Chapter 29 Drugs affecting the upper gastrointestional tract
This chapter reviews the various topical and systemic medications that are used to treat illnesses or disorders affecting the upper gastrointestinal (GI) tract. These include drugs that affect the mouth, stomach, vomiting reflex, small intestine and gallbladder. As many GI disorders—such as peptic ulcers, nausea and vomiting—negatively affect a person’s quality of life, the health-care professional needs to have knowledge of the drugs used in the treatment of GI tract disorders.
Key abbreviations
CTZ chemoreceptor trigger zone
ECL enterochromaffin-like cells
GORD gastro-oesophageal reflux disease
KGF keratinocyte growth factor
Key background
DISORDERS of the upper gastrointestinal tract (GIT), such as indigestion, gastritis and peptic ulcers, are very common problems reported by a large proportion of the population. The causes of many GI diseases remain unclear, and drug treatment is often focussed on relieving symptoms rather than on control or cure.
The digestive system has four main activities—motility, secretion, digestion and absorption—and is made up of the GIT, also called the alimentary canal, and the accessory organs of digestion such as the teeth, tongue, biliary system (liver and gallbladder) and pancreas (Figure 29-1). Mechanical digestion involves processes such as chewing and churning, while chemical digestion relies on secretion of digestive enzymes such as those in the mouth (salivary amylase), stomach (pepsin) and small intestine (pancreatic amylase). Movements by the smooth muscle fibres surrounding the GIT mix the contents by segmental contractions and propel the material through the tract by peristalsis.
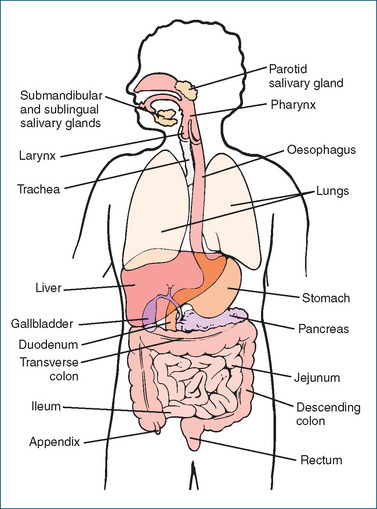
The secretory and muscular activities of the GIT are regulated by both intrinsic and extrinsic neural mechanisms. The enteric nervous system, an interconnecting network of neurons, is located in smooth muscle and secretory cells of the GIT and relays information via the autonomic nervous system and via local reflexes. This intrinsic system is self-regulating and is capable of controlling exocrine gland secretions and muscular contractions independently of the central nervous system. Neurotransmitters in the enteric nervous system include acetylcholine, nitric oxide, 5-hydroxytryptamine (5-HT, serotonin) and substance P.
By contrast, the extrinsic innervation of the GIT is supplied by the divisions of the autonomic nervous system (see Chapter 11). These divisions coordinate activities among different regions of the GIT and also between this system and other parts of the body. The parasympathetic division sends nerve impulses via two branches of the vagus nerve and exerts mostly an excitatory action, which increases digestive secretions and muscular activity. The splanchnic nerves of the sympathetic division are primarily inhibitory nerves and depress digestive secretions and muscular activity. Under normal conditions, the two divisions of the autonomic nervous system and the enteric nervous system maintain a delicate balance of control over the functions of the GIT.
The mouth (buccal cavity) and pharynx
The mouth, or buccal cavity, functions as the starting point of the digestive process. Ingested food is chewed and mixed with saliva that contains the enzymes amylase, which initiates the breakdown of disaccharide sugars and starches (polysaccharides), and lingual lipase, which initiates digestion of dietary triglycerides (fats).
Three pairs of salivary glands secrete saliva via ducts into the mouth. The sublingual and submandibular salivary glands are located beneath the tongue; the largest pair is the parotid glands, which are found in front of and slightly below the ears. When food has been chewed and is reduced to a soft spongy mass in the mouth, it is swallowed. Swallowing (deglutition) is a complex process that begins as a voluntary movement but is continued as an involuntary muscular reflex as the bolus of food is propelled through the pharynx into the oesophagus.
Systemic diseases, nutritional deficiencies and mechanical trauma can cause irritation or inflammation of buccal structures. Dental disorders (e.g. caries and gingivitis) and bacterial, viral or fungal infections (e.g. candidiasis or herpes simplex) can affect the structures of the oral cavity, causing symptoms such as mouth blistering or other lesions, swelling, pain and inflammation. Mumps is an inflammation of the parotid glands by the mumps virus (myxovirus).
The pharynx (throat), which connects the mouth and the oesophagus, is important in swallowing. When food and fluid pass through the pharynx into the oesophagus, the trachea is closed to prevent aspiration into the lungs and respiration is inhibited. The oral and pharyngeal phases of swallowing last less than one second. Like the mouth, the pharynx can be affected by viral infections and become irritated and inflamed (e.g. due to sinusitis or the common cold). Neurological lesions and cerebrovascular accidents involving the medulla and the swallowing centre can result in difficulties in the pharyngeal phase of swallowing.
Drugs affecting the mouth
Good oral hygiene, which includes brushing properly after meals and at bedtime, flossing and gum stimulation, has a major influence on the health of the tissues of the mouth. Many mouth and throat preparations containing antiinflammatory agents, anaesthetics and antiseptics are available for various disorders of the oral cavity, including chapped lips, sun and fever blisters, inflammatory lesions, ulcerative lesions secondary to trauma, gingival lesions, teething pain, toothache, irritation caused by orthodontic appliances or dentures and oral cavity abrasions. Most topical agents that affect the mouth may be purchased over the counter (OTC). In addition, some medications may cause dry mouth as an adverse effect (e.g. muscarinic receptor antagonists like atropine and the anticholinergic drugs used in the treatment of Parkinson’s disease), and prolonged loss of secretions may contribute to poor oral health.
Mouth-washes and gargles
Mouth-washes and gargles are dilute aromatic solutions that often contain a sweetener and an artificial colouring agent. They may also contain an antiseptic (e.g. alcohol, cetylpyridinium chloride or chlorhexidine gluconate), an anaesthetic (e.g. benzocaine, lignocaine hydrochloride), an analgesic (e.g. benzydamine hydrochloride, choline salicylate) and an anticaries agent (sodium fluoride). Use of mouth-washes with high alcohol content may be problematic in some groups of the general population (e.g. children and people with cultural or religious objections to alcohol use). The leading mouth-washes usually contain 7%–30% alcohol and use of mouth-wash in young children is not recommended, as children often swallow the mouth-wash rather than expectorating it.
Products that inhibit plaque formation are available, and clinical trials have demonstrated some success with volatile oils, cetylpyridinium chloride and chlorhexidine. Commercial products that contain at least one of these active ingredients include Cepacol (cetylpyridinium chloride), Listerine (volatile oils) and Plaqacide (chlorhexidine gluconate). A detergent-type product to lessen plaque (Plax) is also available on the market.
Mouth-washes are often used for halitosis, or ‘bad breath’, or as gargles to treat colds or sore throats. They are generally not considered effective for such problems. Mouth-washes can improve mouth odour briefly but if such a problem persists, the underlying cause, such as poor dental hygiene or various gum diseases, needs to be identified and treated.
Sore throats are usually caused by infection, most often viral rather than bacterial. Symptomatic relief may be obtained using lozenges, sprays and gargles. Gargling might not reach the site of infection, which is often deep in the throat tissues. In addition to commercial preparations (e.g. Cepacaine, Betadine Sore Throat Gargle), sodium chloride solution (½ teaspoon of salt in an average-sized glass of warm water) has been commonly used as a gargle and mouth-wash.
Several fluoride-containing preparations, including mouth-wash (e.g. Neutrafluor 900), toothpaste, tablets and solutions, are available for use as anticaries agents. The exact mechanism of action of fluoride in preventing caries is not fully understood; however, fluoride ions appear to exchange for hydroxyl or citrate (anion) ions and then settle in the anionic space in the surface of the enamel. This results in a harder outer layer of tooth enamel (a fluoridated hydroxyapatite) that is more resistant to demineralisation. Fluoridated mouth-washes have been used in communities with both limited fluoridated and unfluoridated water supplies, and their use has been associated with a significant decrease in tooth decay (see Clinical Interest Box 29-1).
Clinical interest Box 29-1 Fluoridated water

Artificial fluoridation of water has been endorsed worldwide by more than 150 scientific and health organisations since the early observations that communities with naturally fluoridated water had lower incidences of dental caries. The minimum concentration to protect against dental carries is 0.5 mg/L. It has been estimated that two-thirds of the Australian population live in areas receiving fluoridated water. Exposure to fluoride is now even more widespread as exposure also occurs through ingestion of food grown in fluoridated areas, use of fluoridated toothpaste and ingestion of fluoride supplements. Fluoride toothpaste is sold widely in Australia and New Zealand.
In Australia, the natural fluoride concentration of surface water is <0.1–0.5 mg/L and all Australian capital cities have implemented water fluoridation. The nominal target level for fluoride in drinking water is 0.7–1.0 mg/L and should not exceed 1.5 mg/L. In New Zealand, the fluoride content recommended for drinking water by the Ministry of Health is 0.7–1.0 mg/L (http://www.moh.govt.nz/fluoride [16 September 2009]).
Increased exposure to fluoride can cause enamel fluorosis, which can vary from whitish striations to pitting and staining of tooth enamel. Factors implicated in the increasing incidence of fluorosis in Australia include changes to tooth-brushing habits, use of fluoridated toothpaste, residence in fluoridated areas, prolonged use of infant formulae and ingestion of fluoride supplements. Use of fluoride supplements is not recommended for children under 3 years old. Compliance with Australian Drinking Water Guidelines ensures the quality of our water, including the level of fluoridation appropriate for conveying health benefits without any health risks. The 2004 Guidelines undergo a rolling revision to ensure they reflect recent scientific evidence (2004 Australian Drinking Water Guidelines; www.nhmrc.gov.au/publications/synopses/eh19syn.htm [16 September 2009]).
