Chapter 38 Drugs Affecting the Female Reproductive System
This chapter reviews the anatomy and physiology of the female reproductive system and its control by higher centres. Drugs that are used to affect the female reproductive system are analogues or antagonists of the pituitary gonadotrophins or oxytocin, or of the ovarian hormones oestrogen and progesterone. They are administered to mimic or suppress the biological effects of endogenous hormones, to supplement inadequate production (e.g. in the menopause), to correct hormonal imbalance (e.g. in the menstrual cycle or in dysfunctional bleeding), to reverse abnormal processes (endometriosis, anovulation and infertility) and for contraception.
The physiology of pregnancy is reviewed briefly and the physiological changes that may impinge on pharmacokinetic parameters of drugs used in pregnancy are discussed, including risks of fetal effects and teratogenesis. Drugs affecting uterine smooth muscle activity include those that induce labour (oxytocics) or inhibit premature labour (tocolytics). These, along with analgesics and anaesthetics, are the typical medications used during the perinatal period in women. Lactation can be affected by drug therapy, as can the infant if drugs pass across into breast milk; guidelines for use of drugs by lactating mothers are described.
Privacy and cultural sensitivities often influence patients’ perceptions of reproductive disorders, so sensitive and appropriate health care, teaching and counselling are required for patients with dysfunctions of the reproductive system.
COC combined oral contraceptive
FSH follicle-stimulating hormone
GnRH gonadotrophin-releasing hormone
hCG human chorionic gonadotrophin
HRT hormone replacement therapy
Key background: the female reproductive system
IN human beings, the reproductive process is highly complex, involving reproductive organs, ducts and supporting structures specialised for producing gametes and hormones, facilitating fertilisation and sustaining the growth of the embryo and fetus. The reproductive system of the human female consists of the ovaries, oviducts (fallopian tubes), uterus and vagina (Figure 38-1). The main functions of the sex hormones are to:
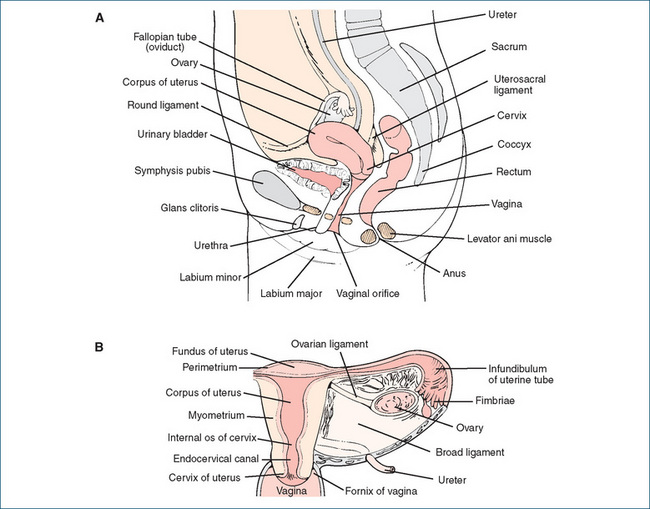
Figure 38-1 The female reproductive system. A Sagittal section of pelvic area. B Anterior section of the uterus, adnexa and upper vagina.
The gonadal hormones concerned with reproduction in females are oestrogens (oestradiol, oestrone, oestriol) and progestogens (progesterone, hydroxyprogesterone).
Hypothalamic and pituitary control
Hypothalamic releasing factors
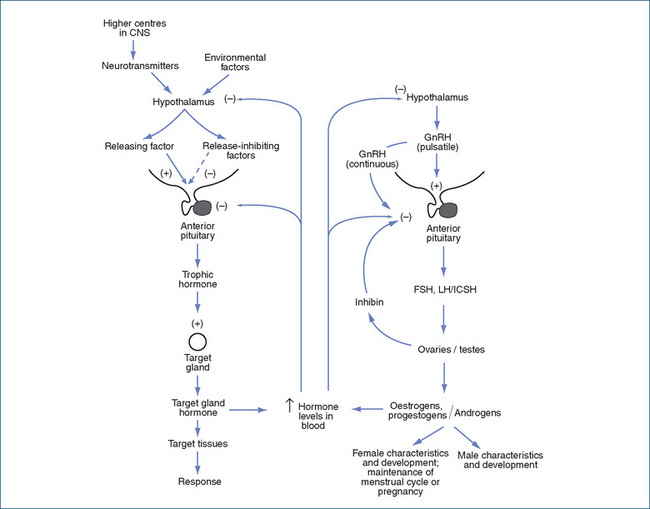
As with most other endocrine glands, there are several levels of control (see Figure 38-2, and compare with Figures 33-2 and 33-4). The hypothalamus secretes a releasing factor (gonadotrophin-releasing hormone [GnRH, gonadorelin]), which stimulates the anterior pituitary gland to release gonadotrophins (follicle-stimulating hormone [FSH] and luteinising hormone [LH]). Other hormones involved in reproduction include prolactin and oxytocin, from the anterior and posterior pituitary gland, respectively; the gonadal hormones; inhibin (which decreases secretion of FSH and LH); and hormones secreted during pregnancy by the placenta (chorionic gonadotrophin and placental lactogen).
Later in life, gonadal function decreases. Women undergo menopause, or cessation of menses. Men have a decrease in sex hormone production, which may lead to physiological and psychological changes sometimes called the male climacteric.1
Gonadotrophin-releasing hormone (GnRH)
GnRH, formerly called FSH-releasing hormone or LH-releasing hormone, is a 10-amino-acid peptide. GnRH has a similar role in the control of sex hormone release to that of corticotrophin-releasing factor (CRF) in the control of adrenal cortex steroids (compare Figures 38-2 and 35-2). The actions of GnRH depend on how it is administered. At low (physiological) doses, and given in a pulsatile intermittent dose schedule, it stimulates pituitary synthesis and release of the pituitary gonadotrophins FSH and LH. By contrast, continuous administration in higher doses results in desensitisation and ultimately in decreased pituitary production of LH and FSH. Thus continuous high doses cause suppression of production of sex hormones in the target glands (ovaries, testes), which can be used to inhibit growth of steroid-dependent tumours.
Gonadorelin, a synthetic form of GnRH identical to the natural hormone, has been used to induce ovulation and as an adjunct to other tests to diagnose hypogonadism in both males and females. There are now synthetic GnRH analogues, with slightly different amino acid sequences, which are more potent and have longer durations of action. At higher continuous doses they are used to suppress the pituitary, as described above (see Table 38-1).
Table 38-1 Characteristics of gnrh analogues
| GnRH analogue | |
| Examples | Goserelin, leuprorelin, nafarelin, triptorelin |
| Administration | Continuous, e.g. SC or IM depot, or nasal spray |
| Rationale | To suppress pituitary gonadotrophin release |
| Indications | Treatment of prostate or breast cancers, endometriosis, uterine fibroids and before controlled ovarian stimulation |
| Half-life (approx) | 3–4 hours |
| Adverse reactions | Impaired fertility, decreased bone density; males: impotence, gynaecomastia; females: hot flushes, headache, pain, menstrual problems, hypercalcaemia |
| Precautions | Pregnancy, lactation |
GnRH inhibitors
Danazol is a compound that inhibits release of GnRH and is used to suppress the pituitary–ovarian axis, suppress menstruation and inhibit ovulation. It has complicated actions: it is an androgen agonist; it also alters sex hormone metabolism, interacts with sex hormone receptors and has weak anabolic effects. Overall, it inhibits ovarian oestrogen production and causes atrophy of endometrial tissue if given in doses sufficient to abolish menstruation. It is mainly used in endometriosis, menorrhagia and fibrocystic breast disease. Common androgenic adverse effects include acne, hirsutism, vaginal dryness and hair loss. Danazol is contraindicated in pregnancy and lactation, cardiovascular disease and liver or kidney impairment.
Another inhibitor of gonadotrophin release, gestrinone, is also used in endometriosis, as it has antiprogesterone actions but no oestrogenic activity (although it does have some partial agonist actions at both progestogen and androgen receptors).
Two GnRH antagonists have recently been released for use by specialist physicians in assisted reproduction programs: cetrorelix and ganirelix. These drugs compete with GnRH for binding sites in the pituitary gland and prevent the surge in gonadotrophins that causes ovulation, so they can allow programmed timing of ovulation and harvesting of follicles.
Pituitary gonadotrophic hormones
The pituitary gonadotrophins, FSH and LH, are glycoprotein hormones responsible for the development and maintenance of sexual gland functions. They can be purified and used clinically in male or female infertility and hypogonadism, and in combination with human chorionic gonadotrophin for stimulation of multiple oocyte development in ovulatory patients who are using other technologies to conceive, such as gamete intrafallopian transfer (GIFT) or in-vitro fertilisation (IVF).
Prescribing of these hormones is usually restricted to specialists, such as endocrinologists and physicians involved in assisted reproduction clinics, as close monitoring of hormone levels and responses is essential.
Follicle-stimulating hormone stimulates the development of the Graafian ovarian follicles up to the point of ovulation in the female, which in turn brings on the characteristic changes of oestrus (menstruation in women). Luteinising hormone (also known in the male as interstitial- cell-stimulating hormone, ICSH) acts in the female to promote the maturation of the follicle, the formation of the corpus luteum and the secretion of oestrogen.
Other natural gonadotrophins include human chorionic gonadotrophin (hCG), secreted by the chorion (the outer membrane enveloping the fetus); this hormone is measurable in the urine of pregnant women within a few days of fertilisation and is the antigen detected in pregnancy tests. hCG has mainly LH-type actions and is used to induce ovulation and to treat hypogonadism (see Drug Monograph 38-1).
Drug monograph 38-1 Human chorionic gonadotrophin (hCG)
The action of human chorionic gonadotrophin is similar to that of pituitary LH, with few or no follicle-stimulating effects. This drug is administered to make up for a deficiency in LH.
INDICATIONS hCG is indicated for:
PHARMACOKINETICS Administered IM, the drug has a half-life of 12–36 hours; in the female, ovulation usually occurs within 32–36 hours of administration. It is excreted by the kidneys within 24 hours. It has no significant drug interactions.
ADVERSE REACTIONS These include mood changes, aggression, headache, tiredness, oedema, precocious puberty, gynaecomastia, ovarian hyperstimulation and arterial thromboembolism; multiple pregnancies can occur.
WARNINGS AND CONTRAINDICATIONS Use with caution in breastfeeding women and in people with epilepsy, migraine headaches, asthma or heart or kidney disease. Avoid use in people with chorionic gonadotrophin hypersensitivity, precocious puberty (and prostate cancer), and in pregnant women.
