13 Interplay of Central and Peripheral Factors that Control the Circulation
1. Describe the sequence of cardiovascular events during exercise.
2. Describe how most cardiovascular functions are integrated in exercise.
3. Describe the effects of blood loss on the cardiovascular system.
4. Explain the various compensatory mechanisms that protect against hemorrhagic shock.
5. Explain the various decompensatory mechanisms that intensify the effects of blood loss.
The primary function of the circulatory system is to deliver the supplies needed for tissue metabolism and growth and to remove the products of metabolism. To explain how the heart and blood vessels serve this function, the circulatory system has been analyzed morphologically and functionally. Furthermore, the mechanisms of the component parts that maintain adequate tissue perfusion under different physiological conditions have been discussed.
Once the functions of the various individual components are understood, it is essential to consider the interrelationships in the overall operation of the circulatory system. Tissue perfusion depends on the arterial pressure and the local vascular resistance. Furthermore, the arterial pressure in turn depends on cardiac output and total peripheral resistance (TPR). Arterial pressure is maintained within a relatively narrow range in normal individuals, a feat accomplished by reciprocal changes in cardiac output and TPR (see Chapter 7). However, cardiac output and peripheral resistance are each influenced by a number of factors, and the interplay among these factors determines the level of these two variables. Acutely, the autonomic nervous system and the baroreceptors play the key role in regulating blood pressure (see Chapter 9). However, from a long-range point of view, the control of fluid balance by the kidneys, adrenal cortex, and central nervous system, along with the maintenance of a constant blood volume, is crucial.
In a well-regulated system, one way to study the extent and sensitivity of the regulatory mechanisms is to disturb the system and then to observe its response in restoring the preexisting steady state. Disturbances in the form of physical exercise and hemorrhage are used to illustrate the effects of the various factors involved in the regulation of the circulatory system.
Exercise
The cardiovascular adjustments in exercise consist of a combination and an integration of neural and local chemical factors. The neural factors consist of (1) central command, (2) reflexes originating in the contracting muscle, and (3) the baroreceptor reflex. Central command is the cerebrocortical activation of the sympathetic nervous system that produces cardiac acceleration, increased myocardial contractile force, and peripheral vasoconstriction (see Chapter 5).
Reflexes can be activated intramuscularly by stimulation of mechanoreceptors (by stretch, tension) and of chemoreceptors (by-products of metabolism) in response to muscle contraction. Impulses from these receptors travel centrally via small myelinated (group III) and unmyelinated (group IV) afferent nerve fibers. The group IV unmyelinated fibers may represent the muscle chemoreceptors; no morphological chemoreceptor has been identified. The central connections of this reflex are unknown, but the efferent limb is composed of the sympathetic nerve fibers to the heart and peripheral blood vessels.
The baroreceptor reflex and the local factors that influence skeletal muscle blood flow (metabolic vasodilators) are described in Chapters 9 and 12. Vascular chemoreceptors play a significant role in regulation of the cardiovascular system in exercise. This assertion is supported by the observation that the pH, Pco2, and Po2 of arterial blood are normal during exercise, and the vascular chemoreceptors are located on the arterial side of the circulatory system.
Mild to Moderate Exercise
In humans and in trained animals, anticipation of physical activity inhibits vagus nerve impulses to the heart; this withdrawal of vagal nerve activity underlies the initial increase in heart rate. Eventually, sympathetic nerve discharge also increases. The concerted inhibition of parasympathetic areas and activation of sympathetic areas of the medulla increase the heart rate and myocardial contractility. The tachycardia and the enhanced contractility increase the cardiac output, in turn raising the arterial pressure.
Exercise can be understood with the help of cardiac function and vascular function curves described in Chapter 10. An example is shown in Figure 13-1. From the interactions described in Chapter 10, Figure 13-1 can be seen as a composite that illustrates the effects of two changes. First, along with tachycardia, there is increased contractility caused by norepinephrine released from cardiac sympathetic nerves (see also Figures 5-23 and 10-15). Second, there is a change in the vascular function curve that results from vasodilation (Figure 13-1; see also Figure 10-13). In this closed circulatory system, TPR is reduced as a result of vasodilation in the skeletal muscle vascular bed. The vascular function curve is shifted upward, as is the contractility curve, because cardiac output and venous return are two measures of the same parameter.
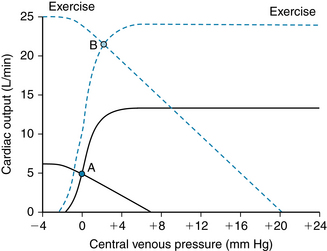
FIGURE 13-1 Cardiac and vascular function curves are greatly altered during strenuous exercise, allowing an increase in cardiac output of fourfold to fivefold. The operating point of the cardiovascular system moves from A to B. The cardiac function curve during strenuous exercise is the result of increased heart rate, stroke volume, and contractility. The vascular function curve reflects greatly decreased total peripheral resistance and increased mean circulatory pressure (see Chapter 10). At the new operating point (B), cardiac output is increased more than fourfold, but filling pressure is increased only slightly.
Peripheral Resistance Declines during Exercise
At the same time that the heart is stimulated, the sympathetic nervous system elicits vascular resistance changes in the periphery. In skin, kidneys, splanchnic regions, and inactive muscle, sympathetically mediated vasoconstriction increases vascular resistance, diverting blood away from these areas (Figure 13-2). This greater resistance in vascular beds of inactive tissues persists throughout the period of exercise.
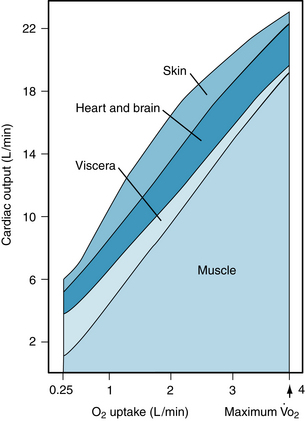
FIGURE 13-2 Approximate distribution of cardiac output at rest and at different levels of exercise up to the maximal O2 consumption (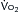 ) in a normal young man.
) in a normal young man.
(Redrawn from Ruch HP, Patton TC: Physiology and biophysics, 12th ed, Philadelphia, Saunders, 1974.)
