Chapter 4 Legal and Ethical Foundations of Pharmacotherapy
Chapter Focus
Health-care professionals who prescribe, dispense or administer drugs are legally accountable for their actions related to drug therapy. This chapter reviews the laws relating to the regulation of prescription and over-the-counter drugs, poisons, controlled substances, proscribed substances and investigational drugs, particularly regulation in Australia and New Zealand. Relevant pieces of Australian Commonwealth and state legislation relating to the regulation, use and testing of drugs in Australia, especially the Therapeutic Goods Act and Drugs, Poisons and Controlled Substances Acts and Regulations, are outlined. The roles of the Commonwealth and states with respect to proscribed drugs are described, and the implications of the relevant Customs, Crimes and Narcotic Drugs Acts. The scheduling of drugs and controlled substances in Australia and New Zealand is compared.
The process of drug discovery and development is a long one; the routes and stages are described, including how drugs are assayed and standardised, and the phases and important elements in clinical trials of investigational drugs.
Many ethical principles also apply to drug use, based on human rights and ethics; these should always be considered and form a basis for decisions related to pharmacology research and clinical practice. Controversy can arise as to how ethical principles are applied in clinical situations.
ADEC Australian Drug Evaluation Committee
ADRAC Adverse Drug Reactions Advisory Committee
APF Australian Pharmaceutical Formulary
CSIRO Commonwealth Scientific and Industrial Research Organisation
CSL Commonwealth Serum Laboratories
CTN Clinical Trial Notification
FDA Food and Drugs Administration
HREC Human Research Ethics Committee
IEC institutional ethics committee
IMMP Intensive Medicines Monitoring Programme
PBS Pharmaceutical Benefits Scheme
RCCT randomised controlled clinical trial
SUSDP Standard for the Uniform Scheduling of Drugs and Poisons
Legal aspects of drug use
BEFORE the 20th century there were few controls on the use of drugs, most of which were natural products, many with low efficacy. There was little information available about drugs compared with what we expect today, such as research studies proving the safety and effectiveness of the preparation, content analysis and strength, drug consistency from one pack to another of the same preparation and information on administration and adverse reactions. As chemical industries developed, more potent and efficacious drugs were synthesised (see Clinical Interest Box 1-3) and trade in drugs of dependence (addictive drugs) increased, it was recognised that controls on drugs were required. The early 1900s marked the beginning of national and international legislation relating to drugs. (See the on-line resource Hot Topics 59: Drugs and the Law [2007] for more information on the history of drug laws.)
International drug controls
Controls on narcotic drugs
Control of drugs in international law began in 1912 when the first Opium Conference was held at The Hague, Netherlands. International treaties were drawn up, calling on governments to:
In 1961, government representatives formulated the United Nations Single Convention on Narcotic Drugs, which became effective in 1964. The convention needs to be ratified and signed by a country before it binds that country, which then has to enact appropriate legislation for the treaty’s provisions to have legal force in the country. This convention consolidated all existing treaties into one document for the control of all narcotic substances,1 except for medical treatment and research, by:
The Convention comes under the auspices of the United Nations Office on Drugs and Crime; the Convention lists drugs in Schedules depending on their liability for abuse and production of adverse effects.
Australia has signed the following international treaties about drugs:
The International Narcotics Control Board (INCB) was established to enforce the Single Convention on Narcotic Drugs; the INCB is ‘the independent and quasijudicial control organ monitoring the implementation of the United Nations drug control conventions’. This board is an international organisation of representatives from governments and the World Health Organization, established to monitor compliance with the Single Convention on Narcotic Drugs and with other United Nations conventions regarding the manufacture and traffic of drugs, international trade in drugs and government control over chemicals used in the illicit manufacture of drugs.
Because enforcement is an immense task, it is impossible to prevent illicit trafficking in drugs. The 2009 UN World Drugs Report (United Nations 2009) noted that the illicit drug market worldwide had become a US$320 billion per year industry. It was estimated that there were in 2007 between 18 and 38 million problem drug users, i.e. those dependent on drugs, who would benefit from treatment and whose level of use adversely affects public health and public order—the report uses the phrase ‘gruesome upsurge of violence in countries like Mexico’ and notes that ‘the criminal justice system is a very blunt instrument for dealing with drug markets’.
Laws need to be frequently updated and strictly enforced, but the unfortunate fact is that the available financial support for regulation and enforcement is not equal to the task and is less than the money to be made by illicit trafficking and pushing of narcotics. New means of distribution such as ‘Internet pharmacies’, most commonly used by consumers in Europe and the United States, pose new problems. Also, international treaties are not automatically binding, even on countries that have signed the treaty, unless those countries introduce local laws and enforce them. For this reason, treaties and international attempts to control illicit drugs are only as strong as the determination of their member countries.
Controls on therapeutic drugs
It is recognised that drugs used therapeutically also need to be controlled, as people cannot assess the safety and efficacy of all drugs. The public want access to drugs but also expect to be protected from harm. Governments generally take a risk assessment role and require that drugs available in their country are assessed for safety, efficacy, quality of manufacture (aspects such as purity, stability and strength), availability and marketing. This provides protection not only for the public but also for drug manufacturers (and for the governments). The principles generally adopted are that most people are not sufficiently knowledgeable about health care and drugs to self-medicate safely in all conditions, that all drugs are inherently potentially dangerous and should be assessed for risks and benefits, that a licence to market a drug is granted for a specified period subject to review, that licences can be revoked and that government guidelines with respect to Good Laboratory Practice (GLP), Good Manufacturing Practice (GMP) and Good Clinical Practice (GCP) should be observed.
As world trade and health practices become ever more based on a global economy, it is increasingly important that requirements for drug registration and licensing be uniform in all developed countries. If, for example, the regulations were different in the major markets of Europe, Asia and the USA–Canada, it would add enormously to the costs of developing and introducing new drugs. Most developed countries require similar standards of preclinical testing, clinical trials and post-marketing surveillance, as described in the later section on drug discovery and development. A tragic situation that developed in the early 1960s, after widespread use of the seemingly safe sedative thalidomide, led to much more rigorous testing and strict controls on the approval and supply of drugs (see Clinical Interest Box 4-1).
Clinical interest Box 4-1 The thalidomide disaster
In the years 1959–1962, it appeared that a new ‘epidemic’ was sweeping England, Europe and other countries, including Australia. Dozens, then hundreds, and eventually thousands of babies were stillborn or born with congenital malformations, commonly with absent or rudimentary limbs and deformities of other organs and systems. The condition was termed phocomelia, meaning ‘seal-like limbs’, and had up until that time been an incredibly rare congenital malformation. Causes were proposed and sought, including viral infections, radiation damage, nutritional deficiencies or environmental contaminants.
Dr W Lenz, of Hamburg in Germany, asked mothers of affected babies to list all the drugs they had taken during pregnancy; Contergan, an apparently safe sedative, appeared in about 29% of the lists. At a meeting of paediatric physicians, Dr Lenz suggested that a drug might have been responsible.
Meanwhile in Sydney, Dr William McBride had been consulted about several babies with phocomelia; all the mothers had taken Distaval, a mild sedative, during pregnancy. McBride wrote to the English journal Lancet, asking if similar cases had been reported in the UK. Lenz replied with his findings and it became apparent that the same drug, thalidomide, was implicated in all the cases. More case reports flooded in and the drug was withdrawn; however, cases kept appearing for many years, partly because the drug was marketed under many trade names and warnings were unheeded, so bottles of tablets lay around for some time.
Other reasons why it took so long for the link between the drug and the adverse effect to be established were that the critical period was so short—between the 37th and 54th days of pregnancy—and the effects were not observed until many months later, and many mothers forgot or denied having taking any drugs in early pregnancy.
The drug had not been released in the USA, as the Food and Drug Administration (FDA) had been concerned, not about its teratogenicity (ability to cause birth defects), but that it appeared to have adverse effects on the nervous system. Overall, it is estimated that in Germany alone about 10,000 babies were affected, of whom half survived, most with severe malformations. Law suits and damages claims against the drug companies concerned were still being pursued in the courts decades later.
Thalidomide had appeared to meet a public need for a safe and effective drug to treat a mild condition—insomnia—and had been submitted to the testing required at the time, yet had set in train a disaster for all affected. A positive benefit was that, after public outcry as to how it could have occurred, regulations regarding the testing, approval and availability of drugs were severely tightened, to the extent that for some years it became very difficult for new drugs to be approved.
For years thalidomide was totally banned in most countries; however, it became apparent that it does have some useful immunosuppressant and anti-inflammatory actions. In Australia it is now an ‘orphan drug’, available for use in very strictly controlled situations: for treatment of some skin conditions in leprosy and for multiple myeloma. An analogue, lenalidomide, is now also available for treatment of multiple myeloma under strict guidelines; interestingly, both drugs show peripheral neuropathy as adverse effects, as noted by the FDA 50 years ago. In some countries thalidomide is less tightly controlled and the warnings on the packs are in English rather than the native language so, tragically, cases of phocomelia still occur.
Adapted from: McBride 1961, Cartwright & Biddiss 1972, McBride 2001, Palumbo et al 2008.
Regulation of drugs in Australia
Australian laws related to drug regulation can be broadly divided into two types: laws that regulate drugs used for medicinal purposes in humans (discussed in this section under ‘Drug regulation’), and laws that prohibit the possession, production and supply of proscribed (i.e. prohibited) drugs (discussed under ‘Drug offences’). Legal non-medicinal drugs such as alcohol and tobacco are also subject to much regulation, which is primarily related to their sale, advertising and packaging (not considered in this chapter).
In Australia, drugs are controlled by Commonwealth, state and territory laws (see Table 4-1). There is no uniform Australian scheme for drug regulation or drug offences, partly because the Commonwealth legislation cannot apply in all situations for Constitutional reasons. However, the Model Criminal Code, developed in 2009, is a set of criminal laws promulgated to facilitate the development of national criminal laws by all states and territories, i.e. ‘harmonisation’ between jurisdictions.
Table 4-1 Principal australian legislation involved in the regulation of drugs
| Jurisdiction | Drug regulation legislation | Additional drug offences acts |
| Commonwealth (Cth) | Therapeutic Goods Act 1989 (Cth) Therapeutic Goods Regulations 1990 (Cth) National Health Act 1953 (Cth) | Customs Act 1901 (Cth) Crimes (Traffic in Narcotic Drugs and Psychotropic Substances) Act 1990 (Cth) Narcotic Drugs Act 1967 (Cth) Criminal Code Act 1995 (Cth) |
| Australian Capital Territory (ACT) | Medicines, Poisons and Therapeutic Goods Act 2008 (ACT) Drugs of Dependence Act 1989 (ACT) Drugs In Sport Act 1999 (ACT) Drugs of Dependence Regulations 2009 (ACT) | Criminal Code 2002 (ACT) |
| New South Wales (NSW) | Poisons and Therapeutic Goods Act 1966 (NSW) Poisons and Therapeutic Goods Regulation 2008 (NSW) | Drug Misuse and Trafficking Act 1985 (NSW) |
| Northern Territory (NT) | Poisons and Dangerous Drugs Act (NT) Therapeutic Goods and Cosmetics Act (NT) Poisons and Dangerous Drugs Regulations (NT) | Misuse of Drugs Act (NT) |
| Queensland (Qld) | Health Act 1937 (Qld) Health (Drugs and Poisons) Regulations 1996 (Qld) | Drugs Misuse Act 1986 (Qld) |
| South Australia (SA) | Controlled Substances Act 1984 (SA) Controlled Substances (Poisons) Regulations 1996 (SA) Controlled Substances (General) Regulations 2000 (SA) | Drugs Act 1908 (SA) |
| Tasmania (Tas) | Poisons Act 1971 (Tas) Poisons Regulations 2008 (Tas) Alcohol and Drug Dependency Act 1968 (Tas) Therapeutic Goods Act 2001 (Tas) Therapeutic Goods Regulations 2002 (Tas) | Misuse of Drugs Act 2001 (Tas) |
| Victoria (Vic) | Therapeutic Goods (Victoria) Act 1994 (Vic) Drugs, Poisons and Controlled Substances Act 1981 (Vic) Drugs, Poisons and Controlled Substances Regulations 2006 (Vic) | |
| Western Australia (WA) | Poisons Act 1964 (WA) Poisons Regulations 1965 (WA) | Misuse of Drugs Act 1981 (WA) |
| New Zealand | Medicines Act 1981 Medicines Regulations 1984 Medicines (Standing Order) Regulations 2002 Medicines (Designated Prescriber: Nurse Practitioner Regulations) Regulations 2005 Misuse of Drugs Regulations 1977 | Misuse of Drugs Act 1975 |
Broadly, state and territory laws control ‘poisons’, and Commonwealth legislation controls ‘therapeutic goods’. Offences related to international drug trafficking are set out in Commonwealth legislation, while the state and territory criminal laws cover the production, possession, use and distribution of proscribed drugs within those jurisdictions. Additional legislation in most states covers drug use related to road safety.
In addition to specific Acts and Regulations relating to drug development and use, there are relevant aspects of Common Law (developed by judicial precedence and interpretation). For example, health professionals are considered to have a ‘duty of care’ to the people with whom they deal, and so are expected to carry out their roles with the best interests of their clients/patients as a priority. Underpinning all health-care-related law are fundamental principles of human rights and ethics (see later section on ‘Ethical principles related to drug use in health care’). Drug availability can also be controlled at the local level, for example by a hospital’s drug committee.
Drug regulation
Dwyer and Newgreen (1998) point to three primary aims of the regulation of medicinal drugs: to control the supply of drugs prone to abuse; to regulate the availability of substances for therapeutic use (to ensure safety and quality); and to include certain products on governmentsponsored assistance schemes. The criteria of quality, safety and efficacy are mentioned as objects of the Therapeutic Goods Act 1989 (Cth).
In Australia, there exist extensive, complex and overlapping pieces of Commonwealth, state and territory legislation regulating drugs. Although the regulation of drugs has traditionally been the domain of the states, the role of the Commonwealth has increased steadily, with the introduction of the Therapeutic Goods Act. Because a substance may be either a poison or a therapeutic good or both, it may be subject to both Commonwealth and state regulation. Note that the term ‘poison’ is used broadly to cover drugs used clinically as well as veterinary, agricultural and domestic chemicals.
Classification in the Standard for the Uniform Scheduling of Drugs and Poisons (SUSDP)
To restrict the availability of drugs of certain types, drugs are classified with others requiring the same level of regulation, into Schedules. Historically, scheduling in Australia was a state responsibility, which led to anomalies such as a drug being available over the counter (OTC) in one state, e.g. in Albury, NSW, whereas in its sister town, Wodonga, on the other bank of the Murray River in Victoria, a prescription might be required.