DOSAGE AND ADMINISTRATION For induction of ovulation, a dose of 5000–10,000 units IM is admin is tered, usually after suppression of endogenous LH levels by GnRH. (The adult dosage for male hypogonadotrophic hypogonadism is 500–1000 units IM 2–3 times a week for several weeks to months or in some cases indefinitely. For boys with prepubertal cryptor chidism, the dose is also 500–1000 units IM 2 times a week.)
New recombinant forms of chorionic gonadotrophin are choriogonadotropin alfa and lutropin alfa; these are used for their LH activities. Other gonadotrophins prepared by recombinant DNA technology are follitropin alfa and beta. They are used for their FSH-type actions.
Puberty
After about the age of 10 years, hormonal changes start to occur in both sexes; during puberty, there is increased output of GnRH, with pulsatile release occurring throughout the day, causing gradual development of secondary sexual characteristics. In girls, the pituitary gland is stimulated to produce increased levels of FSH and LH, and the ratio of LH:FSH secretion stimulates the ovaries to produce oestrogen and progesterone, leading to a surge of LH that causes ovulation. The oestrogenic steroids stimulate maturation of the female reproductive organs, development of secondary sexual characteristics and accelerated growth, followed by closure of the epiphyses of the long bones.
The first menstrual period (menarche) is closely correlated with a skeletal age of 13 years, but may occur some years earlier or later (range 9–15 years; see Figure 33-5). The actual ‘clock’ that switches on reproductive development and function is unknown; the prolonged period of childhood in humans allows a long period of learning before the onset of reproductive life. The ‘clock’ depends on stimulus from the nervous system and can be affected by stress and emotion. After puberty, the female hormones are involved with regulation of the menstrual cycle or of pregnancy until about the age of 50, when levels of oestrogens decrease and menstruation ceases (the menopause).
The menstrual cycle
Uterine changes occurring during the menstrual cycle, and the actions of pituitary gonadotrophins and ovarian hormones, are illustrated in Figure 38-3. The physiological processes of a typical 28-day cycle are summarised below:
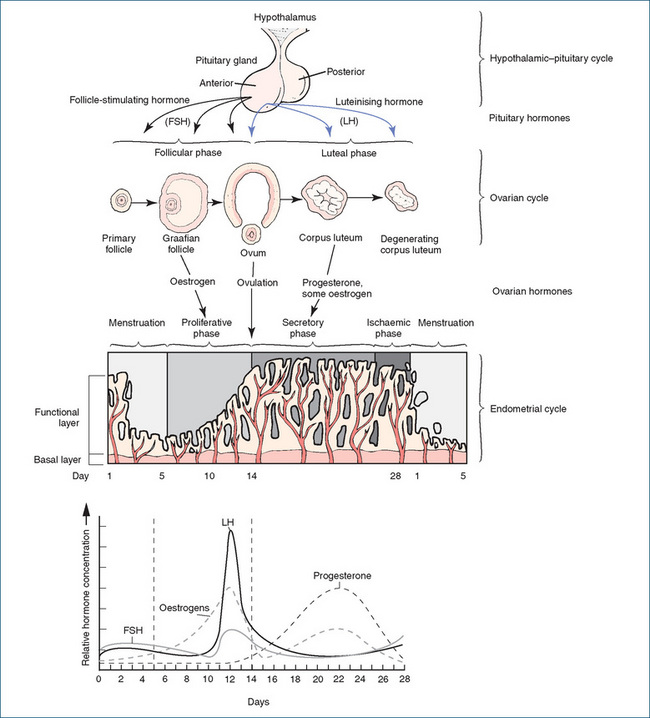
Ovulation
This description of the menstrual cycle is based on a 28-day cycle, but the timing of ovulation varies as does the length of cycles (25–31 days);2 therefore ovulation is not always predictable. (Physiologically, this is the primary reason for the unreliability of the ‘rhythm’ or ‘safe period’ method of contraception, which depends on predicting the day of ovulation, based on previous menstrual cycles. Ovulation most predictably occurs 14 days before the next menstrual period, which requires a very regular cycle to be reliably predicted.) The cyclical nature of secretion of oestrogens and progesterone is critical to normal female reproductive functions, and needs to be mimicked if the hormones are given exogenously, e.g. in the oral contraceptive (OC) pill or post-menopausal hormone replacement therapy (HRT) formulations.
 Disorders of menstruation
Disorders of menstruation
Some disorders of menstruation are listed below, with the recommended treatments. Uterine cramps during menstruation can be very painful, particularly in young women and those who have never been pregnant; non-steroidal anti-inflammatory drugs (NSAIDs) or OC formulations are usually effective. (The debate about whether menstruation should be optional is summarised in Clinical Interest Box 38-1).
Clinical interest Box 38-1 Should menstruation be optional?
Modern women menstruate during their reproductive lives probably hundreds more times overall than did women in earlier centuries, when women reached menarche later, had earlier first births, had more pregnancies, breastfed for longer periods and had shorter life expectancies, often dying during childbirth. Yet menstruation can be easily and safely suppressed by the OC pill (by continuing to take active tablets every day of the month). Why are women not informed of this possibility, and encouraged to try it?
Reasons to suppress menstruation:
Reasons to continue menstruation:
Menstrual control with OC preparations could sub stantially improve the health and quality of life of many women. Women should at least know that this safe, simple and inexpensive option is available if they prefer not to menstruate every month.
Adapted from: Thomas & Ellertson 2000.
The female sexual response
In humans, basic endocrine and neurochemical effects involved in sexual functions are usually overridden by psychological, social, cognitive and emotional influences. In mammalian animals, sexual behaviour and the sexual response (mating) is more likely during oestrus (the recurring period of sexual receptivity in female mammals), which coincides with peak levels of LH and ovulation, implying maximal likelihood of conception.
In women, increasing levels of oestrogen secretion especially during the preovulatory period of the menstrual cycle (Figure 38-3) enhance sexual functioning; there is evidence of increased frequency of intercourse during the middle of the cycle. Erectile tissue in the breasts, vaginal opening and clitoral areas is under autonomic parasympathetic nerve control; acetylcholine acting through muscarinic receptors causes responses such as slow heart rate, dilation of blood vessels and stimulation of secretions. During sexual intercourse dilation of arteries leads to venous compression and engorgement of erectile tissue and increased mucus secretion inside the vagina. Catecholamine neurotransmitters are also involved, both in neuroendocrine integration (dopamine acting as the prolactin release-inhibiting factor) and in vascular control. During orgasm there may be 3–15 rhythmic contractions of the vagina, uterus and perineal muscles, caused by bursts of sympathetic nerve impulses and reflex release of oxytocin from the posterior pituitary gland; the contractions help to promote passage of sperm and seminal fluid and fertilisation of the ovum.
Sexual behaviours in animals may be inhibited by a rise in brain 5-hydroxytryptamine (5-HT, serotonin) levels and by centrally acting muscarinic agonist drugs. These findings have implications for influencing human sexual behaviours with drugs such as selective serotonin reuptake inhibitors and anticholinergic agents.
Female sex hormones
THE ovaries, in addition to producing ova, synthesise and secrete two main types of hormones that control female secondary sex characteristics, the reproductive cycle and the growth and development of the accessory reproductive organs in the female. The oestrogenic, or follicular, hormones (oestrogens)3 are produced by the cells of the developing Graafian follicle; and the progestational, or luteal, hormones (progestogens) are derived from the corpus luteum that is formed in the ovary from the ruptured follicle. Periodic cycling of levels of the female sex hormones results in the menstrual cycle that normally continues throughout reproductive life from menarche until menopause, except during pregnancy.
Oestrogens
Synthesis
Although oestrogens are primarily secreted by the ovarian follicles, some may also be synthesised from androgens secreted by the adrenal glands, and by the corpus luteum, placenta (up to 100 times the pre-pregnancy levels), liver and testes. The chemical structures of representative steroids are shown in Figure 33-3 and the biosynthetic pathways of adrenal cortex steroids from cholesterol are shown diagrammatically in Figure 35-1. It can be seen that the ‘A’ ring of oestrogens is an aromatic ring, unlike that of most other steroid hormones. There are three main endogenous (occurring naturally in women) oestrogens: oestradiol, oestrone and oestriol; oestradiol is metabolised in the body to oestrone and oestriol.
Oestrogens used as drugs are available from natural sources (e.g. the urine of pregnant mares) in conjugated dosage forms known as conjugated equine oestrogens. The three natural oestrogens are rapidly metabolised in the liver, so esterified or semisynthetic derivatives (e.g. oestradiol valerate, ethinyloestradiol (EE), mestranol) have been prepared, which are active orally because they are less rapidly metabolised. The natural oestrogens tend to be used in HRT formulations, which require only low doses, whereas the synthetic hormones, especially EE, are used in oral contraceptive preparations (Table 40-1).
Actions
The main physiological actions of oestrogens are:
Oestrogens appear to be protective against Parkinson’s disease, as evidenced by its lower prevalence in women than men. Overall, oestrogens enhance cell proliferation and the humoural immune response, and women have a higher prevalence of autoimmune disorders such as rheumatoid arthritis and systemic lupus erythematosus.
Mechanism of action
The mechanism of the actions is through activation of oestrogen receptors (ERs) in oestrogen-responsive target tissues, especially in the uterus, vagina and breast. As with other steroid hormones, oestrogens diffuse through the cell membrane, bind to membrane-associated receptors (ERs) and induce a conformational change; ligand-receptor dimers then translocate to the nucleus and, after recruitment of coactivators of the receptor, there is interaction with DNA and activation of gene transcription, leading to altered synthesis of proteins. There are also non-genomic signaltransduction pathways and responses (see review by Ellmann et al [2009]).
Oestrogen receptors
Oestrogen receptors have been classified into alpha- and beta-subtypes (ERα, ERβ), with many different isoform variants; they are members of the nuclear receptor superfamily Class I, which includes receptors for progesterone and adrenocorticosteroids. 17β-oestradiol is the main endogenous ligand bonding to ERα/β; oestrone and oestriol bind with lower affinity. Identification of ER types in various tissues, and characterisation of their acting via different transduction mechanisms or causing specific effects, is not yet complete.
Clinical uses
Clinically, oestrogens are used very commonly. A large number of the world’s female population use them daily for many years, either in oral contraceptive pill preparations, as depot contraceptive implants or devices or as hormone replacement therapy in the menopause (see Drug Monograph 38-2). Other indications include:
Drug monograph 38-2 Oestradiol valerate
Oestradiol valerate is an ester derivative of the natural hormone and has the pharmacological actions of oestrogens on the ovary and uterus. It is used in HRT to prevent and treat oestrogen-deficiency symptoms after ovariectomy or menopause, including hot flushes, osteoporosis and vaginitis.
