Chapter 41 Neoplasia and Treatment of Cancers
Although progress in antineoplastic chemotherapy has helped treat many people diagnosed with cancer, cancers are still a leading cause of death in most countries. In this chapter, an overview is given of tumour cell biology, including the stages involved in cell cycling and its regulation, and the phases in the cell cycle at which checkpoints occur and antineoplastic agents act. The pathways for macromolecular synthesis are described, and the processes of carcinogenesis and growth of cancers.
The main modalities used in treating cancers are outlined, and the principles of antineoplastic chemotherapy and clinical aspects of oncology (such as combination chemotherapy, typical treatment regimens, drug resistance, age-related considerations and safe handling of cytotoxics) are described as background to understanding the types of drugs used in treating cancers.
The mechanisms of action of the main groups of cytotoxic drugs and hormones used in cancer treatment are explained, and potential targets for new drugs are described. The antineoplastic agents have a low therapeutic index; the commonest adverse effects of antineoplastic chemotherapy and their mechanisms are described. (Drug groups are dealt with in more detail in Chapter 42, and sample drug monographs are given there.)
Key background: neoplasia
THIS chapter discusses the principles of antineoplastic therapy and the use of chemotherapeutic drugs in the treatment of cancer. To understand better the mechanisms and sites of action of cancer chemotherapeutic agents, it is important first to have a basic understanding of tumour cell biology and the development of cancers. (Individual antineoplastic agents and adjuncts used in therapy are considered in greater detail in the next chapter). Molecular biology is a rapidly advancing area, and its application to the understanding of cancer and development of new antineoplastic drugs is enormously exciting. This is a highly specialised area of medicine—as evidenced by the fact that whole hospitals such as the Peter MacCallum Cancer Centre in Melbourne are dedicated to cancer research, treatment and education—that we can only outline briefly here; see the relevant chapters in Molecular Cell Biology (Lodish et al 2008) for detailed discussion of relevant molecular mechanisms.
Pathology
Neoplasia (the process of growth of tumours) refers to a group of conditions that are characterised by uncontrolled proliferation and spread of abnormal forms of the body’s cells. The tissue growth is uncoordinated and persists after the end of the stimulus provoking the growth (unlike hypertrophy of muscle in response to increased exercise or growth of the endometrium during the menstrual cycle). Other characteristics of tumours are that their presence is not useful; there may be de-differentiation of cells, leading to loss of specialised functions; and the characteristics of the abnormal cells are inherited indefinitely by successive generations of cells. Benign tumours may cause problems by their excessive growth and may kill by putting pressure on critical adjacent organs.
Malignant tumours (cancers) are more dangerous to the body because, in addition to the above properties, they are generally more rapidly growing and have the ability to spread by invading adjacent tissues and seeding secondary tumours to grow in distant organs (metastasis). Malignant cells may have abnormal or unstable numbers of chromosomes and/or mutated genes (a varied genotype), and their properties may vary over time (varied phenotype). Finding and exploiting differences between normal cells and cancer cells is the goal of much molecular biology research.
Epidemiology
It has been estimated that about 30% of people will develop cancer during their lifetime; cancer is second only to cardiovascular disease as a cause of death in most developed countries. Many people fear cancer because of the likelihood of pain and adverse treatment effects, and because there are many aspects of neoplasia that are still not well understood. Early detection and treatment are imperative to improve success rates in treatment of cancer.
Statistically, the chances of developing cancer and dying from cancer are greater now than ever before. Because diseases that were formerly fatal earlier in life (such as severe malnutrition, childhood infections, complications of childbirth, diabetes and hypertension) have become treatable, most populations are ageing and so a higher proportion will succumb to progressive conditions that occur later in life, such as cancers (see Clinical Interest Box 41-1).
Clinical interest Box 41-1 Cancers in australia
Statistical data from the Australian Institute of Health and Welfare (2008) report ‘Cancer in Australia: an overview, 2008’ show interesting facts about cancer incidence, survival, mortality, screening programs and expenditure in the community. (Note that figures for cancer incidence generally exclude skin cancers other than melanomas, as these are not registrable; it is estimated that non-melanoma skin cancers together account for 4 times the total of all other cancers.)
Website: http://www.aihw.gov.au/publications/index.cfm/title/10607 [29 October 2009].
Tumour cell biology
The cell cycle
Appropriate control in the body of cell division and growth is essential for optimal tissue size and physiological functioning and for replacement of cells as required in response to stimulation and increased demands for function. The replication of each type of cell must be precisely controlled and timed, for normal development of complex tissues such as the brain or kidney. Loss of normal controls on cell replication is the fundamental defect in cancers.
When a cell divides, two daughter cells are produced with identical chromosomes to those of the parent cell. This process of division and proliferation, known as the cell cycle, is essentially the same for normal and cancer cells (Figure 41-1).
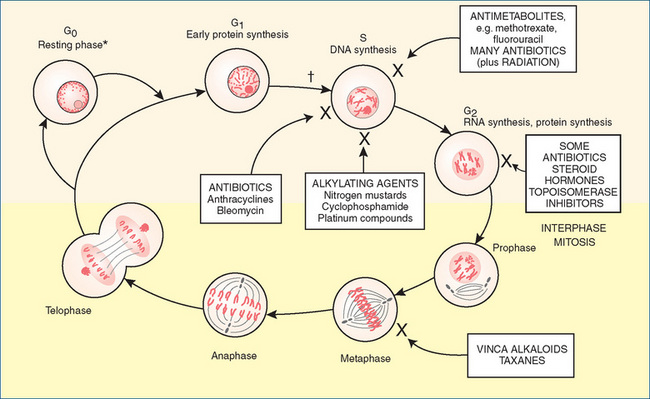
Figure 41-1 Phases of the cell cycle. Drugs (in boxes) are identified, showing the main site at which they exert their effects.
Adapted from: Beare & Myers 1998. * = commitment to cell division: leads to cell enlargement, DNA replication and mitosis; † = first checkpoint: if damaged DNA cannot be repaired, cell undergoes apoptosis; G0 = resting phase, cells not cycling; G1 = first gap, between previous nuclear division and beginning of DNA synthesis (duration highly variable, about 9 hours in rapidly replicating cells); G2 = second gap, between DNA replication and nuclear division (about 4.5 hours); M = mitosis (about 30 minutes); S = period of DNA synthesis (8–20 hours).
In the pre-synthesis ‘gap’ or growth phase (G1), synthesis of RNA and protein occurs in preparation for the next (DNA synthesis) phase. Also during this phase, the decision for cell replication or cell differentiation is determined.
The cell progresses to the synthesis phase (S), during which its genetic material, the DNA in chromosomes, doubles in preparation for cell division.
During the second gap phase (G2), the postsynthesis or premitotic phase, DNA synthesis ceases but RNA and protein synthesis continues, to prepare the cell for mitosis, spindle formation and cell division.
During mitosis (the M phase), cells divide into two new ‘daughter’ cells that may continue to cycle, or may leave the cell cycle to develop into differentiated cells that perform a specialised function (such as neurons or hepatocytes—these cells no longer undergo cell division) or may become either temporarily or permanently nonproliferative (G0 phase).
Cells in the G0 or resting phase, a type of ‘neutral gear’, may remain in this phase (as do fibroblasts until required for healing), may be recruited later to re-enter the cell cycle or may mature and die.
The average time for a rapidly replicating human cell cycle is about 24 hours, depending on the time spent in the S phase.
Regulation of the cell cycle
The phases of the cell cycle occur in the proper order due to tight control of the stages. Regulatory factors determine the progression of cells through the cell cycle by activation of receptors on the cell membrane followed by signal transduction (see Figure 41-2), by which growth stimulatory signals are integrated and passed on to the nucleus in cascades of biochemical reactions, where they trigger activation or repression of various genes required for DNA replication, cell division and proliferation; each regulatory event activates a step and also prepares the cell for the next step in the cycle. Factors that regulate the replication and division of cells include growth factors, tumour suppressor genes and proteins, a family of intracellular proteins called cyclins, regulatory proteins including p53 and ubiquitin, tyrosine kinase enzymes and various types of GTP-binding proteins (such as Ras, Raf, Erk). Genetic aberrations and mutations that result in continuous activation of cell-cycle progression are found in many human tumours. Much research is going into drugs that affect these regulatory processes and thus could be useful in treating cancers.
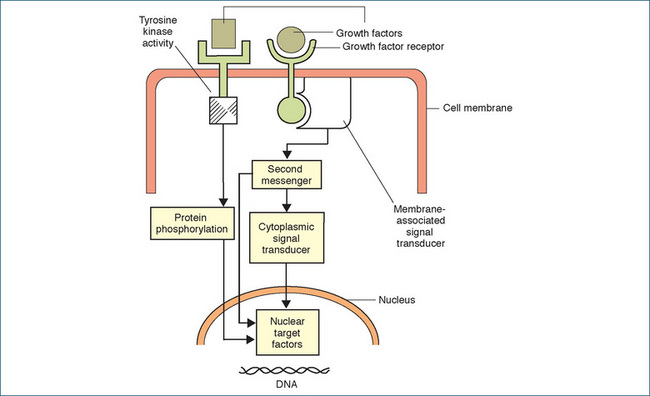
Figure 41-2 Diagram representing simplified mechanisms of growth factors in activation of target factors in the cell nucleus. On the left-hand side, a growth factor is shown binding to a membrane-associated receptor, leading to activation of a tyrosine kinase (a phosphorylating enzyme), which phosphorylates and thus activates a protein that enters the nucleus, where it targets factors involved in cell replication. On the right-hand side, binding of the growth factor activates a signal-transducing protein, which may bind with G-proteins and/or stimulate second messengers, eventually leading to effects inside the nucleus.
