53 Antiprotozoal drugs
Overview
Protozoa are motile, unicellular eukaryotic organisms that have colonised virtually every habitat and ecological niche. They may be conveniently classified, on the basis of their method of locomotion, into four main groups: amoebas, flagellates and sporozoa together with a further group comprising ciliates and other organisms of uncertain affiliation, such as the Pneumocystis jirovecii mentioned in the last chapter. The protozoa have diverse feeding behaviour, with some being parasitic. Many have extremely complex life cycles, sometimes involving several hosts, reminiscent of the helminths discussed in Chapter 54.
As a group, the protozoa are responsible for an enormous burden of disease among humans as well as domestic and wild animal populations. Table 53.1 lists some of these clinically important organisms, together with the diseases that they cause and an overview of anti-infective drugs. In this chapter, we will first discuss some general features of protozoa–host interactions and then discuss the therapy of each group of diseases in turn. In view of its global importance, a discussion of malaria will occupy much of the chapter.
Table 53.1 Principal protozoal infections and common drug treatments

Host–Parasite Interactions
Mammals have developed very efficient mechanisms for dealing with invading parasites but many parasites have, in turn, evolved clever tactics to evade these defensive responses. One common parasite ploy is to take refuge within the cells of the host, where antibodies cannot reach them. Most protozoa do this, for example Plasmodia species take up residence in red cells, Leishmania species infect macrophages exclusively, while Trypanosoma species invade many other cell types. The host deals with these intracellular fugitives by deploying cytotoxic CD8+ T cells and T helper (Th)1 pathway cytokines, such as interleukin (IL)-2, tumour necrosis factor-α and interferon-γ. These cytokines (see Ch. 17) activate macrophages, which can then kill intracellular parasites.
The Th1 pathway responses can be downregulated by Th2 pathway cytokines (e.g. transforming growth factor-β, IL-4 and IL-10). Some intracellular parasites have evolved mechanisms for manipulating the Th1/Th2 balance to their own advantage by stimulating the production of Th2 cytokines. For example, the invasion of macrophages by Leishmania species induces transforming growth factor-β, while the invasion of T cells, B cells and macrophages by trypanosomes induces IL-10 (see Handman & Bullen, 2002, and Sacks & Toben-Trauth, 2002, for further details). Similar mechanisms operate during worm infestations (see Ch. 54).
Toxoplasma gondii has evolved a different ploy: upregulation of some host responses. The definitive (i.e. where sexual recombination occurs) host of this protozoon is the cat, but humans can inadvertently become intermediate hosts, harbouring the asexual form of the parasite. In humans, T. gondii infects numerous cell types and has a highly virulent replicative stage. To ensure that its host survives, it stimulates production of interferon-γ, modulating the host’s cell-mediated responses to promote encystment of the parasite in the tissues.
Improved understanding of host–protozoon relationships has opened up new vistas for the development of antiprotozoal agents. The possibility of using cytokine analogues and/or antagonists to treat disease caused by protozoa is already being investigated (for review, see Odeh, 2001).
Malaria and Antimalarial Drugs
Malaria1 is caused by parasites belonging to the genus Plasmodium. Four main species of plasmodia infect humans: Plasmodium vivax, Plasmodium falciparum, Plasmodium ovale and Plasmodium malariae. The insect vector is the female Anopheles mosquito, which breeds in stagnant water, and the disease it spreads is one of the major killers on our planet.
The statistics are staggering. According to the (2008) World Health Organization (WHO) report, malaria is a significant public health problem in more than 90 countries inhabited by about 50% of the world’s population. In 2006, the disease caused an estimated 880 million acute illnesses each year and nearly 1 million deaths. More than 90% of these occur in sub-Saharan Africa, and it is estimated that the disease kills an African child every 30 seconds. Those who survive may suffer from lasting mental impairment. Other high-risk groups include pregnant women, refugees and labourers entering endemic regions. Malaria also imposes a huge economic burden on countries where the disease is rife.
The symptoms of malaria include fever, shivering, pain in the joints, headache, repeated vomiting, generalised convulsions and coma. Symptoms become apparent only 7–9 days after being bitten by an infected mosquito. By far the most dangerous parasite is P. falciparum.
Malaria was eradicated from most temperate countries in the 20th century, and the WHO attempted to eradicate malaria elsewhere using the powerful ‘residual’ insecticides and the highly effective antimalarial drugs, such as chloroquine, that had become available. By the end of the 1950s, the incidence of malaria had dropped dramatically. However, during the 1970s it became clear that the attempt at eradication had failed, largely owing to the increasing resistance of the mosquito to the insecticides and of the parasite to the drugs. Sadly, malaria has now re-emerged in several countries where it was previously under control or eradicated. International air travel is responsible for sporadic cases in Western Europe and the USA, where the actual risk of transmission is negligible.2 1999 saw the initiation of the Roll Back Malaria programme sponsored by a partnership of transnational organisations including the WHO. While it is unlikely that this programme will achieve all its goals, one encouraging trend has been that the disease has actually begun to decline in some parts of the world following aggressive public health campaigns.
The Life Cycle of the Malaria Parasite
The mosquito, not the human, is the definitive host for plasmodia, and it has been said that the only function of humans is to enable the parasite to infect more mosquitoes so that further sexual recombination can occur. The life cycle of the parasites consists of a sexual cycle, which takes place in the female Anopheles mosquito, and an asexual cycle, which occurs in humans (Fig. 53.1 and the Malaria box).
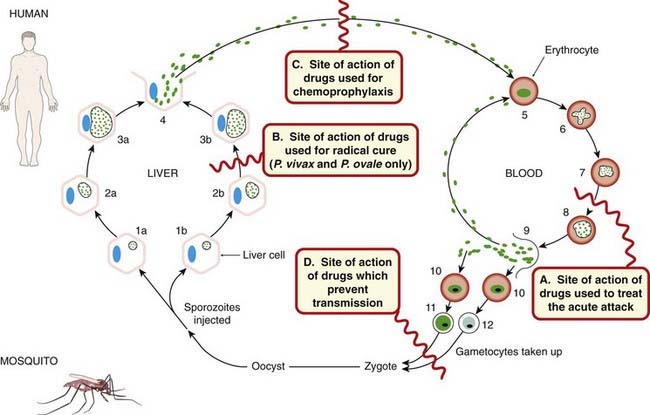
Fig. 53.1 The life cycle of the malarial parasite and the site of action of antimalarial drugs.