PHARMACOKINETICS Oestradiol valerate is a prodrug, a precursor of oestradiol-17β. It is rapidly and completely absorbed and is quickly metabolised in the liver to oestradiol, then to various metabolites, of which oestriol and oestrone have oestrogenic activity.
Oestradiol is highly protein-bound and has a half-life of about 24 hours. Metabolites are excreted in the liver and bile. Bioavailability is about 3%.
DRUG INTERACTIONS See Drug Interactions 38-1 for significant drug interactions.
SIDE EFFECTS AND ADVERSE REACTIONS These include typical oestrogenic effects: breast pain and enlargement, changes in menstrual bleeding, headaches, nausea, vomiting, a change in female libido, oedema of the lower extremities and chloasma (darkened patches of skin on the face). There is an increased risk of thromboembolism. Administration by skin patch can cause irritation and dermatitis.
WARNINGS AND CONTRAINDICATIONS Oestrogens should be used with caution in smokers and in patients with endometriosis, diabetes, migraine and epilepsy. Avoid use in pregnancy (Pregnancy Safety Category B1) and in women with oestrogen hypersensitivity, thromboembolic disorders or severe cardiovascular disease, breast cancer, severe liver disease or hypercalcaemia. Oestrogens reduce lactation and are excreted in breast milk, so administration to nursing mothers is not recommended.
DOSAGE AND ADMINISTRATION The lowest effective dose of oestrogens should be administered for the shortest time period to reduce the possibility of overstimulation of oestrogensensitive tissues. A cyclical dosing schedule of 3 weeks of oestrogen administration (1–4 mg daily) and 1 week off, or the addition of a progestogen for the last 10–13 days of the cycle, will most closely approximate the natural hormonal menstrual cycle. The progestogen must be added to a schedule of HRT in postmenopausal women with an intact uterus to avoid the possibility of endometrial hyperplasia or carcinoma. The prescriber should re-evaluate the patient at least annually.
Transdermal oestradiol is also used for women with oestrogen deficiency, particularly as HRT in menopause. Applied topically to intact skin, 25–100 mcg (0.025–0.1 mg) is released daily from the transdermal patch. It should be replaced twice weekly and is usually worn continuously. Sensitivity reactions at the application site often occur. Oestradiol is also formulated in many combination tablet types and as a nasal spray, vaginal pessary, transdermal gel and implant.
Administration
Oestrogens are administered by many routes: as tablets, implants, injections, patches, creams, nasal inhalations or pessaries. Being lipid-soluble, they are well absorbed orally and pass readily through membranes. They are circulated in the bloodstream bound to albumin and globulin. There is some enterohepatic recycling as is usual with steroids (see under ‘Biliary excretion’ in Chapter 6). They are metabolised rapidly in the liver, with a high first-pass effect; metabolites are excreted in the urine and bile as conjugates. Conjugated oestrogens, a mixture of oestrogenic substances (especially oestrone and equilin), are available in oral, parenteral and vaginal cream dosage forms.
Dosages of oestrogens must be individualised according to diagnosis, clinical use, route of administration and therapeutic response, e.g. for contraception, treatment of breast cancer or vasomotor symptoms associated with menopause. Typical ethinyloestradiol daily doses may be 20–50 mcg in the OC pill and 1–3 mg in breast cancer treatment.
Adverse drug reactions and interactions
Oestrogenic adverse reactions are dose-dependent and include nausea and vomiting, dizziness, fluid retention, irregular menstrual bleeding and breast tenderness. In high doses (such as were present in early formulations of the OC pill), there was increased incidence of thromboembolic disorders such as deep vein thrombosis (DVT) and stroke. Because of this risk, the OC pill is not recommended for women over 35 years who smoke. In previous decades (1950s to 1960s), diethylstilboestrol (DES) was prescribed to pregnant women in an attempt to decrease the risk of spontaneous miscarriage in women for whom this was a problem. It was shown to cause increased incidence of cancer of the vagina or cervix in their daughters 15–20 years after their birth (i.e. the fetus had been affected by DES in utero), so it has been withdrawn from use.
Significant drug interactions with oestrogens are summarised in Drug Interactions 38-1. Most are due to interactions with ethinyloestradiol (EE), which is metabolised by CYP3A4; taking a CPY3A4 inducer with a combined oral contraceptive containing EE can lead to contraceptive failure and pregnancy. Oral contraceptives can increase blood glucose concentrations, hence can interact with any other drugs affecting blood glucose levels (see Table 36-2). The doses of oestrogens in HRT formulations are considerably lower, so fewer drug interactions are expected.
Drug interactions 38-1 Some potentially significant interactions with oestrogens
| Drug | Possible effects and management |
| Enzyme inducers (barbiturates, some antiepileptics, aprepitant, bosentan, modafinil, some antivirals, rifamycins, St John’s wort) | May accelerate metabolism of oestrogens and reduce their activity; additional contraception such as condoms should be used if required |
| Atazanavir, etoricoxib | Increase concentrations of EE and risk of adverse effects; low dose EE (30 mcg) oral contraceptive formulations should be used or a different interacting drug selected |
| Antibiotics (e.g. ampicillin) | May reduce potency of oestradiol because of metabolism by altered gut flora |
| Tobacco (smoking) | Tobacco smoking increases the risk of serious cardiac adverse reactions, such as cerebrovascular accident, transient ischaemic attacks, thrombophlebitis and pulmonary embolism. The risk is higher in women over 35 who smoke; oral contraceptives not recommended |
| Orlistat | Orlistat-induced diarrhoea may reduce oestrogen levels; additional contraception may be necessary |
| Selegiline | COCs may increase selegiline concentration, enhancing its effects and adverse effects; reduce selegiline dose or avoid combination |
| Thyroid hormones | Oestrogens may alter concentrations of thyroid hormones, requiring increased thyroxine dose; thyroid function should be monitored |
COC = combined oral contraceptive.
Selective oestrogen receptor modulators (SERMs)
Knowledge about oestrogen receptors and mechanisms by which drugs may activate or repress oestrogen-dependent gene transcription is helping in the development and clinical use of new anti-oestrogens and selective oestrogen receptor modulators (SERMs). The aim is to find tissuespecific modulators that lower the risk of oestrogendependent cancers (breast and endometrial) yet have positive effects on bone, the cardiovascular system, liver function, CNS function and menopausal symptoms.
The SERM class of drugs, of which raloxifene is the first clinically used example, have agonist actions at some ERs (particularly in bone) and on lipid metabolism and antagonistic actions on other ERs (in breast and uterus). Hence SERMs are potentially useful in the long term for menopausal symptoms, to protect against osteo porosis and cardiovascular disease, while not increasing the risk of breast or uterine cancers. In clinical use raloxifene has been shown to increase bone mineral density, reduce bone resorption, reduce incidence of vertebral fractures and improve the blood lipid profile, with no stimulation of the endometrium or breast tissue; however, the hot flushes associated with menopause may be exacerbated. There is a potential increased risk of venous thromboembolism and stroke. It is contraindicated in pregnancy (category X).
Related drugs such as toremifene, lasofoxifene, basedoxifene and arzoxifene have improved effects in enhancing bone mineral density and reducing cholesterol levels; each SERM appears to have a unique array of clinical activities and thus potential uses.
Tamoxifen, long used as an anti-oestrogen in breast cancer (see below), is now recognised as a SERM. These SERMs have potential uses in many oestrogen-dependent gynaecological conditions, and are being tested in some prostate cancers.
Anti-oestrogens
Tamoxifen binds to most ERs but does not stimulate transcription and so has few oestrogenic actions. It is mainly used as an oestrogen antagonist (anti-oestrogen) in post-menopausal oestrogen-dependent breast cancers4 (see Drug Monograph 42-3). However, it has pro-oestrogenic effects in uterine tissue, so can potentially cause endometrial hyperplasia and cancer.
Fulvestrant, a pure anti-oestrogen, competes for binding with the ER and prevents its dimerisation and nuclear localisation; it thus abolishes all oestrogenic actions including protective effects. It inhibits the growth of ER-positive tumours and is used to treat locally advanced or metastatic breast cancer in post-menopausal women.
Clomiphene acts by rather different mechanisms: it inhibits oestrogen binding in the hypothalamus and pituitary gland (Figure 38-2), reducing the negative feedback effects of circulating oestrogens so that more GnRH is produced, which has the effect of increasing the release of gonadotrophins, especially LH. The raised oestrogen levels from the stimulated ovaries induce ovulation. The drug is therefore useful to treat female infertility. However, there are major risks: multiple pregnancies, ovarian enlargement and oestrogenic adverse effects.
Progesterone and progestogens
Synthesis
Progesterone produced by the ovaries is the main naturally occurring progestogen, along with hydroxyprogesterone. LH secreted from the anterior pituitary stimulates the synthesis and secretion of progesterone from the corpus luteum, mainly during the latter half of the menstrual cycle. Progesterone is also formed during pregnancy (in the placenta, from steroid precursors), when it functions to promote breast development, maintain pregnancy and prevent further ovulation. (Interestingly, it is now known that progesterone and progesterone receptors [PRs] are synthesised in some CNS neurones when stimulated by circulating oestrogens, which partly accounts for the mid-cycle LH surge.)
Actions
The normal physiological actions of progesterone are to:
Progesterone is also thermogenic, i.e. it raises the core body temperature.5 Progestogens are commonly used clinically in hormonal contraception (both combined with an oestrogen and in progestogen-only formulations), in endometriosis (to suppress the ovaries) and in HRT (in women with an intact uterus, to oppose oestrogen’s actions).
Mechanism of action and progesterone receptors
Progesterone receptors (PRs) are also members of the nuclear receptor superfamily Class I; the physiological ligand is (obviously) progesterone. The main signalling pathway via cellular genomic response is as described above for oestrogens on ERs.
Clinical aspects
Progesterone itself is virtually inactive orally, as it is very rapidly metabolised in the liver; it can be administered IM or per vagina to avoid this first-pass effect. When used to treat amenorrhoea caused by hormonal imbalance, the dosage is 5–10 mg IM daily for 6–10 days. Bleeding will usually occur within 2–3 days of the last injection; normal menstrual cycles may then follow. Progesterone is also formulated as a gel or pessary for per vaginal (PV) administration in assisted reproduction technologies.
Orally active derivatives, collectively called progestogens (or progestins), have now been developed and have similar pharmacological effects in the body to those of the natural hormones. The advantages with these synthetic progestogens are the availability of an effective oral or sublingual dosage form and a longer duration of action.
Progestogens commonly used include:
Drug monograph 38-3 Medroxyprogesterone acetate
Medroxyprogesterone acetate (MPA) is a typical synthetic progestogen.