Reproduced from: Souhami & Tobias 2005, with permission.
(Readers wishing not to become embroiled in too much biochemistry or molecular biology can now skip through to ‘Apoptosis’, page 793.)
Growth factors
Growth factors (GFs) involved in these signal transduction pathways include epidermal (or epithelial) GF (EGF), fibroblast GF, transforming GF, insulin-like GF and many more, each with its related receptor. Mammalian cells cultured in the absence of growth factors are arrested in the G0 phase of the cell cycle; addition of GF induces transcription of multiple genes which when expressed can stimulate the cell to start cycling.
An example of an important signalling molecule with a role in cancer is EGF. This factor stimulates proliferation of many types of epithelial cells, including those lining ducts in the mammary glands of women. In about 25% of breast cancers, the tumour cells produce raised levels of an EGF receptor called HER2, which makes the cells hypersensitive to the usual low levels of EGF; consequently the growth of the tumour cells is stimulated. A monoclonal antibody specific against HER2, trastuzumab (Herceptin), is used clinically as an antineoplastic agent in breast cancer.
Angiogenesis
Members of the TGFβ family, particularly vascular endothelial growth factors (VEGFs), are also involved in angiogenesis (development of new blood vessels) in tumours, and may either stimulate or inhibit growth of new vessels. Pathological angiogenesis is one of the characteristics of neoplastic growth, and angiogenesis inhibitors can slow the growth and metastasis of cancers by reducing their blood supply. Antibodies against VEGF or its receptors, or inhibition of VEGF signalling, are being tested as anticancer therapies.
Tumour suppressors
The transforming growth factor β (TGFβ) superfamily contains many extracellular signalling molecules that play widespread roles in regulating development; examples are the bone morphogenetic proteins, BMPs (see Chapter 37). The TGFβ-1 isoforms all potently prevent proliferation by inducing synthesis of proteins (such as p15), that inhibit the cell cycle and thus act as tumour suppressors in early stages. Mutations leading to impairment of receptors or proteins in the TGFβ pathway lead to the enhanced cell proliferation seen in many human cancers, including retinoblastoma, colon, pancreatic and gastric cancers, hepatoma and some T- and B-cell malignancies.
Wnt proteins
Another group of signalling molecules (conserved in many species) are the Wnt proteins, which regulate cell-to-cell interactions during embryogenesis. Mutations in Wnt genes and pathway components lead to developmental defects, and abnormal Wnt signalling is associated with some cancers.1
Cyclins
Cyclins, key regulators of the cell cycle, were discovered in the 1980s during research on marine invertebrates.2Cyclins are the regulatory subunits of protein kinase enzymes; they control mitosis and regulate the activities of a variety of transcription and replication factors, as well as structural proteins and proteins involved in mitosis and chromosome formation. The catalytic parts of these phosphorylating enzymes are called cyclin-dependent kinases (CDKs); they activate or inhibit hundreds of proteins at specific regulatory sites at the appropriate time, and thus govern progression through the cell cycle.
Elevated levels of cyclins are found in a number of tumours. CDKs that are improperly regulated can cause unscheduled proliferation, as well as genomic and chromosomal instability. Selective CDK inhibition is therefore a potential target for new anticancer drugs.
Tyrosine kinases
Kinases are phosphorylating enzymes, and phosphorylation is an essential biochemical reaction in nearly all aspects of cell functions. Thus protein kinases are involved in regulating signalling pathways and help mediate metabolism, transcription, cell cycle progression, differentiation, cell movement, apoptosis and immunological functions. Tyrosine kinases (TKs), enzymes that add phosphate groups to tyrosine residues in proteins, are involved in many signalling activities of growth factors. Thus TKs also help control passage of cells through the cell cycle.
Many receptor tyrosine kinases (RTKs) can activate the Ras protein pathway. Ras is a small intracellular G-protein that promotes formation of signal transduction complexes and thence a cascade of kinase enzymes that activate many target proteins. Mutations in RTKs, Ras proteins or kinase enzymes are found in almost all tumour cell types, hence the RTK–Ras– kinase pathways are of great interest in cancer research.
The RTKs are useful targets for anticancer drugs—for example imatinib, a specific inhibitor of the abl kinase, is highly lethal to chronic myelogenous leukaemia cells (see Drug Monograph 42-4). There are at least 96 TKs encoded in the human genome, so other specific TK inhibitors (the –tinib group of drugs) will prove useful as anticancer agents. As TK inhibitors can be small molecules (potentially able to be administered orally) this is sometimes known as ‘small molecule targeted therapy’.
mTOR protein kinase
A TK important in ageing and cancer is mTOR (the mammalian target of rapamycin)—this is a protein encoded by the FRAP1 gene; it is a serine/threonine PK. The main functions of mTOR appear to be as a sensor of cellular nutrient and energy levels and oxidation status, and in regulating cell growth and controlling protein synthesis, angiogenesis and the cytoskeleton. Rapamycin is a natural bacterial product that can inhibit mTOR. Decreased mTOR activity has been found to slow ageing in some species, and rapamycin increases lifespan in mice. It is hypothesised that some dietary regimes such as calorie (energy) restriction help prolong life by decreasing mTOR activity. Drugs that inhibit mTOR activity are being used in treatment of cancer (e.g. temsirolimus) and of transplant rejection, and may prove useful in treating some age-related diseases.
BRAF gene proteins
Another protein kinase in the raf/mil family, encoded by the BRAF gene, is involved in a signalling pathway affecting cell division, differentiation and secretion. The BRAF gene is mutated in approximately 7% of all human cancers, notably in melanomas which have been difficult to treat; a BRAF inhibitor is currently undergoing clinical trial.
Checkpoints
Checkpoints are additional regulatory mechanisms that control cell cycle events at particular stages in the cell cycle, to ensure completion of phase-specific steps in the biochemical pathways before passage into the next phase of the cycle. After checking at the G1/S transition, S phase, G2/M transition or at mitosis, any mutated DNA can be blocked and the mitotic division of mutated chromosomes inhibited, leading to elimination of mutated cells. Checkpoint surveillance mechanisms are responsible for the exact duplication of chromosomes from parent cell to daughter cells during cell division, and for completion of each stage before initiation of the next stage. The mechanisms of ‘checking’ are mainly through control of activation of cyclin–CDKs. For example, at the intra-Sphase checkpoint, activation of CDK1 is inhibited if DNA synthesis is not yet complete.
Ubiquitin
A small protein with unknown function was identified in 1975 as a component of all eukaryotic (higher than bacteria) cells; because it appeared to be ubiquitously expressed it was named ubiquitin3 (from the Latin ubique: everywhere).
It is now known that ubiquitin is involved in an enormously wide variety of cellular functions, including apoptosis, cell cycle and division, DNA transcription and repair, differentiation and development, modulation of cell surface receptors and ion channels, response to stress and aspects of immune responses. Simply put, it performs its functions through conjugation to a large range of target proteins; this process of ‘marking’ of a target protein by ubiquitin is referred to as ‘ubiquitination’. It occurs through a cascade of reactions ending with formation of a peptide bond between a lysine of the target protein and the C-terminal glycine of ubiquitin. Through such pathways, proteins can be targeted for destruction by proteolysis in proteasomes inside cells or may be marked for removal from membranes or for subcellular localisation.
It is the role of ubiquitin in cell-cycle regulation that concerns us here: ubiquitin–protein ligases are one of the three main types of regulators (the others being cyclin–CDK protein kinases and protein phosphatases). Specific ubiquitin– protein ligases are involved at various stages of the cell cycle; for example, S phase is initiated by ubiquitination of inhibitors of S-phase cyclin CDKs, ensuring that they are degraded. During mitosis another ubiquitin–protein ligase helps degrade a protein that stops the daughter chromatids from separating to opposite spindles. Because these transitions are triggered by regulated degradation of proteins, an irreversible process, the cell cycle can only go in one direction.
p53 protein
The p53 protein in the nucleus, with its associated gene, has been referred to as ‘the guardian of the genome’ (Cavalli et al 2004). p53 is a phosphoprotein with a molecular weight of 53 kD (kilodaltons) and is a major regulator of the G1 checkpoint in response to cellular stress. Cells that have undergone mutations in the p53 gene have a loss of normal ‘braking mechanisms’ and hence there is uncontrolled proliferation of altered cells. The efficiency of the p53 response to stress (shutting down PK pathways including the mTOR pathway) declines significantly with age (in mice), and it is postulated that decline in p53 activity contributes to the dramatic increased incidence of cancers in older people.
If DNA damage has been severe, the p53 protein may activate expression of genes leading to enhanced apoptosis. This normally prevents accumulation of damaged DNA and accumulation of mutations that might lead to cancer. Thus p53 has two important checkpoint roles as a tumour suppressor, explaining why mutations in the p53 gene and associated proteins are commonly associated with cancers.
PI3 enzymes
Phosphoinositide 3-kinases (PI3Ks) are a family of intracellular signal transducer enzymes, in three classes, linked to a great number of cellular functions including cell growth, proliferation, differentiation and survival. Mutations in some PI3Ks cause the enzymes to become more active and contribute to cellular transformation and cancer. Some broad inhibitors of the enzymes have been developed, but they are as yet so general that they are too toxic to be useful medically.
Tumour suppressor genes
The p53 gene and the retinoblastoma (Rb) gene are known as tumour suppressor genes, which can direct synthesis of regulatory proteins that inhibit cyclins and halt the cell cycle at checkpoints to allow for repair of damaged DNA.