The need for reform of the controlled substances system was recognised and a National Competition Review of Drugs, Poisons and Controlled Substances legislation was set in train; an Options Paper and Final Report were published (Galbally 2000, 2001). The Review considered all aspects of the legislation relating to drugs, including how scheduling of drugs determines access, supply and provision of drugs; their labelling, packaging and storage; records and advertising. The controls and restrictions imposed, their effects, costs and benefits were all discussed, and options and alternatives were suggested. The Review strongly supported a uniform regulatory scheme across states and territories, more closely integrated with related legislation, and noted the benefits in terms of quality use of medicines flowing from effective counselling by pharmacists. A uniform system of control also ensures a balance between the many vested interests involved in drug scheduling. For example, drug companies want drugs to be as widely bought and used as possible, governments want to minimise costs and protect the public, while health professionals may wish to maintain their powers and protect their unique roles in the supply of drugs. The Review recommended that funds be provided for further research in the area and that administrative arrangements be amended to streamline functions of the relevant committees. The Review, and the Australian Health Ministers Advisory Council Working Party response, was unanimously approved by the Council of Australian Governments in June 2005.
The SUSDP has attempted to implement the Review’s recommendations by setting uniform schedules and expecting all states to move towards adhering to them. The SUSDP classifies drugs in relation to their safety, appropriate availability and possible therapeutic uses. It is published annually and comprises the recommendations of the National Drugs and Poisons Schedule Committee (NDPSC). The decisions of the NDPSC in relation to the Standard have no force in Commonwealth law but are recommended for incorporation into state and territory drugs and poisons legislation. Most states and territories have adopted the Standard, in whole or in part, to designate which substances are subject to regulation.
The SUSDP also attempts to unify scheduling and control of drugs and poisons between Australia and New Zealand. This is referred to as Trans-Tasman Scheduling Harmonisation and has been largely effective, with a few minor discrepancies still existing. (A planned new trans-Tasman regulatory agency for therapeutic products, The Australia New Zealand Therapeutic Products Authority, had been expected to commence during 2007, to replace the Australian TGA and New Zealand’s Medicines and Medical Devices Safety Authority (MedSafe); however, the NZ government withdrew due to lack of local support.)
Drug Schedules
The SUSDP contains nine Schedules of ‘poisons’ (i.e. drugs and other chemicals) that are subject to varying levels of control; definitions and important aspects of the Schedules are listed in Appendix 5. Schedules 2 (PHARMACY ONLY), 3 (PHARMACIST ONLY), 4 (PRESCRIPTION ONLY) and 8 (CONTROLLED DRUGS) include drugs used medically; Schedules 2 and 3 include OTC drugs, and have been discussed in Chapter 3 (see Tables 3-1 and 3-2). Schedules 5 (CAUTION), 6 (POISON) and 7 (dangerous poison) include mainly non-drug chemicals used domestically or in agriculture. Schedule 9 (PROHIBITED SUBSTANCE) includes some drugs of abuse (such as heroin), and drugs that may be required for research or investigational purposes but are considered too toxic for therapeutic use. Drugs are now labelled with the name of the classification rather than the S number (e.g. ‘Prescription-Only’ rather than ‘Schedule 4’). This change was made to counteract the false perception in the community that higher S numbers necessarily meant higher toxicity; in fact, the Poisons Standard 2009 specifically states that ‘the Schedules listed in order of greatest to least restriction are: 9, 8, 4, 7, 3, 2, 6, 5’. Schedule 1 is intentionally empty; it has been suggested that in future some CAM medicines that require regulation may be moved into it.
The decision to classify a substance into a particular schedule depends not only on the drug’s potential toxicity but also on the purposes for which it is used, the dose in the particular preparation, its potential for abuse, other ingredients present, the formulation (e.g. oral tablet, parenteral injection or topical ointment) and the need for the drug to be readily available in the community. A drug may appear in more than one schedule, e.g. the scheduling of codeine phosphate is summarised as follows2 (there are also restrictions on maximum recommended daily doses):
If a substance does not appear in a Schedule of the SUSDP, i.e. is unscheduled, it is not a poison by definition and can be supplied freely to the public (unless it is subject to other legislative controls). Where a preparation contains two or more poisons included in a schedule, the preparation takes the Schedule that is the most restrictive.
In some Australian states, the SUSDP has been adopted as the basis for determining whether or not a drug is a proscribed drug, so that production or supply of a Schedule 8 or Schedule 9 drug constitutes an indictable offence. In other states there are separate lists in the legislation related to the relevant offences, but these lists correlate closely with the SUSDP Schedules.
Regulation of poisons (drugs and other chemicals)
Drugs are subject to strict regulation in all Australian states and territories from the moment of their manufacture until their administration. Licences determine who can legally make and sell wholesale drugs and poisons. Industry users (health services, industries and others) require a permit to purchase and use drugs.
Usually only registered doctors and dentists are allowed to possess and prescribe Prescription-Only drugs, and pharmacists to dispense them. There are exceptions; for example, in some states of Australia, specially qualified nurse practitioners, optometrists and podiatrists have the right to possess and prescribe a limited number of relevant S4 (Prescription-Only) drugs. (The roles of various health professionals with respect to drugs have been described in Chapter 2.)
Manufacture and wholesaling
All states and territories require manufacturers and wholesalers of Schedule 2, 3, 4 and 8 drugs (and Schedule 9 for wholesaling) to be licensed. The licence may relate exclusively to a chemical of a particular Schedule, or it may be a licence to manufacture chemicals in a particular Schedule or Schedules, classes of substances or specific substances. The security and record-keeping obligations are less rigorous for Schedule 2, 3 and 4 drugs than for S8 or S9. Licences are generally issued by the relevant state or territory Minister or department, for a fee.
The Therapeutic Goods Act also contains provisions covering counterfeiting, recall procedures, reporting of adverse effects and record keeping by manufacturers.
Retailing
In all jurisdictions except Western Australia, poisons legislation authorises pharmacists to possess, manufacture and supply poisons without a licence in the practise of their profession. The circumstances and manner in which the supply can be made, e.g. whether or not a prescription is required, and record-keeping requirements depend on the Schedule of the substance in question.
Sampling
State and territory legislation usually permits representatives of licensed manufacturers and wholesalers to possess and supply samples of certain poisons.
Packaging and labelling
The labelling and packaging of drugs for use in humans are governed by orders and regulations made under the Therapeutic Goods Act and by the SUSDP, as transcribed or adopted by state and territory legislation. It is an offence in Commonwealth and state laws to fail to comply with labelling and packaging standards for therapeutic goods.
Part of the pharmacist’s role is to ensure that medicines are labelled correctly to ensure safe storage and administration. Labels may be advisory, explanatory or reminders, such as:
Many of the requirements for labelling are determined by the Schedule into which a drug or poison is classified (see Appendix 5).
Possession
Certain persons are authorised to possess Schedule 4 and 8 poisons for legitimate commercial, professional, academic, research or emergency purposes. In all states and territories the unauthorised or unlicensed possession of Schedule 8 or 9 poisons is a criminal offence (see ‘Drug offences’, below).
Prescription
Generally, Schedule 4 or 8 poisons can be prescribed by medical practitioners, dentists, optometrists (S4 only) and veterinary surgeons in the lawful practise of their respective professions for the treatment of persons (or animals) under their care. The circumstances in which these poisons can be prescribed differ across the states. There are also special prescribing requirements for patients whom medical practitioners believe are drug-dependent. (The requirements for prescription writing, and sample prescriptions, are discussed in Chapter 2.)
Dispensing
Legally valid prescriptions for Schedule 4 and 8 poisons can be filled by pharmacists or by the prescriber for his or her own patients. For certain poisons in these Schedules, e.g. dextromoramide, hydromorphone and methadone, there are extra controls on dispensing (see Drug Monograph 21–2). In all states and territories except the ACT, pharm acists can prescribe Schedule 4 drugs without a prescription in an emergency; however, a valid written prescrip tion must be provided by the prescriber as soon as is practicable.
Administration
As a general rule, the administration of Schedule 4 and 8 poisons to a person requires the written or verbal authorisation of a medical practitioner or dentist. There are significant exceptions to this rule, such as administration in emergency situations and by certain professionals such as podiatrists and nurses. (Regulations with respect to telephone orders and standing orders are described in Chapter 2.)
Storage and destruction
There are strict security requirements concerning the storage and destruction of Schedule 4 and 8 poisons by manufacturers, wholesalers and pharmacists, and in hospitals. It is usually necessary for some drugs to be stored on hospital wards, to provide ready and out-ofhours access. An ‘imprest’ system of lockable cupboards, trolleys and bedside drawers is used. Strict procedures must be maintained with respect to security of drugs, accuracy of records, ‘loaning’ of drugs between wards, monitoring access by staff, control of keys to drug stores and disposal of unused or expired drugs. The hospital’s director of pharmacy or chief pharmacist should have ultimate responsibility and control.
The storage requirements imposed on doctors, dentists and veterinary surgeons are less stringent, in recognition of the fact that these practitioners are less likely to store large quantities of Schedule 4, 8 or 9 poisons.
Record keeping
People involved in the manufacture, supply, dispensing or administration of Schedule 4 and 8 poisons (except the patient who actually receives the drug) are required to account in writing (or by computer records) for every instance that the poison is dealt with. Thus hospital drug charts (see Figure 2-3) show how each drug was prescribed and when administered.
Therapeutic goods
The Commonwealth Therapeutic Goods Act regulates ‘therapeutic goods’, defined as medicines, blood products and medical devices. As mentioned above, a substance may be a poison and a therapeutic good, in which case both the state poisons legislation and the Commonwealth therapeutic goods legislation may apply.
The regulatory framework under the Therapeutic Goods Act is based on a risk management and quality assurance framework, to ensure the safety, quality and performance of medicines and medical devices. Because of Constitutional restrictions, the Therapeutic Goods Act can regulate therapeutic goods only in certain circumstances. Much of the regulation of therapeutic goods exists in the state poisons legislation, discussed above.
Before a drug (or other therapeutic good) can be marketed in Australia, it must be evaluated by the Therapeutic Goods Administration (TGA), a division of the Commonwealth Department of Health and Ageing, which assesses the product for quality, safety, efficacy and cost-effectiveness, and considers the extent to which the drug should be made available to the public (see Figure 4-1). Sponsors, usually drug companies, are required to submit to the TGA an enormous amount of material relevant to the application for approval, including chemical and manufacturing data and results from pharmacological testing in vitro, in vivo and in clinical trials. This material is examined closely by experienced evaluators, a process that may take some months, and may be referred to an expert committee (the Australian Drug Evaluation Committee, ADEC) for its comments. The TGA makes the final decision on whether or not to register the drug for therapeutic use in Australia and decides into which Schedule it should be put.
The process also applies to non-prescription drugs (OTC and ‘listed’ drugs [see Chapter 3]), complementary and alternative remedies (as discussed in Chapter 3) and to medical devices such as breast implants, diagnostic test kits, dental materials, contact lenses and tampons. There is a ‘Special Access Scheme’ covering approval for supply of unapproved therapeutic goods for a specified purpose.
Manufacture
Irrespective of whether the goods are or are not in a Poisons Schedule, manufacturers of therapeutic goods for supply in Australia for use in humans must hold a licence under the Therapeutic Goods Act. Some types of drug and certain persons are exempt from the licensing requirements. Examples of drugs that are exempt goods are homeopathic remedies, antiperspirants and acne cleansers. Examples of exempt persons are most registered health professionals, such as doctors and pharmacists in the usual practice of their professions, and a wide range of practitioners of alternative health-care modalities.
Supply and wholesaling
It is an offence to supply by wholesale any therapeutic goods that are not on the Australian Register of Therapeutic Goods. Because the Therapeutic Goods Act cannot always apply for Constitutional reasons, New South Wales, Victoria and the Northern Territory have created similar offences under state law.
Advertising
Various state Acts, based on the Therapeutic Goods Regulations (Cth) and the SUSDP, have prohibited the advertising of Schedules 4, 8 and 9 poisons to the general public. Such advertisements are allowed to be included in bona fide professional publications. Unscheduled substances and poisons in Schedules 2 and 3 can be advertised directly to the public in certain circumstances.
Clinical trials
Complicated provisions in the Therapeutic Goods Act and its Regulations govern the use of experimental drugs and testing in animals and humans. These are discussed in subsequent sections of this chapter.
Orphan drugs
Orphan drugs are those used to treat, prevent or diagnose rare diseases and are not otherwise commercially viable. It is recognised that, while such drugs might not be commercially viable, patients with rare conditions have as much right as all others to access drugs that are safe and effective for their treatment.
In 1983 in the USA, the Food and Drug Administration (FDA) established the Orphan Drug Act, which provided grants to encourage research to find drugs for treatment of rare chronic diseases. Because such research was unprofitable, it was very limited before this Act. Among the disorders that benefit from this research are cystic fibrosis, von Willebrand’s disease, leprosy (Hansen’s disease), AIDS and rare cancers.
Australia’s TGA has based its Orphan Drug Program (formerly known as Section 100 Items) on that of the FDA. The program encourages sponsors (drug companies) by reducing the costs associated with drug research, approval and marketing. Fees for application and evaluation processes are waived, exclusive approval of a drug may be given and approval times are shortened. The drugs are usually supplied only through hospitals with appropriate specialist facilities, and funding is subsidised by the government only for the listed indications.
The criteria under which a drug may be considered for ‘orphan status’ are that there are fewer than 2000 affected individuals in Australia or, for vaccines, the vaccine would be given fewer than 2000 times per year, or the costs involved in developing the drug are prohibitive. Most of the conditions involved are serious diseases. The drug does not need to be the only drug for treatment for the condition but, if it is a new drug, it must be clinically superior. Drugs already rejected on safety grounds, or already registered or considered essential drugs, are not considered for the program. If approved and registered, the drug may be considered for listing on the Pharmaceutical Benefits Scheme (PBS) and will be required to be subjected to post-marketing surveillance for safety and efficacy. As at end 2009, approximately 200 drugs were designated as orphan drugs by the TGA; a few examples are listed in Table 4-2.
