11 ALTERATIONS OF ENDOCRINE FUNCTION ACROSS THE LIFE SPAN
INTRODUCTION
The function of the endocrine system involves complex interrelationships and interactions that maintain homeostasis and provide growth and reproductive capabilities. Dysfunction is usually described in terms of excessive or insufficient function of the endocrine gland with alterations in hormone levels. These alterations are caused by either hypersecretion or hyposecretion of the various hormones, leading to abnormal hormone concentrations in the blood.
Most hormones have syndromes or disorders resulting from either too much or too little hormone. However, some of these are quite rare — for example, increased secretions of growth hormone affect a total of 1000 Australians1 and decreased growth hormone affects only 220 Australians.2 In this chapter, we focus on those disorders that have greatest relevance to contemporary Australia and New Zealand. We start by exploring hormone abnormalities that influence fluid and sodium balance, which is of prime focus within healthcare facilities. Then we briefly look at Cushing’s syndrome, which people taking cortisol medications are at risk of developing. Next we introduce diabetes mellitus, and finally we look at disorders relating to the thyroid gland, which are reasonably common.
MECHANISMS OF HORMONAL ALTERATIONS
Significantly elevated or significantly depressed hormone levels may result from various causes (see Figure 11-1). Feedback systems that recognise the need for a particular hormone may fail to function properly or may respond to inappropriate signals. Dysfunction of an endocrine gland may involve its failure to produce adequate amounts of hormone, or a gland may synthesise and release too much hormone. Once hormones are released into the circulation, they may be degraded at an altered rate or inactivated by antibodies before reaching the target cell. Hormones produced by non-endocrine tissues may cause abnormally elevated hormone levels — an example of this is a type of lung tumour that can secrete antidiuretic hormone. This mechanism operates without the benefit of the normal feedback system for hormone control and is an example of ectopic hormone production whereby it is produced from a part of the body where it is not normally made. An additional mechanism of endocrine alteration may result from abnormal receptor function or from altered intracellular response to the hormone at the target cell.
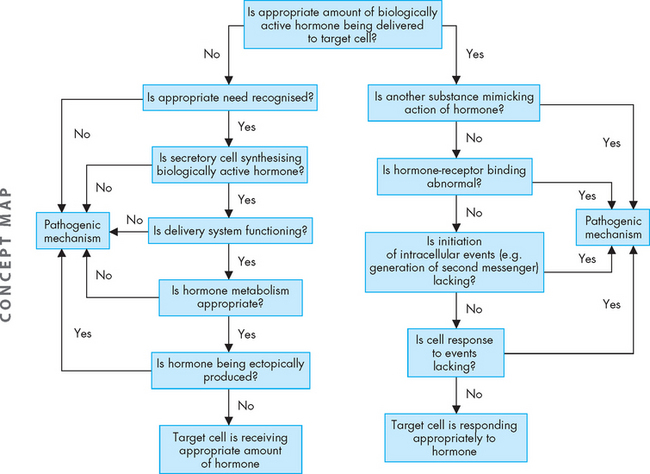
ALTERATIONS OF PITUITARY FUNCTION
Syndrome of inappropriate antidiuretic hormone secretion
Diseases of the posterior pituitary are rare and are usually related to abnormal antidiuretic hormone secretion. Syndrome of inappropriate antidiuretic hormone secretion (SIADH) is characterised by high levels of antidiuretic hormone without the normal physiological stimuli for its release.3 The most common cause is ectopic production, which is associated with cancer, wherein tumour cells secrete antidiuretic hormone. Tumours associated with SIADH include small cell carcinoma of the lung (the most common cause), and other important causes of SIADH include following surgery, brain injury or infection such as trauma, stroke, haemorrhage or meningitis.4 SIADH may also be secreted by a mechanism related to ageing5 or due to antidepressant6 or hypoglycaemic drug therapy.
Postoperative patients can have fluid volume shifts that result in increased antidiuretic hormone secretion for as long as 5–7 days after surgery. SIADH is seen also in individuals with infectious pulmonary diseases, where antidiuretic hormone is produced by infected lung tissue or posterior pituitary secretion is increased in response to a hypoxia-induced decrease in pulmonary perfusion.
PATHOPHYSIOLOGY
The cardinal features of SIADH are symptoms of water intoxication resulting from enhanced renal water retention or increases in total body water, which leads to hyponatraemia (low serum sodium), hypo-osmolality and urine that is inappropriately concentrated with respect to serum osmolality.6 Hyponatraemia is induced due to a relative increase in water without similar increases in sodium concentration. Therefore, hyponatraemia is common in hospitalised patients7 and it is usually caused by this syndrome.7,8 In a subacute geriatric facility in Melbourne, 25% of patients aged over 65 had hyponatraemia, with half of these cases due to SIADH.5 In this syndrome, antidiuretic hormone is released continually. Water retention results from the normal action of antidiuretic hormone on the renal tubules and collecting ducts, increasing their permeability to water and increasing water reabsorption by the kidneys (see Chapter 28).
Due to the retention of water, the extracellular fluid volume expands and a dilutional hyponatraemia develops — this means that sodium is diluted in more fluid. This suppresses renin and therefore aldosterone secretion and decreases reabsorption of sodium by the kidneys. This explains renal sodium loss during hyponatraemia.
In Chapter 6, we examined the role of normal concentrations of sodium and potassium in the normal signalling of neurons. Hyponatraemia can lead to alterations of neural signals, which can have widespread effects throughout the body. It is important to remember that blood concentrations of various substances such as sodium can have powerful effects on body systems.
CLINICAL MANIFESTATIONS
A diagnosis of SIADH requires the following signs: (1) low total concentration of solute in the serum (low osmolality) and hyponatraemia; (2) urine hyperosmolality (that is, urine osmolality is greater than expected for the serum osmolality); (3) urine sodium excretion that matches sodium intake; (4) normal adrenal and thyroid function; and (5) absence of conditions that can alter fluid volume status (e.g. congestive heart failure, hypovolaemia from any cause, or renal insufficiency).
The symptoms of this syndrome result from hyponatraemia and are determined by its severity and sudden onset. Thirst, impaired taste, anorexia, dyspnoea on exertion, fatigue and dulled sensorium (ability to interpret sensory inputs) occur when the serum sodium decreases rapidly from 140 mmol/L to 130 mmol/L. Severe gastrointestinal symptoms, including vomiting and abdominal cramps, occur with a drop in sodium from 130 mmol/L to 120 mmol/L. Peripheral oedema is absent. Symptoms usually resolve with correction of hyponatraemia. Even if hyponatraemia develops slowly, serum sodium levels below 115 mmol/L cause confusion, lethargy, muscle twitching and convulsions, and severe and sometimes irreversible neurological damage may occur. Hyponatraemia in hospitalised patients can contribute to morbidity and mortality.9 Since hospitalised elderly patients are at particular risk,7 the incidence may increase as a result of our ageing population.
