Chapter 19 Central Nervous System Stimulants
The central nervous system (CNS) stimulant drugs such as amphetamine and caffeine may produce dramatic effects by increasing the activity of CNS neurons; however, their therapeutic usefulness is limited because of their many general effects and adverse reactions in the body. Chronic use and misuse occur with these drugs, resulting in patients developing drug tolerance, drug dependence and drug abuse problems. If taken in sufficient doses, all CNS stimulants may cause convulsions. This chapter reviews the CNS-stimulant drugs that are available for clinical use. Their approved indications are for treatment of attention disorders and narcolepsy, and to suppress the appetite. The methylxanthines (caffeine, theophylline and theobromine) are mainly taken in beverages (coffee, tea, cocoa and soft drinks) to increase alertness, while amphetamines have clinical applications in attention deficit hyperactivity disorder (ADHD), narcolepsy and as anorectic agents, but are widely abused for their stimulant effects.
Key abbreviations
ADHD attention deficit hyperactivity disorder
Key background: history and uses of stimulants
THE CNS stimulants (amphetamines and methylxanthines) exert their major effects on the cerebrum, medulla, brainstem and the hypothalamic or limbic regions. Amphetamines are mainly stimulants of the cerebral cortex; whereas anorectic agents suppress the appetite, possibly by a direct stimulant effect on the satiety centres in the hypothalamic and limbic regions; and analeptics (restorative drugs) primarily affect centres in the medulla and the brainstem. CNS stimulants act by increasing the neuronal discharge in excitatory pathways or by blocking inhibitory pathways.
Cerebral stimulants were commonly prescribed in the past for obesity, to counteract CNS-depressant overdosage and to increase alertness in people trying to stay awake during long shift-work or boring tasks, but such use is today considered obsolete. Although CNS stimulants such as phentermine and diethyldepropion suppress appetite, tolerance develops to the anorectic effect, usually before the weight reduction goal is reached. Treating overdosage of CNS depressants with stimulants is discouraged because close monitoring and supportive measures have been found to be successful, avoiding undesirable adverse reactions of stimulants.
These drugs may also affect other parts of the nervous system, including the autonomic nervous system, so adverse effects are common. With their narrow therapeutic range between effectiveness and toxicity, CNS stimulants may induce cardiac arrhythmias, hypertension, convulsions and violent behaviour. They therefore have limited use in practice today and are primarily used for the treatment of ‘alertness disorders’ such as attention deficit hyperactivity disorder (ADHD) and narcolepsy, and as appetite suppressants. They are also being examined for their effectiveness in improving functional recovery after brain injuries such as stroke.
Amphetamines
Pharmacodynamics
Relationships to neurotransmitters
Amphetamine itself (α-methylphenethylamine) is closely related chemically to noradrenaline, adrenaline and many other sympathomimetic amines (see Figure 4-3 and Chapter 12). There are also trace amounts of similar amines in the brain, such as octopamine, tyramine and phenylethylamine, which may act as neuromodulators as well as participating in reactions in the biosynthetic pathways for neurotransmitters. The amphetamine-like analogues have fewer hydroxyl (−OH) groups than do the catecholamines; they thus have higher lipid solubilities and so cross the blood–brain barrier and have CNS activities. The generic term ‘phenylethylamines’ is sometimes used to refer to all the drugs in this group, including the four drugs described in this section: dexamphetamine, methylphenidate, phentermine and diethylpropion; however, as they are all related chemically to the prototype amphetamine, we will refer to the group as the amphetamines.
Central actions
Amphetamine-like drugs have four main effects on the CNS:
Mechanism of action
The proposed mechanism of action for the amphetamines includes the release of noradrenaline, dopamine and other monoamines from storage sites in nerve terminals (hence an indirect sympathomimetic effect), direct stimulating effects on α- and β-adrenoceptor sites and effects on dopamine transmission (see Table 14-1). The primary action centrally appears to be in the cerebral cortex and possibly the reticular activating system. Stimulation results in increased mental alertness and motor function, decreased sense of fatigue and, usually, a euphoric effect. These effects are probably mediated through effects on central adrenoceptors. Amphetamines can also cause stereotyped behaviours in animals (compulsive gnawing and sniffing, and circling) and paranoid psychosis in humans, similar to an acute schizophrenic attack. These effects are most likely related to actions on dopaminergic pathways, as they can be reversed by antipsychotic drugs. Amphetamines can also contribute to serotonin toxicity.
Indirect actions at glutamate receptors have also been implicated in the mechanism of action of amphetamines and related CNS stimulants. Many of the behavioural actions of amphetamines are blocked by antagonists at both types of glutamate receptors, and glutamate-receptor blockers may be useful in treatment of psychostimulant toxicity (Clinical Interest Box 19-1).