The increase in blood flow in the active muscles at the onset of exercise cannot be attributed to a neural mechanism, because a chemical block of the autonomic nervous system does not alter this blood flow response. This increase in muscle blood flow may be caused by the modest elevation of blood pressure or by some unknown mechanism.
As cardiac output and blood flow to active muscles increase with progressive increments in the intensity of exercise, visceral blood flow (splanchnic and renal vasculatures) decreases. Blood flow to the myocardium increases, whereas that to the brain is unchanged (see Figure 13-2). Skin blood flow initially decreases during exercise. It then increases, as body temperature rises with increments in duration and intensity of the exercise. Skin blood flow finally decreases when the skin vessels constrict, as the total body O2 consumption (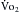 max) nears maximum.
max) nears maximum.
The major circulatory adjustment to prolonged exercise involves the vasculature of the active muscles. Local formation of vasoactive metabolites induces marked dilation of the resistance vessels, which progresses as the intensity level of exercise increases. Potassium is one of the substances released by contracting muscle, and it may be in part responsible for the initial decrease in vascular resistance in the active muscles. Elevated interstitial K+ can cause vasodilation by stimulation of the Na-K pump and by activation of the conductance of inwardly rectifying K+ channels. Either action causes hyperpolarization of vascular smooth muscle membrane, thereby reducing Ca++ entry. Other contributing factors are the release of adenosine and nitric oxide (NO), the activation of adenosine triphosphate–sensitive potassium channels (KATP channels), and a decrease in pH during sustained exercise. All of these factors can act locally to dilate arterioles in contracting muscle. Adenosine release and KATP channel activation are thought to act at more distal arterioles to increase blood flow. The mechanism for the transmission of the dilating effect to distal arterioles is not understood.
The local accumulation of metabolites relaxes the terminal arterioles. Blood flow through the muscle may rise 15 to 20 times above the resting level (see Figure 13-2). This metabolic vasodilation of the precapillary vessels in active muscles occurs very soon after the onset of exercise. Furthermore, the decrease in TPR enables the heart to pump more blood at a lesser load and more efficiently (less pressure work; see Chapter 4) than if TPR were unchanged. Only a small percentage of the capillaries is perfused at rest, whereas in actively contracting muscle, nearly all of the capillaries contain flowing blood (capillary recruitment).
The surface available for exchange of gases, water, and solutes is increased many-fold. Furthermore, the hydrostatic pressure in the capillaries increases because of the relaxation of the resistance vessels. Hence there is a net movement of water and solutes into the muscle tissue. Interstitial pressure rises, and it remains elevated during exercise, as fluid continues to move out of the capillaries and is carried away by the lymphatics. Lymph flow is increased as a result of both the rise in capillary hydrostatic pressure and the massaging effect of the contracting muscles on the valve-containing lymphatic vessels (see Chapter 8).
The contracting muscle avidly extracts O2 from the perfusing blood (increased Ao2–Vo2 difference) (Figure 13-3), and the release of O2 from the blood is facilitated by the nature of oxyhemoglobin dissociation. The reduction in pH caused by the high concentration of CO2 and the formation of lactic acid and the increase in temperature in the contracting muscle all contribute to shifting the oxyhemoglobin dissociation curve to the right, an example of the Bohr effect (see Figure 1-5). At any given partial pressure of O2, less O2 is bound by the hemoglobin in the red blood cells; consequently, O2 removal from the blood is facilitated. Oxygen consumption may increase as much as 60-fold, with only a 15-fold increase in muscle blood flow. Muscle myoglobin serves as a limited O2 store in exercise; it can release attached O2 at very low partial pressures. Hence, it facilitates O2 transport from capillaries to mitochondria by serving as an O2 carrier.
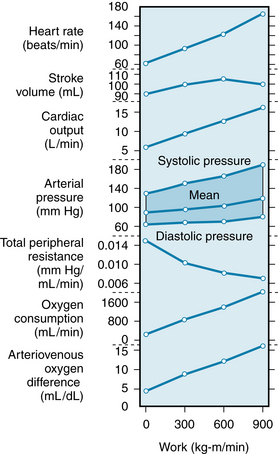
Cardiac Output Can Increase Substantially in Exercise
The enhanced sympathetic drive and the reduced parasympathetic inhibition of the sinoatrial node continue during exercise, and consequently tachycardia persists. If the workload is moderate and constant, the heart rate will reach a certain level and remain there throughout the period of exercise. However, if the workload increases, a concomitant rise in heart rate occurs until a plateau is reached during severe exercise at about 180 beats per minute (beats/min) (see Figure 13-3). With moderate exercise, there is a substantial increase in stroke volume of about 10% to 35% (see Figure 13-3), the larger values occurring in trained individuals. (In very well-trained distance runners, whose cardiac outputs can reach six to seven times the resting level, stroke volume reaches about twice the resting value.) The increase in stroke volume has been ascribed to the Frank-Starling mechanism because left ventricular end-diastolic pressure and end-diastolic volume increase (see Chapter 4). This is seen in the rightward shift of the equilibrium point B in Figure 13-1. (The importance of greater ventricular filling in the response of cardiac output during exercise is underscored by the observation that increasing the heart rate alone through pacing is associated with reduced stroke volume and a constant cardiac output.) Contractility also increases during moderate exercise, an indication of sympathetic activation (see Figure 13-1; see also Chapter 5). When exercise is strenuous, cardiac output increases largely as a result of tachycardia. Stroke volume remains at a plateau or may even diminish slightly (see Figure 13-3) while contractility remains elevated.
Thus, it is apparent that the increase in cardiac output observed with exercise is correlated with an increase in heart rate. If the baroreceptors are denervated, the cardiac output and heart rate responses to exercise are sluggish in comparison with the changes in animals with normally innervated baroreceptors. However, exercise still elicits an increment in cardiac output in the absence of autonomic innervation of the heart, as occurs when a donor heart is prepared for transplantation. Resting heart rate is elevated to about 100 beats/min because parasympathetic innervation is lacking. The increment in cardiac output is less than that observed in normal subjects, and it is achieved principally by means of a gradual increase in heart rate. The stroke volume also increases but is below the level seen in the normal heart. If a β-adrenergic receptor–blocking agent is given, exercise performance is impaired in subjects with transplanted hearts. The β-adrenergic receptor antagonist opposes the cardiac acceleration and enhanced contractility caused by increased amounts of circulating catecholamines. Hence the increase in cardiac output necessary for maximal exercise performance is limited.