INDICATIONS Progesterone and progestogens are indicated for oral contraception (alone or combined with an oestrogen); in long-term contraception (as an IM depot preparation); in postmenopausal HRT; and in the treatment of female hormonal imbalance in amenorrhoea, dysmenorrhoea, endometriosis and specific carcinomas (breast, endometrial, renal cell; specialist oncologist use only).
PHARMACOKINETICS MPA is rapidly absorbed after oral administration and has an apparent half-life of about 30 hours. It is metabolised in the liver, with metabolites excreted by the kidneys.
DRUG INTERACTIONS The effectiveness of oral progestogens, and possibly of the etonogestrel implant, may be reduced by enzyme-inducing drugs, particularly rifamycins, many anticonvulsants and some antivirals; contraceptive failure may result, so alternative contraception may be necessary.
ADVERSE REACTIONS These include menstrual irregularities (amenorrhoea, breakthrough bleeding), breast pain, weight gain, loss of bone mineral density, stomach pain and cramps, oedema, headache and mood alterations including depression, nausea, hyperglycaemia, chloasma and galactorrhoea. Progestogens that are synthetic testosterone derivatives may cause androgenic and adrenocorticoid effects (virilisation, acne, libido changes, fluid imbalance).
WARNINGS AND CONTRAINDICATIONS Should be used with caution in patients with asthma, cardiac insufficiency, epilepsy, hypertension, migraine, kidney or liver function impairment, central nervous system disorders, diabetes mellitus, hyperlipidaemia, thrombophlebitis and uterine, genital or urinary tract bleeding (undiagnosed). Avoid use in women with progestogen hypersensitivity, breast or genital tract cancer (contraceptive use), severe liver disorders or thromboembolic disease, and in pregnancy.
DOSAGE AND ADMINISTRATION Low physiological dosages are used for progestational effects in replacement therapy, and higher doses to suppress ovulation and menstruation and inhibit gonadotrophin production; current package inserts or reference texts need to be consulted for specific recommendations. The following are examples of selected dosing regimens:
Antiprogestogens
Mifepristone6 (RU486) is a partial agonist that binds strongly to PRs and prevents the activity of endogenous progestogens, hence blocks the LH surge and prevents implantation. It also has anti-glucocorticoid activity and sensitises the uterus to PGs. It has been used in treatment of uterine leiomyomas and endometriosis, and also as an emergency contraceptive. Since progesterone inhibits uterine contractions at all gestational stages, the use of an antiprogestogen will cause contractions, and thus mifepristone is used in combination with a PG (gemeprost) for early termination of pregnancy (it is sometimes known as the ‘abortion pill’). It has also been trialled for third trimester induction of labour, for ripening of the cervix and to reduce the need for caesarean section (see later discussion of oxytocic agents). Mifepristone is not marketed in Australia (as at 2010) but may be available through the Special Access Scheme; it is available in New Zealand.
Gestrinone, an antiprogestogen, inhibits ovulation and reduces oestrogen levels, thus causing endometrial atrophy; it is indicated in treatment of endometriosis. It has complex actions including inhibition of gonadotrophin release plus some agonist activity at both androgen and progesterone receptors. The most common adverse effects are virilisation (acne, weight gain, hirsutism, hair loss and voice deepening), which may not be reversible on cessation of treatment. It is contraindicated in pregnancy (category D) as it can cause virilisation of a female fetus.
 Menopause and hormone replacement therapy
Menopause and hormone replacement therapy
Menopause
Menopause is loosely defined as the end of the reproductive period of a woman’s life, when ovulation and menstruation cease; it is a process rather than an event. As with puberty, there is no exact chronological age of initiation, rather a transitional period during which changes occur relatively quickly. The timing varies with ethnicity, geographical regions, socioeconomic status, extremes of body weight, smoking status, amount of heavy exercise and a woman’s menstrual, gynaecological and surgical history. The term ‘climacteric’ is used to refer to the signs, symptoms and consequences of the perimenopause, i.e. the period around the menopause.
The menopausal transition can occur over a wide age range—from 42 to 58 years of age. The average age of the last menstrual period, around 51, has not changed significantly in recorded history (except that it is earlier in women who smoke), despite the increasingly younger onset of menarche and improved living conditions and nutrition. Until about 1900, the median survival age for women was only about 45 years, hence a large proportion of women never reached menopause. The average lifespan of women in developed countries is now over 80, so most women can expect to spend over one-third of their lives in menopause.
Physiological changes
The changes occurring during menopause are considered to be due to the depletion of the stock of ova and follicles (estimated to be 0.2–2 million at birth) by decades of ovulation and atresia. Inhibin B levels fall, and gonadotrophins are still released, but the ovaries are less responsive, so there is a relatively sudden decrease in secretion of oestrogens and hence decreased ovulation and menstruation, decreased progesterone production, atrophy of genital organs and breasts and reduced protein anabolism. Diagnosis is confirmed by low blood levels of oestrogens and progestogens despite high levels of FSH (due to reduced negative feedback from ovarian oestrogens and inhibin), LH and GnRH. There is still some production of oestrone and of testosterone and androstendione, and libido and sexual performance are not necessarily decreased.
Many women suffer some unpleasant symptoms during the early years of menopause: hot flushes (during which there is a sudden rise in temperature and excessive sweating, thought to coincide with bursts of release of GnRH), nausea, insomnia, vaginitis, palpitations, increased risk of ischaemic heart disease, fatigue, depression, breast tenderness and osteoporosis (overall bone calcium content decreases by 1%–2% per year; see Chapter 37 for treatment of osteoporosis). However, for many women, menopause is a relief from decades of menstruation and the possibility of unplanned conception.
Hormone replacement therapy
In some women, menopausal symptoms are so severe as to need treatment. Post-menopausal hormone replacement therapy involves daily low doses of a natural oestrogen (e.g. oestradiol), possibly for many years. In women with an intact uterus, i.e. those who have not undergone surgical hysterectomy, it is important that a dose of a progestogen also be given 10–12 days per month to prevent endometrial hyperplasia and the risk of endometrial cancer; this is known as ‘opposed’ therapy. The adverse reactions, drug interactions and contraindications are generally as for the constituent oestrogen and progestogen, but the oestrogen dose is considerably lower than in OC preparations, so fewer adverse reactions are expected.
The WHI studies
The Women’s Health Initiative (WHI) was a major 15-year research program set up by the USA Department of Health and Human Services to address the most common causes of death, disability and poor quality of life in post-menopausal women—cardiovascular disease, cancer and osteoporosis. The WHI was launched in 1991 and consisted of a set of clinical trials and an observational study, which together involved 161,808 generally healthy post-menopausal women. The clinical trials were designed to test the effects of post-menopausal hormone therapy, diet modification and calcium and vitamin D supplements on heart disease, bone fractures and breast and colorectal cancer.
The hormone trial had two studies: the oestrogen-plusprogestin study of women with a uterus and the oestrogenalone study of women without a uterus. In both, women were randomly assigned to either the hormone medication being studied or to placebo. After the studies ended, many important results were published and disseminated widely (see Writing Group for the Women’s Health Initiative Investigators 2002, and WHI website under ‘On-Line Resources’). The women in these studies are now participating in a follow-up phase, which will last until 2010.
Benefits and risks
The large-scale WHI clinical trials of combined oestrogen plus progestogen in healthy post-menopausal women showed the following results:
HRT also reduced hot flushes and night sweats, urogenital symptoms and arthralgia; demonstrated improvements in sleep problems and sexual problems may have been secondary to other effects.
Prescribing of HRT
After the publication of the results of the WHI trials, prescribing of HRT dropped dramatically around the world, due to the fear of breast cancer (which appears now to have been over-emphasised). Subsequent studies have shown that women who ‘went off’ HRT were often switched instead to clonidine, anxiolytics, sedatives, osteoporosis drugs or antidepressants, all of which also have potential adverse effects.
The results from the WHI studies were quickly absorbed into prescribing guidelines worldwide, including into the Australian Therapeutic Guidelines: Endocrinology and into the Australian Medicines Handbook. The consensus at present appears to be that HRT in low doses is useful in the short term (up to 5 years) to relieve moderate–severe menopausal symptoms and to prevent osteoporosis, but its safety in the long term is less definite and may depend on the woman’s family history of, and predisposition to, cardiovascular disease, osteoporosis and specific cancers (breast, endometrial, colorectal). The need for continued use should be reviewed annually (MacLennan 2003; Anonymous, National Prescribing Service 2009).
Formulations and administration
The natural oestrogens are preferred because of their lower potency and fewer adverse reactions. Oral oestrogen preparations include conjugated equine oestrogens, oestriol and oestradiol valerate (see Drug Monograph 38-2). A low dose is taken daily, e.g. oestradiol valerate 1–2 mg orally. The oral progestogen used most commonly in HRT is MPA (see Drug Monograph 38-3); most of the progestogens used in the OC pill formulations are also used, at similar dosage levels for 12 days/month, e.g. norethisterone 0.5–1 mg, drospirenone 2 mg or dydrogesterone 10 mg. Combination packs are available with various permutations of oestrogen with or without progestogen for varying periods of a cycle, as described in Clinical Interest Box 38-2.
Clinical interest Box 38-2 Hrt—why? why not? how? for how long?
Compared with women not using HRT, those taking oestrogen HRT after the menopause have been shown to have:
HRT is also effective for the treatment of vaginal atrophy and associated sexual problems.
Possible disadvantages of HRT include:
Relative contraindications include liver disease, thromboembolism and hypertension.
Many HRT formulations are available:
There is considerable debate about how long women can continue taking HRT:
Adapted from: MacLennan 2000 and 2003; Australian Medicines Handbook 2010.
Transdermal oestrogen may be preferred, as it bypasses the liver, avoids first-pass metabolism and is considered more physiological in terms of the oestradiol:oestrone ratio. It is more expensive than tablets, however, and many women (10%–20%) suffer unpleasant skin reactions (redness, irritation, itchiness). A combination patch formulation has been released, with four patches containing oestradiol 4 mg and four patches with norethisterone acetate 30 mg and oestradiol 10 mg. The recommended regimen is one of the first type of patch every 3–4 days for 2 weeks, then one of the second type every 3–4 days for 2 weeks. (This mimics the natural menstrual cycle, with low oestrogens in the first 2 weeks, then higher oestrogen and progestogen in the next 2 weeks.) This schedule is indicated for women intolerant of oral oestrogens or who prefer not to take daily tablets.
Other formulation types are a transdermal gel, SC implant, vaginal cream and pessaries. As there are many types and formulations of HRT available, subject to frequent changes, it is recommended that a current reference such as Australian Medicines Handbook be consulted for specific details and comparisons.