Clinical interest box 19-1 Managing psychostimulant poisoning
Drugs from both the legal psychostimulant group (dexamphetamine, methylphenidate, caffeine and some decongestant and weight loss drugs) and the illicit stimulants (other amphetamines, designer drugs including ‘ecstasy’, and cocaine) can cause acute toxicity as well as drug abuse problems.
Signs and symptoms of psychostimulant overdose include tachycardia, dilated pupils, euphoria, insomnia, confusion and tremors through to delirium, convulsions, psychosis, cerebrovascular accidents and death. Neurotoxicity may be irreversible.
There is no specific antidote for an overdose of amphetamines, so symptomatic and supportive measures should be instituted according to the individual patient’s requirement. De-escalation (calming and support) is helpful, with a show of force and then physical restraint if necessary. Excessive stimulation and seizures may be counteracted with diazepam, haloperidol or droperidol (to decrease dopaminergic effects and hyperthermia).
Vital signs, cardiac and respiratory functions, hydration and nutrition should be monitored frequently; cooling and improved nutrition should be considered. Medications usually used are: for hypertension, IV phentolamine or nitrites; for arrhythmias, lignocaine IV; for serotonin toxicity, IV hydration, active cooling and assisted ventilation.
Source: McCormack & Buckley 2006.
Tolerance and dependence
Tolerance develops readily to the peripheral and anorectic effects of amphetamines; indeed, the anorectic effects wear off a few days after taking these drugs, which detracts from their clinical usefulness. Addiction to and dependence on amphetamines can develop, possibly due to users taking more of the drugs to overcome the unpleasant mood swing (depression and tiredness) after the effects of a dose wear off, leading to ‘binge’ drug-taking behaviour. (These aspects of amphetamine abuse are covered in Chapter 21.) Because of their potential for abuse, amphetamines are no longer readily prescribed for use as appetite suppressants; instead, they are indicated only for the treatment of ADHD and in the treatment of narcolepsy. Dexamphetamine and methylphenidate fall under the Australian Poisons and Controlled Substances Regulations into the ‘Controlled Drug’ classification, Schedule 8, for drugs with a high abuse potential. Other proposed clinical uses are in recovery from stroke and from traumatic brain injury.
Adverse drug reactions
The acute toxicity of amphetamines, which causes cellular necrosis and loss of CNS neurons, is thought to be due to the formation of active free radicals and hence mitochondrial malfunction. In chronic abuse of amphetamines there is a strong association with psychoses and especially schizophrenia; whether this is a causal effect (amphetamines causing psychosis) and/or a ‘dual diagnosis’ effect (people with schizophrenia more likely to use or abuse drugs) is at present unclear.
The main central effects are of over-stimulation of the CNS (see Drug Monograph 19-1 for dexamphetamine and Clinical Interest Box 19-1).
Drug monograph 19-1 Dexamphetamine
Dexamphetamine, the (+) or dextro-isomer of amphetamine, is the prototype drug of this group. It is indicated for use in ADHD in children, and in narcolepsy.
Pharmacokinetics
Amphetamines are well absorbed from the gut, with peak plasma concentrations reached 2 hours after oral administration. They are widely distributed to body tissues, with especially high concentrations in the brain and cerebrospinal fluid, lungs and kidneys. Some dexamphetamine is metabolised in the liver, and the remainder is excreted unchanged by the kidneys.
Excretion (and therefore half-life) is pH-dependent; excretion is increased in acidic urine and decreased in more alkaline urine. Approximate half-lives are 6–8 hours in acidic urine with pH <5 (e.g. after taking ammonium chloride); or 15–30 hours in alkaline urine of pH >7.5 (e.g. after taking sodium or potassium citrate).
Adverse reactions
Important adverse reactions include:
With high dosage or prolonged consumption, mood changes, including depression, increased agitation, choreas and psychosis may occur. Drug dependence and tolerance may also develop.
Treating amphetamine overdose consists of symptomatic and supportive care and reversal of CNS stimulation with sedatives, as outlined in Clinical Interest Box 19-1.
Warnings and contraindications
Amphetamines have a high liability for abuse. The CNS stimulation and the rebound depression after withdrawal both impair abilities to drive or operate machinery.
Avoid use in persons with amphetamine hypersensitivity, hyperthyroidism, hypertension, glaucoma, history of drug abuse, cardiovascular disease, severe agitation, severe arteriosclerosis and Tourette syndrome. Amphetamines are contraindicated during pregnancy, as they cause increased risk of malformations, premature delivery and withdrawal symptoms in the infant (Category B3).
