Chapter 412 Chronic Severe Respiratory Insufficiency
Improvements in the treatment of acute respiratory failure and advancements in invasive and noninvasive ventilation have led to an increase in the number of pediatric patients receiving long-term mechanical ventilation. Infants, children, and adolescents with disorders of central control of breathing, disease of the airways, residual lung disease after severe respiratory illness, and neuromuscular disorders may experience hypercarbic and/or hypoxemic chronic respiratory failure. Although it is generally possible to identify a primary cause for the respiratory failure, many children have multiple causative factors. Chronic respiratory failure is pulmonary insufficiency for a protracted period, usually 28 days or longer. Patients are maintained on long-term ventilation until they recover from the initial pulmonary insult. Patients with conditions such as central hypoventilation, progressive neuromuscular disease, and high quadriplegia may need ventilatory support indefinitely. The preferred site for a patient’s care after initial discharge with a ventilator is within the family home. When social circumstances do not allow this placement, patients may be placed in a highly skilled nursing facility.
Less than 1% of patients admitted to pediatric intensive care units require long-term noninvasive or invasive ventilatory assistance. A survey from Massachusetts by Graham, Fleegler, and Robinson (2007) identified 197 children undergoing long-term ventilatory support, a threefold increase over a 15-year period. The majority of the primary diagnoses (54%) were congenital or perinatal-acquired neurologic or neuromuscular disorders. Chronic lung disease of prematurity represented only 7% of the sample, a significant shift downward, presumably a result of improvement in neonatal care over the same period. Seventy percent of the patients were cared for at home.
412.1 Neuromuscular Diseases
Neuromuscular diseases (NMDs) of childhood include muscular dystrophies, metabolic and congenital myopathies, anterior horn cell disorders, peripheral neuropathies, and diseases that affect the neuromuscular junction. Decreases in muscle strength and endurance resulting from neuromuscular disorders can affect any skeletal muscle, including muscles involved in respiratory function. Of particular concern are those muscles mediating upper airway patency, generation of cough, and lung inflation. Acute respiratory insufficiency is often the most prominent clinical manifestation of several acute neuromuscular disorders, such as high-level spinal cord injury, poliomyelitis, Guillain-Barré syndrome (Chapter 608), and botulism (Chapter 202). Although much more insidious in its clinical course, respiratory dysfunction constitutes the leading cause of morbidity and mortality in progressive neuromuscular disorders (e.g., Duchenne muscular dystrophy [Chapter 601], spinal muscular atrophy, congenital myotonic dystrophy, myasthenia gravis [Chapter 604], and Charcot-Marie-Tooth disease [Chapter 605]).
Two of the most common and best understood NMDs of childhood are Duchenne muscular dystrophy (DMD) and spinal muscular atrophy (SMA). Principles utilized to evaluate and treat respiratory insufficiency in these disorders are often based on medical consensus rather than research. Despite this limitation, these principles are useful in the management of DMD and SMA as well as other less common NMDs.
Pathogenesis
Early onset of NMD can lead to chest wall deformity and lung disease due to developmental factors. In infancy the chest wall is very compliant with relatively stiff lungs and small airways. With progressive weakness of the intercostal muscles, the chest wall becomes even more compliant. Small airways have a tendency to become obstructed, leading to micro-atelectasis and decreased functional residual capacity. The compliant chest wall with initial sparing of diaphragm function leads to development of a small bell-shaped chest with depressed sternum, protruding abdomen, and paradoxical breathing, typically seen in SMA type 1. As the disease progresses, chest wall muscles shorten and lose elasticity, costosternal and costovertebral joints contract, and lung volumes decrease as a result of severe hypotonia. Inspiratory and expiratory pressures decrease, expiratory pressures more so than inspiratory, causing ineffective cough and poor airway clearance. As the child with NMD ages, kyphoscoliosis commonly develops, increasing the severity of restrictive lung disease. Although central control of breathing remains normal, response to central chemoreceptors may decrease because of chronic hypercapnia.
Treatment
Even though gene-targeted therapies are being developed for some NMDs, current interventions are primarily supportive rather than curative. Close surveillance through periodic review of the history and physical examination is critical. The development of personality changes, such as irritability, decreased attention span, fatigue, and somnolence, may point to the presence of sleep-associated gas exchange abnormalities and sleep fragmentation. Changes in speech and voice characteristics, nasal flaring, and the use of other accessory muscles during quiet breathing at rest may provide sensitive indicators of progressive muscle dysfunction and respiratory compromise. Although the frequency of periodic re-evaluation needs to be tailored to the individual patient, tentative guidelines were developed for patients with DMD; an abbreviated summary of such recommendations, applicable to all children with NMDs, is provided in Table 412-1.
Table 412-1 PROPOSED GUIDELINES FOR INITIAL EVALUATION AND FOLLOW-UP OF PATIENTS WITH NEUROMUSCULAR DISEASE
| INITIAL EVALUATION | BASIC INTERVENTION/TRAINING |
|---|---|
| History/physical/anthropometrics | Nutritional consultation and guidance |
| Lung function and maximal respiratory pressures (PFTs) | Regular chest physiotherapy |
| Arterial blood gases | Use of percussive devices |
| Polysomnography* | Respiratory muscle training |
| Exercise testing (in selected cases) | Annual influenza vaccine |
| If vital capacity >60% predicted or maximal respiratory pressures >60 cm H2O | Evaluate PFTs every 6 mo |
| CXR and polysomnography every year | |
| If vital capacity <60% predicted or maximal respiratory pressures <60 cm H2O | Evaluate PFTs every 3-4 mo |
| CXR, MIP/MEP every 6 mo | |
| Polysomnography every 6 mo to every year |
CXR, chest x-ray; MEP, maximal expiratory pressure; MIP, maximal inspiratory pressure; PFT, pulmonary function test.
* Please note that if polysomnography is not readily available, multichannel recordings including oronasal airflow, nocturnal oximetry, and end-tidal carbon dioxide levels may provide an adequate alternative.