Table 4-2 Examples of orphan drugs (S100 Items)
| Orphan drug | Rare condition treated |
| Aldesleukin (interleukin-2) | Metastatic renal cell carcinoma, metastatic malignant melanoma |
| Artesunate injection | Severe chloroquine-resistant malaria |
| Blood factors VIII and IX | Haemophilia A and B |
| Caffeine citrate oral solution | Apnoea in very premature babies |
| Human growth hormone | Specific conditions involving short stature, e.g. Prader–Willi syndrome |
| Imatinib | Chronic myeloid leukaemia |
| Mannitol for inhalation | Cystic fibrosis |
| Nandrolone decanoate | Debilitated patients with HIV infection |
| Peritoneal dialysis solutions | End-stage renal disease |
| Rabies vaccine | Contact with rabid animals |
| Thalidomide | Leprous skin reactions, multiple myeloma |
| Tobramycin inhalation solution | Cystic fibrosis patients with respiratory tract infections |
| Tumour necrosis factor α 1-a | Soft tissue sarcoma |
| Various antineoplastic agents | Cancers unresponsive to usual therapy: multiple myeloma, ovarian cancer, metastatic melanoma |
| Various enzymes for replacement | Congenital metabolic diseases, e.g. lipid storage diseases, Fabry’s disease, Gaucher’s disease |
| Various specified antifungals and antivirals | Patients with AIDS-related infections |
Source: TGA website: www.tga.gov.au/docs/html/orphand2.htm.
The Pharmaceutical Benefits Scheme (PBS)
Set up under the National Health Act 1953 (Cth), the Australian PBS is a program in which already registered drugs deemed to be essential to the community but too expensive for individual purchase are subsidised by the government to some extent. In the year to June 2009, the PBS covered approximately 181 million prescriptions, which averages at about eight prescriptions annually for every Australian. The numbers and costs increase every year, causing blow-outs in the health budget.
The overall process is shown diagrammatically in Figure 4-1 (aspects of the PBS related to prescribing are also discussed in Chapter 2). On the advice of an expert committee, the Pharmaceutical Benefits Advisory Committee (PBAC) which evaluates the efficacy, safety and cost-effectiveness of each drug, the government lists recommended drugs for subsidy and negotiates a price. This of course encourages wider use of the drugs, as they are more affordable, and so members of the committee, government and doctors are under considerable pressure from drug companies to ensure that their drugs are PBS-listed. There has been great concern recently as to the pressures put on the committee and the blow-out in costs when new drugs have been subsidised and used to a far greater extent than anticipated—e.g. celecoxib, a COX-2 inhibitor anti-inflammatory agent, bupropion for quitting smoking, and trastuzumab for breast cancer (see editorial by Kaye and Day [2006]).

Figure 4-1 Drug regulation in Australia: from new drug application to the prescription pad.
Source: National Prescribing Service Newsletter 2001: 19; used with permission.
Note that there are separate Schedules for drugs that may be prescribed by dentists (for dental conditions only) and optometrists (eye conditions, by ocular administration). There is now (in Victoria) a list of drugs that especially qualified podiatrists may prescribe; however, these are not yet subsidised by the PBS. Similarly, while qualified nurse practitioners are now legally allowed to prescribe a limited number of drugs depending on their specialty, if the prescription is not covered by the PBS, patients have to pay full amounts for the drug as an unsubsidised ‘private prescription’.
NZ Pharmacology and Therapeutics Advisory Committee (PTAC)
New Zealand has a rather different scheme for subsidising essential drugs, whereby this committee (PTAC) reviews evidence-based processes before recommending subsidies. The co-payments in NZ are considerably lower than those which Australians pay (unless on a special scheme), and the NZ agency contracts with suppliers to purchase medicines. Growth in budgets for medicines has been much faster in Australia than in NZ, partly because many more drugs within a class are subsidised in Australia, whereas NZ keeps the lid on the drug budget by limiting the number of drugs subsidised: of the five most popular drug classes, 81 different drugs products were subsidised in NZ, whereas over 650 were subsidised by the PBS (see Morgan and Boothe [2010]).
Substitution of drugs (generic prescribing)
In an attempt to keep down the escalating costs of drugs to the community, doctors are encouraged to prescribe generic rather than name brand drugs considered equivalent; this allows the pharmacist to supply (and the government to subsidise) the cheapest alternative. This is allowed only if the doctor agrees and if the alternatives are considered ‘bioequivalent’ (see section ‘Generic prescribing’ in Chapter 1 and mock prescription, Figure 2-3B, plus related discussion). Drugs for which bioavailability can vary markedly between different formulations, and which therefore are not allowed to be substituted, are those with a low therapeutic index (safety margin) and low lipid solubility (hence variable absorption). Such drugs include digoxin (a cardiac glycoside used in heart failure), theophylline (a smooth muscle relaxant used for relief of bronchospasm in certain respiratory conditions) and phenytoin (an antiepileptic drug).
Access to drugs outside the PBS
Drugs not subsidised by the PBS may be obtained by other means:
And, of course, many drugs are available over the counter (OTC) in pharmacies, general stores, health shops or supermarkets.
Legal aspects of concern to pharmacists
As the supplier of many OTC and all Pharmacy-, Pharmacist- and Prescription-Only drugs, the final responsibility for safe supply rests with the pharmacist; hence there are many regulations as to the pharmacist’s professional roles and conduct. State and territory Acts, Regulations and Pharmacy Board guidelines cover aspects such as:
Typical day-to-day issues of concern for pharmacists involve:
Medicines in pregnancy
The Australian Drug Evaluation Committee has set up an ‘Australian categorisation of risk of drug use in pregnancy’; the categories are described in detail in Chapter 9 (see Table 9-1). In order of increasing potential risk, they are:
The classification is a warning to users and prescribers of the dangers of drug use in pregnancy (rather than a legally-enforceable regulation), and a reminder that there are two individuals being administered the drug (the woman and the fetus). It also allows the user or prescriber to select the safest drug that will have the desired effects.
Drugs in sport
The use of drugs by athletes and during sporting competition is regulated not so much by the government (although there is some state legislation, e.g. Drugs in Sport Act 1999 [ACT]) as by the International Olympic Movement and its Anti-Doping Rules, and the World Anti-Doping Agency (see Chapter 49). In Australia this is implemented by the Australian Sports Anti-Doping Authority, set up in March 2006 in Canberra. Drugs are classified into groups depending on whether they are allowed, allowed under certain circumstances, prohibited or prohibited in some sports.
Australian drug offences
Historical aspects
Laws relating to drugs are relatively recent, dating from the 1890s in relation to opium and the early 1900s in relation to other drugs. The enactment of drug legislation in Australia has followed the social trends and scientific knowledge of the time. In his book From Mr Sin to Mr Big, Desmond Manderson argued that historically in Australia the selective enactment of drug laws has also been influenced by racism, powerful international pressures and the vested interests of the medical profession, bureaucrats and politicians. He used the example of opium:
In nineteenth-century Australia, opium was the preserve of neither the creative few nor the urban poor. It was freely available and freely used. Furthermore, perhaps partly as a consequence of the weakness of the medical profession, the line which is now seen to divide medical ‘use’ from non-medical ‘abuse’ was not yet apparent.
By the late 1880s, however, opium was seen as a ‘pollutant, moral as well as physical’ and was associated with Chinese ‘opium dens’. Soon after, its use was criminalised.
Foreign trends and international law have also influenced Australian drug policy. Two pieces of Commonwealth legislation that relate to certain dealings in drugs, both within and outside Australia, the Crimes (Traffic in Narcotic Drugs and Psychotropic Substances) Act 1990 (Cth) and the Narcotic Goods Act 1967 (Cth), were introduced pursuant to United Nations Conventions.
Controversies over criminalisation
There has been much debate about whether the criminalisation of certain drugs reduces or increases the harms associated with drug use. The 2009 UN World Drugs Report noted that ‘illicit drugs continue to pose a health danger to humanity’, so some drugs should remain controlled. However, it also noted ‘a growing chorus’ among politicians, the press and the public that ‘drug control is not working’ (United Nations 2009).
The negative consequences of criminalisation are said to include increased social problems, related criminal offences and the high cost of enforcement. Proponents of decriminalisation argue that, without the adverse reactions associated with their illegal character, drug offences would be ‘victimless’: consumers, producers and suppliers share an interest in continued production and supply of drugs. In this way, drug offences are unlike other serious crimes. Some commentators believe that certain drugs should be decriminalised (e.g. marijuana) or have their regulation modified (e.g. heroin) to reduce the associated harm.
Legislation
Like the laws regulating poisons and therapeutic goods in Australia, drug offences are set out in Commonwealth, state and territory legislation. Dealings with drugs can be an offence under both Commonwealth and state and territory laws. Responsibility for the policing of drug laws is shared by the Commonwealth, states and territories.
Commonwealth legislation proscribes the importation and exportation of narcotic drugs, the possession of drugs that have been illegally imported and certain dealings in drugs within and outside Australia. Commonwealth laws also regulate the manufacture of certain proscribed drugs.
State legislation proscribes the possession, production and distribution of certain drugs. Because of the historical origins of these offences, they appear in the same legislation that governs poisons and, in some states and territories, also appear in the state or territory’s Crimes Act or Criminal Code.
Generally, for offences for which possession of the drug is a necessary part, it will be a defence to show that the possession was authorised—for example, the defendant holds a licence or is authorised under the relevant state law to deal with the drug as part of his or her profession.
Which drugs are proscribed (illegal)?
The drugs that are proscribed in the states, territories and the Commonwealth are very similar, although the means of definition and classification of proscribed drugs varies across the jurisdictions. Drugs that are proscribed are usually set out in an authoritative list, which is often based on the SUSDP.
Commonwealth offences
The principal piece of Commonwealth legislation containing drug offences is the Criminal Code Act 1995 (Cth). The Criminal Code is supplemented by the Customs Act 1901 (Cth) and the Narcotic Goods Act. Part 9.1 of the Criminal Code addresses the trafficking, illegal manufacture, supply and possession of controlled drugs and plants. Prohibited conduct under the Criminal Code includes: the cultivation of certain plants (e.g. opium poppy) to produce narcotic drugs; making narcotic drugs or psychotropic substances; and the sale, supply or possession of a narcotic drug or psychotropic substance. Offences under the Criminal Code relate to conduct wholly or partly in Australia, dealing in drugs on board an Australian aircraft in flight or an Australian ship at sea and dealings outside Australia in various circumstances.
The Customs Act also details offences relating to importing or exporting narcotic goods. The Narcotic Goods Act forbids the manufacture of a drug unless the manufacturer holds a licence to do so. It contains other restrictions on the manufacture of drugs, including constraints on the premises, conditions of licence, labelling requirements and permission to destroy drugs, narcotic preparations or by-products.
State and territory offences
In all Australian states and territories it is an offence to possess, produce, use, sell, distribute or supply drugs that are proscribed, unless the act in question was otherwise authorised. In some jurisdictions there exist related offences, such as the possession of equipment for use in relation to proscribed drugs. In most jurisdictions it is an offence, unless authorised, to:
A range of related offences exists in certain states and territories, such as the theft of proscribed drugs, the possession of property derived from drug dealing, possessing instructions or equipment for producing proscribed drugs and various offences related to prescriptions for proscribed drugs. State and Federal legislation also provides for the seizure of assets obtained through serious drug offences.
The practices of health professionals are usually governed by relevant Acts of Parliament (such as the Nurses Act) and by regulations of the appropriate professional board (e.g. the Podiatrists Registration Board of Victoria). These are specific to the profession concerned, and details of their functions are beyond the scope of this book.
While drug offences are dealt with in state and territory criminal justice systems, some states have set up ‘drug courts’ and diversion programs to divert illicit drug users from prisons into treatment programs.
New Zealand drug regulations
Scheduling and subsidisation of drugs
In New Zealand, legislation relevant to drugs is contained in the Medicines Act 1981 and Regulations (1984). Drugs are scheduled, more simply than in Australia, into three main categories:
All other products are for general sales. There are no registration numbers assigned to products to show that they have been approved for sale, as there are in the Australian system.
Despite efforts at trans-Tasman harmonisation and simplification, there are still some differences in the scheduling of drugs. Single-agent sedating (old) antihistamines, for example, are S3 (Pharmacist-Only) in Australia, except when used as antiemetics against travel sickness (S2), but Pharmacy-Only in New Zealand, except in small packs as ‘sleeping aids’, when they are Pharmacist-Only. Salbutamol metered-dose inhalers are Pharmacist-Only in Australia but require a prescription in New Zealand, possibly reflecting the concern over some decades about higher than expected mortality rates due to asthma there. Omeprazole is Pharmacist-Only in small dose packs in New Zealand but a prescription medicine in Australia.
Nurses registered as Nurse Practitioners in New Zealand also have limited prescribing rights, as do midwives, and New Zealand allows direct-to-consumer advertising of drugs, which is usually prohibited in Australia. With respect to categories of safety of drugs in pregnancy, New Zealand follows the Australian guidelines and categories.
PHARMAC is a New Zealand Crown body with the responsibility for managing the pharmaceutical budget. It tenders for the subsidised drugs and its committees set the access criteria applying to expensive items. In the year 2000/01, it negotiated subsidy reductions worth about NZ$50 million. The top six expenditure groups of drugs in New Zealand recently were lipid-modifying agents, antiulcerants, antipsychotics, agents affecting the renin–angiotensin system, antidepressants and inhaled corticosteroids. This list can be compared with the lists of the top 10 drugs in Australia (Table 1-4); despite the differences in how the lists are compiled, the similarities are striking.
Controlled drugs (drugs of dependence)
The New Zealand Dangerous Drugs Act 1927 dealt with the controls required for opium and non-opiate drugs for which regulation was required by the League of Nations. Before this, opium was readily available in many pharmaceutical preparations; however, the main drugs causing problems (then as now) were alcohol and tobacco.
After World War II, cannabis and amphetamines began to appear as problem drugs, and in the ‘hippy’ days of the 1960s and 1970s people also experimented with Datura (containing the plant alkaloids atropine and hyoscine), amphetamines, hallucinogens and solvent sniffing. The Narcotics Act 1965 included controls on mescaline, cocaine and LSD as well as opiates. The Misuse of Drugs Act 1975 and subsequent Regulations (1977) classified controlled drugs into different schedules to allow different penalties depending on the severity of the abuse. Alcohol and tobacco are excluded from the Acts; alcohol is subject to the Sale of Liquor Act 1989 and amendments.
The United Nations Single Convention on Narcotic Drugs (1961, 1972) imposed wider controls on possession and use as well as production and trafficking in drugs, including marijuana. Countries signatory to this Convention are constrained to abide by its international agreements. There is some debate in New Zealand as to the wisdom of including marijuana along with drugs such as heroin and cocaine; drug trafficking and abuse of the latter two cause much more devastating consequences (Fastier 1998).
Standardisation of drugs
Medicines (formulations of drugs) may vary considerably in strength and activity depending on the amounts of active drug(s) they contain. Drugs obtained from plants such as opium and digitalis may fluctuate in strength depending on where the plants are grown, the season during which they are harvested and how they are preserved or extracted. Because accurate dosage and reliability of a drug’s effect depend on uniformity of strength and purity, standardisation and publication of standards are necessary. (Drug information resources, pharmacopoeias and formularies are reviewed in Chapter 1.)