Dosage and administration
Dosage depends on the indications for which the drug is prescribed and is adjusted individually to the lowest effective dose, not taken in the evenings because of the CNS excitation effects. Typical doses for school-age children are initially 2.5–10 mg daily, increasing to a maximum of 40 mg/day in ADHD, taken in the morning and early afternoon.
In the peripheral nervous system, amphetamines and related phenylethylamines such as pseudoephedrine have indirect sympathomimetic actions by causing the release of noradrenaline; hence they have vasoconstrictor and hypertensive effects. Adverse drug interactions are common (Drug Interactions 19-1). Related drugs such as xylometazoline and phenylephrine are used topically in the nose and eye for their decongestant effects (see Drug Monograph 28-6).
Drug interactions 19-1 Amphetamines
| Drug or drug group | Likely effects and management |
| Tricyclic antidepressants; other | CNS stimulants; sympathomimetics including inotropes Effects of these drugs are enhanced, which may result in adverse cardiovascular and CNS effects, such as arrhythmias, tachycardia or severe hypertension. Avoid or a potentially serious drug interaction may occur. Chronic users of amphetamines may require higher doses of sympathomimetic inotropes in an emergency, due to tolerance |
| Non-selective MAO inhibitors, and reversible inhibitors of MAO-A (RIMAs) | Avoid concurrent usage because effects of catecholamines are increased; headaches, arrhythmias, vomiting, sudden severe hypertension or hyperpyrexia may result. Avoid or a potentially serious adrenergic crisis may occur |
| α-Adrenergic blocking drugs (systemic and ophthalmic) and other autonomic antihypertensive agents | Amphetamines may overcome adrenoceptor antagonism of α- or β-blockers, causing sympathomimetic effects resulting in loss of blood pressure control and hypertension |
| Digitalis glycosides | May result in an increase in cardiac arrhythmias |
| Thyroid hormones | Concomitant administration may result in enhanced effects of thyroid hormones or amphetamines |
Drug interactions with amphetamines
Typical interactions and possible outcomes are shown in Drug Interactions 19-1.
Clinical uses
Attention deficit hyperactivity disorder (ADHD)
The syndrome of ADHD is considered a psychiatric disorder of childhood (previously these children were probably just considered naughty or unmanageable). It is characterised by distractibility, a short attention span, impulsive behaviour, hyperactivity and learning disabilities; and the child may be moody and irritable and have low self-esteem. Most children with ADHD also have other concurrent disorders, such as conduct, anxiety, learning or depressive disorders. Improper functioning of the monoamine neurotransmitter systems (noradrenergic, dopaminergic and serotonergic) have been implicated in ADHD. Paradoxically, CNS-stimulant medications tend to decrease the distractibility and hyperactivity, resulting in a lengthened attention span and improved cognitive performance and social behaviour.
The prevalence of ADHD in the community is about 1% and may be greater in more socially disadvantaged groups. Symptoms may present from infancy, and ADHD usually becomes apparent between the ages of 3–7 years, with boys affected more often than girls by a 10:1 ratio. Usually professional intervention is unnecessary until the child enters the school setting, when symptoms may start to cause functional impairment. ADHD may persist into adulthood, with higher incidences of substance abuse, antisocial personality disorders, anxiety and depression being observed in comparisons with control groups.
Behavioural management
Managing this disorder requires a behavioural modification program (family support, directed activities, special educational programs, speech and/or occupational therapy and psychotherapy) with use of pharmacological therapy as an adjunct if necessary. Around 15%–20% of children do not respond or their symptoms increase with the stimulant drugs; in these cases, therapy with tricyclic antidepressants or with clonidine may be tried. To promote the child’s proper psychosocial development, the distractibility and hyperactivity must be managed during school hours and at other times (e.g. for participation in clubs, music lessons or social events).
Other non-stimulant therapies (for which there is little hard evidence of efficacy) include behavioural modification techniques in children and cognitive behavioural therapy in adolescents and adults; other drugs used include antidepressants, α2-adrenoceptor agonists (clonidine) and cholinergic agents.
Use of stimulants
The two amphetamine-related drugs approved for treatment of ADHD in Australia are dexamphetamine (see Drug Monograph 19-1) and methylphenidate, which is more selective at blocking dopamine transporters. Use of these psychostimulants helps improve academic performance, vocational success and social and emotional development. Response is usually rapid and obvious. Doses are started low and gradually increased to a maximum of 30 mg dexamphetamine per day, provided effective responses are obtained. The prescriber needs to work closely with the child, the parents, carers and school staff in evaluating results and planning dosages. There are as yet no clear guidelines as to how long therapy should be continued; drug-free trials are recommended at yearly intervals. Widespread use of the drugs increases their availability for abuse, and diversion of the drugs from the school playground to the black market is becoming a problem, in both Australia and New Zealand (see Clinical Interest Box 19-2). New extended-release, long-acting formulations of methylphenidate are now available; these have many advantages including once-daily dosing, which improves the privacy of patients taking them and minimises the likelihood of the drugs being diverted or abused.