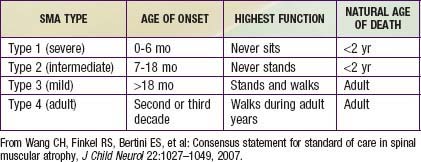
Guidelines for evaluation and management of patients with SMA were developed on the basis of expert consensus. Four classifications of SMA, based on age of onset and level of function, are recognized (Table 412-2). Treatment of SMA is focused on level of function (non-sitter, sitter, or walker) rather than SMA type. Unlike patients with DMD, patients with SMA do not demonstrate correlation between pulmonary function and need for mechanical ventilatory support. Rather, longitudinal monitoring for signs and symptoms of sleep-disordered breathing and ineffective airway clearance should be utilized to direct patient care.
Annane D, Orlikowski D, Chevret S, et al. Nocturnal mechanical ventilation for chronic hypoventilation in patients with neuromuscular and chest wall disorders. Cochrane Database Syst Rev. 2007;4:1-34.
Kennedy JD, Martin AJ. Chronic respiratory failure and neuromuscular disease. Pediatr Clin North Am. 2009;56:261-273.
Simonds AK. Recent advances in respiratory care for neuromuscular disease. Chest. 2006;130:1879-1886.
Wang CH, Finkel RS, Bertini ES, et al. Consensus statement for standard of care in spinal muscular atrophy. J Child Neurol. 2007;22:1027-1049.
412.2 Congenital Central Hypoventilation Syndrome
Congenital central hypoventilation syndrome (CCHS) is a clinically complex disorder of respiratory and autonomic regulation. In the classic case of CCHS, symptoms of alveolar hypoventilation are manifest during sleep only, but in more severe cases, the symptoms are manifest during sleep and wakefulness. The classic syndrome is further characterized by ventilatory failure to respond to hypercarbia and hypoxemia during wakefulness and sleep as well as physiologic and/or anatomic autonomic nervous system (ANS) dysregulation (ANSD). The physiologic ANSD may include all organ systems affected by the ANS, specifically the respiratory, cardiac, sudomotor, ophthalmologic, neurologic, enteric systems, and more. The anatomic or structural ANSD in CCHS includes Hirschsprung disease and tumors of neural crest origin (neuroblastoma, ganglioneuroma, or ganglioneuroblastoma). Most patients with CCHS present in the neonatal period, although later-onset CCHS (LO-CCHS) may manifest in infancy, childhood, and even adulthood. The initial symptoms include diminished tidal volume and a typically monotonous respiratory rate with cyanosis and hypercarbia. Diagnosis and management of children with CCHS have improved considerably, owing to greater knowledge in genetic testing, comprehensive care, and availability of technology for the home.
Genetics
In 2003, the paired-like homeobox 2B (PHOX2B) gene was identified as the disease-defining gene for CCHS. This gene, which is essential to the embryologic development of the ANS from the neural crest, is expressed in key regions that explain much of the CCHS phenotype. Individuals with CCHS are heterozygous for either a polyalanine repeat expansion mutation (PARM) in exon 3 of the PHOX2B gene (normal number of alanines is 20 with normal genotype 20/20), such that individuals with CCHS have 24-33 alanines on the affected allele (genotype range is 20/24-20/33), or a non–polyalanine repeat expansion mutation (NPARM) resulting from a missense, nonsense, or frameshift mutation. Roughly 90% of the cases of CCHS have PARMs and the remaining ≈10% of cases have NPARMs. The specific type of PHOX2B mutation is clinically significant as it can help with anticipatory guidance in patient management.
The majority of CCHS cases occur because of a de novo PHOX2B mutation, but 5-10% of children with CCHS inherit the mutation from an asymptomatic parent who is mosaic for the PHOX2B mutation. CCHS is inherited in an autosomal dominant manner. Therefore, an individual with CCHS has a 50% chance of transmitting the mutation, and resulting disease phenotype, to each child. Mosaic parents have up to a 50% chance of transmitting the PHOX2B mutation to each offspring. Genetic counseling is essential for family planning and for preparedness in the delivery room for an anticipated CCHS birth. PHOX2B testing is advised for both parents of a child with CCHS to anticipate risk of recurrence in subsequent pregnancies. Further, prenatal testing for PHOX2B mutation is clinically available (www.genetests.org) for families with a known PHOX2B mutation.
Ventilator Dependence
A correlation between the PHOX2B genotype and ventilator dependence has been reported. The greater the number of extra alanines, the more likely the need for continuous ventilatory support. Thus patients with the 20/25 genotype seldom require awake ventilatory support, although they do require support during sleep. Patients with the 20/26 genotype have variable awake support needs, and patients with 20/27-20/33 genotypes and those with NPARMs are likely to need continuous ventilatory support.
Hirschsprung Disease (Chapter 324.3)
Overall, 20% of children with CCHS also have Hirschsprung disease, and any infant or child with CCHS who presents with constipation should undergo rectal biopsy to screen for absence of ganglion cells. The type of PHOX2B mutation can help the primary physician anticipate which children are at higher risk. The frequency of Hirschsprung disease seems to increase with the longer polyalanine tracts (genotypes 20/27-20/33) and in those with NPARMs. Thus far no children with the 20/25 genotype have been reported to have Hirschsprung disease.
Tumors of Neural Crest Origin
Tumors of neural crest origin are more frequent in patients with NPARMs (50%) than in those with PARMs (1%). These extracranial tumors are more often neuroblastomas in individuals with NPARMs, rather than ganglioneuromas and ganglioneuroblastomas, which have been reported in patients with longer PARMs (20/29 and 20/33 genotype only).
Cardiac Asystole
Transient, abrupt, and prolonged sinus pauses have been identified in patients with CCHS, necessitating implantation of cardiac pacemakers when the pauses are ≥3 seconds. Among patients with the PHOX2B genotypes, 19% of those with the 20/26 genotype and 83% of those with the 20/27 genotype have pauses in the heartbeat of 3 sec or longer. Children with the 20/25 genotype have not been noted to have prolonged asystole, though one adult diagnosed with LO-CCHS has demonstrated a prolonged asystole.