The Rb family proteins are thought to act as an ‘emergency brake’ to prevent cell-cycle progression when activated. Mutations that impair this signalling pathway and inactivate the Rb function, thus allowing unscheduled progression of cells from the G1 to the S phase, have been shown to occur in nearly every type of adult cancer. Inheritance by a fetus of a mutant allele of the RB gene is likely to cause a cell to become cancerous; this most frequently happens in the retina, leading to highly malignant retinoblastomas that usually become manifest very early in infancy.
Another example is the repressor protein that is encoded by the Wilms’ tumour gene (WT1); it is expressed preferentially in the developing kidney. Children who inherit two mutated WT1 genes produce no functional repressor WT1 protein, and inevitably develop kidney tumours.
The PTEN gene is a tumour suppressor gene that is deleted in many advanced human cancers, allowing abnormal cells to proliferate. Cells lacking the PTEN gene have elevated levels of PI3 and of protein kinase B activity, and reduced ability to undergo apoptosis. Restoring PTEN functions is another potential mechanism for cancer therapy.
Apoptosis
If DNA repair fails during a cell-cycle checkpoint, an altered cell normally undergoes programmed cell death or apoptosis. This is a useful process that eliminates damaged, redundant or abnormal cells via a series of genetically programmed biochemical reactions. Apoptosis involves rounding up of the cell, shrinkage, then fragmentation and digestion. This process occurs constantly, for example, in the shedding of intestinal mucosal cells or skin cells and in the expiry of red blood cells, and is also an important defence against malignant cells. It is thought that apoptosis is the ‘fallback’ position, automatically triggered at cell-cycle checkpoints unless inhibited by the many regulatory factors, hormones and growth factors necessary for cell cycling and survival.
The p53 gene helps mediate apoptosis via complicated pathways involving signalling proteins, growth factors, ‘death receptors’, specialised enzymes called caspases and all their related genes (including PIGs: p53-inducible genes). The precise mechanisms and details of these pathways are still being elucidated. This fascinating area of molecular biology will be crucial in the design of new anticancer drugs, but details are beyond the scope of this chapter (see Lodish et al [2008]).
Macromolecular synthesis (synthesis of proteins and nucleic acids)
For cells to proliferate, the genetic material deoxyribonucleic acid (DNA) must be replicated once every cell cycle. DNA, a large double-stranded helical molecule, is composed of four kinds of serially repeating nucleotide bases: pyrimidines (cytosine and thymine) and purines (adenine and guanine); see Figure 41-3. Particular nucleotide sequences make up the genes, the biological units of inheritance that occupy precise positions on a chromosome. When genes are expressed, DNA is ‘transcribed’ into messenger ribonucleic acid copies, which are transported out of the nucleus into the cytoplasm and there act as templates to direct the amino acid sequence (translation) in the synthesis of proteins such as enzymes and structural proteins. Many anticancer agents act at different stages in the pathways of macromolecular synthesis; these are described briefly later (under types of antineoplastic agents) and in more detail in Chapter 42.
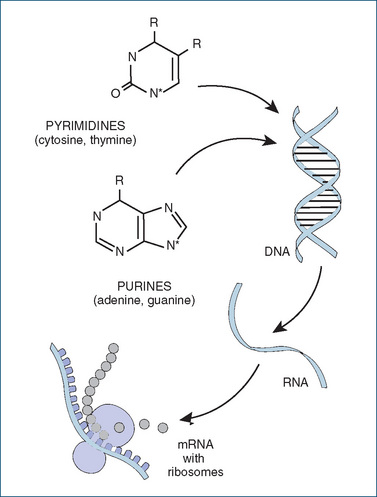
Figure 41-3 Synthesis of macromolecules (nucleic acids and proteins). In the general structures shown for the purine and pyrimidine bases, R stands for an oxygen (=O) or amine (–NH2) group. In the polymers of DNA or RNA, each base is linked via the N* nitrogen atom to a sugar molecule (deoxyribose or ribose), and the sugar molecules are linked via phosphate groups to form long chains. In DNA, two complementary strands are twisted into the famous double-helix shape.
DNA replication and topoisomerases
Packaging of DNA into the chromosomes visible in the nucleus during mitosis involves ‘supercoiling’ of the DNA fragments, i.e. regions where the double-stranded DNA helix is twisted on itself. This requires the actions of enzymes known as topoisomerases, one type of which was previously called gyrase. Topoisomerases have the ability to control the number and amount of twist in the supercoils, by cutting one or both strands, twisting them about each other and resealing the ends. These actions are essential to the complete replication of DNA and controlled growth of cells. Agents that selectively inhibit these enzymes have become useful anticancer drugs.
Chromosome duplication and telomerase
Normal cells appear to be able to undergo only a limited number of divisions; the telomeres (the sections of DNA forming the ends of chromosomes) can be replicated only a limited number of times because a small fragment of ‘junk’ DNA is shaved off each time. Without some special repair mechanism, the daughter DNA strand is shortened at each cell division.
An enzyme called telomerase (telomere terminal transferase) can re-form the telomeres, thus preventing the shortening of the chromosome. The gene for telomerase and its associated RNA are active in germ cells and stem cells, but are usually switched off in fully differentiated cells of adult tissues. However, this enzyme is present in about 90% of human cancers (and in immortal cell lines), so it is thought that the presence of the enzyme may confer immortality to cancer cells. The prognosis for neuroblastoma, a paediatric tumour of the peripheral nervous system, can be estimated by measuring levels of telomerase activity—high enzyme levels predict a poor response to therapy. Inhibition of telomerase activity is thus another possible target for action of anticancer agents.
Carcinogenesis and cancer growth
Development of cancer cells
As described above, normal cells grow and divide in an orderly fashion, with the body’s homeostatic mechanisms and regulatory signalling pathways controlling the entire cell growth process, the process of cell adhesion inhibiting the movement of the newly formed cells and apoptosis removing expendable cells. Exposure of a cell to a carcinogenic stimulus may result in malignancy if apoptosis or the immune system fails to remove a cell with altered genome.
Carcinogenesis (the development of cancers, also known as oncogenesis) is known to be a multicausal, multi-step process (see Clinical Interest Box 41-2). Cancer cells may arise from a hereditary or genetic predisposition or from contact with certain environmental conditions. Many carcinogens (agents that cause the development of or increase the incidence of cancer) have been identified, e.g. the Epstein–Barr virus in Burkitt’s lymphoma, cigarette smoking in many cancers and asbestos in mesothelioma (see review by Irigaray et al [2007]).
Genetic inheritance also plays a role in some cancers (e.g. breast or colon cancer); however, the concordance rate for cancers between identical twins is rather low, indicating that environmental influences predominate. Impairment of functions of tumour suppressor genes is involved in many cancers, as described above for the p53 and Rb genes; when there is loss of the ‘brake’ effect, the oncogenic potential of other proteins cannot be suppressed, and tumours result. Physical effects such as ionising radiation from UV or gamma-rays may cause mutations, and wherever there are higher rates of mitosis, e.g. during the monthly cycle of growth and regression of cells in breast and endometrial tissue, or during repeated physical injury, there is higher risk of mutations and potentially an increased frequency of cancer.
Carcinogenic chemicals
Many carcinogenic chemicals have been identified, including tars and other polycyclic hydrocarbons, phorbol esters, nitrosamines, aflatoxins, organochlorine pesticides, dioxins, hair-dyes and asbestos. These may act synergistically, with some chemicals acting as initiators of tumours and others as promoters. Chemicals, such as benzopyrene and aflatoxin from fungi, and UV radiation, act as mutagens and carcinogens by activating p53 signalling, also causing crosslinks and mutations in the p53 gene and thus impairing checkpoint functions. (Cells with defective checkpoint functions may, however, be more sensitive to anticancer agents; thus checkpoints may become new targets for anticancer drugs.) Mutagens, agents that induce a genetic mutation or increase the mutation rate, may increase the risk of development of cancers.
Oncogenic viruses
Certain viruses are capable of integrating into the genome of the host cell and immortalising it to ensure their own replication. The expressed viral genes induce cell growth and proliferation and prevent checkpoint functions and apoptosis. Known oncogenic viruses are the Epstein-Barr virus, human papilloma virus (HPV, for which an effective vaccine has been developed and gone into large-scale use), hepatitis B and C viruses, Kaposi’s sarcoma virus and human T-cell leukaemia virus. Research into mechanisms of oncogenic viruses led to the discovery of oncogenes and of tumour suppressor genes.
Viruses are known to contribute worldwide to about 16% of cancers, ranging from <10% in high-income countries to >25% in Africa.
Oncogenes
Oncogenes are altered genes that are able to transform normal cells to malignant cells. They may initially occur as part of an oncogenic virus or may derive from an altered cellular counterpart of a viral oncogene (a proto-oncogene). They are named with a 3-letter italic designation, e.g. src, ras or jun; the related proto-oncogene is designated with an initial c-, thus c-ras or c-jun.
Oncogenes are probably all derived from growthcontrolling genes, as oncogene proteins are known to interact with four types of growth-controlling systems in cells:
To anchor proteins to the plasma membrane, the proteins may have a covalently bound lipid group. This is known as prenylation of the protein, and the lipid group may be a geranyl or farnesyl molecule. For example, the ras proteins have a farnesyl group attached (farnesyl is a 15-carbon unit normally involved in the synthesis of steroids and carotenoids). This ‘farnesylation’ is an essential lipidation step that allows the oncogene proteins to transform normal cells into tumour cells. The enzyme involved, farnesyl transferase, is a potential target for anticancer agents.