Clinical interest box 19-2 Methylphenidate—its (mis)use in new zealand
Medicines with legitimate medical indications are being increasingly diverted for illicit purposes. Methylphenidate is used for the treatment of narcolepsy and for ADHD in children and is regarded as a first choice pharmacological agent in the treatment of ADHD. It is available as an immediate-release (10 mg) or sustained-release (20 mg) tablet.
Methylphenidate stimulates the CNS and has a calming effect on ADHD children and allows them to focus on schoolwork. Patients with ADHD do not get addicted to the stimulant drug. However, when misused as an illicit drug, being snorted like cocaine or injected like heroin, it has powerful stimulant effects and can cause serious health risks. The street names include ‘Rits’ and ‘poor man’s coke’. In New Zealand, from 1 February 2000, methylphenidate prescriptions were restricted to specialist recommendation, reflecting concerns that parents and children were obtaining methylphenidate too readily and then supplying it to others. Up till March 2002, more than 72 000 prescriptions for methylphenidate had been written, an increase of more than 30% over 2000. The street value of methylphenidate tablets is about $5, considerably less than in the USA where the street value is about NZ$20.
Atomoxetine
A non-stimulant drug now recommended as second-line treatment for ADHD is atomoxetine. This compound inhibits the reuptake of noradrenaline (as do cocaine, amphetamines and both TCA and SSRI antidepressants); however, it appears not to cause CNS stimulation and does not cause dependence, hence is not a controlled (S8) drug. It is well absorbed but has variable bioavailability; the half-life varies from 5 to 22 hours. Clinical placebocontrolled trials in children showed its efficacy in reducing ADHD symptoms; it was approximately equiactive with (but not better than) methylphenidate. As expected, autonomic side effects are common, including dry mouth, decreased appetite and raised pulse rate and blood pressure. It is metabolised by CYP2D6 enzyme, so interactions with drugs that inhibit or induce this enzyme are common.
Clinical experience has shown that, in children aged 12 years old or younger taking atomoxetine regularly, there is increased risk of suicidal ideation; children should be closely monitored for clinical worsening or changes in behaviour. Precautions need to be taken in patients with cardiovascular disease, glaucoma and history of seizures. There are potential drug interactions with other drugs that raise monoamine levels, including MAOI and SSRI antidepressants.
Narcolepsy
Narcolepsy is a condition characterised by excessive drowsiness and uncontrollable sleep attacks during the daytime, even while eating, driving or talking.2 In addition, the patient may exhibit a sleep paralysis (inability to move that occurs immediately on falling asleep or on awakening), cataplexy (stress-induced generalised muscle weakness) and hypnagogic illusions or hallucinations (vivid auditory or visual dreams occurring at onset of sleep). It is a specific, permanent neurological disorder, coming on in early adulthood and causing great distress to the sufferers.
Although narcolepsy is essentially incurable, education about the condition assists the patient to recognise the symptoms and adapt the daily schedule. CNS stimulants such as methylphenidate are useful in controlling the daytime drowsiness and excessive sleep patterns, whereas TCAs are being tested in conjunction with the stimulants for cataplexy and sleep paralysis.
Modafinil
A non-amphetamine drug, modafinil, has been shown to be clinically effective in treating narcolepsy, with significantly increased scores on tests for maintenance of wakefulness and sleep latency. Its mechanism of action is unclear: it does not appear to bind with receptors for any monoamine transmitters. It improves alertness and opposes the impaired cognitive functioning caused by lack of sleep, while not affecting appetite, behaviour, nocturnal sleep or the autonomic nervous system. It is indicated in treatment of excessive sleepiness associated with narcolepsy, obstructive sleep apnoea and disturbed sleep patterns due to shift work changes. A single dose taken in the morning is slowly absorbed and eliminated mainly by metabolism in the liver to inactive metabolites which are excreted via the kidneys. The elimination half-life is approximately 10–12 hours. There is little risk of dependence.
There are potential drug interactions with other drugs metabolised by CYP3A4; in women, combined oral contraceptives may be inactivated faster so other contraception should be used. Main adverse effects are central: headache, nausea, nervousness and possibly euphoria, hence the drug might be abused. The standard dose is 200–400 mg each morning. Modafinil may also prove useful in other disorders of sleep/wakefulness such as obstructive sleep apnoea and in shift-work.