The pre- or exoerythrocytic cycle in the liver and the erythrocytic cycle in the blood are shown: 1a Entry of sporozoite into liver cell (the parasite is shown as a small circle containing dots, and the liver cell nucleus as a blue oval). 2a and 3a Development of the schizont in liver cell. 4 Rupture of liver cell with release of merozoites (some may enter liver cells to give resting forms of the parasite, hypnozoites). 5 Entry of merozoites into a red cell. 6 Trophozoite in red cell. 7 and 8 Development of schizont in red cell. 9 Rupture of red cell with release of merozoites, most of which parasitise other red cells. 10–12 Entry of merozoites into red cells and development of male and female gametocytes. 1b Resting form of parasite in liver (hypnozoite). 2b and 3b Growth and multiplication of hypnozoites. Sites of drug action are as follow. A Drugs used to treat the acute attack (also called ‘blood schizonticidal agents’ or ‘drugs for suppressive or clinical cure’). B Drugs that affect the exoerythrocytic hypnozoites and result in a ‘radical’ cure of P. vivax and P. ovale. C Drugs that block the link between the exoerythrocytic stage and the erythrocytic stage; they are used for chemoprophylaxis (also termed causal prophylactics) and prevent the development of malarial attacks. D Drugs that prevent transmission and thus prevent increase of the human reservoir of the disease.
Malaria 
 The cycle in the mosquito involves fertilisation of the female gametocyte by the male gametocyte, with the formation of a zygote, which develops into an oocyst (sporocyst). A further stage of division and multiplication takes place, leading to rupture of the sporocyst with release of sporozoites, which then migrate to the mosquito’s salivary glands and a few enter the human host with the mosquito’s bite.
The cycle in the mosquito involves fertilisation of the female gametocyte by the male gametocyte, with the formation of a zygote, which develops into an oocyst (sporocyst). A further stage of division and multiplication takes place, leading to rupture of the sporocyst with release of sporozoites, which then migrate to the mosquito’s salivary glands and a few enter the human host with the mosquito’s bite.
The sporozoites are thus inoculated into the bloodstream. Within 30 min, they disappear from the blood and enter the parenchymal cells of the liver where, during the next 10–14 days, they undergo a pre-erythrocytic stage of development and multiplication. At the end of this stage, the parasitised liver cells rupture, and a host of fresh merozoites are released. These bind to and enter the red cells of the blood and form motile intracellular parasites termed trophozoites. The development and multiplication of the plasmodia within these cells constitutes the erythrocytic stage. During maturation within the red cell, the parasite remodels the host cell, inserting parasite proteins and phospholipids into the red cell membrane. The host’s haemoglobin is transported to the parasite’s food vacuole, where it is digested providing a source of amino acids. Free haem, which would be toxic to the plasmodium, is rendered harmless by polymerisation to haemozoin. Some antimalarial drugs act by inhibiting the haem polymerase enzyme responsible for this step (see below).
Following mitotic replication of its nucleus, the parasite in the red cell is termed a schizont, and its rapid growth and division, schizogony. Another phase of multiplication results in the production of further merozoites, which are released when the red cell ruptures. These merozoites then bind to and enter fresh red cells, and the erythrocytic cycle begins again. In certain forms of malaria, some sporozoites entering the liver cells form hypnozoites, or ‘sleeping’ forms of the parasite, which can be reactivated months or years later to continue an exoerythrocytic cycle of multiplication.
Malaria parasites can multiply in the body at a phenomenal rate—a single parasite of P. vivax can give rise to 250 million merozoites in 14 days. To appreciate the action required of an antimalarial drug, note that destruction of 94% of the parasites every 48 h will serve only to maintain equilibrium and will not further reduce their number or their propensity for proliferation. Some merozoites, on entering red cells, differentiate into male and female forms of the parasite, called gametocytes. These can complete their cycle only when taken up by the mosquito, when it sucks the blood of the infected host.
The periodic episodes of fever that characterise malaria result from the synchronised rupture of red cells with release of merozoites and cell debris. The rise in temperature is associated with a rise in the concentration of tumour necrosis factor-α in the plasma. Relapses of malaria are likely to occur with those forms of malaria that have an exoerythrocytic cycle, because the dormant hypnozoite form in the liver may emerge after an interval of weeks or months to start the infection again.
The characteristic presentations of the different forms of human malaria are as follows (see Fig. 53.1 for details):
Individuals living in areas where malaria is endemic may acquire a natural immunity, but this may be lost if the individual is absent from the area for more than 6 months.
The best way to deal with malaria is to prevent mosquito bites and travellers to infected areas are advised to wear clothes that cover much of the skin and use insect repellents in living, and especially sleeping, areas, because mosquitoes tend to bite between dusk and dawn. Bed nets sprayed with insecticides such as permethrin can be very effective.
Antimalarial therapy and the parasite life cycle
Drugs used in the treatment of malaria may have several sites of action:
Antimalarial Drugs
Some drugs can be used prophylactically to prevent malaria (see Table 53.2), while others are directed towards treating acute attacks. In general, antimalarial drugs are classified in terms of the action against the different stages of the life cycle of the parasite (Fig. 53.1).
Table 53.2 Summary of drugs used for treatment and chemoprophylaxis of malariaa
| Infections | Typical drug choices for acute attacks | Typical drug choices for chemoprophylaxis |
|---|---|---|
| Infection with chloroquine-resistant Plasmodium falciparum or with unknown or mixed organisms | Oral quinine plus: proguanil + atovoquone;b or artemether + lumefantrinec | Short term (weeks): atovoquone + proguanil or doxycycline Long term (months/years): chloroquine + proguanil or atovoquone + proguanil |
a It must be appreciated that this is only a summary, not a definitive guide to prescription, as the recommended drug combinations vary depending on the patient, the area visited, the overall risk of infection, the presence of resistant forms of the disease and so on. This information is based on current UK recommendations (source: British National Formulary 2008).
b Malarone is a proprietary combination of atovoquone and proguanil hydrochloride.
c Riamet is a proprietary combination of artemether and lumefantrine.