Diet and cancer
Much research effort has gone into studying any relationship between diet and cancer, particularly the Western diet, which is generally high in energy and animal fats and low in fibre. In the early 21st century it has been accepted that there is very little evidence for any relationship between diet and cancer, except for proven carcinogens such as aflatoxins, nitrosamines and salted fish, and associations with alcohol and obesity in increasing the risk of many cancers.
Environmental and lifestyle factors
Lifestyle factors known to increase the risk of developing cancers include obesity, heavy drinking of alcohol, smoking and exposure to radiation and pollutants such as combustion products of plastics. Obesity may contribute due to adipose tissue acting as a long-term reservoir for lipid-soluble molecules that may be mutagenic or carcinogenic. Alcohol mainly acts as a co-carcinogen, enhancing the actions of mutagens or promoters; it is associated in particular with cancers of the oropharynx, oesophagus, liver and larynx.
Smoking tobacco is known to contribute to about 33% of all cancer deaths (compared to infection with viruses to 15%–20%). A review of cancer epidemiology (Peto 2001) concluded that ‘Avoidance of overweight and prevention or treatment of oncogenic infections are the most important aims for non-smokers; but it is absurd for smokers in the West to worry about anything except stopping smoking’. Reduction of smoking in a population takes decades before the effects are manifest in reduction of cancer deaths (see later Clinical Interest Box 41-7).
Radiation is another common cause of cancers. The discovery of the radioactive isotopes polonium and radium by Marie Curie was followed some years later by her death from aplastic anaemia, almost certainly contracted from exposure to radiation. The atomic bombs dropped on the Japanese cities of Hiroshima and Nagasaki in August 1945 are estimated to have killed over 200,000 people, many dying late from radiation burns, radiation sickness, leukaemias or solid cancers. Closer to home, the high prevalence of skin cancers in Australia is attributed to the habit of fair-skinned people in a tropical country exposing their skin to the damaging effects of the sun’s UV rays (see Chapter 48). Other damaging radiation may come from medical use of X-rays and nuclear power plant accidents; electromagnetic radiation from power lines and cellular (mobile) phones is being studied as a possible contributor to higher incidence of brain tumours.
Chromosomal abnormalities
Chromosomal abnormalities are very common in tumours. Tumour cells may have an abnormal number of chromosomes (aneuploid); may contain translocations of parts of genes, which modify or activate proto-oncogenes; or may have localised duplications (gene amplification) or deletions of DNA fragments. The Philadelphia chromosome, which is diagnostic for chronic myelogenous leukaemia (CML), was discovered in 1960. It is due to a translocation that gener ates the bcr-abl oncogene, which in bone marrow causes neoplastic proliferation of white blood cells. Imatinib, the tyrosine kinase inhibitor effective in CML, binds directly to the Abl kinase active site. Many other cases of leukaemia are also due to chromosome rearrangements that create new fusion genes and proteins.
Amplification of the HER2 gene occurs in approximately 25% of breast cancer patients; this makes the cells sensitive to growth stimulation by low levels of EGF, hence gives the patients a worse prognosis and shortened survival time.
Growth of cancers
Neoplastic cells may lack the cellular differentiation of the tissues in which they originate and therefore may be unable to function like the normal cells around them. Cancer growth is enhanced by an increased rate of cell proliferation due to lack of regulating mechanisms. Because of the genetic differences, cancer cells lack the cell–cell or cell– basement membrane adhesive properties of normal cells, which may lead to metastasis, or spread of the cancer.
A tumour cell burden of 109 cells is usually the smallest (quantitative size) that is physically detectable (palpable) in a solid tumour. At this point, the tissue has about 1 billion cancer cells, which is equivalent to a tumour about the size of a small grape and weighing around 1 g. This is the point at which clinical symptoms usually first appear. If untreated, a malignant tumour may continue to grow in size until it is fatal.
Hypoxia and angiogenesis
The growth of a cancer is usually rapid and exponential in the early stages but, as the tumour enlarges, the centre may outgrow its blood and nutrient supply and become hypoxic and eventually necrotic. To continue to grow, the tumour must develop new blood vessels (angiogenesis) and requires the actions of specialised enzymes (metalloproteinases), which allow space for new cells and blood vessels. New blood vessels developed may be aberrant and have limited blood flow. However, tumour cells can undergo adaptive changes that allow them to survive and proliferate in a hypoxic environment. A key transcription factor increased by hypoxia is called hypoxia-inducible-factor-1α (HIF-1α). Pathways that are influenced by hypoxia include angiogenesis, glycolysis, growth factor signalling, immortalisation of cell lines, genetic instability, tissue invasiveness and metastasis. In many tumours, low oxygen tension in tumour cells is associated with clinical resistance to radiation therapy and chemotherapy, and with increased metastasis and poor prognosis. Most of these pathways enhance the growth of tumours; however, apoptosis can also be increased by hypoxia, so the balance between tumourenhancement and apoptosis is critical to the overall growth of the tumour.
Tumour promotion induced by hypoxia is a potential target for anticancer drugs. A technique that has been used for many years is to correct hypoxia before radiation therapy by use of high concentrations of inhaled oxygen, blood transfusions or erythropoietin to correct anaemia. New prodrugs that are activated only in hypoxic situations will have tumour-specific toxicity. An example is tirapazamine, which when activated by hypoxia inhibits DNA repair initiated by radiation or chemotherapy; it improves survival of patients with non-small-cell lung cancer. Drugs that inhibit HIF-1α activity are being studied in animal models of cancers; rapamycin and the effect ive antineoplastic drug herceptin both inhibit HIF-1α. Inhibition of angiogenesis is another target for new anticancer drugs; inhibitors of the specific metalloproteinase enzymes reduce development of new blood vessels (see reviews by Harris [2002] and Grimm et al [2009]).
Inflammation and cancer
The association between cancer and inflammation has been known for hundreds of years: back in 1863 the famous pathologist Virchow hypothesised that the origin of cancer was at the sites of chronic inflammation, where irritants and tissue injury cause enhanced cell proliferation. It is now known that inflammation itself does not cause cancers, but an environment rich in inflammatory cells, growth factors, activated cell stroma and DNA-damaging agents increases the risk of neoplasia. Wound healing is usually self-limited; however, dysregulation of healing can lead to progression to neoplasia. As described earlier, viral or chemical carcinogens can cause irreversible DNA alterations that initiate cancers, which are promoted by later factors such as chemical irritants, hormones or factors released from sites of wounding, chronic irritation or inflammation. Inflammation associated with a tumour produces many inflammatory cells and mediators such as cytokines, interleukins, interferons, reactive oxygen species, angiogenic growth factors, tumour necrosis factor α (TNFα) and protease enzymes all of which can enhance growth of cells and promote angiogenesis. Later, inflam mation may actually be protective against neoplastic growth, by enhancing apoptosis and immune responses. The balance between these effects is critical (as with hypoxia). Tumour cells can also use adhesion molecules, chemokines and receptors to enhance migration and metastasis to distant tissues.
Cancers associated with inflammation include those caused by viruses such as human papilloma virus, hepa titis B virus and Epstein Barr virus. Gastric carcinoma is associated with infection by Helicobacter pylori and peptic ulcers. Cancers are also associated with many chronic inflammatory conditions, e.g. colon cancer with ulcerative colitis and Crohn’s disease, and liver carcinoma with hepatitis C.
The best evidence for association between inflammation and cancers is that long-term use of non-steroidal antiinflammatory drugs (NSAIDs) is protective against some cancers and may reduce risk of colon cancers by up to 50%. The mechanism of this protection may be through inhibition of COX enzymes and hence reduced production of inflammatory mediators, and also through induction of apoptosis and stimulation of immune surveillance. This use of NSAIDs is being studied in attempts to reduce risk of other cancers (see review by Coussens and Werb [2002]).
Staging of tumours
Tumours are not homogeneous, even tumours of the same cell type in the same organ. They may vary in cell-cycling time, the proportion of cells cycling, their vascularity, susceptibility or resistance to the actions of particular drugs, and their size and extent of spread. Tumours are commonly ‘staged’ by the ‘TNM’ method to indicate the size of the primary tumour and extent of spread to lymph nodes and distant metastases. In some cases, the growth of the tumour can be monitored by measuring levels of a (relatively) specific marker, such as prostate specific antigen (PSA) for prostate cancers or monoclonal immunoglobulins for multiple myeloma.
Treatment of cancers
Treatment modalities
The three main treatment modalities in cancer are surgery, irradiation and drugs (and, in slowly growing cancers, ‘watchful waiting’). If it is possible to remove all of a cancer surgically, this is obviously the first choice and may be curative. When this is not possible (e.g. with a large inoperable tumour in a vital organ, a tumour that is difficult to access or non-solid tumours such as leukaemias or lymphomas), then irradiation and/or drugs are used. Other adjuvant therapies include immunotherapy to enhance the immune response against neoplastic cells; drug treatment of adverse effects and related conditions such as pain, depression or anxiety; palliative care and complementary and alternative therapies such as massage, behavioural therapy and counselling.
Radiotherapy
Radiation therapy uses X-rays, targeted to minimise damage to non-cancer cells, or radiopharmaceuticals, which are drugs or chemicals containing radioactive isotopes emitting gamma rays that damage cells. Radiation therapy has been shown to be important in reducing the risk of metastases (e.g. of leukaemias to the head and neck areas).
Other radiopharmaceuticals, used in radiographic diagnostic procedures rather than treatment of cancers, are those containing the isotopes technetium-99, gallium-67 and thallium-201 (see Clinical Interest Box 41-3).
Clinical interest Box 41-3 Radiopharmaceuticals from ansto
In Australia there is only one nuclear reactor, operated by the Australian Nuclear Science and Technology Organisation (ANSTO) at its facility at Lucas Heights, south of Sydney (see http://www.ansto.gov.au/). ANSTO is the centre of Australia’s nuclear science capabilities, offering a wide range of scientific and technical services and expertise to governments, medical research, treatment facilities and industry groups in Australia and around the world.