Venous Return Is Enhanced in Exercise
In addition to the contribution made by sympathetically mediated constriction of the capacitance vessels in both exercising and non-exercising parts of the body, venous return is aided by the working skeletal muscles and the muscles of respiration (see Figure 13-1). The intermittently contracting muscles compress the vessels that course through them (see Figure 12-2). In the case of veins with their valves oriented toward the heart, blood is pumped back toward the right atrium. The flow of venous blood to the heart is also aided by the increase in the pressure gradient developed by the more negative intrathoracic pressure produced by deeper and more frequent respirations. In humans, there is little evidence that blood reservoirs such as skin, lungs, and liver contribute much to circulating blood volume. In fact, blood volume is usually reduced slightly during exercise, as evidenced by a rise in the hematocrit ratio, because of both water lost externally by sweating and enhanced ventilation, and movement of fluid into the contracting muscle.
The fluid loss from the vascular compartment into contracting muscle reaches a plateau as interstitial fluid pressure rises and opposes the increased hydrostatic pressure in the capillaries of the active muscle. The fluid loss is partially offset by movement of fluid from the splanchnic regions and inactive muscle into the bloodstream. This influx of fluid occurs as a result of a decrease in hydrostatic pressure in the capillaries of these tissues and an increase in plasma osmolarity due to movement of osmotically active particles into the blood from the contracting muscle. In addition, reduced urine formation by the kidneys helps to conserve body water.
Arterial Pressure Increases Slightly during Exercise
If the exercise involves a large proportion of the body musculature, as in running or swimming, the reduction in total vascular resistance can be considerable (see Figure 13-3). Nevertheless, arterial pressure starts to rise with the onset of exercise, and the increase in blood pressure roughly parallels the intensity of the exercise performed (see Figure 13-3). Therefore the increase in cardiac output is proportionally greater than the decrease in TPR. The vasoconstriction produced in the inactive tissues by the sympathetic nervous system (and to some extent by the release of catecholamines from the adrenal medulla) is important for maintaining normal or increased blood pressure because sympathectomy or drug-induced block of the sympathetic adrenergic nerve fibers results in a decrease in arterial pressure (hypotension) during exercise.
Some sympathetically mediated vasoconstriction occurs in active muscle and increases during strenuous exercise, when more than half of the total body musculature is contracting. In experiments in which one leg is working at maximal levels and then the other leg starts to work, blood flow decreases in the first working leg. Furthermore, blood levels of norepinephrine rise significantly in exercise, and most is derived from the sympathetic nerve endings in the active muscles.
As body temperature rises during exercise, the skin vessels dilate in response to thermal stimulation of the heat-regulating center in the hypothalamus, and TPR decreases further (see Figure 13-3). This would result in a decline in blood pressure were it not for the increasing cardiac output and constriction of arterioles in the renal, splanchnic, and other tissues.
In general, mean arterial pressure rises during exercise, as a result of the increase in cardiac output (see Figure 13-3). However, the effect of enhanced cardiac output is offset by the overall decrease in TPR. Hence, the mean blood pressure rise is relatively small. Vasoconstriction in the inactive vascular beds contributes to the maintenance of a normal arterial blood pressure for adequate perfusion of the active tissues. The actual pressure attained represents a balance between cardiac output and TPR (see p. 140). Systolic pressure usually increases more than diastolic pressure, resulting in an increase in pulse pressure (see Figure 13-3). The larger pulse pressure is primarily attributable to a greater stroke volume and, to a lesser degree, to a more rapid ejection of blood by the left ventricle. Peripheral runoff is less during the brief ventricular ejection period.
Severe Exercise
When severe exercise is taken to the point of exhaustion, the compensatory mechanisms begin to fail. Heart rate attains a maximal level of about 180 beats/min, and stroke volume reaches a plateau. The stroke volume often decreases, resulting in a fall in blood pressure. Dehydration occurs, and sympathetic vasoconstrictor activity supersedes the vasodilator influence on the cutaneous vessels. The neural activity has the hemodynamic effect of a slight increase in effective blood volume. However, cutaneous vasoconstriction also reduces the rate of heat loss. Body temperature is normally elevated in exercise, and reduction in heat loss through cutaneous vasoconstriction can, under these conditions, lead to very high body temperatures, with associated feelings of acute distress. The tissue and blood pH values decrease as a result of greater lactic acid and CO2 production. The reduced pH is probably the key factor that determines the maximal amount of exercise a given individual can tolerate because of muscle pain, subjective feelings of exhaustion, and inability or loss of the will to continue. A summary of the neural and local effects of exercise on the cardiovascular system is schematized in Figure 13-4.
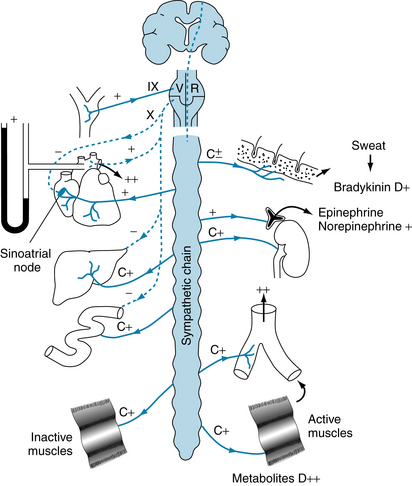
Postexercise Recovery
When exercise stops, sympathetic activity to the heart declines, and the heart rate and cardiac output decrease. Peripheral sympathetic activity also decreases, and coupled with resistance vessel dilation (caused by the accumulated vasodilator metabolites), arterial pressure falls, often below the pre-exercise level. This hypotension is brief, and the baroreceptor reflexes restore the blood pressure to normal levels.
Limits of Exercise Performance
The two main forces that can limit skeletal muscle performance in humans are the rate of O2 utilization by the muscles and the O2 supply to the muscles. Muscle O2 usage is probably not critical, because maximum O2 consumption (max) during exercise by a large percentage of the body muscle mass either is unchanged or increases slightly when additional muscles are activated. In fact, during exercise of a large muscle mass, as in vigorous bicycling, commencement of bilateral arm exercise without change in the cycling efforts produces only a small increment in cardiac output and max. However, it causes a decrease in blood flow to the legs. This centrally mediated (baroreceptor reflex) vasoconstriction during maximal cardiac output prevents a fall in blood pressure. This drop in blood pressure would otherwise be caused by metabolically induced vasodilation in the active muscles. If muscle O2 utilization were limiting, recruitment of more contracting muscle would use much more O2 to meet the enhanced O2 requirements (an amount about equal to the sum of O2 consumption of the arms and legs exercised alone).
