells’ environment. This may be precipitated by a persistent irritant to the cells, such as cigarette smoking. Metaplasia occurs when normal epithelial cells lining the upper airways in the lungs (columnar cells) are replaced by stratified squamous epithelial cells (see Figure 4-9). The newly formed cells do not secrete mucus or have cilia (projections that protect the lining of the airway), causing loss of a vital protective mechanism. Metaplasia can be reversed if the inducing stimulus, cigarette smoking in this example, is removed. With prolonged exposure to the inducing stimulus, however, dysplasia and cancerous transformation may occur.
Dysplasia
Dysplasia refers to abnormal changes in the size, shape and organisation of mature cells (see Figure 4-9). Dysplasia is not considered a true adaptive process but is related to hyperplasia. Dysplastic changes are often encountered in epithelial tissue of the cervix and respiratory tract, where they are strongly associated with cancer development. However, it should be noted that dysplastic cell changes are likely to be adaptive in nature, but have gone off course, meaning that if the inciting stimulus is removed early enough, dysplastic changes are often reversible.
REVERSIBLE AND IRREVERSIBLE CELL INJURY
All cells in the body require oxygen and nutrients to provide fuel for cellular metabolism. There must be a constant flow of blood with oxygen and the necessary molecules that cells use as raw materials to be able to produce ATP, as well as for other chemical reactions. In addition, cells produce waste products, such as carbon dioxide, that need to be removed from the cell, the space surrounding the cell and ultimately out of the body. To achieve these processes, the intracellular and extracellular fluids interact in a homeostatic balance to facilitate cellular function. This is considered to be the normal state and it operates using a steady state — that is, slight variations in function are allowed, but essentially homeostasis is maintained.
However, we have seen that a decrease in blood flow (ischaemia) or hypoxia severely affects the cell’s normal functions. In fact, any alteration to the extracellular environment will eventually affect inside the cell and possibly lead to a disruption in cell function. If the noxious stimulus is severe and is not removed, the cell may undergo adaptations in an attempt to cope with the change in environmental conditions. If the stimulus ceases, over time the cell will return to a normal state, thereby reversing its injury — this is known as reversible cell injury. However, if the insult continues, it is very likely that a critical point will be reached and irreversible cell injury occurs. This is shown in Figure 4-10. In the following sections we explore these two concepts in more detail.
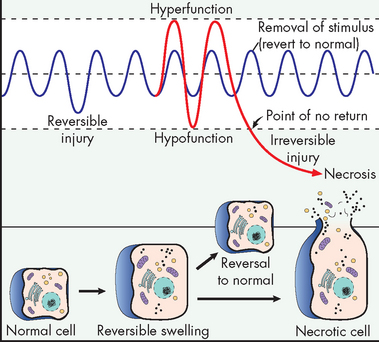
FIGURE 4-10 Changes in cellular function that can lead to reversible and irreversible cell damage.
Source: Damjanov I. Pathology for the health professions. 3rd edn. St Louis: Saunders; 2006.
Reversible cell injury
Cellular injury occurs if the cell is unable to maintain homeostasis (see Figure 4-11). However, not all injured cells die — some may recover. The exact mechanisms for why some cells survive and recover and others progress to cell death is not fully understood. This feature has been termed the ‘point of no return’ and is the defining feature of reversible cell injury. If this point is not passed, the cell can recover.
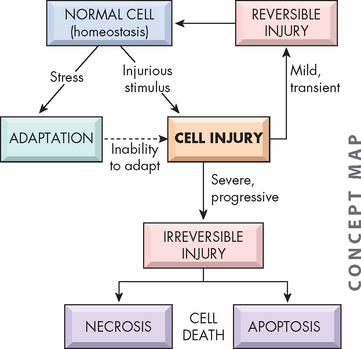
FIGURE 4-11 Stages of cellular response to stress and injurious stimuli.
Source: Kumar V. Robbins & Cotran pathological basis of disease. 8th edn. Philadelphia: Saunders; 2010.