The use of drugs for the treatment of malaria has changed considerably during the last half-century mainly because resistance developed to chloroquine and other successful early drug combinations. Today monotherapy has been abandoned in favour of artemisinin-based combination therapy (ACT; see Table 53.3). Only antimalarial drugs in common use are described in this chapter. For a brief summary of currently recommended treatment regimens, see the Antimalarial drugs box and Table 53.1. Newton & White (1999) and Baird (2005) give a more detailed coverage of the treatment of malaria around the world.
Drugs used to treat the acute attack
Blood schizonticidal agents (Fig. 53.1, site A) are used to treat the acute attack but also produce a ‘suppressive’ or ‘clinical’ cure. They act on the erythrocytic forms of the plasmodium. In the case of P. falciparum or P. malariae, which have no exoerythrocytic stage, these drugs effect a cure; however, with P. vivax or P. ovale, the drugs suppress the actual attack but exoerythrocytic forms can re-emerge later to cause relapses.
Combinations of these agents are frequently used. Some antibiotics, such as the tetracycline doxycycline and clindamycin (see Ch. 50), have proved useful when combined with the above agents. They have an antiparasite effect in their own right but also control other concomitant infections.
Drugs that effect a radical cure
Tissue schizonticidal agents effect a ‘radical’ cure by eradicating P. vivax and P. ovale parasites in the liver (Fig. 53.1, site B). Only the 8-aminoquinolines (e.g. primaquine and tafenoquine) have this action. These drugs also destroy gametocytes and thus reduce the spread of infection.
Drugs used for chemoprophylaxis
Drugs used for chemoprophylaxis (also known as causal prophylactic drugs: see Table 53.2) block the link between the exoerythrocytic stage and the erythrocytic stage, and thus prevent the development of malarial attacks. True causal prophylaxis—the prevention of infection by the killing of the sporozoites on entry into the host—is not feasible with present drugs, although it may be feasible in the future with vaccines. Clinical attacks can be prevented by chemoprophylactic drugs that kill the parasites when they emerge from the liver after the pre-erythrocytic stage (Fig. 53.1, site C). The drugs used for this purpose are mainly artemisinin derivatives, chloroquine, lumefantrine, mefloquine, proguanil, pyrimethamine, dapsone and doxycycline. They are often used in combinations.
Chemoprophylactic agents are given to individuals who intend travelling to an area where malaria is endemic. Administration should start at least 1 week before entering the area and should be continued throughout the stay and for at least a month afterwards. No chemoprophylactic regimen is 100% effective, and unwanted effects may occur. A further problem is the complexity of the regimens, which require different drugs to be taken at different times, and the fact that different agents may be required for different travel destinations. For a brief summary of currently recommended regimens of chemoprophylaxis, see Table 53.2.
Drugs used to prevent transmission
Some drugs (e.g. primaquine, proguanil and pyrimethamine) can also destroy gametocytes (Fig. 53.1, site D), preventing transmission by the mosquito and thus diminishing the human reservoir of the disease, although they are rarely used for this action alone.
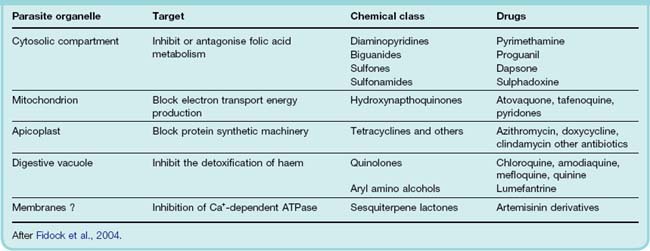
Table 53.3 summarises what is known about the molecular targets of these drugs and Figure 53.2 shows chemical structures of some significant drugs.
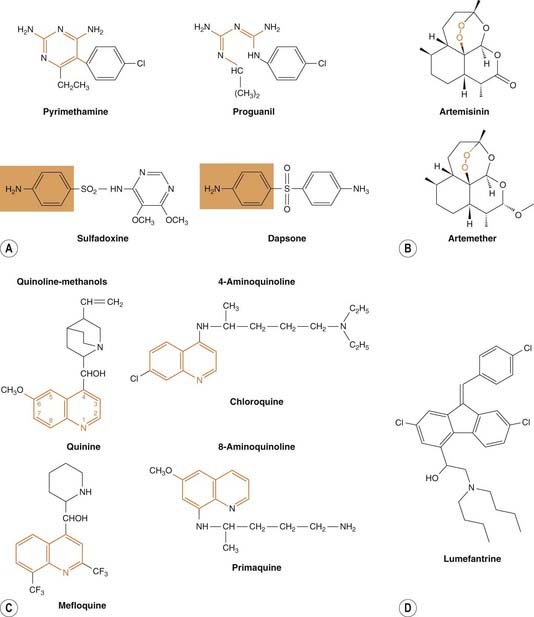
Fig. 53.2 Structures of some significant antimalarial drugs.
[A] Drugs that act on the folic acid pathway of the plasmodia. Folate antagonists (pyrimethamine, proguanil) inhibit dihydrofolate reductase; the relationship between these drugs and the pteridine moiety is shown in orange. Sulfones (e.g. dapsone) and sulfonamides (e.g. sulfadoxine) compete with p-aminobenzoic acid for dihydropteroate synthetase (relationship shown in orange box; see also Ch. 49). [B] Artemisinin and a derivative artemether. Note the endoperoxide bridge structure (in orange box) that is crucial to their action. [C] Some quinolone antimalarials. The quinoline moiety is shown in orange. [D] The aryl amino alcohol lumefantrine.
Chloroquine
The 4-aminoquinoline chloroquine dates from the 1940s but is still widely used as a blood schizonticidal agent (Fig. 53.1, site A), effective against the erythrocytic forms of all four plasmodial species (where resistance is not an issue), but it does not have any effect on sporozoites, hypnozoites or gametocytes. It is uncharged at neutral pH and can therefore diffuse freely into the parasite lysosome. At the acid pH of the lysosome, it is converted to a protonated, membrane-impermeable form and is ‘trapped’ inside the parasite. Its chief antimalarial action derives from an inhibition of haem polymerase, the enzyme that polymerises toxic free haem to haemozoin. This poisons the parasite and prevents it from utilising the amino acids from haemoglobin proteolysis. Chloroquine is also used as a disease-modifying antirheumatoid drug (Ch. 26) and also has some quinidine-like actions on the heart. The clinical use of chloroquine is summarised in Tables 53.2 and the Antimalarial drugs box.
