23 ALTERATIONS OF CARDIOVASCULAR FUNCTION ACROSS THE LIFE SPAN
INTRODUCTION
Cardiovascular diseases are conditions and diseases that affect the heart and vasculature (blood vessels). There are variations in the definition of cardiovascular diseases, with some classifications including heart disease, vascular disease, stroke and circulatory disease. The most common forms of cardiovascular diseases are hypertension, coronary heart disease, heart failure and cerebrovascular disease. Cerebrovascular disease arises from pathological processes in blood vessels of the brain, with stroke being the most frequent manifestation of cerebrovascular disease. Although stroke is classified as a cardiovascular disease, it is discussed in Chapter 9 to consider the effects on the brain.
In Western countries, cardiovascular disease is an epidemic and major health problem. Approximately 18% of Australians (3.5 million people) are reported to have a long-term cardiovascular condition, with the prevalence of disease increasing with age (see Figure 23-1). In addition, cardiovascular disease remains a major contributor to mortality, accounting for 36% and 40% of all deaths in Australia and New Zealand, respectively. In more recent years, there has been a reduction in the mortality rate attributable to cardiovascular disease due to improvements in cardiovascular disease management and a lowering of some risk factors (such as smoking).1 Unfortunately, these reductions are somewhat offset by the increased prevalence of cardiovascular disease in the elderly, combined with increasing rates of obesity and diabetes mellitus in the population (see Chapter 35). In addition, most people are afflicted with more than one cardiovascular condition and many have multiple cardiovascular risk factors. Furthermore, in both Australia and New Zealand cardiovascular disease is more prevalent in the Indigenous population than in the non-Indigenous population.2,3
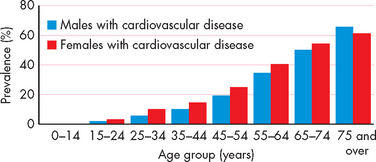
FIGURE 23-1 Reported prevalence of cardiovascular conditions in Australia across age groups.
Source: Australian Bureau of Statistics. National Health Survey Australia, 2004–05. Canberra: ABS. Note: 2007–08 data unavailable at time of writing.
It is vital that you have a comprehensive understanding of the pathophysiology of cardiovascular conditions, due to the high prevalence of cardiovascular disease in the community. Nurses are more actively involved than they have been previously in the management of cardiovascular conditions such as hypertension and heart failure, and your comprehension of the pathophysiology will aid your ability to care for individuals with cardiovascular conditions.
ALTERATIONS OF BLOOD FLOW AND PRESSURE
Pathophysiological alterations to arteries and veins include hypertension, atherosclerosis and peripheral vascular disease, and all of these conditions can lead to other cardiovascular diseases. The damage to the arteries in particular can lead to coronary heart disease, cerebrovascular disease or heart failure — the top three causes of death due to cardiovascular disease in Australia and New Zealand.1 This section details the formation of arterial and venous alterations, which will aid your understanding of the primary cardiovascular diseases. We start with the most common cardiovascular condition worldwide, hypertension.
Hypertension
Hypertension, or high blood pressure, is consistent elevation of systemic arterial blood pressure. It considerably increases the individual’s risk of developing coronary heart disease, heart failure and strokes. It is the most prevalent cardiovascular condition and is estimated to afflict about one billion people worldwide — just over one-quarter of the world’s adult population.4 Approximately 3.7 million Australians over the age of 25 years (30% of adults) have high blood pressure or are on medication to treat high blood pressure.1 Unfortunately, evidence suggests that a large number of adults and children have undiagnosed hypertension.5,6 The prevalence of hypertension increases in the elderly and in Aboriginal and Torres Strait Islander peoples and Maori and Pacific Islander peoples compared to the non-Indigenous population.1,3
The diagnosis of hypertension is based on repeated blood pressure (BP) measurements at different times, when systolic blood pressure is equal to or greater than 140 mmHg or diastolic pressure is 90 mmHg or greater (see Table 23-1).7 Normal blood pressure is associated with the lowest cardiovascular risk, whereas those who fall in the ‘high–normal’ range are at risk for developing hypertension unless they institute lifestyle modifications (in fact, more than 90% will develop hypertension8). All categories of hypertension are associated with an increased risk of myocardial infarction, kidney disease and stroke. Systolic hypertension, even when not accompanied by an increase in diastolic pressure, is the most significant factor in causing organ damage (heart, kidney and brain). Table 23-1 also indicates the grades of hypertension, which reflect the severity of increased blood pressure.
Table 23-1 CLASSIFICATION OF BLOOD PRESSURE LEVELS IN ADULTS
| DIAGNOSTIC CATEGORY* | SYSTOLIC (mmHg) | DIASTOLIC (mmHg) |
|---|---|---|
| Normal | < 120 | < 80 |
| High–normal | 120–139 | 80–89 |
| Grade 1 (mild) hypertension | 140–159 | 90–99 |
| Grade 2 (moderate) hypertension | 160–179 | 100–109 |
| Grade 3 (severe) hypertension | ≥ 180 | ≥ 110 |
| Isolated systolic hypertension | ≥ 140 | < 90 |
| Isolated systolic hypertension with widened pulse pressure | ≥ 160 | ≤ 70 |
* When a patient’s systolic and diastolic blood pressure levels fall into different categories, the higher diagnostic category and recommended action/s apply.
Source: National Heart Foundation of Australia. Guide to management of hypertension. National Blood Pressure and Vascular Disease Advisory Committee; 2008.
Individuals may have combined systolic and diastolic hypertension or isolated systolic hypertension. Most cases of combined systolic and diastolic hypertension are diagnosed as primary hypertension (also called essential or idiopathic hypertension) and account for approximately 90–95% of cases of hypertension. Secondary hypertension is caused by an underlying disease process such as kidney disease, hormone imbalances and drugs, and accounts for approximately 5–10% of cases. Ultimately, hypertension results from a sustained increase in peripheral resistance (vasoconstriction of the arterioles) or an increase in circulating blood volume (cardiac output), or both.
Factors associated with primary hypertension
A specific cause for primary hypertension has not been identified, but a combination of genetic and environmental factors is thought to be responsible for its development. Genetic predisposition to hypertension is thought to be polygenic; that is, there is more than one gene involved (see Chapter 37). A range of environmental factors are associated with primary hypertension — see the box ‘Risk factors: primary hypertension’. You may notice that many of these factors are also risk factors for other cardiovascular disorders; this is a recurring feature of cardiovascular disease.
Although populations with a high dietary sodium intake have long been shown to have an increased incidence of hypertension, studies indicate that low dietary potassium, calcium and magnesium intakes are also risk factors, because, without their intake, sodium is retained in the blood, rather than being excreted in the urine.9 The nicotine in cigarette smoke is a potent vasoconstrictor that can elevate both systolic and diastolic blood pressure acutely. The incidence of hypertension is higher among heavy drinkers of alcohol (more than three drinks per day) than among non-drinkers, but moderate drinkers (two to four drinks per week) appear to have lower blood pressures, as well as lower cardiovascular mortality. Obesity is recognised as an important risk factor for hypertension and is discussed in Chapter 35.
Primary hypertension
Primary hypertension is the result of an extremely complicated interaction of genetics and environmental or lifestyle factors causing neural and hormonal effects. Multiple pathophysiological mechanisms mediate these effects, including the sympathetic nervous system, the renin-angiotensin-aldosterone system (see Chapter 28) and natriuretic peptides (peptides consist of small numbers of amino acids). The term natriuresis refers to the excretion of large amounts of sodium in the urine, which in an otherwise healthy individual would be accompanied by loss of water in the urine, and hence a decrease in the total blood volume. Inflammation, endothelial dysfunction and insulin resistance also contribute to both an increase in peripheral resistance and blood volume. Increased vascular volume is related to a decrease in renal excretion of sodium, often referred to as a shift in the pressure–natriuresis relationship (see Figure 23-2). This means that individuals with hypertension tend to excrete less sodium in their urine.10
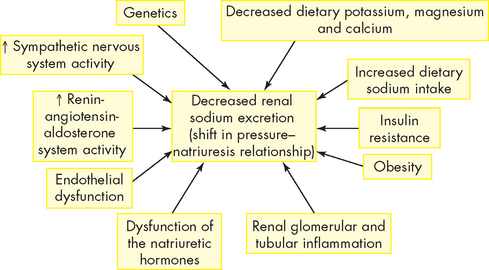
FIGURE 23-2 Factors that cause a shift in the pressure–natriuresis relationship.
Numerous factors have been implicated in the pathogenesis of sodium retention in individuals with hypertension. These factors cause less renal excretion of sodium than would normally occur with increased blood pressure.
The sympathetic nervous system has been implicated in both the development and the maintenance of elevated blood pressure. Increased sympathetic nervous system activity causes increased heart rate and systemic vasoconstriction, thus raising blood pressure. Structural changes in blood vessels, called vascular remodelling, which result in permanent increases in peripheral resistance, are induced by sympathetic nervous system activity. In addition, renal sodium retention, insulin resistance, increased renin and angiotensin levels and procoagulant effects are all induced by the sympathetic nervous system (see Figure 23-3).11
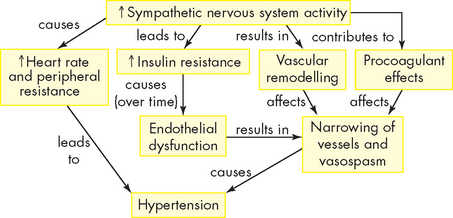
FIGURE 23-3 The role of the sympathetic nervous system in the pathogenesis of hypertension.
Increased activity of the sympathetic nervous system causes increases in heart rate, peripheral resistance and vascular remodelling, with narrowing and spasm of the arteries. The sympathetic nervous system contributes to insulin resistance, which is associated with endothelial dysfunction and decreased production of vasodilators. The sympathetic nervous system has procoagulant (tendency to clot) properties, making vascular spasm and thrombosis more likely.
The renin-angiotensin-aldosterone system plays an important role in blood pressure regulation by moderating vascular tone and influencing sodium and water retention by the kidneys. Furthermore, angiotensin II mediates arteriolar remodelling and is associated with end-stage organ effects of hypertension, including renal disease and cardiac hypertrophy.
Natriuretic hormones promote the excretion of sodium in the urine. They include atrial natriuretic peptide, B-type natriuretic peptide, C-type natriuretic peptide and urodilatin. The function of these hormones can be affected by excessive sodium intake; inadequate dietary intake of potassium, magnesium and calcium; and obesity.12 Dysfunction of these hormones, along with alterations in the renin-angiotensin-aldosterone system and the sympathetic nervous system, cause an increase in vascular tone and a change in the pressure–natriuresis relationship. Sodium retention leads to water retention, causing an increase in blood volume, which contributes to elevations in blood pressure. Subtle renal injury results, with renal vasoconstriction and tissue ischaemia. Tissue ischaemia causes inflammation of the kidneys, and contributes to dysfunction of the internal structure of the kidneys, namely the glomeruli and tubules, which actually promotes additional sodium retention. This vicious cycle leads to increases in blood pressure at rest and eventually hypertension.
Inflammation plays a role in the pathogenesis of hypertension. Endothelial injury and tissue ischaemia result in the release of vasoactive inflammatory cytokines (see Chapter 13). Although many of these cytokines (for instance, histamine) have vasodilatory actions in acute inflammatory injury, chronic inflammation contributes to vascular remodelling and smooth muscle contraction. Endothelial injury and dysfunction in primary hypertension are further characterised by a decreased production of vasodilators, such as nitric oxide, and increased production of vasoconstrictors, such as endothelin.
Finally, insulin resistance (see Chapter 35) is common in hypertension, even in individuals without clinical diabetes mellitus.13 Insulin resistance is associated with decreased endothelial release of nitric oxide and other vasodilators. It also affects renal function and causes the kidneys to retain sodium and water. Insulin resistance is associated with overactivity of the sympathetic nervous system and the renin-angiotensin-aldosterone system. The pathophysiology of primary hypertension is summarised in Figure 23-4.
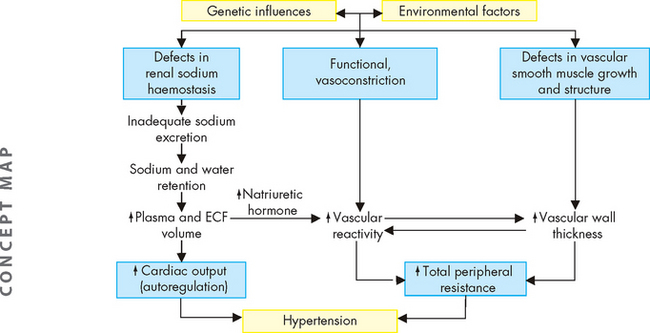
FIGURE 23-4 The pathophysiology of primary hypertension.
A hypothetical scheme for the pathogenesis of essential hypertension, implicating genetic defects in renal excretion of sodium, functional regulation of vascular tone and structural regulation of vascular calibre. Environmental factors, especially increased sodium intake, may potentiate the effects of genetic factors. The resultant increases in cardiac output and peripheral resistance contribute to hypertension. ECF = extracellular fluid.
Source: Modified from Kumar V. Robbins & Cotran pathologic basis of disease. 7th edn. Philadelphia: Saunders; 2004.
Secondary hypertension
Secondary hypertension is caused by an underlying disease process or medication that raises peripheral vascular resistance or cardiac output. The condition is more prevalent in younger people (< 30 years of age) and those over 50 years of age.14 If the cause is identified and removed before permanent structural changes occur, blood pressure returns to normal. Examples include kidney disease due to the retention of sodium and water (see Chapter 30), adrenocortical hormonal imbalances such as primary hyperaldosteronism (see Chapter 11), and drugs (oral contraceptives, corticosteroids, antihistamines).
Isolated systolic hypertension
Isolated systolic hypertension is typically defined as a sustained systolic BP ≥ 140 mmHg and diastolic BP below 90 mmHg. Isolated systolic hypertension accounts for a substantial proportion of hypertension in individuals older than 65 years of age and is strongly associated with cardiovascular and cerebrovascular events.
An increased pulse pressure (systolic minus diastolic pressure) indicates reduced vascular compliance of large arteries. Pulse pressure is always increased in isolated systolic hypertension and is related to either an increase in cardiac output (heart valve disease) or peripheral resistance (caused by atherosclerosis). Pharmacological management of isolated systolic hypertension is required because the systolic blood pressure is greater than 140 mmHg.
Complicated hypertension
Cardiovascular complications of sustained hypertension include left ventricular hypertrophy, angina pectoris, heart failure, coronary heart disease, myocardial infarction and sudden death. Myocardial hypertrophy in response to hypertension is mediated by several neurohormonal substances, including catecholamines from the sympathetic nervous system (adrenaline and noradrenaline) and angiotensin II.15 In addition, the increased size of the heart muscle increases demand for oxygen delivery over time, contractility of the heart is impaired, and the individual is at increased risk for heart failure. Vascular complications include the formation, dissection and rupture of aneurysms (outpouchings in vessel walls) and atherosclerosis leading to vessel occlusion. Microalbuminuria (small amounts of protein in the urine) occurs in 10–25% of individuals with essential hypertension and is now recognised as an early sign of impending renal dysfunction and significantly increased risk for cardiovascular events. The pathological effects of sustained essential hypertension are summarised in Table 23-2.
Table 23-2 THE PATHOLOGICAL EFFECTS OF SUSTAINED PRIMARY HYPERTENSION
| SITE OF INJURY | MECHANISM OF INJURY | POTENTIAL PATHOLOGICAL EFFECTS |
|---|---|---|
| Heart | ||
| Myocardium | Increased workload combined with diminished blood flow through coronary arteries | Left ventricular hypertrophy, myocardial ischaemia, left heart failure |
| Coronary arteries | Accelerated atherosclerosis (coronary artery disease) | Myocardial ischaemia, myocardial infarction, sudden death |
| Kidneys | Renin and aldosterone secretion stimulated by reduced blood flow | Retention of sodium and water, leading to increased blood volume and continuation of hypertension |
| Reduced oxygen supply | Tissue damage that compromises filtration | |
| High pressures in renal arterioles | Renal failure | |
| Brain | Reduced blood flow and oxygen supply; weakened vessel walls, accelerated atherosclerosis | Transient ischaemic attacks, cerebral thrombosis, aneurysm, haemorrhage, acute brain infarction |
| Eyes (retinas) | Reduced blood flow | Retinal vascular sclerosis |
| High arteriolar pressure | Exudation, haemorrhage | |
| Aorta | Weakened vessel wall | Dissecting aneurysm |
| Arteries of lower extremities | Reduced blood flow and high pressures in arterioles, accelerated atherosclerosis | Intermittent claudication, gangrene |
CLINICAL MANIFESTATIONS
The early stages of hypertension have no clinical manifestations other than elevated blood pressure. Most importantly, there are usually no signs and symptoms; thus, hypertension is often called a silent disease. Some hypertensive individuals never have signs, symptoms or complications, whereas others become very ill. Still other individuals have anatomical and physiological damage caused by past hypertensive disease, despite current blood pressures being within normal ranges.