Intracellular accumulations, also known as infiltrations, occur not only when injury is sub-lethal and sustained in injured cells, but also in normal cells. Such accumulations can may or may not be toxic to the cell and consist of substances that are normally present, such as fluids and electrolytes, lipids, glycogen, calcium and proteins. Abnormal accumulations of these substances can occur in the cytoplasm (often in the lysosomes) or in the nucleus if: (1) the normal, endogenous substance is produced in excess or at an increased rate; (2) a substance (normal or abnormal) is not effectively catabolised, usually because of a lack of a vital lysosomal enzyme; or (3) harmful exogenous materials, such as heavy metals or microorganisms, accumulate because of inhalation, ingestion or infection.
Two types of intracellular accumulation are commonly associated with reversible cell injury:
Water swelling. Cellular swelling, the most common degenerative change, is caused by the shift of extracellular water into the cells. In hypoxic injury, movement of fluid and ions into the cell is associated with acute failure of metabolism and loss of ATP production. Normally, the pump that transports sodium ions out of the cell is maintained by the presence of ATP (the sodium–potassium pump). In metabolic failure caused by hypoxia, reduced ATP allows sodium to accumulate in the cell while potassium diffuses outwards. This is not normal, as sodium should be in greater concentrations outside the cell and potassium greater inside the cell. The increased intracellular sodium increases osmotic pressure, drawing more water into the cell. The endoplasmic reticulum becomes distended, ruptures and forms large vacuoles (enclosed compartments) that isolate the water from the cytoplasm. If cellular swelling affects all the cells in an organ, the organ increases in weight and becomes distended (stretched) and pale. Increased deposition of lipids. Although lipids sometimes accumulate in heart and kidney cells, the most common site of intracellular lipid accumulation, or fatty change, is liver cells. Because hepatic (liver) metabolism and secretion of lipids are crucial to proper body function, imbalances and deficiencies in these processes lead to major pathological changes. Lipid accumulation in liver cells causes fatty liver or fatty change (see Figure 4-12). As lipids fill the cells, vacuolation pushes the nucleus and other organelles aside. Lipid accumulation in liver cells occurs after cellular injury. Grossly, the liver looks yellowish and greasy.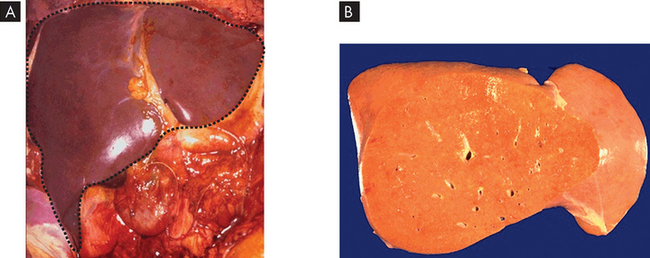
A Normal liver in the upper abdomen (dashed black line). It has a shiny, smooth appearance and is a deep red colour. B Fatty liver due to accumulation of lipid intracellularly. The liver is pale yellow and smooth (the front or anterior part of the liver has been removed).
Source: Klatt EC. Robbins & Cotran atlas of pathology. 2nd edn. Philadelphia: Saunders; 2010.
Irreversible cell injury
If the stimulus is severe and sustained and the ‘point of no return’ is passed, irreversible cell injury ensues and the cell dies. However, there are two types of irreversible cell injury and their mechanisms are quite different (see Figure 4-11):
Necrosis is a common form of cellular death with severe cellular swelling and breakdown of the organelles in response to sustained, severe, noxious stimuli.The defining feature between these two forms is that apoptosis is usually a normal cellular function that is not associated with any form of cellular insult, whereas necrosis always arises in response to pathophysiological processes.
Apoptosis
Apoptosis (‘dropping off’) is an important, distinct type of cell death that differs from necrosis in several ways (see Figure 4-13). Apoptosis is an active process of cellular self-destruction called programmed cell death. Why would cells be programmed to die after a set amount of time? The answer is very simple: cells need to die otherwise endless proliferation would lead to gigantic bodies. In addition, cells become ‘worn out’ and need to be replaced. Every day the average adult may create 10 billion new cells — and kill off the same number.11 A specific set of enzymes and genes are activated to cause apoptosis. These genes are sometimes called suicide cells because their activation by the nucleus inactivates so-called life-sustaining genes and promotes pathways leading to killer genes.