Resistance
P. falciparum is now resistant to chloroquine in most parts of the world. Resistance appears to result from enhanced efflux of the drug from parasitic vesicles as a result of mutations in plasmodia transporter genes (Baird, 2005). Resistance of P. vivax to chloroquine is also a growing problem in many parts of the world.
Administration and pharmacokinetic aspects
Chloroquine is generally administered orally, but severe falciparum malaria may be treated by frequent intramuscular or subcutaneous injection of small doses, or by slow continuous intravenous infusion. Following oral dosing, it is completely absorbed, extensively distributed throughout the tissues and concentrated in parasitised red cells. The free base form of the drug is trapped in the acidic environment in the food vacuole of the malaria parasites where it disrupts the haemoglobin digestion pathway (Ch. 8). Release from tissues and infected erythrocytes is slow. The drug is metabolised in the liver and excreted in the urine, 70% as unchanged drug and 30% as metabolites. Elimination is slow, the major phase having a half-life of 50 h, and a residue persists for weeks or months.
Unwanted effects
Chloroquine has few adverse effects when given for chemoprophylaxis. However, unwanted effects, including nausea and vomiting, dizziness and blurring of vision, headache and urticarial symptoms, can occur when larger doses are administered to treat acute attacks of malaria. Large doses have also sometimes resulted in retinopathies and hearing loss. Bolus intravenous injections of chloroquine may cause hypotension and, if high doses are used, fatal dysrhythmias. Chloroquine is considered to be safe for use by pregnant women.
Amodiaquine has very similar action to chloroquine. It was withdrawn several years ago because of the risk of agranulocytosis, but has now been reintroduced in several areas of the world where chloroquine resistance is endemic.
Quinine
The methanol quinolone, quinine, is derived from cinchona bark. It has been used for the treatment of ‘fevers’ since the 16th century, when Jesuit missionaries bought the bark to Europe from Peru. It is a blood schizonticidal drug effective against the erythrocytic forms of all four species of plasmodium (Fig. 53.1, site A), but it has no effect on exoerythrocytic forms or on the gametocytes of P. falciparum. Its mechanism of action is the same as that of chloroquine, but quinine is not so extensively concentrated in the plasmodium as chloroquine, so other mechanisms could also be involved. With the emergence and spread of chloroquine resistance, quinine is now the main chemotherapeutic agent for P. falciparum. Pharmacological actions on host tissue include a depressant action on the heart, a mild oxytocic effect on the uterus in pregnancy, a slight blocking action on the neuromuscular junction and a weak antipyretic effect. The clinical uses of quinine are given in Table 53.2 and in the Antimalarial Drugs box.
Some degree of resistance to quinine is developing because of increased expression of plasmodial drug efflux transporters.
Pharmacokinetic aspects
Quinine is well absorbed and is usually administered orally as a 7-day course, but it can also be given by slow intravenous infusion for severe P. falciparum infections and in patients who are vomiting. A loading dose may be required, but bolus intravenous administration is contraindicated because of the risk of cardiac dysrhythmias. The half-life of the drug is 10 h; it is metabolised in the liver and the metabolites are excreted in the urine within about 24 h.
Unwanted effects
Quinine has a bitter taste, and oral compliance is often poor.3 It is irritant to the gastric mucosa and can cause nausea and vomiting. ‘Cinchonism’—characterised by nausea, dizziness, tinnitus, headache and blurring of vision—is likely to occur if the plasma concentration exceeds 30–60 µmol/l. Excessive plasma levels may also cause hypotension, cardiac dysrhythmias and severe CNS disturbances such as delirium and coma.
Other, infrequent, unwanted reactions that have been reported are bone marrow depression (mainly thrombocytopenia) and hypersensitivity reactions. Quinine can stimulate insulin release. Patients with marked falciparum parasitaemia can have low blood sugar for this reason and also because of glucose consumption by the parasite. This can make a differential diagnosis between a coma caused by cerebral malaria and hypoglycaemia difficult. A rare result of treating malaria with quinine, or of erratic and inappropriate use of quinine, is Blackwater fever, a severe and often fatal condition in which acute haemolytic anaemia is associated with renal failure.
Mefloquine
Mefloquine (Fig. 53.2) is a blood schizonticidal compound active against P. falciparum and P. vivax (Fig. 53.1, site A); however, it has no effect on hepatic forms of the parasites, so treatment of P. vivax infections should be followed by a course of primaquine (see below) to eradicate the hypnozoites. Mefloquine acts in the same way as quinine, and is frequently combined with pyrimethamine.
Resistance has occurred in P. falciparum in some areas—particularly in South-east Asia—and is thought to be caused, as with quinine, by increased expression in the parasite of drug efflux transporters. The clinical use of mefloquine is given in Table 53.2 and the Antimalarial drugs box.
Pharmacokinetic aspects and unwanted effects
Mefloquine is given orally and is rapidly absorbed. It has a slow onset of action and a very long plasma half-life (up to 30 days), which may be the result of enterohepatic cycling or tissue storage.
When mefloquine is used for treatment of the acute attack, about 50% of subjects complain of gastrointestinal disturbances. Transient CNS side effects—giddiness, confusion, dysphoria and insomnia—can occur, and there have been a few reports of aberrant atrioventricular conduction and serious, but rare, skin diseases. Rarely, mefloquine may provoke severe neuropsychiatric reactions. Mefloquine is contraindicated in pregnant women or in those liable to become pregnant within 3 months of stopping the drug, because of its long half-life and uncertainty about its teratogenic potential. When used for chemoprophylaxis, the unwanted actions are usually milder, but the drug should not be used in this way unless there is a high risk of acquiring chloroquine-resistant malaria.
Lumefantrine
This aryl amino alcohol drug is related to an older compound, halofantrine, which is now seldom used. Lumefantrine is never used alone but in combination with artemether. Its mode of action is probably to prevent parasite detoxification of haem. The pharmacokinetics of the combination is complex and the reader is referred to Ezzet et al., 1998, for more details. Unwanted effects of the combination may include gastrointestinal and CNS symptoms.