EVALUATION AND TREATMENT
Serum electrolyte levels, serum osmolality, urine volume, urine electrolyte levels and urine osmolality are adequate measures of the presence of SIADH. A chest X-ray is necessary to assist with diagnosis to rule out respiratory conditions. The treatment of this syndrome involves the correction of any underlying causal problems and careful fluid restriction to allow the sodium levels to increase slowly. Daily fluid intake may be restricted to 500–1000 mL.7 Resolution usually occurs within 3 days, with a 2–3 kg weight loss and correction of hyponatraemia and sodium loss. Demeclocycline, which causes the renal tubules to develop resistance to antidiuretic hormone, may be used to treat resistant or chronic SIADH. Although demeclocycline is not easily available in Australia, it can be obtained under the Special Access Scheme.
Diabetes insipidus
Diabetes insipidus is related to an insufficiency of antidiuretic hormone, leading to polyuria and polydipsia. It is actually quite rare, affecting approximately 6 in 100,000 people. Note that this is a different condition to diabetes mellitus (usually referred to simply as diabetes), which is becoming increasingly common in our community. (Refer to the later section on alterations of pancreatic function.) The term diabetes means ‘overflow’, or an increased urine volume. The two forms of diabetes insipidus are as follows (see Figure 11-2):
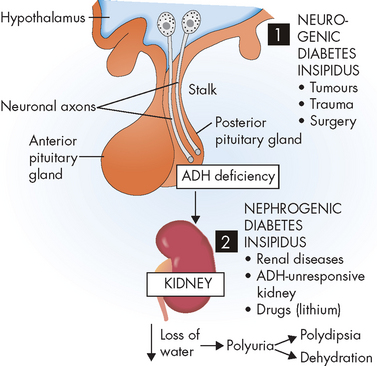
FIGURE 11-2 Diabetes insipidus.
The origin can be neurogenic (1) or nephrogenic (2).
Source: Based on Damjanov I. Pathophysiology. Philadelphia: Saunders; 2009.
There is also a psychogenic form, caused by chronic ingestion of extremely large quantities of fluid. It resolves with effective management.
PATHOPHYSIOLOGY
Diabetes insipidus usually has an acute onset. Individuals with diabetes insipidus have a partial or total inability to concentrate urine. Insufficient antidiuretic hormone secretion causes polyuria (excretion of large volumes of dilute urine), leading to increased plasma osmolality. In conscious individuals, the thirst mechanism is stimulated and induces polydipsia (increased thirst — usually a craving for cold drinks). The urine output varies and may be more than 12 L/day, with a low specific gravity. Dehydration develops rapidly without ongoing fluid replacement.
CLINICAL MANIFESTATIONS
The clinical manifestations of diabetes insipidus include polyuria, nocturia, continuous thirst, polydipsia, low urine specific gravity, low urine osmolality10 and high–normal plasma osmolality. Plasma osmolality is always higher than urine osmolality after 8 hours of water deprivation.
EVALUATION AND TREATMENT
The diagnosis of diabetes insipidus is generally established by correlating the clinical presentation with serum osmolality and plasma antidiuretic hormone levels. The diagnosis of psychogenic polydipsia can be extremely difficult, and differentiation from nephrogenic diabetes insipidus is based on plasma antidiuretic hormone levels.
Treatment of neurogenic diabetes insipidus is based on the extent of the hormone deficiency and on age, endocrine and cardiovascular status, and lifestyle. Oral hydration often is adequate, but aggressive intravenous fluid resuscitation may be required to match urine output. Some individuals require antidiuretic hormone replacement, including administration of desmopressin (a synthetic form of antidiuretic hormone), which acts on the kidneys to increase water retention.
ALTERATIONS OF ADRENAL FUNCTION
Disorders of the adrenal cortex are most commonly related to hyperfunction. Hyperfunction that causes increased levels of aldosterone leads to hyperaldosteronism, and that which causes hypercortisolism leads to Cushing’s syndrome.
Hyperaldosteronism
Hyperaldosteronism is characterised by excessive aldosterone secretion by the adrenal glands. In primary hyperaldosteronism, excessive secretion of aldosterone is caused by an abnormality of the adrenal cortex. In secondary hyperaldosteronism, excessive aldosterone secretion results from an extra-adrenal stimulus, most often a renin-angiotensin mechanism.
Primary hyperaldosteronism (also known as Conn’s syndrome) presents with hypertension, renal potassium wasting, hypokalaemia and neuromuscular manifestations.11 The most common cause of primary hyperaldosteronism is a benign adrenal adenoma. The incidence of primary hyperaldosteronism is estimated to be 2–10% of all hypertensive individuals.12 This condition therefore is of particular interest to those with hypertension, which has a high incidence in Australia and New Zealand (see Chapter 23).
Because aldosterone secretion normally is stimulated by the renin-angiotensin system (see Chapter 28), secondary hyperaldosteronism results from sustained elevated renin release and activation of angiotensin II. This occurs in various situations, including decreased circulating blood volume (e.g. in dehydration, shock or hypoalbuminaemia) and decreased delivery of blood to the kidneys (e.g. heart failure or hepatic cirrhosis). Here, the activation of the renin-angiotensin system and subsequent aldosterone secretion may be seen as compensatory, although in some instances (e.g. congestive heart failure) the increased circulating volume further worsens the condition.
PATHOPHYSIOLOGY
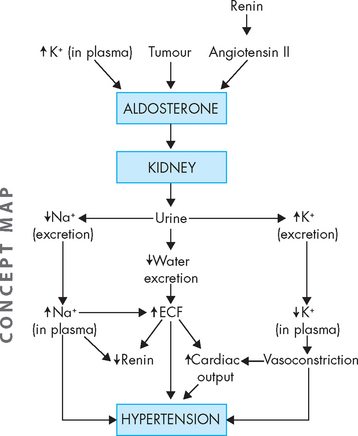
In primary hyperaldosteronism, pathophysiological alterations are caused by excessive aldosterone secretion and the fluid and electrolyte imbalances that ensue. Hyperaldosteronism promotes: (1) increased renal sodium and water reabsorption with corresponding hypervolaemia (low blood volume, see Chapter 29) and hypertension; and (2) renal excretion of potassium. The extracellular fluid volume overload, hypertension and suppression of normal feedback mechanisms of renin secretion are characteristic of primary disorders. Oedema usually does not occur with primary hyperaldosteronism.