Standards for drug quality and actions
Drug standards in Australia
The main standards for drugs in Australia are those published in the British Pharmacopoeia3 by the British Pharmacopoeia Commission and those in the Australian Pharmaceutical Formulary (APF). The BP gives detailed, legally accepted standards for hundreds of drugs, with chemical information and the approved formulations that contain the substance, and lists criteria for purity, chemical methods for identification and assay (measurement), tests for likely contaminants and maximum levels allowed for impurities. Preparations shown to meet these standards are then referred to as the BP preparation; for example, ‘Morphine Sulphate BP’ must contain 98%–102% of the stated amount; methods for identification and assay are given and limits set for the levels of iron, codeine, pseudomorphine and hydroxymorphine.
The BP also has monographs for natural substances. For opium BP it notes that: ‘Raw opium is the air-dried latex obtained by incision from the unripe capsules of Papaver somniferum … it contains not less than 10.0% of morphine and not less than 2% of codeine … Raw opium has a characteristic odour and a blackish-brown colour’. Test methods are given for its identification (under microscope and by thin-layer chromatography) and assay by liquid chromatography. The maximum limit for the contaminant thebaine is 3%.
The APF, by contrast, is more a reference and ‘recipe book’ for pharmacists. As well as much useful medical information and dispensing practice guidelines, it gives the standard formulae for many formulations. For Calamine Lotion APF, for example, it lists the six ingredients and their amounts, gives the method for preparation of the lotion and describes its uses.
Assays
The technique, either chemical or biological, by which the strength and purity of a drug are measured is known as an assay. Chemical assay is a chemical analysis to determine the ingredients present and their amounts. Opium, for example, is known to contain certain alkaloids and these may vary greatly in different preparations; the BP’s official standard gives details of assay methods for these alkaloids.
Bioassays
For some drugs, either the active ingredients are not completely identified or there are no available chemical methods of analysing and standardising them. These drugs may be standardised by biological methods, or bioassay. A bioassay is a biological test method for measuring the amount of a pharmacologically active substance in a preparation (tissue extract or pharmaceutical formulation). Bioassays are typically performed by determining the amount of a preparation required to produce a defined effect on a suitable laboratory animal or tissue under certain standard conditions, and then comparing the response to that produced by a standard preparation in the same bioassay. Examples of early bioassays were for the potency of a sample of insulin measured by its ability to lower the blood glucose levels of rabbits or for the strength of digoxin preparations by their effects on human electrocardiogram tracings.
Bioassays are especially applicable to:
Bioassays require setting up a model system on which pharmacological activities can be measured. The test method may be in vitro (in glass), e.g. using a suspension of an enzyme, a cell or tissue culture, a microbiological culture, a standard preparation of an antibody or an isolated organ or tissue; or in vivo (in the living organism), e.g. testing the effect of a drug on blood pressure or behaviour.4 Some drug actions are virtually impossible to test in animals either in vitro or in vivo, particularly effects of centrally acting agents on mood, perception and thought processes.
Clinical trials (see later section) are essentially bioassays in humans: the new drug (unknown) is tested against the best currently available therapy (standard) and compared for safety and efficacy.
Isolated organ experiments
In these pharmacological experiments, a small piece of animal tissue (such as a strip of intestinal smooth muscle) or an entire organ (such as a heart) is ‘isolated’ from the animal’s body and kept alive in warmed, oxygenated physiological saline solution, set up so that responses of the tissue (e.g. contractions of muscle, beats of the heart) can be monitored following administration of a drug solution into the organ bath. The classic is the isolated guinea-pig ileum smooth muscle preparation, which responds to stimulation by several neurotransmitters and other endogenous mediators; a great deal of classical pharmacology can be learned by this simple technique.
The use of isolated tissues to assay responses reached an extraordinarily sophisticated level in the classic experiments of Sir John Vane at the Royal College of Surgeons in London in the 1960s. A set of five organ-baths was set up in vertical series such that the physiological saline solution from the top bath flowed down over (superfused) the next bath, and so on down the cascade (see Figure 4-2). Small samples of GIT smooth muscle from four different species were set up in the baths, and the pattern of contraction or relaxation responses to seven endogenous mediators, including noradrenaline, bradykinin, prostaglandins and antidiuretic hormone, was studied. Using this technique, Vane discovered the mech anism of action of aspirin and other non-steroidal anti-inflammatory drugs, viz. inhibition of the synthesis of prostaglandins; for this he was subsequently awarded the 1982 Nobel Prize for Medicine (and knighted by the Queen).
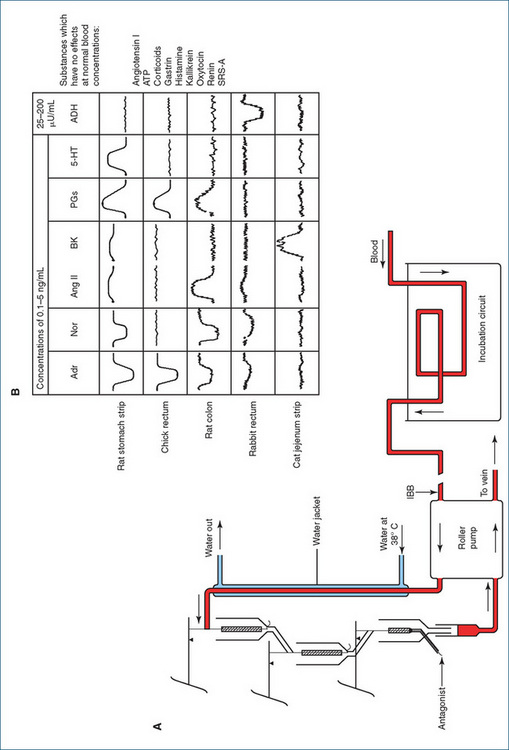
Figure 4-2 Parallel assay by the cascade superfusion technique. A Blood is pumped continuously from the anaesthetised test animal over a succession of test organs, whose responses are measured by a simple transducer system. B The response of these organs to a variety of test substances (at 0.1–5 ng/mL) is shown. Each active substance produces a distinct pattern of responses, enabling unknown materials present in the blood to be identifi ed and assayed. ADH = antidiuretic hormone; Adr = adrenaline; Ang II = angiotensin II; ATP = adenosine triphosphate; BK = bradykinin; IBB=into the bathing blood; Nor = noradrenaline; PG = prostaglandin; SRS-A=slow-reacting substance of anaphylaxis; 5-HT = 5-hydroxytryptamine. From: Vane 1969 and 1971, reproduced with permission, thanks to Wiley-Blackwell, Oxford.
Bioassays in the BP
Because of biological variability there may be variations in results and lack of precision in quoting the absolute amount of biologically active material. Bioassays are no longer used as frequently as previously because techniques such as radioimmunoassay (RIA, itself a type of bioassay) and high-performance liquid chromatography (HPLC) have allowed very low levels of chemicals to be measured accurately without using animals. The BP 2000 gave methods for bioassay of several drugs, including:
There are also BP biological tests for biological products such as blood-clotting factors, cytokines (e.g. interferons), vaccines, antibiotics and pyrogens (substances that cause fever), and tests for microbiological sterility or contamination.
The design of bioassays usually involves comparison of two preparations, a standard and an unknown, by testing many concentrations of each on the same model and constructing log dose–response curves. If the substances act by similar mechanisms, the curves will be roughly parallel in their mid-sections and so the potency ratio can be determined, allowing the strength of the unknown to be calculated compared to the known standard (see Figure 5-5).
International Units of Activity
The strength of extracts of natural substances for which the purity is not 100% cannot be expressed in absolute terms such as grams or milligrams, as it cannot be assumed that the whole weight is due to the active ingredient. Such preparations are assayed biologically, and a unit of pharmacological activity must be defined to compare the unknown with the standard. In the past a particular standard, e.g. of an animal hormone, enzyme preparation or plant alkaloid, was designated the International Standard preparation, against which other national standard preparations were assayed, and then these latter were used to standardise all preparations. In Australia, for example, the Commonwealth Serum Laboratories (CSL) in Melbourne maintained the national standards for insulins, and all CSL insulin preparations were compared to them. The strengths of preparations were expressed in terms of International Units of Activity (IU), for whatever activity was measured in the particular bioassay (see Clinical Interest Box 36-7).
Statistical methods in bioassays
Variability in responses to drugs may be due to many causes, especially errors in measurement and inherent biological variability both within and between individuals. In bioassays the same dose repeated may therefore give differing responses, and the dose required to give the same response varies. (Just as the heights of a large number of adults are normally distributed, and when plotted as a frequency distribution will assume a bell-shaped curve, so it can be expected that responses to a drug will also be normally distributed about a mean value.) Consequently, biological experiments need to be repeated many times to get an average or mean result, which is taken to represent the true value. Variability in responses can be partly reduced by refining methods and using a very homogeneous population of animals or very similar subjects, but this reduces the wide applicability of the results.
Statistical methods are then applied to deal with random variations and to extrapolate from the sample mean to the population; such techniques are the province of a text on biostatistics rather than pharmacology. In the pharmacological context, statistical methods are typically applied to bioassays studying dose–response relationships, cause–effect correlations, differences between groups of subjects treated in different ways and clinical trials (see Clinical Interest Box 4-3). Usually a ‘null hypothesis’ is defined (i.e. that there is no statistically significant difference between the groups being studied), and when results are analysed the null hypothesis is either accepted or rejected. The probability level (P) at which the results are accepted as being due to a real difference rather than occurring by chance is usually set at 0.05, i.e. there is only 5% chance (1 in 20) that the results could have occurred by chance. Typical statistical tests employed are either parametric (assuming a normal distribution of results) such as Student’s t-test, analysis of variance or variance ratio; or non-parametric (when normality cannot be assumed), e.g. the sign test or Wilcoxon rank-sum test.
Drug discovery and development
Drug discovery
There are several main ways in which drugs are ‘discovered’, i.e. ways in which the potential therapeutic uses of chemicals are determined. The routes to drug discovery are not mutually exclusive and an eclectic approach is often the most successful. This has been summarised as ‘three steps: understand the science, unravel the story, and … apply the technology’ (Handen 2005).
Development from herbal or traditional remedies
For thousands of years people have been trying natural products—plants, minerals and parts of animals—to see if they were useful as foods or in treating disease (these sources of drugs have been discussed in detail in Chapters 1 and 3; see Table 1-2, Figures 1-2 and 1-3, Tables 3-3 and 3-4, Clinical Interest Box 3-5). Examples of drugs developed from natural sources include morphine and codeine from the opium poppy Papaver somniferum, atropine from the belladonna lily Atropa belladonna, growth hormone from extracts of pituitary gland, insulin from beef and pig pancreas and iron and iodine from mineral resources. The natural products may be used as crude extracts, such as raw opium or herbal teas, or purified and/or synthesised and then formulated as pharmaceutical preparations, such as tablets and injections.
This route to new drugs is sometimes called the ‘reefs and rainforests’ approach, as it is recognised that there are millions of natural chemicals out there needing to be identified and tested before exploitation and despoilation of the world’s resources permanently terminate our chances of finding novel anticancer or antibiotic agents (for example).
Serendipity (sheer good luck)
While luck plays a part in some drug discoveries, such as Fleming’s bacterial culture plate becoming contaminated with a growth of the fungus Penicillium notatum, it usually takes lateral thinking (e.g. questioning why growth of the bacterial culture was inhibited near the fungus), intelligence and years of hard work (extracting the natural antibacterial agent or antibiotic, determining its structure and developing methods of mass production) to exploit the lucky find (producing enough penicillin to treat people with bacterial infections).
Other examples of serendipity in pharmacological discovery are the finding that patients treated with the first safe oral antibacterial agents, sulfonamides, had a lowering in their blood glucose levels, which had a ‘spin-off’ to the development of the sulfonylurea oral hypoglycaemic agents; the finding that hypertensive patients treated with the vasodilator minoxidil tended to grow more hair (the drug is now mainly used as a hair restorer); and the finding that sildenafil, undergoing clinical trial as a vasodilator in cardiovascular disease, was unexpectedly popular with the male subjects in the trial, leading to its use in treating impotence (see Drug Monograph 40–3 and Clinical Interest Box 40-5).
Empirical chemistry plus pharmacological studies
As chemical techniques developed in the 20th century, the chemical structures of pharmacologically active substances could be determined and similar substances synthesised and then tested for activities. These structure–activity relations led to the development of many drug groups; for example, the various formulations of insulin all based on bovine or porcine insulins, and the second- and third-generation penicillins were adapted from the first penicillin.
All the sympathomimetic amines were initially noradrenaline ‘look-alikes’: studies of Ephedra sinica, long known in traditional Chinese medicine to be useful in respiratory conditions (asthma), led to the purification of the active ingredient, ephedrine, then synthesis of the related compounds isoprenaline (a useful antiasthmatic drug) and salbutamol (a β2-adrenoceptor agonist with fewer cardiovascular adverse reactions). Then β-blockers were designed to act as ligands at the receptor without activating it, such as propranolol, a non-selective blocker at both β1- and β2-receptors, and later atenolol, a selective β1-receptor antagonist useful in cardiovascular diseases and with less likelihood of causing asthma (the chemical structures of these compounds are shown in Figure 4-3).
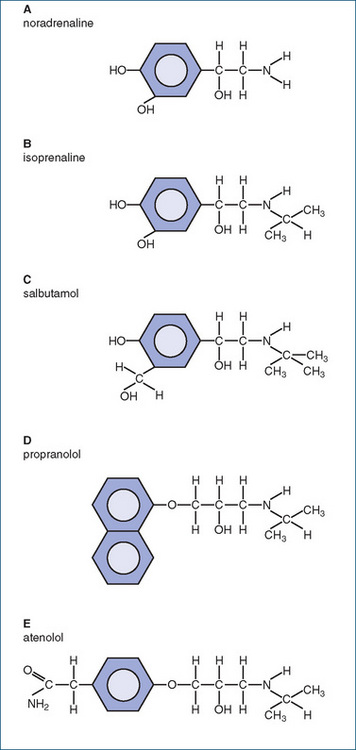
Figure 4-3 Structure–activity relationships for drugs based on the sympathetic neurotransmitter noradrenaline (A); B isoprenaline, a non-selective β-adrenoceptor agonist; C salbutamol, a selective β2-adrenoceptor agonist; D propranolol, a non-selective β-adrenoceptor antagonist; E atenolol, a selective β1-adrenoceptor antagonist. It can be seen that increasing the ‘bulkiness’ of the substituents at the catechol end or the amine end may select for ligand-binding affinity and agonist/antagonist activity at the different receptors.
Active metabolites of existing drugs
Sometimes drugs have been found to be more active after metabolism in the body and so the metabolites are tested as drugs. Paracetamol is one of the metabolites of phenacetin, an early antipyretic analgesic agent (see Figure 15-6), and is much safer. Many of the benzodiazepine antianxiety agents have pharmacologically active metabolites, some of which are marketed as distinct drugs in their own right.