Drugs Affecting Folate Metabolism
Sulfonamides and sulfones, used as antibacterial drugs (see Ch. 50) inhibit the synthesis of folate by competing with p-aminobenzoic acid. Pyrimethamine and proguanil inhibit dihydrofolate reductase, which prevents the utilisation of folate in DNA synthesis. Used together, they block the folate pathway at different points, and thus act synergistically.
The main sulfonamide used in malaria treatment is sulfadoxine, and the only sulfone used is dapsone (see Fig. 53.3). Details of these drugs are given in Chapter 50. The sulfonamides and sulfones are active against the erythrocytic forms of P. falciparum but are less active against those of P. vivax; they have no activity against the sporozoite or hypnozoite forms of the plasmodia. Pyrimethamine–sulfadoxine has been extensively used for chloroquine-resistant malaria, but resistance to this combination has developed in many areas.
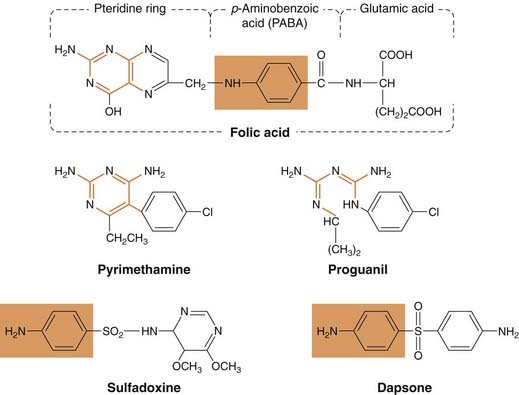
Fig. 53.3 Structures of some antimalarial drugs that act on the folic acid pathway of the plasmodia.
Folate antagonists (pyrimethamine, proguanil) inhibit dihydrofolate reductase; the relationship between these drugs and the pteridine moiety is shown in orange. Sulfones (e.g. dapsone) and sulfonamides (e.g. sulfadoxine) compete with p-aminobenzoic acid for dihydropteroate synthetase (relationship shown in orange box).
Pyrimethamine (see Fig. 53.3) is similar in structure to the antibacterial drug trimethoprim (see Ch. 50). Proguanil has a slightly different structure (see Fig. 53.3) but its metabolite can assume a similar configuration. Both drugs have a greater affinity for the plasmodial enzyme than for the human enzyme. They have a slow action against the erythrocytic forms of the parasite (Fig. 53.1, site A), and proguanil is believed to have an additional effect on the initial hepatic stage (1a to 3a in Fig. 53.1) but not on the hypnozoites of P. vivax (Fig. 53.1, site B). Pyrimethamine is used only in combination with either a sulfone or a sulfonamide.
Pharmacokinetic aspects
Both pyrimethamine and proguanil are given orally and are well, although slowly, absorbed. Pyrimethamine has a plasma half-life of 4 days, and effective ‘suppressive’ plasma concentrations may last for 14 days; it is taken once a week. The half-life of proguanil is 16 h. It is a prodrug and is metabolised in the liver to its active form, cycloguanil, which is excreted mainly in the urine. It must be taken daily.
Unwanted effects
These drugs have few untoward effects in therapeutic doses. Larger doses of the pyrimethamine–dapsone combination can cause serious reactions such as haemolytic anaemia, agranulocytosis and lung inflammation. The pyrimethamine–sulfadoxine combination can cause serious skin reactions, blood dyscrasias and allergic alveolitis; it is no longer recommended for chemoprophylaxis. In high doses, pyrimethamine may inhibit mammalian dihydrofolate reductase and cause a megaloblastic anaemia (see Ch. 25) and folic acid supplements should be given if this drug is used during pregnancy. Resistance to antifolate drugs arises from single-point mutations in the genes encoding parasite dihydrofolate reductase.
Primaquine
Primaquine is an 8-aminoquinoline drug, which is (almost uniquely among clinically available antimalarial drugs) active against liver hypnozoites (see Fig. 53.2). Etaquine and tafenoquine are more active and slowly metabolised analogues of primaquine. These drugs can effect a radical cure of P. vivax and P. ovale malaria in which the parasites have a dormant stage in the liver. Primaquine does not affect sporozoites and has little if any action against the erythrocytic stage of the parasite. However, it has a gametocidal action and is the most effective antimalarial drug for preventing transmission of the disease in all four species of plasmodia. It is almost invariably used in combination with another drug, usually chloroquine. Resistance to primaquine is rare, although evidence of a decreased sensitivity of some P. vivax strains has been reported. The pharmacology of primaquine and similar drugs has been reviewed by Shanks et al. (2001).
Pharmacokinetic aspects
Primaquine is given orally and is well absorbed. Its metabolism is rapid, and very little drug is present in the body after 10–12 h. The half-life is 3–6 h. Tafenoquine is metabolised much more slowly and therefore has the advantage that it can be given on a weekly basis.
Unwanted effects
Primaquine has few unwanted effects in most patients when used in normal therapeutic dosage. Dose-related gastrointestinal symptoms can occur, and large doses may cause methaemoglobinaemia with cyanosis.
Primaquine can cause haemolysis in individuals with the X chromosome-linked genetic metabolic condition, glucose 6-phosphate dehydrogenase deficiency, in red cells (Ch. 11). When this deficiency is present, the red cells are not able to regenerate NADPH, which is depleted by the oxidant metabolic derivatives of primaquine. As a consequence, the metabolic functions of the red cells are impaired and haemolysis occurs. The deficiency of the enzyme occurs in up to 15% of black males and is also fairly common in some other ethnic groups. Glucose 6-phosphate dehydrogenase activity should be estimated before giving primaquine.
Artemesinin and Related Compounds
These sesquiterpene lactones are derived from the herb qing hao, a traditional Chinese remedy for malaria. The scientific name, conferred on the herb by Linnaeus, is Artemisia.4 Artemisinin, a poorly soluble chemical extract from Artemisia, is a fast-acting blood schizonticide effective in treating the acute attack of malaria (including chloroquine-resistant and cerebral malaria). Artesunate, a water-soluble derivative, and the synthetic analogues artemether and artether, have higher activity and are better absorbed. The compounds are concentrated in parasitised red cells. The mechanism of action is probably through inhibition of a parasite Ca+-dependent ATPase (Eckstein-Ludwig et al., 2003) and it is likely that the ‘endoperoxide bridge’ of this drug (see Fig. 53.2) has to be ‘activated’ in the presence of intracellular iron before it can exert its effects. These drugs are without effect on liver hypnozoites. Artemisinin can be given orally, intramuscularly or by suppository, artemether orally or intramuscularly, and artesunate intramuscularly or intravenously. They are rapidly absorbed and widely distributed, and are converted in the liver to the active metabolite dihydroartemisinin. The half-life of artemisinin is about 4 h, of artesunate 45 min and of artemether 4–11 h.