The chance of developing primary hypertension increases with age. Although hypertension is usually thought to be an adult health problem, it is important to remember that hypertension does occur in children and is being diagnosed with increasing frequency (see Paediatrics box). Usually, however, increased peripheral resistance and early hypertension develop in the second, third and fourth decades of life. If elevated blood pressure is not detected and treated, it becomes established and may begin to accelerate its effects on tissues when the individual is 30–50 years of age. This sets the stage for the complications of hypertension that begin to appear during the fourth, fifth and sixth decades of life.
Most clinical manifestations of hypertensive disease are caused by complications that damage organs and tissues outside the vascular system. Besides elevated blood pressure, the signs and symptoms therefore tend to be specific for the organs or tissues affected. Evidence of heart disease, renal insufficiency, central nervous system dysfunction, impaired vision, impaired mobility, vascular occlusion or oedema can all be caused by sustained hypertension.
EVALUATION AND TREATMENT
A single elevated blood pressure reading does not indicate hypertension. Diagnosis requires the measurement of blood pressure on at least two separate occasions. The individual should be seated and relaxed, preferably in a quiet room prior to measurement, the arm supported at heart level and free of clothing that could impede blood flow. After 30 seconds, repeat the procedure on the same arm and average the readings if the systolic blood pressure difference is less than 10 mmHg and the diastolic blood pressure difference is less than 6 mmHg.7 In addition, the person should have a physical examination, with investigations such as 24-hour blood pressure monitoring in selected individuals, blood analysis (testing for sodium, potassium, chloride, bicarbonate, urea, creatinine, uric acid, haemoglobin, fasting glucose, total cholesterol, LDL cholesterol (see ‘Dyslipidaemia and atherosclerosis-promoting diet’ below), HDL cholesterol, triglycerides, liver function), urinalysis (testing for blood and protein) and an electrocardiogram.7 Individuals who have elevated blood pressure are assumed to have primary hypertension unless their history, physical examination or investigations indicates secondary hypertension.
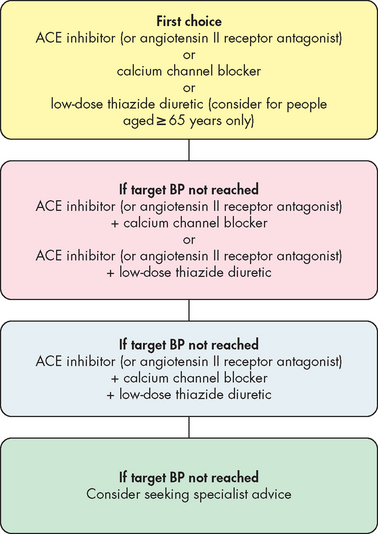
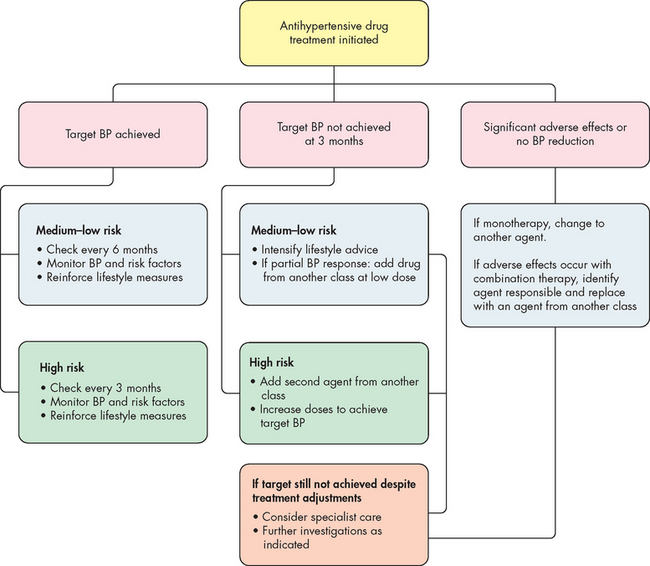
Treatment of primary hypertension depends on its severity. Lifestyle modification is important for preventing hypertension in those individuals who fall into the high–normal category (see Table 23-1) and for treating hypertension. Important lifestyle modifications include increasing exercise levels, making dietary modifications, ceasing smoking, limiting alcohol intake and losing weight. Pharmacological interventions are required when lifestyle modifications and systolic or diastolic blood pressure is not controlled (see Figure 23-5). The decision to commence antihypertensive drugs should be based on the severity of the hypertension and the extent of end-organ damage. A variety of drugs are used to manage high blood pressure and these drugs are grouped in Table 23-3. An understanding of how these drugs work can be derived from reviewing the location of α (alpha)- and β (beta)-adrenergic receptors (refer to Table 6-8). Also, referring to Figure 28-14 will give an understanding of how aldosterone is released and its functions. This will explain why angiotensin-converting enzyme (ACE) inhibitors may be useful: they decrease the formation of angiotensin II and the release of aldosterone. The National Heart Foundation of Australia, using the latest evidence, has provided guidelines for the initiation of antihypertensive medication and the type of antihypertensive for newly diagnosed hypertension (see Figure 23-6).7 The continuation of long-term pharmacological management is outlined in Figure 23-7, as it is necessary to re-evaluate treatment strategies, depending on the success of maintaining appropriate blood pressure.
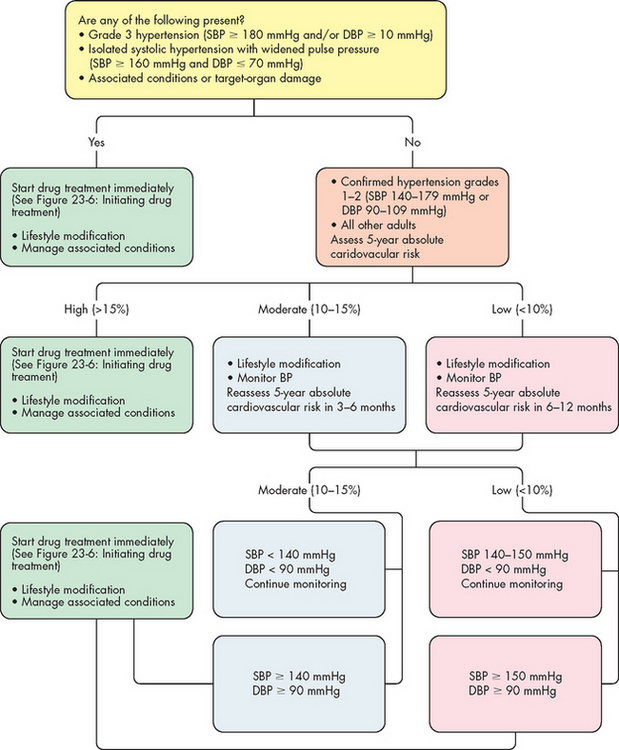
FIGURE 23-5 Determination of non-pharmacological and pharmacological management of high blood pressure.
BP = blood pressure; SBP = systolic blood pressure; DBP = diastolic blood pressure.
Source: National Heart Foundation of Australia. Guide to management of hypertension. National Blood Pressure and Vascular Disease Advisory Committee; 2008.
Table 23-3 DRUG CLASSIFICATIONS USED TO TREAT HYPERTENSION AND THE VARIABLES THEY AFFECT
| REDUCE STROKE VOLUME | REDUCE SYSTEMIC VASCULAR RESISTANCE | DECREASE HEART RATE |
|---|---|---|
α = alpha; β = beta.
Source: Based on Copstead-Kirkhorn L-EC, Banasik JL. Pathophysiology. 4th edn. St Louis: Saunders; 2010.
Orthostatic hypotension
Orthostatic hypotension, or postural hypotension, refers to a decrease in both systolic and diastolic arterial blood pressure on standing. Normally when an individual stands up, the gravitational changes on the circulation are compensated by mechanisms such as reflex arteriolar and venous constriction controlled by the baroreceptors and increased heart rate. Furthermore, mechanical factors such as the closure of valves in the venous system, pumping of the leg muscles and a decrease in intrathoracic pressure assist in increasing venous return in the heart. Collectively, these maintain blood pressure.
Orthostatic hypotension is often accompanied by dizziness, blurring or loss of vision and syncope (fainting) caused by insufficient vasomotor compensation and reduction of blood flow through the brain. This occurs because the normal or compensatory vasoconstrictor response to standing is absent so that there is blood pooling in the muscle vasculature, as well as in the splanchnic and renal beds.
Orthostatic hypotension may be acute and temporary or chronic:
 Acute orthostatic hypotension is caused when the normal regulatory mechanisms are sluggish as a result of (1) altered body chemistry, (2) drug action (e.g. antihypertensives, antidepressants), (3) prolonged immobility caused by illness, (4) starvation, (5) physical exhaustion, (6) any condition that produces volume depletion (e.g. dehydration, diuresis, potassium or sodium depletion) or (7) venous pooling (e.g. pregnancy, extensive varicosities of the lower extremities). The elderly are particularly susceptible to this type of orthostatic hypotension. Chronic orthostatic hypotension may be (1) secondary to a specific disease or (2) idiopathic or primary. The diseases that cause secondary orthostatic hypotension are endocrine disorders (e.g. adrenal insufficiency, diabetes mellitus), metabolic disorders (e.g. porphyria) or diseases of the central or peripheral nervous systems (e.g. intracranial tumours, cerebral infarcts, Wernicke’s encephalopathy, peripheral neuropathies). It is more prevalent in the aged population and may be attributable to an increase in mortality due to secondary effects of orthostatic hypotension, such as falls.17 In addition to cardiovascular symptoms, associated impotence and bowel and bladder dysfunction are common.
Acute orthostatic hypotension is caused when the normal regulatory mechanisms are sluggish as a result of (1) altered body chemistry, (2) drug action (e.g. antihypertensives, antidepressants), (3) prolonged immobility caused by illness, (4) starvation, (5) physical exhaustion, (6) any condition that produces volume depletion (e.g. dehydration, diuresis, potassium or sodium depletion) or (7) venous pooling (e.g. pregnancy, extensive varicosities of the lower extremities). The elderly are particularly susceptible to this type of orthostatic hypotension. Chronic orthostatic hypotension may be (1) secondary to a specific disease or (2) idiopathic or primary. The diseases that cause secondary orthostatic hypotension are endocrine disorders (e.g. adrenal insufficiency, diabetes mellitus), metabolic disorders (e.g. porphyria) or diseases of the central or peripheral nervous systems (e.g. intracranial tumours, cerebral infarcts, Wernicke’s encephalopathy, peripheral neuropathies). It is more prevalent in the aged population and may be attributable to an increase in mortality due to secondary effects of orthostatic hypotension, such as falls.17 In addition to cardiovascular symptoms, associated impotence and bowel and bladder dysfunction are common.Although no curative treatment is available for orthostatic hypotension, often it can be managed adequately with a combination of non-pharmacological and pharmacological therapies. For both acute and chronic forms, hypotension resolves when the underlying disorder is corrected.
 PAEDIATRICS
PAEDIATRICSHypertension
The normal range of blood pressure in children is dependent on body size and age, with systolic and diastolic pressure increasing until adolescence. Hypertension in children is defined as systolic and diastolic blood pressure levels greater than the 95th percentile for age and gender on at least three occasions (see Table 23-4).
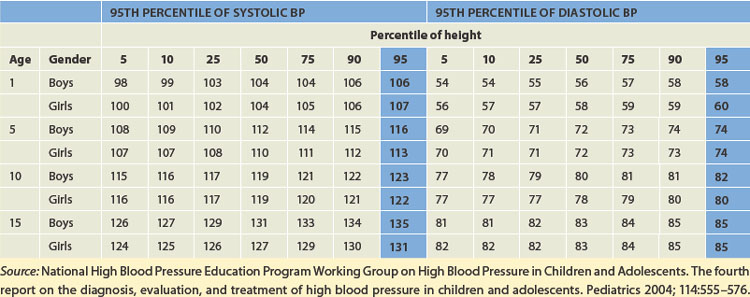
The incidence of hypertension in children is low, approximately 2–5%, but with increasing rates of childhood obesity and diabetes mellitus it is thought that signs of high blood pressure may be evident in children before diagnosis.5,16 Hypertension is classified the same as adults: primary and secondary hypertension. However, hypertension in children differs from adult hypertension in aetiology and presentation because in the majority of cases some underlying disease can be found, such as renal disease or coarctation (narrowing) of the aorta (see Table 23-5).
Table 23-5 COMMON CAUSES OF CHRONIC SUSTAINED HYPERTENSION
| AGE GROUP | CAUSES |
|---|---|
| Newborn | Renal artery thrombosis, renal artery stenosis, congenital renal malformation, coarctation of the aorta |
| < 6 years | Renal parenchymal disease, coarctation of the aorta, renal artery stenosis |
| 6–10 years | Renal artery stenosis, renal parenchymal disease, primary hypertension |
| > 10 years | Primary hypertension, renal parenchymal disease |
Source: Park MK. Pediatric cardiology for practitioners. 4th edn. St Louis: Mosby; 2002. See also National High Blood Pressure Education Program Working Group on High Blood Pressure in Children and Adolescents. The fourth report on the diagnosis, evaluation, and treatment of high blood pressure in children and adolescents. Pediatrics 2004; 114:555–576.
PATHOPHYSIOLOGY
The pathophysiology of primary hypertension in children is not clearly understood, but may result from a complex interaction of a strong predisposing genetic component with disturbances in sympathetic vascular smooth muscle tone, hormones (angiotensin and the catecholamines adrenaline and noradrenaline), renal sodium excretion and cardiac output. Ultimately these factors impair the ability of the peripheral vascular bed to relax.
CLINICAL MANIFESTATIONS
Most children with hypertension are asymptomatic. It is necessary that a thorough history and physical examination be obtained, with blood pressure measurements over three different occasions.
EVALUATION AND TREATMENT
In children, the history and physical examination should be directed at determining the aetiology of hypertension. If primary hypertension is found, non-pharmacological therapy is used initially. For example, moderate weight loss and exercise can decrease systolic and diastolic pressures in overweight or inactive children. Appropriate diet and regular physical activity have been shown to be effective in reducing blood pressure. Drug therapy is controversial in children with primary hypertension; however, when non-pharmacological approaches fail, drug therapy, such as angiotensin-converting enzyme (ACE) inhibitors or angiotensin receptor blocker medication, may be used. When secondary hypertension arises, treatment of the underlying cause, such as surgical correction of coarctation of the aorta, usually alleviates the high blood pressure.
Arteriosclerosis
Arteriosclerosis is a chronic disease of the arterial system characterised by abnormal thickening and hardening of the vessel walls. Smooth muscle cells and collagen fibres migrate into the tunica intima (internal layer of the arterial wall), causing it to stiffen and thicken, gradually narrowing the arterial lumen (see Figure 23-8). Changes in lipid, cholesterol and phospholipid metabolism within the tunica intima also contribute to arteriosclerosis. Although these changes may be part of normal ageing, pathophysiological conditions such as hypertension, insufficient perfusion (blood flow) of tissues or weakening and outpouching of arterial walls can be exacerbated by the changes to the arterial walls brought about by arteriosclerosis.