Rational molecular design
Structure–activity studies can lead to speculation as to the shape of the active site of a receptor and to the design and synthesis of drugs that may be agonists or antagonists at that receptor. The early antihistamines were modelled on the histamine molecule, and subsequent brilliant pharmacology by Sir James Black led to the discovery of histamine H2-receptors and development of specific H2-antagonists that revolutionised the treatment of peptic ulcer.
Computer-aided design
Drug receptors, enzymes, ion channels and transporters are no longer simply ‘black boxes’ referred to by pharmacologists wishing to explain (or bluff that they understand) drug mechanisms, but are proteins with known amino acid sequences and tertiary structures (three-dimensional shapes), able to be cloned and genetically engineered. Computer modelling of the active sites of such proteins assists drug design, as chemical structures can be modelled and tested for virtual affinity for binding to the active sites. Using such techniques, angiotensin-converting enzyme (ACE) inhibitors were designed for use in hypertension, dopa-decarboxylase inhibitors were designed for administration with levodopa in Parkinson’s disease, the anti-flu drug zanamivir was designed to inactivate the flu virus and potential anticancer drugs are designed to inhibit various stages in the pathways of macro-molecular synthesis.
Monitoring the activity of the scientific community
Research carried out by pharmacologists, biochemists and chemists in universities and research institutes may lead to the discovery of new drugs in unexpected ways. The pharmaceutical industry monitors such research via the scientific literature, patent applications and scientific conferences.
Drug development
Development of new drugs is regulated by government legislation and administered by government authorities—the TGA in Australia. Regulation is necessary because consumers need protection so that only safe and effective drugs are approved, and the sponsoring drug companies need protection for their investment in terms of intellectual property, patents and copyright. The main processes of drug regulation in Australia are summarised in Figure 4-1; see also review by Barnes (2006).
The pharmaceutical industry
The pharmaceutical industry is constantly screening substances with potential to market as new drugs; worldwide the industry is estimated to be worth about US$250 billion/year.
Prospective drugs take years and large amounts of capital for basic and clinical studies and for the costs of application and promotion. The increasing emphasis on ‘lifestyle drugs’, which may be taken for decades and hence require studies of long-term safety; the prevalence of polypharmacy, with its inherent risks of drug interactions; and the insistence of governments on proof of costeffectiveness all contribute to the enormous costs of testing drugs. In addition, drug companies carry out research aimed at new drug discovery. Because of the huge commitment and risks involved, drug companies have been merging over the past few years, for economies of scale and to combine their research and development efforts and achievements, with the result that there are only a few major drug companies left worldwide.
It is interesting to note that, while drug companies insist that the high costs of new drugs are to recoup money spent on research, on average drug companies allocate 20% of their expenditure to marketing and only 1% to research. The major markets are of course the USA, Europe and Japan; Australia takes only 1%–2% of world sales of pharmaceuticals.
The stages of drug development
Drug development has traditionally been described as occurring in several clearly defined phases:
Clinical interest Box 4-2 High-throughput screening
High-throughput screening (HTS) refers to the process whereby millions of chemicals from ‘chemical libraries’ can be put through automated biochemical tests for activity, in minute amounts, very rapidly. The aim is to discover ‘lead compounds’—those that lead to potentially useful drugs. Such screens have been described as ‘fishing trips’, trawling through millions of compounds in the hope of catching something interesting.
The tests are carried out in an array of hundreds of tiny ‘cells’ (minute test-tubes) or on silicon chips, leading to the term ‘in silico’ for this type of assay, as distinct from in vitro and in vivo. Most tests involve a particular protein (e.g. receptor, enzyme, ion channel or antibody), gene or RNA fragment in the cell or on the chip, and the amount of binding of the new compounds to the protein or gene is determined. Tests may be based on enzyme-linked immuno-sorbent assays (ELISA) and commonly use high-performance liquid chromatography–mass spectrometry technologies (HPLC–MS) and/or fluorescence methods.
Binding is picked up electrically by a voltage-sensing detector and registers as a ‘blip’ signal. Positive binding can be followed up in laboratory assays to determine the nature and strength of the binding, e.g. whether the compound is an agonist or antagonist at a receptor or whether a particular enzyme activity is enhanced or inhibited.
Gene fragments and RNA molecules from the huge databases of the Human Genome Project can be checked in different disease states, to test for under- or over-expression of encoded proteins, identify genes implicated in disease or diagnose inherited diseases or predisposition to cancers. HTS methods can also be applied to pharmacokinetic aspects of new compounds, checking for enzymes responsible for metabolism, rapidity of metabolism and inhibition or induction of drug-metabolising enzymes.
Pharmaceutical characteristics of the compounds can be tested, such as solubility and permeability, and potential for crossing the blood–brain barrier. HTS procedures are also applied to toxicological analysis, e.g. detection simultaneously of several different compounds in forensic samples in criminal cases or doping control in sport.
The procedures are highly automated, using robot technology, and are computer-controlled. In an HTS facility, millions of compounds can be screened per month. Techniques are becoming both higher-capacity and better focussed. Using HTS, drug companies hope not only to discover new drugs but also to reduce the drop-out rate of compounds in the later, more expensive stages of animal and human testing.
See Walter et al 2001, Mayr & Bojanic 2009.
The costs in time, money and effort
The development of a drug takes a prodigious amount of time, money and effort. It has been estimated that drug development from idea to market takes 12–24 years, including the time from active compound to clinical trials taking 7–9 years. Once the idea, chemical or process is patented (to protect the developers from other companies stealing their ideas) the clock starts ticking! In most countries, the duration of a patent is usually 15–17 years, with a possible short extension. When the patent expires, other companies can manufacture and market the drug as a ‘generic’ product under their own trade names. Consequently, companies need to minimise the time taken to get their drug onto the market.5
The financial costs involved in bringing a drug to market are so great now that very few companies can afford to carry out the research and development. It is estimated that every new drug costs around A$1.2 billion and that a drug company needs 1–2 new drugs every 3–4 years to remain financially viable. While the spin-offs from research into the human genome have been incredibly exciting in terms of potential targets for drug actions and disease treatment, this research is very expensive to exploit and there are estimated to be 3000–10,000 protein targets to be explored, of which the G-protein receptors are likely to be especially important to pharmacology (see later section on ‘Future drug development and the new genetics’).
The costs in terms of effort involved are also immense and may eventually be wasted. It was formerly estimated that only one out of every 10,000 chemicals synthesised made it to the marketplace; the proportion would be even smaller now in the era of combinatorial chemistry and high-throughput screening. This is because drug development may be abandoned at any stage if the drug is ‘thrown out’ because of problems with safety, efficacy, changes in fashion or a better competitor drug. Drug companies are trying to streamline testing procedures and get early information on toxicity or pharmacokinetic problems so as to waste as little time and money as possible.
Clinical trials of drugs
A clinical trial is a prospective study carried out in humans to determine whether a treatment that is believed to benefit a patient actually does benefit; thus it is a type of human experiment. The treatment being tested may be investigational (new) or a new version of an established treatment; it may be a drug, diet, medical device, surgical or physical procedure6 or other modality. In the context of testing a new drug, the trial provides scientific data on safety (by rate or severity of adverse drug reactions) and efficacy (by statistically significant evidence of difference between treated and control groups). The ‘gold standard’ of clinical trials is the randomised controlled clinical trial (RCCT).
Characteristics
Typically, each subject enrolled in the trial is randomly allocated into a treatment group, to be administered the new drug under test, or to a control group, usually given the current best therapy or a placebo if there is no available treatment. It must be noted that the treatments are considered equivalently beneficial before the trial (otherwise it is unethical to deny one group the better treatment) and that the results are applicable only to this treatment regimen and cannot be widely extrapolated to other similar drugs or patient groups. All tests in humans must be approved by a local institutional ethics committee (IEC see later section).
Clinical trials are generally required for all new drugs and for new uses or formulations of old drugs; however, there are exceptions.
The objectives of RCCTs need to be realistic, efficient, compatible, valid and specific, yet allow for generalisation. The criteria for efficiency and validity require that statisticians become involved in planning, to ensure that enough subjects are involved for the results to be statistically acceptable (see Clinical Interest Box 4-3). Clinical trials are a staged process with distinct phases, which allows for lower risk (few patients in early phases) and stepwise decisions (trials can be stopped if clear differences or toxicities become apparent); but are slow, expensive and put pressure on the participants involved.
Clinical interest Box 4-3 Streptomycin, smoking and statistics
Before the 1950s, medical practice was based on accumulated wisdom acquired through everyday experience, passed down to succeeding generations of doctors. In 1950, however, two events occurred that caused a paradigm shift in medicine, such that statistical proof of efficacy and randomised controlled clinical trials became the criteria for scientific evidence on which practice should be based (evidence-based health care).
Both events were due to Professor Austin Bradford Hill, of the Medical Statistics Department of the London School of Hygiene and Tropical Medicine. Bradford Hill had intended to follow his father into medical practice but contracted tuberculosis (TB) during World War II, and so could not enter clinical medicine. He studied economics and statistical methods, and then applied these studies to epidemiology.
Bradford Hill’s first major contribution to medicine was the demonstration that two drugs together, streptomycin and paraaminosalicylic acid (PAS), given over a period of several months, resulted in marked clinical improvement in patients with TB and reduced the development of resistance to the antibiotic. When invited to join the Tuberculosis Trial Committee, Bradford Hill realised that streptomycin, an antibiotic discovered in 1943, had to be given for several months before any improvement became obvious; that the British supplies of the drug were very limited; and that patients with TB sometimes underwent spontaneous remission, making proof of drug efficacy difficult. He argued persuasively that investigation of the two drugs in a clinical trial could be conclusive only if a control group were used and if the patients were randomly allocated into the control group, ‘treated’ with bed-rest and collapse of the lung, or the treatment group on streptomycin (and in later trials, PAS as well). The trials demonstrated conclusively that the drug combination considerably reduced both deaths from TB and the risk of development of streptomycin-resistant TB.
The second achievement was the convincing proof that smoking causes lung cancer. Because a very large proportion of the population smoked cigarettes during and after World War II and air pollution levels in cities were very high, the association of tobacco smoking with respiratory disease was not obvious. By applying statistical methods to this epidemiological issue and separating subjects into groups based on their smoking habits, Bradford Hill was able to demonstrate convincingly that the more cigarettes people smoked, the greater their risk of lung cancer.
Bradford Hill is credited with showing that ‘the detached objectivity of statisticians inherent in the notion of randomisation is more likely to get at the truth than the subjective impressions generated from clinical experience’. The gold standard of the RCCT was to prove indispensable in evaluating the enormous number of new drugs that were produced in the 1950s and 1960s, and medical statistics proved invaluable in demonstrating associations between lifestyle factors and conditions such as cancers and cardiovascular disease.
Adapted from: Le Fanu 1999.
Phase I: the first tests in humans
After extensive testing in vitro and in animals, the drug is administered initially in very low doses to a small number of healthy volunteers, e.g. in a research centre or institution, under close medical and scientific supervision. The objectives are to determine pharmacological activities in humans, pharmacokinetic parameters, a safe dosage range and any acute toxicity. Few Phase I trials are done in Australia, mainly because relatively little drug development work is done here (however, see Table 4-3).
Table 4-3 Some drugs developed and/or trialled in Australia
| Drugs or compounds | Research institute or drug company |
| PI-88, a potential anticancer drug targeting the enzyme heparanase, important in the growth and spread of cancers | Progen Industries and John Curtin School of Medical Research at the Australian National University |
| Novel crystalline carbohydrates, with applications in preventing adverse gut reactions with antibiotics, and graft rejection after transplant surgery | Alchemia P/L and the University of Queensland’s Centre for Drug Design and Development |
| Blood products: vaccines, sera, factors for bleeding disorders, antibodies for preventing severe infections; interferon β-1a for treating multiple sclerosis | CSL Ltd, Melbourne |
| Thebaine, used to synthesise the opioids oxycodone and buprenorphine, produced in high yields from new strains of opium poppy | Tasmanian Alkaloids P/L |
| Zanamivir (Relenza): treats influenza by stopping production of neuraminidase protein in flu virus (see Clinical Interest Box 4-5) | Victorian College of Pharmacy, CSIRO, Australian National University and Biota Holdings Ltd |
| A unique chemical from a marine organism, that kills malaria parasites | Eskitis Institute at Griffith University, Brisbane |
| A new formulation of mannitol in inhaled nanospheres, for treatment of cystic fibrosis and bronchiectasis | Pharmaxis, Sydney |
| Three anti-angiogenesis products that target growth factors in blood vessels | Progen and subsidiary PharmaSynth, Queensland |
Adapted from: Harrison 2000 and other sources.
Phase II: the first administration to patients
These are the initial efficacy studies to test if the drug is indeed effective in treating the condition of interest, in a small number of closely supervised patients (10–200) in tertiary hospitals (usually major metropolitan teaching hospitals). The tests are ‘single-blind’, which means that the patients do not know which treatment they are getting but the investigators do. The investigators are specialists in the appropriate field of medicine, such as endocrinologists, psychiatrists, anaesthetists or rheumatologists. Phase II studies give an indication of the therapeutic range of doses, maximum tolerated dose and common adverse reactions in patients with the disease.
After the Phase II trial the drug company may apply for approval to conduct a RCCT. In Australia, data are sent to the Australian Drug Evaluation Committee and its specialist subcommittees, which assess formulation, production, efficacy, adverse reactions, protocols in the proposed trials and ethical aspects. If the drug appears to be safe and efficacious, with likely benefits outweighing risks, it progresses to the next phase.
Phase III: the full-scale randomised controlled clinical trial
This is ‘the clinical trial’, in which the drug is administered to numerous patients under the guidance of experienced clinical investigators. The aim is to ascertain whether, under the defined conditions, the drug shows clinical benefit for the disease state, with acceptably low rate and severity of adverse drug reactions. The trial is usually ‘multicentre’, i.e. carried out simultaneously in several medical centres in different countries, to increase the number of subjects and investigators, achieve quicker results and enrol different ethnic groups; many are carried out in Australia. Statistically significant results able to be extrapolated to a wide range of populations are desirable. An RCCT may cost up to US$5 million, so it is important that it be designed carefully to ensure valid results. Other aspects that may be included in the study are the drug’s pharmacokinetics, effects of other concurrent diseases or drugs and costs of therapy.
Important elements of the RCCT are as follows:
It is essential that the trial be planned well to determine in advance parameters such as the maximum length, end-points, justification (who benefits?), information to be given to patients, consent forms, protocols, inclusion criteria, withdrawal procedures and follow-up schedules. Ethical aspects must be considered (discussed later) and an application made to the IEC for approval to run the trial.