Limitation of O2 supply may be caused by inadequate oxygenation of blood in the lungs or by limitation of the supply of O2-laden blood to the muscles. Failure to fully oxygenate blood by the lungs can be excluded, because even with the most strenuous exercise at sea level, arterial blood is fully saturated with O2Therefore O2 delivery (or blood flow because arterial blood O2 content is normal) to the active muscles appears to be the limiting factor in muscle performance. This limitation could be caused by the inability to increase cardiac output beyond a certain level, as the result of a limitation of stroke volume, which might occur because heart rate reaches maximal levels before max is reached. Hence the major factor is the pumping capacity of the heart.
Physical Training and Conditioning
The response of the cardiovascular system to regular exercise is to increase its capacity to deliver O2 to the active muscles, and to improve the ability of the muscle to utilize O2. The max is quite reproducible in a given individual, and it varies with the level of physical conditioning. Training progressively increases the max, which reaches a plateau at the highest level of conditioning. Highly trained athletes have a lower resting heart rate, greater stroke volume, and lower peripheral resistance than they had before training or will have after deconditioning (i.e., becoming sedentary). The low resting heart rate is caused by a higher vagal tone and a lower sympathetic tone. With exercise, the maximal heart rate in the trained individual is the same as that in the untrained person, but it is attained at a higher level of exercise.
The trained athlete also exhibits a low vascular resistance that is inherent in the muscle. For example, if an individual exercises one leg regularly over an extended period and does not exercise the other leg, the vascular resistance is lower, and the max higher, in the “trained” leg than in the “untrained” leg. Physical conditioning is associated with a greater extraction of O2 from the blood (greater Ao2–Vo2 difference) by the muscles. With long-term training, capillary density and the numbers of mitochondria increase, as does the activity of the oxidative enzymes in the mitochondria. Also, it appears that ATPase activity, myoglobin, and enzymes involved in lipid metabolism increase with physical conditioning.
Endurance training, such as running or swimming, produces proportionate increases of left ventricular wall thickness (t) and of left ventricular chamber radius (r) such that the ratio, t/r, remains constant. In contrast, strength exercises, such as weight lifting, appear to produce some increase in left ventricular wall thickness (hypertrophy) with little effect on ventricular chamber radius. However, this increase in wall thickness is small relative to that observed in hypertension, in which there is a persistent elevation of afterload because of the high peripheral resistance.
Hemorrhage
In an individual who has lost a large quantity of blood, the principal system affected is the cardiovascular system. The arterial systolic, diastolic, and pulse pressures diminish, and the arterial pulse is rapid and feeble. The cutaneous veins collapse and fill slowly when compressed centrally. The skin is pale, moist, and slightly cyanotic. Respiration is rapid, but the depth of respiration may be shallow or deep.
Hemorrhage Evokes Compensatory and Decompensatory Effects on the Arterial Blood Pressure
Cardiac output decreases as a result of blood loss (see Chapter 10). The amount (10%) of blood removed in a donation of blood is well tolerated; there is little change in mean arterial blood pressure. This is not the case when greater amounts are lost from the circulation. The changes in mean arterial pressure evoked by an acute hemorrhage in experimental animals are illustrated in Figure 13-5. If sufficient blood is withdrawn rapidly from a subject to bring mean arterial pressure to about 50 mm Hg, the pressure tends to rise spontaneously toward control over the subsequent 20 or 30 minutes. In some animals (curve A, Figure 13-5), this trend continues, and normal pressures are regained within a few hours. In other animals (curve B), after an initial pressure rise, the pressure begins to decline, and it continues to fall at an accelerating rate until death ensues. This progressive deterioration of cardiovascular function is termed hemorrhagic shock. At some point the deterioration becomes irreversible; a lethal outcome can be retarded only temporarily by any known therapy, including massive transfusions of donor blood.
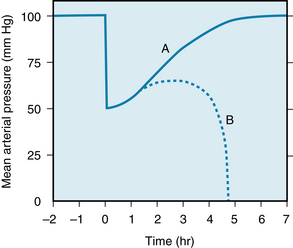
FIGURE 13-5 Changes in mean arterial pressure after a rapid hemorrhage. At time 0, the animal is bled rapidly to a mean arterial pressure of 50 mm Hg. After a period in which the pressure returns toward the control level, some animals continue to improve until the control pressure is attained (curve A). However, in other animals the pressure will begin to decline until death ensues (curve B).
The Compensatory Mechanisms are Neural and Humoral
The changes in arterial pressure immediately after an acute blood loss (see Figure 13-5) indicate that certain compensatory mechanisms must be operating. Any mechanism that senses the level of blood pressure and that raises the pressure toward normal in response to the reduction in pressure may be designated a negative feedback mechanism. It is termed “negative” because the direction of the secondary change in pressure is opposite to that of the initiating change. The following negative feedback responses are evoked: (1) baroreceptor reflexes, (2) chemoreceptor reflexes, (3) cerebral ischemic responses, (4) reabsorption of tissue fluids, (5) release of endogenous vasoconstrictor substances, and (6) renal conservation of salt and water.
Baroreceptor Reflexes
The reductions in mean arterial pressure and in pulse pressure during hemorrhage decrease the stimulation of the baroreceptors in the carotid sinuses and aortic arch (see Chapter 9). Several cardiovascular responses are thus evoked, all of which tend to restore the normal level of arterial pressure. Reduction of vagal tone and enhancement of sympathetic tone raise the heart rate and enhance myocardial contractility.
The increased sympathetic discharge also produces generalized venoconstriction, which has the same hemodynamic consequences as a transfusion of blood (see Chapter 10). Sympathetic activation constricts the vasculature in certain blood reservoirs. This vasoconstriction provides an autotransfusion of blood into the circulation. In humans, the cutaneous, pulmonary, and hepatic vasculatures probably constitute the principal blood reservoirs.