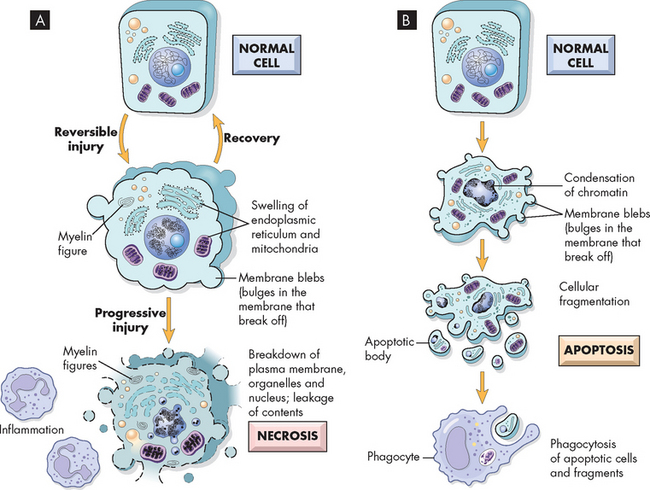
FIGURE 4-13 Schematic illustration of the changes in cell injury causing necrosis and apoptosis.
A If the cellular injury is reversible, the cell may recover and return to normal. If the injury is irreversible, the cell progresses through to necrosis. B On the other hand, apoptosis is an intentional program to cause cell suicide.
Source: Based on Kumar V. Robbins & Cotran pathological basis of disease. 8th edn. Philadelphia: Saunders; 2010.
Apoptosis affects scattered, single cells; however, there are examples of it occurring in widespread areas. The process of apoptosis consists of nuclear and cytoplasmic shrinkage of a cell. This is followed by fragmentation into membrane-bound fragments and subsequent phagocytosis (literally cell eating — specialised cells (phagocytes) perform this task, and these are discussed in detail in Chapter 12) by neighbouring healthy cells. As a controlled process in normal development, apoptosis determines the size, patterning and function of many tissues.12,13 Apoptosis occurs throughout the life span — from birth to old age. It can be activated by outside factors — for example, a long-lasting viral infection — or triggered internally by the absence of certain growth factors (these normally stimulate tissues to grow). Accordingly, apoptosis can be classified as physiological or pathological:
Physiological apoptosis is important in the development of body tissue. It is responsible for local deletion of cells during tissue turnover and normal embryonic development. One important example of widespread areas of apoptosis is in the central nervous system during childhood and adolescence. We are born with far more neurons than are required in the adult brain and apoptosis in the early years of life removes the unnecessary neurons.Necrosis
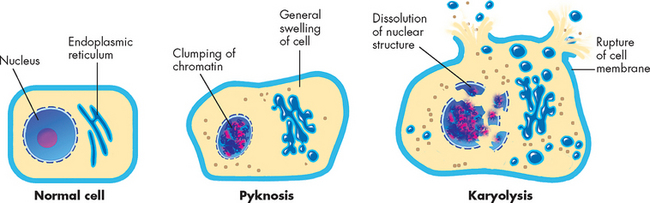
Cellular death eventually leads to cellular dissolution or necrosis. Necrosis is the sum of cellular changes after local cell death and the process of cellular self-digestion known as autodigestion, or autolysis (see Figure 4-13). Cells die long before any necrotic changes are noted by light microscopy.14 The structural signs that indicate irreversible injury and progression to necrosis are dense clumping and progressive disruption of genetic material, and disruption of the cellular and organelle membranes. In later stages of necrosis, most organelles are disrupted and karyolysis (dissolving of the nucleus due to enzymes) is under way. In some cells, the nucleus shrinks and becomes a small, dense mass of genetic material (pyknosis). The pyknotic nucleus eventually dissolves by karyolysis as a result of the action of lysosomal enzymes on DNA. These processes are shown in Figure 4-14.