In secondary hyperaldosteronism, the effect of increased extracellular volume on renin secretion may vary. If renin secretion is abnormal, increased circulating blood volume may not decrease renin secretion through feedback mechanisms.
Potassium secretion is promoted by aldosterone, so that with excessive aldosterone, hypokalaemia occurs (see Chapter 29). Hypokalaemic alkalosis, changes in myocardial conduction and skeletal muscle alterations may be seen, particularly with severe potassium depletion (refer to Chapter 6 for the role of potassium in neuron signalling and muscle contraction). The renal tubules may become insensitive to antidiuretic hormone, thus promoting excessive loss of water. In this situation, hypernatraemia may also occur because water is not able to follow the sodium that is reabsorbed.
CLINICAL MANIFESTATIONS
Hypertension and hypokalaemia are the hallmarks of primary hyperaldosteronism (see Figure 11-3). With sustained hypertension, the chronic effects of elevated arterial pressure become evident; for example, left ventricular dilation and hypertrophy and progressive atherosclerosis. Aldosterone-stimulated potassium loss can be substantial, resulting in typical manifestations of hypokalaemia. Hypokalaemic alkalosis may develop (see Chapter 29).
EVALUATION AND TREATMENT
Various clinical and laboratory measurements are useful in assessing hyperaldosteronism. Tests include the following:
Imaging techniques, including CT scans and nuclear MRI, may be used to localise an aldosterone-secreting adenoma.
Treatment includes management of hypertension and hypokalaemia, as well as correction of any underlying causes. If an aldosterone-secreting adenoma is present, it must be surgically removed.13
Hypercortisolism
Hypercortisolism is caused by excessive anterior pituitary secretion of adrenocorticotropic hormone, which results in increased adrenal cortex secretion of cortisol. Cushing’s syndrome is a collection of hormonal disorders due to hypercortisolism from any condition. It is an uncommon disorder affecting only 1 in 50,000 people14 and occurs whenever there is an excessive level of cortisol, regardless of the cause. Cushing’s syndrome is more common in adults and is two to three times more common in women than in men. Hypercortisolism can occur at any age, but usually occurs between the ages of 30 and 50 years. In older adults, it usually results from ectopic adrenocorticotropic hormone secretion, whereas in children it is usually the result of adrenal tumours. Importantly, a Cushing-like syndrome may develop as a result of the pharmacological administration of glucocorticoids15 — this requires caution for patients who use these drugs. Cushing’s disease occurs when the specific cause of hypercortisolism is related to tumours of the pituitary or adrenal gland, and it accounts for approximately 70–85% of cases of Cushing’s syndrome.16
PATHOPHYSIOLOGY
Individuals with Cushing’s syndrome do not have diurnal or circadian secretion patterns of adrenocorticotropic hormone and cortisol, and do not increase secretion of these hormones in response to a stressor.17 When the secretion of cortisol by the tumour exceeds normal cortisol levels, symptoms of hypercortisolism develop.
CLINICAL MANIFESTATIONS
Weight gain is the most common feature and results from the accumulation of adipose tissue in the trunk, facial and cervical areas. These characteristic patterns of fat deposition have been described as ‘truncal obesity’, ‘moon face’ and ‘buffalo hump’ (see Figures 11-4 and 11-5). Transient weight gain from sodium and water retention also may be present.
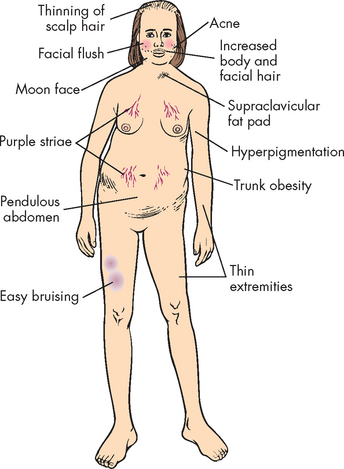
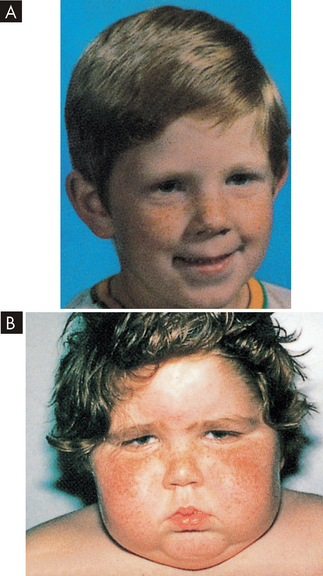
FIGURE 11-5 Cushing’s syndrome.
A Patient before onset of Cushing’s syndrome. B Patient four months later: ‘moon face’ is clearly demonstrated.
Source: Zitelli BJ, Davis HW. Atlas of pediatric physical diagnosis. 3rd edn. London: Gower; 1997.
Glucose intolerance occurs because of cortisol-induced insulin resistance and overt diabetes mellitus develops in approximately 20% of individuals with hypercortisolism (see Chapter 35). Polyuria is a manifestation of hyperglycaemia and resultant glycosuria.
Protein wasting is caused by the catabolic (breaking-down) effects of cortisol on peripheral tissues. Muscle wasting leads to muscle weakness. In bone, loss of the protein matrix leads to osteoporosis, with pathological fractures, vertebral compression fractures, bone and back pain, kyphosis and reduced height. Cortisol interferes with the action of growth hormone in long bones — children who present with short stature may be experiencing growth retardation related to Cushing’s syndrome rather than growth hormone deficiency. Bone disease may contribute to hypercalciuria and resulting renal stones, which are experienced by approximately 20% of individuals with disease. Loss of collagen also leads to thin, weakened integumentary tissues, through which capillaries are more visible and which are easily stretched by adipose deposits. Together, these changes account for the characteristic purple striae in the trunk area. Loss of collagenous support around small vessels makes them susceptible to rupture, leading to easy bruising, even with minor trauma. Thin, atrophied skin is also easily damaged, leading to skin breaks and ulcerations. Hyperpigmentation in Cushing’s syndrome can occur and involves the mucous membranes, hair and skin, all of which acquire a characteristic brownish or bronze colour.
With elevated cortisol levels, vascular sensitivity to catecholamines (adrenaline and noradrenaline) increases significantly, leading to vasoconstriction and hypertension. Mineralocorticoid (aldosterone) effects promote sodium retention and hypokalaemia. Elevated blood pressure occurs in most individuals, and suppression of the immune system and increased susceptibility to infections also occur.