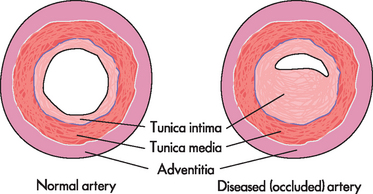
Atherosclerosis
Atherosclerosis is the most common form of arteriosclerosis. It is characterised by soft deposits of intra-arterial fat and fibrin in the vessels walls that harden over time. Atherosclerosis is not a single disease entity but rather a pathological process that can affect vascular systems throughout the body, resulting in ischaemic syndromes that can vary widely in their severity and clinical manifestations. It is the leading cause of coronary heart and cerebrovascular disease. (Atherosclerosis of the coronary arteries is described later in this chapter, and atherosclerosis of the cerebral arteries leading to cerebrovascular disease is described in Chapter 9.)
PATHOPHYSIOLOGY
Inflammation plays a fundamental role in mediating all of the steps in the initiation and progression of atherosclerosis formation.18–20 4 6 Atherosclerosis begins with injury to the endothelial cells that line the artery walls. Possible causes of endothelial injury include the common risk factors for atherosclerosis, such as smoking, hypertension, diabetes mellitus, increased levels of low-density lipoprotein (LDL) cholesterol and decreased levels of high-density lipoprotein (HDL) cholesterol. Other causes of endothelial injury are called the ‘novel’ risk factors, such as elevated C-reactive protein (CRP), increased serum fibrinogen, insulin resistance, oxidative stress, infection and periodontal disease. There is recent evidence that individuals with a defect in the production of precursor endothelial cells in the bone marrow are at greater risk for atherosclerotic disease because these precursor cells are not available to repair injured endothelium.21,22
Injured endothelial cells become inflamed and cannot make normal amounts of antithrombic and vasodilating substances. When the endothelium is injured, it loses the ability both to prevent clotting and to vasodilate. This results in platelets aggregating when thromboxane A2 increases (refer to Chapter 6), and the release of serotonin and endothelin combines to cause vasoconstriction. This leads to a decrease in blood flow and, ultimately, ischaemia. At the same time, sympathetic nervous system activation causes vasoconstriction when noradrenaline is released. The enzyme ACE in the endothelium also converts angiotensin I to angiotensin II (Figure 23-9 summarises these events). Collectively, this leads to vasoconstriction and increased clotting.
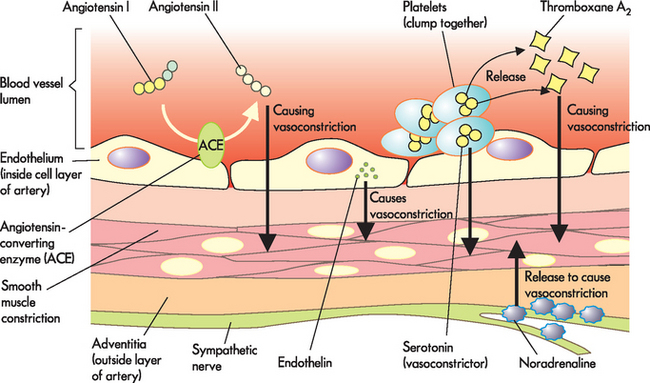
FIGURE 23-9 Endothelium regulation of vasoconstriction, vasodilation and platelet aggregation.
Endothelial cell damage, such as that associated with atherosclerosis formation, causes vasoconstriction and platelet aggregation to dominate over vasodilation.
Source: Modified from Stern S (ed.). Silent myocardial ischemia. St Louis: Mosby; 1998.
The next step in the formation of atherosclerosis occurs when inflamed endothelial cells express adhesion molecules that bind macrophages and other inflammatory and immune cells. Macrophages adhere to the injured endothelium and release numerous inflammatory cytokines (e.g. tumour necrosis factor-alpha [TNF-α], interferons, interleukins and C-reactive protein) and enzymes that further injure the vessel wall.23 Toxic oxygen radicals generated by the inflammatory process cause oxidation (i.e. addition of oxygen) of LDL. Oxidised LDL is engulfed by macrophages, which then penetrate into the intima of the vessel. These lipid-laden macrophages are now called foam cells and when they accumulate in significant amounts, they form a lesion called a fatty streak (see Figures 23-10 and 23-11). Even small-sized lesions can be found in the walls of arteries of most people, including young children. Once formed, fatty streaks produce more toxic oxygen radicals and cause immunological and inflammatory changes resulting in progressive damage to the vessel wall.
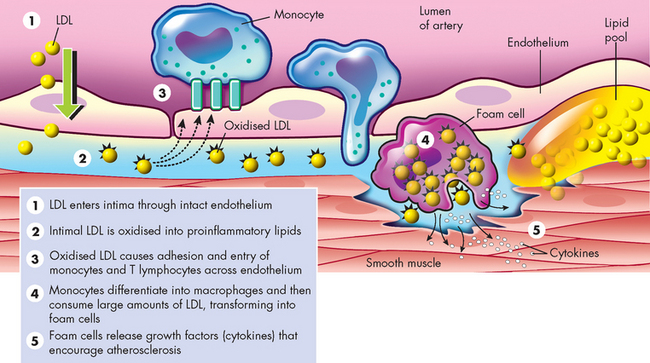
FIGURE 23-10 Low-density lipoprotein (LDL) oxidation.
Source: Crawford MH, DiMarco JP (eds). Cardiology. London: Mosby; 2001.
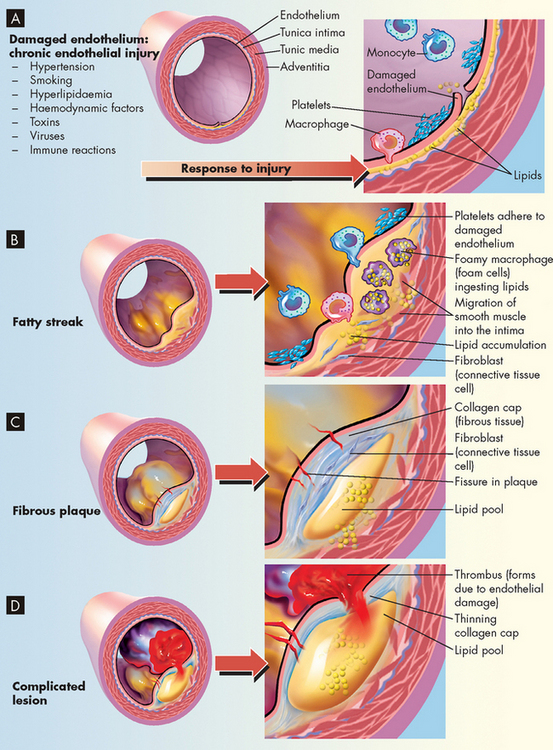
FIGURE 23-11 The progression of atherosclerosis.
A Damaged endothelium. B Diagram of fatty streak and lipid core formation. C Diagram of fibrous plaque. Raised plaques are visible: some are yellow; others are white. D Diagram of complicated lesion; thrombus is red; collagen is blue. Plaque is complicated by red thrombus deposition.
Macrophages also release growth factors that stimulate smooth muscle cell proliferation. Smooth muscle cells in the region of endothelial injury proliferate, produce collagen and migrate over the fatty streak forming a fibrous plaque (see Figure 23-11). The fibrous plaque may calcify, protrude into the vessel lumen and obstruct blood flow to distal tissues (especially during exercise), which may cause symptoms such as angina or intermittent claudication (leg pain with exercise) due to poor blood circulation.
Many plaques, however, are ‘unstable’, meaning they are prone to rupture even before they affect blood flow significantly and are clinically silent until they rupture. Plaques that have ruptured are called complicated plaques. Once rupture occurs, exposure of underlying tissue results in platelet adhesion, initiation of the coagulation (or clotting) cascade and rapid thrombus formation. The thrombus may suddenly occlude the affected vessel, resulting in ischaemia and infarction. Aspirin or other antithrombotic agents are used to prevent this complication of atherosclerotic disease.
CLINICAL MANIFESTATIONS
Atherosclerosis presents with symptoms and signs that result from inadequate perfusion of tissues because of obstruction of the vessels that supply them. Partial vessel obstruction may lead to transient ischaemic events, often associated with exercise or stress. As the lesion becomes complicated, increasing obstruction with superimposed thrombosis may result in tissue infarction. Obstruction of peripheral arteries can cause significant pain and disability. Coronary heart disease caused by atherosclerosis (see Figure 23-12) is the major cause of myocardial ischaemia and is one of the most important health issues in Western countries, including Australia and New Zealand.1–3, 4 66 Atherosclerotic obstruction of the vessels supplying the brain is the major cause of strokes. Similarly, any part of the body may become ischaemic when its blood supply is compromised by atherosclerotic lesions.
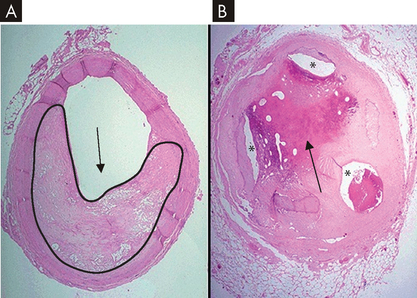
FIGURE 23-12 Coronary artery atherosclerosis.
A A coronary artery with 60–70% occlusion (black outline and arrow demonstrate narrowing of lumen) due to atherosclerosis. This degree of occlusion may lead to angina. B Severe coronary artery disease and evidence of past thrombus (black arrow), with only three small lumens (*) providing blood flow to the myocardium.
Source: Modified from Klatt EC. Robbins and Cotran atlas of pathology. 2nd edn. Philadelphia: Saunders; 2010.
EVALUATION AND TREATMENT
In evaluating individuals for the presence of atherosclerosis, a complete history (including risk factors), physical examination and laboratory data are considered. The use of X-rays, electrocardiography, ultrasonography, nuclear scanning and angiography may be necessary to identify affected vessels, particularly coronary vessels.
The primary goal in the management of atherosclerosis is to restore adequate blood flow to the affected tissues. If an individual presents with acute ischaemia, interventions are specific to the diseased area (e.g. acute myocardial infarction due to occlusion of the coronary vessel). In situations where the disease process does not require immediate intervention, management focuses on removing the initial causes of vessel damage and preventing lesion progression. This includes lifestyle modifications, such as increasing exercise levels, ceasing smoking and controlling hypertension and diabetes mellitus where appropriate, and reducing LDL levels by diet or medications, or both.
Coronary heart disease
Coronary heart disease refers to conditions that affect the coronary blood vessels that supply the heart with nutrients and oxygen (see Chapter 22). We have chosen to use the term coronary heart disease to align with the terminology used by the Australian Institute of Health and Welfare, the New Zealand Ministry of Health and the peak cardiac bodies in both countries, the National Heart Foundation of Australia and the Cardiac Society of Australia and New Zealand. In clinical practice you may see a variety of terms used for coronary heart disease, such as ischaemic heart disease, coronary artery disease and heart disease. These terms are essentially synonymous and are often used interchangeably. However, one of the most important aspects to remember is that more than 90% of all cases of myocardial ischaemia (a local state in which the cardiac cells are temporarily deprived of blood supply) result from obstruction of the coronary arteries due to atherosclerosis. Persistent ischaemia or complete occlusion of a coronary artery causes the acute coronary syndromes including infarction or irreversible myocardial damage. Acute myocardial infarction constitutes the often-fatal event known within the community as a heart attack.
The development of coronary heart disease
Approximately 3.2% of Australians (more than 600,000 people) have coronary heart disease. It is associated with the elderly, as the prevalence of coronary heart disease increases with age (see Figure 23-1): for example, it afflicts about 7.5% of Australians aged 55–64 years but 20.3% of those aged 75 years and over.1 Moreover, coronary heart disease is the single largest killer in Australia and New Zealand compared to all other diseases, accounting for about 20% of all deaths.24,25
Numerous types of genetic susceptibilities to coronary heart disease have been identified in individuals with a family history of heart disease. Risk factors can be categorised as conventional (major) versus non-traditional (novel) and modifiable versus non-modifiable. Much new information has been obtained about the conventional risk factors, which have markedly improved prevention and management of the disease. Conventional risk factors for coronary heart disease that are non-modifiable include:
We now focus our attention on discussing these modifiable risk factors.
Dyslipidaemia and atherosclerosis-promoting diet
The term lipoprotein refers to lipids, phospholipids, cholesterol and triglycerides bound to carrier proteins. These carrier molecules assist in transporting lipid-based molecules through the water-based environment of the blood. Lipids (cholesterol in particular) are required by most cells for the manufacture and repair of plasma membranes. Cholesterol is also a necessary component for the production of such essential substances as bile acids and steroid hormones. Although cholesterol can easily be obtained from dietary fat intake, most body cells can also produce cholesterol.
The cycle of lipid metabolism is complex. Dietary fat is packaged into particles known as chylomicrons in the small intestine. Chylomicrons are required for absorption of fat; they function by transporting lipid from the small intestine to the liver and peripheral cells. Chylomicrons are the least dense of the lipoproteins and primarily contain triglyceride. Some of the triglyceride may be removed from the blood and either stored by adipose tissue or used by muscle as an energy source. The chylomicron remnants, composed mainly of cholesterol, are taken up by the liver. A series of chemical reactions in the liver results in the production of several lipoproteins that vary in density and function. These include very-low-density lipoproteins (VLDL), primarily triglyceride and protein; low-density lipoproteins (LDL), mostly cholesterol and protein; and high-density lipoproteins (HDL), mainly phospholipids and protein.
Dyslipidaemia refers to abnormal concentrations of serum lipoproteins. The National Heart Foundation of Australia and the Cardiac Society of Australia and New Zealand (2005) have classified the recommended target levels of lipoproteins in the blood (see Table 23-6).
Table 23-6 RECOMMENDED TARGET LEVELS
| LIPOPROTEIN | mmol/L |
|---|---|
| Low-density lipoprotein | < 2.0 |
| High-density lipoprotein | > 1.0 |
| Triglycerides | < 1.5 |
| Total cholesterol | < 4.0 |
Source: National Heart Foundation of Australia and the Cardiac Society of Australia and New Zealand. Position statement on lipid management. Heart Lung Circ 2005; 14:275–291.
An increased serum concentration of LDL is a strong indicator of coronary risk.26,27 Serum levels of LDL are normally controlled by hepatic receptors that bind LDL and limit liver synthesis of this lipoprotein. High dietary intake of cholesterol and fats, often in combination with a genetic predisposition to accumulations of LDL, results in high levels of LDL in the bloodstream. Oxidation of LDL, its migration into the vessel wall and phagocytosis by macrophages are key steps in the pathogenesis of atherosclerosis (see Figure 23-10). LDL cholesterol also plays a role in endothelial injury, inflammation and immune responses that have been identified as being important in atherosclerosis formation.28 Aggressive reduction of LDL with diet and cholesterol-lowering drugs, such as HMG-CoA reductase inhibitors, more commonly referred to as statins, is associated with a dramatic decrease in risk for coronary heart disease.29
Low levels of HDL are also a strong indicator of coronary risk and high levels of HDL may be more protective for the development of atherosclerosis than low levels of LDL.30 HDL is responsible for ‘reverse cholesterol transport’, which returns excess cholesterol from the tissues to the liver for metabolism — in this way cholesterol is cleared out of the blood. HDL also participates in endothelial repair and decreases thrombosis.31 Exercise, weight loss, fish oil consumption and moderate alcohol use can result in modest increases in HDL. You will often hear reference to LDL as ‘bad’ cholesterol (atherogenic — promoting development of atherosclerosis), while HDL is referred to as ‘good’ cholesterol (for protection from atherosclerosis).
Other lipoproteins associated with increased cardiovascular risk include elevated serum VLDL (triglycerides) and increased lipoprotein (a). Triglycerides are associated with an increased risk for coronary heart disease, especially in combination with other risk factors such as diabetes mellitus. Lipoprotein (a) is a genetically determined molecular complex between LDL and a serum glycoprotein called apolipoprotein A and has been shown to be an important risk factor for atherosclerosis, especially in women.
In order to understand how modification of the diet can impact on the risk of developing atherosclerosis, it is important to consider the relationship between dietary fats and the lipid profile in the blood. The main types of dietary fats that are more atherogenic are the saturated fats, with the trans-fats also contributing. In contrast, the dietary fats that can assist in protection from atherosclerosis are the unsaturated fats (polyunsaturated and monounsaturated; see the box ‘Health alert: the basics on fats’). The National Heart Foundation of Australia recommends that intake of saturated fats should be reduced and that the total energy of the diet obtained from saturated and polyunsaturated fats should be 8% and 8–10%, respectively. However, currently the average Australian dietary intake comprises 12.7% saturated fats and only 4.9% polyunsaturated fats.32
The basics on fats
Unsaturated fats consist of two types: monounsaturated and polyunsaturated. Both contain essential fatty acids (EFAs), but polyunsaturated fats have more.