Different types of necroses tend to occur in different organs or tissues and sometimes can indicate the mechanism or cause of cellular injury. The four major types of necroses are coagulative, liquefactive, caseous and fatty. Another type, gangrenous necrosis, is not a distinctive type of cell death but refers instead to larger areas of tissue death. These necroses are summarised as follows:
Coagulative necrosis. Occurs primarily in the kidneys, heart and adrenal glands and commonly results from hypoxia caused by severe ischaemia. Coagulation is caused by protein denaturation (a process that modifies the molecular structure of protein), which causes the protein albumin to change from a gelatinous, transparent state to a firm, opaque state (see Figure 4-15). Liquefactive necrosis. Commonly results from ischaemic injury to nerve cells in the brain (see Figure 4-16). Dead brain tissue is readily affected by liquefactive necrosis because brain cells are rich in digestive enzymes and lipids and the brain contains little connective tissue. Cells are digested by their own enzymes, so the tissue becomes soft, liquefies and is walled off from healthy tissue. Caseous necrosis. Usually results from a lung infection that caused tuberculosis (see Figure 4-17). It is a combination of coagulative and liquefactive necroses. The dead cells disintegrate, but the debris is not completely digested by enzymes. Tissues resemble clumped cheese — hence the name caseous, in that they are soft and granular. Fat necrosis. Fat necrosis is cellular dissolution caused by powerful enzymes, called lipases, that occur in the pancreas and other abdominal structures (see Figure 4-18). Lipases break down triglycerides, releasing free fatty acids, which then combine with calcium, magnesium and sodium ions, creating soaps (called saponification). The necrotic tissue appears opaque and chalk-white. Gangrenous necrosis. Refers to the death of tissue and results from severe hypoxic injury. This commonly occurs because of blockages of arteries, particularly those in the lower leg (see Figure 4-19). With hypoxia and subsequent bacterial invasion, the tissues can undergo necrosis. In dry gangrene the skin becomes very dry and shrinks, resulting in wrinkles, and its colour changes to dark brown or black. Wet gangrene develops when the main white blood cells of the body, neutrophils, invade the site, causing liquefactive necrosis. This usually occurs in internal organs, causing the site to become cold, swollen and black. A foul odour is present, and if systemic symptoms become severe, death can ensue.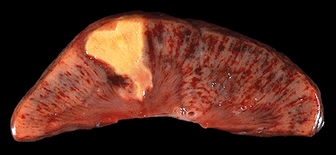
FIGURE 4-15 Coagulative necrosis.
This picture of a kidney shows a wedge shape of dead cells (yellow).
Source: Kumar V. Robbins & Cotran pathological basis of disease. 8th edn. Philadelphia: Saunders; 2010.
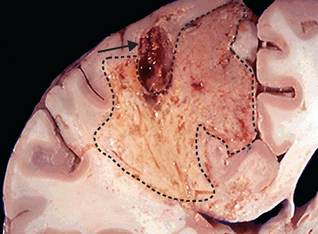
FIGURE 4-16 Liquefactive necrosis.
An area in the brain of necrosis, showing dissolution of the tissue (dashed line). The arrow indicates a blood clot that caused the cell death.
Source: Kumar V. Robbins & Cotran pathological basis of disease. 8th edn. Philadelphia: Saunders; 2010.
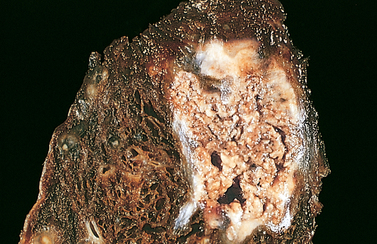
Tuberculosis of the lung, with a large area of caseous necrosis containing yellow-white and cheesy debris.
Source: Kumar V. Robbins & Cotran pathological basis of disease. 8th edn. Philadelphia: Saunders; 2010.
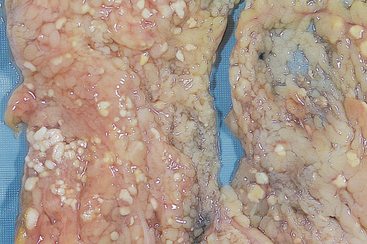
The areas of white chalky deposits throughout the tissue represents fat necrosis with soap formation (saponification) at sites of lipid breakdown.
Source: Kumar V. Robbins & Cotran pathological basis of disease. 8th edn. Philadelphia: Saunders; 2010.
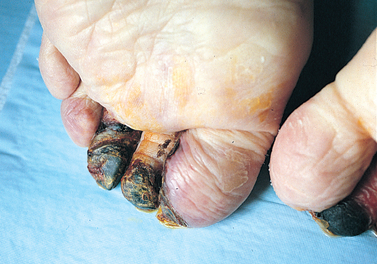
FIGURE 4-19 Gangrenous necrosis.