Radiopharmaceuticals prepared and distributed from ANSTO include products labelled with isotopes chromium-51, gallium-67, iodine-123 and iodine-131, molybdenum-99, technetium-99 and thallium-201, plus the associated generators, elution pots and vials.
A new plant to produce molybdenum-99 is being commissioned; molybdenum-99 is used in 80% of nuclear medicine procedures performed around the world. It is formed by the fission of uranium-235 formed by irradiation of low enriched uranium (LEU) in ANSTO’s reactor. The molybdenum-99 is used to make technetium-99, which in turn is placed in small ‘generators’. The product technetium-99 is ‘milked’ into a sterile saline solution that can then be injected into patients for radio-imaging techniques, in which a computer-enhanced image provides information about the patient’s internal organs.
Radioactive iodine, formulated as sodium iodide-I-131, is used to treat thyroid cancers by its ionising radiation producing γ- and β-rays (see Drug Monograph 34-3). It is produced at ANSTO in a form >99% pure, and is calibrated at 0900 hours on Mondays, then despatched on the same day as ordered. As the radioactive half-life is only 8.04 days, it must be used soon after arrival in the receiving nuclear medicine facility; the product expires 14 days after calibration. The dose must be calculated using the radioactive decay curve, to determine the actual amount of radioactivity left in the sample at the time of administration.
This is a highly specialised area of hospital pharmacy, and is carried out only in hospitals with expert staff and adequate facilities for safe dispensing of radioactive products, such as lead shielding of dedicated cabinets, lead aprons for staff and regular monitoring of body and laboratory radioactivity.
Chemotherapy
Drugs used in therapy of cancer may include antineoplastic agents, hormones, immunostimulating agents, radiopharmaceuticals, specific drugs to treat adverse effects of anticancer drugs and other adjuncts. However, the term chemotherapy4 is generally used in the more limited sense to refer to drugs used specifically to treat cancers, i.e. antineoplastic agents. (Details of drugs are given in Chapter 42.)
Principles of cancer chemotherapy
Selective toxicity
Chemotherapy is defined as the clinical use of drugs that, in low concentrations, inhibit the growth of neoplastic (or microbial) cells. Chemotherapy usually relies on finding and exploiting a difference in the biochemical pathways between the normal cell and the neoplastic cell (or host cell and the microbial cell); this is referred to as selective toxicity.
Drug delivery
Cancer chemotherapy is most effective against small tumours because they usually have an efficient blood supply and therefore drug delivery to the cancer site is increased. Also, small tumours generally have a higher percentage of proliferating cells against which the antineoplastic drug will act. Specialised techniques may be used to deliver the drug directly to the cancer site, e.g. intra-arterial injection, or use of pro-drugs that are activated preferentially in neoplastic cells.
Cell-kill fraction
Studies in animals with leukaemias and lymphomas have shown that chemotherapeutic drugs given in adequate doses tend to kill a constant proportion of the cancer cells; this is known as the cell-kill fraction. A drug or drug combination capable of killing 99.9% of the cells would leave 0.1% surviving and thus would only reduce a 1010 cell burden to 107 cancer cells (the smallest detectable tumour, at an early stage when clinical symptoms first appear, might weigh approximately 1 g and contain about 109 cells). Hence several courses of chemotherapy may be required before the cancer cell burden is very low.
Growth fraction
Solid tumours can be comprised of both actively cycling and non-cycling cell populations; this is proved by radioisotope labelling experiments, which show that the fraction of labelled mitotic cells always exceeds that of interphase cells. The term ‘growth fraction’ was coined to distinguish the cell population, in the tumour, that is actively engaged in cell cycling from the fraction that is not (i.e. cells in G0 phase). Tumours with a high growth fraction are relatively more susceptible to treatment with antineoplastic agents.5
Courses of treatment
Each course of chemo- or radiation therapy may reduce cancer cells to levels that can eventually be controlled by the patient’s immune system (Figure 41-4). This reduction may produce an apparent remission (the patient gets well again) but, if further therapies are not instituted or the immune system response is inadequate, the remaining cells may multiply and grow into another detectable tumour, i.e. cause recurrence and relapse. The aim of each cycle of cancer therapy is therefore to achieve total cancer cell kill if possible.
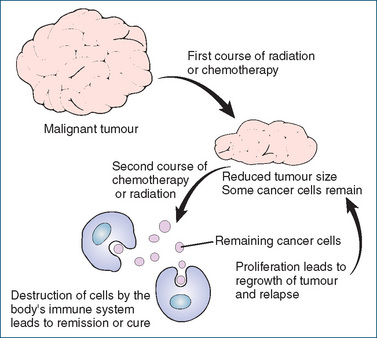
Figure 41-4 Response of cancer cells to therapy. The aim of the initial treatment (surgery, radiation and/or chemotherapy) is to reduce the cancer cell burden sufficiently low that the body’s immune system can deal with the remaining cancer cells.
Adapted from: Beare & Myers 1998.
Adjuvant therapy
Removing large localised tumours by surgery reduces the tumour cell burden. The subsequent use of chemotherapy is to help eradicate the remaining cancer cells and micrometastases (the migration of cancer cells via the bloodstream or lymphatic system to grow in organs, bone or tissues far from the primary site) of cancer after surgery or irradiation. Other adjuvant drugs include immunostimulants, analgesics, laxatives, antibiotics etc.
Cell cycle specificity
The cytotoxic agents have different sites of action on the dividing cell cycle (see Figure 41-1), but they all inhibit cell replication and are thus antiproliferative. Agents that are most effective in one specific phase are referred to as cellcycle- phase-specific agents. Antimetabolites, for example, are most active impairing DNA synthesis in the S-phase of the cell cycle, so these drugs are considered S-phase cellcycle- specific agents. The podophyllotoxins, which inhibit topoisomerase II activity, are S- and G2-phase-specific; the topoisomerase I inhibitors, the -tecans, are S-phasespecific. The vinca alkaloids generally act against mitosis and so are M-phase-specific.
Some antineoplastic agents that act on cycling cells, but not on a specific phase, are called cell-cycle-specific agents; examples are most alkylating agents and some antibiotics, which may inhibit at many sites such as DNA, RNA and protein synthesis.
Antineoplastic agents that are active against both proliferating and resting cells are called cell-cycle-nonspecific agents. The platinum compounds, some alkylating agents (the nitrosoureas), the anthracycline antibiotics and agents that enhance apoptosis are examples of this group (see Table 42-1).
Antineoplastic classifications are an important consideration in selecting the appropriate drug or drugs for a specific cancerous state. Methotrexate, for example, an agent active predominantly in the S-phase of the cell cycle, is not very effective in treating large tumour masses, which generally have slowly dividing cancer cells. Some oncologists consider it important to select and combine chemotherapeutic agents that act at different phases, in order to have synergistic effects and to reduce the development of drug resistance. (However, these terms tend not to be used as frequently now as formerly, perhaps because drugs with very specific mechanisms of action targeted against particular enzymes or growth factors are being developed.)
Types of antineoplastic agents
Cytotoxic agents
The formation of macromolecules (the nucleic acids DNA and RNA and, ultimately, proteins) requires pyrimidines and purines (nitrogen-containing ‘bases’) as the buildingblock materials for nucleic acids (see Figure 41-3). Antineoplastic agents known as cytotoxic agents (drugs that are toxic to cells, specifically used to kill cancer cells) act by inhibiting the synthesis of macromolecules. Some drugs have more than one specific site of action.
Antimetabolites have structures similar to those of necessary cofactors or building blocks for the formation of DNA, e.g. similar to folic acid or to a purine or pyrimidine base. This ‘false building block’ is accepted by the enzyme or cell as the necessary ingredient for cell growth but, because it is an impostor, it interferes with the normal production of DNA. Examples are methotrexate (Drug Monograph 42-2), fluorouracil and mercaptopurine.
Alkylating agents are drugs that substitute an alkyl chemical group for a hydrogen atom in DNA. This results in a cross-linking between strands of DNA, preventing cell division. Alkylator-like drugs may also cross-link DNA similar to the action of alkylating agents. Examples are cyclophosphamide (Drug Monograph 42-1), lomustine and dacarbazine.
Antitumour antibiotics are naturally occurring agents that interfere with DNA functioning by blocking the transcription of DNA to RNA. In addition, they may delay or inhibit mitosis. Examples are bleomycin and dactinomycin.
Mitotic inhibitors, such as the vinca alkaloids or the taxanes, are plant alkaloids that block cell division in metaphase. Examples are vincristine and paclitaxel.
Hormone anticancer agents
The growth of some tumours is dependent on the stimulation of neoplastic cells by particular hormones. For example, breast cancer is stimulated by oestrogens, prostate cancer by androgens and thyroid cancer by thyroidstimulating hormone. These cancers may be effectively suppressed by antihormones (hormones having opposite effects to those stimulating the tumour, such as tamoxifen, an anti-oestrogen), by drugs that suppress synthesis or secretion of the hormone or by surgical removal or irradiation of the gland producing the hormone.
Other antineoplastic agents
Some miscellaneous antineoplastics do not conveniently fit into only one classification of mechanism of action, e.g. platinum compounds, topisomerase inhibitors, the enzyme crisantaspase (aka asparaginase), the hydroxyureas, tyrosine kinase inhibitors (e.g. imatinib, Drug Monograph 42-4), DNA-intercalating agents and interferons.