Trans-fats are primarily found in artificially solidified (hydrogenated) oils (e.g. margarine and vegetable shortening). By becoming more solid they lose EFAs. They can raise LDL levels and lower HDL levels. They also can raise lipoprotein (a) levels, which increases the risk of heart disease. Trans-fats raise blood-sugar levels and contribute to more weight gain than the same amount of other fats. ‘Partially hydrogenated’ or ‘hydrogenated’ on a food label means the food contains trans-fatty acids (e.g. cakes, biscuits, crackers, processed cheese).
Saturated fats are found in animal fats (butter, cheese, beef, pork, lamb, chicken) and some tropical oils (e.g. palm kernel). All saturated fats are not the same; some are stickier than others. They consist of a long chain of atoms that take a longer time to burn than shorter chained fats. The longer the fat takes to burn, the stickier it becomes. Those fats that become stickiest are more conducive to weight gain and heart disease.
Hypertension
Hypertension is responsible for a twofold to threefold increased risk of atherosclerotic cardiovascular disease. It contributes to endothelial injury and can lead to myocardial hypertrophy, which increases myocardial demand for coronary flow. This risk factor was discussed fully at the start of this chapter.
Cigarette smoking
Both direct and passive (environmental) smoking increase the risk of coronary heart disease. Nicotine stimulates the release of catecholamines (adrenaline and noradrenaline), which increase heart rate and peripheral vasoconstriction. As a result, blood pressure increases, as do cardiac workload and oxygen demand. Cigarette smoking is also associated with an increase in LDL and a decrease in HDL, and contributes to damage of the blood vessel endothelial lining, blood vessel inflammation and thrombosis. It is likely that additional mechanisms by which smoking increase atherosclerosis also occur.
Diabetes mellitus and insulin resistance
Diabetes mellitus is an extremely important risk factor for coronary heart disease. Insulin resistance and diabetes have multiple effects on the cardiovascular system, including endothelial damage, thickening of the vessel wall, increased inflammation, increased thrombosis and decreased production of endothelial-derived vasodilators such as nitric oxide.33 Diabetes mellitus is also associated with dyslipidaemia. Diabetes mellitus is discussed in Chapter 35.
Obesity and sedentary lifestyle
The prevalence of people who are overweight or obese is increasing steadily in Western countries, and Australia and New Zealand are following this trend. Approximately, 60% of Australians (7.5 million people) are classified as overweight or obese, and the prevalence of obesity has doubled over the last two decades. For older Australians the rate of obesity has trebled over the last 20 years: one in five seniors are now obese.34 In New Zealand, one in four adults are now classified as obese.35 In both countries the incidence of overweight and obesity is higher in the Indigenous than non-Indigenous population.1,34,35
The metabolic syndrome is a combination of central (abdominal) obesity, abnormal glucose tolerance or impaired glucose tolerance, raised triglycerides, decreased HDL, elevated blood pressure and insulin resistance (see Chapter 35). It has been calculated that almost 30% of Australian adults have the metabolic syndrome.6 The combination of these risk factors considerably increases the development of cardiovascular disease and confers an even higher risk for coronary heart disease.36,37 Abdominal obesity has the strongest link with increased coronary heart disease risk and is related to insulin resistance, decreased HDL, increased blood pressure and decreased levels of a recently described cardioprotective protein called adiponectin.38 Physical activity and weight loss offer substantial reductions in risk factors for coronary heart disease.39
Research is emerging that some non-traditional risk factors may also contribute to the development of coronary heart disease— the major one is inflammation. This is discussed briefly in the box ‘Health alert: inflammatory markers for cardiovascular risk’.
Coronary heart disease, myocardial ischaemia and acute myocardial infarction form a pathophysiological continuum that impairs the pumping ability of the heart by depriving the heart muscle of blood-borne oxygen and nutrients.40 We now explore how coronary heart disease results in myocardial dysfunction and possible cardiac cell death.
Inflammatory markers for cardiovascular risk
Atherosclerosis is an inflammatory disease. A number of serum markers of inflammation have been found to be excellent predictors of cardiovascular risk, especially C-reactive protein (CRP). Other inflammatory markers found to be predictive of cardiovascular risk include fibrinogen, erythrocyte sedimentation rate, von Willebrand’s factor, interleukin-6, interleukin-1 and tumour necrosis factor-α. CRP is made by the liver in response to inflammatory stimuli and has been demonstrated convincingly to be a good predictor of coronary heart disease. However, problems remain in determining its use in clinical practice. CRP is a nonspecific marker of inflammation. It can therefore be elevated in many other inflammatory states, and its use is limited to identifying high-risk individuals and for following individuals with known coronary disease. It should not be used to screen the general population.
Myocardial ischaemia
PATHOPHYSIOLOGY
The coronary arteries supply blood flow sufficient to meet the demands of the myocardium during normal levels of cardiac activity, as well as when the heart is working harder (such as during exercise). Oxygen is extracted from these vessels with maximal efficiency. If demand increases, healthy coronary arteries dilate to increase the flow of oxygenated blood to the myocardium. Various pathological mechanisms can interfere with blood flow through the coronary arteries, giving rise to myocardial ischaemia. Narrowing of a major coronary artery by more than 50% impairs blood flow enough to interfere with cellular metabolism (see Figures 23-12 and 23-13).
Myocardial ischaemia develops if blood flow or oxygen content of coronary blood is insufficient to meet the metabolic demands of myocardial cells. Imbalances between coronary blood supply and myocardial demand can result from a number of conditions. The most common cause of decreased coronary blood flow and myocardial ischaemia is the formation of atherosclerotic plaques in the coronary circulation. As the plaque increases in size, it may partially occlude the vessel, thus limiting coronary flow and causing ischaemia (see Figure 23-14). This is common when metabolic demand increases, such as during exercise. Some plaques are ‘unstable’, meaning they are prone to ulceration or rupture. When this occurs, underlying tissues of the vessel wall are exposed, resulting in platelet adhesion and thrombus formation. This can suddenly cut off blood supply to the heart muscle, resulting in acute myocardial ischaemia and, if the vessel obstruction cannot be reversed rapidly, ischaemia will progress to infarction (death of the cells). Myocardial ischaemia can also result from other causes of decreased blood and oxygen delivery to the myocardium, such as coronary spasm, hypotension, arrhythmias and decreased oxygen-carrying capacity of the blood, such as anaemia. Common causes of increased myocardial demand for blood include tachycardia, exercise and valvular disease.
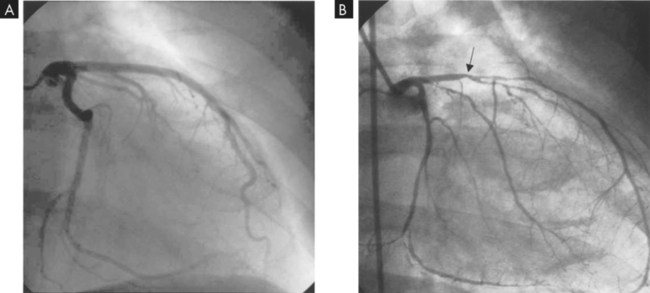
FIGURE 23-14 Angiogram of coronary artery disease.
A Normal left coronary artery angiogram. B Left coronary artery angiogram with decreased blood flow due to atherosclerosis. Note the position of the arrow in B; atherosclerosis significantly decreases blood flow from that point.
Source: Drake RL, Volg W, Mitchell AWM. Gray‘s anatomy for students. Churchill Livingstone; 2004.
In coronary heart disease, ischaemia can develop within 10 seconds of coronary occlusion. Myocytes (cardiac muscle cells) do not have the capacity to store a large amount of adenosine triphosphate (ATP). Therefore, myocytes need a constant supply of blood that carries oxygen and nutrients, so that ATP can be manufactured continually. If coronary artery blood flow is impeded, after several minutes, the heart cells lose the ability to contract, thus hampering pump function and depriving the myocardium of a glucose source necessary for aerobic metabolism. Anaerobic processes take over and lactic acid accumulates. Cardiac muscle cells remain viable for approximately 20 minutes under ischaemic conditions. If blood flow is restored, aerobic metabolism resumes, contractility is restored and cellular repair begins. If perfusion is not restored, then myocardial infarction occurs (see Figure 23-13), which causes death to those deprived cardiac cells. These cells do not regenerate and remain as non-contractile scar tissue, no longer able to contribute to cardiac function.
CLINICAL MANIFESTATIONS
Individuals with reversible myocardial ischaemia can present clinically in several ways. Chronic coronary obstruction results in recurrent predictable chest pain called angina pectoris (commonly referred to as angina). Abnormal vasospasm of coronary vessels results in unpredictable chest pain called Prinzmetal’s angina (also known as variant angina). Myocardial ischaemia that does not cause obvious and detectable symptoms is called silent ischaemia.
Angina
Angina is chest pain caused by myocardial ischaemia. The discomfort is usually transient, lasting approximately 3–5 minutes. If blood flow is restored, no permanent change or damage results. Angina is typically experienced as substernal chest discomfort, ranging from a sensation of heaviness or pressure to moderately severe pain. Individuals often describe the sensation by clenching a fist over the left sternal border. Discomfort may radiate to the neck, lower jaw, left arm and left shoulder or, occasionally, to the back or down the right arm. Discomfort is commonly mistaken for indigestion. The pain is presumably caused by the build-up of lactic acid or abnormal stretching of the ischaemic myocardium that irritates myocardial nerve fibres. These afferent sympathetic fibres enter the spinal cord from levels C3 to T4, accounting for the variety of locations and radiation patterns of angina. Pallor, diaphoresis (excessive sweating) and dyspnoea may be associated with the pain. Stable angina is caused by gradual luminal narrowing and hardening of the arterial walls, so that affected vessels cannot dilate in response to increased myocardial demand associated with physical exertion or emotional stress.
Prinzmetal’s angina is chest pain attributable to transient ischaemia of the myocardium that occurs unpredictably and often at rest. Pain is caused by vasospasm of one or more major coronary arteries, with or without associated atherosclerosis. The pain often occurs at night during rapid eye movement sleep and may have a cyclic pattern of occurrence. The angina may result from hyperactivity of the sympathetic nervous system, increased calcium flux in arterial smooth muscle or endothelial dysfunction with impaired production or release of prostaglandin or thromboxane and abnormal responses to acetylcholine.41
Myocardial ischaemia may not cause detectable symptoms such as angina. Ischaemia may be totally asymptomatic, referred to as silent ischaemia. Individuals may complain only of fatigue, dyspnoea or a feeling of unease. Silent ischaemia and atypical symptoms are more common in women (see the box ‘Health alert: women and coronary heart disease’). In addition, individuals who experience angina often have additional silent episodes of myocardial ischaemia.
EVALUATION AND TREATMENT
Many individuals with reversible myocardial ischaemia will have a normal physical examination between events. However, in those with chronic ischaemia, the examination may disclose rapid pulse or extra heart sounds, indicating impaired left ventricular function during ischaemia. The presence of xanthelasmas (small fat deposits; see Figure 23-15) around the eyelids or arcus senilis of the eyes (a yellow lipid ring around the cornea) suggests dyslipidaemia and possible atherosclerosis.
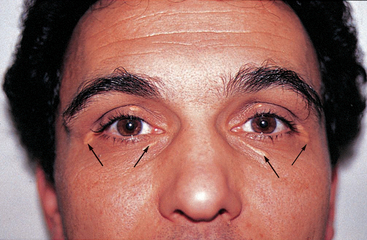
FIGURE 23-15 Xanthelasmas around the eyes (arrows) are indicative of high blood lipid levels and possible atherosclerosis.
Source: Talley NJ, O’Connor S. Clinical examination: a systematic guide to physical diagnosis. 6th edn. Sydney: Elsevier; 2010.
Women and coronary heart disease
Currently, more women die from coronary heart disease and stroke than from all cancers combined. Women have a higher rate of mortality than men, in part because of underdiagnosis and treatment. Nearly two-thirds of women who die from coronary heart disease have no prior warning symptoms, and symptoms that do occur are often different from those classically seen in men. One postulate is that women have more microvascular coronary disease than men, which can cause fewer and less recognisable symptoms. Women also have more avoidable risk factors than men, especially elevated cholesterol and physical inactivity, and women are less likely to receive counselling about nutrition, exercise and weight control. In addition, coronary heart disease risk rises dramatically after menopause. Although many studies suggest that endogenous oestrogen is protective of vascular function, several large prospective studies have determined that oestrogen replacement regimens do not reduce the risk of coronary heart disease in postmenopausal women. In addition to lifestyle changes, the most effective interventions to reduce coronary heart disease risk in women are found to be the statin drugs, which lower cholesterol and exert both anti-inflammatory and plaque-stabilising effects.
Source: D’Antono B et al. Angina symptoms in men and women with stable coronary artery disease and evidence of exercise-induced myocardial perfusion defects. Am Heart J 2006; 151(4):813–819; Gauri AJ et al. Disparities in the use of primary prevention and defibrillator therapy among blacks and women. Am J Med 2006; 119(2):167.e17–167.e21; Kuller LH. Women’s Health Initiative: hormone replacement therapy and risk of cardiovascular disease: implications of the results of the Women’s Health Initiative. Arterioscler Thromb Vasc Biol 2003; 23(1):11–16; Morise AP. Assessment of estrogen status as a marker of prognosis in women with symptoms of suspected coronary artery disease presenting for stress testing. Am J Cardiol 2006; 97(3):367–371; Shaw LJ et al. Insights from the NHLBI-sponsored Women’s Ischemia Syndrome Evaluation (WISE) study. Part I: gender differences in traditional and novel risk factors, symptom evaluation, and gender-optimized diagnostic strategies. J Am Coll Cardiol 2006; 47(suppl): 1s–71s; Vaccarino V et al. Sex and racial differences in the management of acute myocardial infarction, 1994 through 2002. N Engl J Med 2005; 353(7):671–682; Dale KM et al. Impact of gender on static efficacy. Curr Med Res Opin 2007; 23(3):565–574.
Electrocardiography is a critical tool for the diagnosis of myocardial ischaemia. Because many individuals have normal electrocardiograms when there is no pain, diagnosis requires that electrocardiography be performed during an episode of angina or during stress testing. The ST segment and T wave segment of the electrocardiogram correlate with ventricular contraction and relaxation. Transient ST segment depression and T wave inversion are characteristic signs of subendocardial ischaemia. ST elevation, indicative of transmural ischaemia, is seen in individuals with Prinzmetal’s angina and transmural myocardial infarction (see Figure 23-16). The ECG may also indicate which coronary artery is involved. Exercise stress testing is useful in differentiating angina from other types of chest pain, as well as detecting ischaemic changes that occur in the absence of angina pain.
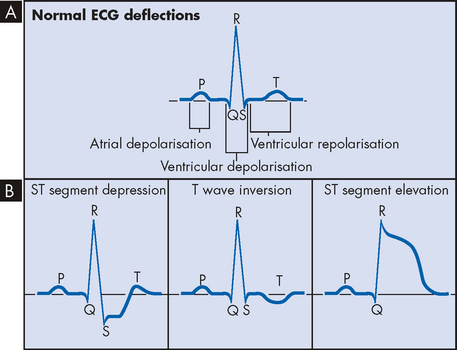
FIGURE 23-16 Electrocardiogram and ischaemic changes.
A Normal ECG rhythm. B Associated ECG changes with ischaemia.
Coronary angiography helps determine the anatomical extent of coronary heart disease. The procedure involves threading a small aperture catheter into the coronary arteries, via arterial cannulation (usually using the femoral artery), and injecting dye directly into the coronary arteries. Sophisticated X-rays allow visualisation of the vessels and the extent of disease can be determined.