Autonomic Nervous System Dysregulation
A higher number of polyalanine repeats among the PARMs is associated with an increased number of physiologic symptoms of ANSD. A higher frequency of anatomic ANSD findings is seen in individuals with CCHS who have NPARMs than in those who have PARMs. In addition, there is a spectrum of physiologic ANSD symptoms, including decreased heart rate variability, esophageal/gastric/colonic dysmotility, decreased pupillary response to light, reduced basal body temperature, altered distribution and amount of diaphoresis, lack of perception of shortness of breath, and altered perception of anxiety.
Facial Phenotype
Children with CCHS and PARMs have a characteristic facies that is boxy in appearance, flattened on profile, and short relative to its width. The following five variables correctly predict 86% of CCHS cases: upper lip height, binocular width, upper facial height, nasal tip protrusion, and inferior inflection of the upper lip vermillion border (lip trait).
Neuropathology
Anatomic findings in the brains of individuals with CCHS from early MRI studies were unremarkable, and those from autopsies were inconsistent. In a small cohort of adolescents with suspected CCHS, though without PHOX2B mutation confirmation, neuropathologic brainstem changes were identified by diffusion tensor imaging (DTI) in structures known to mediate central chemosensitivity and to link a network of cardiovascular, respiratory, and affective responses. The neuroanatomic defects in CCHS are likely the result of focal PHOX2B (mis)expression coupled with, in the suboptimally managed patient, sequelae of recurrent hypoxemia/hypercarbia. On the basis of rodent studies and functional MRI (fMRI) in humans, the following regions pertinent to respiratory control show PHOX2B expression in the pons and medulla of the brainstem: locus coeruleus, dorsal respiratory group, nucleus ambiguus, parafacial respiratory group, among other areas. Physiologic evidence suggests that the respiratory failure in these children is mostly based on defects in central mechanisms, but peripheral mechanisms (mainly carotid bodies) are also important.
Patients with CCHS have deficient carbon dioxide sensitivity during wakefulness and sleep; they do not respond with increased ventilation or arousal to hypercarbia during sleep. During wakefulness, a subset of patients may respond sufficiently to avoid hypercarbia, but most individuals with CCHS have hypoventilation that is severe enough that hypercarbia is apparent in the resting awake state. Children with CCHS also have altered sensitivity to hypoxia while awake and asleep. A key feature of CCHS is the lack of respiratory distress or sense of asphyxia with physiologic compromise. This lack of responsiveness to hypercarbia and/or hypoxemia with subsequent respiratory failure does not improve with advancing age. A subset of older children with CCHS may show an increase in ventilation (specifically increase in respiratory rate rather than increase in tidal volume) when they are exercised at various work rates, a response that is possibly secondary to neural reflexes from rhythmic limb movements—although the increase in minute ventilation is often insufficient to avoid physiologic compromise.
Clinical Manifestations
Patients with CCHS usually present in the first few hours after birth. Most children are the products of uneventful pregnancies and are term infants with appropriate weight for gestational age; Apgar scores have been variable. They do not show signs of respiratory distress, but their shallow respirations and respiratory pauses (apnea) evolve to respiratory failure with apparent cyanosis in the first day of life. In neonates with CCHS, the PaCO2 accumulates during sleep to very high levels, sometimes >90 mm Hg, and may decline to normal levels after the infants awaken. This problem becomes most apparent with failure of multiple attempts at extubation in an intubated neonate (who appears well with ventilatory support but in whom respiratory failure develops after removal of the support). However, the more severely affected infants hypoventilate awake and asleep; thus the previously described difference in PaCO2 between states is not apparent. Often, the respiratory rate is higher in rapid eye movement (REM) sleep than in non-REM sleep in individuals with CCHS.
LO-CCHS should be suspected in infants, children, and adults who have unexplained hypoventilation, especially subsequent to the use of anesthetic agents, sedation, acute respiratory illness, and potentially treated obstructive sleep apnea. These individuals may have other evidence of chronic hypoventilation, including pulmonary hypertension, polycythemia, and elevated bicarbonate concentration.
Besides treatment for these respiratory symptoms, children with CCHS require comprehensive evaluation and coordinated care to optimally manage associated abnormalities such as Hirschsprung disease, tumors of neural crest origin, symptoms of physiologic ANSD, and cardiac asystole, among other findings.
Differential Diagnosis
Studies should be performed to rule out primary neuromuscular, lung, and cardiac disease as well as an identifiable brainstem lesion that could account for the constellation of symptoms characteristic of CCHS. Introduction of clinically available PHOX2B genetic testing allows for early and definitive diagnosis of CCHS. Because CCHS mimics many treatable and/or genetic diseases, the following disorders should be considered: X-linked myotubular myopathy, multiminicore disease, congenital myasthenic syndrome, altered airway or intrathoracic anatomy (diagnosis made with bronchoscopy and chest CT), diaphragm dysfunction (diagnosis made with diaphragm fluoroscopy), congenital cardiac disease, a structural hindbrain or brainstem abnormality (diagnosis made with MRI of the brain and brainstem), Möbius syndrome (diagnosis made with MRI of the brain and brainstem and neurologic examination), and specific metabolic diseases, such as Leigh syndrome, pyruvate dehydrogenase deficiency, and discrete carnitine deficiency.