EVALUATION AND TREATMENT
The diagnosis of Cushing’s syndrome is challenging and various laboratory tests must be used, including urinary cortisol and a dexamethasone (a cortisol drug) suppression test. Visualising procedures include pituitary MRI and abdominal scanning.18
Without treatment, approximately 50% of individuals with Cushing’s syndrome die within 5 years of onset as a result of overwhelming infection, complications from generalised arteriosclerosis and hypertensive disease. Treatment is specific for the cause of hypercortisolism and includes medication, radiation and surgery. Therefore, differentiation among pituitary, adrenal and ectopic causes of hypercortisolism is essential for effective treatment.
ALTERATIONS OF PANCREATIC FUNCTION
Diabetes mellitus is not a single disease but a group of disorders with glucose intolerance in common. The term diabetes mellitus describes a syndrome characterised by chronic hyperglycaemia and other disturbances of carbohydrate, protein and fat metabolism. While diabetes indicates an increased urine output, mellitus is Latin for honey — hence the urine is sweet (contains glucose) and copious. There are three main categories of diabetes mellitus:19
In this chapter, we consider type 1 and gestational diabetes. Type 2 diabetes is one of the main chronic health complications in Australia and New Zealand, with strong links to obesity, and has had a relatively recent surge in incidence — thus, it is discussed fully in Chapter 35 in the part on contemporary health issues. Importantly, general features that are common to type 2 and other forms of diabetes are reserved for Chapter 35.
Type 1 diabetes mellitus
Type 1 diabetes mellitus accounts for approximately 8% of diabetes mellitus in Australia, affecting some 35,500 people over the age of 25.20 Type 1 diabetes mellitus results from autoimmune destruction of beta cells in the islets of Langerhans and is thought to be the result of a gene–environment interaction, with the strongest genetic risk markers in the human leucocyte antigen (HLA) region of chromosome 6. Genetic factors may increase susceptibility to environmental causes of diabetes.21,22 There is a 50% concordance rate in twins. Between 10% and 13% of individuals with newly diagnosed type 1 diabetes have a first-degree relative (parent or sibling) with type 1 diabetes. Diagnosis peaks at 12 years of age.
Historically, type 1 diabetes mellitus has been thought to have an abrupt onset. More recently, however, prospective studies show a distinctive natural history involving genetic susceptibility; a long preclinical period; immunologically mediated destruction of beta cells, eventually leading to insulin deficiency; and hyperglycaemia.
PATHOPHYSIOLOGY
Type 1 diabetes results from a severe, absolute lack of insulin caused by loss of beta cells. Destruction of islet cells is related to genetic susceptibility, autoimmunity and environmental factors.21,22 It results from beta cell autoantibodies (as well as antibodies to insulin) that participate in damage to islet cells. Autoantibodies (antibodies produced against the body’s own tissues; see Chapter 15) are often detected long before symptoms appear. Environmental factors that may trigger autoimmune injury are summarised in Box 11-1. Non-immune type 1 diabetes can occur secondarily to other diseases such as pancreatitis.
BOX 11-1 SPECIFIC ENVIRONMENTAL FACTORS TO TYPE 1 DIABETES
Viruses
Mumps and Coxsackie — type 1 diabetes does occur rarely as a complication of viral infections, but no evidence of substantial relationship exists.
Rubella — 40% of individuals with congenital rubella infection later develop type 1 diabetes.
Cytomegalovirus (CMV) — persistent CMV infections appear to be relevant to pathogenesis of some cases of type 1 diabetes.
Source: Akerblom HK, Knip M. Putative environmental factors in type 1 diabetes. Diabetes Metab Rev 1998; 14(1): 31–67; Knip M, Akerblom HK. Environmental factors in the pathogenesis of type 1 diabetes mellitus. Exp Clin Endocrinal Diabetes 1999; 107(suppl 3): S93–S100.
Before hyperglycaemia occurs, 80–90% of the insulin-secreting beta cells of the islets of Langerhans must be destroyed. This is because the remaining cells can increase their production of insulin to compensate, but with so few beta cells remaining, insulin production is no longer adequate. Beta cell abnormalities are present long before the acute clinical onset of type 1 diabetes. In addition to the decline in insulin secretion, the production of amylin, another beta cell hormone, also falls; amylin is co-secreted with insulin. One of the critical actions of amylin is to suppress glucagon release from the alpha cells.
Regardless of cause, a disequilibrium of hormones produced by the islets of Langerhans occurs in diabetes mellitus. Both beta cell function and alpha cell function are abnormal, with a lack of insulin and a relative excess of glucagon (produced by alpha cells). Hyperglycaemia and ketonaemia can result from insulin deficiency alone, but a relative excess of glucagon clearly facilitates the metabolic alterations seen in diabetes — elevated blood glucose levels fail to suppress the production of glucagon.
CLINICAL MANIFESTATIONS
Type 1 diabetes mellitus affects the metabolism of fat, protein and carbohydrates. Hyperglycaemia allows glucose to appear in the urine as the renal threshold for glucose is exceeded (see Chapter 28). In addition, proteins and fats break down because of the lack of insulin, resulting in weight loss. Initial clinical manifestations of type 1 diabetes are generally acute, with polyuria, polydipsia and polyphagia (see Table 11-1). Weight loss and wide fluctuations in blood glucose levels occur.
Table 11-1 CLINICAL MANIFESTATIONS AND MECHANISMS FOR TYPE 1 DIABETES MELLITUS
| MANIFESTATION | RATIONALE |
|---|---|
| Polydipsia | Because of elevated blood sugar levels, water is osmotically attracted from body cells, resulting in intracellular dehydration and stimulation of thirst in the hypothalamus. |
| Polyuria | Hyperglycaemia acts as an osmotic diuretic; the amount of glucose filtered by the glomeruli of the kidneys exceeds that which can be reabsorbed by the renal tubules; glycosuria results, accompanied by large amounts of water lost in the urine. |
| Polyphagia | Depletion of cellular stores of carbohydrates, fats and proteins results in cellular starvation and a corresponding increase in hunger. |
| Weight loss | Weight loss occurs because of fluid loss in osmotic diaeresis and the loss of body tissue as fats and proteins are used for energy. |
| Fatigue | Metabolic changes result in poor use of food products, contributing to lethargy and fatigue. |
Ketoacidosis is caused by increased metabolism of fats and proteins resulting in high levels of circulating ketones. The pH drops, triggering the buffering systems associated with metabolic acidosis (see Chapter 29). Acetone (a volatile form of ketones) is then blown off, giving the breath a sweet or ‘fruity’ odour. Occasionally, diabetic coma is the initial symptom of the disease. Further details that are common with type 2 diabetes mellitus are detailed in Chapter 35.
EVALUATION AND TREATMENT
The diagnosis of diabetes is not difficult when the symptoms of polydipsia, polyuria, polyphagia, weight loss and hyperglycaemia are present in fasting and postprandial states.