Other menopause therapies
SERMs and phytoestrogens
Selective oestrogen receptor modulators (SERMs, see earlier notes under ‘Oestrogens’), also known as tissuespecific oestrogens, are currently being tested for their effects in menopause. It is hoped that, because of their selectivity on ERs in bone and on metabolic processes, they will have an advantageous spectrum of pharmacological activities. Clinical trials have shown that they effectively decrease bone resorption and improve the blood lipid picture, while not stimulating breast or endometrial tissue growth.
Phytoestrogens (plant compounds with oestrogenic activity) are also of great interest, especially as they are advertised as being more ‘natural’ than white tablets in calendar packs (see Clinical Interest Box 38-3). They are isoflavones, coumestans and lignans occurring in plants and seeds, especially in clovers and soya beans.7 Pharmacologically, they are best classified as SERMs, as they have varying agonistic and antagonistic activities in oestrogen-responsive tissues and also have oestrogen enzyme-modulating and antioxidant activities. They could potentially be useful in control of menopausal symptoms but clinical trials so far have shown little if any benefit over placebo. In Australian Indigenous medicine, berries of the ‘kangaroo apple’, Solanum aviculare, have been used for contraception. Active extracts contain steroids, alkaloids and solasadine, and have been used in the synthesis of OCs and cortisone.
Clinical interest Box 38-3 Hrt—why? why not? how? for how long?
In menopausal symptoms, including hot flushes, various results have been obtained in clinical studies of comple mentary and alternative medicine methods:
In nausea and vomiting of pregnancy, acupressure, ginger and vitamin B6 have been shown to be effective. The herb dong quai (Angelica sinensis) has long been used in traditional Chinese medicine for treating menstrual disorders, among other conditions. It has a volatile oil constituent that relaxes the uterus, and a non-volatile con stituent that produces strong uterine contractions. Overall, the effects depend on the functional state of the uterus; there is no good evidence of efficacy in menopausal symptoms.
In breast cancer, some relief of symptoms (nausea, pain, depression) has been obtained with acupuncture, mind–body therapies and mistletoe and other herbs that may enhance immune functions.
In premenstrual syndrome, symptoms of breast tenderness and neuropsychological symptoms such as anxiety and depression have been shown in some studies to be reduced with use of black cohosh, chaste tree berries, evening primrose oil or Gingko biloba; however, the evidence is not strong.
Adapted from: Spencer & Jacobs 1999; Braun & Cohen 2007; Duffy et al 2007; Nedrow et al 2006.
‘Bioidentical’ hormone therapy (natural hormones such as oestriol, progesterone and testosterone, com pounded extemporaneously by a pharmacist) are promoted as being more natural and safer than the same hormones produced in a laboratory by a drug company, and ‘customised’ to an individual woman’s needs. However there is little scientific evidence to support such claims, and any chemicals with oestrogenic or androgenic actions are liable to have the same adverse effects and interactions as the same hormones in synthetic formulations.
Tibolone
Tibolone is an interesting drug found useful in treating menopausal symptoms, especially in protecting against bone resorption and in elevating mood and libido. It is neither an oestrogen nor a SERM but is classified as a ‘gonadomimetic’, with oestrogenic, anti-oestrogenic, progestogenic and androgenic effects.
Tibolone has a steroid structure that is rapidly metabolised, after oral administration, to active metabolites that have oestrogenic effects in the vagina, bone and thermoregulatory centre, and progestogenic and androgenic actions in the breast and bloodstream. It appears not to stimulate the endometrium, so it is potentially a good alternative to HRT in women with an intact uterus. Overall, tibolone is useful in treating menopausal symptoms of hot flushes and vaginal dryness, and in preventing reduced bone mineral density. Adverse reactions are mild and rare and include headache, dizziness, excess hair growth and acne. In older women, stroke risk was increased; recurrence of breast cancer was increased. Precautions are required in women with a history of hormone-dependent tumours or cardiovascular or hepatic diseases. Results from studies of long-term administration of tibolone are still being accumulated.
Non-hormonal therapies
Useful lifestyle modifications include regular light exercise, reducing stress, wearing layered clothing and practising ‘sleep hygiene’ (see Chapter 16). Factors to be avoided are smoking and excessive caffeine, alcohol and spicy foods.
Antidepressants of the selective serotonin or noradrenaline reuptake inhibitor types (SSRIs, SNRIs) have a small effect in reducing hot flushes. 5-HT is known to be involved in many CNS functions including emotions and mood, control of body temperature, endocrine regulation and sleep. High doses of gabapentin, an anticonvulsant, also reduce frequency of hot flushes by approximately two per day. Gabapentin binds to particular subunits of calcium channels, and inhibits the release of various neurotransmitters and modulators. Its main clinical uses are in treatment of epilepsy, anxiety and neuropathic pain and prophylaxis of migraine. Clonidine, a partial agonist at β2-adrenoceptors and a centrally-acting antihypertensive agent, also reduces frequency of hot flushes; its mechanism of action in this condition is unclear.
Treatment of gynaecological disorders
Many gynaecological disorders can be treated with combinations of the sex hormones, to either stimulate or inhibit normal endocrine functions. The main drug groups—oestrogens, progestogens, hormone inhibitors, hypothalamic factors and pituitary hormones—have already been considered in detail.
Endometriosis
Endometriosis is the location of endometrial tissue at unusual (ectopic) locations, e.g. within the oviducts, ovaries, myometrium or pelvis; the aetiology is complex and multifactorial, and may involve altered ERs or metabolism. It is relatively common, occurring in about 10% of women during their lifetime. As all endometrial tissue is subject to stimulation by gonadotrophic and ovarian hormones, ectopic tissue will also undergo cyclical changes during the menstrual period and produce menstrual fluid that cannot escape the abdominal cavity or other location. This can lead to ovarian cysts, pain and chronic inflam mation, scar formation, infertility, dysmenorrhoea and painful intercourse. First-line treatment is surgical, to remove ectopic endometrial tissue. Pharmacological treatments tried include progestogens, OC preparations, danazol (to inhibit GnRH and block LH surge) or GnRH analogues such as gestrinone (continuous dose for gonadal suppression), with NSAIDs to reduce inflammation and pain.
Dysfunctional uterine bleeding
Dysfunctional uterine bleeding (DUB) occurs in about 5% of menstruating women and may be related to ovulation or to anovulatory cycles. It is relatively common around puberty and menopause, and in association with platelet dysfunction, cervical cancer, endocrine disorders or in women with low body-fat mass or who do excessive exercise. The primary causes should be sought and anaemia monitored. Pharmacological treatment of DUB associated with ovulation may include antianaemic drugs (iron, folic acid), NSAIDs (e.g. naproxen) to relieve symptoms, tranexamic acid (an antifibrinolytic agent), the OC pill or depot progestogen. Less frequently, danazol or GnRH agonists are used. Treatment of anovulatory DUB depends on the cause but usually involves balancing progestogen and oestrogen levels with OC or HRT preparations. Surgical treatments include dilatation and curettage of the uterus, laser endometrial ablation and, as a last resort, hysterectomy.
Dysmenorrhoea and premenstrual syndrome are treated similarly with NSAIDs, OCs, a diuretic and sometimes also vitamin B6, or an SSRI such as fluoxetine.
Polycystic ovary syndrome
Polycystic ovary syndrome is present in approximately 5%–10% of women of child-bearing age; it is a common endocrine cause of infertility, menstrual irregularities, hirsutism, acne and male pattern alopecia in women. It may be associated with obesity, metabolic syndrome, diabetes mellitus, ovarian dysfunction and hyperandrogenic anovulation. Adipocytes are now known to synthesise and release mediators (adipokines), including leptin and tumour necrosis factor α, involved in cardiovascular risk and insulin resistance (see review by Radosh [2009]).
The most common therapies are COC tablets, antiandrogens and hypoglycaemic agents, especially insulin sensitisers such as metformin and thiazolidinediones (glitazones, see Figure 36-3). In women hoping to conceive a baby, ovulation inducers such as clomiphene citrate (Drug Monograph 40-1) and aromatase inhibitors are used (see Chapter 40 for treatment of infertility).
Hirsutism
Hirsutism, or excessive hair growth in women, can be distressing and require treatment. It may be an inherited trait or an adverse reaction to certain drugs, including phenytoin, minoxidil or steroids such as the progestogens based on the testosterone structure. Hirsutism can also be due to a relative androgen excess at the hair follicles, especially on the lower face and midline of the trunk. The androgens may come from hyperactive ovaries (e.g. polycystic ovaries), from the adrenal cortex or from other virilising endocrine disorders.
Possible treatments include suppression of ovarian functions, e.g. with OC preparations; suppression of the adrenal cortex, e.g. with dexamethasone; or administration of antiandrogens such as cyproterone or spironolactone.
Drugs during pregnancy, the perinatal period and lactation
Drugs during pregnancy
Hormone levels in pregnancy
If implantation of a fertilised ovum occurs during the second stage of a woman’s cycle, i.e. if she becomes pregnant, the chorionic gonadotrophin (see Drug Monograph 38-1) secreted ‘rescues’ the corpus luteum, which continues to secrete progesterone (the pro-pregnancy hormone), so that uterine lining is not shed, menstruation does not occur and all the changes of pregnancy commence. By the second and third month, the placenta is maturing and acts as an autonomous endocrine gland, secreting large amounts of progesterone, oestrogens, chorionic gonadotrophin and human chorionic somatomammotrophin (also called human placental lactogen). Other hormones, including growth hormone, adrenocorticotrophic hormone and thyroid-stimulating hormone, can also be secreted by the placenta. Various hormone actions are important in pregnancy, and are summarised below.
Progesterone levels from the corpus luteum and placenta continue to rise sharply throughout pregnancy, to about 10 times the amount in late menstrual cycle. Progesterone relaxes the myometrium, ensures the cervix remains closed, assists oestrogens in development of breast tissue and has an immunosuppressant effect, ensuring that the maternal immune system does not reject the (immunologically ‘non-self’) fetus.
Oestrogens (oestradiol, oestriol and oestrone) are secreted in increasing amounts, to about 10 times pre-pregnancy levels. Oestrogens stimulate breast development, encourage a 20-fold increase in weight of the uterus (excluding the conceptus) and cause skin changes. There is a further sharp rise in oestriol levels in the last few weeks of pregnancy, which enhances haemostatic mechanisms and may mediate dilation of the birth canal early in labour.
Human chorionic gonadotrophin is measurable in the urine within a few days of fertilisation and is the antigen detected in pregnancy tests. Levels remain very high for the first 2–3 months, then decrease rapidly. This hormone has effects similar to those of LH (the chorion is the outermost of the two membranes that enclose the embryo; the inner membrane is the amnion).