Generalized arteriolar vasoconstriction is a prominent response to the diminished baroreceptor stimulation during hemorrhage. The reflex increase in peripheral resistance minimizes the fall in arterial pressure that results from the reduction of cardiac output. Figure 13-6 shows the effect of an 8% blood loss on mean aortic pressure in a group of dogs. If both vagi are cut to eliminate the influence of the aortic arch baroreceptors, and only the carotid sinus baroreceptors are operative (left panel), this hemorrhage reduces mean aortic pressure by 14%. This pressure change does not differ significantly from the pressure decline (12%) evoked by the same hemorrhage before vagotomy (not shown). When the carotid sinuses are denervated and the aortic baroreceptor reflexes are intact, the 8% blood loss decreases mean aortic pressure by 38% (middle panel in Figure 13-6). Hence the carotid sinus baroreceptors are more effective than the aortic baroreceptors in attenuating the fall in pressure. The aortic baroreceptors must also be operative, however, because when both sets of afferent baroreceptor pathways are interrupted, an 8% blood loss reduces arterial pressure by 48% (right panel in Figure 13-6).
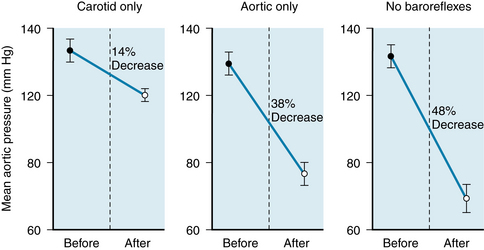
FIGURE 13-6 Changes in mean aortic pressure (mean +/- SE) in response to an 8% blood loss in a group of eight dogs. Left panel, The carotid sinus baroreceptor reflexes were intact and the aortic reflexes were interrupted. Middle panel, The aortic reflexes were intact and the carotid sinus reflexes were interrupted. Right panel, All sinoaortic reflexes were abrogated.
(From Shepherd JT: Lewis A. Conner Memorial Lecture: The cardiac catheter and the American Heart Association. Circulation 50:418, 1974; derived from the data of Edis AJ: Aortic baroreflex function in the dog. Am J Physiol 221:1352, 1971.)
Although arteriolar vasoconstriction is widespread during hemorrhage, it is by no means uniform. Vasoconstriction is most severe in the cutaneous, skeletal muscle, and splanchnic vascular beds, and it is slight or absent in the cerebral and coronary circulations. In many instances the cerebral and coronary vascular resistances are diminished. Thus the reduced cardiac output is redistributed to favor flow through the brain and the heart.
In the early stages of mild to moderate hemorrhage, the changes in renal resistance are usually slight. The tendency for increased sympathetic activity to constrict the renal vessels is counteracted by autoregulatory mechanisms (see Chapter 12). With more prolonged and severe hemorrhage, however, renal vasoconstriction becomes intense. The reductions in renal circulation are most severe in the outer layers of the renal cortex. The inner zones of the cortex and outer zones of the medulla are spared.
The severe renal and splanchnic vasoconstriction during hemorrhage favors the heart and brain. However, if such constriction persists too long, it may be detrimental. Frequently, patients survive the acute hypotensive period only to die several days later from kidney failure because the prolonged renal ischemia causes acute tubular necrosis. Intestinal ischemia may also have dire effects. In the dog, for example, intestinal bleeding and extensive sloughing of the mucosa occur after only a few hours of hemorrhagic hypotension. Furthermore, the diminished splanchnic flow swells the centrilobular cells in the liver. The resulting obstruction of the hepatic sinusoids raises the portal venous pressure, intensifying the intestinal blood loss. Fortunately, the pathological changes in the liver and in the intestine are usually much less severe in humans than in dogs.
Chemoreceptor Reflexes
Reductions in arterial pressure below about 60 mm Hg do not evoke any additional responses through the baroreceptor reflexes, because this pressure level constitutes the threshold for stimulation (see Chapter 9). However, low arterial pressure may stimulate peripheral chemoreceptors, because inadequate local blood flow produces hypoxia in the chemoreceptor tissue. Chemoreceptor excitation enhances the already existing peripheral vasoconstriction evoked by the baroreceptor reflexes. Also, respiratory stimulation assists venous return by the auxiliary pumping mechanism described in Chapter 10.
Cerebral Ischemia
When the arterial pressure is below about 40 mm Hg, the resulting cerebral ischemia activates the sympathoadrenal system. The sympathetic nervous discharge is several times greater than the maximal neural activity that occurs when the baroreceptors cease to be stimulated. Therefore the vasoconstriction and facilitation of myocardial contractility may be pronounced. With more severe degrees of cerebral ischemia, however, the vagal centers also become activated. The resulting bradycardia may aggravate the hypotension that initiated the cerebral ischemia.
Reabsorption of Tissue Fluids
The arterial hypotension, arteriolar constriction, and reduced venous pressure during hemorrhagic hypotension lower the hydrostatic pressure in the capillaries. The balance of transmural forces promotes the net reabsorption of interstitial fluid into the vascular compartment (see Chapter 8). The rapidity of this response is displayed in Figure 13-7. In a group of cats, 45% of the estimated blood volume was removed over a 30-minute period. The mean arterial blood pressure declined rapidly to about 45 mm Hg. The pressure then returned quickly, but only temporarily, to near the control level. The plasma colloid osmotic pressure declined markedly during the bleeding and continued to decrease more gradually for several hours. The reduction in colloid osmotic pressure reflects the dilution of the blood by reabsorption of tissue fluids that contain little protein. The hematocrit is accordingly reduced, as is the O2-carrying capacity of the blood.
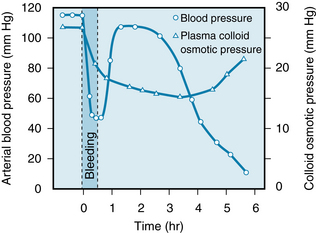
FIGURE 13-7 Changes in arterial blood pressure and plasma colloid osmotic pressure in response to withdrawal of 45% of the estimated blood volume over a 30-minute period, beginning at time 0. The data are the average values for 23 cats.
(Redrawn from Zweifach BW: Mechanisms of blood flow and fluid exchange in microvessels: hemorrhagic hypotension model. Anesthesiology 41:157, 1974.)