There have been few unwanted effects reported to date. Transient heart block, decrease in blood neutrophil count and brief episodes of fever have been reported. In animal studies, artemisinin causes an unusual injury to some brain stem nuclei, particularly those involved in auditory function; however, there have been no reported incidences of neurotoxicity in humans. So far, resistance has not been a problem, but recent reports suggest that it is developing in some countries.
In rodent studies, artemisinin potentiated the effects of mefloquine, primaquine and tetracycline, was additive with chloroquine and antagonised the sulfonamides and the folate antagonists. For this reason, artemisinin derivatives are frequently used in combination with other antimalarial drugs; for example, artemether is often given in combination with lumefantrine.
In randomised trials, the qinghaosu compounds have cured attacks of malaria, including cerebral malaria, more rapidly and with fewer unwanted effects than other antimalarial agents. Artemisinin and derivatives are effective against multidrug-resistant P. falciparum in sub-Saharan Africa and, combined with mefloquine, against multidrug-resistant P. falciparum in South-east Asia.
Atavoquone
Atavoquone is a hydroxynaphthoquinone drug used prophylactically to prevent malaria, and to treat cases resistant to other drugs. It acts primarily to inhibit the parasite’s mitochondrial electron transport chain, possibly by mimicking the natural substrate ubiquinone. Atavoquone is usually used in combination with the antifolate drug proguanil, because they act synergistically. The mechanism underlying this synergism is not known, but it is specific for this particular pair of drugs, because other antifolate drugs or electron transport inhibitors have no such synergistic effect. When combined with proguanil, atavoquone is highly effective and well tolerated. Few unwanted effects of such combination treatment have been reported, but abdominal pain, nausea and vomiting can occur. Pregnant or breastfeeding women should not take atavoquone. Resistance to atavoquone alone is rapid and results from a single point mutation in the gene for cytochrome b. Resistance to combined treatment with atavoquone and proguanil is less common.
Potential New Antimalarial Drugs
Several new drugs are currently under test for antimalarial activity, with positive results in animals and in preliminary trials in humans. One of these, pyronaridine, has shown encouraging results. It is active against P. falciparum and P. vivax, and is also active in chloroquine-resistant P. falciparum. It is effective orally and has low toxicity. The mechanism of action is unknown. Other novel agents are reviewed by Lanteri et al. (2007).
In 2002, the results from the malaria genome sequencing project were published and it is highly likely that this information will eventually yield new candidate drugs with novel actions. A group of cysteine proteases used by the parasite to digest haem seems to be one attractive target that is currently receiving attention.
Given the extraordinary lifestyle of the malarial parasite with its many forms both inside and outside cells, the challenges facing vaccine developers are enormous. Nevertheless, there is cause for optimism, and a large-scale trial of the first candidate vaccine began in 2009. Discussion of this topic is beyond this chapter but further details may be found in Greenwood et al. (2008).
Antimalarial drugs
Amoebiasis and Amoebicidal Drugs
The main organism in this group to concern us here is Entamoeba histolytica, the causative agent of amoebiasis, which may manifest as a severe colitis (dysentery) and, sometimes, liver abscesses.
The infection is encountered around the world, but more often in warmer climates. Approximately 500 million people are thought to harbour the disease, with 40 000–100 000 deaths occurring each year as a result (Stanley, 2003). It is considered to be the second leading cause of death from parasitic diseases worldwide.
The organism has a simple life cycle, and humans are the chief hosts. Infection, generally spread by poor hygiene, follows the ingestion of the mature cysts in water or food that is contaminated with human faeces. The infectious cysts pass into the colon, where they develop into trophozoites. These motile organisms adhere to colonic epithelial cells, utilising a galactose-containing lectin on the host cell membrane. Here, the trophozoites feed, multiply, encyst and eventually pass out in the faeces, thus completing their life cycle. Some individuals are symptomless ‘carriers’ and harbour the parasite without developing overt disease, but cysts are present in their faeces and they can infect other individuals. The cysts can survive outside the body for at least a week in a moist and cool environment.
The trophozoite lyses the colonic mucosal cells (hence ‘histolytica’) using amoebapores (peptides that form pores in cell membranes) and proteases or by inducing host cell apoptosis. The organism then invades the submucosa, where it secretes factors that modify the host response, which would otherwise prove lethal to the parasite. It is this process that produces the characteristic bloody diarrhoea and abdominal pain, although in many subjects a chronic intestinal infection may be present in the absence of dysentery. In some subjects, an amoebic granuloma (amoeboma) may be present in the intestinal wall. The trophozoites may also migrate through the damaged intestinal tissue into the portal blood and hence the liver, giving rise to the most common extraintestinal symptom of the disease—amoebic liver abscesses.
The use of drugs to treat this condition (see Drugs used in amoebiasis box) depends largely on the site and type of infection. The drugs of choice for the various forms of amoebiasis are:
These agents are often used in combination.
Metronidazole
Metronidazole kills the trophozoites of E. histolytica but has no effect on the cysts. It is the drug of choice for invasive amoebiasis of the intestine or the liver, but it is less effective against organisms in the lumen of the gut. Metronidazole is activated by anaerobic organisms to a compound that damages parasite DNA, leading to parasite apoptosis.
Metronidazole is usually given orally and is rapidly and completely absorbed. Rectal and intravenous preparations are also available. It is distributed rapidly throughout the tissues, reaching high concentrations in the body fluids, including the cerebrospinal fluid. Some is metabolised, but most is excreted in urine.
Unwanted effects are mild. The drug has a metallic, bitter taste in the mouth but causes few unwanted effects in therapeutic doses. Minor gastrointestinal disturbances have been reported, as have central nervous system (CNS) symptoms (dizziness, headache, sensory neuropathies). Metronidazole causes a disulfiram-like reaction to alcohol (see Ch. 48), which should be strictly avoided. Metronidazole should not be used in pregnancy.