Fluoridated mouth-washes are generally used once a day (rinsed for a minute and expectorated), preferably after brushing and flossing. Eating and drinking should be avoided for about 30 minutes after use.
Dentifrices
A dentifrice is a substance used to aid in cleaning teeth. Ordinary dentifrice contains one or more mild abrasives, a foaming agent and flavouring materials made into a powder or paste (toothpaste) to be used as an aid in the mechanical cleansing of accessible parts of the teeth. Fluoride dentifrices are effective anticaries agents.
Dentifrices are also available for the treatment of hypersensitive teeth. This usually occurs from exposed root areas at the cement–enamel junction, allowing access to nerve fibres in the pulp area of the tooth. Dentists often suggest desensitising dentifrices that contain potassium nitrate, such as Sensodyne toothpaste.
Saliva substitutes
Saliva substitutes such as Aquae spray, Biotene, Oralbalance gel and Oralube spray are used for the relief of dry mouth caused by factors such as salivary gland dysfunction or occurring as a result of drug administration (e.g. anticholinergics and tricyclic antidepressants). Available as solutions and as pump sprays, saliva substitutes contain electrolytes (potassium, magnesium, calcium and sodium chloride), potassium phosphate, saccharin, sorbitol solution and carboxymethylcellulose as the base.
Drugs used to treat mouth blistering
Acute viral diseases such as herpes simplex, herpes zoster and varicella are treated symptomatically with antipyretic analgesics such as paracetamol and aspirin. In worsening cases and in recurrent herpetic infection, treatment with the antiviral drug aciclovir should be considered. Aciclovir acts to reduce viral shedding, time to crusting, duration of local pain and severity of symptoms, and is available in oral and parenteral dosage forms. Aciclovir and other antiviral agents are covered in Chapter 45. In addition to viral infections, mouth lesions can be caused by local irritation, medications, radiation, dental manipulations or systemic disease. Instituting proper treatment involves initial identification of the causative factor.
Drugs used to treat oral candidiasis
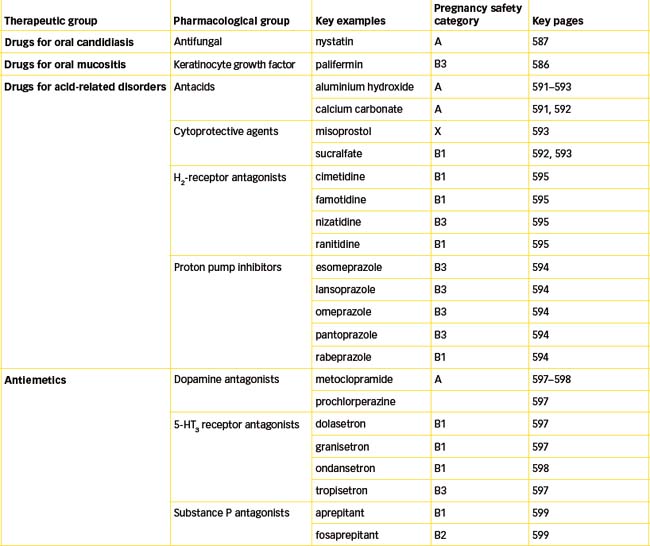
The term candidiasis (‘thrush’) is used commonly to refer to a superficial fungal infection; in the case of the mouth, rarely are fungi other than Candida involved. Local factors predisposing to an outbreak of visible oral fungal lesions include smoking, the wearing of dentures, decreased salivation and the use of inhaled corticosteroids. In some individuals, however, the precipitating factor may be associated with systemic antibiotic or corticosteroid use or cancer chemotherapeutic treatment regimens. Although there are various antifungal agents, amphotericin (lozenge), miconazole (oral gel) and nystatin (pastilles and oral suspension) are the most commonly used drugs for oral candidiasis. In contrast, in severely immunocompromised patients, oral antifungal drugs such as fluconazole and ketoconazole are preferred. Only nystatin will be reviewed in this section (see Drug Monograph 29-1), as the azole antifungal agents are discussed in Chapter 45.
Nystatin is a polyene antifungal product of Streptomyces noursei and was one of the first antifungal drugs to be discovered and used clinically. It exhibits fungicidal activity against a broad spectrum of fungal pathogens (e.g. Candida albicans, Candida glabrata, Candida kruse and Candida tropicalis), including azole-resistant strains of Candida and, in some cases, amphotericin B-resistant strains of Candida albicans. It is used for the treatment of cutaneous, intestinal, oropharyngeal and vulvovaginal candidiasis.
Mechanism of action
It is thought to exert its effect by interacting with the sterol moiety of ergosterols primarily in the cell membrane. This interaction leads to the formation of pores or channels that result in leakage of essential intracellular components such as ions, amino acids and sugars.
Pharmacokinetics
Nystatin is not absorbed from mucous membranes of the mouth, GIT and vagina or from the skin. Due to systemic toxicity, its use is limited to the treatment of mucocutaneous and intestinal candidiasis.
Adverse reactions
The most common adverse reactions of nausea, vomiting and diarrhoea occur more frequently with higher doses.
Warnings and contraindications
Nystatin is contraindicated in people with a previous history of hypersensitivity to the drug. As GI absorption is negligible, use in pregnancy is considered safe.
Dosage and administration
The dose ranges from 100,000 to 1,000,000 units 3–4 times daily and is taken as a suspension, tablet or capsule. In general, the preparation should be held within the mouth for as long as possible to increase contact time with the mucosa. The oral liquid should be swallowed, as expectorating the drug may lead to failure to treat infections of the mucosa of the posterior pharynx or oesophagus.
Drugs used to treat oral mucositis
Inflammation of or injury to the mucous membrane of the mouth and throat may result from a number of factors including infections, use of chemotherapeutic agents (e.g. high-dose methotrexate) and irradiation. In some cases mucositis is associated with an increased risk of severe or life-threatening infections. Treatments vary depending on the cause and include basic oral care (e.g. brushing, flossing etc) and use of mouth-washes, topical antifungal drugs, topical and systemic analgesics and the recombinant human keratinocyte growth factor palifermin.
Keratinocyte growth factor (KGF) is a member of the family of fibroblast growth factors that was originally isolated from pulmonary fibroblasts. KGF binds to KGF receptors, which are located on epithelial cells of the tongue, buccal mucosa, salivary glands, oesophagus, stomach and various other tissues of the GIT. Activation of KGF receptors leads to proliferation, differentiation and migration of epithelial cells, which is a factor for consideration in some malignancies as KGF receptors occur on some tumour cells but are not located on haemopoietic cancer cells.
Palifermin is a recombinant truncated version of endogenous KGF and has similar biological activity to the native protein, but with increased stability. The exact mechanism of action of palifermin has not been elucidated but it binds to KGF receptors resulting in an increase in the thickness of oral epithelium, decreased ulcer formation and a reduction in atrophy of the oral mucosa. In addition, palifermin downregulates the production of pro-inflammatory cytokines, increases production of the anti-inflammatory cytokine IL-13 and reduces the production of reactive oxygen species through activation of the transcription factor Nrf2 that regulates the expression of genes involved in the antioxidant response element (Schmidt et al 2008).
The oesophagus and stomach
The oesophagus is a collapsible muscular structure about 25 cm long that extends from the pharynx to the upper region (cardia) of the stomach. It passes through the diaphragm at the oesophageal hiatus into the abdominal cavity. Oesophageal disorders are characterised by retrosternal pain (heartburn) and difficulty in swallowing (dysphagia). The sources of the pain are numerous; potential causes include diffuse oesophageal spasm, achalasia (failure of lower oesophageal sphincter to relax), pyloric or duodenal ulcers, postural changes (bending forward) and excessive alcohol ingestion. Heartburn commonly results from reflux oesophagitis (backflow of gastric contents into the oesophagus) or from hiatal hernia (protrusion of a part of the stomach through the diaphragm). Dysphagia can be a symptom, for example, of oesophageal obstruction, mechanical interference with or paralysis of the muscles of deglutition, carcinoma of the oesophagus, anxiety states or hysteria. Inflammation of the oesophagus can have many causes (e.g. reflux oesophagitis associated with hiatal hernia, irritant ingestion, infection, peptic ulceration or prolonged gastric intubation).
The stomach is a J-shaped pouch-like structure lying below the diaphragm and has four divisions: the cardia, the fundus, the body and the pylorus. The pyloric sphincter controls communication between the stomach and the duodenum. The stomach wall is composed of the mucosa, which contains the gastric glands responsible for the secretion of pepsinogen, gastric lipase, hydrochloric acid and intrinsic factor, and the submucosa, which connects the mucosa to the underlying muscularis mucosae. The muscularis has three layers of smooth muscle: longitudinal, circular and oblique layers, which allow the stomach to churn and mix the contents. In general, vagal stimulation (parasympathetic) increases the force and frequency of contractions, while input from the sympathetic nervous system decreases both activities. When the stomach is empty, the mucosa forms large folds or rugae, which tend to flatten out when the stomach distends.
The stomach functions as a temporary storage site for food as it is being digested and is capable of holding 1500–2000 mL. When a bolus of food arrives, the proximal region of the stomach relaxes to accommodate the ingested meal. Gentle peristaltic movements pass over the stomach and the food is mixed with the gastric secretions to form a liquid called chyme. More vigorous mixing movements occur in the body of the stomach, and the velocity and force of contractions increase as the chyme is moved towards the pylorus. The contractions, which last 2–20 seconds and occur at a rate of 3–5 per minute, are responsible primarily for the mixing of the chyme with the digestive juices but they also assist the propulsion of the chyme into the duodenum. As the peristaltic contraction approaches the pylorus, a small volume of the chyme is forced into the duodenum but the majority is forced back into the body of the stomach, where further mechanical and chemical digestion occurs. Only a limited amount of nutrient and drug absorption takes place in the stomach (see Chapter 6).