Considerable quantities of fluid may thus be drawn into the circulation during hemorrhage. About 0.25 mL of fluid per minute per kilogram of body weight may be reabsorbed. Approximately 1 L of fluid per hour might be autoinfused into the circulatory system of an average individual from the interstitial spaces after an acute blood loss.
Considerable quantities of fluid may also be slowly shifted from intracellular to extracellular spaces. Thus fluid exchange is probably mediated by secretion of cortisol from the adrenal cortex in response to hemorrhage. Cortisol appears to be essential for a full restoration of plasma volume after hemorrhage.
Endogenous Vasoconstrictors
The catecholamines, epinephrine, and norepinephrine are released from the adrenal medulla in response to the same stimuli that evoke widespread sympathetic nervous discharge. Blood levels of catecholamines are high during and after hemorrhage. When animals are bled to an arterial pressure level of about 40 mm Hg, the concentration of catecholamines increases as much as 50 times.
Epinephrine comes almost exclusively from the adrenal medulla, whereas norepinephrine is derived from both the adrenal medulla and the peripheral sympathetic nerve endings. These humoral substances reinforce the effects of sympathetic nervous activity listed previously.
Vasopressin, a potent vasoconstrictor, is actively secreted by the posterior pituitary gland in response to hemorrhage. The plasma concentration of vasopressin rises progressively as the arterial blood pressure diminishes (Figure 13-8). The receptors responsible for the augmented release are the sinoaortic baroreceptors and stretch receptors in the left atrium.
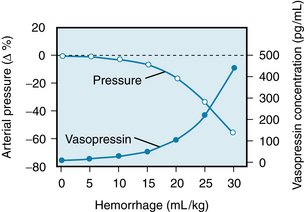
FIGURE 13-8 Mean percentage changes in arterial blood pressure and in plasma vasopressin concentration in response to blood loss (0.5 mL/kg/min) in a group of 12 dogs; the maximal volume of blood withdrawn was 30 mL/kg.
(Redrawn from Shen Y-T, Cowley AW Jr, Vatner SF: Relative roles of cardiac and arterial baroreceptors in vasopressin regulation during hemorrhage in conscious dogs. Circ Res 68:1422, 1991.)
The diminished renal perfusion during hemorrhagic hypotension leads to the secretion of renin from the juxtaglomerular apparatus (see Chapters 9 and 12). This enzyme acts on a plasma protein, angiotensinogen, to form angiotensin I, from which angiotensin II, a very powerful vasoconstrictor, is produced.
Renal Conservation of Salt and Water
During hemorrhage, the kidneys conserve fluid and electrolytes in response to various stimuli, including the increased secretion of vasopressin (antidiuretic hormone) noted previously (see Figure 13-8). The lower arterial pressure decreases the glomerular filtration rate and thus curtails the excretion of water and electrolytes. Also, the diminished renal blood flow raises the blood levels of angiotensin II, as previously described. This polypeptide accelerates the release of aldosterone from the adrenal cortex. Aldosterone, in turn, stimulates sodium reabsorption by the renal tubules, and water accompanies the sodium that is actively reabsorbed.
The Decompensatory Mechanisms are Mainly Humoral, Cardiac, and Hematologic
In contrast to the negative feedback mechanisms just described, latent positive feedback mechanisms are also evoked by hemorrhage. Such mechanisms exaggerate any primary change initiated by the blood loss. Specifically, positive feedback mechanisms aggravate the hypotension induced by blood loss, and they tend to initiate vicious circles, which may lead to death. The result of the operation of positive feedback mechanisms is illustrated in curve B of Figure 13-5.
Whether a positive feedback mechanism will lead to a vicious circle depends on the gain of that mechanism. Gain is defined as the ratio of the secondary change evoked by a given mechanism to the initiating change itself. A gain greater than 1 induces a vicious circle; a gain less than 1 does not. For example, consider a positive feedback mechanism with a gain of 2. If, for any reason, mean arterial pressure were to decrease by 10 mm Hg, the positive feedback mechanism with a gain of 2 would then evoke a secondary pressure reduction of 20 mm Hg, which in turn would cause a further decrement of 40 mm Hg; that is, each change would induce a subsequent change that was twice as great. Hence mean arterial pressure would decline at an ever-increasing rate until death supervened, much as is depicted by curve B in Figure 13-5.
Conversely, a positive feedback mechanism with a gain of 0.5 would indeed exaggerate any change in mean arterial pressure, but it would not necessarily lead to death. For example, if arterial pressure suddenly decreased by 10 mm Hg, the positive feedback mechanism would initiate a secondary, additional fall of 5 mm Hg. This in turn would provoke a further decrease of 2.5 mm Hg. The process would continue in ever-diminishing steps, with the arterial pressure approaching an equilibrium value asymptotically.
Some of the more important positive feedback mechanisms are (1) cardiac failure, (2) acidosis, (3) central nervous system depression, (4) aberrations of blood clotting, and (5) depression of the reticuloendothelial system.
Cardiac Failure
The role of cardiac failure in the progression of shock during hemorrhage is controversial. All investigators agree that the heart fails terminally, but opinions differ concerning the importance of cardiac failure during earlier stages of hemorrhagic hypotension. Shifts to the right in ventricular function curves (Figure 13-9) constitute experimental evidence of a progressive depression of myocardial contractility during hemorrhage.
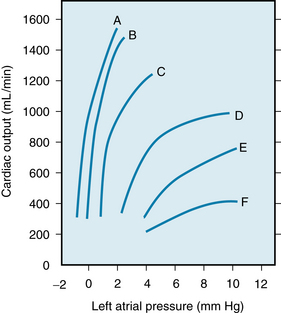
FIGURE 13-9 Ventricular function curves for the left ventricles during the course of hemorrhagic shock. Curve A represents the control function curve. Curve B represents 117 min, curve C 247 min, curve D 280 min, curve E 295 min, and curve F 310 min after the initial hemorrhage.
(Redrawn from Crowell JW, Guyton AC: Further evidence favoring a cardiac mechanism in irreversible hemorrhagic shock. Am J Physiol 203:248, 1962.)
The hypotension induced by hemorrhage reduces the coronary blood flow and therefore depresses ventricular function. The consequent reduction in cardiac output leads to a further decline in arterial pressure, a classic example of a positive feedback mechanism. Furthermore, the reduced blood flow to the peripheral tissues leads to an accumulation of vasodilator metabolites. These substances decrease peripheral resistance and therefore aggravate the fall in arterial pressure.