Dry gangrene of the toe from severe ischaemia to the lower leg.
Source: Damjanov I. Pathology for the health professions. 3rd edn. St Louis: Saunders; 2006.
AGEING
Ageing usually is defined as a normal physiological process that is both universal and inevitable — that means all of us will get older! Ageing traditionally has not been considered a disease because it is ‘normal’; and disease is usually considered ‘abnormal’. Conceptually, this distinction seems clear until the concept of injury is introduced. Some pathologists have defined disease as the result of injury. Ageing has been defined as the time-dependent loss of structure and function that proceeds slowly and in such small increments that it appears to be the result of the accumulation of small, imperceptible injuries — a gradual result of ‘wear and tear’.
However, as individuals age, they are continually exposed to agents that may cause cellular injury. These may result from unavoidable aspects of life — like joint movement, which contributes to the wear of the cellular structures that comprise these tissues. In addition, microinsults (little insults) caused by continuous bombardment by ultraviolet light, environmental temperature changes, infectious agents and chemical reactions in the body that can produce substances like free radicals all may alter cellular structure and function.15 In this context, the distinction between ageing and disease becomes unclear. For example, some degree of atrophy of the brain is considered normal in old age until it proceeds far enough to cause clinically significant disability and is called a disease. Likewise, all human beings have atherosclerosis (gradual thickening and hardening of the arterial walls due to deposition of fats called plaques) and the plaques progress with age, but at what point in this progression is atherosclerosis considered abnormal?
These conceptual distinctions have given rise to two general categories of theories of ageing. The first category proposes that ageing is the result of the accumulation of random injuries and events — general ‘wear and tear’. The second category proposes that ageing is the result of a genetically controlled developmental program or built-in self-destructive processes. There is an ongoing debate in the scientific community as to which is more likely or if there is another reason or possibly a combination of both theories. To date, evidence exists both for and against any particular theory of ageing.
Genetic and environmental factors
Cellular ageing results from wear and tear that causes functional changes and eventual cellular death. Cells may become damaged during replication as a result of factors within the cell, such as DNA and protein mechanisms, or factors outside the cell. Cells may already be programmed at birth or injured during life so as to cause errors in mitosis and in the replication of genetic material, eventually leading to either cellular atrophy or death. Atrophy is common in the testes, ovaries, uterus and breasts of aged individuals, although these organs age differently.
One genetic mechanism of ageing is programmed ageing. Regardless of damaging environmental factors, some investigators think that each normal cell may have a finite life span during which it can replicate. This may be in the form of a genetic program that progressively slows or shuts down physiological mechanisms, including mitosis, and so cells are not replaced.
Extracellular factors (those outside of the cell) that affect the ageing process include the binding of connective tissue, which makes it stiffer; the increase in free radicals’ effects on cells; the structural alterations of tendons, ligaments, bones and joints; and diseases of the blood vessels, particularly athersclerosis (see Chapter 23).
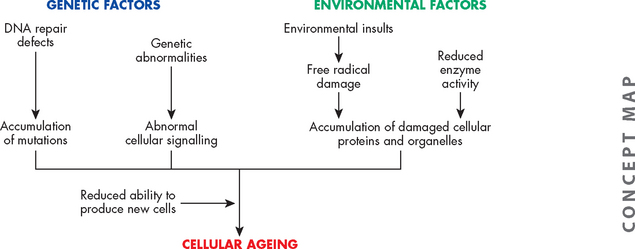
Reactive oxygen species, produced during aerobic metabolism, damage tissues during the ageing process. These oxygen products are extremely reactive and can damage nucleic acids, destroy carbohydrates, damage proteins and fatty acids, and kill cells. Oxidant effects on target cells can also cause DNA damage. That progressive and cumulative damage from oxygen radicals may lead to harmful alterations in cellular function is consistent with those alterations of ageing. Because these oxygen-reactive species not only can permanently damage cells but also may lead to cell death, there is new support for their role in the ageing process. The combination of genetic and environmental factors on ageing is summarised in Figure 4-20.