Tinidazole is similar to metronidazole in its mechanism of action and unwanted effects, but is eliminated more slowly, having a half-life of 12–14 h.
Diloxanide
Diloxanide or, more commonly an insoluble ester, diloxanide furoate, are the drugs of choice for the asymptomatic infected patient, and are often given as a follow-up after the disease has been reversed with metronidazole. Both drugs have a direct amoebicidal action, affecting the parasites before encystment. Diloxanide furoate is given orally, and acts without being absorbed. Unwanted gastrointestinal or other effects may be seen but it has an excellent safety profile.
Other drugs that are sometimes used include the antibiotic paromomycin.
Drugs used in amoebiasis
Amoebiasis is caused by infection with E. histolytica, which causes dysentery and liver abscesses. The organism may be present in motile invasive form or as a cyst. The main drugs are:
Trypanosomiasis and TRYPANOCIDAL Drugs
Trypanosomes belong to the group of pathogenic flagellate protozoa. The three main species that cause disease in humans are Trypanosoma gambiense and Trypanosoma rhodesiense, which cause sleeping sickness in Africa, and Trypanosoma cruzi, which causes Chagas’ disease in South America. About 50–60 million people are thought by the WHO to be at risk of contracting sleeping sickness each year. The disease caused by T. rhodesiense is the more aggressive form. Civil unrest, famine and AIDS encourage the spread of the disease by reducing the chances of distributing medication or because patients are immunocompromised, but despite this, improved surveillance has resulted in a recent reduction in the total number of new cases reported. Related trypanosome infections also pose a major risk to livestock and thus have a secondary impact on human health and well-being.
The vector is the tsetse fly. In both types of disease, there is an initial local lesion at the site of entry, which may (in the case of T. rhodesiense) develop into a painful chancre (ulcer or sore). This is followed by bouts of parasitaemia and fever as the parasite enters the haemolymphatic system. The parasites and the toxins they release during the second phase of the disease cause organ damage. This manifests as ‘sleeping sickness’ when parasites reach the CNS causing somnolence and progressive neurological breakdown, or ‘Chagas’ disease’ when parasites damage the heart, muscles and sometimes liver, spleen, bone and intestine. Left untreated, such infections are fatal.
The main drugs used for African sleeping sickness are suramin, with pentamidine as an alternative, in the haemolymphatic stage of the disease, and the arsenical melarsoprol for the late stage with CNS involvement and eflornithine (see Burchmore et al., 2002; Burri & Brun, 2003). All are toxic. Nifurtimox, eflornithine and benznidazole are used in Chagas’ disease: however, there is no totally effective treatment for this form of trypanosomiasis.
Suramin
Suramin was introduced into the therapy of trypanosomiasis in 1920. The drug binds firmly to host plasma proteins, and the complex enters the trypanosome by endocytosis from where it is liberated by lysosomal proteases. It inhibits key parasite enzymes inducing gradual destruction of organelles, such that the organisms are cleared from the circulation after a short interval.
The drug is given by slow intravenous injection. The blood concentration drops rapidly during the first few hours and then more slowly over the succeeding days. A residual concentration remains for 3–4 months. Suramin tends to accumulate in mononuclear phagocytes, and in the cells of the proximal tubule in the kidney.
Unwanted effects are common. Suramin is relatively toxic, particularly in a malnourished patient, the main organ affected being the kidney. Many other slowly developing adverse effects have been reported including optic atrophy, adrenal insufficiency, skin rashes, haemolytic anaemia and agranulocytosis. A small proportion of individuals have an immediate idiosyncratic reaction to suramin injection that may include nausea, vomiting, shock, seizures and loss of consciousness.
Pentamidine
Pentamidine has a direct trypanocidal action in vitro. It is rapidly taken up in the parasites by a high-affinity energy-dependent carrier and is thought to interact with their DNA. The drug is administered intravenously or by deep intramuscular injection, usually daily for 10–15 days. After absorption from the injection site, it binds strongly to tissues (especially the kidney) and is eliminated slowly, only 50% of a dose being excreted over 5 days. Fairly high concentrations of the drug persist in the kidney, the liver and the spleen for several months, but it does not penetrate the blood–brain barrier. It is also active in Pneumocystis pneumonia (Ch. 50). Its usefulness is limited by its unwanted effects—an immediate decrease in blood pressure, with tachycardia, breathlessness and vomiting, and later serious toxicity, such as kidney damage, hepatic impairment, blood dyscrasias and hypoglycaemia.
Melarprosol
This is an organic arsenical compound that is used mainly when the CNS is involved. It is given intravenously and enters the CNS in high concentrations where it is able to kill the parasite. It is a highly toxic drug that produces many unwanted effects including encephalopathy and, sometimes, immediate fatality. As such, it is only administered under strict supervision.
Eflornithine
A relatively new drug, eflornithine inhibits the parasite ornithine decarboxylase enzyme. It shows good activity against T. gambiense and is used as a back-up for melarsoprol, although unfortunately it has limited activity against T. rhodesiense. Side effects are common and may be severe, but are readily reversed when treatment is discontinued.
There is an urgent need for new agents to treat trypanasome infections, partly because of the toxicity of existing drugs and partly because of developing drug resistance. The recent publication of the complete genome sequence of some trypanasome species has led to optimism that new agents may be forthcoming in the medium term. The interested reader is referred to Gehrig & Efferth, 2008; Kennedy, 2008; and Myler, 2008 for recent accounts of these opportunities.
Other Protozoal Infections and Drugs Used To Treat Them
Leishmaniasis
Leishmania organisms are flagellate protozoa that cause disease (sometimes fatal). Some 350 million people in 90 countries are at risk, mainly in tropical and subtropical regions. The disease afflicts about 12 million people: there are about 2 million new cases each year of which some 60 000 die. With increasing international travel, leishmaniasis is being imported into new areas and opportunistic infections are now being reported (particularly in AIDS patients).
The vector in this case is the sandfly, and the parasite exists in two forms, a flagellated form (promastigote) found in the gut of the infected insect, and a non-flagellated intracellular form (amastigote) that occurs in the infected mammalian host, where it is harboured by mononuclear phagocytes. Within this cell, the parasites thrive in modified phagolysosomes and protect themselves from the usual intracellular killing mechanisms by modifying the macrophage’s microbiocidal systems, apparently by deploying a lipophosphoglycan on their surface (Handman & Bullen, 2002). The amastigotes multiply, and eventually the infected cell releases a new crop of parasites into the haemolymphatic system, where they can infect further macrophages and possibly other cells.