The time required for digestion in the stomach depends on the amount and type of food eaten. Normal gastric emptying time is 2–6 hours but certain drugs, physical activity of the individual and body position during digestion may affect this. Liquids empty more rapidly, whereas solids must be reduced in size to particles less than 2 mm3 in volume before emptying occurs. Neural and hormonal reflexes control the balance between the gastric emptying rate and the processing capacity of the small intestine.
Gastric secretions
The major stimulant to gastric acid secretion is protein. Gastric juice comprises pepsin, hydrochloric acid, mucus and intrinsic factor. The secretion of hydrochloric acid by parietal cells kills bacteria in food, denatures protein and converts inactive pepsinogen into active pepsin, which aids further in protein degradation. Mucus and bicarbonate ions secreted by superficial mucosal cells form a gel-like layer that serves to protect the stomach from the acid environment and provides lubrication between the superficial cells and bulky undigested material. The parietal cells also secrete intrinsic factor, a protein essential for the binding of vitamin B12 before its absorption in the ileum. The stimuli for gastric secretion and the cells involved are illustrated in Figure 29-2.
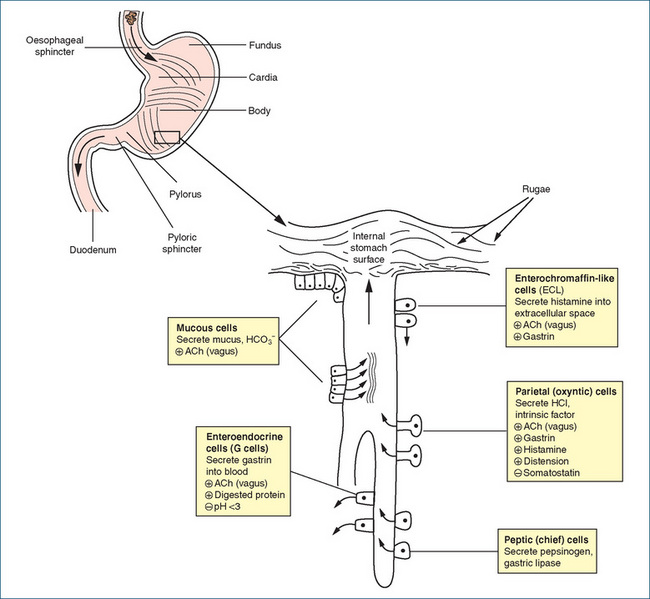
Figure 29-2 Schematic diagram of stomach, gastric gland and secretory cells. ACh = acetylcholine; HCl = hydrochloric acid; HCO3- = bicarbonate ions; + = stimulated by; − = inhibited by.
The hormone gastrin, the neurotransmitter acetylcholine and the local hormone histamine directly stimulate acid secretion by parietal cells. In contrast, prostaglandins E2 and I2 inhibit acid secretion. The process of gastric acid production and secretion is illustrated in Figure 29-3. The exact mechanism is not clearly established but, in general, gastrin and acetylcholine (ACh) stimulate histamine release from enterochromaffin-like (ECL) cells. Histamine then acts via histamine (H2) receptors on parietal cells to increase acid secretion. Acetylcholine and gastrin activate calcium-dependent pathways via their respective receptors while histamine binding to H2 receptors activates cAMPdependent pathways. The calcium-dependent and the cAMP-dependent pathways activate the H+, K+ ATPase (the proton pump), which exchanges hydrogen and potassium across the parietal cell membrane. The proton pump is responsible for maintaining the intracellular pH at ∼7.3 and the intracanalicular pH at ∼0.8.
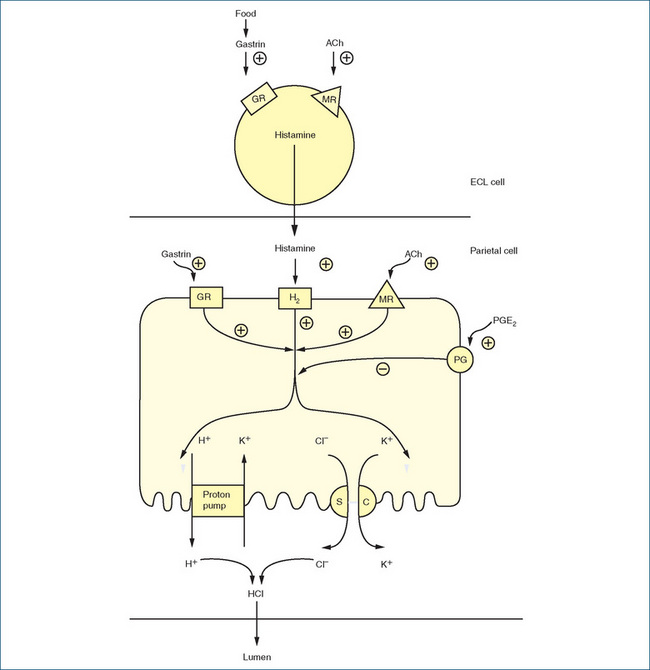
Figure 29-3 Schematic diagram of gastric acid secretion and interrelationship of histamine secretion from enterochromaffin-like cell (ECL) and the acid-secreting parietal cell. Pathways may be stimulated (+) or inhibited (−). ACh = acetylcholine; GR = gastrin receptor; H2 = histamine (H2) receptor; M = muscarinic receptor; PG = prostaglandin E2 (PGE2) receptor; proton pump = H+, K+ ATPase; SC = symport carrier.
Adapted from: Rang HP, Dale MM, Ritter JM, Moore PK. Pharmacology, 5th edn, Edinburgh: Churchill Livingstone, 2003.
Parietal cells secrete 1000–2000 mL hydrochloric acid per day, so maintenance of the gastric mucosal barrier is essential to prevent ulceration. Changes in mucosal blood flow, decreased secretion of protective mucus, bacterial infection and damage by agents such as alcohol and aspirin may all lead to weakening of the mucosal barrier and ulceration. Understanding the role of prostaglandins, the various hormones and neurotransmitters in regulating acid secretion provides the basis for the pharmacological management of peptic ulcer disease.
Disorders affecting the stomach
Acute gastritis is an inflammatory response of the stomach lining to ingestion of irritants, such as ethanol (alcohol) or non-steroidal anti-inflammatory drugs (NSAIDs), including aspirin. Symptoms include epigastric discomfort, nausea, abdominal tenderness and GI haemorrhage. Treatment consists of lifestyle modifications and drugs such as antacids.
Chronic gastritis is a long-term inflammation of the stomach lining, generally with degeneration of the gastric mucosa, but its causes are not well established. It is more common in women, and the incidence increases with age, excessive smoking and ethanol use. Symptoms are non-specific but may include flatulence, epigastric fullness after meals, diarrhoea and bleeding. Treatment is the same as for acute gastritis. Iron deficiency anaemia and pernicious anaemia may result from chronic gastritis. Treatment of symptoms and elimination of possible causative or aggravating factors (e.g. aspirin use) comprise the usual therapeutic regimen.
Peptic ulcer disease is a broad term encompassing both gastric and duodenal ulcers. Although both types of ulcers produce a break in the gastric mucosa, the causes differ. With gastric ulcers, the ability of the gastric mucosa to protect and repair itself seems to be defective; in duodenal ulcers, hypersecretion of acid and pepsin is responsible for the erosion of the duodenal mucosa. Gastric colonisation with Helicobacter pylori, a common Gram-negative bacillus, has been identified as a major causative agent in individuals with peptic ulcer disease not caused by NSAIDs (see Clinical Interest Box 29-2). Treatment with various drug combinations results in healing and a low peptic ulcer recurrence rate (Bytzer & O’Morain 2005).
Clinical interest Box 29-2 Helicobacter pylori
H. pylori is a Gram-negative bacterium that commonly infects around 30%–40% of Australian-born adults. It is more common in persons older than 40 years (<10% incidence in children) and in persons of Middle Eastern, Asian and Eastern European origin. There appears to be no difference between males and females in the frequency of infection (Australian Gastroenterology Institute; http://www.nevdgp.org.au/info/gastro/Helicobacterpylori.htm). It is a spiral (helical)-shaped organism that has evolved to inhabit the highly acidic environment of the stomach, particularly the pylorus. Once the bacterium adheres to the gastric epithelial cells, it breaks down endogenous urea, creating a protective cloud of ammonia and bicarbonate that enables it to protect itself against the effects of gastric acid. The ability of the bacterium to degrade urea and release carbon dioxide forms the basis of the H. pylori breath test, which is used to detect infestation and also to monitor the effectiveness of drug-mediated eradication (see the section ‘Helicobacter pylori treatment regimens’ below). H. pylori has been established as a causal agent in the development of chronic gastritis, duodenal and gastric ulcers and gastric cancer.
Duodenal ulcers are more common than gastric ulcers, accounting for nearly 80% of all peptic ulcers, and usually occur more frequently in younger persons. Overall, the reported incidence of peptic ulcers is much lower in females. In addition to various drug treatment regimens, diet and lifestyle modifications are equally important. Hereditary factors, use of some drugs (e.g. aspirin and corticosteroids), psychological factors, stress and diet have also been implicated in the development of peptic ulcer disease.
Drugs that neutralise or inhibit gastric acid secretion
Conditions of the stomach requiring drug therapy include hyperacidity, ulcer disease, nausea, vomiting and hypermotility. Drugs used include antacids, anticholinergics, antidepressants, anxiolytics, H2-receptor antagonists, proton pump inhibitors and cytoprotective agents (substances that protect cells from damage) such as sucralfate and the prostaglandin analogue misoprostol. The following sections are limited to those drugs not covered elsewhere in this book: antacids, cytoprotective agents, proton pump inhibitors and H2-receptor antagonists.