Rapid-Onset Obesity with Hypothalamic Dysfunction, Hypoventilation, and Autonomic Dysregulation
Previously referred to as late-onset central hypoventilation syndrome with hypothalamic dysfunction (LO-CHS/HD), rapid-onset obesity with hypothalamic dysfunction, hypoventilation, and autonomic dysregulation (ROHHAD) is a very rare disorder that was renamed in 2007 to clarify that it is distinct from LO-CCHS. The acronym is intended to describe the general sequence of presenting symptoms, which can evolve over years. Most dramatic is that these children are seemingly normal, and then demonstrate rapid weight gain (often >20 lb), which occurs within 6-12 mo. The diagnosis is based on clinical criteria that include onset of obesity and alveolar hypoventilation after the age of 1.5 yr (typically between 1.5 and 7 yr) and evidence of hypothalamic dysfunction as defined by ≥1 of the following findings: rapid-onset obesity, hyperprolactinemia, central hypothyroidism, disordered water balance, failure of response to growth hormone stimulation, corticotropin deficiency, and delayed/precocious puberty. Though it may not be apparent early in the course, all children with ROHHAD have hypoventilation. Considering the high prevalence of cardiorespiratory arrest and multisystem involvement, children with this disorder require coordinated comprehensive care with attention to development of hypoventilation (such as with repeated sleep studies and initiation of supported ventilation), bradycardia (via Holter monitor), treatment of hypothalamic dysfunction (with involvement of an endocrinologist), tumors of neural crest origin (often ganglioneuromas or ganglioneuroblastomas), and behavioral/intellectual decline (with annual neurocognitive testing).
Although children with ROHHAD can present with obstructive sleep apnea after development of obesity, it is distinct from obstructive sleep apnea hypoventilation syndrome (OSAHS) and obesity hypoventilation syndrome (OHS). In children, the existence of OHS is controversial and is often referred to as OSAHS because it describes chronic obstructive sleep apnea with resulting overnight hypercarbia, hypoxemia, and frequent arousals that lead to an altered set point of the central control of breathing (insensitivity to hypercarbia). Therefore, children with OSAHS experience awake hypoventilation and daytime sleepiness. The management is simply relief of the obstruction, such as tonsillectomy and/or initiation of noninvasive ventilation. If these interventions are unsuccessful, tracheostomy must be considered. In children with OSAHS, treatment of the upper airway obstruction would be expected to result in complete resolution of hypoventilation and daytime sleepiness. In contrast, in children with ROHHAD, relief of the upper airway obstruction unveils the central alveolar hypoventilation that requires lifelong ventilatory support. Both are distinguished from LO-CCHS by the absence of PHOX2B mutation and the presence of (often morbid) obesity.
Management
Supported Ventilation—Diaphragm Pacing
Depending on the severity of respiratory control deficit, the child with CCHS can have various means of ventilatory support: noninvasive positive pressure ventilation or mechanical ventilation via tracheostomy (see later section on long-term mechanical ventilation). Diaphragm pacing offers another mode of supported ventilation; it involves bilateral surgical implantation of electrodes beneath the phrenic nerves, with connecting wires to subcutaneously implanted receivers. The external transmitter, which is much smaller and lighter than a ventilator, sends a signal to donut-shaped antennae that are placed on the skin, over the subcutaneously implanted receivers. A signal can now travel from the external transmitter, ultimately, to the phrenic nerve to stimulate contraction of the diaphragm. A tracheostomy is typically required, at least initially, because the pacers induce a negative pressure on inspiration as a result of the contraction of the diaphragm being unopposed by pharyngeal dilatation. Individuals with CCHS who are ventilator dependent 24 hr/day are ideal candidates for diaphragm pacing, which provides increased ambulatory freedom (without the “ventilator tether”) while they are awake and mechanical ventilator support while they are asleep. This balance between awake pacing and asleep mechanical ventilation allows for a rest from phrenic nerve stimulation at night.
Monitoring in the Home
Home monitoring for children with CCHS is distinctly different from and more conservative than that for other children requiring long-term ventilation because children with CCHS lack innate ventilatory and arousal responses to hypoxemia and hypercarbia. In the event of physiologic compromise, other children likely are able to show clinical signs of respiratory distress. By contrast, for children with CCHS, the only means of determining adequate ventilation and oxygenation is with objective measures from a pulse oximeter, end-tidal carbon dioxide monitor, and close supervision of these values by a trained registered nurse (RN). At a minimum, it is essential that individuals with CCHS have continuous monitoring with pulse oximetry and end-tidal carbon dioxide with RN supervision during all sleep time, but ideally 24 hr per day. While awake, they are not able to sense or adequately respond to a respiratory challenge as may occur with ensuing respiratory illness, increased activity, or even the simple activity of eating.
Bougneres P, Pantalone L, Linglart A, et al. Endocrine manifestations of the rapid-onset obesity with hypoventilation, hypothalamic, autonomic dysregulation, and neural tumor syndrome in early childhood. J Clin Endocrinol Metabol. 2008;93:3971-3980.
Carroll MS, Pallavi PP, Weese-Mayer DE. Carbon dioxide chemoreception and hypoventilation syndromes with autonomic dysregulation. J Appl Physiol. 2010;108:979-988.
Gronli JO, Santucci BA, Leurgans SE, et al. Congenital central hypoventilation syndrome: PHOX2B genotype determines risk for sudden death. Pediatr Pulmonol. 2008;43:77-86.
Ize-Ludlow D, Gray J, Sperling MA, et al. Rapid onset obesity with hypothalamic dysfunction, hypoventilation, and autonomic dysregulation presenting in childhood. Pediatrics. 2007;120:e179-e188.
Katz ES, McGrath S, Marcus CL. Late-onset central hypoventilation with hypothalamic dysfunction: a distinct clinical syndrome. Pediatr Pulmonol. 2000;29:62-68.
Kinney HC. Structural abnormalities in the brainstem and cerebellum in congenital central hypoventilation syndrome. Pediatr Res. 2008;64:226-227.
Kumar R, Lee K, Macey P, et al. Mammillary body and fornix injury in congenital central hypoventilation syndrome. Pediatr Res. 2009;66:429-434.
Kumar R, Macey PM, Woo MA, et al. Diffusion tensor imaging demonstrates brainstem and cerebellar abnormalities in congenital central hypoventilation syndrome. Pediatr Res. 2008;64:275-280.
Marazita ML, Maher BS, Cooper ME, et al. Genetic segregation analysis of autonomic nervous system dysfunction in families of probands with congenital central hypoventilation syndrome. Am J Med Genet. 2001;100:229-236.