Currently, treatment regimens are designed to avoid high and low levels of glucose and insulin.23 Management requires individual planning according to type of disease, age and activity level, but all individuals require some combination of insulin, meal planning and exercise. Glycated haemoglobin A1c (abbreviated to HbA1c) testing is useful in confirming the diagnosis and in monitoring the effectiveness of treatment and preventing complications (refer to Chapter 35). When assessing blood glucose control, the blood glucose levels that are determined by a glucometer (by the patient) or a blood test indicate the glucose at a particular point in time, while the HbA1c testing gives a longer term view of glucose control over recent weeks. Both types of information are used to determine the effectiveness of the blood glucose control.
Islet cell transplantation is being explored for treatment of type 1 diabetes and early results are promising.24 Pancreatic transplant is generally reserved for those individuals with type 1 diabetes and associated end-stage renal failure.25 The acute and chronic complications, evaluation and treatment of type 1 diabetes mellitus are similar with those seen in type 2 (refer to Chapter 35 for full discussion).
Gestational diabetes mellitus
Gestational diabetes mellitus develops when glucose intolerance appears during pregnancy, and pregnant women at risk should be screened. Risk factors include a family history of diabetes, membership in a high-risk ethnic group, advanced maternal age (> 25 years of age), a history of delivering large babies, a prior history of gestational diabetes or polycystic ovary syndrome, being overweight before pregnancy (a body mass index, or BMI, > 25 kg/m2) and a history of obstetric complications associated with gestational diabetes. In Australia, 4–5% of pregnant women are affected with gestational diabetes.20 Approximately one-third of these women are over the age of 35, although only 20% of total births are in this age group — this indicates a higher proportion of women with gestational diabetes in this age group compared with younger mothers.
While the screening and diagnosis of gestational diabetes varies substantially between countries, in Australia and New Zealand it is recommended that all pregnant women be screened, not just those at risk (using the oral glucose tolerance test if necessary).26 Aggressive treatment is required to prevent morbidity and fetal mortality.27 Treatment with metformin may be more successful in fetal outcomes and controlling maternal weight compared with insulin treatment;28 however, at this stage current guidelines from a range of countries continue recommending insulin.26 Gestational diabetes can progress to type 2 diabetes, particularly within the first 5 years, with the fasting glucose levels during pregnancy being the main risk factor associated with this future risk.29
ALTERATIONS OF THYROID FUNCTION
Hyperthyroidism
Hyperthyroidism is one of the more common endocrine disorders, as approximately two out of every 100 women will experience some hyperthyroidism.30 Although males can have hyperthyroidism, it is more common in females.
Graves’ disease
Graves’ disease is the most common cause of hyperthyroidism and is an autoimmune condition. Normally, thyroid-stimulating hormone binds to receptors on the thyroid gland, resulting in the production of thyroid hormone. In Graves’ disease, the thyroid gland is stimulated by antibodies against the thyroid-stimulating hormone receptor. The antibodies stimulate the thyroid cells to produce high concentrations of triiodothyronine (T3) and thyroxine (T4) (see Figure 11-6). The combined action of the antibodies and increased serum levels of thyroid hormone produce the symptoms of Graves’ disease: diffuse thyroid enlargement (goitre), ophthalmopathy (effects on the eyes), dermopathy (pretibial myxoedema) and other effects on the extremities.
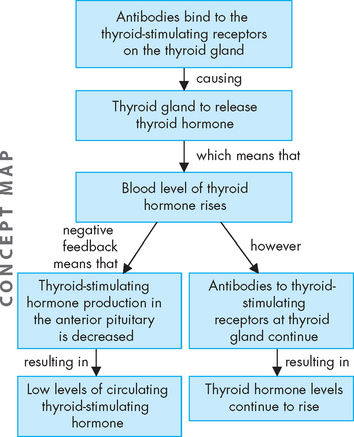
The many signs and symptoms of Graves’ disease can be divided into three components: (1) adrenergic stimulation (tachycardia, palpitations, nervousness, depression, tremor, lid lag, increased systolic blood pressure, increased cardiac contractility); (2) excess thyroid hormone (increased oxygen consumption, metabolic changes in protein metabolism); and (3) immunological stimulation of diffuse goitre (enlarged thyroid gland).31
Two categories of ocular manifestations are associated with Graves’ disease (see Figure 11-7):
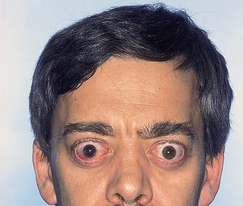
Note the large and protruding eyeballs.
Source: Belchetz P, Hammond P. Mosby’s color atlas and text of diabetes and endocrinology. Edinburgh: Mosby; 2003.
Elevated thyroid hormone levels and suppressed serum thyroid-stimulating hormone levels are diagnostic for hyperthyroidism. Unfortunately, current treatment for Graves’ disease does not reverse the ocular changes and is palliative.31
Hyperthyroidism resulting from nodular thyroid disease
The thyroid gland normally enlarges in response to the increased demand for thyroid hormone that occurs in puberty, pregnancy, iodine deficiency and immunological, viral or genetic disorders. When the condition requiring increased thyroid hormone resolves, thyroid-stimulating hormone secretion normally subsides and the thyroid gland returns to its original size.
Irreversible changes may occur in some follicular cells, however, so that they then function autonomously. Hyperthyroidism may or may not result from these irreversible changes. Autonomously functioning cells may produce less thyroid hormone than the body requires. The remainder of the gland then functions to supply the remainder of the body’s need and a euthyroid state (normal thyroid hormone level) is achieved and maintained. If the autonomously functioning cells produce sufficient or excessive thyroid hormone for the usual body requirements, the remainder of the gland undergoes involution, becoming normal but inactive tissue. This condition may result in euthyroidism or hyperthyroidism, depending on the amount of thyroid hormone produced.