Antacids
Antacids are chemical compounds that buffer or neutralise hydrochloric acid in the stomach and thereby raise the gastric pH. They have been used for centuries, often in the form of ‘baking soda’ (sodium bicarbonate), and are indicated for the relief of symptoms associated with peptic ulcer disease, gastritis, gastro-oesophageal reflux disease (GORD, see Clinical Interest Box 29-3) and dyspepsia. The major ingredients in antacids include aluminium hydroxide, calcium carbonate, magnesium salts and sodium bicarbonate, alone or in combination. Heartburn, indigestion and stomach upset are common and most antacids may be purchased as OTC preparations. These include Gastrogel, Gaviscon and Mylanta.
Clinical interest Box 29-4 Milk–alkali syndrome
The earliest record of a reaction to milk and alkali was described by Hippocrates more than 2000 years ago, but during the 1920s and 1930s, when ‘Sippy’ (antacid) powders were widely used for heartburn and indigestion, cases of milk–alkali syndrome were widespread. Reports continue to appear, and the characteristic features of the syndrome arising from prolonged and excessive intake of milk and antacids are irritability, distaste for milk, occasional nausea and vomiting, headache, mental confusion, anorexia, muscle ache, weakness and malaise. Impairment of renal function ensues, with elevated plasma calcium, phosphorus and bicarbonate. Calcium and phosphate precipitate in the kidney tubules, contributing to the renal damage. The syndrome has a reported mortality of around 5%.
Clinical interest Box 29-3 Gastro-oesophageal reflux disease (Gord)
GORD characterised by reflux of acidic gastric contents into the oesophagus is extremely common and as many as 25% of the population experience symptoms. Considered a disease with a high relapse rate the prevention of recurrences of symptoms, mucosal lesions and complications is the main aim of therapy. The pathogenesis of GORD is multifactorial and includes impaired function of the lower oesophageal sphincter and delayed gastric emptying. Approximately 30% of patients with GORD have erosive oesophagitis while the remaining 70% have non-erosive reflux disease (NERD). Use of PPIs results in successful symptom control of GORD reducing night-time heartburn and improving sleep-related disturbances in patients. NERD is a more heterogeneous disorder overlapping with functional dyspepsia and current evidence indicates that the response to PPIs is less consistent than in patients with clearly established erosive disease (Savarino et al 2009).
GORD can also occur in infants due to oesophageal sphincter immaturity and a predominantly fluid dietary intake. Pharmacological management is reserved for infants presenting with symptoms of irritability, disrupted sleep patterns, anorexia, frequent vomiting, respiratory problems or poor weight gain. Acid-suppressing medication such as proton pump inhibitors, H2 antagonists and to a lesser extent antacids are used as first-line therapy (Tighe et al 2009). GORD resolves in most infants by 2–3 years of age.
Although there are many antacid preparations on the market, the magnesium–aluminium combinations (e.g. Mylanta, Gaviscon) are among the most common antacids selected by individuals and health-care professionals. Combination antacids have been formulated to reduce the risk of diarrhoea or constipation as an adverse effect. In some formulations, alginic acid or simethicone may also be included. Gaviscon contains alginic acid, which forms a viscous cohesive foam; this is thought to be beneficial in reflux oesophagitis by increasing adherence of mucus to the lower oesophageal mucosa. Simethicone, a defoaming agent, relieves flatulence by dispersing and preventing the formation of mucus-surrounded gas pockets in the GIT.
Dosage and administration
The amount of antacid needed to neutralise hydrochloric acid depends on the individual, the condition being treated and the buffering capability of the preparation used. The acid-neutralising property of antacids varies and is defined as the quantity (milliequivalents [mEq]) of hydrochloric acid brought to a pH of 3.5 in 15 minutes (Table 29-1). The maximum dosages listed on antacid packages should be followed; however, many individuals exceed the recommendations, thus increasing the potential for producing many of the adverse reactions.
Table 29-1 Antacids: Acid-neutralising capacity
| Antacid | Primary ingredients | Acid-neutralising capacity |
| Liquid preparations | (mEq/5 mL) | |
| Gelusil | Aluminium hydroxide, magnesium hydroxide, simethicone | 12 |
| Mylanta Original | Aluminium hydroxide, magnesium hydroxide, simethicone | 12.7 |
| Mylanta Double Strength | Aluminium hydroxide, magnesium hydroxide, simethicone | 25.4 |
| Tablet preparations | (mEq/tablet) | |
| Gelusil | Aluminium hydroxide, magnesium hydroxide, simethicone | 11 |
| Mylanta Original | Aluminium hydroxide, magnesium hydroxide, simethicone | 11.5 |
| Mylanta Double Strength | Aluminium hydroxide, magnesium hydroxide, simethicone | 23 |
| Andrews | Tums Calcium carbonate | 10 |
Antacids are considered either rapid-acting (e.g. sodium bicarbonate) or less rapid-acting (e.g. aluminium hydroxide). When administered in a fasting state, the antacid effect lasts 20–40 minutes. If administered 1 hour after meals, the effects may be extended for up to 3 hours. Liquid and powder dosage forms have been found to be more effective antacids than the tablet dosage form. Most tablets require chewing before swallowing to ensure complete dissolution of the antacid in the stomach. Absorption of antacids varies, and those that contain aluminium, calcium or magnesium are absorbed to a lesser extent than those containing sodium bicarbonate. Most of the unreacted insoluble antacids are excreted in the faeces.
In people with normal renal function, absorption of cations (e.g. Al3+, Mg2+, Ca2+) causes little in the way of systemic problems; however, in the presence of renal insufficiency, absorption of, for example, Ca2+ may cause hypercalcaemia. As sodium bicarbonate is also absorbed in the intestine, prolonged use of this antacid should be avoided, particularly in people with heart failure or hypertension and those on sodium-restricted diets. Additionally, antacids should be avoided in the presence of coexisting conditions such as constipation (worsened by aluminium) or diarrhoea (aggravated by magnesium). Antacids are generally considered safe for use in pregnancy if prolonged or high doses are avoided.
Adverse reactions
A concise list of adverse reactions is given in Table 29-2.
Table 29-2 Adverse reactions associated with antacids
| Constituent of antacid | Adverse reactions* | Contraindication/coexisting conditions |
| Aluminium hydroxide | Constipation, chalky taste, phosphate depletion, faecal impaction, intestinal obstruction, encephalopathy | Chronic renal failure because of increased risk of aluminium toxicity |
| Calcium carbonate | Belching, flatulence, constipation, abdominal distension, hypercalcaemia, alkalosis, phosphate depletion, renal calculi, milk–alkali syndrome (see Clinical Interest Box 29-4). | Hypercalcaemia, hyperparathyroidism, renal impairment (increased risk of hypercalcaemia) |
| Sodium bicarbonate | Belching, abdominal distension, metabolic alkalosis (high doses), hyperventilation, hypokalaemia, hyperirritability, tetany, volume overload, pulmonary oedema | Metabolic or respiratory alkalosis, chloride depletion, hypoventilation, oedema associated with heart failure, renal failure or cirrhosis, renal impairment (increased risk of sodium retention) |
| Magnesium salts | Diarrhoea, chalky taste, belching, elevated plasma magnesium | Diarrhoea (may be aggravated), renal impairment (increased risk of raised plasma magnesium) |
* Adverse reactions are listed in order of most common through to rare (AMH 2010).
Drug interactions
Antacid–drug interactions depend on the composition of the antacid used; in general, antacids have been reported most frequently to reduce or delay the absorption of many drugs. In some instances the reverse occurs and, in particular, antacids containing magnesium hydroxide can increase the absorption of some hypoglycaemic drugs, thus potentially placing the person at risk of hypoglycaemia. Health-care professionals should be aware of the need for careful scheduling of antacids, as most medications need to be separated by at least 2 hours from an antacid. There are multiple drug interactions which include but are not limited to:
Cytoprotective agents
Protection of the gastric and duodenal mucosa is aided by secretion of bicarbonate ions into a mucus layer that protects the underlying epithelial cells against erosion from gastric acid. Cytoprotective agents enhance the protection afforded by the mucus layer by providing a physical barrier over the ulcerated surface. The two main agents are sucralfate and the prostaglandin analogue misoprostol.
Sucralfate is composed of sulfated sucrose and aluminium hydroxide. It is a non-absorbable drug that in the presence of acid undergoes a chemical reaction that results in formation of a sticky, yellow–white gel that forms a protective, acid-resistant shield in the ulcer crater. This barrier hastens the healing of the ulcer by protecting the mucosa for up to 6 hours. The binding to the ulcer crater is thought to be the main therapeutic effect, but sucralfate also stimulates angiogenesis, production of mucus and protective prostaglandins. It is administered orally with minimal systemic absorption (up to 5%) and is excreted primarily by the faecal route. This product is indicated for short-term (up to 8 weeks) peptic ulcer treatment and for the prevention of stress-induced ulcers.
The most common adverse reaction is constipation, which occurs in 1%–15% of patients. Infrequently, there are reports of nausea, vomiting, dry mouth, dizziness, back pain, rash and headache.
The adult dose for peptic ulcer disease is 1 g four times daily (1 hour before each meal and at bedtime; maximum 8 g daily) for 4–8 weeks, with a maintenance regimen of 1 g twice daily on an empty stomach, which favours binding of sucralfate in the low pH environment. For prophylaxis of stress ulcers, administer 1 g every 4 hours.