The primary aim of therapy for myocardial ischaemia and angina is to reduce myocardial oxygen consumption by favourably altering its various determinants. The factors most amenable to pharmacological manipulation are blood pressure, heart rate, contractility and left ventricular volume. Medications that reduce vasospasm, lower cholesterol and prevent clotting are also useful. These drugs include nitrates, β-adrenergic blocking agents, calcium channel blockers, ACE inhibitors, lipid-lowering agents (statins) and antiplatelet agents. Such drugs are recommended by the National Heart Foundation of Australia and the Cardiac Society of Australia and New Zealand.42 More recently, a large meta-analysis has shown that long-acting calcium channel blockers reduce the risk of stroke, angina pectoris and heart failure in patients with coronary heart disease.43
One of the main treatment options for cardiac tissue that is affected by myocardial ischaemia includes percutaneous (through the skin) coronary intervention, which involves accessing the coronary arterial system in a similar manner to performing coronary angiography as described above. Percutaneous coronary intervention includes angioplasty and stenting.
Percutaneous transluminal coronary angioplasty (PTCA) is a procedure whereby stenotic (narrowed) coronary vessels are dilated with a balloon. As the balloon is inflated (temporarily) a number of times, the plaque becomes compressed, thereby increasing the blood vessel lumen. Several different types of catheters with balloons can be used to open the blocked vessel. PTCA is generally used to treat single-vessel disease, but it can be effective with multiple-vessel disease or restenosis (or reocclusion) of a coronary artery bypass graft. Restenosis of the artery is the major complication of the procedure.44–46 4 6
Improved outcomes following PTCA have been achieved by the use of coronary stenting. Following balloon treatment during PTCA, a small cylinder of metal called a stent is inserted into the artery — this remains permanently within the coronary vessel. Multiple stents are often used where a few blockages are treated. Although the stent initially maintains the vessel lumen, stents have been thrombogenic (tending to produce a thrombus [clot]) in nature and have tended to promote hypertrophy of vessel endothelium as well as platelet plug formation.45 Use of antiplatelet drugs such as abciximab (pronounced ‘ab-sick-see-mab’) has improved outcomes.44 Furthermore, stents can be coated with drugs that are slowly released to reduce the restenosis.46
Coronary heart disease and associated ischaemic events can be surgically treated using coronary artery bypass graft. This technique uses grafts from blood vessels (for example, the internal mammary artery or saphenous vein from the leg) to oversew the diseased coronary artery portion, such that blood flow to the myocardium can be restored. The surgery can be performed either using cardiopulmonary bypass (the heart is stopped and blood is circulated and oxygenated external to the body using a bypass circuit and artificial pump) or without bypass. The requirement for this surgical treatment is based on the degree of symptoms arising from myocardial ischaemia (angina), the extent of coronary heart disease in multiple vessels and which coronary arteries are affected. This treatment option would be used when coronary occlusions are severe (only a small lumen remaining open) or widespread (affecting a number of vessels).
The acute coronary syndromes
The acute coronary syndromes are one of the most common causes of acute medical admissions to Australian hospitals.47 The syndromes are a continuum of clinical presentations that encompasses unstable angina without myocyte death through to myocardial infarction with and without ST segment elevation.48 However, the terminology used to describe the acute coronary syndromes is changing to match clinical approaches that are more appropriate to the patient’s condition, rather than forming a definitive diagnosis upon presentation.47 Figure 23-17 shows how initial presentation of the acute coronary syndromes may change over time.
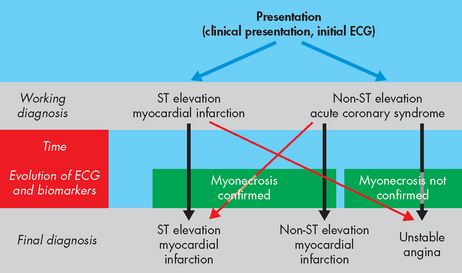
FIGURE 23-17 The acute coronary syndromes: clinical presentation and possible changes to the initial working diagnosis.
Source: Aroney CN, Aylward P, Kelly A-M, Chew DPB, Clune E on behalf of the Acute Coronary Syndrome Guidelines Working Group. Guidelines for the management of acute coronary syndromes. MJA 2006; 184(8); S1–S30.
The process of atherosclerotic plaque progression can be gradual, taking several decades before angina manifests. Eventually plaque formation will ensue, which can be either stable and result in stable angina, or unstable and prone to rupture and thrombus formation. When there is sudden coronary obstruction caused by thrombus formation over a ruptured or ulcerated atherosclerotic plaque, the acute coronary syndromes result. Thrombus formation on a ruptured plaque that disperses in less than 20 minutes leads to transient ischaemia and unstable angina. If vessel obstruction is sustained, myocardial infarction with inflammation and necrosis of the myocardium results (see Figure 23-18).
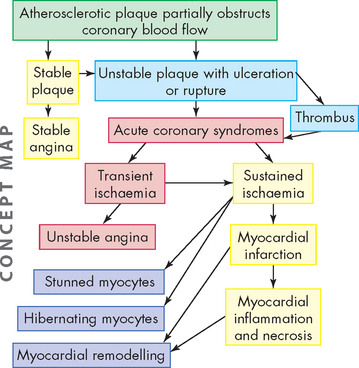
Unstable angina is the result of reversible myocardial ischaemia and is a strong indicator of impending myocardial infarction. Acute myocardial infarction (AMI) results when there is prolonged ischaemia causing irreversible damage to the myocytes (heart muscle cells). Plaque disruption occurs because of shear forces, inflammation with release of multiple inflammatory mediators, secretion of macrophage-derived degradative enzymes, immune cell activation and apoptosis of cells at the edges of the lesions (see Figure 23-19).23,40,49,50 The underlying layer of plaque is then exposed and this causes activation of the coagulation cascade. The resulting thrombus can form quickly. The thrombus may break up before permanent myocyte damage has occurred (unstable angina) or it may cause prolonged ischaemia by impeding blood flow past the thrombus and causing lack of oxygen and nutrients to the myocardium, resulting in infarction of the heart muscle (acute myocardial infarction: see Figure 23-20). Acute myocardial infarction can be further subdivided into non-ST elevation MI (non-STEMI) and ST elevation MI (STEMI). Sudden cardiac death can occur as a result of any of the acute coronary syndromes.
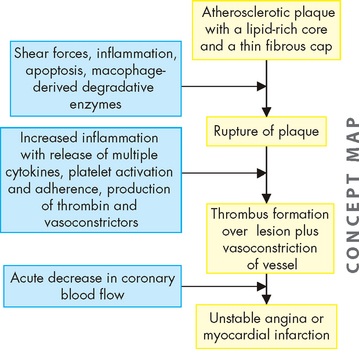
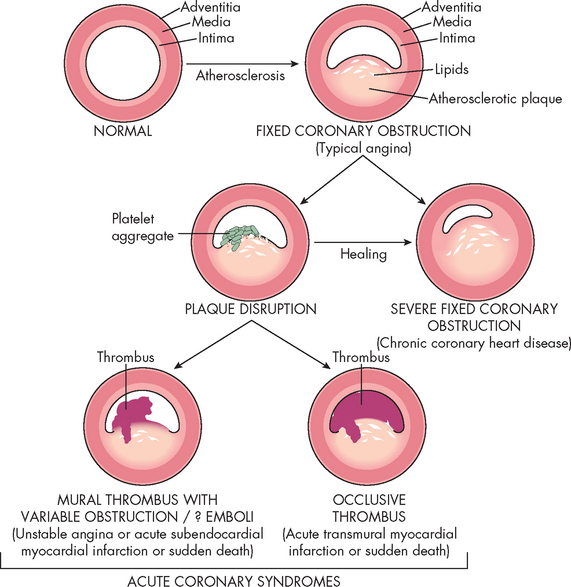
FIGURE 23-20 Acute coronary syndromes and thrombus formation resulting in angina, myocardial infarction or sudden death.
Schematic representation of sequential progression of coronary artery lesion morphology, beginning with stable chronic plaque responsible for typical angina and leading to the various acute coronary syndromes.
Source: Schoen FJ, Banasik JL. Interventional and surgical cardiovascular pathology: clinical correlations and basic principles. Philadelphia: Saunders; 1989.
Unstable angina
Unstable angina is a form of acute coronary syndrome that results from reversible myocardial ischaemia. It is important that you can recognise this syndrome, because it signals that the atherosclerotic plaque has become complicated and infarction may soon follow. Unstable angina occurs when a fairly small fissuring or superficial erosion of the plaque leads to transient episodes of thrombotic vessel occlusion and vasoconstriction at the site of plaque damage. This thrombus is unstable and may occlude the vessel for no more than 10–20 minutes, with return of perfusion before significant myocardial necrosis occurs. Unstable angina presents as new-onset angina, angina occurring at rest or angina that is increasing in severity or frequency. Individuals may experience increased dyspnoea (difficulty breathing), diaphoresis (excessive sweating) and anxiety as the angina worsens. Physical examination may reveal evidence of ischaemic myocardial dysfunction such as tachycardia. The ECG most commonly reveals ST segment depression and T wave inversion during pain that resolves as the pain is relieved (see Figure 23-16). Approximately 20% of individuals with unstable angina will progress to acute myocardial infarction or death. Management of unstable angina requires some form of antithrombotic therapy. In most cases, individuals are given aspirin and clopidogrel when PTCA is anticipated — these drugs are used to prevent platelet aggregation and therefore avoid platelet plug formation.
Acute myocardial infarction
When coronary blood flow is interrupted for an extended period of time, myocyte necrosis occurs.51 This results in acute myocardial infarction. Pathologically, there are two major types of myocardial infarction: subendocardial infarction and transmural infarction. Plaque progression, disruption and subsequent clot formation are the same for acute myocardial infarction as they are for unstable angina (see Figures 23-18, 23-19 and 23-20).52 In this case, however, the thrombus is lodged in the coronary artery and occludes the vessel for a prolonged period, such that myocardial ischaemia progresses to myocyte necrosis and death.
If the thrombus breaks up before complete distal tissue necrosis has occurred, the infarction will involve only the myocardium directly beneath the endocardium (subendocardial infarction). If the thrombus lodges permanently in the vessel, the infarction will extend through the myocardium all the way from the endocardium to the pericardium, resulting in severe cardiac dysfunction (transmural infarction).Clinically, it is important to identify those individuals with transmural infarction, who are at highest risk for serious complications and should receive definitive intervention without delay. These individuals usually have marked elevations in the ST segments on ECG and are categorised as having STEMI. Those without T segment elevation are more likely to have subendocardial infarction and are said to have non-STEMI (see Figure 23-16).
PATHOPHYSIOLOGY
Cellular injury
Cardiac cells can withstand a lack of oxygen and blood flow for about 20 minutes before cellular death takes place. After only 30–60 seconds of hypoxia (low oxygen levels in the tissues), electrocardiograph changes are visible. Yet even if cells are metabolically altered and non-functional, they can remain viable if blood flow is restored within 20 minutes.
After 8–10 seconds of decreased blood flow, the affected myocardium becomes cyanotic and cooler. Myocardial oxygen reserves are used quickly (within about 8 seconds) after complete cessation of coronary flow. Glycogen stores decrease as anaerobic metabolism begins. Unfortunately, glycolysis can supply only 65–70% of the total myocardial energy requirement and produces much less ATP than aerobic (oxygen-dependent) processes. Hydrogen ions and lactic acid accumulate. Because myocardial tissues have poor buffering capabilities and myocardial cells are sensitive to low cellular pH (build-up of acid), accumulation of these products further compromises the myocardium. Acidosis may make the myocardium more vulnerable to the damaging effects of lysosomal enzymes and may suppress impulse conduction and contractile function, thereby leading to heart failure.
Oxygen deprivation is also accompanied by electrolyte disturbances, specifically the loss of potassium, calcium and magnesium from inside the cell. Myocardial cells deprived of necessary oxygen and nutrients lose contractility, thereby diminishing the pumping ability of the heart. Normally, the myocardium takes up varying quantities of catecholamines (adrenaline and noradrenaline). Significant coronary artery occlusion causes the myocardial cells to release catecholamines, predisposing the individual to serious imbalances of sympathetic and parasympathetic function, irregular heartbeat (arrhythmia) and heart failure (see Figure 23-21). Catecholamines mediate the release of glycogen, glucose and stored fat from body cells. Therefore, plasma concentrations of free fatty acids and glycerol rise within 1 hour after the onset of acute myocardial infarction. Excessive levels of free fatty acids can have a harmful effect on cell membrane structure. Noradrenaline elevates blood glucose levels through stimulation of liver and skeletal muscle cells and suppresses pancreatic beta-cell activity, which reduces insulin secretion and elevates blood glucose further. Not surprisingly, hyperglycaemia is noted approximately 72 hours after acute myocardial infarction.53
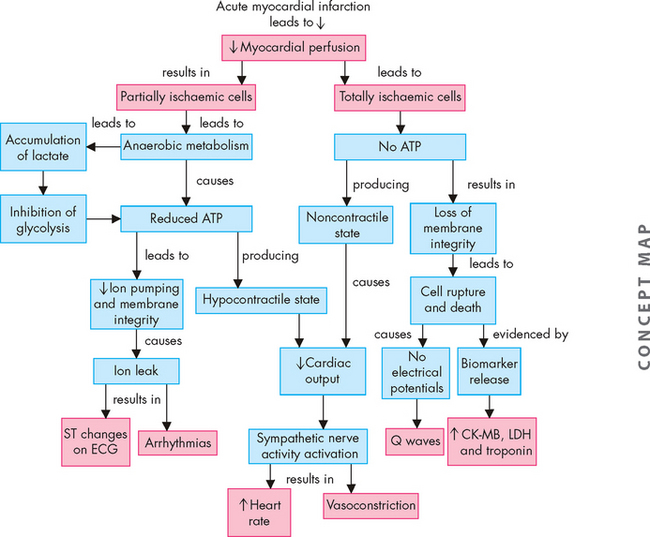
FIGURE 23-21 Cellular and systemic events occurring after acute myocardial infarction.
ATP = adenosine triphosphate; CK-MB = myocardial band of creatine kinase; LDH = lactate dehydrogenase.
Source: Copstead-Kirkhorn L-EC. Pathophysiology. 8th edn. Philadelphia: Saunders; 2010.
Angiotensin II is released during myocardial ischaemia and contributes to the pathogenesis of myocardial infarction in several ways. First, it promotes catecholamine release and causes coronary artery spasm. Second, it results in peripheral vasoconstriction and fluid retention. Finally, it is a growth factor for vascular smooth muscle cells, myocytes and cardiac fibroblasts, resulting in structural changes in the myocardium called ‘remodelling’.54
Cellular death
After about 20 minutes of myocardial ischaemia, irreversible hypoxic injury causes cellular death and tissue necrosis. This lack of blood flow deprives myocytes of oxygen and nutrients, and ultimately damage to the sarcolemma (cell membrane in myocytes) and coagulation and necrosis develop (see Chapter 4). This results in the release of intracellular enzymes such as creatine kinase and myocyte proteins such as the troponins through the damaged cell membranes into the interstitial spaces, and the lymphatic vessels pick up the enzymes and transport them into the bloodstream, where they can be detected by blood tests.
Structural and functional changes
With infarction, ventricular function is abnormal and the ejection fraction (the percentage of blood ejected from the ventricles with each heart beat, usually about 65%) falls, which results in increases in ventricular end-diastolic volume. If the coronary obstruction involves the perfusion to the left ventricle, pulmonary venous congestion ensues; if the right ventricle is ischaemic, increases in systemic venous pressures occur.
Myocardial infarction results in both structural and functional changes of cardiac tissues (see Figures 23-22 and 23-23). Gross tissue changes at the area of infarction may not become apparent for several hours, despite the almost immediate onset (within 30–60 seconds) of electrical conduction changes. Cardiac tissue surrounding the area of infarction also undergoes changes that can be categorised into:
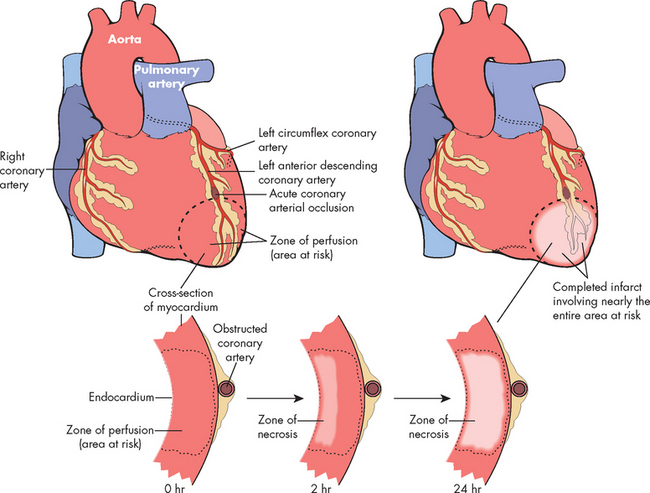
FIGURE 23-22 A schematic representation of myocardial infarction after acute coronary artery occlusion.