Patwari PP, Carroll MS, Rand CM, et al. Congenital central hypoventilation syndrome and the PHOX2B gene: a model of respiratory and autonomic dysregulation. Respir Physiol Neurobiol. 2010. Jun 30 [Epub ahead of print]
Todd ES, Weinberg SM, Berry-Kravis EM, et al. Facial phenotype in children and young adults with PHOX2B-determined congenital central hypoventilation syndrome: quantitative pattern of dysmorphology. Pediatr Res. 2006;59:39-45.
Weese-Mayer DE, Berry-Kravis EM, Ceccherini I, et al. An official ATS clinical policy statement. Congenital central hypoventilation syndrome: genetic basis, diagnosis, and management. Am J Respir Crit Care Med. 2010;181:626-644.
Weese-Mayer DE, Marazita ML, Berry-Kravis EM. Congenital central hypoventilation syndrome. Pagon RA, Bird TC, Dolan CR, et al, editors. GeneReviews. University of Washington, Seattle, 1993. www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=ondine.
Weese-Mayer DE, Rand CM, Berry-Kravis EM, et al. Congenital central hypoventilation syndrome from past to future: model of translational and transitional autonomic medicine. Pediatr Pulmonol. 2009;44:521-535.
Weese-Mayer DE, Silvestri JM, Huffman AD, et al. Case/control family study of ANS dysfunction in idiopathic congenital central hypoventilation syndrome. Am J Med Genet. 2001;100:237-245.
412.3 Other Conditions
Myelomeningocele with Arnold-Chiari Type II Malformation
Arnold-Chiari type II malformation (Chapter 585.11) is associated with myelomeningocele, hydrocephalus, and herniation of the cerebellar tonsils, caudal brainstem, and the fourth ventricle through the foramen magnum.
Sleep-disordered breathing, including obstructive sleep apnea and hypoventilation, has been reported. Direct pressure on the respiratory centers or brainstem nuclei, or increased intracranial pressure because of the hydrocephalus may be responsible. Vocal cord paralysis, apnea, hypoventilation, and bradyarrhythmias have also been reported.
Patients with Arnold-Chiari type II malformation have blunted responses to hypercapnia, and to a lesser degree, hypoxia.
Management
An acute change in the ventilatory state of a patient with this malformation requires immediate evaluation. Consideration must be given to posterior fossa decompression and/or treatment of the hydrocephalus. If this treatment is unsuccessful in resolving central hypoventilation or apnea, tracheostomy and long-term mechanical ventilation should be considered.
Rapid-Onset Obesity, Hypothalamic Dysfunction, and Autonomic Dysregulation (Chapter 412.2)
Obesity Hypoventilation Syndrome
As its name implies, obesity hypoventilation syndrome is a syndrome of central hypoventilation during wakefulness in obese patients with sleep-disordered breathing. Although it was initially described mainly in adult obese patients, obese children have also demonstrated the syndrome. Sleep-disordered breathing is a combination of obstructive sleep apnea, hypopnea, and/or sleep hypoventilation syndrome. Patients are hypercapnic with cognitive impairment, morning headache, and hypersomnolence during the day. Chronic hypoxemia may lead to pulmonary hypertension and cor pulmonale.
Obesity is associated with reduced respiratory system compliance, increased airway resistance, reduced functional residual capacity, and increased work of breathing. Affected patients are unable to increase their respiratory drive in response to hypercapnia. Leptin may have a role in this syndrome. The sleep-disordered breathing leads to compensatory metabolic alkalosis. Because of the long half-life of bicarbonate, its elevation causes compensatory respiratory acidosis during wakefulness with elevated PaCO2.
Acquired Alveolar Hypoventilation
Traumatic, ischemic, and inflammatory injuries to the brainstem, brainstem infarction, brain tumors, bulbar polio, and viral paraneoplastic encephalitis may also result in central hypoventilation.
Obstructive Sleep Apnea
Epidemiology
Habitual snoring during sleep is extremely common during childhood, and up to 27% of children are affected. The current obesity epidemic has affected the epidemiology of this condition. Peak prevalence is at 2-8 yr. The ratio between habitual snoring and obstructive sleep apnea (OSA) is 4 : 1 to 6 : 1.
Pathophysiology
OSA occurs when the luminal cross-sectional area of the upper airway is significantly reduced during inspiration. With increased airway resistance and reduced activation of pharyngeal dilators, negative pressure leads to upper airway collapse. The site of upper airway closure in children with OSA is at the level of tonsils and adenoids. The size of tonsils and adenoids increases throughout childhood up to 12 yr of age. Environmental irritants such as cigarette smoke or allergic rhinitis may accelerate the process. Reports now suggest that early viral infections may affect adenotonsillar proliferation.
Clinical Presentation
Snoring during sleep, behavioral disturbances, learning difficulties, excessive daytime sleepiness, metabolic issues, and cardiovascular morbidity may alert the parent or physician to the presence of OSA.
Diagnosis is made with the help of a polysomnogram and airway radiograms.
Treatment
When adenotonsillar hypertrophy is suspected, a consultation with an ear, nose, and throat specialist for adenoid tonsillectomy may be indicated. For patients who are not candidates for this treatment or who persist with OSA, CPAP or BiPAP during sleep may alleviate the obstruction (Chapter 17).
Spinal Cord Injury (SCI)
Epidemiology
Spinal cord injury (SCI) occurs at a rate of 30-40 per million population per year, resulting in 10,000 new cases each year. Spinal cord injury is relatively rare in pediatric patients, with an incidence of 1-13% of all SCI patients. The incidence in infancy and early childhood is similar for boys and girls. There is male preponderance in children >13 yr of age. Motor vehicle accidents, falls, sports injuries, and assaults are the main causes. SCI usually leads to lifelong disability.
Pathophysiology
In children with SCI there is disproportionately higher involvement of the upper cervical spine, a high frequency of spinal cord injury without radiographic abnormality, delayed onset of neurologic deficits, and a higher proportion of complete injury. Sixty percent of pediatric SCI cases involve C1-C3 and 30-40% involve C4-C7. Thus, there is a high likelihood in a child with SCI of quadriplegia with intercostal muscle and/or diaphragmatic paralysis leading to respiratory failure.