Human chorionic somatomammotrophin levels rise steadily throughout pregnancy. This hormone is related to growth hormone: it helps prepare the mammary glands for lactation, enhances maternal growth and alters protein, fat and glucose metabolism so that more glucose and amino acids are available for the fetus (a consequence is exacerbation or development of diabetes mellitus in the mother).
Prolactin levels from the anterior pituitary rise steadily throughout pregnancy and prepare the breasts for lactation. Relaxin, a peptide hormone produced by the corpus luteum and later the placenta, increases the flexibility of the pelvic bones and joints during pregnancy and helps dilate the uterine cervix during labour and delivery. Corticotrophin-releasing factor is secreted by the placenta, especially late in pregnancy, and may help control the timing of childbirth. It also increases the secretion of adrenal cortex hormones, which help maturation of the fetal lungs and production of surfactant (see Chapter 28).
Hormone levels can be readily measured during pregnancy (e.g. in maternal urine), which allows for close monitoring of how the pregnancy is progressing, and treatment if required.
Common conditions in pregnancy
Maternal adaptations to pregnancy include anatomical changes due to the expanding fetus putting pressure on the mother’s internal organs and weight gain due to the fetus, placenta, amniotic fluid and enlarged uterus. There are also physiological adaptations, which may affect how the mother responds to drugs or how her body handles drugs. In the cardiovascular system, for example, cardiac output, stroke volume, heart rate and blood volume all increase by 15%–30%. Similarly, pulmonary function increases to meet the demands of the fetus for oxygen, and pressure on the urinary bladder increases frequency of urination. Decreased gastrointestinal tract motility and increased risk of nausea and vomiting may delay absorption of nutrients and drugs.
Common conditions occurring in pregnancy include nausea, heartburn and ‘morning sickness’. These can be severe enough to threaten the health of the mother and fetus, so antiemetic drugs may be required. Anaemias are common because of the increased demand for iron and blood cell functions, so iron and folic acid are frequently prescribed. Urinary tract infections are also more common. Other chronic conditions that the mother suffered before the pregnancy, such as diabetes, epilepsy, asthma, hyper tension, peptic ulcer, migraine, depression, thyroid disorder or urinary tract infection, will need careful monitoring and treatment to optimise the mother’s (and fetus’s) health.
Miscarriage, or spontaneous abortion, before 20 weeks may be a natural rejection of an abnormal embryo or fetus. Threatened abortion (miscarriage) later in pregnancy can sometimes be successfully delayed by tocolytic drugs. Therapeutic abortion, sometimes carried out if there is a threat to the life or health of the mother, is assisted by oxytocic drugs (see later sections).
Pre-eclampsia
Pre-eclampsia (toxaemia of pregnancy) is a potentially dangerous combination of hypertension, proteinuria and oedema, thought to be due to renal ischaemia. It can occur at any time in the second half of pregnancy and complicates 2%–8% of pregnancies. Treatment is with magnesium sulfate by IV infusion or slow IM injection, to control convulsions. Large doses (several grams of magnesium sulfate over a period of hours) are given and can lead to magnesium toxicity, manifest by loss of reflexes and respiratory and cardiovascular collapse. (Magnesium is being used in treatment of many other varied conditions, from migraine, arrhythmias, severe asthma, dysmenorrhoea, leg cramps and metabolic syndrome to dyspepsia and constipation.)
Pre-eclampsia is a warning sign for the dangerous complication eclampsia, a condition carrying a high maternal and fetal mortality. The only safe treatment of eclampsia, which can cause convulsions, coma and death, is delivery of the baby.
Drug use in pregnancy
Because most drugs taken by a pregnant woman can cross the placenta (see Chapter 9) and affect the fetus, it is important to know the relative risks of known harmful effects of medicines on the developing fetus, and balance these against the need of the pregnant woman for drug therapy. The Australian Drug Evaluation Committee (ADEC) has categorised drugs on the basis of their potential for harmful effects during pregnancy (see Table 9-1), so that the safest effective drug can be prescribed.
Drugs in the perinatal period
Drugs given to the mother in the perinatal period (i.e. the period around childbirth, defined as from week 28 of pregnancy to the end of the first week of the infant’s life) may affect the fetus or infant, due to drug passage across the placenta or into breast milk. Infants are born with immature hepatic and renal functions, so drugs given to them are cleared more slowly than in older people and may reach toxic concentrations. Another problem is that the immature blood–brain barrier of a premature or very young infant may allow passage into the CNS of drugs (and normal metabolites including bilirubin), which may have central effects or adverse effects. (These pharmacokinetic aspects are discussed in Chapter 9; the pharmacodynamic and clinical aspects are covered in chapters on the drug groups.)
Drugs affecting the uterus
The uterus is a highly muscular organ: the smooth muscle fibres extend longitudinally, circularly and obliquely in the organ. The uterus has a rich blood supply; however, blood flow is diminished when the uterine muscle contracts. The myometrium undergoes rhythmical contraction; the contractions originate in muscle, with the myometrial cells in the fundus (the inner surface of the dome of the uterus) acting as pacemakers. During the menstrual cycle, contractions are weak on days 6–14, then become gradually larger and more prolonged, until during menstruation (days 1–5) the contractions are strong and coordinated and can lead to cramping pain. Drugs that act on the uterus include oxytocics, which increase uterine contractility, and tocolytics, which decrease it (from the Greek stem tokos: birth).
Profound changes occur in the uterus during pregnancy: it increases in weight from about 50 g to around 1000 g, its capacity increases 10-fold in length and new muscle fibres may be formed. The rhythmical contractions of the myometrium are depressed during pregnancy by the high concentrations of progesterone and oestrogens. These changes may be accompanied by changes in response to drugs.
Control of uterine activity is via excitatory and inhibitory sympathetic fibres: noradrenaline (acting at α2 receptors) causes stimulation of muscle contraction, and adrenaline (at β2-adrenoceptors) causes inhibition, i.e. relaxation of uterine muscle, hence the use of β2-receptor stimulants as tocolytics. Adrenoceptor antagonists appear to have no effect on uterine muscle activity.
Hormones during parturition
During the last few weeks of pregnancy, painless uterine contractions become increasingly frequent and the lower uterine segment and cervix become softer and thinner, preparing the uterus for parturition (childbirth). The key event stimulating labour in women is still not determined; it appears to involve complex interactions of several placental, fetal and maternal hormones. Suggested factors causing initiation of labour include:
After labour has been initiated, regular uterine contractions moving downwards from the fundus of the uterus help expel the fetus. The process of birth usually takes several hours, with the stages of dilation of the cervix (6–12 hours) and expulsion (delivery of the baby, 10 minutes to several hours) taking much longer than the third stage, delivery of the placenta (10–30 minutes). The birth process is stressful on both mother and baby. The fetal adrenal medulla responds by secreting high amounts of catecholamines, and the adrenal cortex by secreting corticosteroids, all of which help prepare the infant for independent existence, clear the lungs, provide surfactant for breathing, mobilise nutrients and promote increased blood flow to the brain and heart.
Drugs during parturition
Because many drugs are available for use during labour and delivery, it is important to consider the benefit versus risk to both the mother and the fetus. The pharmacokinetics of drugs may be altered during labour and delivery: e.g. during labour, gastric emptying is delayed and vomiting may result, which would alter drug absorption (see Chapter 8). Vomiting may also be exacerbated by the use of opioid analgesics. Because oral drug absorption is unpredictable at this time, parenteral routes should be used. Drug metabolism and excretion may be altered and prolonged during labour and, although clinical data are currently sparse, the potential for inducing adverse or undesirable effects is always a concern. If a drug such as an opioid analgesic or sedative is potentially harmful to the fetus, then the lowest effective dose for the mother should be used.
Drugs commonly used during childbirth include:
Other drugs used by the mother may have the predictable effects in the neonate, e.g. β-blockers depress the infant’s cardiovascular system, insulin causes hypoglycaemia and antithyroid drugs can cause goitre. ‘Social drugs’ can also affect the infant; for example, babies born to women who smoke are smaller and have an increased incidence of jaundice, and babies born to women dependent on narcotic analgesics such as heroin suffer a withdrawal syndrome after birth, which can be alleviated by administration of an opioid (Clinical Interest Box 21-7). Alcohol consumption during pregnancy can cause severe abnormalities; see Clinical Interest Box 9-1.
Oxytocics
Agents that stimulate contraction of the smooth muscle of the uterus, resulting in contractions and labour, are oxytocics. The most commonly used oxytocics are synthetic oxytocin, alkaloids of the plant fungus ergot and PGs of the E and F series. Many other drugs may have some effect on the contractility of uterine smooth muscle, but their effects are too non-specific to be clinically useful. Oxytocics are used to induce labour, to reduce postpartum haemorrhage and to terminate pregnancy.
Oxytocin
Oxytocin is one of two hormones secreted by the posterior pituitary; the other hormone is vasopressin, or antidiuretic hormone (see Chapter 33). Oxytocin means ‘rapid birth’, a term derived from its ability to contract the pregnant uterus. It also facilitates milk ejection during lactation. The non-pregnant uterus is relatively insensitive to oxytocin, but uterine sensitivity to oxytocin gradually increases during pregnancy, with the uterus being most sensitive at term. Oxytocin secretion may precede and possibly trigger delivery of the fetus. Large amounts of oxytocin have been detected in the blood during the expulsive phase of delivery. A positive feedback mechanism may be operant: more forceful contractions of uterine muscle and greater stretching of the cervix and vagina result in more oxytocin release, until birth of the baby. Oxytocin acts directly on the myometrium, having a stronger effect on the fundus than on the cervix, thus helping propel the fetus down the birth canal. Oxytocin also transiently impedes uterine blood flow and stimulates the mammary gland to increase milk excretion from the breast, although it does not increase the production of milk. The clinical use of oxytocin is described in Drug Monograph 33-4; it is administered IM or by IV infusion.
Carbetocin, a new oxytocin analogue, has longer duration of action (41 minutes compared to 1–5 minutes); it is administered as a single slow IV injection, 100 mcg dose. While a single IV injection may be preferable to a long infusion, the overall efficacy and adverse effects appear to be similar to those of the natural hormone.
Ergot alkaloids
The ergot alkaloids are naturally occurring compounds that have varied effects in the body through their actions on several types of receptors (see Clinical Interest Box 38-4). Ergometrine produces prolonged, strong contractions of the uterus, especially postpartum, and has vasoconstrictor actions, so is useful in treatment of postpartum haemorrhage (see Box 8Drug Monograph 38-4).
Clinical interest Box 38-4 Hrt—why? why not? how? for how long?