Necrosis begins in a small zone of the myocardium beneath the endocardial surface in the centre of the ischaemic zone. This entire region of myocardium (shaded) depends on the occluded vessel for perfusion and is the area at risk. Note that a very narrow zone of myocardium immediately beneath the endocardium is spared from necrosis because it can be oxygenated by diffusion from the ventricle. The end result is necrosis of the muscle that was dependent on perfusion from the obstructed coronary artery. Nearly the entire area at risk loses viability.
Source: Kumar V. Robbins & Cotran pathologic basis of disease. 7th edn. Philadelphia: Saunders; 2004.
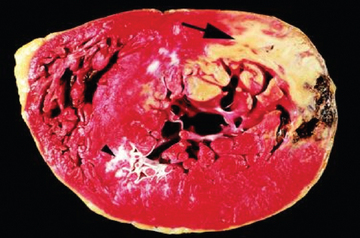
FIGURE 23-23 Evidence of acute myocardial infarction of the left ventricle.
The large arrow at the top indicates an area of necrosis, coloured yellow. The far edge (right side) of the myocardial infarction contains a myocardial haemorrhage that was associated with cardiac rupture. An old infarct is also evident (arrowhead).
Source: Kumar V. Robbins & Cotran pathologic basis of disease. 8th edn. Philadelphia: Saunders; 2010.
The severity of functional impairment depends on the size of the lesion and the site of infarction. Functional changes can include (1) decreased cardiac contractility with abnormal wall motion, (2) altered left ventricular compliance, (3) decreased stroke volume, (4) decreased ejection fraction, (5) increased left ventricular end-diastolic pressure and (6) sinoatrial node malfunction. Life-threatening arrhythmias and heart failure often follow myocardial infarction.
Repair
Acute myocardial infarction causes a severe inflammatory response that ends with wound repair. Damaged cells undergo degradation, fibroblasts proliferate and scar tissue is synthesised. Many cell types, hormones and nutrient substrates must be available for optimal healing to proceed. Within 24 hours, leucocytes infiltrate the necrotic area and proteolytic enzymes from scavenger neutrophils degrade necrotic tissue. The collagen matrix that is deposited is initially weak, mushy and vulnerable to re-injury. Unfortunately, it is at this time in the recovery period (10–14 days after infarction) that individuals may feel like increasing their activities and so may stress the newly formed scar tissue. After 6 weeks, the necrotic area is completely replaced by scar tissue, which is strong but cannot contract and relax like healthy myocardial tissue.
CLINICAL MANIFESTATIONS
The first symptom of acute myocardial infarction is usually sudden, severe chest pain. The pain is similar to angina pectoris but more severe and persistent and is not relieved by medication or rest. It may be described as heavy and crushing. Pain radiating to the neck, jaw, back, shoulder or left arm is common. Some individuals, especially those who are elderly or have diabetes mellitus, experience no pain, thereby having a ‘silent’ infarction. Infarction often simulates a sensation of unrelenting indigestion. Nausea and vomiting may occur because of reflex stimulation of vomiting centres by pain fibres. Reflexes from the area of the infarcted myocardium may also result in stimulation of the gastrointestinal tract via parasympathetic nervous system reflexes. Catecholamine release results in sympathetic stimulation, producing diaphoresis (excessive sweating) and peripheral vasoconstriction that cause the skin to become cool and clammy.
Various cardiovascular changes are found on physical examination:
The number and severity of post-infarction complications depend on the location and extent of necrosis, the physiological condition of the individual before infarction and the availability of rapid therapeutic intervention, such as thrombolysis. Sudden cardiac death may occur in individuals with myocardial ischaemia even if infarction is absent or minimal and is a multifactorial problem. Risk factors for sudden death are related to three factors: ischaemia, left ventricular dysfunction and electrical instability. These factors interact with each other (see Figure 23-24). Table 23-7 lists the most common complications from acute myocardial infarctions.
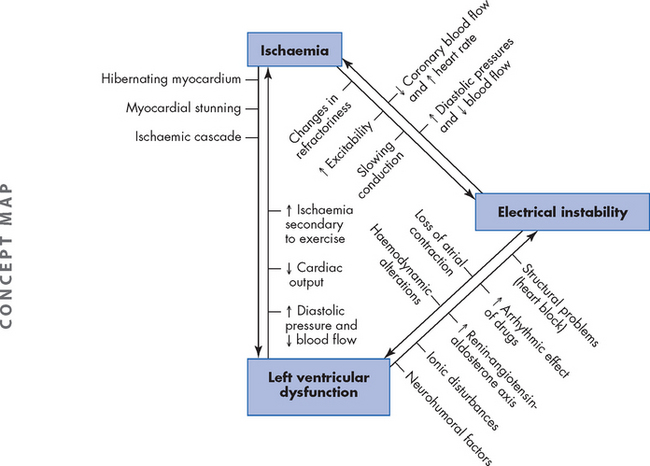
FIGURE 23-24 Three interacting factors relating to sudden cardiac events that leads to death.
The three factors are ischaemia, left ventricular dysfunction and electrical instability.
Table 23-7 COMPLICATIONS FROM ACUTE MYOCARDIAL INFARCTIONS
| TYPE | CHARACTERISTICS |
|---|---|
| Arrhythmias | |
| Left ventricular failure | |
| Inflammation of the pericardium (pericarditis) | |
| Organic brain syndrome | Can occur if brain blood flow is impaired secondary to acute myocardial infarction |
| Transient ischaemic attacks or stroke | Occur if thromboemboli break loose from clots that form in the cardiac chambers or on cardiac valves |
| Rupture of heart structures | |
| Rupture of wall of infarcted ventricle | Can be caused by aneurysm formation when pressure becomes too great |
| Left ventricular aneurysm | Late (month to years) complication of acute myocardial infarction that can contribute to heart failure and thromboemboli |
| Infarctions around septal structures | |
| Systemic thromboembolism | May disseminate from debris and clots that collect inside dilated aneurysmal sacs or from infarcted endocardium |
| Pulmonary thromboembolism | |
| Sudden death |
EVALUATION AND TREATMENT
The diagnosis of acute myocardial infarction is made on the basis of the history, physical examination, ECG and serial enzyme alterations. The cardiac enzymes, troponin I (cTnI) and troponin T (cTnT), are the most specific indicators of an acute myocardial infarction. A transient rise in these plasma enzyme levels can confirm the occurrence of acute myocardial infarction and indicate its severity. Other enzymes released by myocardial cells include creatine kinase-myocardial band (CK-MB) and lactate dehydrogenase. Cardiac troponins will always be within normal levels when tissue other than cardiac muscle is damaged, therefore, troponin I and troponin T are more specific and are elevated soon after infarction and remain elevated for longer than other enzymes (see Figure 23-25). Clinically, repeated measures of elevated troponin and isoenzymes (enzymes with slightly different structures) over time are essential to determine if an acute myocardial infarction has occurred. Elevation of troponin, CK-MB and lactate dehydrogenase may be noted at characteristic times and laboratory confirmation that an infarction has occurred may be delayed up to 12 hours.
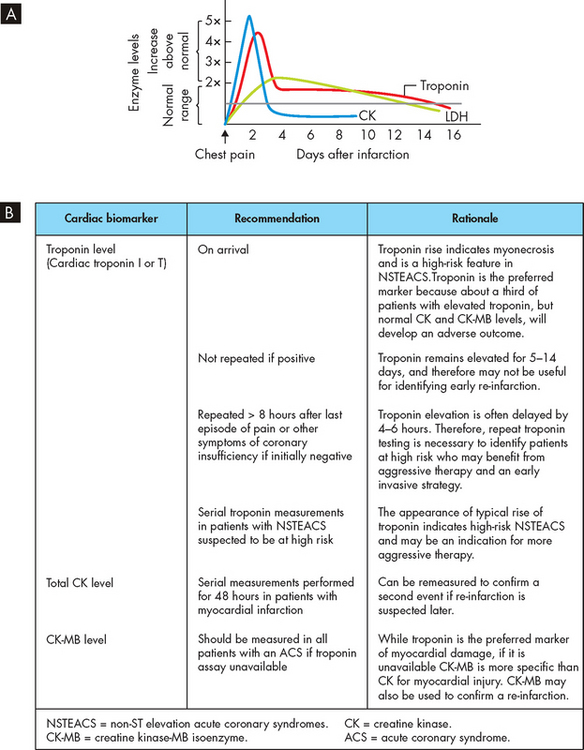
FIGURE 23-25 Serum cardiac enzymes (biomarkers) after acute myocardial infarction.
A Graphical representation of changes in enzymes (cardiac specific and nonspecific) over time. B Recommended times for measuring cardiac enzymes (biomarkers) when acute myocardial infarction is suspected.
Source: A Pagana KD. Mosby’s diagnostic and laboratory test reference. 9th edn. St Louis: Mosby; 2008. B Aroney CN et al on behalf of the Acute Coronary Syndrome Guidelines Working Group (2006). Guidelines for the management of acute coronary syndromes MJA 2006; 184(8);S1–S30.
Myocardial infarction can occur in various regions of the heart wall and may be described as anterior, inferior, posterior, lateral, subendocardial or transmural, depending on the anatomical location and extent of tissue damage from infarction. Twelve-lead electrocardiograms help localise the affected area through identification of Q waves and changes in ST segments and T waves (see Figure 23-26). The infarcted myocardium is surrounded by a zone of hypoxic injury, which may progress to necrosis or return to normal. Adjacent to this zone of hypoxic injury is a zone of reversible ischaemia. Ischaemic and injured myocardial tissue causes ST and T wave changes. If the thrombus breaks up before complete distal tissue necrosis has occurred, the infarction will involve only the myocardium directly beneath the endocardium. This type of myocardial infarction most often presents with no elevation of the ST segment on ECG and therefore is termed non-STEMI.58,59 It is especially important to recognise this form of acute coronary syndrome because recurrent clot formation on the disrupted atherosclerotic plaque is likely, with resultant infarct expansion. If the thrombus lodges more permanently in the vessel, the infarction will extend through the myocardium from endocardium to pericardium, resulting in severe cardiac dysfunction. This usually presents with significant ST segment elevation on ECG (STEMI). Often a characteristic Q wave will develop on ECG some hours later. An ST elevated myocardial infarction (STEMI) requires rapid intervention to prevent serious complications and sequelae.
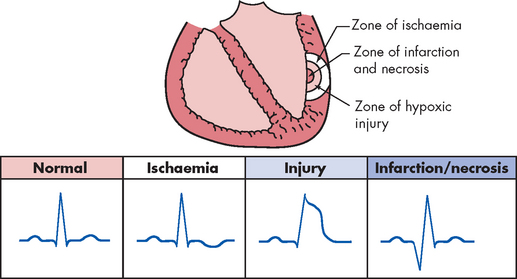
Additional laboratory data may reveal leucocytosis, elevated sedimentation rate and C-reactive protein, all of which indicate inflammation. The blood glucose level is usually elevated and the glucose tolerance level may remain abnormal for several weeks.53 Hypoxaemia may also accompany heart failure.
Immediate treatment
Acute myocardial infarction requires admission to hospital, often directly into a coronary care unit. The individual should be placed on supplemental oxygen and given aspirin immediately. Pain relief is of the utmost importance and involves the use of sublingual glyceryl trinitrate and intravenous morphine. Continuous monitoring of cardiac rhythms and enzymatic changes is essential, because the first 24 hours after onset of symptoms is the time of highest risk for sudden death. Both non-STEMI and STEMI are managed with the urgent administration of thrombolytics or by percutaneous coronary intervention along with antithrombotics.47 Further management may include ACE inhibitors and β-blockers. Individuals who are in shock require aggressive fluid resuscitation, inotropic drugs (drugs that increase contraction of the myocardium) and possible emergency invasive procedures (see the section on shock later in this chapter).
Ongoing treatment
Bed rest, followed by gradual return to the activities of daily living, reduces the myocardial oxygen demands of the compromised heart. Individuals not receiving thrombolytic or heparin infusion must receive prophylaxis (preventative medication) for deep venous thrombosis as long as their activity is significantly limited. Stool softeners are given to eliminate the need for straining, which can precipitate bradycardia and can be followed by increased venous return to the heart, causing possible cardiac overload. Treatment of dyslipidaemia with statins (e.g. simvastatin, atorvastatin) can reduce the risk of future cardiovascular events.42 Drugs that decrease lipidaemia should be administered before the patient is discharged from hospital. Education on diet, caffeine intake, smoking cessation, exercise and other aspects of risk factor reduction is crucial for secondary prevention of recurrent myocardial ischaemia.42
Aneurysm
An aneurysm is a localised dilation or outpouching of a vessel wall or cardiac chamber. Aneurysms form in arteries when there is disruption of the wall of the vessel associated with changes in collagen and elastin, which make the vessel more vulnerable to intravascular pressures. The aorta is particularly susceptible to aneurysm formation because of constant stress on the vessel wall. Some 75% of all aneurysms occur in the abdominal aorta (see Figure 23-27). Atherosclerosis is the most common cause of arterial aneurysms because plaque formation erodes the vessel wall and contributes to inflammation that can further weaken the vessel. Hypertension also contributes to aneurysm formation by increasing wall stress.
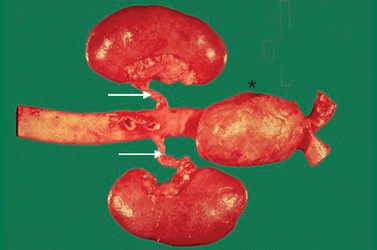
An abdominal aortic aneurysm (*) is distal to the renal arteries (white arrows). The large pouching of the artery occurs due to atherosclerosis weakening the wall, and the high pressure of the arterial system causes the bulging of the vessel. Aortic aneurysms tend to enlarge over time and if undiscovered or treated can rupture, often leading to death.
Source: Klatt E. Robbins & Cotran atlas of pathology. 2nd edn. Philadelphia: Saunders; 2010.
Aortic aneurysms are often asymptomatic until they leak and possibly rupture, when they become painful. Symptoms of dysphagia (difficulty swallowing) and dyspnoea (breathlessness) are caused by the pressure of a thoracic aneurysm on surrounding organs. An aneurysm that impairs flow to an extremity causes symptoms of ischaemia. Cerebral aneurysms are associated with signs and symptoms of increased intracranial pressure and can relate to stroke. (Cerebral aneurysms are described in Chapter 9.)
The diagnosis of an aneurysm is usually confirmed by ultrasonography, CT scan, MRI or angiography. The goals of medical treatment of aneurysms are to maintain a low blood volume and low blood pressure to decrease the mechanical forces thought to contribute to vessel wall dilation. Medical treatment is indicated for slow-growing aortic aneurysms, particularly in the early stages, and includes smoking cessation, reducing blood pressure and blood volume. For those aneurysms that are dilating rapidly, surgical treatment is often indicated. Surgery should be done when aortic aneurysms become large and usually includes replacement with a prosthetic graft. New endovascular surgical techniques make aneurysm repair possible for more individuals.60
Thrombus formation
As in venous thrombosis, arterial thrombi tend to develop when intravascular conditions promote activation of coagulation or when there is stasis of blood flow. These conditions include those in which there is intimal irritation or roughening (such as in surgical procedures), inflammation, traumatic injury, infection, low blood pressures or obstructions that cause blood stasis and pooling within the vessels. Inflammation of the endothelium leads to activation of the coagulation cascade causing platelets to adhere readily. An anatomical change in an artery can contribute to thrombus formation, particularly if the change results in a pooling of arterial blood. Thrombi also form on heart valves altered by calcification or bacterial vegetation. Valvular thrombi are most commonly associated with inflammation of the endocardium (endocarditis) and rheumatic heart disease. Shock (circulatory failure), particularly shock resulting from septicaemia, can also activate the intrinsic and extrinsic pathways of coagulation. The impaired cellular metabolism that occurs with all types of shock activates the extrinsic pathway of coagulation, whereas blood stasis caused by very low blood pressures activates the intrinsic pathway.