Potential new drugs and targets
Relatively new agents, and those currently under investigation and development, include chemicals and processes that target specific sites in the cell cycle and in tumour cell functions (described earlier), such as:
Clinical aspects
Cure and remission
In the context of cancer, the term ‘cure’ is used rather differently from the way it is understood in acute conditions that can be totally eradicated. Cancer cure may be defined as the disappearance of any evidence of tumour for several years, with a high probability of a normal lifespan. This definition, referring as it does to a statistical probability, implies the difficulty of removing every malignant cell and recognises the possibility of recurrence of cancer cell proliferation.
Patients are said to go into remission if the tumour and signs and symptoms of its presence are no longer detectable, whereas if the patient relapses, the tumour and/or its signs and symptoms are again detectable. Hence there may be remission after a course of chemotherapy, then a relapse some months or years later, and further remission after another course of therapy (see again Figure 41-4).
Tumours with a high growth fraction are most responsive to chemotherapy, as most of their cells are undergoing active cycling and can therefore be targeted by drugs that impair macromolecular synthesis or cell division. Examples are choriocarcinoma, a malignant tumour of fetal origin that is usually sensitive to methotrexate or dactinomycin, and Burkitt’s lymphoma, usually sensitive to cyclophosphamide. Other tumours, including large solid tumours of internal organs such as lung, kidney and colon, may be more difficult to treat, although antineoplastic drugs may prolong and improve the quality of life.
Treatment regimens
As cancer treatment is a highly specialised and rapidly advancing area of medicine, patients are usually treated in a specialised oncology (cancer) unit or clinic, attached to a major hospital. Patients are often enrolled in clinical trials, as new drugs are continually being developed and can best be trialled in patients with the specific condition. Multicentre trials are usually coordinated from one large cancer institute but with investigators, physicians and patients in many centres around the world. This increases the number of patients enrolling in trials and, it is hoped, shortens the time taken for results (safety and efficacy of the new treatment) to become apparent. For some cancers (hepatoma, renal cell carcinoma) there may be no definitive optimal therapies, and patients are given adjuvant therapy and enrolled in clinical trials of new agents as appropriate.
Treatment should always be individualised to the patient, with careful monitoring of efficacy and toxicity of therapies administered. The aims of the treatment (curative or palliative) need to be discussed with the patient (see Clinical Interest Box 41-4), and adjuvant therapies added as required (e.g. analgesics, laxatives, antimicrobials, antianxiety agents and sedatives).
Clinical interest Box 41-4 What patients want to know
The information patients want or need to know about any prescribed drug can be summed up in three questions. In the context of antineoplastic agents, the following sample answers help provide information and counselling.
Combination chemotherapy
In the late 1960s, combination chemotherapy, the use of two or more anticancer drugs at the same time, was initiated for treating acute lymphoblastic leukaemia and Hodgkin’s disease. When the complete response rates for single agents were compared with those for combination drugs, it was evident that drugs can have synergistic therapeutic effects, with less development of resistance. In general, combinations of cancer chemotherapeutic agents help bring about prolonged survival times, due to their higher cancer cell-kill fraction than treatment with a single drug agent, and specific drug toxicities may be reduced. Table 41-1 lists some commonly prescribed drug combinations. The toxicity of anticancer drugs needs to be anticipated and, if possible, organs need to be protected and treatment for adverse effects administered.
Table 41-1 Examples of combination chemotherapeutic regimens
| Cancer | Acronym | Drugs |
| Breast* | CMF(P) | Cyclophosphamide–methotrexate–fluorouracil (+/− prednisolone) |
| CFPT | Cyclophosphamide–5-fluorouracil–prednisone–tamoxifen | |
| Colon | FOLFOX4 | Leucovorin (folinic acid)–5-fluorouracil (2 dose levels)–oxaliplatin |
| Lung | CAV | Cyclophosphamide–adriamycin (doxorubicin)–vincristine |
| CE | Cyclophosphamide–etoposide | |
| Stomach | ECF | Epirubicin–cisplatin–5-fluorouracil |
* Note: Chemotherapy of breast cancer is complicated, and choice of drug(s) depends on whether the woman is pre- or postmenopausal, whether or not there is lymph node involvement, what histological cell type is involved, whether the tumour tests positive or negative for oestrogen receptors and progesterone receptors, whether the woman has undergone mastectomy or lumpectomy, and whether there are any cardiovascular risk factors.
Treatment regimens are often complicated, with patients being administered 2–4 cytotoxic agents on various days for 2 weeks, followed by a drug-free period (to allow white cells to recover), then another course of therapy, possibly with surgery and/or radiation therapy (see Clinical Interest Box 41-5). Regimens recommended have been designed for optimum efficacy in specific cancers and take into account potential drug interactions. Adjuvant drugs may also be given to treat adverse effects, and fluids administered to rehydrate the patient and ‘flush out’ the kidneys, where cytotoxic agents may concentrate.
Clinical interest Box 41-5 Chemotherapy regimens for stomach and colon cancers
A typical regimen of chemotherapy for advanced stomach cancer is ECF:
ECF is recommended as first-line therapy for patients with metastatic disease. Patients with poor performance status or other contraindication to intensive therapy may be considered for 5-fluorouracil alone.
For colon cancer advanced disease, FOLFOX6 is recommended:
Adjuvant chemotherapy is generally recommended for patients with Stage III colorectal cancer. It may also be considered in patients with high-risk Stage II disease on an individual basis.
The MOSAIC study showed an improved 3-year survival with the addition of oxaliplatin to LV5FU2 (FOLFOX4) as compared to LV5FU2 (a bolus + infusional 5FU/LV regimen) alone.
Personal communication: with thanks to Associate Professor Dr Michael Michael, Division of Haematology and Medical Oncology, Peter MacCallum Cancer Centre, Melbourne 2009; see also MOSAIC study group: Andre et al (2006).
The following principles are used to select the drugs for combination chemotherapy:
The response rates for the treatment of advanced Hodgkin’s disease with mustine, Oncovin (vincristine), procarbazine and prednisone (MOPP) are a good illustration:
| Drug | Complete response rates |
| M (mustine) | 20% |
| O (Oncovin [vincristine]) | <10% |
| P (procarbazine) | <10% |
| P (prednisone) | <5% |
| MOPP combination | 80% |
When the principles are applied to MOPP drug therapy, it can be seen that each drug is effective against Hodgkin’s disease, the sites of major activity for each antineoplastic agent are believed to be different, and both cell-cyclespecific and cell-cycle-non-specific agents are included.
Mustine is an alkylating agent that can interfere with the replication, transcription and translation of DNA. Vincristine inhibits mitosis by interfering with the mitotic spindle. Procarbazine inhibits the synthesis of DNA, RNA and protein and also interferes with mitosis; its antineoplastic action is believed to occur during the S-phase (it is also a weak monoamine oxidase inhibitor and central nervous system depressant). Prednisone has lympholytic properties and so is useful in white-cell tumours, and may produce an antifibrotic effect that would be useful in treating cancer metastases surrounded by fibrous materials. It also improves appetite and general feelings of wellbeing.
The fourth principle, that of different organ toxicity or toxicities that occur at different times, has also been substantiated for the MOPP combination. The doselimiting toxicity of bone marrow suppression is a property of both mustine and procarbazine, but the nadir (the lowest depression point for this effect) occurs about 10 days after drug administration for mustine and 21 days after for procarbazine. The additive myelosuppressant effects from this combination are thus essentially avoided. Also, vincristine does not have bone marrow-suppressing effects but does exhibit a dose-limiting neurotoxicity. Prednisone does not demonstrate neurotoxicity.
Dosage
Due to the low therapeutic index of most anticancer drugs, there may be a fine line between minimum effective dose and maximum tolerated dose, so determining the correct dosage for cytotoxic agents presents a challenge. Several methods have been used.
By body surface area
Potent and potentially toxic anticancer drugs are frequently dosed in units of mg drug/m2body surface area (see explanation in Chapter 8) rather than the more common mg drug/kg body weight. The surface area of cells in the target tumour (across the membranes of which antineoplastic drugs would be absorbed) presumably relates more closely to the patient’s body surface area than it does to the patient’s weight.6 However, large interpatient variabilities in drug clearance and response still occur.
By maximum tolerated dose (MTD)
Traditionally, the dose of cytotoxic agents has been considered to be the maximum amount that a patient could take before intolerable adverse effects occurred, such as life-threatening depression of the bone marrow or unbearable vomiting, and ‘dose-limiting toxicities’ have been quoted. However, with the advent of highly targeted drugs, such as growth factor antibodies or cyclin-dependent tyrosine kinase inhibitors, the MTD may be substantially greater than the required therapeutic dose, i.e. lower doses may be effective without reaching intolerable adverse effects. The ‘minimum effective dose and duration’ may be a better guideline (see Olver and Haines [2009]).
By pharmacogenetic and -genomic differences
Genetic differences in drug-metabolising enzymes are being studied in an effort to explain pharmacokinetic and -dynamic variabilities between anticancer drugs. Pharmacogenetic studies can determine differences in specific metabolising enzymes, and thus allow personalisation of dosing of particular drugs; for example, levels of thiopurine methyltransferase (TPMT) can predict the metabolism and potential toxicity of mercaptopurine in acute lymphoblastic leukaemia (ALL) in children. Broader-based pharmacogenomic studies may elucidate the significance of specific genes and their expression in the disease; several sub-types of ALL have been identified, and their genetic translocations and T-cell phenotypes, which help determine prognosis and optimise treatment. (See reviews by Cheok, Lugthart and Evans [2006] and Walko and McLeod [2009].)