Ergot is a fungus (Claviceps purpurea) that grows on the grain rye. Poisoning (ergotism) occurs from eating bread made from ergot-infected rye. Symptoms of ergotism are mainly related to ergot’s vasocon strictor actions: dry cold skin, ischaemic pain in muscles, gangrene of the extremities, dizziness and CNS depression or convulsions; powerful contractions of the uterus can cause abortion.
In Europe in the middle ages, victims of ergotism travelled to St Anthony’s Shrine near Vienne, in France, to pray for cure, as St Anthony was considered to have special powers to protect against fire, infections and epilepsy. The intense pain from ischaemic limbs was thus referred to as ‘St Anthony’s fire’. Often the pilgrims were healed, possibly because they had removed themselves from the area where the grains were infected with ergot. The infamous ‘bewitchings’ that occurred in Salem, Massa chusetts, in December 1691 are also thought to have been due to ergotism, as the previous summer had been hot and wet, conducive to good growth of fungi on grain, causing convulsions and miscarriages attributed to witchcraft.
An early British pharmacologist, HH Dale, studied the activity of ergot extracts in the early 1900s. Dale showed that ergot extracts contain, in addition to active amines such as histamine, acetylcholine and tyramine, some alkaloids that antago nise actions of adrenaline. The ergot alkaloids (i.e. nitrogencontaining active prin ciples of ergot) were identified as derivatives of lysergic acid amide. They have varied (and confusing) effects on many receptors and tissues:
Other ergot derivatives include methysergide (a powerful 5-HT antagonist used prophylactically in migraine), bromo criptine (a dopamine agonist used to inhibit prolactin secretion and in treatment of Parkinson’s disease) and lysergic acid diethylamide, the psychedelic hallucinogen better known as LSD. (See Chapter 21 and Figure 21Figure 21-4.)
Drug monograph 38-4 Medroxyprogesterone acetate
Ergometrine increases the force and frequency of uterine contractions by direct stimulation of the smooth muscle of the uterine wall. The increased contractions and muscle tone and the vasoconstriction of bleeding vessels at the placental site arrest haemorrhage. It is indicated to prevent and treat postpartum haemorrhage. Ergometrine is contraindicated to induce labour or during the first two stages of labour because the cervix of the uterus would be contracted, as well as the fundus, causing fetal distress and compression.
PHARMACOKINETICS Ergometrine has unpredictable bioavailability, so is given parenterally. It is formulated for IM administration (or, in emergency, IV). It has a rapid onset of action, within 1–3 minutes. The duration of uterine contractions after IM injection is about 3 hours. The drug is metabolised in the liver and excreted mainly in the faeces.
DRUG INTERACTIONS Ergometrine has significant interactions with many drugs that also affect receptors for noradrenaline or 5-HT. The effects of other vasoconstrictors are enhanced, whereas those of antianginal vasodilators are antagonised. Drugs that reduce the metabolism of ergometrine (e.g. erythromycin, clarithromycin and some antivirals) may cause ergotism and ischaemia.
ADVERSE REACTIONS These include nausea, vomiting, hypertension, headache and, rarely, infarction, pulmonary oedema and gangrene. A dose-related effect is abdominal cramping.
WARNINGS AND CONTRAINDICATIONS Use with caution in patients with hypocalcaemia. Avoid use in women with ergot alkaloid hypersensitivity, cardiac or vascular disease, eclampsia or pre-eclampsia, sepsis or liver or kidney function impairment. Contraindicated in pregnancy, during the first two stages of labour or to induce labour.
DOSAGE AND ADMINISTRATION Parenterally, 0.5 mg is administered IM or IV; the IV route is recommended only in emergencies or in patients with excessive uterine bleeding. Ergometrine (0.5 mg/mL) is also formulated in combination with oxytocin (5 IU/mL) for active management of the third stage of labour.
Prostaglandins
Prostaglandins are produced in the endometrium and myometrium; PGF2 is a vasoconstrictor, while PGE2 and prostacyclin are vasodilators. Both PGEs and PGFs contract uterine smooth muscle and are implicated in disorders of menstruation. The uterus becomes increasingly sensitive to PGs during pregnancy, so they can be used to produce abortion.
In obstetrics, PGs are used to soften and dilate the cervix (‘uterine priming’) and to stimulate uterine contractions, as follows:
The PG analogues are used only where emergency gynaecological care is available. (They are also used to prevent dyspepsia and peptic ulceration induced by NSAIDs, in the treatment of erectile dysfunction in men and topically as eye-drops to treat glaucoma; see Drug Monograph 31-1.)
Premature labour inhibitors (tocolytics)
Preterm labour, or labour that occurs before the 37th week of pregnancy, is a major problem in obstetrics, occurring in 10%–15% of all pregnancies. Premature birth increases the possibility of neonatal morbidity and mortality. Drugs that relax the uterus, and hence delay labour or inhibit threatened abortion, are described as tocolytics. They may be administered in order to prolong the time of the fetus in utero by 2–7 days. This allows time for maturation of the fetus or for administration of corticosteroids to the mother to facilitate fetal production of lung surfactant, and/or to allow transport of the mother to a special centre for delivery of a significantly preterm infant.
Tocolytics are not used in conditions in which prolongation of pregnancy is hazardous for the fetus or mother. Most tocolytics are effective in stopping labour for 2–3 days; however, many studies have shown that they do not themselves improve perinatal outcomes or decrease the rate of preterm delivery. Responses of both fetus and mother (heart rate, glycaemia) must be carefully monitored.
Nifedipine and salbutamol
The main drugs used as tocolytics are the calcium channel blocker nifedipine, the β2-adrenoceptor agonist salbutamol and the oxytocin antagonist atosiban (not available in Australia but available in New Zealand). Other drugs that may relax uterine smooth muscle include progestogens, general anaesthetics, alcohol, magnesium sulfate, direct vasodilators and NSAIDs by their actions inhibiting prostaglandin synthesis).
The preferred drug for delaying labour is now nifedipine; this is a new use for the calcium channel blocker, which is usually indicated in the treatment of hypertension and angina. In threatened preterm labour, standard 20 mg tablets can be given every 3–8 hours until contractions cease or labour becomes too well established to stop, or up to 34 weeks gestation.
The clinical use of β2-adrenoceptor agonists as bronchodilators in asthma has been described (Drug Monograph 28-2). When used as a tocolytic, salbutamol is usually administered by IV infusion or IV injection, for management of uncomplicated preterm labour (24–34 weeks’ gestation). The drug is infused at a slowly increasing rate up to 45 microgram/minute until contractions cease, maintained for 1 hour, then gradually reduced over 6 hours when oral administration is started.
Drugs during lactation
Lactation, i.e. secretion and ejection of milk from the mammary glands (breasts), is initiated and maintained by the anterior pituitary hormone prolactin (see Chapter 33). During the late months of pregnancy, high levels of progesterone and oestrogens inhibit the actions of prolactin, so lactation commences when levels of progesterone and oestradiol drop rapidly after birth. Suckling at the breast by the infant increases secretion of prolactin by neural reflexes, which act via the hypothalamus and pituitary gland, and triggers the milk let-down reflex, with oxytocin as a mediator.
Prolactin release is inhibited by the hypothalamic prolactin release-inhibitory factor (PRIF, now known to be the neurotransmitter dopamine), hence dopamine agonist drugs may decrease lactation, whereas dopamine antagonists such as the phenothiazine psychotropic agents may cause gynaecomastia and galactorrhoea (see Clinical Interest Box 33-5).
Lactation promoters
Prolactin, as its name implies, is the natural lactation promoter, and suckling or mechanical stimulation of the nipple are the best stimulators of prolactin secretion. The dopamine antagonists domperidone and metoclopramide, normally prescribed as antiemetics or to stimulate motility of the upper GI tract, have been used to stimulate lactation.
Lactation inhibitors
Oestrogens in combination contraceptive pill formulations are known to inhibit lactation, hence the use of progestogenonly minipill formulations for contraception in breastfeeding women. In the past, oestrogens such as chlorotrianisene (a prodrug with long-acting oestrogenic effects) have been used to treat postpartum breast engorgement. This use of oestrogens has declined over the years, mainly because the incidence of painful engorgement is considered low, and studies have indicated that analgesics and other conservative measures such as breast binding and ice packs are effective. A prescriber must also weigh the benefit of using oestrogens for this purpose against the risks, particularly of inducing thromboembolism or uterine bleeding.
It may be necessary, for clearly defined medical reasons, to inhibit lactation, e.g. if the mother must take essential drugs that would be harmful to the infant when ingested in breast milk, or if the infant has died. The dopamine agonists bromocriptine and cabergoline inhibit the release of prolactin from the anterior pituitary gland, resulting in suppression of lactation.
Drugs and breastfeeding
Drugs can enter breast milk and be absorbed by the infant in sufficient amounts to cause pharmacological or toxic effects. In particular, very lipid-soluble drugs will partition into the lipid component of milk and so may cause problems, e.g. CNS depressants may sedate the baby and depress suckling (see Chapter 9). Alternatively, drugs given to the mother may depress lactation. It is essential for the welfare of the infant, however, that the mother’s health be maintained, so drug therapy may need to be instituted or continued. Psychotropic drugs, being lipid-soluble, are likely to partition into breast milk, and hence need caution on prescribing. In particu lar, antidepressants may be needed to treat postpartum depression; sertraline and paroxetine appear to be the firstline medications with respect to safety.