As anorectic agents
Anorectic drugs (also called appetite-suppressants or anorexiants) include some indirectly acting sympath omimetics and phenylethylamine-like or amphetamine-like drugs used for the short-term treatment of obesity (see Chapter 50). Their exact mechanism of action is unknown but they appear to reduce hunger by effects in the hypothalamus and limbic areas of the brain. In the past, many such drugs were readily available to treat obesity by decreasing appetite; however, the amphetamines are liable to be abused because of their dependence potential, and tolerance develops rapidly, so amphetamine and dexamphetamine are no longer prescribed for obesity. Two other amphetamine analogues, fenfluramine and dexfenfluramine, were withdrawn in Australia because of their tendency to cause adverse cardiovascular effects (particularly pulmonary hypertension).
Diethylpropion and phentermine
The only two remaining amphetamine-related compounds indicated as anorectics are diethylpropion and phentermine. These act mainly on adrenergic pathways and, while they do cause some CNS stimulation and mild euphoria, they are less liable to lead to dependence than are other amphetamines. Actions, adverse effects and drug interactions are generally similar to those of dexamphetamine.
Careful selection and dosing are necessary to minimise the unwanted effects. As appetite suppressants they are recommended only for the short term because tolerance to the anorectic effect may occur within a few weeks. They are used as adjuncts to other obesity treatment regimens such as reducing absorption of fats, reducing energy intake, modifying diet, increasing physical activity, behavioural therapy and surgery (see Chapter 50 for pharmacological aspects of obesity).
Sibutramine
Sibutramine, recently released in Australia, is a serotonin and noradrenaline reuptake inhibitor (SNRI) that induces the sensation of satiety (fullness), hence is used to treat obesity, e.g. in adults with a body mass index >30 kg/m2. Autonomic adverse effects are common. Some other selective serotonin reuptake inhibitors (SSRIs, e.g. fluoxetine) used as antidepressants have also been shown to reduce appetite.
Methylxanthines
Caffeine
The methylxanthines—caffeine (Drug Monograph 19-2), theophylline, theobromine and the herbal medicine Paullinia cupana (commonly known as guarana)—are naturally occurring chemicals found in beverages such as coffee, tea, cocoa and cola drinks. Caffeine is also present in many foods, over-the-counter drugs and prescription drugs; it is probably the most commonly used stimulant worldwide. A large daily intake of caffeine-containing products may increase alertness but may also induce insomnia and heart arrhythmias in some people, especially the elderly. (Aspects of caffeine related to the social use of, and dependence on, caffeine-containing products are discussed in Chapter 21; the clinical use of methylxanthines as bronchodilators is considered in Chapter 28).
Indications
Caffeine is used in the treatment of fatigue or drowsiness and as an adjunct to analgesics to enhance relief of pain; it is sometimes used as a respiratory stimulant in infants with respiratory difficulties.
Pharmacokinetics
Caffeine is rapidly and totally absorbed after oral administration. It is only 35%–40% protein-bound and is distributed to all body compartments. It crosses the blood– brain barrier and enters the CNS, and passes readily through the placenta. The peak plasma level is achieved within 50–75 minutes, with therapeutic plasma levels at 5–25 mg/mL.
Caffeine is metabolised in the liver. In adults it is metabolised to theophylline and theobromine, and thence via xanthine derivatives to uric acid; in the neonate only a small portion is metabolised to theophylline. Caffeine’s half-life is 3–10 hours (average 4 hours) in adults and 65–130 hours in neonates. In adults, caffeine metabolites are excreted by the kidneys, with only 1%–2% excreted unchanged; in neonates it is excreted by the kidneys, with about 85% excreted unchanged.
Drug interactions
The following effects may occur when caffeine is taken with other drugs: caffeine antagonises the antiarrhythmic actions of adenosine, so larger doses of adenosine may be needed, and actions of dipyridamole when used in cardiac stress testing. When caffeine is taken along with other CNS-stimulating drugs, or other caffeine-containing medications or drinks, increased CNS stimulation, nervousness and arrhythmias can occur.
Adverse reactions
Common adverse reactions include increased nervousness or anxiety and irritation of the gastrointestinal tract, resulting in dyspepsia and nausea. Adverse reactions in neonates include abdominal swelling or distension, vomiting, body tremors, tachycardia or nervousness, feed intolerance, irritability and reduced weight gain.
Signs of overdose include raised temperature, headache, confusion, increased irritability and sensitivity to pain or touch, tinnitus, insomnia, palpitations, fine tremor, increased urination, dehydration, nausea and vomiting, abdominal pain and convulsions. A withdrawal syndrome of irritability, head ache and increased weakness has been reported when users of more than 600 mg/day (about six cups of coffee) decrease or eliminate their intake.