Cellular, tissue and systemic ageing
The maximal life span of humans is between 80 and 100 years. In Western societies, many individuals attain the maximal life span, which is primarily due to improvements in nutrition, housing, water quality, hygiene, sanitation and, importantly and relevant to you as a student of health science, health care. Although the maximal life span has not changed significantly over time, the average life span, or life expectancy, has increased steadily in Western countries over the last 100 years. It is anticipated that this trend will continue,16 although this has been disputed due to increasing obesity and diabetes mellitus epidemics.17 In addition, the death rate in the elderly population — those 65 years of age and older — has declined significantly, largely as a result of decreased incidence and superior management of cardiovascular disease.
Cellular changes characteristic of ageing include atrophy, decreased function and loss of cells, possibly caused by apoptosis. Loss of cellular function from any of these causes initiates the compensatory mechanisms of hypertrophy and hyperplasia of remaining cells, which can lead to metaplasia, dysplasia and cancers. In the aged cell, DNA, RNA, cellular proteins and membranes are most susceptible to injurious stimuli. DNA is particularly vulnerable to such injuries, which can cause breaks, deletions and additions in the DNA code. Lack of DNA repair increases the cell’s susceptibility to mutations that may be lethal or promote the development of cancer (see Chapters 36 and 37).
It is probably safe to say that every physiological process functions less efficiently with increasing age. The most characteristic tissue change with age is a progressive stiffness or rigidity that affects many systems, including the arterial, pulmonary (lungs) and musculoskeletal systems. A consequence of blood vessel and organ stiffness is a progressive increase in the resistance to blood flow, which means that cells and tissues may not receive enough blood supply. Changes in the endocrine and immune systems affect the elderly individual’s ability to combat invasions of foreign substances, like bacteria. In women, the reproductive system loses oocytes, and in men sperm production decreases. The stomach experiences decreases in the rate of emptying and secretion of hormones and acid. Muscular atrophy diminishes mobility by decreasing motor tone and contractility. Sarcopenia, loss of muscle mass and strength, can occur into old age. The skin of the aged individual is affected by atrophy and wrinkling of the epidermis and alterations in the underlying dermis, fat and muscle. We consider each of these changes as we progress through the chapters on individual body systems.
Total body changes include a decrease in height; a reduction in circumference of the neck, thighs and arms; widening of the pelvis; and lengthening of the nose and ears. Several of these changes are the result of tissue atrophy and decreased bone mass. Body composition changes with age. With middle age, there is an increase in body weight (men gain weight until about 50 years of age and women until 70 years) and fat mass, followed by a decrease in stature, weight, fat-free mass (which includes all minerals, proteins and water plus all other constituents except lipids) and body cell mass at older ages. As fat increases, total body water decreases, so older individuals have less body water and are more prone to dehydration. Increased body fat and centralised fat distribution (abdominal) are associated with diabetes mellitus and heart disease. Some of these changes are summarised in Figure 4-21.
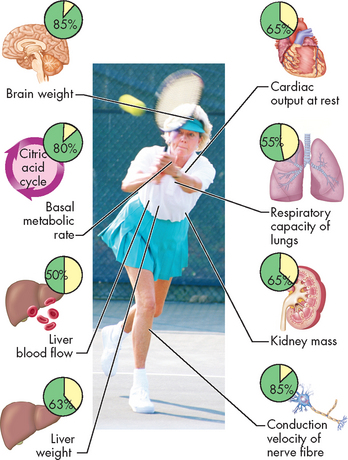
FIGURE 4-21 Biological changes associated with ageing.
Insets show the proportion of remaining function in the organs of an elderly person compared with that of a 20-year-old. These data are for illustration only; individuals exhibit highly variable changes over the life span.
Source: Based on Patton KT, Thibodeau GA. Anatomy & physiology. 7th edn. St Louis: Mosby; 2010.
Although some of these alterations are probably inherent in ageing, others represent consequences of the process. Advanced age increases susceptibility to disease, and death occurs after an injury or insult because of diminished cellular, tissue and organic function.
DEATH
The word somatic refers to the body, so technically we call the death of an individual somatic death. Unlike the changes that follow cellular death in a live body, post-mortem changes after somatic death are widespread. Within minutes after death, post-mortem changes appear, eliminating any difficulty in determining that death has occurred. The most notable manifestations are complete cessation of breathing and circulation, meaning there is no heart beat. The surface of the skin usually becomes pale and yellowish.