Management
Immobilization and stabilization of the spine must be accomplished simultaneously with resuscitation and stabilization. Patients with high SCI in many instances require lifelong ventilation. Depending on the patient’s age and general condition, tracheostomy with mechanical ventilation or diaphragmatic pacing may be indicated. Often patients with diaphragmatic pacing need tracheostomy placement if there is no coordination between pacing and glottis opening. Muscle spasms occur frequently in these patients and are treated with muscle relaxants. Occasionally the muscle spasms involve the chest and present a serious impediment to ventilation. Continuous intrathecal infusion of muscle relaxant may be indicated (Chapter 598.5).
Metabolic Disease
Mucopolysaccharidoses (Chapter 82)
Mucopolysaccharidoses (MPSs) are a group of progressive hereditary disorders that lack the lysosomal enzymes that degrade glycosaminoglycans. Incompletely catabolized mucopolysaccharides accumulate in connective tissue throughout the body. The incidence is 1 : 30,000 to 1 : 150,000 live births. The inheritance is autosomal recessive except for Hunt syndrome, which is X-linked. The diagnosis is suggested by a pattern of glycosaminuria and is confirmed by a lysosomal enzyme assay.
I-cell disease mucolipidosis type II is an inherited lysosomal disorder with accumulation of mucolipids. Phenotypically, it is similar to MPS but the age of onset is earlier and there is no mucopolysacchariduria.
Mucopolysaccharide deposits are frequently found in the head and neck and cause airway obstruction. Typically, the affected patient has a coarse face and large tongue. Significant deposits are found in the adenoids, tonsils, and cartilage. A polysomnogram and airway radiograms may help define the severity of the upper airway obstruction.
Treatment has been attempted with enzyme replacement therapy and stem cell transplantation, with limited success. Adenoidotonsillectomy may be indicated but seldom solves the problem. Noninvasive positive end expiratory pressure or ventilation may be helpful. Tracheostomy may be indicated with CPAP or ventilatory support.
Dysplasias
Camptomelic dysplasia and thanatophoric dysplasia affect rib cage size, shape, and compliance, leading to respiratory failure. Most patients with these disorders do not survive beyond early infancy. Tracheostomy and ventilation may prolong life.
Lung Disease
Common metabolic lung conditions include bronchopulmonary dysplasia (BPD) and recuperation from acute respiratory distress syndrome (ARDS). Former premature infants recuperating from respiratory distress syndrome may experience BPD (Chapters 95.3 and 410). Volutrauma, barotrauma, and air leak syndromes incurred during mechanical ventilation contribute to lung injury. When extreme, BPD may progress to respiratory failure. Increased pulmonary vascular resistance, pulmonary hypertension, cor pulmonale, and lower airway obstruction are known complications.
Treatment of patients with these conditions may include mechanical ventilation, diuretics, bronchodilators, inhaled steroids, intermittent systemic steroids, and pulmonary vasodilators.
Glycogenosis Type II (Chapter 81.1)
Glycogenosis type II is an autosomal recessive disorder. Clinical manifestations include cardiomyopathy and muscle weakness. Cardiac issues may include heart failure and arrhythmias. Muscle weakness leads to respiratory insufficiency and sleep-disordered breathing. Treatment includes emerging therapies such as enzyme replacement therapy, chaperone molecules, and gene therapy. Supportive therapy may consist of either noninvasive ventilation, or tracheostomy and mechanical ventilation. Cardiac medications, protein-rich nutrition, and judicious physical therapy are additional measures that can be utilized.
Severe Tracheomalacia and/or Bronchomalacia (Airway Malacia)
Conditions associated with airway malacia include tracheoesophageal fistula, innominate artery compression, and pulmonary artery sling after surgical repair (Chapter 381). Patients with tracheobronchomalacia present with cough, lower airway obstruction, and wheezing. Diagnosis is made via bronchoscopy, preferably with the patient breathing spontaneously. Positive end-expiratory pressure (PEEP) titration during the bronchoscopy helps identify the airway pressure required to maintain airway patency and prevent tracheobronchial collapse.
Neuromyopathy of Severe Illness
Children recuperating from severe illness in the intensive care unit often have neuromuscular weakness from suboptimal nutrition. This neuromuscular weakness can be devastating when coupled with the catabolic effects of severe illness and the residual effects of sedatives, analgesics, and muscle relaxants, particularly if corticosteroids were administered. Children with neuromuscular disease have limited ability to increase ventilation and usually do so by increasing respiratory rate. Because of weakness, sternal retractions may not be observed. In severe illness, some of these children respond to increased respiratory load by becoming apneic. A look of panic, a change in vital signs such as significant tachycardia or bradycardia, and cyanosis may be the only signs of respiratory failure.
Bembi B, Cerini E, Danesino C, et al. Management and treatment of glycogenosis type II. Neurology. 2008;71:S12-S36.
Davin Miller J, Waldemar AC. Pulmonary complications of mechanical ventilation in neonates. Clin Perinatol. 2008;35:273-281.
Dayyat E, Kheeirandish-Gozal L, Gozal D. Childhood obstructive sleep apnea: one or two distinct disease entities? Sleep Med Clin. 2007;2:433-444.
Lee JH, Sung IY, Kang JY, et al. Characteristics of pediatric onset spinal cord injury. Pediatr Int. 2009;51:254-257.
Muzumdar H, Arens R. Central alveolar hypoventilation syndromes. Sleep Med Clin. 2008;3:601-615.
Young AH, Cowan MJ, Horn B, et al. Airway management in children with mucopolysaccharidoses. Arch Otolaryngol Head Neck Surg. 2009;135:73-79.
412.4 Long-Term Mechanical Ventilation
Some children with chronic severe respiratory insufficiency benefit from long-term ventilatory support. The goals of such support are to maintain normal oxygenation and ventilation and to minimize the work of breathing. Long-term ventilation in the home is a complex, physically demanding, emotionally taxing, and expensive process for the family and for society. It changes the family’s way of life, priorities, and relationships. It may adversely affect intrafamilial and extrafamilial relationships.