During the execution of the trial, subjects are monitored closely and the trial is regularly audited for safety, ethics and quality control. If it becomes apparent that one group is benefiting far more (statistically significantly) than the other,7 or suffering more adverse reactions, the trial is halted. Usually the statistical basis for the trial is the null hypothesis—that there is in fact no difference between the two treatments (i.e. the new drug is as good as the current therapy); results are analysed for statistical equivalence and the null hypothesis is accepted or rejected.
Phase IV: post-marketing studies
Assuming that the clinical trial process is concluded and the new drug is shown to be safe, efficacious and costeffective, it may be approved for marketing. It is recognised, however, that there are several limitations in the testing and trialling processes. The number of people studied and the time allotted to the study are limited. Also, certain types of individuals may be excluded from the clinical trial, such as children, pregnant women, persons with multiple disease states or taking other drugs and the elderly. If a drug is considered safe and effective during the time of study, with the previously mentioned limitations, it is marketed when approval has been granted.
Once marketed, the drug is used in much greater numbers of patients and probably for longer periods; extended monitoring of safety and efficacy (pharmacovigilance) is then required. It is inevitable that the drug will be reported to produce additional effects (possibly therapeutic but often adverse) that were not noted during the trial studies, such as rare adverse reactions, effects in subgroups of the population and effects in patients taking other drugs with which the new drug may interact. The DoTS system is recommended as a way of classifying and monitoring ADRs based on the most relevant factors: the Dose, the Time course over which it appears and the Susceptibility of the patient (see Aronson and Ferner [2003]). Studies in older people are especially important, as they often have many co-morbidities, may be on many drugs, may require drugs for prolonged periods and may not have ready access to regular medical care.
Post-marketing surveillance by the TGA also involves laboratory investigations of products on the market and ongoing monitoring of products to ensure compliance with legislation. Later, meta-analysis (or overview analysis) may be carried out to combine and analyse data from all similar clinical trials. This has the advantage of increasing the statistical power of analysis by increasing the numbers of subjects, making significant results more likely; however, meta-analysis suffers inevitably from ‘publication bias’, as negative results are less likely to be published than positive results. (Some regulating authorities and journals require authors to advise in advance when trials are to be carried out, to ensure that results of all trials are published.) Sometimes, different trials will produce conflicting results; the choice of which drug to prescribe then becomes one based on the clinical judgement of the prescriber.
The ‘blue form’
Through its Advisory Committee on the Safety of Medicines (ACSOM; formerly the Adverse Drug Reactions Advisory Committee, ADRAC), the TGA has instituted a voluntary program to enhance the reporting by health-care professionals of adverse reactions they suspect are related to medications and medical devices. The verification of an adverse reaction is not necessary. It is important to inform the agency of medication- or medical-devicerelated events suspected to have resulted in adverse reaction. Confidentiality is maintained in the reporting. The paperwork is a one-page form (the ‘blue form’; see Figure 4-4) that is readily available and is regularly sent to prescribers of drugs, along with copies of the Adverse Drug Reactions bulletin; it can also be filled in electronically (at http://www.tga.gov.au/adr/bluecard.htm). Reports of adverse reactions are reviewed, coded, entered into a database and analysed for patterns. Some are forwarded to ADRAC, which updates and informs health professionals about adverse reactions and can recommend actions ranging from no action required, change of aspects of prescribing or dispensing, through to withdrawal of a drug from the market. (New Zealand has a similar Intensive Medicines Monitoring Programme; see Clinical Interest Box 4-4.)

Figure 4-4 The ‘blue form’, on which suspected adverse reactions to drugs and vaccines are reported. Published by the Australian Adverse Drug Reactions Advisory Committee, Woden, ACT; reproduced with permission (see also http://www.tga.gov.au/adr/bluecard.htm).
Clinical interest Box 4-4 The intensive medicines monitoring programme
The Intensive Medicines Monitoring Programme (IMMP) was initiated in New Zealand in 1977, as part of post-marketing surveillance of new drugs, to identify adverse reactions early, see who is particularly at risk and ensure appropriate actions for safe use of medicines.
IMMP intensively monitors new medicines using a method known as ‘prescription event monitoring’. New Zealand has made valuable contributions to this worldwide programme by identifying previously unrecognised adverse effects. For example, New Zealand took the lead in taking regulatory action over agranulocytosis caused by mianserin and the liver toxicity that resulted from nefazodone (now discontinued in New Zealand, Australia and most other countries). It is the only drug monitoring system that can help to protect groups of people (such as Maori and Pacific Islanders) in whom the metabolic pathways of drugs are genetically affected, by collating post-marketing data.
In 2004 there were reports that the NZ Government was withdrawing its funding of the IMMP. This raised concerns worldwide, some of which were published in the British Medical Journal. However, the Ministry of Health responded by stating that the NZ Government was committed to strengthening its pharmacovigilance services, but felt that the labour-intensive manner in which IMMP had operated in the past required reviewing. The Ministry is using an innovative approach to monitor the use of a new vaccine which had been developed to deal with the meningococcal epidemic in New Zealand.
Source: Matheson D. British Medical Journal 2004; 329: 460; see http://www.medsafe.govt.nz/Profs/adverse/IMMP.asp.
ACSOM has introduced Medicines Safety Updates (to be published regularly in the Australian Prescriber), Medicines Risk Management Plans and a new alert system for recently introduced drugs. In Australia, adverse reactions can now be reported on-line, via the TGA’s website, following links to the Online Services. Consumers can also report their own adverse reactions, via a 1300 telephone number designated the Adverse Medicine Events (AME) Line (1300 134 237).
Drug development in Australia
There is little basic research carried out in the ‘big pharma’ drug companies in Australia or New Zealand, where most companies are offshoots of multinationals based overseas. Scientific work carried out in their Australian companies is mainly pharmaceutical work on formulations suitable for Australian conditions and preparation of submissions for the marketing in Australia of drugs that have been developed and trialled overseas. Australian companies such as Fauldings, GlaxoWellcome Australia and AMRAD try to carve out niches for themselves in areas such as parenteral formulations or developing products of local biotechnology companies. Overall, it is estimated that the pharmaceutical industry in Australia employs about 40,000 people. In 2007 it allocated about A$860 million to research and development (R&D), and it exported products worth nearly A$4 billion in 2008. However, this is small in comparison to the ‘big pharma’ players: Australia’s share of global R&D is only about 1.3%; so Australia relies on the rest of the world to create 98% of advances in knowledge and progress.
Biotechnology in Australia
Australia has about 500 biotechnology companies. CSL (formerly Commonwealth Serum Laboratories, set up over 100 years ago to develop ‘immune sera’ (vaccines), and later a major producer of antibiotics and blood products) employs more than 10,000 people in 27 countries, and in 2007/08 its revenue totalled $3.79 billion. The costs of developing a biotech product to approved drug stage are estimated at $1.2 billion, with costs roughly equally allocated to preclinical and clinical development. One of the multinationals recently damned Australian efforts with faint praise: ‘Australia’s growing biotechnology industry does not have the finances or the expertise to bring a medicine to the global market’.
Clinical trials in Australia and New Zealand
In Australia the TGA has overall control of therapeutic goods via pre-market evaluation and approval of products, licensing of manufacturers and post-market surveillance. Therapeutic goods not yet registered may be accessed for clinical trials by application to the TGA, which thus regulates clinical trials in Australia. Details of the relevant regulations and guidelines are covered in the TGA booklet Australian Clinical Trial Handbook 2006 (see details in ‘On-line resources’ at the end of this chapter). Use of a registered or listed product in a clinical trial beyond the conditions for which registration/listing has already been granted also requires approval by the TGA.
There are two main schemes under which drugs (and medical devices) may be trialled. The first is application for approval under the Clinical Trial Exemption (CTX) scheme. An application to conduct a trial is submitted to the TGA, whose delegate reviews the data and may object to the trial or comment on the proposal. When any objections have been satisfactorily met and the local Human Research Ethics Committee (HREC) has approved it, the trial may go ahead without further assessment from the TGA. Early Phase I and II studies and trials of medical devices most commonly come under the CTX scheme. The scheme is complex, and few trials now come under these rules.
The second approach is notification under the Clinical Trial Notification (CTN) scheme, under which data are submitted to the local HREC of the institution where the trial will be conducted. The HREC reviews the data and the trial design and advises the institution if it approves the trial. A CTN form must be submitted to notify the TGA of the trial. Phase III and IV trials and bioequivalence studies are best suited to the CTN scheme. The HREC can refer the application to the CTX scheme if it is uncomfortable with making its decision based on the data available.
The TGA has strict regulations relating to the roles of HRECs, trials involving gene therapy and related therapies, preventing or stopping a trial and indemnity and compensation. The Code of Good Clinical Practice must be followed. This covers aspects such as the responsibilities of the chief investigator and of the drug company; drug product handling, storage and accounting; reporting of adverse effects; and keeping and archiving of records. There are potential problems relating to delaying or withholding of negative results, applying ‘spin’ to make drugs look better than they really are, participating doctors accepting funding or gifts from sponsoring drug companies and lack of transparency about procedures.
In New Zealand, approval to trial a new drug not yet registered is submitted to the Standing Committee on Therapeutic Trials (SCOTT). Quite a few clinical trials are carried out in NZ, as it is a small, closed, not too mixed population. The New Zealand Regulatory Guidelines for Medicines, Volume 3, contains the Interim Good Clinical Research Practice Guidelines, which aim to make those involved in the design, performance and analysis of clinical studies aware of the minimum requirements for highquality research. The guidelines are based on the European Union, UK, Nordic, Australian and World Health Organization guidelines and codes for Good Clinical Research Practice.
The New Zealand guidelines outline the need to evaluate the risks and benefits, requirements for obtaining informed consent, quality control, audit, data recording, analysis, interpretation of the results and reporting of adverse events for studies conducted in New Zealand. Pharmaceutical companies conducting clinical research have to comply with the principles contained in the guidelines. It is essential that doctors are familiar with Good Clinical Research Practice requirements and assess the proposed research for compliance before participating.
Drugs developed in Australia
Australian medical schools and medical research institutes have had an enviable reputation worldwide for health-care research; however, commercial exploitation and ‘valueadding’ of the research usually happens overseas. Some drugs that have recently been developed and/or trialled in Australia are summarised in Table 4-3, and a case study of the discovery and development of zanamivir is given in Clinical Interest Box 4-5 (clinical aspects of this drug are described in Drug Monograph 28-8).
Clinical interest Box 4-5 An australian drug discovery: relenza
More than 40 million people have died from influenza in the past 100 years. Although vaccines are effective, they need to be continually updated, as the virus has the ability to mutate frequently to new strains that are not inactivated by old vaccines. Research carried out by Dr Peter Colman, a scientist with the CSIRO, led to the development of a novel anti-flu drug, zanamivir (Relenza). The history of the discovery and development of the drug is as follows.
| 1978 | Dr Peter Colman and Dr Jose Varghese, at CSIRO’s Division of Protein Chemistry in Parkville, Melbourne, recognised that the only invariant (unchanging) part of the flu virus when it mutates is part of the neuraminidase protein on the viral cell surface. (Neuraminidase is an enzyme that hydrolyses sialic acid residues in sugar groups in biological membranes.) Working with Dr Graeme Laver at the Australian National University, they set out to determine the three-dimensional structure of the protein. |
| 1983 | The structure of the protein was elucidated, and the next step was to find funds to set up a team to synthesise chemicals that might inhibit the functions of the protein and thus inactivate the virus. |
| 1985 | A local entrepreneur, Mark Crosling, formed Biota Holdings Ltd to back the development, and a licensing agreement was signed. Biota subsequently funded research at both CSIRO and the Victorian College of Pharmacy (VCP), Parkville. |
| 1986 | A synthetic chemistry program was started in the Pharmaceutical Chemistry department at the VCP, led by Dr Mark von Itztein, to custom-design an organic molecule that would inactivate the viral neuraminidase enzyme. |
| 1989 | The first potent inhibitor was synthesised. As much more funding was required to develop the drug and submit it to clinical trials, the drug company GlaxoWellcome, which had earlier shown interest, was contacted and collaboration initiated. |
| 1990 | A formal agreement was signed between Biota and Glaxo, allowing Glaxo access to Biota’s intellectual property and the marketing rights to the drug. |
| 1992–93 | Glaxo took the lead compound into exploratory development, and the efficacy of the most active compound in animal trials (code-named GG167) was established. |
| 1994–97 | Phase I, II and III clinical trials. |
| 1998 | Registration applications in Australia, Europe, Canada and USA for zanamivir (Relenza, GG167). |
| 1999 | Market release of zanamivir in Australia, Europe and USA; shares in Biota Holdings soar! |
It is estimated that the Australian investment in the research and development that led to the development of zanamivir totalled A$26.3 million in 1994/95 dollars. This included funding by CSIRO and Biota and extra government top-up grants. The costs of clinical trials, manufacturing scale-up and marketing were funded by the sponsoring company, GlaxoWellcome.
Note: Another neuraminidase inhibitor, oseltamivir (Tamiflu), a competitor drug, was released successfully just a few months after zanamivir, with the advantage that it could be taken orally, whereas zanamivir was administered by inhalation due to its low oral bioavailability.
Based on information from: Warrick Glynn, CSIRO Parkville, and CSIRO (1998).
Future drug development
The new genetics
Drugs are still being discovered by the old methods, including structure–activity studies on drugs binding to receptors and ‘fishing’ for interesting leads in reefs and rainforests (and other natural sources). There has been great interest and excitement in the hope for new drugs from ‘the new genetics’—the application of molecular biology and its techniques to human physiology and pathology. This has led to reports of genes to combat cancer, ageing and arthritis, and hope for cures after discovery of genes associated with cystic fibrosis, breast cancer, type 1 diabetes and various anaemias. While there have been some successes, as yet the promises of the new genetics have not been extensively fulfilled.
The terms ‘pharmacogenetics’ and ‘pharmacogenomics’ tend to be used interchangeably; a distinction may be that pharmacogenomics is a broader-based field where studies may elucidate the significance of specific genes and their expression in disease, their genetic translocations and related phenotypes, which helps determine prognosis and optimise treatment. Pharmacogenetics more commonly refers to studies of genetic differences in drug-metabolising enzymes or receptors to explain pharmacokinetic and -dynamic variabilities between responses to drugs, and thus allow personalisation of dosing of particular drugs. (These topics are discussed in more detail in Chapter 7, and some applications given in Chapter 41 under ‘Dosage of antineoplastic agents’.)