Warnings and contraindications
Use with caution in persons with insomnia, nervousness and tachycardia. Avoid use in patients with caffeine or xanthine hypersensitivity; severe anxiety, including agoraphobia or panic attacks; severe cardiac disease; liver function impairment; or hypertension.
Dosage and administration
The adult dose is 100–200 mg orally, repeated in 3–4 hours if necessary to a maximum of 500 mg daily. (A standard cup of coffee contains 50–150 mg caffeine.) Caffeine is not recom mend ed for use in children up to 12 years of age (except in neonatal respiratory distress). It is present in combination with ergotamine in several form ulations for treatment of migraine (see Chapter 20), in caffeinated beverages and in some ‘tonic’ preparations in combination with vitamins and glucose; the usual dose of caffeine in these formulations is 100 mg.
Mechanism of action
The mechanism of action of caffeine was initially postulated to involve raising of cyclic adenosine monophosphate (cAMP) levels through blocking of the enzyme phosphodiesterase (Figure 28-4), leading to smooth muscle relaxation and other effects. However, it is now recognised that the concentrations required for this action are probably not reached in clinical (or social) doses.
Recent studies indicate that caffeine’s effects are primarily due to antagonism of adenosine receptors (adenosine is an endogenous nucleoside and a neuromodulator that is structurally similar to caffeine; see Table 14-1). Adenosine mediates CNS depression, has cardiac depressant and bronchoconstrictor effects, inhibits platelet aggregation and is an important regulator of blood flow (vasodilator in most regions, including the coronary circulation, but vasoconstrictor in the renal and cerebral circulations). Adenosine is used clinically in supraventricular tachycardias: rapid IV injection decreases atrioventricular conduction and effectively converts the arrhythmia to sinus rhythm.
By antagonising adenosine A1 and A2A receptors, methylxanthines oppose these effects and indirectly lead to increased cAMP levels. This ‘second messenger’ is involved in activating many protein kinases, which may cause variations in energy metabolism, cell division and differentiation; changes in ion transport and ion channel functions; and contraction of cardiac and smooth muscle—hence the pharmacological effects described.
Some of the behavioural effects of caffeine may be mediated by dopamine. By antagonising inhibitory effects of adenosine on dopamine receptors, caffeine may indirectly stimulate dopamine activity. This mechanism could explain the similarities between the behavioural effects of caffeine, amphetamines and cocaine; antagonists at A2A receptors are being trialled in Parkinson’s disease.
Pharmacological effects of caffeine
Because caffeine has effects on many body functions and is so widely used, both its short-term and possible long-term effects are important. Overall, moderate habitual coffee intake is not a health hazard.
Central nervous system
Although all levels of the CNS may be affected, regular doses of caffeine (100–150 mg) will stimulate the cortex and produce increased alertness but decreased motor reaction time to both visual and auditory events (see Clinical Interest Box 19-3). Drowsiness and fatigue generally disappear. Larger doses may affect the medullary, vagus, vasomotor and respiratory centres, resulting in slowing of the heart rate, vasoconstriction and increased respiratory rate.
Clinical interest box 19-3 Smart drugs?
An article in an Australasian science journal (Pockley 2000) reported that drugs were increasingly being used by school students in attempts to enhance their concentration and improve marks. Students have long used caffeine to stay awake to study and were also turning to ‘pep’ drinks with massive caffeine levels and to other drugs such as amphetamines. The trend was seen to have ethical implications and to increase competitiveness among students. Journalists and lecturers were questioning whether candidates might have to submit to urine testing before entering examination venues.
Ten years later, ‘cognitive enhancers’ (also known as smart drugs, memory enhancers or nootropic agents) are being used clinically to treat people with cognitive difficulties in dementias, neurodegenerative disorders such as Parkinson’s disease, ADHD, and to aid recovery after stroke and traumatic brain injury, as well as to improve memory, motivation and attention.
While there is little hard evidence for efficacy, a wide variety of drugs are being tried as smart drugs, acting on every imaginable neurotransmitter: old favourites like amphetamines and caffeine; antidepressants including noradrenaline or serotonin reuptake inhibitors and monoamine oxidase inhibitors; piracetam and related drugs; modafinil and atomoxetine, cholinergics (choline, galantamine, nicotine), amino acids (phenylalanine, tyrosine, tryptophan), fish in the diet, herbal products (yohimbine, rosemary, sage, St John’s wort, passion flower) and even kava.
Sources: Pockley 2000; Greely et al 2008.