Arterial thrombi pose two potential threats to the circulation. First, the thrombus may grow large enough to occlude the artery, causing ischaemia in tissue supplied by the artery. Second, the thrombus may dislodge, becoming a thromboembolus that travels through the vascular system until it occludes flow into a distal systemic vascular bed.
Diagnosis of arterial thrombi is usually accomplished through the use of Doppler ultrasonography and angiography. Pharmacological treatment involves the administration of heparin, warfarin derivatives, thrombin inhibitors or thrombolytics. A balloon-tipped catheter can also be used to remove or compress an arterial thrombus. Various combinations of drug and catheter therapies are sometimes used concurrently.
Embolism
Embolism is the obstruction of a vessel by an embolus (a bolus of matter circulating in the bloodstream). The embolus may consist of a dislodged thrombus; an air bubble; an aggregate of amniotic fluid; an aggregate of fat, bacteria or cancer cells; or a foreign substance (see Table 23-8 for more details). An embolus travels in the bloodstream until it reaches a vessel through which it cannot fit. No matter how tiny it is, an embolus will eventually lodge in a systemic or pulmonary vessel determined by its source. Pulmonary emboli originate on the venous side (mostly from the deep veins of the legs) of the systemic circulation or in the right side of the heart; systemic (or arterial) emboli most commonly originate in the left side of the heart and are associated with thrombi after myocardial infarction, valvular disease, left heart failure, endocarditis and arrhythmias. Embolism causes ischaemia or infarction in tissues distal to the obstruction.
| TYPE | CHARACTERISTICS |
|---|---|
| Arteries | |
| Arterial thromboembolism | Dislodged thrombus; source is usually from the heart; most common sites of obstruction are lower extremities (femoral and popliteal arteries), coronary arteries and cerebral vasculature |
| Veins | |
| Venous thromboembolism | Dislodged thrombus; source is usually from the lower extremities; obstructs branches of the pulmonary artery |
| Air embolism | Bolus of air displaces blood in the vasculature; source is usually room air entering the circulation through intravenous cannula; trauma to the chest may allow air from lungs to enter vascular space |
| Amniotic fluid embolism | Bolus of amniotic fluid; extensive intra-abdominal pressure attending labour and delivery can force amniotic fluid into the maternal bloodstream; introduces antigens, cells and protein aggregates that trigger inflammation, coagulation and immune responses |
| Bacterial embolism | Aggregates of bacteria in bloodstream; source is subacute bacterial endocarditis or abscess |
| Fat embolism | Globules of fat floating in the bloodstream associated with trauma to long bones; the lungs in particular are affected |
| Foreign substances | Small particles introduced during trauma or through an intravenous cannula; the coagulation cascade is initiated and thromboemboli form around the particles |
Peripheral artery disease
Peripheral artery disease refers to atherosclerotic disease of the arteries that perfuse the limbs, especially the lower extremities. The risk factors for peripheral artery disease are the same as those previously described for atherosclerosis and it is especially prevalent in individuals with diabetes mellitus.
Lower extremity ischaemia resulting from arterial obstruction in peripheral artery disease can be gradual or acute. In most individuals, gradually increasing obstruction to arterial blood flow to the legs caused by atherosclerosis in the iliofemoral vessels results in pain with ambulation called intermittent claudication. If a thrombus forms over the atherosclerotic lesion, perfusion can cease acutely with severe pain, loss of pulses and skin colour changes in the affected extremity.
Peripheral artery disease is often asymptomatic; therefore, evaluation for peripheral artery disease requires a careful history and physical examination that focuses on looking for evidence of atherosclerotic disease and non-invasive Doppler measurement of blood flow. Treatment includes risk factor reduction (smoking cessation and treatment of diabetes mellitus, hypertension and dyslipidaemia) and antiplatelet therapy. Symptomatic peripheral artery disease should be managed with vasodilators in combination with antiplatelet or antithrombotic medications (aspirin or clopidogrel) and exercise rehabilitation.61 If acute symptoms occur, percutaneous or surgical revascularisation may be indicated.
Alterations to veins
Venous thromboembolus
The insidious nature of venous thromboembolus is often overlooked in hospital patients. In Western countries, venous thromboembolism is both a common cause of morbidity and one of the most preventable causes of hospital mortality. It has been calculated that approximately 17,400 episodes of venous thromboembolism occur in Australia every year and the incidence is greater in the elderly.62 In addition, venous thromboembolism has been shown to be responsible for 7% of all deaths in Australian hospitals.63 Therefore, an understanding of the pathophysiology and treatment options is vital for all healthcare workers.
Venous thrombi are more common than arterial thrombi because flow and pressure are lower in the veins than in the arteries. The two main presentations of venous thromboembolus are deep vein thrombosis (DVT) and pulmonary embolism (see Figure 23-28). In this section, we discuss DVT; pulmonary embolism is discussed in detail in Chapter 25.
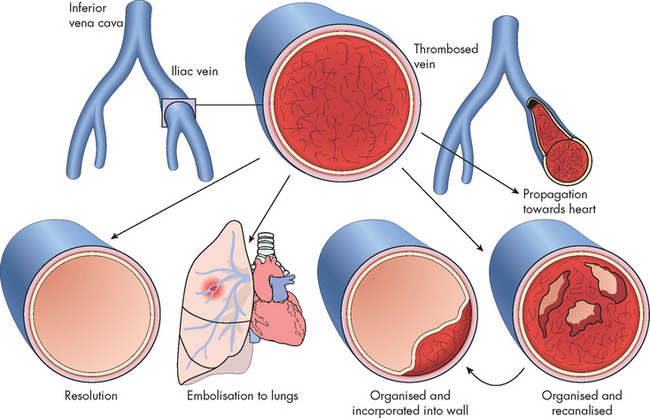
FIGURE 23-28 Venous thromboembolism: potential outcomes.
Source: Kumar V. Robbins & Cotran pathologic basis of disease. 7th edn. Philadelphia: Saunders; 2004.
DVT occurs primarily in the lower extremities. Three factors promote venous thrombosis: (1) venous stasis (e.g. immobility, age, heart failure); (2) venous endothelial damage (e.g. trauma, medications); and (3) hypercoagulable states (e.g. inherited disorders, malignancy, pregnancy, oral contraceptives, hormone replacement). Orthopaedic trauma (such as hip or leg fractures) or surgery, spinal cord injury and major trauma and general surgery can be associated with an extreme likelihood of DVT.63 Genetic abnormalities are also associated with an increased risk for venous thrombosis primarily related to states of hypercoagulability. Accumulation of clotting factors and platelets leads to thrombus formation in the vein, often near a venous valve. Inflammation around the thrombus promotes further platelet aggregation and the thrombus propagates or grows proximally. This inflammation may cause pain and redness, but because the vein is deep in the leg, it is usually not accompanied by clinical symptoms or signs. If the thrombus creates significant obstruction to venous blood flow, increased pressure in the vein behind the clot may lead to oedema of the extremity. Most thrombi eventually dissolve without treatment, but untreated DVT is associated with a high risk of an embolus forming and lodging in the lungs (pulmonary embolism).
Prevention is important in at-risk individuals. DVT prophylaxis (preventative therapies) is commonly used in hospital and out-of-hospital settings. This includes pharmacological therapies and mechanical devices to prevent formation of a DVT. Heparin and warfarin are the two most common anticoagulants used to prevent or decrease the incidence of DVT.64 Mechanical devices include graduated compression stockings of which there are two types: those used to prevent DVT (e.g. thromboembolic deterrent stockings (TED)) and those used for chronic venous insufficiency. Pneumatic devices that rhythmically compress the calf muscle are also effective; however, patients have limited mobilisation with these devices as they do not facilitate walking. One effective and simple measure is early ambulation (walking around) after a period of being bedridden, such as after surgery. This facilitates blood flow and decreases venous stasis in peripheral veins. If thrombosis does occur, best practice guidelines indicate that venous duplex ultrasound scanning for DVT and pulmonary imaging for pulmonary embolism are the most accurate. Diagnosis using blood coagulation tests, such as D-dimer measurement, is not recommended.64
Travel-related venous thromboembolus
In more recent times the frequency of air travel has increased dramatically: more people are flying and the length of time in-flight has increased. There have been many reports in the media about the increased risk of DVT formation with long-haul flights, once termed ‘economy class syndrome’.65 Contributing factors include immobility, cramped seating, lower atmospheric pressure and inadequate fluid intake, and combined it was thought that these considerably increased an individual’s risk of developing a DVT. However, more recent evidence indicates that the incidence of DVT formation and associated death following long-haul flights is actually low.66 Nonetheless, the risk is considerably increased in people with a pre-existing haematological disorder67 and others who have had recent surgery, a history of venous thromboembolism or a significant medical illness.64 In addition, endurance athletes may be prone to DVT formation related to air travel. For these individuals it is suggested that thromboembolic deterrent stockings and heparin be administered to mitigate the risk.64
Varicose veins
A varicose vein is a vein in which blood has pooled, producing distended, tortuous and palpable vessels. Veins are thin-walled, highly distensible vessels with valves to prevent backflow and pooling of blood. If a valve is damaged, a section of the vein is subjected to the pressure of a larger volume of blood under the influence of gravity. The vein becomes engorged with blood, which increases hydrostatic pressure and increases movement of plasma through the vessel wall, resulting in interstitial oedema (see Figure 23-29).
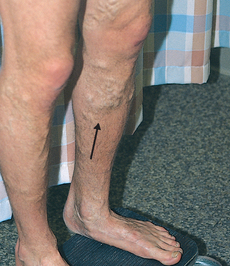
FIGURE 23-29 Varicose veins, indicated by the arrow and above the knee.
Varicosities are best observed when the patient is standing because standing increases the pressure and causes the tortuous veins to become more visible.
Source: Black JM et al. Medical-surgical nursing: clinical management for positive outcomes. 8th edn. Philadelphia: Saunders; 2008.
Venous distension can develop over time in individuals who habitually stand for long periods, wear constricting garments or cross their legs at the knees, which diminishes the action of the skeletal muscle pump (which assists venous return). Risk factors also include age, being female, family history, obesity, pregnancy, phlebitis and previous leg injury.68 Eventually, the pressure in the vein damages venous valves, rendering them incompetent and unable to maintain normal venous pressure. Hydrostatic pressure increases, further distending the vein and making it tortuous; oedema then develops in the extremity.
Chronic venous insufficiency is inadequate venous return over a long period. Venous hypertension, circulatory stasis and tissue hypoxia lead to an inflammatory reaction in vessels and tissue, resulting in fibrosclerotic remodelling of the skin and then ulceration.69 Symptoms include chronic pooling of blood in the veins of the lower extremities and hyperpigmentation of the skin of the feet and ankles. Oedema in these areas may extend to the knees.
Circulation to the extremities can become so sluggish that the metabolic demands of the cells for oxygen, nutrients and waste removal are barely met. Any trauma or pressure can therefore lower the oxygen supply and cause cell death and necrosis (venous stasis ulcers) (see Figure 23-30). Patients with venous ulcers typically have a poor quality of life. Infection can occur because poor circulation impairs the delivery of the cells and biochemicals for the immune and inflammatory responses. This same sluggish circulation makes infection following reparative surgery a significant risk. Varicose veins and chronic venous insufficiency may be associated with DVT in up to 15% of affected individuals because of changes in collateral flow and shared risk factors; therefore, anyone with new-onset varicose veins should be evaluated for the possibility of underlying DVT.70
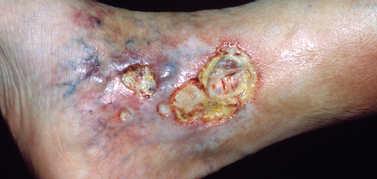
FIGURE 23-30 Venous ulcer on the medial aspect of the lower leg.
The venous ulcer has an irregular margin, pale surrounding neo-epithelium (new skin) and a pink base of granulation tissue. The skin is warm and oedema is often present.
Source: Talley NJ, O’Connor S. Clinical examination: a systematic guide to physical diagnosis. 6th edn. Sydney: Elsevier; 2010.
Treatment of varicose veins and chronic venous insufficiency begins conservatively and excellent wound-healing results have followed non-invasive treatments such as leg elevation, compression stockings and physical exercise.
Superior vena cava syndrome
Superior vena cava syndrome is a progressive occlusion of the superior vena cava that leads to venous distension in the upper extremities and head. Causes include bronchogenic cancer (75% of cases), lymphomas (15%) and metastasis of other cancers (7%). The superior vena cava is easily compressed as it is a relatively low-pressure vessel that lies in the closed thoracic compartment.
Clinical manifestations of superior vena cava syndrome are oedema and venous distension in the upper extremities and face, including the ocular beds. Other symptoms include a feeling of fullness in the head or tightness of shirt collars, necklaces and rings. Respiratory distress is a late symptom of compression of the bronchus by carcinoma.
Diagnosis is made by chest X-ray, Doppler studies, CT scans, MRI and ultrasound. Treatment of the carcinoma includes radiation therapy, surgery, chemotherapy and the administration of diuretics, steroids and anticoagulants, as necessary.
ALTERATIONS OF CARDIAC FUNCTION
Congenital heart disease
Congenital heart disease (present at birth) accounts for approximately one-third of all congenital defects and is the major cause of death in the first year of life other than prematurity. The incidence varies according to the particular defect; however, the overall rate is about 5 per 1000 live births.1,71 Several environmental and genetic risk factors are associated with the incidence of different types of congenital heart disease. Among the environmental factors are:
Table 23-9 MATERNAL CONDITIONS AND ENVIRONMENTAL EXPOSURES AND THE ASSOCIATED CONGENITAL HEART DEFECTS
| CAUSE | CONGENITAL HEART DEFECT |
|---|---|
| Infection | |
| Intrauterine | Patent ductus arteriosus, pulmonary stenosis, coarctation of aorta |
| Systemic viral | Patent ductus arteriosus, pulmonary stenosis, coarctation of aorta |
| Rubella | Patent ductus arteriosus, pulmonary stenosis, coarctation of aorta |
| Metabolic disorders | |
| Diabetes | Ventricular septal defect, cardiomegaly, transposition of the great vessels |
| Phenylketonuria (PKU) | Coarctation of aorta, patent ductus arteriosus |
| Drugs | |
| Alcohol | Tetralogy of Fallot, atrial septal defect, ventricular septal defect |
| Peripheral conditions | |
| Prematurity | Patent ductus arteriosus, ventricular septal defect |
Genetic factors also have been implicated in the incidence of congenital heart disease, although the mechanism of causation is often unknown. The incidence of congenital heart disease is three to four times higher in siblings of affected children and chromosomal defects account for about 6% of all cases of congenital heart disease. However, the cause of most defects is multifactorial.72,73
Congenital heart defects can be described with respect to three principal areas:
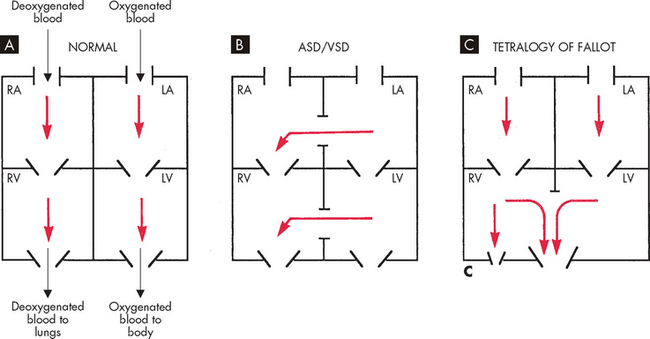
FIGURE 23-31 Shunting of blood in congenital heart diseases.
A Normal. B Acyanotic defect. C Cyanotic defect. ASD = atrial septal defect; VSD = ventricular septal defect; RA = right atrium; LA = left atrium; RV = right ventricle; LV = left ventricle.
Source: Hockenberry MJ, Wilson D. Wong’s essentials of pediatric nursing. 8th edn. St Louis: Mosby; 2009.
One way to categorise congenital heart defects is according to (a) whether they cause cyanosis, (b) whether they increase or decrease blood flow into the pulmonary circulation and (c) whether they obstruct blood flow from the ventricles. Figure 23-32 categorises congenital heart defects based on these three characteristics. In the following sections we examine the most common defects.