The prognosis of the disease is a critical factor in the decision to initiate long-term ventilation. The discharge process for a child undergoing ventilatory support should start as soon as the child is medically stable and supported by equipment that can be maintained in the home. Children with degenerative neuromuscular disease, such as type I SMA, suffer from respiratory failure very early in life, often triggered by the first respiratory illness. Although some parents decide to provide only palliative end-of-life care (Chapter 40) for the child with SMA, others choose long-term invasive or noninvasive ventilatory support. Young children with chronic lung disease and airway malacia have the potential to improve their pulmonary function and to wean successfully off ventilator support if provided with adequate ventilation, good nutrition, and measures to promote development and prevent further lung injury.
Successful home discharge of a patient receiving mechanical ventilation depends on whether there are adequate resources in the community to support the family. Some hospital programs that transition children home on ventilators utilize professional nurses in the home to assist with round-the-clock care. This measure depends on funding as well as availability of nursing agencies in the community. Housing can be a significant barrier to home discharge because there must be adequate space for the child and caretakers, equipment, and supplies, environmental safety, including compliance with building and electrical codes, and home modifications for mobility, including ramping and lifts.
Funding for home care is usually a difficult issue for this population of children. Even if they have private insurance, coverage for home care benefits is frequently limited. In the USA, for children eligible for public aid, most states have funds available to meet the special needs of children who are ventilator dependent, although the extent of coverage varies considerably among geographic areas.
Respiratory Equipment for Home Care
Modes of mechanical ventilation support are discussed in Chapter 65.1.
Noninvasive Equipment
Supplemental oxygen and positive pressure can be administered by nasal cannula. This system delivers heated, supersaturated, high-flow gases. A number of devices are available for the delivery of CPAP and biPAP. These machines attach to nasal and full-face masks or to nasal pillows and are best suited for the treatment of obstructive sleep apnea. Long-term use of these devices in small children may result in midface dysplasia or pressure wounds. This type of ventilation has also been used in less severely affected patients with recurrent atelectasis and/or nocturnal hypoventilation, as well as for palliation in more severely affected patients.
Rocker Bed
A rocker bed moves in a longitudinal seesaw motion at a set rate. The child is secured to the bed with a strap. Movement of the bed promotes diaphragm movement. The bed may be an option for children with mild neuromuscular weakness, for instance, during recuperation from Guillain-Barré syndrome. This device should not be placed in a home with toddlers or young children, who may get trapped in its mechanism.
Cuirasse
The cuirasse is a negative-pressure device that resembles a turtle shell. It is designed to fit over the anterior chest and provide a tight seal. Cycled negative pressure is applied to the child’s chest through a hole in the cuirasse. The device is suitable only for infants and children with mild neuromuscular weakness. A plastic bag–like device that fits snugly around the chest applies the same principle.
Iron Lung
The iron lung is a device that applies negative pressure to the child’s body. The child is placed in the iron lung cylinder with the head extending outside the device. A cuff is placed around the neck to minimize air leaks. Negative pressure is cycled within the iron lung, facilitating chest wall movement. Ventilation is disrupted when the device is opened to deliver care. This device is suitable for children with muscular weakness who require ventilation for part of the day. Its main advantage is that it does not require a tracheostomy; however, upper airway obstruction may occur, and this risk requires ongoing evaluation. A smaller, lightweight version of this device is available for travel.
Diaphragmatic Pacing
Detailed in the management section of Ch. 412.2, diaphragm pacers may also be considered in children with spinal cord injury involving a level above C3, though the immediate advantages are less apparent than in CCHS.
Positive Pressure Ventilation
Ideally, a ventilator intended for home use is lightweight and small, is able to entrain room air, preferably has continuous flow, and has a wide range of settings (pressure, volume, pressure support, and rate) that would allow ventilation from infancy to adulthood. Battery support for the ventilator, both internal and external, should be sufficient to permit unrestricted portability in the home and community. The equipment must also be impervious to electromagnetic interference and must be relatively easy to understand and troubleshoot. A variety of ventilators that can be used in the home are available, and familiarity with these devices is necessary to choose the best option for the child.
Airway Clearance
Thick, copious secretions may contribute to increased airway resistance and may provide substrate for bacterial and fungal growth. Respiratory infections in turn lead to an increase in the amount of secretions and may increase viscosity, contributing to problems in airway clearance. Patients with neuromuscular weakness often have discoordination or absence of swallow, putting them at risk for aspiration of oral secretions or food. Reflux resulting in aspiration is also common. Additionally, many patients have poor or nonexistent cough; some of them may have ciliary dysfunction.
Modalities that help with clearance of secretions include postural drainage, manual or mechanical percussion or vibration, and vest or wrap percussion therapy. Cough effort may be enhanced with a cough assist device and/or abdominal binder. In addition, oropharyngeal or tracheal suctioning to remove secretions may promote airway clearance.
Control of oral secretions can be achieved pharmacologically with anticholinergic drugs or by localized injection of botulinum toxin (Botox) or surgical ligation of selected salivary ducts. In extreme cases, surgical tracheolaryngeal separation may be indicated. If thick, tenacious secretions are problematic, patient hydration and dosing of anticholinergic medication should be reviewed. Administration of DNAase or N-acetylcysteine may be considered to thin secretions. In selected cases, bronchoscopy may be indicated for the removal of inspissated secretions and/or re-expansion of atelectatic pulmonary lobe or segment.
Physical Therapy, Occupational Therapy, and Speech Therapy
Therapies are very important in chronic respiratory failure. Potential goals for physical therapy are mobilization of the patient and strengthening of muscles, particularly truncal and abdominal muscles essential to pulmonary rehabilitation. Occupational therapy goals revolve around achieving or maintaining developmental milestones. Child life/developmental therapy focuses on provision of developmentally appropriate environmental stimulation and age-appropriate play. Speech therapy goals deal with oromotor skills for feeding and communication. Evaluation of swallow is a key component of therapy for children with chronic respiratory failure. Sign language is frequently utilized for communication, because of delayed speech or hearing loss. Audiology specialists should be involved in the assessment of hearing, as there is a higher incidence of hearing loss in the patients undergoing long-term ventilation.