Acidosis
The inadequate blood flow during hemorrhage affects the metabolism of all cells in the body. The resulting stagnant anoxia accelerates the production of lactic acid and other acid metabolites by the tissues. Furthermore, impaired kidney function prevents adequate excretion of the excess H+, and generalized metabolic acidosis ensues (Figure 13-10). The resulting depressant effect of acidosis on the heart (see Chapter 5) further reduces tissue perfusion, thus aggravating the metabolic acidosis. Acidosis also diminishes the reactivity of the heart and resistance vessels to neurally released and circulating catecholamines and thereby intensifies the hypotension.
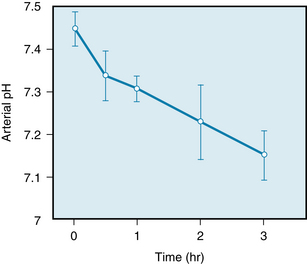
FIGURE 13-10 Reduction in arterial blood pH (mean ± SD) in a group of 11 dogs whose blood pressure had been held at a level of 35 mm Hg by bleeding into a reservoir, beginning at time 0.
(Modified from Markov AK, Oglethorpe N, Young DB, et al: Irreversible hemorrhagic shock: treatment and cardiac pathophysiology. Circ Shock 8:9, 1981.)
Central Nervous System Depression
The hypotension in shock reduces cerebral blood flow. Moderate degrees of cerebral ischemia induce a pronounced sympathetic nervous stimulation of the heart, arterioles, and veins, as stated previously. With severe degrees of hypotension, however, the cardiovascular centers in the brainstem eventually become depressed because of inadequate blood flow to the brain. The resulting loss of sympathetic tone then reduces cardiac output and peripheral resistance. The consequent reduction in mean arterial pressure intensifies the inadequate cerebral perfusion.
Various endogenous opioids, such as enkephalins and β-endorphin, may be released into the brain substance and into the circulation in response to hemorrhage. Opioids exist, along with catecholamines, in secretory granules in the adrenal medulla and sympathetic nerve terminals. Also, they are released together in response to stress. Similar stimuli release β-endorphin and adrenocorticotropic hormone (ACTH) from the anterior pituitary gland. The opioids depress the medullary vasomotor center in the brainstem. Such centers mediate some of the compensatory autonomic adaptations to blood loss, endotoxemia, and other shock-provoking stresses. Conversely, the opioid antagonist naloxone improves cardiovascular function and survival in various forms of shock.
Aberrations of Blood Clotting
The alterations of blood clotting after hemorrhage are typically biphasic—an initial phase of hypercoagulability followed by a secondary phase of hypocoagulability and fibrinolysis. In the initial phase, platelets and leukocytes adhere to the vascular endothelium, and intravascular clots, or thrombi, develop within a few minutes of the onset of severe hemorrhage. Coagulation may be extensive throughout the minute blood vessels.
Thromboxane A2 (TxA2) may be released from various ischemic tissues. It aggregates platelets, and more TxA2 is released from the trapped platelets, serving to trap additional platelets. This form of positive feedback intensifies and prolongs the clotting tendency. The mortality from certain standard shock-provoking procedures has been reduced considerably by anticoagulants such as heparin.
In the later stages of hemorrhagic hypotension, the clotting time is prolonged and fibrinolysis is prominent. As stated previously, hemorrhage into the intestinal lumen is common after several hours of hemorrhagic hypotension in dogs. Blood loss into the intestinal lumen would aggravate the hemodynamic effects of the original hemorrhage.
Depression of the Reticuloendothelial System
During the course of hemorrhagic hypotension, reticuloendothelial system (RES) function becomes depressed. The phagocytic activity of the RES is modulated by an opsonic protein. The opsonic activity in plasma diminishes during shock; this process may account in part for the depression of RES function. As a consequence, antibacterial and antitoxic defense mechanisms are impaired. Endotoxins from the normal bacterial flora of the intestine constantly enter the circulation. Ordinarily they are inactivated by the RES, principally in the liver. When the RES is depressed, these endotoxins invade the general circulation. Endotoxins produce profound, generalized vasodilation, mainly by inducing the abundant synthesis of an isoform of NO synthase in the smooth muscle of blood vessels throughout the body. The profound vasodilation aggravates the hemodynamic changes caused by blood loss.
In addition to their role in inactivating endotoxin, macrophages release many of the mediators that are associated with shock. These mediators include acid hydrolases, neutral proteases, oxygen free radicals, certain coagulation factors, and arachidonic acid derivatives: namely prostaglandins, thromboxanes, and leukotrienes. Macrophages also release certain monokines that modulate temperature regulation, intermediary metabolism, hormone secretion, and the immune system.
The Positive and Negative Feedback Mechanisms Interact
Hemorrhage provokes a multitude of circulatory and metabolic derangements. Some of these changes are compensatory, and others are decompensatory. Certain feedback mechanisms possess a high gain, and others a low gain. Furthermore, the gain of any specific mechanism varies with the severity of the hemorrhage. For example, with only a slight loss of blood, mean arterial pressure is within the range of normal, and the gain of the baroreceptor reflexes is high. With greater losses of blood, during which mean arterial pressure is below about 60 mm Hg (i.e., below the threshold for the baroreceptors), further reductions of pressure have no additional influence through the baroreceptor reflexes. Hence, below this critical pressure, the baroreceptor reflex gain is zero or near zero.
As a general rule, with minor degrees of blood loss, the gains of the negative feedback mechanisms are high, whereas those of the positive feedback mechanisms are low. The converse is true with more severe hemorrhages. The gains of the various mechanisms are additive algebraically. Therefore whether a vicious circle develops depends on whether the sum of the various gains exceeds 1. Total gains in excess of 1 are more likely when blood losses are severe. To avert a vicious circle, serious hemorrhages must be treated quickly and intensively, preferably by whole blood transfusions, before the process becomes irreversible.
Summary
Exercise
 In anticipation of exercise, the vagus nerve impulses to the heart are inhibited and the sympathetic nervous system is activated by central command. The result is an increase in heart rate, myocardial contractile force, cardiac output, and arterial pressure.
In anticipation of exercise, the vagus nerve impulses to the heart are inhibited and the sympathetic nervous system is activated by central command. The result is an increase in heart rate, myocardial contractile force, cardiac output, and arterial pressure.