Adverse drug reactions
Drug toxicities or adverse drug reactions (ADRs) may be divided into adverse reactions common to antineoplastic agents generally and dose-limiting effects specific to particular drugs. A dose-limiting effect is a response to a drug that indicates that the maximum tolerable dose has been reached and that the drug dose should be decreased or discontinued (but see also discussion above).
Antiproliferative effects
For most of the currently available antineoplastic agents, the biochemical mechanism on which the drug acts is basically similar in both normal and malignant cells. This lack of tumour-cell specificity is a major limitation of cancer drugs. Because the target of most cytotoxic drugs is cell proliferation, it follows that normal tissues in the body are also vulnerable when their cells are proliferating. The most rapidly dividing cells in the body, which are in the bone marrow, hair follicles, skin and gastrointestinal tract, are therefore generally most adversely affected by anticancer drugs. The commonest adverse effects are alopecia (hair loss), gastrointestinal distress (nausea, vomiting, anorexia, diarrhoea, ulceration) and mucositis and stomatitis (inflammation of the mucous membranes of the gastrointestinal tract and mouth).
Long-term toxicities that need to be considered and monitored follow from cytotoxic agents impairing various stages in cell division and proliferation; they may thus cause infertility or possibly be mutagenic, carcinogenic or teratogenic. Patients need to be warned of these potential dangers, as do health professionals handling the drugs or patients’ excreta, since the drugs may be absorbed through the skin (see Clinical Interest Box 41-6).
Clinical interest Box 41-6 Safe handling of cytotoxic agents
Cytotoxic agents are inherently toxic and, if lipid-soluble, may be readily absorbed through the skin of people handling the drugs or coming into contact with the excreta and vomitus of patients who have been administered such agents. Consequently, safe handling guidelines have been developed to protect pharmacists, doctors and nurses and also patients’ families and carers. Suggested precautions include:
Adapted from: the Peter MacCallum Cancer Institute 1999.
 Nausea and vomiting
Nausea and vomiting
The nausea and vomiting can be so severe as to become dangerous and discourage the patient from continuing with chemotherapy (see Table 41-2); for those with high emetogenic potential, potent antiemetic drugs such as dexamethasone plus a 5-HT3 antagonist (e.g. ondansetron) or aprepitant may be required for both prophylaxis and relief.
Table 41-2 Emetic potentials of some chemotherapeutic agents
| High emetic potential | Cisplatin, cyclophosphamide (IV), dacarbazine (DTIC), dactinomycin, nitrogen mustards, nitrosoureas (BCNU, CCNU), streptozotocin |
| Intermediate emetic potential | Carboplatin, cyclophosphamide (oral), daunorubicin, doxorubicin, etoposide, gemcitabine, ifosfamide, irinotecan, methotrexate (>250 mg/m2), mitomycin, teniposide, topotecan |
| Low emetic potential | Busulfan, chlorambucil, fluorouracil, 6-mercaptopurine, methotrexate (<250 mg/m2), bleomycin, monoclonal antibodies, taxanes, tyrosine kinase inhibitors, vinblastine, vincristine |
BCNU = 1,3-bis-(2-chloroethyl)-1-nitrosurea;
CCNU = N-(2-chloroethyl)-N’-cyclohexyl-N-nitrosurea;
DTIC = dimethyltriazenyl imidazole carboxamide, aka dacarbazine.
Note that emetic potential may depend on doses administered.
Adapted from: Peter MacCallum Cancer Institute Melbourne 2000; AMH 2010.
Myelosuppression
Bone marrow suppression (myelosuppression) is the major dose-limiting adverse reaction most often encountered in cancer chemotherapy: suppression of white cells (leucopenia or neutropenia) or of platelets (thrombo cytopenia) can lead to serious and even life-threatening infections and haemorrhage, respectively. For this reason, cytotoxic agents are usually dosed to the limit of tolerance, with subsequent drug-free periods to allow recovery of white cell functions.
Other ADRs
Drugs that can produce hepatotoxicity include methotrexate, mercaptopurine, lomustine and carmustine (CCNU, BCNU), dacarbazine and doxorubicin (Adriamycin).
Renal problems can occur from the high levels of these toxic agents that can accumulate in the kidney and renal tubules during drug excretion. Because dehydration increases the likelihood of high concentrations of the drugs in the renal tubules, adequate fluid intake is important when these agents are administered. Haemorrhagic cystitis is associated with cyclophosphamide, and renal tubular necrosis with methotrexate, cisplatin and related drugs.
Cardiac toxicity is reported with both doxorubicin and daunorubicin. Cardiotoxicity increases in patients who receive more than 550 mg/m2 body surface (total accumulated dosage given throughout therapy). Toxicity is also greater in elderly patients and in children under two years. As this effect is cumulative if either drug is given, the amount of one drug already received by the person must be considered when planning therapy with the other drug.
Neurological toxicity may range from tingling of the hands and feet and loss of deep tendon reflexes to ataxia, footdrop, confusion and personality changes. Drugs reported to produce neurological effects include vincristine and the other vinca alkaloids and methotrexate. Ototoxicity and peripheral neuropathy have been reported with cisplatin and related compounds.
Tumour lysis syndrome refers to the massive release of breakdown products from tumour cells killed by chemotherapeutic agents; it occurs most commonly in leukaemias and lymphomas. In particular, urate may accumulate and precipitate in the renal distal tubules and in joints; to prevent this occurring, the urine may be alkalinised and/or hydration increased and allopurinol administered prophylactically. Hyperkalaemia, hyperphosphataemia and hypocalcaemia also occur.
Hand–foot syndrome (palmar–plantar erythrodysaesthesia) is associated with high doses of a variety of agents, including capecitabine, doxorubicin, docetaxel and sunitinib. There may be altered sensations in the hands and feet, with swelling, pain, desquamation and blistering. Cold compresses and anti-inflammatory drugs have been tried in prevention and treatment.
Large molecule drugs such as the monoclonal antibodies (immunoglobulins) can elicit immune reactions and infusion reactions. Hormones and antihormones are likely to cause their specific adverse reactions, e.g. oestrogenic effects or impotence.
Other general adverse effects include fever, extravasation (dispersal of IV solution out of blood vessels into the tissues, with subsequent necrosis reactions) and delayed healing.
Treatment of ADRs
Most adverse effects are treated symptomatically, e.g. nausea with antiemetics, infections with appropriate antimicrobial agents, mouth ulcers with mouth washes and zinc solutions and fever with antipyretic analgesics. Many patients find the loss of hair most distressing; this can be helped with sensitive encouragement and use of hairpieces, wigs and scarves; usually the hair regrows after cessation of cytotoxic chemotherapy. (Adjunctive agents used as more specific treatments of adverse drug reactions are discussed in Chapter 42.)
Development of drug resistance
Just as microorganisms can develop resistance to antimicrobial drugs (see Figure 43-1 and related text) and pass the genes for drug resistance to other organisms, leading, for example, to the spread of multi-resistant strains of Staphylococcus aureus, so neoplastic cells can develop resistance to anticancer drugs and pass this acquired characteristic on to daughter cells. The cancer then becomes resistant to specific drugs, leading to declining therapeutic benefits in successive treatment cycles.
Mechanisms of drug resistance
Some of the mechanisms by which cells develop resistance are:
Overcoming drug resistance
As with antimicrobial drug therapy, optimising anticancer drug usage will help to minimise development of drug resistance. Methods include use of the most specific drugs available to target the particular neoplastic cell, use of chemotherapeutic agents in rotation or cyclic patterns and use of combinations of drugs acting at different stages of the cell cycle. Inhibitors of drug-efflux pumps are an emerging group of potentially useful agents; strategies to overcome cells’ resistance to apoptosis are also being studied. Functional genomics studies will help identify target pathways in particular patients in order to optimise therapy (see review by O’Connor [2009]).
Age-related considerations
Cancer in children
Cancer in children is relatively uncommon, with fewer than 1% of new cancers occurring in children under 15 years (in Victoria, Australia).7 The commonest cancers occurring in children under 15 in Victoria are leukaemias and lymphomas, then brain and central nervous system cancers. Carcinomas (malignant tumours of epithelial tissues such as breast, gastrointestinal tract, lung and skin), which are common in adults, are rare in children, while sarcomas (malignant tumours of connective tissues such as bone and muscle) are more common in children than adults. Because tumours in children grow rapidly, childhood cancer is generally more responsive to chemotherapy than is cancer in an adult. Children also tend to tolerate the acute adverse effects of chemotherapy better than adults. Of all children with cancer, 50% become long-term survivors or are actually cured.
Cancer in those of reproductive age
Antineoplastic agents may have deleterious effects on reproduction, at different stages of life; factors to be considered carefully are as follows:
Cancer in older adults
The incidence of cancer increases sharply with age, with about 70% of new cancers occurring in people aged 60 and over (see also Clinical Interest Box 41-1). Cancer risk is clearly related to duration of exposure to exogenous carcinogens, such as radiation or chemicals, or endogenous mutations; hence older people have a greater chance of having accumulated the critical number of mutations required for cell transformation and therefore of cancer (see Clinical Interest Box 41-7).
Clinical interest Box 41-7 Prevention is better than cure
While predisposition to some cancers is inherited, and thus unavoidable, there are many controllable environmental and lifestyle factors and screening technologies that can reduce the risk of development of cancers. Some milestones in development of possible prevention methods are:
Source: Lippman and Hawk, 2009inter alia.
Elderly people have more concurrent illnesses and less efficient homeostatic mechanisms than younger cancer victims, which may decrease their ability to withstand the effects of cancer or of antineoplastic therapies. Other factors to be considered when managing regimens for elderly persons are the possibility of impaired liver or kidney functions that can reduce drug clearance, concurrent administration of many other drugs (polypharmacy), reduced independence and income and loss of friends and family support.