Misoprostol, a synthetic analogue of prostaglandin E1, is indicated for the treatment of peptic ulcers and the prevention of gastric ulcers associated with the use of NSAIDs. Normally, prostaglandins of the E and I series (see Chapter 47) protect the stomach by decreasing gastric acid secretion and increasing gastric cytoprotective mucus and bicarbonate. The NSAIDs inhibit prostaglandin synthesis, which reduces the effectiveness of the protective mechanisms and can result in gastric ulcer formation. Misoprostol suppresses gastric acid secretion and thus helps to heal gastric ulcers.
Misoprostol is rapidly absorbed after oral administration and is metabolised by fatty acid oxidising systems to an active metabolite, misoprostol acid, which is further metabolised. Less than 1% of the dose is excreted as unchanged drug; renal elimination accounts for 75% and faecal elimination for about 15%. No significant drug interactions have been reported. Infrequent adverse reactions reported include constipation, gas, headache, nausea and vomiting. In about 30% of people, diarrhoea limits its usefulness.
As misoprostol can cause hypotension in patients with cerebrovascular or coronary artery disease, it should be used with caution in these groups. Importantly, as misoprostol can induce premature labour and may be teratogenic in large doses, it should not be used in pregnant women or in those contemplating pregnancy.
Proton pump inhibitors
Proton pump inhibitors (PPIs) suppress gastric acid secretion by inhibiting the proton pump (H+, K+ ATPase enzyme system) at the secretory surface of the gastric parietal cells (see Figure 29-3). These drugs consist of a benzimidazole and a pyridine ring and are weak bases. Following administration these drugs accumulate in the highly acidic environment (pH ∼0.8) of the secretory canaliculi of the parietal cells. There the drugs are converted to a thiophilic sulfonamide (a permanent cation), which interacts covalently with H+, K+ ATPase involved in hydrogen ion transport. When sufficient drug molecules bind to the proton pump, they block the final step of acid production, suppressing both basal and stimulated gastric acid secretion. The covalent (irreversible) interaction with H+, K+ ATPase explains why the duration of action of PPIs exceeds the plasma half-life of these drugs. PPIs are the most potent inhibitors of gastric acid secretion available and dosing once daily inhibits maximal acid output by ∼ 65%. When administration of a PPI is ceased acid secre tion is restored following synthesis of new H+, K+ ATPase.
The first of these drugs to be developed was omeprazole (see Drug Monograph 29-2), which binds irreversibly to the proton pump; others now available include lansoprazole, pantoprazole, rabeprazole and esomeprazole (the S-isomer of omeprazole).
Drug monograph 29-2 Omeprazole
Omeprazole, the prototype drug, is a racemate comprising R-omeprazole and S-omeprazole (or esomeprazole). It is indicated for the treatment of peptic ulcer disease, treatment and prevention of NSAID-induced peptic ulceration, severe erosive oesophagitis that occurs with GORD, and long-term treatment of hypersecretory gastric conditions such as Zollinger–Ellison syndrome.
Pharmacokinetics
After a single oral dose, the onset of action of omeprazole as indicated by decreased gastric acid secretion is within 1 hour, its peak effect in 2 hours and its duration of action is 3–5 days (time needed for return of secretory activity). Bioavailability is in the range of 30%–40% and the plasma half-life is ∼0.5 hours. It is extensively metabolised in the liver by CYP2C19 and CYP3A4, and the metabolites are excreted in urine (about 80%) and faeces (about 20%). Esomeprazole is eliminated more slowly accounting for its slightly longer half-life (∼0.7–1.0 hours).
Drug interactions
In view of its metabolism by CYP2C19 and CYP3A4, interactions with other drugs should be expected. Omeprazole is an inhibitor of CYP2C19 and concurrent administration of omeprazole with diazepam or phenytoin can lead to increased plasma concentrations of diazepam and phenytoin, and dosage reduction might be necessary. Similarly, coadministration with warfarin can lead to increased anticoagulation, and monitoring of the INR should be considered (for a review of drug interactions see Andersson [1991]). Additionally, PPIs can decrease the bioavailability of drugs that rely on an acidic environment for absorption e.g. atazanavir (HIV-protease inhibitor), ampicillin, iron and digoxin.
Adverse reactions
Omeprazole is generally well tolerated. Minor adverse effects include abdominal pain, dizziness, headache, nausea, vomiting, diarrhoea, flatulence and skin rash. Rare adverse reactions include agranulocytosis, pancytopenia, thrombocytopenia and increased liver enzymes. Decreased absorption of vitamin B12 has also been observed with chronic treatment.
Warnings and contraindications
Avoid use in people with omeprazole hypersensitivity. Care should be exercised in patients with impaired hepatic function because of risk of accumulation of the drug when high doses are used.
Dosage and administration
The adult oral dose for gastrooesophageal reflux is 20–40 mg daily for 4–8 weeks. For gastric hypersecretory conditions, the maintenance dose is 20–120 mg daily, adjusting as necessary. The capsule formulation should not be opened, crushed or chewed, as the drug will degrade in the acidic environment of the stomach.
Helicobacter pylori treatment regimens

It is now well established that infection with Helicobacter pylori causes chronic active gastritis, is associated with the development of gastric and duodenal ulcers and is a recognised risk factor in the development of gastric carcinoma. Eradication of H. pylori is considered first-line treatment because it vastly improves the odds of non-recurrence of the ulcer. Early therapies involved use of a single drug such as an antibiotic, a PPI or bismuth, but monotherapy was found to be effective in less than 30% of people so combinations were developed. The first ‘triple therapy’, which included bismuth, metronidazole and tetracycline, was effective in eradicating H. pylori in about 90% of people, but adverse effects were common.
Undoubtedly the most successful therapy is three drugs administered twice a day for 1 week. However, there is still debate as to whether the duration of therapy for successful eradication (while limiting both drug resistance and adverse effects) should be increased to 10–14 days.
Current triple therapy regimens include:
Combining the individual agents in a single packet helps simplify a complicated drug schedule, and omeprazole, clarithromycin and amoxycillin are available in Australia and New Zealand as a ‘single script’ combination pack (Klacid Hp7 and Nexium Hp7).
The success of H. pylori eradication hinges on adherence to therapy and susceptibility of the bacterium to the antibiotics. The 1-week regimens with lower instances of adverse reactions have a high adherence rate (>95%), whereas therapies over 10 days tend to have much higher discontinuation rates. Bacterial resistance is an ever-increasing problem, and resistance of strains to clarithromycin hinders success in about 10% of the population in the United States, southwestern Europe and Japan, and in about 20% of the Australian population. If two courses that include clarithromycin and metronidazole fail to eradicate H. pylori, patients are likely to have at least single resistance and more frequently double resistance (Gisbert & Pajares 2005). Continued maintenance acid suppressant treatment may also be necessary in people where eradication treatment either fails or is contraindicated, in those with gastric ulcers, ulcers >1 cm in diameter or if peptic ulcers recur in the absence of reinfection (AMH 2010).
H2-receptor antagonists
Histamine is produced in ECL cells of the oxyntic mucosa by decarboxylation of L-histidine by histidine decarboxylase. Released histamine then acts on histamine (H2) receptors to increase gastric acid secretion. The H2-receptor antagonists include cimetidine, ranitidine, famotidine and nizatidine. They competitively block histamine from stimulating the H2 receptors located on the gastric parietal cells, thus reducing (∼70%) gastric acid secretion, particularly nocturnal secretion (from stimuli such as food, histamine, caffeine and insulin).
These drugs are indicated for treatment of peptic ulcer disease, GORD and dyspepsia, and for stress ulcer prophylaxis. These drugs have similar structural and pharmacokinetic characteristics, which are summarised in Table 29-3. All of these drugs are well absorbed achieving maximal plasma concentration in ∼1–3 hours.
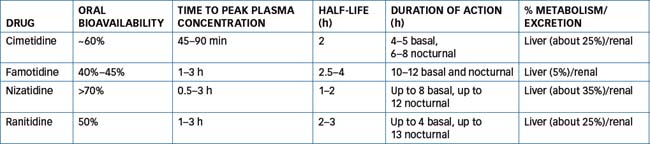
Drug interactions and adverse reactions
Cimetidine, unlike the other H2-receptor antagonists, inhibits the metabolism of other drugs by CYP1A2, CYP2C9, CYP2D6 and CYP3A4 (weak inhibitor) and hence multiple drug interactions have been noted. These include interactions with benzodiazepines, oral anticoagulants, phenytoin, theophylline, nifedipine, flecainide, metoprolol, lignocaine and tricyclic antidepressants. Current drug information sources should be consulted before administering cimetidine concomitantly with other drugs. In addition, all the H2-receptor antagonists reduce the bioavailability of drugs that require an acidic environment for absorption.
In general, these drugs are well tolerated. Common adverse reactions include diarrhoea, constipation, headache, dizziness, rash and confusion in the elderly. The acute confusional state usually resolves with discontinuation of the therapy. Cimetidine can cause breast swelling, impotence and decreased libido. The less common and rare adverse effects include hypotension, hepatitis, agranulocytosis, thrombocytopenia and bradyarrhythmias.
Dosage varies, depending on the condition being treated (peptic ulcer disease, GORD, dyspepsia, stress ulcer prophylaxis). Consult the relevant drug information sources for dosing recommendations. If impaired renal function is a coexisting condition, dosage reduction may be necessary as these drugs are excreted predominantly as unchanged drug (30%–60%).