Body temperature falls gradually immediately after death and then more rapidly until, after 24 hours, body temperature equals that of the environment.18 After death caused by certain infective diseases, body temperature may continue to rise for a short time. Post-mortem reduction of body temperature is called algor mortis, from the Latin, meaning coolness of death.
The pupils become dilated and do not react to light. The face, nose and chin become sharp or peaked-looking as blood and fluids drain away.19 Gravity causes blood to settle in the lowest tissues, which develop a purple discolouration. Incisions made at this time usually fail to cause bleeding. The skin loses its elasticity and transparency.
Within 6 hours after death, acidic compounds accumulate within the muscles because of the breakdown of carbohydrate and depletion of ATP. This interferes with proteins in the muscle that are responsible for muscle contraction, causing muscle stiffening, or rigor mortis. The smaller muscles are usually affected first, particularly the muscles of the jaw. Within 12–14 hours, rigor mortis usually affects the entire body. Rigor mortis gradually diminishes and the body becomes flaccid at 36–62 hours.
Causes of cellular injury
Mechanisms of cellular injury
Biochemical changes that are important to cell injury are: (a) ATP depletion; (b) oxygen and oxygen-derived free radicals; and (c) intracellular calcium and loss of calcium steady state. The sequence of events leading to cell death is commonly decreased ATP production, failure of active transport mechanisms (the sodium–potassium pump), cellular swelling, detachment of ribosomes from the endoplasmic reticulum, cessation of protein synthesis, mitochondrial swelling as a result of calcium accumulation, vacuolation, leakage of digestive enzymes from lysosomes, autodigestion of intracellular structures, lysis of the cell membrane and death. The initial insult in hypoxic injury is usually ischaemia (the cessation of blood flow into vessels that supply the cell with oxygen and nutrients). Free radicals cause cell injury because they have an unpaired electron that makes the molecule unstable. To stabilise itself, the molecule gives up an electron to another molecule or steals one. In so doing it forms injurious chemical bonds with proteins, lipids and carbohydrates — key molecules in membranes and nucleic acids.Cellular adaptation
Cellular adaptation is an alteration that enables the cell to maintain a steady state despite adverse conditions. Atrophy is a decrease in cellular size caused by ageing, disuse or lack of blood supply, hormonal stimulation or neural stimulation. The amounts of endoplasmic reticulum and mitochondria decrease. Hypertrophy is an increase in the size of cells caused by increased work demands or hormonal stimulation. The amounts of protein in the cell membrane, endoplasmic reticulum and mitochondria increase.Reversible and irreversible cell injury
Cell injury occurs if the cell is unable to maintain homeostasis. Injured cells may recover (reversible injury) or die (irreversible injury). Reversible cell injury is usually associated with cellular swelling. This occurs when there is an accumulation of excessive water in the cell caused by the failure of transport mechanisms. Irreversible cell injury occurs when the ‘point of no return’ is passed and the cell dies. There are two types — apoptosis and necrosis — which are physiologically different. Apoptosis is a process of selective cellular self-destruction that occurs in both normal and pathological tissue changes. Cellular death is manifested as cellular dissolution, or necrosis. Necrosis is the sum of the changes after local cell death and includes the process of autolysis, or cellular self-destruction. There are four major types of necroses: coagulative, liquefactive, caseous and fat necroses. Different types of necroses occur in different tissues.Ageing
It is difficult to determine normal physiological changes of ageing from the pathological changes of ageing. Humans have an inherent maximal life span (80 to 100 years) that is dictated by currently unknown intrinsic mechanisms.Jonathon, who is 20 years old, and his 71-year-old father, Terry, are discussing ageing. Jonathon has an active lifestyle, including playing sport, but his diet is poor — he frequently eats fast-food from take-away restaurants. He has had no major illnesses. Jonathon notices that his dad’s skin contains many wrinkles and is stiff looking, and that he has lost muscle mass from his legs and arms but gained weight around his abdomen.
In contrast, Terry, although remaining active, developed colon cancer when he was 67 and has recently been diagnosed with diabetes mellitus. Terry observes that his son’s skin contains few wrinkles, he is taller than him and his energy levels are high.