Infections
Infections—tracheitis (Chapter 377.2), bronchitis (Chapter 383.2), pneumonia (Chapter 392)—are common in patients with chronic respiratory failure. They may be due to community-acquired viruses (adenovirus, influenza, respiratory syncytial virus, parainfluenza) or community- or hospital-acquired bacteria. Many of the latter organisms are gram-negative, highly antimicrobial-resistant pathogens that cause further deterioration in pulmonary function. Bacterial infection is most likely in the presence of fever, deteriorating lung function (hypoxia, hypercarbia, tachypnea, retractions), leukocytosis, and mucopurulent sputum. The presence of leukocytes and organisms on Gram stain of tracheal aspirate, as well as the visualization of new infiltrates on radiographs, may be consistent with bacterial infection. Infection must be distinguished from tracheal colonization, which is asymptomatic and associated with normal amounts of clear tracheal secretions. If infection is suspected, it must be treated with antibiotics, based on the culture and sensitivities of organisms recovered from the tracheal aspirate. Inhaled tobramycin, started early, may avert more serious infection. Infections should be prevented by appropriate immunizations (influenza, pneumococcus, Haemophilus influenzae type b), passive immunity (respiratory syncytial virus), and good tracheostomy care. Antibiotics should be used judiciously to prevent further colonization with drug-resistant organisms. However, some patients who have recurrent infections may benefit from prophylaxis with inhaled antibiotics.
Monitoring in the Home
A patient who is ventilated in the home must be monitored at all times. The patient must be under direct observation of the caregivers. Continuous monitoring of O2 saturation and heart rate is recommended during sleep, and either continuous or intermittent monitoring during the daytime, depending on patient stability. Patients with CCHS or pulmonary hypertension are particularly vulnerable to episodes of hypoxemia and/or hypercarbia. Patients with pulmonary hypertension may experience a rapid drop in O2 saturation.
Patients followed up in pulmonary clinics should be monitored at each clinic visit for heart rate, O2 saturation, and transcutaneous and/or end-tidal CO2. Pulmonary function tests should be considered for those patients who are old enough and able to cooperate, usually after 5 yr of age.
Enhanced monitoring and surveillance are recommended for patients whose pulmonary status has improved and are in the process of being weaned completely off ventilator support. A polysomnogram may be useful when total liberation from mechanical ventilation is being contemplated. In addition to physiologic parameters, patients must be monitored for signs of stress, agitation, and fatigue. Often these signs appear one or more days after the ventilator parameter changes.
Discharge Process
The initial discharge process for a child going home on a ventilator is complex. A multidisciplinary, coordinated team approach is needed to develop a comprehensive plan that addresses medical, psychosocial, developmental, educational, and safety issues. The ventilated child must demonstrate medical stability that can be safely managed at home; interventions to maintain stability should be minimal before discharge. The child should be transitioned to a ventilator suitable for home use that allows portability as well as adequate ventilation. Medical management should also focus on transitioning oxygen and ventilator parameters to settings appropriate for home care. Depending on the type of ventilation employed, a tracheostomy may be placed to promote comfort and a stable airway as soon as the decision for long-term ventilation is made.
Nutrition should be optimized to promote growth yet minimize excessive weight gain and carbon dioxide production. The nutritional requirements of a ventilated child are frequently decreased due to the supported work of breathing. The ventilated child often has problems with swallowing from discoordination and oral aversion secondary to intubation. Speech therapy should be introduced early to begin oromotor therapy and return of swallow. Many children require gastrostomy tube placement to replace or supplement oral intake. Evaluation and management of reflux and the risk of aspiration should also be considered. Some children with severe reflux may require jejunal feeding. Communication devices to augment speech and introduction of sign language for speech and hearing impaired should be part of the planning.
Training of caregivers should be initiated early in the discharge process and should be provided by nurses, respiratory care practitioners, and physical, occupational, and speech therapists. Caregivers must be trained in all aspects of the child’s care, including tracheostomy care, ventilator management, and cardiopulmonary resuscitation. Their independence in delivery of care at the bedside and while transporting the child should be emphasized. Special focus should be placed on safety and the appropriate response in the event of an emergency. An emergency bag containing critical supplies should accompany the patient at all times. Caregivers must demonstrate their proficiency before the child is discharged.
Community agencies should be identified for provision of home support services. They may include a nursing agency to provide private duty nursing services. It is ideal to train home care nurses about the ventilator and the child’s care before home discharge. An equipment vendor who can provide the ventilator equipment, supplies, and service should be selected. A care conference involving the hospital team, funding agency, home nursing agency, equipment vendor, and family caregivers should take place before discharge. The conference is important for coordination of last-minute details and facilitation of a smooth transition to home.
Providing continued support to the child and family after discharge is essential. The pediatrician in the community has a central role in providing coordination of care, well child care, and all other medical services, with the possible exception of ventilatory management. Equally important is the establishment of lines of communication to the medical center and the provision of timely access for advice and troubleshooting during the intervals between multidisciplinary clinic visits.
Fischer JR. Home ventilator guide. International Ventilator Users Network, affiliate of Post-Polio Health International, St Louis, 2008. www.post-polio.org/ivun/index.html.
Graham RJ, Fleegler EW, Robinson WM. Chronic ventilator need in the community: a 2005 pediatric census of Massachusetts. Pediatrics. 2007;119:1280-1287.
O’Brien JE, Dumas HM, Haley SM, et al. Ventilator weaning outcomes in chronic respiratory failure in children. Int J Rehab Res. 2007;30:171-174.
Siner JM, Manthous CA. Liberation from mechanical ventilation: what monitoring matters? Crit Care Clin. 2007;23:613-638.