Vomiting reflex
The induction of vomiting involves a complex coordinated response between two areas, an area of sensory nerve cells called the chemoreceptor trigger zone (CTZ), located in the floor of the fourth ventricle of the brain, and the vomiting centre, or emetic centre, located in the medulla. The emetic centre receives inputs from:
In the absence of the blood–brain barrier the CTZ (Figure 29-4) is activated by both cerebrospinal fluid-borne and blood-borne emetics, such as chemical toxins and drugs, and by the neurotransmitter 5-HT, released from afferent nerve pathways from the stomach and small intestine. The CTZ itself is not able to induce vomiting but is stimulated by smells, strong emotion, severe pain, raised intracranial pressure, labyrinthine disturbances (motion sickness), endocrine disturbances, toxic reactions to drugs, GI disease, radiation treatments and chemotherapy. The CTZ then relays messages to the emetic centre through actions of the neurotransmitters acetylcholine, 5-HT, histamine and dopamine. Antagonism of transmission through these pathways forms the basis for the antiemetic effects of several drugs used clinically. Because the CTZ is close to the respiratory centre in the brain, it is difficult to completely control vomiting initiated from this site without affecting respiration. Discharge from both the sympathetic and parasympathetic nervous systems often leads to the accompanying symptoms of salivation, sweating, rapid breathing and cardiac arrhythmias.
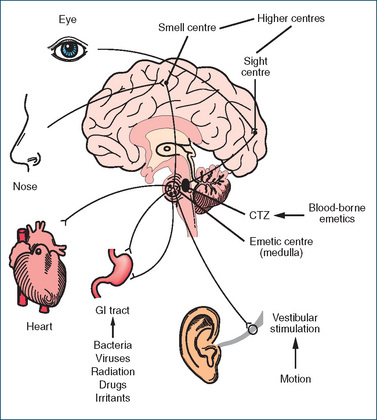
Vomiting is characterised by forceful expulsion of the contents of the stomach (and sometimes that of the duodenum) through the mouth. This occurs as a result of impulses sent via efferent nerves from the emetic centre to the upper GIT, diaphragm and abdominal muscles. Strong contraction of the abdominal muscles then forces the contents past the oesophageal sphincter and into the mouth. Relaxation of the abdominal muscles allows any material remaining in the oesophagus to empty back into the stomach. This cycle may be repeated many times. Although vomiting in many instances is a protective mechanism to rid the body of toxic substances, it may in severe cases lead to fluid and electrolyte disturbances. The cerebral cortex is also involved in anticipatory nausea and vomiting, a conditioned response caused by a stimulus connected with a previous unpleasant experience
Drugs for nausea and vomiting

There are numerous causes of nausea and vomiting, and treatment differs for acute situations such as pregnancy (see Clinical Interest Box 29-5) and gastroenteritis, chronic situations such as gastric or metabolic diseases and psychogenic vomiting such as that occurring with bulimia. Control of vomiting is important and at times it can be very difficult, which can be distressing to the individual concerned.
Clinical interest Box 29-5 Nausea and vomiting in pregnancy
Nausea, often described as ‘morning sickness’, affects about 80% of pregnant women and as many as 50% experience vomiting. In severe cases of ‘hyperemesis gravidarum’ (1%–2%), women are hospitalised for correction of dehydration and electrolyte imbalance. This phase of nausea and vomiting tends to last 7–12 weeks, and in many cases dietary changes are sufficient (low-fat, high-carbohydrate, small meals) and pharmacological intervention is not necessary. Where possible avoid drug therapy and ensure adequate hydration.
In the wake of the thalidomide disaster of the early 1960 s, most antiemetics are contraindicated in pregnancy. If drug therapy is considered because of impaired quality of life, health-care professionals should refer to the category of risk for an individual drug. Drugs used include metoclopramide, pyridoxine (vitamin B6), prochlorperazine, promethazine and vitamin B12 (Magee et al 2002).
Antiemetics
Vomiting is a complex process involving multiple nerve pathways and neurotransmitters (e.g. ACh acting on muscarinic receptors, histamine on H1 receptors, dopamine on D2 receptors, substance P acting on NK1 receptors and 5-hydroxytryptamine acting on 5-HT3 receptors). Antiemetics act principally by blocking these neurotransmitters in the vomiting centre, the cerebral cortex, the CTZ or the vestibular apparatus. A variety of miscellaneous drugs are also used to control vomiting; these include corticosteroids (dexamethasone and methylprednisolone); benzodiazepines used primarily for their sedative and anxiety-relieving actions (e.g. lorazepam used for anticipatory nausea and vomiting associated with chemotherapy); and the common spice ginger (Zingiber officinale).
The neurotransmitters and drugs used to control and prevent nausea and vomiting are summarised in Table 29-4.
Table 29-4 Drugs for controlling nausea and vomiting, and the associated neurotransmitters
| Neurotransmitter and receptor | Drug class | Antiemetic agent |
| Dopamine acting via (D2) receptors located in the stomach and CTZ | Dopamine antagonists | Domperidone, droperidol, haloperidol, metoclopramide, prochlorperazine |
| Acetylcholine acting on muscarinic receptors in the vestibular and vomiting centres. Overstimulation of the labyrinth (inner ear) results in the nausea and vomiting of motion sickness | Muscarinic receptor antagonists (anticholinergics) | Hyoscine hydrobromide |
| Histamine (H1) receptors in vestibular and vomiting centres | H1 receptor antagonists (antihistamines) | Dimenhydrinate, promethazine |
| 5-hydroxytryptamine (5-HT3) receptors in the GIT, CTZ and vomiting centres | 5-HT3 receptor antagonists | Dolasetron, granisetron, ondansetron, tropisetron |
| Substance P acting via neurokinin-1 (NK1) receptors located in CNS | NK1 receptor antagonist | Aprepitant, fosaprepitant |
Cancer chemotherapy-induced vomiting
Vomiting caused by cancer chemotherapy and radiotherapy can be severe enough that treatment can be delayed, and many individuals vehemently refuse further treatment. Often when cancer chemotherapeutic agents are used in combination, the emetogenic potentials of the agents are additive. Typically, vomiting starts within 4 hours of treatment, peaks towards 10 hours and subsides over the following 12–24 hours. Delayed vomiting can occur with high-dose cisplatin and can last 3–5 days. It is not surprising that anticipation of therapy and the sight and smell of the hospital can trigger nausea and vomiting in as many as 25% of individuals. Because antiemetics are usually more effective in preventing vomiting than they are in treating it, they should be administered (often in high doses) prophylactically before cytotoxic therapy. Chemotherapy-induced vomiting may also require several antiemetic agents with different sites of action for effectiveness (e.g. metoclopramide and lorazepam, metoclopramide and dexamethasone, or prochlorperazine and dexamethasone). In addition to drug therapy, behavioural and psychological support should be provided.
Dopamine antagonists
Drugs within this class include prochlorperazine, domperidone and metoclopramide (see Drug Monograph 29-3). Prochlorperazine is a phenothiazine derivative with antiemetic effects, probably by an inhibitory action on the CTZ and vomiting centre. Phenothiazines are thought to act mainly as D2-receptor antagonists but they also have antihistamine and antimuscarinic properties. Only their actions relevant to nausea and vomiting are discussed here; other information on phenothiazines and their use as antipsychotic drugs can be found in Chapter 18.
Drug monograph 29-3 Metoclopramide
Metoclopramide is used for diabetic gastroparesis, gastrooesophageal reflux and, parenterally, for the prevention of nausea and vomiting secondary to emetogenic cancer chemotherapeutic agents, radiation and opioid medications. It is also used as an adjunct for GI radiological examinations because it hastens barium’s transit through the upper GIT by its stimulation of gastric emptying and acceleration of intestinal transit. Parenteral metoclopramide may be used to facilitate small-intestinal intubation.
Mechanism of action
Metoclopramide has both central and peripheral actions in preventing or relieving nausea and vomiting. Centrally it blocks dopamine (D2) receptors in the CTZ (in high doses 5-HT3 antagonism may be observed), while peripherally it accelerates gastric emptying, reduces reflux from the duodenum and stomach into the oesophagus and enhances motility of the upper GIT. These latter effects may be mediated through an action on muscarinic cholinergic systems within the GIT.
Pharmacokinetics
Metoclopramide is almost completely absorbed following oral dosing, and peak plasma concentrations occur 30–180 min after oral administration, 10–15 min after an IM dose and within 5–20 min of an IV dose. The half-life in plasma is 2.5–5 h. Metoclopramide is extensively metabolised by the liver (about 70%) and excreted in urine.
Drug interactions
An additive CNS depressant effect is observed with a combination of metoclopramide and CNS depressant drugs. Avoid this combination or a potentially serious drug interaction could occur. Changes in absorption affect the plasma concentrations of cyclosporin and digoxin, which should be monitored and dosage adjustments made if indicated. In surgical patients, metoclopramide can reduce inactivation of succinylcholine and hence prolong neuromuscular blockade.
Adverse reactions
These include diarrhoea, sleepiness, restlessness, dizziness, headache, extrapyramidal (parkinsonian) effects, hypotension, tachycardia and, rarely, agranulocytosis and tardive dyskinesia (see Chapter 20).
Warnings and contraindications
Metoclopramide is contraindicated where a previous reaction to dopamine antagonists has been reported and in phaeochromocytoma because of a risk of a hypertensive crisis. The drug should be used with caution in Parkinson’s disease and depression, as it can worsen the symptoms. Dosage reduction (25%–50%) should be considered in situations of severe renal impairment, and low doses used in children because of an increased risk of extrapyramidal adverse effects.
Dosage and administration
To treat diabetic gastroparesis or gastro-oesophageal reflux in an adult, the oral dose of metoclopramide is 10 mg four times daily. Nausea and vomiting in children (<10 kg) is treated with a dose of 0.1 mg/kg (maximum 1 mg) twice daily. The maximum dose is 0.5 mg/kg/day for all age groups.
Source: AMH 2010.
Prochlorperazine is indicated for the treatment of nausea and vomiting due to causes such as migraine and vertigo, as in Ménière’s syndrome. Use is contraindicated where there is evidence of previous hypersensitivity to phenothiazines and in situations of CNS depression. Adverse reactions are common and include constipation, dry mouth, sleepiness, dizziness, blurred vision and extrapyramidal effects (parkinsonism in the elderly and dystonia in younger people). Less common reactions include skin rash, hypotension, peripheral oedema, agranulocytosis and cholestatic jaundice. Prochlorperazine is considered safe for use during lactation. (For additional information, including phenothiazine warnings and contraindications, see Chapter 18.)