Hyperthyroidism can lead to a hypermetabolic state known as thyrotoxicosis or toxic multinodular goitre. If only one nodule is hyperfunctioning, it is termed toxic adenoma. Symptoms usually develop slowly and consist of rapid heart action; tremors; elevated basal metabolic rate; enlarged, multinodular goitre or a single, large nodule; and weight loss. Lid lag and proptosis (forward projection of the eyeball) may be seen, but exophthalmos and pretibial myxoedema do not occur. All forms of thyrotoxicosis share some common characteristics.32
CLINICAL MANIFESTATIONS
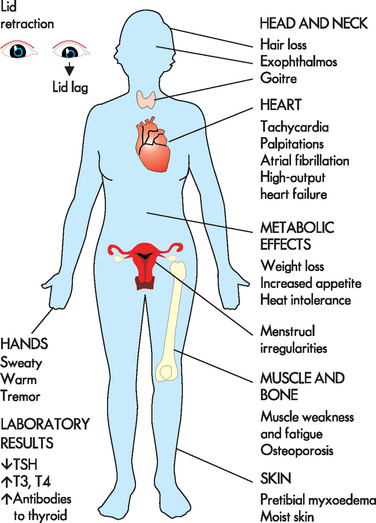
The metabolic effects of increased circulating levels of thyroid hormones cause clinical symptoms. The metabolic rate increases with heat intolerance and increased tissue sensitivity to sympathetic stimulation. The major manifestations include intolerance to heat, fatigue, insomnia, increased appetite, weight loss, increased cardiac output, tachycardia and eyelid retraction (see Figure 11-8). Minimal or atypical symptoms are common in the elderly population.33 Goitre (enlarged thyroid) is usually present.
EVALUATION AND TREATMENT
Elevated serum thyroxine (T4) and triiodothyronine (T3) are characteristic of hyperthyroid conditions. By contrast, thyroid-stimulating hormone–secreting pituitary tumours are characterised by normal to increased thyroid-stimulating hormone levels in the face of elevated thyroid hormone concentrations. Radioactive iodine is used to test for increased uptake in hyperthyroidism.34 Treatment is directed at controlling excessive thyroid hormone production, secretion or action and employs drug therapy, radioactive iodine therapy and surgery. Although complete surgical removal of the thyroid gland is preferable to partial removal to treat hyperthyroidism,35 it also causes removal of the parathyroid gland (see the later section on alterations of parathyroid function).
Thyrotoxic crisis
Thyrotoxic crisis (thyroid storm) is a rare but dangerous worsening of the thyrotoxic state, in which death occurs within 48 hours without treatment. The condition may develop spontaneously, but it usually occurs in individuals who have undiagnosed or partially treated Graves’ disease and are subjected to excessive stress, such as infection, pulmonary or cardiovascular disorders, emotional distress, physical stress, dialysis, plasmapheresis or inadequate preparation for thyroid surgery.
The systemic symptoms of thyrotoxic crisis include hyperthermia, tachycardia, high-output heart failure, agitation or delirium, and nausea, vomiting or diarrhoea contributing to fluid volume depletion. The symptoms may be attributed to increased sympathetic nervous system stimulation. The treatment is designed to: (1) reduce circulating thyroid hormone levels by inducing a block of thyroid hormone synthesis (using propylthiouracil) and thereby reducing their effects to eliminate the precipitating disorder; and (2) provide symptomatic and supportive care.
Hypothyroidism
Deficient production of thyroid hormone by the thyroid gland results in hypothyroidism. Primary causes include: (1) congenital defects; (2) defective hormone production resulting from autoimmune thyroiditis, iodine deficiency or anti-thyroid drugs; or (3) iatrogenic (inadvertent) loss of thyroid tissue after surgical or radioactive treatment for hyperthyroidism. Causes of secondary hypothyroidism are less common and are related to either pituitary or hypothalamic failure. Hypothyroidism is the most common disorder of thyroid function, occurs more commonly in women than men and affects 6–10% of women.36
Primary hypothyroidism
Primary hypothyroidism results from several disorders: acute thyroiditis, subacute thyroiditis, autoimmune thyroiditis, painless thyroiditis and postpartum thyroiditis. Acute thyroiditis is caused by a bacterial infection of the thyroid gland and is rare. Subacute thyroiditis is a nonbacterial inflammation of the thyroid often preceded by a viral infection. Both conditions are accompanied by fever, tenderness and enlargement of the thyroid gland. Symptoms may last for 2–4 months and corticosteroids usually resolve symptoms. Autoimmune thyroiditis (Hashimoto’s disease) results in destruction of thyroid tissue by circulating thyroid antibodies and infiltration of lymphocytes. Autoimmune thyroiditis also may be caused by an inherited immune defect. Goitre formation is common. Painless thyroiditis has a course similar to subacute thyroiditis but is pathologically identical to Hashimoto’s disease. Postpartum thyroiditis generally occurs up to 6 months after delivery with a course similar to Hashimoto’s disease. Spontaneous recovery occurs in 95% of these hypothyroid conditions.
PATHOPHYSIOLOGY
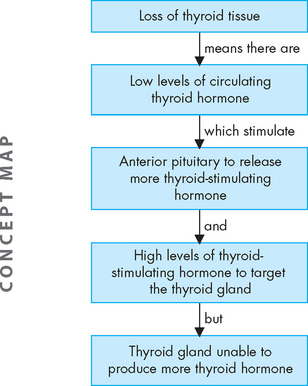
In primary hypothyroidism, loss of thyroid tissue leads to decreased production of thyroid hormone, increased secretion of thyroid-stimulating hormone and goitre (see Figure 11-9). Secondary hypothyroidism is usually caused by the pituitary’s failure to synthesise adequate amounts of thyroid-stimulating hormone. Pituitary tumours, or the results of their treatment, are the most common causes of secondary hypothyroidism.
CLINICAL MANIFESTATIONS
Hypothyroidism generally affects all body systems and occurs insidiously over months or years. The decrease in thyroid hormone lowers energy metabolism and heat production. The individual develops a low basal metabolic rate, cold intolerance, lethargy, tiredness and slightly lowered basal body temperature. The decrease in thyroid hormone can lead to excessive thyroid-stimulating hormone production and goitre (see Figure 11-10).
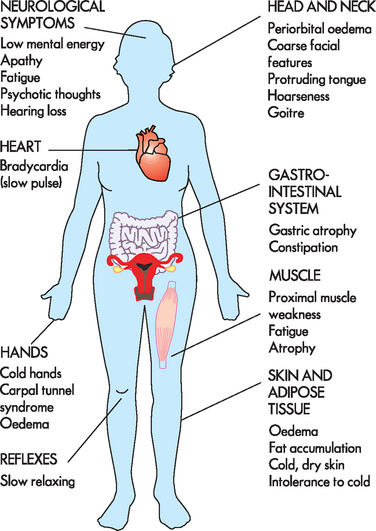
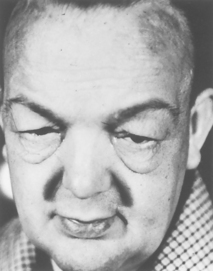
The characteristic sign of severe or long-standing hypothyroidism is myxoedema, which results from the altered composition of the dermis and other tissues. The connective fibres are separated by large amounts of protein and other substances that bind water, producing non-pitting, boggy oedema, especially around the eyes (see Figure 11-11), hands and feet and in the supraclavicular fossae. The tongue and laryngeal and pharyngeal mucous membranes thicken, producing thick, slurred speech and hoarseness.