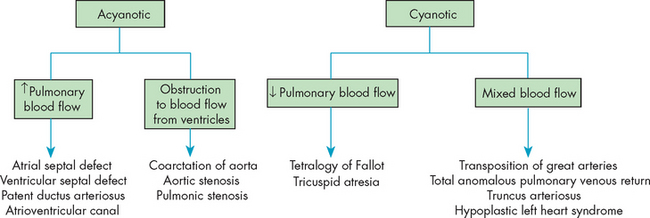
Defects with increased pulmonary blood flow
Ventricular septal defect
A ventricular septal defect (VSD) is an opening of the septal wall between the ventricles (see Figure 23-33). VSDs are the most common type of congenital heart defect and are classified by location, either high in the septal wall of the ventricle underneath the aortic valve or low in the septal wall. They can also be located in the inlet or outlet portion of the ventricle. VSDs shunt blood from left to right. Depending on the size and location, VSDs can spontaneously close, most often within the first 2 years of life.
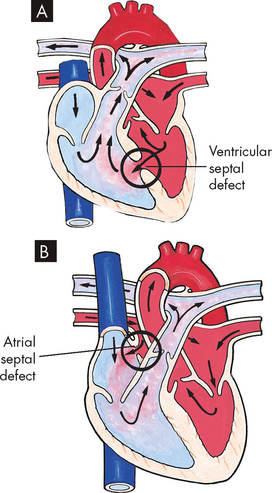
FIGURE 23-33 Ventricular septal defect (A) and atrial septal defect (B).
Note the colour of the oxygenated (red) and deoxygenated (blue) blood, and the mixing of blood in the right ventricle and pulmonary artery. This is an example of a left-to-right shunt.
Source: Hockenberry MJ, Wilson D. Wong’s essentials of pediatric nursing. 8th edn. St Louis: Mosby; 2009.
Depending on the size, location and degree of pulmonary vascular resistance, children may have no symptoms or they may have clinical effects from excessive pulmonary blood flow. Clinically, children with large left-to-right shunts present with poor growth (failure to thrive) and tachypnoea (rapid breathing). If the degree of shunting is significant and not corrected, the child is at risk for developing pulmonary hypertension. Children with VSD are also at increased risk of developing endocarditis.
Diagnosis is confirmed by echocardiography. Cardiac catheterisation may be needed to calculate the degree of left-to-right shunting. Depending on the size of the VSD and the degree of symptoms, management may be minimal. Small VSDs may close completely or become small enough that surgical closure is not required. If the infant has severe heart failure or failure to thrive that is unmanageable with medical therapy, early surgical repair is performed.
Atrial septal defect
An atrial septal defect (ASD) is an opening in the septal wall between the two atria (see Figure 23-33). This opening allows blood to shunt from the higher pressure left atrium to the lower pressure right atrium.
Children with an ASD are usually asymptomatic. Infants with a large ASD may, in rare cases, develop pulmonary overcirculation and slow growth. Some older children and adults will experience shortness of breath with activity as the right ventricle becomes less compliant with age. Pulmonary hypertension and stroke are associated rare complications. A systolic ejection murmur and a widely split second heart sound are the expected findings on physical examination.
Diagnosis is confirmed by echocardiography. The ASD may be closed surgically with primary repair (sutured closed) or with a patch. Surgical repair involves open-heart surgery with cardiopulmonary bypass. Interventional catheterisation closure involves placement of a closure device. Long-term follow-up finds atrial arrhythmias (10%) in both groups after closure.
Patent ductus arteriosus
Patent ductus arteriosus is failure of the fetal ductus arteriosus (the artery connecting the aorta and pulmonary artery) to close within the first weeks of life (see Figure 23-34). The continued patency of this vessel allows blood to flow from the higher pressure aorta to the lower pressure pulmonary artery, causing a left-to-right shunt.
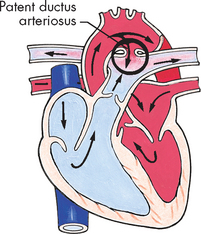
Infants may be asymptomatic or show signs of pulmonary overcirculation, such as dyspnoea, fatigue and poor feeding. There is a characteristic machinery-like murmur. Children are at risk for bacterial endocarditis and, rarely, may develop pulmonary hypertension in later life from chronic excessive pulmonary blood flow.
Diagnosis is confirmed by echocardiography. Administration of indomethacin (a prostaglandin inhibitor) has proved successful in closing a patent ductus arteriosus in premature infants and some newborns. Surgical division of the patent ductus arteriosus needs to be performed when pharmacological therapies are unsuccessful. Closure with an occlusion device during cardiac catheterisation is performed for mostly older children. Both surgical and nonsurgical procedures can be considered low risk.
Atrioventricular canal defect
Atrioventricular canal (AVC) defect, also known as atrioventricular septal defect (AVSD) or by the traditional term of endocardial cushion defect, is the result of incomplete fusion of the endocardial cushions (see Figure 23-35). AVC defect consists of an ASD and VSD with associated abnormalities of the atrioventricular valve tissue. These valve abnormalities range from a cleft in the mitral valve to a common mitral and tricuspid valve. The directions and pathways of flow are determined by pulmonary and systemic resistance, left and right ventricular pressures, and the compliance of each chamber. Flow is generally from left to right.
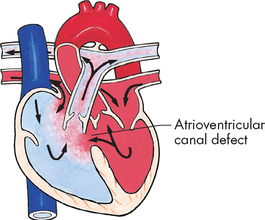
Infants with this defect often display moderate to severe heart failure due to left-to-right shunting. Infants with pulmonary hypertension and high pulmonary resistance have less shunting and therefore minimal signs of heart failure. There may be mild cyanosis that increases with crying. Those with a large left-to-right shunt will have a murmur and those with minimal shunt may not have a murmur.
Atrioventricular canal defect is one of the most frequent diagnoses made with fetal echocardiography. Cardiac catheterisation is usually not needed. Infants are followed closely for signs or symptoms of failure to thrive. Complete surgical repair is performed between 3 and 6 months of age to prevent irreversible pulmonary hypertension.
Defects with decreased pulmonary blood flow
Tetralogy of Fallot
The classic form of tetralogy of Fallot includes four defects: (1) VSD, (2) pulmonary stenosis, (3) overriding aorta and (4) right ventricular hypertrophy (see Figure 23-36). The pathophysiology varies widely, depending not only on the degree of pulmonary stenosis but also on the pulmonary and systemic vascular resistance to flow. If total resistance to pulmonary flow is higher than systemic resistance, the shunt is from right to left. If systemic resistance is higher than pulmonary resistance, the shunt is from left to right. Pulmonary stenosis decreases blood flow to the lungs and, consequently, the amount of oxygenated blood that returns to the left heart. Physiological compensation to chronic hypoxia includes production of more red blood cells, development of collateral bronchial vessels and enlargement of the nail beds (clubbing).
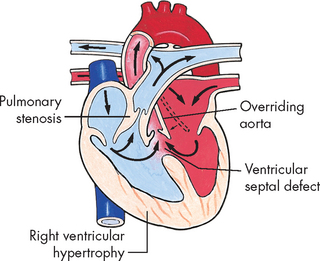
Some infants may be acutely cyanotic at birth. In others, progression of hypoxia and cyanosis may be more gradual over the first year of life as the pulmonary stenosis worsens. Chronic cyanosis may cause clubbing of the fingers, poor growth and squatting. Without being instructed to do so, these children squat in compensation — the squatting position traps blood in the legs and allows for greater oxygenation of blood in the central organs. Children with unrepaired tetralogy of Fallot are at risk for emboli, cerebrovascular disease, brain abscess, seizures and loss of consciousness or sudden death.
Diagnosis is confirmed with echocardiography. Elective surgical repair is usually performed in the first year of life. Indications for earlier repair include increasing cyanosis or the development of hypercyanotic spells. Complete repair involves closure of the VSD, resection of the stenosis and enlargement of the right ventricular outflow tract.
Obstructive defects
Coarctation of the aorta
Coarctation of the aorta is a localised narrowing of the aorta near the insertion of the ductus arteriosus, resulting in increased blood pressure proximal to the defect (head and upper extremities) and decreased blood pressure distal to the obstruction (torso and lower extremities; see Figure 23-37). Prior to birth, the ductus arteriosus allows blood to flow from the pulmonary artery into the distal aorta. However, once the ductus closes after birth, blood flow to the lower extremities is limited by the coarctation of the aorta.
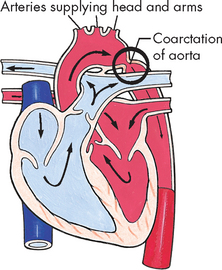
The location and severity of the coarctation of the aorta determines whether an infant will become symptomatic after the ductus arteriosus closes. If the coarctation of the aorta is severe, infants will present with low cardiac output, acidosis and hypotension. Physical examination of the infant will reveal weak or absent femoral pulses with poor perfusion. Some infants with coarctation of the aorta will remain asymptomatic after the closure of the ductus arteriosus. As they grow, older children with undiagnosed coarctation of the aorta will present with unexplained hypertension.
Physical examination and measurement of upper and lower extremity blood pressure will often suggest the diagnosis. Echocardiography, MRI and cardiac catheterisation may be needed to confirm the diagnosis. Initial treatment in the symptomatic newborn consists of continuous intravenous infusion of prostaglandin E1 to reopen and maintain the ductus arteriosus. Once the symptomatic newborn is stabilised, surgical correction is indicated.74,75
Aortic stenosis
Aortic stenosis is a narrowing or stricture of the aortic outlet, causing resistance of blood flow from the left ventricle into the aorta. If severe, there may be decreased cardiac output and pulmonary vascular congestion (see Figure 23-38). The physiological consequence of severe aortic stenosis is hypertrophy of the left ventricular wall, which eventually leads to increased end-diastolic pressure, resulting in pulmonary venous and pulmonary arterial hypertension. Left ventricular hypertrophy also impedes coronary artery perfusion and may result in subendocardial ischaemia and associated papillary muscle dysfunction causing mitral insufficiency.
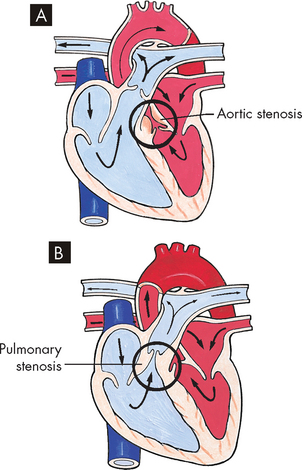
FIGURE 23-38 Aortic (A) and pulmonary (B) stenosis.
Note the irregularity of the respective valves compared to the atrioventricular valves.
Source: Hockenberry MJ, Wilson D. Wong’s essentials of pediatric nursing. 8th edn. St Louis: Mosby; 2009.
Valvular aortic stenosis occurs as a consequence of malformed or fused cusps resulting in a unicuspid or bicuspid valve. Valvular aortic stenosis is a serious defect because: (1) the obstruction tends to be progressive; (2) sudden episodes of myocardial ischaemia, or low cardiac output, on rare occasions can result in sudden death in late childhood or adolescence; and (3) surgical repair will not result in a normal valve. This is one of the rare instances in which strenuous physical activity may be curtailed because of the cardiac condition.74
Subvalvular aortic stenosis is a stricture caused by a fibrous ring below a normal valve. It can also be caused by a narrowed left ventricular outflow tract in combination with a small aortic valve annulus. Supravalvular stenosis, a narrowing of the aorta just above the valve, occurs infrequently.
Infants with significant aortic stenosis demonstrate signs of decreased cardiac output with faint pulses, hypotension, tachycardia and poor feeding. Children may also have complaints of exercise intolerance and chest pain. Children are at risk for bacterial endocarditis, coronary insufficiency, ventricular dysfunction and, rarely, sudden death.
Valvular aortic stenosis is diagnosed by echocardiography. Mild to moderate valvular aortic stenosis does not usually require intervention or restriction of activity. Treatment of severe valvular aortic stenosis varies. Nonsurgical therapy such as balloon angioplasty has a high morbidity and mortality rate. Surgical correction for subvalvular aortic stenosis depends on the extent of alteration, but may require multiple approaches.
Pulmonary stenosis
Pulmonary stenosis is a narrowing or stricture of the pulmonary valve causing resistance to blood flow from the right ventricle to the pulmonary artery (see Figure 23-38). Generally moderate to severe stenosis causes right ventricular hypertrophy.
Mixing defects
Transposition of the great arteries
In transposition of the great arteries (TGA) the pulmonary artery leaves the left ventricle and the aorta exits from the right ventricle (see Figure 23-39). Associated defects, such as ASD, VSD or patent ductus arteriosus, permit mixing of saturated and desaturated blood, which maintains adequate tissue oxygenation.
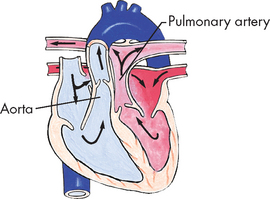
Clinical manifestations depend on the type and size of the associated defects. Children with limited communication between cardiac chambers are severely cyanotic, acidotic and ill at birth. Those with large septal defects or a patent ductus arteriosus may be less severely cyanotic but may have symptoms of pulmonary overcirculation.
Diagnosis is suspected by physical examination and confirmed with echocardiography. Administration of intravenous prostaglandin E1 may be initiated to temporarily increase oxygen delivery. Surgical repair for transposition of the great arteries is complex and needs to be performed early (the first 2 weeks of life); it is directed to correcting the vascular anatomy.
Truncus arteriosus
Truncus arteriosus is failure of normal septation and division of the embryonic outflow track into a pulmonary artery and an aorta, resulting in a single vessel that exits the heart. There is always an associated VSD with mixing of the systemic and arterial circulations (see Figure 23-40) causing cyanosis. Blood ejected from the heart flows preferentially to the lower pressure pulmonary arteries, causing increased pulmonary blood flow.
Acquired cardiovascular disorders
Acquired heart diseases refer to disease processes or abnormalities that occur after birth. They result from various causes, such as infection, genetic disorders, autoimmune processes in response to infection, environmental factors or autoimmune diseases. Examples of acquired heart diseases include myocarditis, viral endocarditis, rheumatic heart disease, cardiomyopathy and Kawasaki disease. Endocarditis, myocarditis, rheumatic heart disease and cardiomyopathy are discussed elsewhere within the chapter, but Kawasaki disease pathophysiology is briefly discussed here.
Kawasaki disease
Kawasaki disease (formerly known as mucocutaneous lymph node syndrome) is an acute, usually self-limiting systemic vasculitis that may result in cardiac sequelae. It is the most common acquired heart disease in children in developed countries and was first identified in Japan in 1967.72 This reflects the genetic component of Kawasaki disease: the case rate is highest among Asians, with most cases seen in toddlers and children up to 5 years of age. Current aetiological theories centre on an immunological response to an infectious, toxic or antigenic substance.76
Kawasaki disease progresses through three stages: acute, subacute and convalescent. In the acute stage, small blood vessels (capillaries, arterioles and venules) become inflamed, as does the heart itself. During the subacute stage, inflammation spreads to larger vessels and aneurysms of the coronary arteries develop. In the convalescent stage, medium-sized arteries begin the granulation process, causing coronary artery thickening with increased risk for thrombosis. After the convalescent stage, inflammation wanes, leaving scarring of the affected vessels, calcification and stenosis.
In the acute stage, the child has fever, conjunctivitis, oral changes (‘strawberry’ tongue), rash and lymphadenopathy, and is often irritable. Myocarditis may also develop. Progression to the subacute stage occurs when the fever ends and continues until the clinical signs have resolved. It is at this time that the child is most at risk for coronary artery aneurysm development. The convalescent stage is marked by the continued elevation of the erythrocyte sedimentation rate and platelet count. This stage continues until all laboratory values return to normal — usually about 6–8 weeks after onset.
Diagnosis is based on criteria and an echocardiogram as a baseline to assess for coronary aneurysms or inflammation. Treatment includes oral administration of aspirin and intravenous infusion of gamma globulin. Aspirin is continued until the manifestations of inflammation are resolved. Most children recover completely from Kawasaki disease, including regression of aneurysms. The most common cardiovascular sequela is coronary thrombosis.76