Domperidone is used as a second-line treatment for GORD in symptomatic infants; however there are limited randomised controlled trials of its use in infants and findings to date suggest variable efficacy (Pritchard et al 2005).
Muscarinic receptor antagonists (anticholinergics)
Hyoscine hydrobromide is a competitive antagonist of the actions of ACh at muscarinic receptors and is used to prevent motion-induced (sea, air, car, train) nausea and vomiting by depressing conduction in the labyrinth of the inner ear. Overstimulation in this area is responsible for the nausea and vomiting of motion sickness common in ocean yacht races. Hyoscine is partially metabolised in the liver and excreted by the kidneys. The adverse effects of hyoscine are related to its anticholinergic effects; these include dry mouth, tachycardia, blurring of vision and, less commonly, constipation, mental confusion, fatigue and restlessness and irritability. Administration is recommended 30 minutes prior to travel. Travacalm contains dimenhydrinate, hyoscine hydrobromide and caffeine.
5-HT3-receptor antagonists
Ondansetron, dolasetron, granisetron and tropisetron are selective 5-hydroxytryptamine (5-HT, serotonin) antagonists (see Drug Monograph 29-4). 5-HT3 receptors are located peripherally on the vagus nerve terminal and centrally in the CTZ. One theory is that cancer chemotherapeutic agents cause the release of stored 5-HT from the enterochromaffin cells of the GIT, which stimulates 5-HT3 receptors located in the vagus nerve in the GIT (Veyrat-Follet et al 1997). Stimulation of vagal afferents via 5-HT3 receptors results in the CTZ initiating the vomiting reflex. When ondansetron is administered before antineoplastic therapy, 5-HT3 receptors in the brainstem and GIT are blocked. As a result, 5-HT released in response to the administration of antineoplastic agents cannot bind to 5-HT3 receptors and thus vomiting is prevented.
Drug monograph 29-4 Ondansetron
Ondansetron was the first of a new class of 5-HT receptor (5-HT3) antagonists approved for the prevention of nausea and vomiting associated with the use of cytotoxic agents and radiotherapy.
Pharmacokinetics
Absorption of ondansetron is maximal after 1–1.5 hours, oral bioavailability is ∼60% and its plasma halflife is 3–4 hours. It is extensively metabolised in the liver (about 90%) by CYP1A2, CYP2D6, and CYP3A4 and less than 10% is excreted as unchanged drug in urine.
Drug interactions
The combination of apomorphine and ondansetron should be avoided because of the risk of severe hypotension and bradycardia. Rifampicin, an inducer of CYP1A2 and CYP3A4, increases metabolism of ondansetron decreasing its efficacy, which may necessitate an increase in dose. The combination with tramadol can decrease tramadol’s analgesic effect.
Adverse reactions
Common adverse reactions include constipation, headache, anxiety and dizziness. Chest pain, hypotension and rash are infrequent and, rarely, anaphylaxis, extrapyramidal effects and seizures have been reported.
Warnings and contraindications
Ondansetron should be used with caution in patients with impaired liver function as plasma clearance can be reduced and dosage adjustment may be necessary.
Dosage and administration
To prevent cancer chemotherapy-induced nausea and vomiting in children over >1 year, ondansetron is administered at a dosage of 5 mg/m2 (maximum 8 mg) IV immediately before the start of chemotherapy, followed by 5 mg/m2 (maximum 8 mg) every 8–12 hours during chemotherapy and for 24 hours afterwards. The oral adult dose is 24 mg 1–2 hours before the start of cancer chemotherapy (AMH 2010).
Aprepitant and fosaprepitant
Substance P, a neurotransmitter that acts on neurokinin-1 (NK1) receptors, is widely distributed in the CNS. It is thought to be involved in pain transmission and emetic pathways. Aprepitant and fosaprepitant (the prodrug of aprepitant) are oral NK1 receptor antagonists that act centrally to control, in particular, chemotherapy-induced vomiting. They are most effective when used in combination with a 5-HT3 receptor antagonist and dexamethasone. Currently, use in combination with other antiemetic drugs has not been fully investigated. As a consequence of aprepitant metabolism by CYP3A4, potential drug interactions are likely with agents such as ketoconazole (an inhibitor of CYP3A4) and dexamethasone (a substrate for CYP3A4). When aprepitant is used concomitantly with oral dexamethasone, the dose of dexamethasone is halved. At present there are no data for its use in severe hepatic impairment; in children; or in pregnancy and lactation. Common adverse effects include diarrhoea, fatigue, headache, hiccoughs and, rarely, angio-oedema and urticaria.
Corticosteroids
Corticosteroids have been reported to be effective for chemotherapy-induced nausea and vomiting, either alone or when used in combination with other antiemetics. The mechanism of action is unknown, but it has been proposed that these drugs may inhibit prostaglandin synthesis and decrease 5-HT turnover in the CNS, which might be involved in cancer chemotherapy-induced vomiting. Research has indicated that certain prostaglandins (especially the E series) can induce nausea and vomiting.
Many studies with corticosteroids have involved the use of dexamethasone and methylprednisolone. Their effectiveness as antiemetics was a serendipitous discovery—it was noticed patients receiving various chemotherapeutic regimens had less nausea and vomiting when corticosteroids were one of the agents administered. It has since been established that the addition of corticosteroids to an antiemetic regimen enhances overall the antiemetic effect and can diminish the severity of some of the adverse reactions, e.g. diarrhoea. A full discussion of the pharmacology of corticosteroids can be found in Chapter 35.
The pancreas
Continued digestion and absorption of food in the small intestine relies on secretions from the accessory organs—the pancreas, liver and gallbladder. The pancreas (see Chapter 36) secretes about 1500 mL liquid daily, comprising water, sodium bicarbonate and enzymes such as pancreatic amylase, trypsin and chymotrypsin. The acidic duodenal chyme is neutralised by the aqueous component, which brings the pH within range for further digestion of nutrients by the pancreatic enzymes. Regulation of pancreatic secretion is complex; for example, to prevent erosion of the duodenal mucosa, the rate of delivery of acidic chyme into the duodenum is equalled by the rate of secretion of bicarbonate ions, which neutralise the chyme.
With the exception of diabetes mellitus (see Chapter 36), many pancreatic diseases have symptoms that are not readily diagnosed. Inflammation of the pancreas may be acute or chronic. Among the many causes are blockage of the pancreatic ducts, trauma to the pancreas, excessive alcohol consumption, drug use and tumours, cysts or abscesses. Symptoms are non-specific but ultimately include severe pain. Carcinoma of the pancreas is as difficult to diagnose as other pancreatic disorders.
Pancreatic enzyme supplements
The pancreas releases digestive enzymes and bicarbonate into the duodenum to help in the digestion of fats, carbohydrates and proteins. Bicarbonate neutralises acid and thus helps to protect the enzymes from both acid and pepsin. When acid chyme enters the duodenum, vagal stimulation regulates pancreatic secretion, and enzyme replacement therapy may be necessary for patients who have had the vagal fibres surgically severed or who have had surgical procedures that cause food to bypass the duodenum. In addition, replacement therapy is usually necessary in exocrine pancreatic enzyme deficiency states, chronic pancreatitis, cystic fibrosis, pancreatic tumours and pancreatic obstruction.
The supplements are all of porcine origin and contain principally lipase with protease and amylase. If possible, use enteric-coated products because the microsphere formulation resists gastric inactivation, so the enzymes reach the duodenum to hydrolyse fats into glycerol and fatty acids, proteins into peptides and starch into dextrins and sugars.
The most common adverse reactions include nausea, vomiting and abdominal pain. Hyperuricaemia and intestinal obstruction occur rarely. Dosage should be adjusted as necessary to suit the individual and is guided by the quality and quantity of stools. Use of these products should be avoided in people with hypersensitivity to pork proteins.
The gallbladder
Hepatocytes secrete into the bile canaliculi 800–1000 mL bile per day, which flows into the gallbladder, a pear-shaped organ 7–10 cm long and 2.5–3.5 cm wide, lying on the undersurface of the liver. Bile, a yellowish-green liquid (pH 7.6–8.6), contains water, bile acids, bile salts, cholesterol, phospholipids and bile pigments such as bilirubin. The gallbladder stores and concentrates the bile and, after a meal, contracts rhythmically, expelling bile into the duodenum. The bile salts aid in the emulsification and absorption of lipids in the small intestine.
Cholecystitis (i.e. inflammation of the gallbladder) is often associated with the presence of gallstones. The stones lodge in the gallbladder neck or ducts, causing congestion and oedema as bile builds up. This may be an acute or a chronic condition. Malignant tumours of the gallbladder are infrequent.
Drugs that affect the biliary system
Ursodeoxycholic acid is a minor constituent of human bile. Its administration results in a change in bile acid composition and an increase in bile acid output and bile flow. It is indicated for the treatment of chronic cholestatic liver disease and cholestasis related to cystic fibrosis. There are limited data on its use in children and pregnant women. Drugs such as cholestyramine, colestipol, charcoal and antacids can bind ursodeoxycholic acid, resulting in reduced absorption of the drugs. In contrast, ursodeoxycholic acid may increase the absorption of cyclosporin, leading to an increased cyclosporin plasma concentration. Diarrhoea is a common adverse reaction.