Often, compromises in treatment are made because of a person’s advanced age; however, data suggest that a dosage reduction of chemotherapy based on age alone is not always appropriate and that the efficacy of chemotherapy is not age-dependent. A treatment approach should be based on the individual cancer and the physiological parameters noted in the elderly person (see review by Balducci [2009]). Because new drugs are usually not tested in children or elderly persons, more clinical trials are needed to examine the relation between cancer chemotherapy responsiveness and the person’s age.
Key points


 Tumours are a common cause of morbidity and mortality in the community. Cells in malignant tumours are usually rapidly dividing, dedifferentiated and have the ability to invade and metastasise. Research in tumour cell biology is elucidating the detailed biochemical pathways involved in cell cycling, regulation of cycling, checkpoints, stimulators and inhibitors of cell growth, tumour suppressor genes, synthesis of macromolecules, duplication of genetic material and apoptosis. In the development of cancers, normal cells may be transformed to neoplastic cells by viruses, carcinogenic chemicals, genetic influences, radiation or oncogenes and their products. Small tumours and those undergoing rapid cycling are most responsive to treatment. The main modalities for cancer treatment are surgery, radiation and chemotherapy. In many cases, cure or remission is possible; however, cancers may relapse. Chemicals targeting specific enzymes, proteins, receptors, growth factors, genes and cytokines in relevant biochemical pathways may prove useful as new anticancer drugs. The cytotoxic agents used in chemotherapy generally have antiproliferative effects by impairing macromolecular synthesis (antimetabolites, alkylating agents, antitumour antibiotics) or by disrupting mitosis (vinca alkaloids, taxanes). Tumours that depend on hormones for growth may be treated with antihormones or by suppressing secretion of the trophic hormone. Chemotherapeutic drug regimens usually combine cytotoxic agents acting by different mechanisms at different phases of the cell cycle, with different specific adverse effects; this minimises toxicity and the development of drug resistance. Typical ‘chemo’ regimens involve several courses of therapy, with drug-free weeks to allow bone marrow recovery. Adverse drug reactions common to cytotoxic agents include damage to other rapidly dividing cells (bone marrow, hair, skin, gastrointestinal mucosa), severe nausea and vomiting, kidney tubule damage and hyperuricaemia. Treatment of these adverse effects involves use of specific drugs and hydration with IV fluids.
Tumours are a common cause of morbidity and mortality in the community. Cells in malignant tumours are usually rapidly dividing, dedifferentiated and have the ability to invade and metastasise. Research in tumour cell biology is elucidating the detailed biochemical pathways involved in cell cycling, regulation of cycling, checkpoints, stimulators and inhibitors of cell growth, tumour suppressor genes, synthesis of macromolecules, duplication of genetic material and apoptosis. In the development of cancers, normal cells may be transformed to neoplastic cells by viruses, carcinogenic chemicals, genetic influences, radiation or oncogenes and their products. Small tumours and those undergoing rapid cycling are most responsive to treatment. The main modalities for cancer treatment are surgery, radiation and chemotherapy. In many cases, cure or remission is possible; however, cancers may relapse. Chemicals targeting specific enzymes, proteins, receptors, growth factors, genes and cytokines in relevant biochemical pathways may prove useful as new anticancer drugs. The cytotoxic agents used in chemotherapy generally have antiproliferative effects by impairing macromolecular synthesis (antimetabolites, alkylating agents, antitumour antibiotics) or by disrupting mitosis (vinca alkaloids, taxanes). Tumours that depend on hormones for growth may be treated with antihormones or by suppressing secretion of the trophic hormone. Chemotherapeutic drug regimens usually combine cytotoxic agents acting by different mechanisms at different phases of the cell cycle, with different specific adverse effects; this minimises toxicity and the development of drug resistance. Typical ‘chemo’ regimens involve several courses of therapy, with drug-free weeks to allow bone marrow recovery. Adverse drug reactions common to cytotoxic agents include damage to other rapidly dividing cells (bone marrow, hair, skin, gastrointestinal mucosa), severe nausea and vomiting, kidney tubule damage and hyperuricaemia. Treatment of these adverse effects involves use of specific drugs and hydration with IV fluids.Review exercises
Andre T., Tournigand C., Achille E., et al. Adjuvant treatment of colon cancer MOSAIC study’s main results. Bulletin du Cancer. 2006;93(Suppl 1):S5-S9.
Australian Institute of Health and Welfare (AIHW) & Australasian Association of Cancer Registries (ACCR). Cancer in Australia: An Overview 2008. AIHW cat. no. CAN 42. Canberra: AIHW (Cancer Series No. 46),;1; 2008. Available: http://www.aihw.gov.au/publications/index.cfm/title/10607 [29 October 2009].
Australian Medicines Handbook 2010. Adelaide, AMH, 2010.
Balducci L. Supportive care in elderly cancer patients. Current Opinion in Oncology. 2009;21(4):310-317.
Beare P.G., Myers J.L. Adult Health Nursing, 3rd edn. St Louis: Mosby; 1998.
Cavalli F., Hansen H.H., Kaye S.B., editors. Textbook of Medical Oncology, 3rd edn., London: Taylor & Francis, 2004.
Cheok M.H., Evans W.E. Acute lymphoblastic leukemia: a model for the pharmacogenomics of cancer therapy. Nature Reviews: Cancer Feb. 2006;6:117-129.
Cheok M.H., Lugthart S., Evans W.E. Pharmacogenomics of acute leukemia. Annual Review of Pharmacology & Toxicology. 2006;46:317-353.
Consultative Council on Obstetric and Paediatric Mortality Morbidity. Annual Report for the Year 2004. Melbourne: Victorian Department of Human Services; 2005.
Coussens L.M., Werb Z. Inflammation and cancer. Nature. 2002;420(6917):860-867.
Dawkins R. The Ancestor’s Tale. London: Phoenix; 2004.
Grimm D., Bauer J., Schoenberger J. Blockade of neoangiogenesis, a new and promising technique to control the growth of malignant tumors and their metastases. Current Vascular Pharmacology. 2009;7(3):347-357.
Harris A.L. Hypoxia: a key regulatory factor in tumour growth. Nature Reviews: Cancer. 2002;2(1):38-47.
Irigaray P., Newby J.A., Clapp R., et al. Lifestyle-related factors and environmental agents causing cancer: an overview. Biomedicine & Pharmacotherapy. 2007;61:640-658.
Javier R.T., Butel J.S. The history of tumor virology. Cancer Research. 2008;68(19):7693-7706.
Johansson M., Persson J.L. Cancer therapy: targeting cell cycle regulators. Current Medicinal Chemistry: Anti-Cancer Agents. 2008;8(7):723-731.
Kitigawa K., Kotake Y., Kitagawa M. Ubiquitin-mediated control of oncogene and tumor suppressor gene products. Cancer Science. 2009;100(8):1374-1381.
Le Fanu J. The Rise and Fall of Modern Medicine. London: Little, Brown; 1999.
Lippman S.M., Hawk E.T. Cancer prevention: from 1727 to milestones of the past 100 years. Cancer Research. 2009;69(13):5269-5284.
Liu W., Zhou Y., Reske S.N., Shen C. PTEN mutation: many birds with one stone in tumorigenesis. Anticancer Research. 2008;28(6A):3613-3619.
Lodish H., Berk A., Kaiser C.A., et al. Molecular Cell Biology, 6th edn. New York: Freeman; 2008.
Malumbres M., Barbacid M. Cell cycle. CDKs and cancer: a changing paradigm. Nature Reviews: Cancer. 2009;9(3):153-166.
O’Connor R. A review of mechanisms of circumvention and modulation of chemotherapeutic drug resistance. Current Cancer Drug Targets. 2009;9(3):273-280.
Olver I.N., Haines I.E. What changes are needed to the current direction and interpretation of clinical cancer research to meet the needs of the 21st century? Medical Journal of Australia. 2009;190(2):74-77.
Palliative Care Expert Group. Therapeutic Guidelines: Palliative Care, version 3. Melbourne: Therapeutic Guidelines; 2010.
Pawlik T.M., Keyomarsi K. Role of cell cycle in mediating sensitivity to radiotherapy. International Journal of Radiation Oncology, Biology, Physics. 2004;59(4):928-942.
Peter MacCallum Cancer Institute. Guidelines for the Safe Handling of Cytotoxic Agents, 5th edn. Melbourne: Peter MacCallum Cancer Institute; 1999.
Peter MacCallum Cancer Institute. Standard Treatment Guidelines, January 2000. Melbourne: Medical Oncology Unit; 2000.
Peto J. Cancer epidemiology in the last century and the next decade. Nature. 2001;411:390-395.
Souhami R., Tobias J. Cancer and its Management, 5th edn. Malden, MA: Blackwell; 2005.
Stern M., Herrmann R. Overview of monoclonal antibodies in cancer therapy: present and promise. Critical Reviews in Oncology/Hematology. 2005;54(1):11-29.
Walko C.M., McLeod H. Pharmacogenomic progress in individualized dosing of key drugs for cancer patients. Nature Clinical Practice Oncology. 2009;6(3):153-162.
Zalatnai A. Potential role of cell cycle synchronizing agents in combination treatment modalities of malignant tumours. In Vivo. 2005;19(1):85-91.
Australian Nuclear Science and Technology Organisation (ANSTO): www.ansto.gov.au/
Australian Institute of Health and Welfare: www.aihw.gov.au/
The Wnt homepage: www.stanford.edu/∼rnusse/wntwindow.html
More weblinks at: http://evolve.elsevier.com/AU/Bryant/pharmacology