Genetic engineering
Genetic engineering, commonly referred to as biotechnology, is basically another route for the synthesis of proteins and other complex molecules using recombinant DNA techniques. Once the gene that codes for a particular protein is discovered, it can be inserted into a microorganism such as a strain of bacteria, so that the bacteria will now synthesise the protein. The first drug to be made this way was human insulin (in 1978), which previously had been synthesised by changing the amino acid sequence of pig insulin. Now, human insulin is readily produced by the bacterium Escherichia coli and the yeast Saccharomyces cerevisiae (see Drug Monograph 36–2).
Other ‘biotech’ products rapidly came into therapeutic use, including interferon-alpha, human growth hormone, erythropoietin, interleukin-2, factor VIII (antihaemophilic factor) and other growth-stimulating factors and monoclonal antibodies. Many products are now produced by transgenic animals which have been genetically-engineered to produce human or other useful proteins, including proteins in human milk and antibacterial polypeptides (cathelicidins). Twenty cows can now provide the world’s requirements of human growth hormone (Drug Monograph 33–2).
Genetic screening
There are about 4000 diseases resulting from the mutation of single genes; most of them are very rare except for cystic fibrosis, Huntington’s chorea, muscular dystrophy and some congenital blood disorders such as sickle-cell anaemia, thalassaemia and haemophilia. New genetic techniques in the 1980s allowed the identification of many of these genes, so that some of these disorders can now be diagnosed prenatally by genetic testing of an ‘at-risk’ fetus in the uterus. (However, this does not help people in whom the disorder arises spontaneously by mutation.)
These conditions can therefore now be diagnosed in utero, making it possible for parents to decide whether to terminate the pregnancy or allow it to continue. Genes that predispose to cancers are also being hunted; however, cancers closely linked to a particular gene are very much in the minority, e.g. only about 5% of breast cancers are related to the two main breast cancer genes BRCA1 and BRCA2.
Gene therapy
This refers to the concept that if a normal copy of a gene can be inserted into a cell carrying a genetic error, the error can be corrected and the genetic disease cured by physically changing the gene itself. This leads to the dream that humans might no longer be constrained by their genetic make-up. For gene therapy to be feasible, first the gene for a condition must be identified, then a copy of the normal gene must be inserted via a ‘vector’ into certain cells, which use the gene to make the missing proteins. The vector is usually a virus that has been inactivated so as to be no longer pathogenic. Gene therapy has been carried out in several situations, including inserting the gene for the enzyme adenosine deaminase into T lymphocytes of patients with a deficiency in this enzyme; in cystic fibrosis, administering the normal gene via a nasal solution into the airways of children with the disease; and in muscular dystrophy, injecting primitive muscle cells containing the normal gene into the muscles of boys with muscular dystrophy.
Unfortunately, none of these methods has worked for an extended period; the main problems appear to be that the vectors are not sufficiently effective in taking the gene into the appropriate cells, that the genes require many other helper genes to regulate their actions and that there may be immunological reactions to the inserted genes and their proteins. So dreams for gene therapy on a wide scale have not yet been realised.
Human genome targets: pharmacogenomics
The exciting work that led to the publication in 2001 of the genetic code for the whole human body—the human genome8—has provided a huge amount of information that can potentially be exploited for the benefit of the human race. Even just in the field of pharmacology it is estimated that there are 3000–10,000 protein targets that may be important in terms of drug actions. This field is now known as ‘pharmacogenomics’, and research may lead to drugs targeted to genetically identifiable subsets of the population.
Likely to be of particular interest are proteins related to G-protein receptors, and genes with sequences similar to those coding for receptors. It is conceivable that researchers may study a protein similar to a receptor, find a ligand (a chemical that binds to the protein) and study the binding or substances that inhibit binding, all without knowing whether the receptor look-alike is implicated in any biological function or disease. Other genes of particular interest are those encoding proteins involved in regulation of the cell cycle and cell division, as these are targets for drugs useful in cancers (see discussion in Chapters 41 and 42 of genes encoding tyrosine kinases and mTor, Wnt and BRAF proteins). Systems biology methods are being used to analyse protein–protein interactions, quantify binding of ligands to target proteins, predict transplant rejection, identify enzymes involved in drug metabolism and select molecules with specific mechanisms of action. The possibilities are endless and the costs enormous, which is why drug companies are merging to pool their research efforts and resources in this exciting new field of pharmacology.
Some examples of recent research and drug development in pharmacogenomics are:
Nanomedicines
The term ‘nanotechnology’ refers to the study of controlling matter at the nanometre (nm) level; a nanometre is onebillionth of a metre, or 10−9m. Thus nanotech is about matter on the scale of atoms and molecules—1 nm is about 7 or 8 carbon–carbon bond lengths and a DNA doublehelix has a diameter of about 2 nm. Very different physical properties can be expected of particles at the nano-scale, where surface tension is more important than gravity and the particles are small enough to pass readily into cells. Innovative medicinal products are being manufactured at the nano-level, particularly in the area of drug delivery in the treatment of brain disorders and cancers (see Clinical Interest Box 42-4). Nanomedicines include polymeric particles, micelles, liposomes, magnetic particles, sunscreens, particles of silver and gold and multifunctional carriers. Different toxicity issues can be expected from administration of drugs in such tiny forms.
Ethical principles related to drug use in health care
Human rights, the basis for bioethics
The basic human rights, acknowledged by the United Nations and accepted by most countries, are the rights for life, security, health, dignity, privacy, autonomy, marriage and procreation, and freedom of thought and religion. Codes of bioethics are based on these human rights and date back as far as the Hippocratic Oath (5th century BC). More recently, bioethics are based on the Declaration of Geneva (1948) and the International Code of Ethics (1949), agreed to by the United Nations after World War II; modern versions of the Hippocratic Oath have been devised (see Clinical Interest Box 4-6).
Clinical interest Box 4-6 A hippocratic oath for the 21st century
Hippocratic Oaths, or modernised versions, are still proclaimed at graduation or medical registration ceremonies at approximately half of Australia’s medical schools (McNeill & Dowton 2002). This is not just for sentimental reasons (although there is hardly a dry eye among the parents), but for the purposes of imparting a professional bond, stating high aims and a moral commitment, for continuity in the medical tradition, as a reminder of professional standards and to affirm core values publicly. Medical graduates in the faculty usually join in, to re-affirm their commitment. As an example, below is the ‘Declaration of Ethical Intention’ written and proclaimed by the graduating class of 2003 from Monash University in Melbourne:
(With thanks to the Faculty of Medicine, Nursing and Health Sciences, Monash University; and Dr Philippa Shilson, Melbourne.)
Medical ethics
An ethic is defined as ‘a set of moral principles’, and ethics as ‘the science of morals in human conduct; moral principles’. The consideration of ethical principles is to answer the question, ‘What ought I to do’ in this situation? Medical ethics are the principles and values that define the manners, modes and morals of medical practitioners and guide their decisions, and by extension apply to all health practitioners. They are usually listed as:
Responsibilities of health practitioners
The principles of bioethics put responsibilities on health practitioners to practise ethically. This implies that they will remain competent and up-to-date in their practice, that they will use all appropriate resources in the best interests of their patients and will accord their patients all basic human rights, including observing confidentiality of information. What constitutes unethical conduct may be hard to determine; in the health-care context, it has at times been taken to mean serious misconduct compared with what would reasonably be expected by a general body of colleagues.
Current issues in bioethics
Bioethics issues arise frequently, and are often hard to resolve. Typically these involve professional secrecy, consent to treatment (must be valid, informed and specific) and procedures with legal problems (sterilisation, abortion, assisted pregnancies, maiming and experimentation). This section cannot attempt to cover all aspects of medical ethics relevant to pharmacology, but some aspects of selected current issues are summarised below.
Consent to treatment
The principle of requiring informed consent from a patient before any medical intervention is an implementation of the person’s right to autonomy. Some consent is taken as implied—e.g. for routine examinations or medications— but for more serious procedures, formal consent is required. In fact, a signed form has little standing in law. Consent is better viewed as a continuing process of communication and decision making between doctor and patient. The patient must be mentally competent, and consent must be given voluntarily after adequate information has been understood. Issues arise especially in emergency situations; when the patient is confused or subject to coercion; when families intervene or when there are issues relating to custody of children.
Warnings of risks
Patients need enough information on which to base their consent to, or choice of, treatment or participation in a trial; however, if every possible adverse effect or drug interaction is explained in detail, they may never take any drug, thus putting their health further at risk. In realistic practice, it is usual for doctors and pharmacists to discuss with the person all real risks, considering how much information the person wishes to be told; however, the fear that extensive information will put the person off treatment altogether does not justify withholding information. The High Court of Australia has said that the patient must be informed about ‘material’ (i.e. significant) risks. Equally, health-care professionals have an ethical obligation not to recommend inappropriately risky treatments.
Animal rights
It is now generally recognised that animals should be used in testing of drugs and medical devices or procedures only when absolutely necessary. While results from animal tests cannot automatically be extrapolated to humans, such tests do protect humans. When it comes to the decision, few people would be prepared to take drugs in a lifethreatening condition, or allow drugs to be administered to their children, if the drugs had not previously been tested in some animal or human. The Australian and New Zealand Council for the Care of Animals in Research and Teaching (ANZCCART) works diligently to protect animal rights, minimise the use of animals in testing and promote ‘the three Rs’: replacement of animals wherever possible, reduction in the numbers of animals used and refinement of techniques to minimise harm and use.
Privacy
Increasingly, information about individuals is becoming generally available—indeed much information is now linked to Australian Medicare cards and numbers. Some personal information is required to be given to the government—such as when doctors apply for authority to prescribe a restricted drug for a patient with a particular condition. In an attempt to prevent people from ‘shopping around’ for multiple prescriptions, Medicare Australia is allowed to identify patients whose records show that they are ‘prescription shopping’, and alert doctors before more scripts are written.
Information technology/‘telemedicine’
The advent of the Internet and availability of instantaneous communication and information have raised new issues. Should medical information and consultations be available to all people, e.g. on the World Wide Web, should doctors be allowed to prescribe on-line and pharmacists supply drugs by mail? Does this erode the doctor’s role or the clinical relationship between health professional and patient, or is it the patient’s right to know about and obtain drugs? Does it disperse responsibilities from the doctor to the patient or to whoever publishes information on the web?
Population studies and off-label prescribing
During the normal procedures for clinical trials or epidemiological studies, many groups may be excluded from participation, e.g. children, pregnant women, elderly patients or people of particular indigenous or ethnic groups. On the other hand, trials that would not be approved in countries with strong regulations regarding clinical trials are sometimes carried out in under-developed countries.9 Some of the relevant ethical issues are those of informed consent, the ownership of intellectual property of the data, access of all people to equally high standards of health care and the exploitation and/or stereotyping of people.
In particular, children have been described as ‘therapeutic orphans’ (see Gazarian [2003]). Children are rarely included as subjects in clinical trials, so if the drug is subsequently approved for use, this will usually be only in adults; hence children will be denied access to many new medications. Also, drugs commonly used in children may never be tested for safety and efficacy. Extrapolating results from adult studies to children poses many risks, and new drugs are unlikely to be formulated into forms suitable for children. Some authorities are now requiring that drugs likely to be used in children are subject to clinical trials in children; some drug studies are now being carried out in children overseas, but only in potentially profitable drugs.
‘Off-label’ prescribing is another situation in which drugs are used for conditions for which they have not been trialled or approved, or for which there is no good evidence of efficacy. This is risky for the patient and doctor, and costly to the patient (or parents), as it is not subsidised. The drugs most commonly used ‘off-label’ are psychotherapeutics: antidepressants, antipsychotics and anxiolytics/sedatives (see Walton et al [2008]).
Institutional ethics committees (IECs)
The TGA requires that all institutions in which human or animal testing is carried out, including universities, research institutions and hospitals, have properly constituted institutional ethics committees to respond to and judge ethical questions in the specific environment. There are many potential problems, including:
Equal access to drugs or medical care
The increasing costs of medical technologies and new drugs, the demands of an ageing population, patients’ expectations, doctors’ fear of litigation and governments’ need for tight budget controls all make the rationing of health care a difficult ethical issue. The agenda on public health is often set by the commercial demands of the pharmaceutical industry. (Some of these aspects of pharmacoeconomics have been discussed in Chapter 2.) Principles of equity and fairness need to be applied or high-quality care will only be available to the wealthy. Hospital drug and therapeutics committees may need to draw up guidelines in advance to prioritise needs and allocation of resources. The TGA’s Pharmaceutical Benefits Advisory Committee has to juggle demands from the public for cheap access to all (safe) drugs with those from drug companies wanting subsidies for their products and from other interests competing for scarce government funds (see Kaye and Day [2006]).
Promotion or advertising of medicines
The World Health Organization has a code of ‘ethical criteria for medical drug promotion’, both to health professionals and to the public. Drug companies obviously consider promotion of drugs to doctors as being effective in increasing the prescribing and use of particular products, otherwise it would not be carried out. Advertising adds to the already heavy cost of new drugs. In Australia, ‘detailing’ of drugs to doctors is regulated by TGA legislation, guidelines and the Code of Conduct of Medicines Australia (formerly the Australian Pharmaceutical Manufacturers Association), and by complaints from consumers and ‘watchdogs’. Breaches of the code can require withdrawal of promotional material and heavy fines, plus exposure in professional journals (for example, annually in the Australian Prescriber10). Advertising needs to be monitored to check that it is objective and not biased; it can often be noted that the benefits of a drug are emphasised rather than the risks and that adverse reactions are mentioned only in the finest print. Internet advertising can target consumers and/or prescribers, and is difficult to monitor or regulate.
Advertising of prescription medicines to the public is not permitted in Australia but advertising of OTC drugs is allowed. ‘Direct-to-consumer’ drug advertising is legal in NZ and the USA, and is very effective in increasing demand for prescription medicines. Drug companies can also boost the demand for their products by defining common, mild problems (such as mild dyspepsia, headache, baldness or anxiety) as diseases requiring drug treatment and running ‘disease-awareness’ campaigns. This adds to the heavy costs of drugs to governments.
Advertisements generally, whether in medical journals, glossy magazines, on television or the Internet, should be monitored for superficial or misleading information, shock tactics, insidious comparisons and stereotyping. It is instructive, for example, to check for advertisements aimed at the middle-aged male doctor, with subtle references to anxious housewives, children who may ‘need’ antibiotics for a cold, confused elderly women or stressed hypertensive male business executives. Advertisements should emphasise the information supplied, rather than aiming to catch the eye or shock with sexist or racist images.
Relationships between health practitioners and the pharmaceutical industry
Doctors and other health professionals in a position to prescribe or encourage drug use are often ‘wooed’ by representatives from drug companies to increase their prescribing of a particular drug. Incentives may range from equipment for the desk and lunches after seminars to subsidisation of trips to overseas conferences or funding for research. While doctors generally maintain that they can resist such pressures, studies have shown that even subconsciously their prescribing patterns are affected by pressure from drug company ‘reps’. As more health professionals (nurse practitioners, optometrists, podiatrists) gain the right to prescribe drugs, they will be subject to similar pressures. There is a range of positions that prescribers can adopt, from refusing all gifts,11 so as to avoid all compromise, through to acceptance of lavish gifts while hoping to maintain independence.