Key points


 The primary functions of the GIT system are digestion and absorption. These processes are facilitated by the motile and secretory properties of the GIT and associated organs. The secretory and muscular activities of the GIT are regulated by both intrinsic and extrinsic neural mechanisms. Drugs used for maintaining oral hygiene include mouthwashes, gargles, dentifrices and topical antifungals and antiviral agents. The term candidiasis (‘thrush’) is used commonly to refer to a superficial fungal infection; in the case of the mouth, rarely are fungi other than Candida involved. Antifungal agents including miconazole (oral gel) and nystatin (pastilles and oral suspension) are most commonly used for oral candidiasis. The oesophagus extends from the pharynx to the upper region (cardia) of the stomach and passes through the diaphragm at the oesophageal hiatus into the abdominal cavity. Oesophageal disorders are characterised by retrosternal pain (heartburn) and difficulty in swallowing (dysphagia). Gastric acid secretion is regulated by neural (parasympathetic) and hormonal (gastrin, histamine) mechanisms. Drugs that neutralise or inhibit gastric acid secretion include antacids, anticholinergics, antidepressants, anxiolytics, H2-receptor antagonists, proton pump inhibitors and cytoprotective agents. Antacids are chemical compounds that buffer or neutralise hydrochloric acid in the stomach and thereby raise the gastric pH. Antacid–drug interactions depend on the composition of the antacid used; in general, antacids have been reported most frequently to reduce or delay the absorption of many drugs. The drugs used in the treatment of peptic ulcer include cytoprotective agents, H2-receptor antagonists and the proton pump inhibitors. Cytoprotective agents enhance the protection afforded by the mucus layer by providing a physical barrier over the ulcerated surface. The two main agents are sucralfate and the prostaglandin analogue misoprostol. Histamine is produced in ECL cells of the oxyntic mucosa by decarboxylation of L-histidine by histidine decarboxylase. Released histamine then acts on histamine (H2) receptors to increase gastric acid secretion. The H2-receptor antagonists include cimetidine, ranitidine, famotidine and nizatidine. Proton pump inhibitors (PPIs) suppress gastric acid secretion by inhibiting the hydrogen–potassium adenosine triphosphatase (ATPase) enzyme system at the secretory surface of the gastric parietal cells. It is now well established that infection with Helicobacter pylori causes chronic active gastritis, is associated with the development of gastric and duodenal ulcers and is implicated in the development of gastric carcinoma. Use of H. pylori eradication regimens improves the odds of non-recurrence of peptic ulcers and success hinges on adherence to therapy and susceptibility of the bacterium to the antibiotics. The induction of vomiting involves a complex coordinated response between the chemoreceptor trigger zone (CTZ), located in the floor of the fourth ventricle of the brain, and the vomiting or emetic centre, located in the medulla. The process of vomiting involves multiple nerve pathways and neurotransmitters (e.g. acetylcholine, histamine, dopamine, substance P and 5-hydroxytryptamine). Antiemetics, which include dopamine antagonists, muscarinic receptor antagonists, 5-HT3 receptor antagonists and NK1 receptor antagonists, are given for the relief of nausea and vomiting, including that associated with cancer chemotherapy.
The primary functions of the GIT system are digestion and absorption. These processes are facilitated by the motile and secretory properties of the GIT and associated organs. The secretory and muscular activities of the GIT are regulated by both intrinsic and extrinsic neural mechanisms. Drugs used for maintaining oral hygiene include mouthwashes, gargles, dentifrices and topical antifungals and antiviral agents. The term candidiasis (‘thrush’) is used commonly to refer to a superficial fungal infection; in the case of the mouth, rarely are fungi other than Candida involved. Antifungal agents including miconazole (oral gel) and nystatin (pastilles and oral suspension) are most commonly used for oral candidiasis. The oesophagus extends from the pharynx to the upper region (cardia) of the stomach and passes through the diaphragm at the oesophageal hiatus into the abdominal cavity. Oesophageal disorders are characterised by retrosternal pain (heartburn) and difficulty in swallowing (dysphagia). Gastric acid secretion is regulated by neural (parasympathetic) and hormonal (gastrin, histamine) mechanisms. Drugs that neutralise or inhibit gastric acid secretion include antacids, anticholinergics, antidepressants, anxiolytics, H2-receptor antagonists, proton pump inhibitors and cytoprotective agents. Antacids are chemical compounds that buffer or neutralise hydrochloric acid in the stomach and thereby raise the gastric pH. Antacid–drug interactions depend on the composition of the antacid used; in general, antacids have been reported most frequently to reduce or delay the absorption of many drugs. The drugs used in the treatment of peptic ulcer include cytoprotective agents, H2-receptor antagonists and the proton pump inhibitors. Cytoprotective agents enhance the protection afforded by the mucus layer by providing a physical barrier over the ulcerated surface. The two main agents are sucralfate and the prostaglandin analogue misoprostol. Histamine is produced in ECL cells of the oxyntic mucosa by decarboxylation of L-histidine by histidine decarboxylase. Released histamine then acts on histamine (H2) receptors to increase gastric acid secretion. The H2-receptor antagonists include cimetidine, ranitidine, famotidine and nizatidine. Proton pump inhibitors (PPIs) suppress gastric acid secretion by inhibiting the hydrogen–potassium adenosine triphosphatase (ATPase) enzyme system at the secretory surface of the gastric parietal cells. It is now well established that infection with Helicobacter pylori causes chronic active gastritis, is associated with the development of gastric and duodenal ulcers and is implicated in the development of gastric carcinoma. Use of H. pylori eradication regimens improves the odds of non-recurrence of peptic ulcers and success hinges on adherence to therapy and susceptibility of the bacterium to the antibiotics. The induction of vomiting involves a complex coordinated response between the chemoreceptor trigger zone (CTZ), located in the floor of the fourth ventricle of the brain, and the vomiting or emetic centre, located in the medulla. The process of vomiting involves multiple nerve pathways and neurotransmitters (e.g. acetylcholine, histamine, dopamine, substance P and 5-hydroxytryptamine). Antiemetics, which include dopamine antagonists, muscarinic receptor antagonists, 5-HT3 receptor antagonists and NK1 receptor antagonists, are given for the relief of nausea and vomiting, including that associated with cancer chemotherapy.Review exercises
References and further reading
Andersson T. Drug interactions with omeprazole. Clinical Pharmacokinetics. 1991;21:195-212.
Australian Medicines Handbook 2010. Adelaide: AMH, 2010.
Bytzer P., O’Morain C. Treatment of Helicobacter pylori. Helicobacter. 2005;10(Suppl 1):40-46.
Gastrointestinal Expert Group. Therapeutic Guidelines: Gastrointestinal. In version 3. Melbourne: Therapeutic Guidelines; 2002.
Gisbert J.P., Pajares J.M. Helicobacter pylori ‘rescue’ therapy after failure of two eradication treatments. Helicobacter. 2005;10:363-372.
Habib A.S., Gan T.J. Evidence-based management of postoperative nausea and vomiting: a review. Canadian Journal of Anesthetics. 2004;51:326-341.
Hoogerwerf W.A., Pasricha P. Pharmacotherapy of gastric acidity, peptic ulcers, and gastroesophageal reflux disease. In Brunton L.L., Lazo J.S., Parker K.L., editors: Goodman & Gilman’s The Pharmacological Basis of Therapeutics, 11th edn, New York: McGraw-Hill, 2006. [ch 36]
Kovac A.L. Benefits and risks of newer treatments for chemotherapy-induced and postoperative nausea and vomiting. Drug Safety. 2003;26(4):227-259.
Lambert J. Testing for Helicobacter pylori. Australian Prescriber. 1997;20(4):96-97.
Magee L.A., Mazzotta P., Koren G. Evidence-based view of safety and effectiveness of pharmacologic therapy for nausea and vomiting of pregnancy (NVP). American Journal of Obstetrics and Gynecology. 2002;186:S256-S261.
Maton P.N., Burton M.E. Antacids revisited: a review of their clinical pharmacology and recommended therapeutic use. Drugs. 1999;57:855-870.
Mazzotta P., Magee L.A. A risk–benefit assessment of pharmacological and nonpharmacological treatment for nausea and vomiting of pregnancy. Drugs. 2000;59:781-800.
Mejia A., Kraft W.K. Acid peptic diseases: pharmacological approach to treatment. Expert Review Clinical Pharmacology. 2009;2:295-314.
Neuvonen P.J., Kivisto K.T. Enhancement of drug absorption by antacids: an unrecognised drug interaction. Clinical Pharmacokinetics. 1994;27:120-128.
Olver I.N. Aprepitant in antiemetic combinations to prevent chemotherapy-induced nausea and vomiting. International Journal of Clinical Practice. 2004;58P:201-206.
Podolsky D.K. Inflammatory bowel disease. New England Journal of Medicine. 1991;325(13):928-937.
Pritchard D.S., Baber N., Stephenson T. Should domperidone be used for the treatment of gastro-oesophageal reflux in children? Systematic review of randomized controlled trials in children aged 1 month to 11 years old. British Journal of Clinical Pharmacology. 2005;59(6):725-729.
Savarino V., Di Mario F., Scarpignato C. Proton pump inhibitors in GORD: an overview of their pharmacology, efficacy, safety. Pharmacological Research. 2009;59:135-153.
Schmidt E., Thoennissen N.H., Rudat A., et al. Use of palifermin for the prevention of high-dose methotrexate-induced oral mucositis. Annals of Oncology. 2008;19:1644-1649.
Stieler J.M., Reichardt P., Riess H., Oettle H. Treatment options for chemotherapy-induced nausea and vomiting: current and future. American Journal of Cancer. 2003;2:15-26.
Tighe M.P., Afzal N.A., Bevan A., Beattie R.M. Current pharmacological management of gastro-esophageal reflux in children. Pediatric Drugs. 2009;11(3):185-202.
Veyrat-Follet C., Farinotti R., Palmer J.L. Physiology of chemotherapy-induced emesis and antiemetic therapy. Drugs. 1997;53:206-234.