Caffeine is thus useful for counteracting fatigue in shift-workers (and students) and as a cognitive enhancer, but can cause anxiety. The mechanism is thought to be via antagonism of adenosine receptors and consequent enhancement of dopamine activity. Caffeine also lifts the mood and may have antidepressant effects; it has been shown to reduce the risk of suicide. Caffeine withdrawal leads to headaches, fatigue, decreased alertness and irritability;3 it has therefore been used clinically to relieve postoperative withdrawal symptoms and for post-duralpuncture headaches.
Caffeine is used in analgesic products and in combination with ergotamine for treating migraine and other headaches, to enhance pain relief. The enhanced effect of ergotamine may be a result of better absorption of the ergotamine in the presence of caffeine; caffeine itself may also have some direct anti-migraine action.
Respiratory effects
Although the mechanism of action is not clearly defined, caffeine appears to stimulate the medullary respiratory centre and normalise autonomic function. Thus it may be useful for treating apnoea in preterm infants and Cheyne–Stokes respiration in adults, as an adjunct to non-drug measures and as an alternative to theophylline. The methylxanthines are an important group of bronchodilator agents; in particular, aminophylline, a derivative of theophylline (Drug Monograph 28-3), is used in childhood asthma.
Cardiovascular system
In low doses caffeine is thought to enhance vagal stimulation and thus slow the heart. In higher doses, caffeine stimulates the myocardium, increasing both heart rate and cardiac output. Overstimulation may cause tachycardia and cardiac irregularities.
Depending on the dose, caffeine may cause either vasodilation or a reflex increase in systemic vascular resistance and vasoconstriction, which can cause a rise in blood pressure. This latter effect may be secondary to stimulation of the sympathetic nervous system and blockade of adenosine-induced vasodilation. Overall, caffeine has a weak vasodilator action, with little effect on blood pressure. Theophylline causes potent cerebral vasoconstriction and has been trialled in ischaemic stroke, on the rationale that a decrease in blood flow in perfused areas in the brain may enhance development of collateral vessels in ischaemic areas after stroke. Clinical evidence for benefit is not yet convincing.
Musculoskeletal system
Caffeine affects voluntary skeletal muscles to increase the force of contraction and decrease muscle fatigue. These effects are via activation of the ‘ryanodine receptor’ family, activation of which opens calcium channels in the sarcoplasmic reticulum of skeletal muscle cells, causing calcium release and contraction of the muscle (see Clinical Interest Box 19-4 and Chapter 14, under ‘Adverse effects and toxicity of general anaesthetics’). Caffeine also has a general thermogenic action, increasing heat production, possibly via the hypothalamus or by enhancing catecholamine effects.
Clinical interest box 19-4 Caffeine, anaesthetics and malignant hyperpyrexia
About one in 6000–20,000 persons in the population has an autosomal-dominant inherited tendency to malignant hyperthermia after being administered general anaesthetics combined with skeletal muscle relaxants.
These people appear to have mutation(s) in the ‘ryanodine receptors’ involved in excitation–contraction coupling in skeletal muscle and so have increased sensitivity to calcium releasers, including caffeine. They are thus predisposed to excessive contractile responses and a markedly increased rate of muscle metabolism.
During anaesthesia with general anaesthetics and skeletal muscle relaxants there may be muscle contracture, acidosis, hyperkalaemia and a rapid rise in temperature by 2°C per hour. The mortality of the condition is very high (25%).
This inherited tendency can be diagnosed by testing the effectiveness of caffeine in causing calcium release in a small sample of skeletal muscle removed at biopsy. Skeletal muscle relaxants are contraindicated in people with this condition.
Recently, interesting effects of methylxanthines have been demonstrated in bone: in animals abnormalities of fetal bone and joint development have been shown. In humans there is some evidence that high caffeine intake may increase urinary excretion of calcium, decreasing bone mineral density. This could have important implications for the development of osteoporosis, especially in post-menopausal women.
Other actions
In the gastrointestinal tract, caffeine increases secretion of pepsin and hydrochloric acid from the parietal cells; hence coffee may cause dyspepsia, and intake is restricted in patients who have a gastric or duodenal ulcer.
The methylxanthines produce a mild diuretic effect by increasing renal blood flow and glomerular filtration rate and by decreasing the tubular reabsorption of sodium and water. Theophylline is the only xanthine still used for this diuretic effect; however, the effect is well known to coffee drinkers and is additive with the diuretic effects of alcohol (see Figure 19-1).