Guidelines
For these reasons, it is often necessary to know how a drug affects lactation and whether it is safe for a breastfeeding mother and child. Guidelines have been drawn up, based on clinical experience in breastfeeding mothers (see Drugs and Breastfeeding, from the Royal Women’s Hospital, Melbourne), and including drugs. In general, it is suggested that:
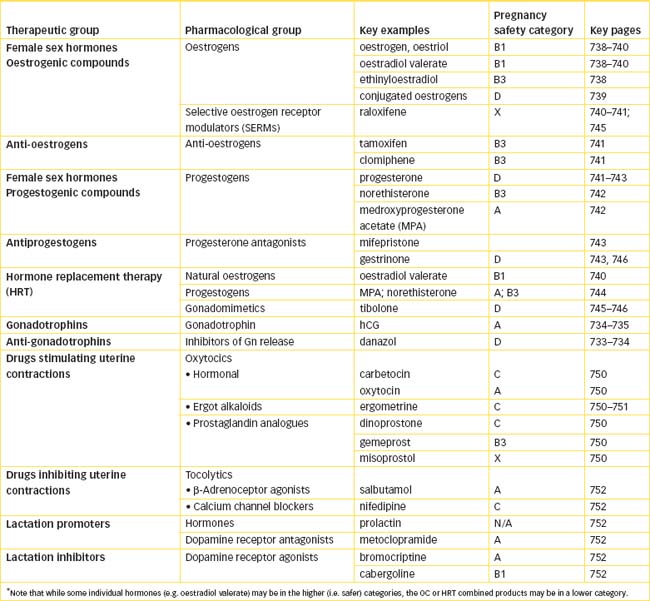
Key points


 The production of the sex hormones is under the control of gonadotrophins released from the anterior pituitary gland when stimulated by gonadotrophin-releasing hormone (GnRH) from the hypothalamus. GnRH can be used clinically to stimulate or suppress the release of pituitary gonadotrophins, depending on whether it is given in low pulsatile doses or high continuous doses. The pituitary gonadotrophic hormones FSH and LH regulate the development and maintenance of sex gland functions. Oestrogens and progesterone are the natural ovarian hormones and are very commonly used in medicine. Oestrogens stimulate and maintain the regular menstrual cycle, have metabolic actions and inhibit the release of pituitary gonadotrophins. Female reproductive functions are regulated by the menstrual cycle, in which cyclical changes in ovarian and pituitary hormone levels lead to monthly periods of regeneration and destruction of the uterine lining (if fertilisation and pregnancy do not occur). Oestrogens are used in oral contraceptive preparations; in hormone replacement therapy after menopause; for disorders of menstruation, treating breast (and prostatic) carcinomas; hirsutism; and for preventing osteoporosis in post-menopausal women. Anti-oestrogens and selective oestrogen receptor modulators are used in breast cancers, infertility and menopausal problems. Progesterone stimulates the secretory phase of the menstrual cycle and facilitates and maintains pregnancy. Synthetic progestogens are orally active and are indicated for hormonal replacement therapy, treating endometriosis and specific carcinomas, and for preventing pregnancy. Cessation of menstruation at the menopause may be associated with symptoms of low oestrogen levels and brings increased risk of cardiovascular disease and osteoporosis. Hormone replacement therapy, i.e. regular use of low-dose natural oestrogen with cyclical progesterone, suppresses menopausal symptoms and protects against some cancers and bone disease. HRT may be administered orally or transdermally. Long-term use of HRT may cause a slightly increased risk of some conditions (e.g. stroke, breast cancer, venous thromboembolism) and reduced risk of others (osteoporotic fractures, colorectal cancer, dementia and diabetes). Other gynaecological disorders (endometriosis, dysfunctional bleeding, polycystic ovary syndrome and hirsutism) are treated with agonists or antagonists of oestrogens and progestogens. Drugs taken by the mother pass across into the fetal circulation and may cause adverse pharmacological effects in the fetus; precautions must be taken with drug use during pregnancy. Drugs affecting uterine smooth muscle activity include those that induce labour (oxytocics), such as oxytocin. Ergometrine induces strong sustained uterine contraction that is useful in controlling postpartum haemorrhage, and is only ever given after delivery. Tocolytics inhibit premature labour and include nifedipine and salbutamol.
The production of the sex hormones is under the control of gonadotrophins released from the anterior pituitary gland when stimulated by gonadotrophin-releasing hormone (GnRH) from the hypothalamus. GnRH can be used clinically to stimulate or suppress the release of pituitary gonadotrophins, depending on whether it is given in low pulsatile doses or high continuous doses. The pituitary gonadotrophic hormones FSH and LH regulate the development and maintenance of sex gland functions. Oestrogens and progesterone are the natural ovarian hormones and are very commonly used in medicine. Oestrogens stimulate and maintain the regular menstrual cycle, have metabolic actions and inhibit the release of pituitary gonadotrophins. Female reproductive functions are regulated by the menstrual cycle, in which cyclical changes in ovarian and pituitary hormone levels lead to monthly periods of regeneration and destruction of the uterine lining (if fertilisation and pregnancy do not occur). Oestrogens are used in oral contraceptive preparations; in hormone replacement therapy after menopause; for disorders of menstruation, treating breast (and prostatic) carcinomas; hirsutism; and for preventing osteoporosis in post-menopausal women. Anti-oestrogens and selective oestrogen receptor modulators are used in breast cancers, infertility and menopausal problems. Progesterone stimulates the secretory phase of the menstrual cycle and facilitates and maintains pregnancy. Synthetic progestogens are orally active and are indicated for hormonal replacement therapy, treating endometriosis and specific carcinomas, and for preventing pregnancy. Cessation of menstruation at the menopause may be associated with symptoms of low oestrogen levels and brings increased risk of cardiovascular disease and osteoporosis. Hormone replacement therapy, i.e. regular use of low-dose natural oestrogen with cyclical progesterone, suppresses menopausal symptoms and protects against some cancers and bone disease. HRT may be administered orally or transdermally. Long-term use of HRT may cause a slightly increased risk of some conditions (e.g. stroke, breast cancer, venous thromboembolism) and reduced risk of others (osteoporotic fractures, colorectal cancer, dementia and diabetes). Other gynaecological disorders (endometriosis, dysfunctional bleeding, polycystic ovary syndrome and hirsutism) are treated with agonists or antagonists of oestrogens and progestogens. Drugs taken by the mother pass across into the fetal circulation and may cause adverse pharmacological effects in the fetus; precautions must be taken with drug use during pregnancy. Drugs affecting uterine smooth muscle activity include those that induce labour (oxytocics), such as oxytocin. Ergometrine induces strong sustained uterine contraction that is useful in controlling postpartum haemorrhage, and is only ever given after delivery. Tocolytics inhibit premature labour and include nifedipine and salbutamol.Review exercises
References and further reading
Anonymous. Therapeutic choices for menopausal symptoms. National Prescribing Service Newsletter. 2009;64:1-7.
Australian Medicines Handbook 2010. Adelaide: AMH, 2010.
Braun L., Cohen M. Herbs and Natural Supplements: An Evidence-Based Guide, 2nd edn. Sydney: Elsevier Mosby; 2007.
Burger H. The menopausal transition: endocrinology. Journal of Sexual Medicine. 2008;5(10):2266-2273.
Chandraharan E., Arulkumaran S. Acute tocolysis. Current Opinion in Obstetrics and Gynecology. 2005;17(2):151-156.
Clark S.L., Simpson K.R., Knox G.E., Garite T.J. Oxytocin: new perspectives on an old drug. American Journal of Obstetrics and Gynecology. 2009;200(1):35. e1–6
Duffy C., Perez K., Partridge A. Implications of phytoestrogen intake for breast cancer. CA: A Cancer Journal for Clinicians. 2007;57(5):260-277.
Ellmann S., Sticht H., Thiel F., Beckmann M.W., Strick R., Strissel P.L. Estrogen and progesterone receptors: from molecular structures to clinical targets. Cellular and Molecular Life Sciences. 2009;66:2405-2426.
Endocrinology Expert Group. Therapeutic Guidelines: Endocrinology, version 4. Melbourne: Therapeutic Guidelines Limited; 2009.
Finn M., Bowyer L., Carr S., et al, editors. Women’s Health: A Core Curriculum. Sydney: Elsevier Mosby, 2005.
Gentile S. Use of contemporary antidepressants during breastfeeding: a proposal for a specific safety index. Drug Safety. 2007;30(2):107-121.
Guerrera M.P., Volpe S.L., Mao J.J. Therapeutic uses of magnesium. American Family Physician. 2009;80(2):157-162.
Howland R.H. Evaluating the safety of medications during pregnancy and lactation. Journal of Psychosocial Nursing and Mental Health Services. 2009;47(3):19-22.
Ilett K.F., Kristensen J.H. Drug use and breastfeeding. Expert Opinion on Drug Safety. 2005;4(4):745-768.
MacLennan A.H. Long-term hormone replacement therapy. Australian Prescriber. 2000;23(5):90-92.
MacLennan A.H. Hormone replacement therapy: where to now? Australian Prescriber. 2003;26(1):8-10.
Mann J. Murder, Magic and Medicine. Oxford: Oxford University Press; 1992.
Menon S.J. Psychotropic medication during pregnancy and lactation. Archives of Gynecology and Obstetrics. 2008;277(1):1-13.
Nedrow A., Miller J., Walker M., et al. Complementary and alternative therapies for the management of menopauserelated symptoms: a systematic evidence review. Archives of Internal Medicine. 2006;166:1453-1465.
Nelson H.D. Menopause. Lancet. 2008;371(9614):760-770.
Ozkan S., Arici A. Advances in treatment options of endometriosis. Gynecologic and Obstetric Investigations. 2009;67(2):81-91.
Radosh L. Drug treatments for polycystic ovary syndrome. American Family Physician. 2009;79(8):671-676.
Rosenfield R.L. What every physician should know about polycystic ovary syndrome. Dermatologic Therapy. 2008;21(5):354-361.
Royal Women’s Hospital Melbourne. Drugs and Breastfeeding 2004. Melbourne: Royal Women’s Hospital; 2004.
Shelly W., Draper M.W., Krishnan V., Wong M., Jaffe R.B. Selective estrogen receptor modulators: an update on recent clinical findings. Obstetrical and Gynecological Survey. 2008;63(3):163-181.
Spencer J.W., Jacobs J.J. Complementary/Alternative Medicine: An Evidence-Based Approach. St Louis: Mosby; 1999.
Stevenson J.C. Hormone replacement therapy: review, update and remaining questions after the Women’s Health Initiative Study. Current Osteoporosis Reports. 2004;2(1):12-16.
Therapeutic Goods Administration. Prescribing Medicines in Pregnancy: An Australian Categorisation of Risk of Drug Use in Pregnancy, 4th edn. Canberra: Therapeutic Goods Administration; 1999. (and subsequent updates)
Thomas S.L., Ellertson C. Nuisance or natural and healthy: should monthly menstruation be optional for women? Lancet. 2000;355:922-924. (March 11)
Writing Group for the Women’s Health Initiative Investigators. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the Women’s Health Initiative Randomized Controlled Trial. Journal of the American Medical Association. 2002;288(3):321-333.
Drugs and breastfeeding: www.kemh.health.wa.gov.au/health/breastfeeding/drugs_breastfeeding.htm
Endometriosis support group: www.endometriosis.org.au/
Jean Hailes Foundation for Women’s Health: www.jeanhailes.org.au/
Medications in Mother’s Milk, by T. Hale: www.iBreastfeeding.com
New Zealand Medicines and Medical Devices Safety Authority: www.medsafe.govt.nz
RANZCOG. Advice to medical practitioners regarding the use of postmenopausal hormone therapy. Consensus statement, August 2004 www.jeanhailes.org.au/issues/hrt_benefits_con.htm
Women’s Health Initiative (USA Department of Health and Human Services): www.nhlbi.nih.gov/health/women/index.htm
More weblinks at: http://evolve.elsevier.com/AU/Bryant/pharmacology