With exercise, vascular resistance increases in skin, kidneys, splanchnic regions, and inactive muscles and decreases in active muscles.
The increase in cardiac output is accomplished mainly by the rise in heart rate. Stroke volume increases only slightly. In well-trained endurance athletes, stroke volume increases substantially during exercise.
During exercise total peripheral resistance decreases, O2 consumption and blood O2 extraction increase, and systolic and mean blood pressures rise slightly.
As body temperature rises during exercise, the skin blood vessels dilate. However, when the heart rate becomes maximal during severe exercise, the skin vessels constrict. This enlarges the effective blood volume but causes greater increases in body temperature and a feeling of exhaustion.
The limiting factor in exercise performance is the delivery of blood to the active muscles.
Hemorrhage
Acute blood loss induces tachycardia, hypotension, generalized arteriolar vasoconstriction, and generalized venoconstriction.
Acute blood loss invokes a number of negative feedback (compensatory) mechanisms, such as baroreceptor and chemoreceptor reflexes, responses to moderate cerebral ischemia, reabsorption of tissue fluids, release of endogenous vasoconstrictors, and renal conservation of water and electrolytes.
Acute blood loss also induces a number of positive feedback (decompensatory) mechanisms, such as cardiac failure, acidosis, central nervous system depression, aberrations of blood coagulation, and depression of the reticuloendothelial system.
The outcome of acute blood loss depends on the gains of the various feedback mechanisms and on the interactions between the positive and negative feedback mechanisms.
Bengel F.M., Ueberfuhr P., Karja J., et al. Sympathetic reinnervation, exercise performance and effects of β-adrenergic blockade in cardiac transplant recipients. Eur Heart J. 2004;25:1726.
Buckwalter J.B., Clifford P.S. Autonomic control of skeletal muscle blood flow at the onset of exercise. Am J Physiol. 1999;277:H1872.
Calbet J.A.L., Jensen-Urstad M., van Hall G., et al. Maximal muscular vascular conductances during whole body upright exercise in humans. J Physiol. 2004;558:319.
Clark M.G., Wallis M.G., Barrett E.J., et al. Blood flow and muscle metabolism: A focus on insulin action. Am J Physiol. 2003;284:E241.
Clifford P.S. Skeletal muscle vasodilatation at the onset of exercise. J Physiol. 2007;583:825.
Evans R.G., Ventura S., Dampney R.A., Ludbrook J. Neural mechanisms in the cardiovascular responses to acute central hypovolaemia. Clin Exp Pharmacol Physiol. 2001;28:479.
Foëx B.A. Systemic responses to trauma. Br Med Bull. 1999;55:726.
Hickner R.C., Fisher J.S., Ehsani A.A., et al. Role of nitric oxide in skeletal muscle blood flow at rest and during dynamic exercise in humans. Am J Physiol. 1997;273:H405.
Kulecs J.M., Collins H.L., Dicarlo S.E. Postexercise hypotension is mediated by reductions in sympathetic nerve activity. Am J Physiol. 1999;276:H27.
Laughlin M.H., Korthuis R.J., Duncker D.J., et al. Control of blood flow to cardiac and skeletal muscles during exercise. In: Rowell L.B., Shepherd J.T., eds. Handbook of Physiology, Section 12: Exercise: Regulation and Integration of Multiple Systems. Bethesda, Md: Oxford University Press, 1996.
Michelini L.C., Morris M. Endogenous vasopressin modulates the cardiovascular responses to exercise. Ann N Y Acad Sci. 1999;897:198.
O’Sullivan S.E., Bell C. The effects of exercise and training on human cardiovascular reflex control. J Auton Nerv Syst. 2000;81:16.
Potts J.T. Exercise and sensory integration. Role of the nucleus tractus solitarius. Ann N Y Acad Sci. 2001;940:221.
Prabhakar N.R., Peng Y.J. Peripheral chemoreceptors in health and disease. J Appl Physiol. 2004;96:359.
Prior B.M., Yang H.T., Terjung R.L. What makes vessels grow with exercise training? J Appl Physiol. 2004;97:1119.
Putnam R.W., Filosa J.A., Ritucci N.A. Cellular mechanisms involved in CO2 and acid signaling in chemosensitive neurons. Am J Physiol. 2004;287:C1493.
Rowland T.W. The circulatory response to exercise: Role of the peripheral pump. Int J Sports Med. 2001;22:558.
Squires R.W. Exercise therapy for cardiac transplant recipients. Prog Cardiovasc Dis. 2011;53:429.
Tipton C.M. The autonomic nervous system. In: Tipton C.M., ed. Exercise Physiology: People and Ideas. New York: Oxford University Press; 2003:189–254.
Tschakorsky M.E., Hughson R.L. Ischemic muscle chemoreflex response elevates blood flow in nonischemic exercising human forearm muscle. Am J Physiol. 1999;277:H635.
CASE 13-1
History
A 23-year-old male track star decided to enter the Boston Marathon. He had only run in short distance events, up to 10 K (6.21 miles), before entry in the marathon. At the 15-mile mark, he was leading the race, but he was soon passed by one of his competitors. This inspired him to make a strong effort to retake the lead, but he was unable to increase his speed. At the 20-mile mark he began to feel faint, and within the next mile he became sick to his stomach and somewhat disoriented, and he finally staggered and fell to the ground, exhausted.
CASE 13-2
History
A 47-year-old woman had an acute episode of severe abdominal pain, and she suddenly vomited a large amount of bloody material. Her husband called for an ambulance to take her to the hospital. The emergency room physician learned that the patient had had frequent, severe episodes of upper abdominal pain over the past 6 weeks. Physical examination revealed that the patient’s skin was very pale and cold, her heart rate was 110 beats/ min, and her blood pressure was 85/65 mm Hg. Examination of the patient’s blood revealed that the hematocrit ratio (the ratio of red blood cell volume to whole blood volume) was 40%. The physician made the tentative diagnosis of a bleeding peptic ulcer.
Questions
1. The patient’s skin was pale and cold because:
2. The patient’s arterial blood pressure, 85/65 mm Hg, indicates that:
3. If the patient’s bleeding had stopped before she arrived at the hospital, which of the following changes would be expected in the patient’s blood 1 hour after she had arrived at the hospital?