EVALUATION AND TREATMENT
In addition to the clinical symptoms of hypothyroidism, a decrease in serum T4 is nearly always present. Thyroid-stimulating hormone concentration increases from loss of negative feedback from thyroid hormone.37 When hypothyroidism is caused by pituitary deficiencies, serum thyroid-stimulating hormone levels and basal metabolic rate decrease. Hormone replacement therapy is the treatment of choice. The restoration of normal thyroid hormone levels should be timed appropriately; a regimen of hormonal therapy depends on the individual’s age, the duration and severity of the hypothyroidism, and the presence of other disorders, particularly cardiovascular disorders.
ALTERATIONS OF PARATHYROID FUNCTION
Hyperparathyroidism
Hyperparathyroidism is characterised by greater than normal secretions of parathyroid hormone. Hyperparathyroidism is classified as primary or secondary.
PATHOPHYSIOLOGY
Estimates suggest that primary hyperparathyroidism occurs in 0.2–0.3% of the adult population, with twice as many cases in women. It is generally found in older adults.38 Because postmenopausal women are at risk for developing osteoporosis, the effects of increased levels of parathyroid hormone (which breaks down bone to release calcium to the blood) on bone disease can be significant. In primary hyperparathyroidism, parathyroid hormone secretion is increased and is not under the usual feedback control mechanisms. Most cases of hyperparathyroidism (approximately 80%) result from chief cell adenoma with an increased secretion of parathyroid hormone.39
Secondary hyperparathyroidism is a compensatory response of the parathyroid glands to chronic hypocalcaemia, which can be associated with decreased renal activation of vitamin D (renal failure) or malabsorption. Secretion of parathyroid hormone is elevated, but normal calcium levels cannot be achieved because of insufficient levels of activated vitamin D.
Hyperplasia (increase in the number of cells) of the parathyroid glands and loss of sensitivity to circulating calcium levels can cause autonomous secretion of parathyroid hormone, even with normal calcium levels. It often occurs in individuals with chronic renal failure. Signs and symptoms are similar to those of primary hyperparathyroidism.
CLINICAL MANIFESTATIONS
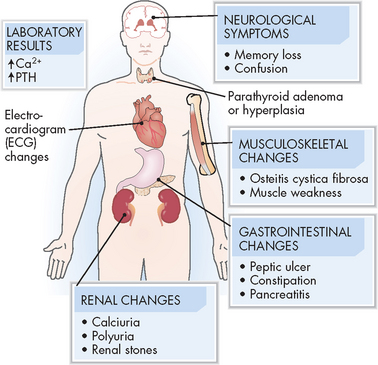
Parathyroid hormone hypersecretion causes hypercalcaemia and may be asymptomatic or present with excessive osteoclastic and osteocytic activity, resulting in bone resorption.40 (Bone resorption is discussed in Chapter 20.) Pathological changes include fractures, kyphosis of the dorsal spine and compression fractures of the vertebral bodies. The increased renal filtration load of calcium leads to hypercalciuria.
Hypercalcaemia also affects proximal renal tubular function, causing metabolic acidosis and production of an abnormally alkaline urine. Parathyroid hormone hypersecretion enhances renal phosphate excretion and results in hypophosphataemia and hyperphosphaturia. The combination of hypercalciuria, alkaline urine and hyperphosphaturia predisposes the individual to the formation of calcium stones, particularly in the renal pelvis or renal collecting ducts. These may be associated with infections. Both kidney stones and renal infection can lead to impaired renal function. Hypercalcaemia also impairs the concentrating ability of the renal tubule by decreasing its response to antidiuretic hormone.
Chronic hypercalcaemia of hyperparathyroidism is associated with mild insulin resistance, necessitating increased insulin secretion to maintain normal glucose levels. Hypercalcaemia also affects the muscular, nervous and gastrointestinal systems, causing fatigue, headache, depression, anorexia, and nausea and vomiting (see Figure 11-12).41
EVALUATION AND TREATMENT
Hyperparathyroidism is generally diagnosed by excluding all other possible causes of hypercalcaemia. A definitive diagnosis must be supported by at least a 6-month history of symptoms associated with hypercalcaemia, including kidney stones, hypophosphataemia, hyperchloraemia and increased urinary calcium levels. With continued improvements in the ability to measure parathyroid hormone, the evaluation of hyperparathyroidism has become simplified. Simultaneous measurements of serum parathyroid hormone and calcium will document elevations of both, and diagnosis is confirmed by measuring urinary calcium excretion.
Definitive treatment involves surgical removal of the solitary adenoma or, in the case of hyperplasia, complete removal of three and partial removal of the fourth hyperplastic parathyroid glands. Observation of asymptomatic individuals with mild hypercalcaemia is also an option. These individuals are advised to avoid dehydration and limit dietary calcium intake.42
Hypoparathyroidism
Hypoparathyroidism is most commonly caused by damage to the parathyroid glands during thyroid surgery. This occurs because of the anatomical proximity of the parathyroid glands to the thyroid.
PATHOPHYSIOLOGY
A lack of circulating parathyroid hormone causes depressed serum calcium levels and increased serum phosphate levels. In the absence of parathyroid hormone, resorption of calcium from bone and regulation of calcium reabsorption from the renal tubules are impaired. Phosphate reabsorption by the renal tubules is therefore increased, causing hyperphosphataemia.
Hypoparathyroidism may also result from hypomagnesaemia. Once serum magnesium levels return to normal, parathyroid hormone secretion does likewise. Hypomagnesaemia may be related to chronic alcoholism, malnutrition, malabsorption, increased renal clearance of magnesium caused by use of aminoglycoside antibiotics or certain chemotherapeutic agents, or prolonged magnesium-deficient parenteral nutritional therapy.
CLINICAL MANIFESTATIONS
Symptoms associated with hypoparathyroidism are primarily those of hypocalcaemia. Hypocalcaemia causes a lowered threshold for nerve and muscle excitation so that a nerve impulse may be initiated by a slight stimulus anywhere along the length of a nerve or muscle fibre (see Chapter 6). This creates muscle spasms, hyperreflexia, convulsions, laryngeal spasms and, in severe cases, death by asphyxiation. Other symptoms of hypocalcaemia include dry skin, loss of body and scalp hair, hypoplasia of developing teeth, horizontal ridges on the nails, cataracts, basal nuclei calcifications (may be associated with a parkinsonian syndrome) and bone deformities.
Phosphate retention caused by increased renal reabsorption of phosphate is also associated with hypoparathyroidism. Hyperphosphataemia results from parathyroid hormone deficiency and, in turn, hyperphosphataemia further lowers calcium by inhibiting the activation of vitamin D, thereby lowering gastrointestinal absorption of calcium.