The chief concerns about these practices are that commercial objectives override properly prioritised health care, education and research; that advertising inevitably increases prescribing of the company’s drugs; and that there may be distortions in scientific evidence, evaluations and publications. Most biomedical journals now require that authors declare all conflicts of interest. Institutions and learned colleges expect there will be minimal acceptance of gifts or support, and that research and publication will be guided by scientific and ethical values.
As part of marketing strategies, sample ‘starter packs’ of drugs are often given to doctors by drug companies, with the obvious hope that these will be passed out to patients who will then expect continuing prescriptions for the drug. While this may be useful in some cases, there have been problems reported, e.g. inadequate information as to dosage, administration, storage and possible adverse effects, and some patients have been given excessive quantities of drugs without adequate monitoring.
Ethical aspects of clinical trials
The Declaration of Helsinki, recognised internationally, outlines ethical considerations related to clinical trials:
Every biomedical research project involving human subjects should be preceded by careful assessment of predictable risks in comparison with foreseeable benefits to the subject or to others. Concern for the interests of the subject must always prevail over the interests of science and society.
Many of the general issues in medical ethics discussed above also apply to the situation of a clinical trial. Some ethical issues particularly relevant to clinical trials are:
Key points



 Drugs are controlled at many levels: international, national and state. Laws apply to the classification and control of chemicals, poisons, drugs and other therapeutic goods. Some chemicals are proscribed and criminal law relates to offences under these Acts. The road to market of a new drug product requires an initial drug discovery or design, drug development, animal and human testing and approval before a new drug is marketed and reaches the consumer. During this process, legal requirements for drug regulation apply to remove or eliminate unsafe or ineffective drugs from reaching the marketplace; drugs are classified into various schedules to protect the consumer. Special arrangements exist for ‘orphan drugs’ to treat very rare diseases, for classifying drugs for safe use in pregnancy and with respect to drug use in sport. Post-marketing surveillance is important in identifying adverse reactions or serious problems not previously identified. This voluntary program requires the cooperation of health-care professionals. During the process of testing drugs, assays are carried out to determine their strength and purity; these can be chemical or biological assays. Bioassays may be of various types: in vitro, in vivo or in silico; drugs need to be tested in animals and humans before being approved as safe and effective. Drug testing in humans, both preclinical testing in volunteers and testing in patients in clinical trials, is closely controlled and monitored. Randomised controlled clinical trials are the best method of objectively gathering information as to the safety and efficacy of proposed treatments.
Drugs are controlled at many levels: international, national and state. Laws apply to the classification and control of chemicals, poisons, drugs and other therapeutic goods. Some chemicals are proscribed and criminal law relates to offences under these Acts. The road to market of a new drug product requires an initial drug discovery or design, drug development, animal and human testing and approval before a new drug is marketed and reaches the consumer. During this process, legal requirements for drug regulation apply to remove or eliminate unsafe or ineffective drugs from reaching the marketplace; drugs are classified into various schedules to protect the consumer. Special arrangements exist for ‘orphan drugs’ to treat very rare diseases, for classifying drugs for safe use in pregnancy and with respect to drug use in sport. Post-marketing surveillance is important in identifying adverse reactions or serious problems not previously identified. This voluntary program requires the cooperation of health-care professionals. During the process of testing drugs, assays are carried out to determine their strength and purity; these can be chemical or biological assays. Bioassays may be of various types: in vitro, in vivo or in silico; drugs need to be tested in animals and humans before being approved as safe and effective. Drug testing in humans, both preclinical testing in volunteers and testing in patients in clinical trials, is closely controlled and monitored. Randomised controlled clinical trials are the best method of objectively gathering information as to the safety and efficacy of proposed treatments.Review exercises
References and further reading
Anonymous. Medicines Australia Code of Conduct: breaches. Australian Prescriber, 2009;32(6):160-161.
Aronson J.K., Ferner R.E. Joining the DoTS: new approach to classifying adverse drug reactions. British Medical Journal. 2003;327:1222-1225.
Australian and New. Zealand Council for the Care of Animals in Research and Teaching. Promoting awareness of the three Rs. ANZCCART News. 1998;11(1):1-5.
Australian Medicines Handbook 2009. Adelaide: AMH, 2009.
Australian Pharmaceutical Manufacturers Association (APMA). The Australian Pharmaceutical Industry Code of Conduct. Sydney: APMA, c. 2001.
Barnes D. How prescription drugs are developed. Australian Prescriber. 2006;29(6):159-161.
Bochner F., Burgess N.G., Martin E.D. Approaches to rationing drugs in hospitals: an Australian perspective. Pharmacoeconomics. 1996;10(5):467-474.
British Pharmacopoeia Commission. British Pharmacopoeia 2010. London: Her Majesty’s Stationery Office, 2010.(5 volumes printed, also available as CD-ROM and in on-line format).
Cartwright F.F., Biddiss M.D. Disease and History. Man-Made Problems of the Present and Future. London: Hart-Davis; 1972. [ch 9]
Cheok M.H., Evans W.E. Acute lymphoblastic leukemia: a model for the pharmacogenomics of cancer therapy. Nature Reviews: Cancer. 2006;6:117-129.
Davies C.A. Keeping advertisers honest—an overview of the regulation of the advertising of medicines and medical devices in Australia. Australian Prescriber. 2004;27(5):124-127.
Drews J. Drug discovery: a historical perspective. Science. 2000;287(17):1960-1964. March
Dwyer P, Newgreen DB. Regulation of drugs. In: Linden-Laufer S (ed). Ch 20 Health and guardianship. In: The Laws of Australia. Sydney: Law Book Company, 1998.
Fastier F.N. Drugs and the Law in New Zealand. Dunedin: Amidine Publications; 1998.
Galbally R. Review of Drugs, Poisons & Controlled Substances Legislation: Options Paper and Final Report. Woden, ACT: Therapeutic Goods Administration, 2000 and 2001.
Gazarian M. Why are children still therapeutic orphans? Australian Prescriber. 2003;26(6):122-123.
Handen J.S., editor. Industrialization of Drug Discovery: From Target Selection Through Lead Optimization. Boca Raton: Taylor & Francis, 2005.
Harrison R., editor. Biomedical, Biotechnology and Pharmaceutical Innovation: Australia’s Opportunities. Sydney: CL Creations & Health Media, 2000.
Kaye K.I., Day R.O. Can we deny patients expensive drugs? Australian Prescriber. 2006;29(6):146-147.
Kerridge I.H., Lowe M., McPhee J. Ethics and Law for the Health Professions. Sydney: Federation Press; 2005.
Komesaroff P.A. Relationships between health professionals and industry: maintaining a delicate balance. Australian Prescriber. 2007;30(6):150-153.
Komesaroff P.A., Cohen A. The growth of ethics in medicine over the past 50 years. Medical Journal of Australia. 2001;174:41-44.
Kubler P. New drugs for old. Australian Prescriber. 2006;29(6):148-149.
Laurence D.R., Bennett P.N. Clinical Pharmacology, 5th edn. Edinburgh: Churchill Livingstone; 1980.
Le Carre J. The Constant Gardener. UK: Hodder & Stoughton; 2001.
Le Fanu J. The Rise and Fall of Modern Medicine. London: Little Brown; 1999. [ch 3]
McBride W. Thalidomide and congenital abnormalities. Lancet 1961; ii: 1358.
McBride W. Specialist life—William McBride. European Journal of Obstetrics & Gynecology and Reproductive Biology. 2001;95(1):139-140.
Mackay K. Showing the blue card: reporting adverse reactions. Australian Prescriber. 2005;28(6):140-142.
McEwen J. What does TGA approval of medicines mean? Australian Prescriber. 2004;27(6):156-158.
McNeill P.M., Dowton S.B. Declarations made by graduating medical students in Australia and New Zealand. Medical Journal of Australia. 2002;176:123-125.
Manderson D. From Mr Sin to Mr Big: A History of Australian Drug Laws. Oxford: Oxford University Press; 1993.
Mayr L.M., Bojanic D. Novel trends in high-throughput screening. Current Opinion in Pharmacology. 2009;9(5):580-588.
Meunier J.C. Utilizing functional genomics to identify new pain treatments: the example of nociceptin. American Journal of Pharmacogenomics. 2003;3(2):117-130.
Moran M. Why are global drug prices so high … and other questions. Australian Prescriber. 2003;26(2):26-27.
Morgan S., Boothe K. Prescription drug subsidies in Australia and New Zealand. Australian Prescriber. 2010;33(1):2-4.
Moulds RFW. How do we obtain consent for surgery? Study Guide 7, problem 2.10, 2008; Fiji School of Medicine, unpublished material.
Muthu M.S., Singh S. Targeted nanomedicines: effective treatment modalities for cancer, AIDS and brain disorders. Nanomedicine. 2009;4(1):105-118.
National Drugs and Poisons Schedule Committee. Standard for the Uniform Scheduling of Drugs and Poisons no. 16. Woden ACT: National Drugs and Poisons Schedule Committee, 2001.
National Health and Medical Research Council. NHMRC Statement on Human Experimentation and Supplementary Notes 1992. Canberra: NHMRC, 1992.
Palumbo A., Facon T., Sonneveld P., et al. Thalidomide for treatment of multiple myeloma: 10 years later. Blood. 2008;111(8):3968-3977.
Patounas M.P. Starter packs: a good start to therapy? Australian Prescriber. 2007;30(1):14-16.
Petrie A., Sabin C. Medical Statistics at a Glance, 2nd edn. Malden, MA: Blackwell; 2005.
Pharmaceutical Society of Australia. Australian Pharmaceutical Formulary and Handbook, 21st edn. Deakin, ACT: PSA; 2009.
Scott I. Interpreting risks and ratios in therapy trials. Australian Prescriber. 2008;31(1):12-16.
Steinbock B., Arras J.D., London A.J. Ethical Issues in Modern Medicine, 6th edn. Boston: McGraw–Hill; 2003.
Sweet M. Pharmaceutical marketing and the Internet. Australian Prescriber. 2009;32(1):2-4.
Therapeutic Goods Administration, Dept of Health and Ageing. Drugs Designated as Orphan Drugs. Woden ACT: Australian Government; 2006.
Turner M.B. Safety of fish oil and omega-3 fatty acids. Medicines Safety Update in Australian Prescriber. 2010;33(2):48-51.
United Nations. United Nations Convention Against Illicit Traffic in Narcotic Drugs and Psychotropic Substances. Vienna: 20 December 1988 [1993] ATS 4.
United Nations. United Nations Single Convention on Narcotic Drugs. New York 30 March 1961 [1967] ATS 31.
United Nations. Office on Drugs and Crime. In World Drug Report 2009. New York: United Nations; 2009.
Vane J.R. The release and fate of vaso-active hormones in the circulation. British Journal of Pharmacology. 1969;35:209-242.
Vane J.R. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nature New Biology. 1971;231:232-239.
Vitry A. Is Australia free from direct-to-consumer advertising? Australian Prescriber. 2004;27(1):4-6.
Vitry A. Time for transparency at the TGA. Australian Prescriber. 2008;31(5):114-115.
Walter G., Konthur Z., Lehrach H. High-throughput screening of surface displayed gene products. Combinatorial Chemistry & High Throughput Screening. 2001;4(2):193-205.
Walton S.M., Schumock G.T., Lee K.V., et al. Prioritizing future research on off-label prescribing: results of a quantitative evaluation. Pharmacotherapy. 2008;28(12):1443-1452.
Zhang L., Demain A.L., editors. Natural Products: Drug Discovery and Therapeutic Medicine. Totowa, NJ: Humana Press, 2005.
Adverse Drug Reactions Advisory Committee (ADRAC): www.tga.gov.au/adr/adrac.htm.
Adverse Medicines Event Line: www.nps.org.au/consumers/ask_an_expert/contact_a_pharmacist/medicines_line
Australian Adverse Drug Reactions Bulletin. Canberra: Therapeutic Goods Administration (published 4 times per year): www.tga.gov.au/adr/aadrb.htm
Australian and New Zealand Council for the Care of Animals in Research and Teaching (ANZCCART): www.adelaide.edu.au/ANZCCART/
Australian Drug Evaluation Committee: www.tga.gov.au/docs/html/adec/adec.htm
Australian Pharmaceutical Formulary and Handbook (21st edn), 2009 (APF): www.psa.org.au/site.php?id=1805
British Pharmacopoeia: www.pharmacopoeia.co.uk/
Drugsand the law: www.legalanswers.sl.nsw.gov.au/hot_topics/pdf/drugs_59.pdf
Hot Topics 59: Drugs and the Law (2007): www.infocus.sl.nsw.gov.au/res/resdesc.cfm?res_code=2710&sres=1
International Narcotics Control Board: www.incb.org/
Medicines Australia: www.medicinesaustralia.com.au/pages/index.asp
Medicines regulation and the TGA: www.tga.gov.au/subject/index.htm
New Zealand Drug Regulatory Information: www.medsafe.govt.nz/reg.htm
New Zealand Intensive Medicines Monitoring Programme (IMMP): www.medsafe.govt.nz/Profs/adverse/IMMP.asp
New Zealand Standing Committee on Therapeutic Trials: www.hrc.govt.nz/root/pages_regulatory/Standing_Committee_on_Therapeutic_Trials.html
Orphan drugs: www.tga.gov.au/docs/html/orphand2.htm
Pharma Phacts: www.healthyskepticism.org/pharmaphacts/
Pharmaceutical Benefits Advisory Committee: www.health.gov.au/internet/main/publishing.nsf/Content/health-pbsgeneral-listing-committee3.htm
Pharmaceutical Benefits Scheme: www.pbs.gov.au/html/home
Prescription Shopping Program: www.medicareaustralia.gov.au/provider/pbs/prescription-shopping/index.jsp
Standard for the Uniform Scheduling of Drugs and Poisons (SUSDP): www.tga.health.gov.au/ndpsc/susdp.htm
Therapeutic Goods Administration. Access to unapproved therapeutic goods: clinical trials in Australia. Canberra: TGA, 2001: www.tga.gov.au/DOCS/HTML/clintrials.htm
TGA Australian Clinical Trial Handbook 2006 www.tga.gov.au/ct/cthandbook.htm
TGA ‘Blue Form’: www.tga.gov.au/adr/bluecard.htm
United Nations Office on Drugs and Crime: www.unodc.org/unodc/
More weblinks at: http://evolve.elsevier.com/AU/Bryant/pharmacology/