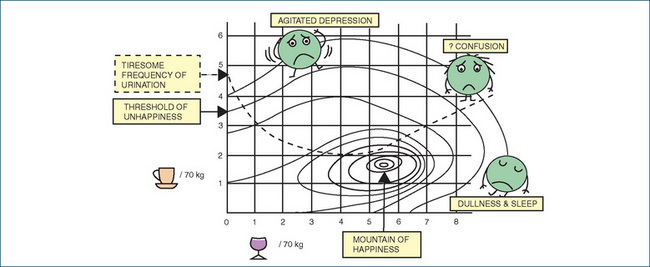
Figure 19-1 Drug interactions between caffeine and alcohol. The scenario is a dinner party or nightclub: alcohol is consumed during the evening, and coffee before leaving. Doses of alcohol (measured in glasses of wine per 70 kg adult) are plotted along the X-axis, and doses of caffeine (measured in cups of coffee per 70 kg adult) up the Y-axis. The CNS-depressant effect of alcohol taken alone leads to dullness and sleep, whereas the CNS-stimulant effect of caffeine alone causes agitation and depression. These effects are antagonistic, causing CNS confusion at high doses of both taken together. Unfortunately, the diuretic effects of the two drugs are additive, leading to a tiresome frequency of urination. Figure courtesy of Dr Andrew Herxheimer (in Laurence 1973); used with permission.
Caffeine also increases metabolic activity, inhibits uterine contractions, transiently raises glucose levels by stimulating glycolysis and raises catecholamine levels in plasma and urine. It is a marker drug for activities of various enzymes, including CYP1A2, N-acetyltransferase and xanthine oxidase (the latter enzyme is involved in the metabolism of the anticancer drug 6-mercaptopurine, in which it competes with the enzyme TPMT [thiopurine methyltransferase]; genetically-determined levels of these enzymes can determine the efficacy or toxicity of 6-MP—an interesting application of pharmacogenomics to drug dosing, discussed in Chapter 41).
Key points
 The CNS-stimulant drugs have a limited use in clinical practice today, as mild stimulants, appetite suppressants and in treating ‘alertness disorders’. The amphetamines and related drugs have sympathomimetic actions (indirect and direct) and may also act through effects on dopamine and glutamate receptors. The main actions of the amphetamines are to cause euphoria, locomotor stimulation, anorexia and stereotyped movements. In overdose or chronic use they may lead to the development of tolerance, dependence and psychoses, as well as acute cardiovascular and neurological toxicity. The amphetamine-related stimulants dexamphetamine and methylphenidate are approved for use in treatment of attention deficit hyperactivity disorder (ADHD) and narcolepsy.
The CNS-stimulant drugs have a limited use in clinical practice today, as mild stimulants, appetite suppressants and in treating ‘alertness disorders’. The amphetamines and related drugs have sympathomimetic actions (indirect and direct) and may also act through effects on dopamine and glutamate receptors. The main actions of the amphetamines are to cause euphoria, locomotor stimulation, anorexia and stereotyped movements. In overdose or chronic use they may lead to the development of tolerance, dependence and psychoses, as well as acute cardiovascular and neurological toxicity. The amphetamine-related stimulants dexamphetamine and methylphenidate are approved for use in treatment of attention deficit hyperactivity disorder (ADHD) and narcolepsy.Review exercises
References and further reading
Australian Medicines Handbook 2010. Adelaide: AMH, 2010.
Cauli O., Morelli M. Caffeine and the dopaminergic system. Behavioural Pharmacology. 2005;16(2):63-77.
Greely H., Sahakian B., Harris J., et al. Towards responsible use of cognitive-enhancing drugs by the healthy. Nature. 2008;456:702-705.
Laurence D.R. Clinical Pharmacology, 4th edn. Edinburgh: Churchill Livingstone; 1973.
McCormack D., Buckley N.A. Psychostimulant poisoning. Australian Prescriber. 2006;29(4):109-111.
Pockley P. Memory drugs flood the classroom. Australasian Science. 2000. Nov/Dec: 28
Psychotropic Expert Group. Therapeutic Guidelines: Psychotropics, version 6. Melbourne: Therapeutic Guidelines Limited; 2008.
Selikowitz M. ADHD: The Facts. Oxford: Oxford University Press; 2004.
Sulzer D., Sonders M.S., Poulsen N.W., Galli A. Mechanisms of neurotransmitter release by amphetamines: a review. Progress in Neurobiology. 2005;75(6):406-433.
Vance A. A current treatment approach for attention deficit hyperactivity disorder. Australian Prescriber. 2008;31(5):129-132.
Waxmonsky J.G. Nonstimulant therapies for attention-deficit hyperactivity disorder (ADHD) in children and adults. Essential Pharmacology. 2005;6(5):262-276.
New Zealand Medicines and Medical Devices Safety Authority: www.medsafe.govt.nz
More weblinks at: http://evolve.elsevier.com/AU/Bryant/pharmacology