EVALUATION AND TREATMENT
A low serum calcium and high phosphorus level in the absence of renal failure, intestinal disorders or nutritional deficiencies is diagnostic of hypoparathyroidism.
The parathyroid glands are removed with thyroid gland removal. One of the newer means of maintaining parathyroid gland function that is being used increasingly in Australia and New Zealand is parathyroid autotransplantation, whereby the patient’s own parathyroid glands are transplanted back during thyroidectomy.43 However, short-term hypocalcaemia may still result until the parathyroid glands return to normal function.
Treatment is directed towards alleviation of the hypocalcaemia. In acute states, this involves parenteral administration of calcium, which corrects serum calcium within minutes. Maintenance of serum calcium is achieved with pharmacological doses of an active form of vitamin D and oral calcium. Hypoplastic dentition, cataracts, bone deformities and basal nuclei calcifications do not respond to the correction of hypocalcaemia, but the other symptoms of hypocalcaemia are reversible.
Mechanisms of hormonal alterations
 Abnormalities in endocrine function may be caused by elevated or depressed hormone levels. This may result from: (a) faulty feedback systems, whereby all of the components of a negative-feedback loop are not working appropriately; (b) dysfunction of the gland, in that it produces too much or too little of the hormone, despite normal levels of stimulus; (c) altered metabolism of hormones, such that they are metabolised faster or slower than normal; and (d) production of hormones from non-endocrine tissues such as cancerous growths.
Abnormalities in endocrine function may be caused by elevated or depressed hormone levels. This may result from: (a) faulty feedback systems, whereby all of the components of a negative-feedback loop are not working appropriately; (b) dysfunction of the gland, in that it produces too much or too little of the hormone, despite normal levels of stimulus; (c) altered metabolism of hormones, such that they are metabolised faster or slower than normal; and (d) production of hormones from non-endocrine tissues such as cancerous growths.Alterations of pituitary function
Disorders of the posterior pituitary include syndrome of inappropriate antidiuretic hormone secretion (SIADH) and diabetes insipidus. SIADH is characterised by abnormally high antidiuretic hormone secretion; while in diabetes insipidus, antidiuretic hormone secretion is abnormally low. In SIADH, high antidiuretic hormone levels interfere with renal water excretion. As a result, plasma sodium is diluted in larger quantities of water, leading to hyponatraemia and hypo-osmolality. This condition usually arises due to brain injury or with certain forms of cancer, apparently because of secretion of antidiuretic hormone by tumour cells.Alterations of adrenal function
Excessive aldosterone secretion causes hyperaldosteronism, which may be primary or secondary. Primary hyperaldosteronism is caused by an abnormality of the adrenal cortex. Secondary hyperaldosteronism involves an extra-adrenal stimulus, often angiotensin. Hyperaldosteronism promotes increased sodium reabsorption by the kidneys, so that sodium remains in the blood. This corresponds with hypervolaemia (as water is retained with the sodium), increased extracellular volume (which is variable) and hypokalaemia (as potassium is excreted by the kidneys when sodium is reabsorbed).Alterations of pancreatic function
Diabetes mellitus is a group of disorders characterised by glucose intolerance, chronic hyperglycaemia (raised blood glucose levels) and disturbances of carbohydrate, protein and fat metabolism. Type 1 diabetes mellitus is characterised by a gradual process of autoimmune destruction of pancreatic beta cells in genetically susceptible individuals. This leads to a lack of insulin.Alterations of thyroid function
In general, hyperthyroidism has a range of manifestations related to the endocrine, reproductive and gastrointestinal systems, as well as affecting the eyes. These are caused by increased circulating levels of thyroid hormone and by stimulation of the sympathetic division of the autonomic nervous system. Graves’ disease, the most common form of hyperthyroidism, is caused by an autoimmune mechanism that overrides normal mechanisms for control of thyroid hormone secretion and is characterised by thyrotoxicosis, ophthalmopathy and circulating thyroid-stimulating immunoglobulins. Thyrotoxicosis is a general condition in which elevated thyroid hormone levels cause greater than normal physiological responses. The condition can be caused by a variety of specific diseases, each of which has its own pathophysiology and course of treatment. Thyrotoxic crisis (thyroid storm) is a severe form of hyperthyroidism that is often associated with physiological or psychological stress. Without treatment, death occurs quickly. Primary hypothyroidism is caused by deficient production of thyroid hormone by the thyroid gland. Secondary hypothyroidism is caused by hypothalamic or pituitary dysfunction. Symptoms depend on the degree of thyroid hormone deficiency. Common manifestations include decreased energy metabolism, decreased heat production and myxoedema. Acute thyroiditis is inflammation of the thyroid gland, often caused by bacteria, which can result in hypothyroidism. Subacute thyroiditis is a self-limiting nonbacterial inflammation of the thyroid gland. The inflammatory process damages follicular cells, causing leakage of T3 and T4. Hyperthyroidism is then followed by transient hypothyroidism, which is corrected by cellular repair and a return to normal levels in the thyroid. Autoimmune thyroiditis is associated with infiltration or fibrosis of the thyroid, circulating thyroid antibodies and gradual loss of thyroid function. Autoimmune thyroiditis occurs in those individuals with genetic susceptibility to an autoimmune mechanism that causes thyroid damage and eventual hypothyroidism.Alterations of parathyroid function
Hyperparathyroidism, which may be primary or secondary, is characterised by greater than normal secretion of parathyroid hormone. Primary hyperparathyroidism is caused by an interruption of the normal mechanisms that regulate calcium and parathyroid hormone levels. Manifestations include chronic hypercalcaemia, increased bone resorption and hypercalciuria. Secondary hyperparathyroidism is a compensatory response to hypocalcaemia and often occurs with chronic renal failure.Susan is a 51-year-old information technology professional. She recently started losing weight without trying, which she found to be quite satisfying! At the same time, she also noticed that she feels quite hot when others around her do not complain of the heat. Susan thought she was probably getting the menopause, as she knows that this is characterised by hot flushes. She tires easily, but put this down to working long hours and getting older. None of these symptoms gave Susan any real cause for alarm. However, what made her think that there might be something wrong is the increasing amount of heart palpitations she has — she can feel her heart racing quite quickly. She has been feeling more anxious and suspects that this is because her fast heart rate makes her quite worried. So she decides to seek medical attention.
Physical examination reveals tachycardia (a high heart rate of 105 beats per minute), hypertension (high blood pressure of 145/85) and a high respiratory rate (22 breaths per minute). Her eyes appear somewhat wide open and there is an enlargement in the anterior (front) of her neck.