The different species of Leishmania occur in different geographical zones and cause different clinical manifestations (see Table 53.1). Typical presentations include:
The main drugs used in visceral leishmaniasis are pentavalent antimony compounds such as sodium stibogluconate, pentamidine (see above) and amphotericin (see Ch. 52), which is sometimes used as a follow-up treatment. Miltefosine, an antitumour drug, which has been used with success to treat the disease, is also used in some countries (not UK), as is meglumine antimoniate.
Sodium stibogluconate is given intramuscularly or by slow intravenous injection in a 10-day course. It is rapidly eliminated in the urine, 70% being excreted within 6 h. More than one course of treatment may be required.
Unwanted effects include anorexia, vomiting, bradycardia and hypotension. Coughing and substernal pain may occur during intravenous infusion. Treatment may also be associated with increased incidence of herpes zoster. The mechanism of action of sodium stibogluconate is not clear, but the drug may increase production of toxic oxygen free radicals in the parasite.
Miltefosine (hexadecylphosphocholine) is also effective in the treatment of both cutaneous and visceral leishmaniasis. The drug may be given orally and is well tolerated. Side effects are mild and include nausea and vomiting. In vitro, the drug induces DNA fragmentation and apoptosis in the parasites (Verma & Dey, 2004).
Other drugs such as antibiotics and antifungals may be given concomitantly with the above agents. They may have some action on the parasite in their own right, but their main utility is to control the spread of secondary infections. Current drug usage and possible future approaches to the treatment of leishmaniasis are discussed by Mishra et al. (2007). The publication of complete leishmania genomes will initiate a major effort to discover novel parasite-specific pathways that could serve as useful drug targets (see Kumari et al., 2008).
Trichomoniasis
The principal Trichomonas organism that produces disease in humans is T. vaginalis. Virulent strains cause inflammation of the vagina and sometimes of the urethra in males. The main drug used in therapy is metronidazole (Ch. 50), although resistance to this drug is on the increase. High doses of tinidazole are also effective, with few side effects.
Giardiasis
Giardia lamblia colonises the upper gastrointestinal tract in its trophozoite form, and the cysts pass out in the faeces. Infection is then spread by ingestion of food or water contaminated with faecal matter containing the cysts. It is encountered worldwide, and epidemics caused by bad sanitation are not uncommon. Metronidazole is the drug of choice, and treatment is usually very effective. Tinidazole or mepacrine may be used as an alternative.
Toxoplasmosis
Toxoplasma organisms belong to the group of pathogenic Sporozoa. The cat is the definitive host of Toxoplasma gondii (i.e. it is the only host in which the sexual cycle can occur), and expels the infectious cysts in its faeces; humans can inadvertently become intermediate hosts, harbouring the asexual form of the parasite. Ingested oocysts develop into sporozoites, then to trophozoites, and finally encyst in the tissues. In most individuals, the disease is asymptomatic or self-limiting, although intrauterine infections can severely damage the developing fetus and it may cause fatal generalised infection in immunosuppressed patients or those with AIDS, in whom toxoplasmic encephalitis may occur. In humans, T. gondii infects numerous cell types and has a highly virulent replicative stage.
The treatment of choice is pyrimethamine–sulfadiazine (to be avoided in pregnant patients); trimethoprim–sulfamethoxazole (co-trimoxazole, see Ch. 50) or combinations of pyrimethamine with clindamycin, clarithromycin or azithromycin (see Ch. 50) have shown promise.
Pneumocystis
First recognised in 1909, Pneumocystis carinii (now known as P. jirovecii; see also Ch. 52) was presumed to belong to the protozoa, but recent studies have shown that it shares structural features with both protozoa and fungi, leaving its precise classification uncertain. Previously considered to be a widely distributed but largely innocuous microorganism, it is now recognised as a cause of opportunistic infections in immunocompromised patients. It is common in AIDS, where P. carinii pneumonia is often the presenting symptom as well as a leading cause of death.
High-dose co-trimoxazole (Ch. 49) is the drug of choice in serious cases, with parenteral pentamidine (see above) as an alternative. Treatment of milder forms of the disease (or prophylaxis) can be effected with atovaquone, trimethoprim–dapsone, or clindamycin–primaquine combinations.
Future Developments
This field is a huge challenge, with each protozoa species posing its own distinct problems to the would-be designer of new antiprotozoal drugs. Where appropriate in this chapter, we have indicated possible future avenues for research and development, but the interested reader is referred to the reading list and Web sites listed below for further information.
It is abundantly clear that the diseases caused by the protozoa constitute a major global challenge, but the problems of provision and distribution of new drugs are daunting. Managing the costs of research and development in this area is complex. Transnational initiatives (e.g. Medicines for Malaria Venture and Institute for OneWorld Health) are now major players in the development of new medicines for protozoal diseases. But it is not simply a lack of new drugs that is the problem: for economic reasons, the countries and populations most affected often lack an efficient infrastructure for the distribution and safe administration of the drugs that we already possess. Cultural attitudes, civil wars, famine, the circulation of counterfeit or defective drugs, drought and natural disasters also exacerbate this problem.
References and Further Reading
Brenier-Pinchart M.-P., Pelloux H., Derouich-Guergour D., et al. Chemokines in host–parasite interactions. Trends Parasitol.. 2001;17:292-296. (Good review of role of immune system)
Ashley E.A., White N.J. Artemisinin based combinations. Curr. Opin. Infect. Dis.. 2005;18:531-536. (This review details the results of successful clinical trials of artemisinin combinations in South-east Asia)
Baird J.K. Effectiveness of antimalarial drugs. N. Engl. J. Med.. 2005;352:1565-1577. (An excellent overview covering many aspects of drug therapy, drug resistance and the socioeconomic factors affecting the treatment of this disease—thoroughly recommended)
Eckstein-Ludwig U., Webb R.J., Van Goethem I.D., et al. Artemisinins target the SERCA of Plasmodium falciparum. Nature. 2003;424:957-961. (Research paper that elucidates the site of action of artemisinin drugs)
Ezzet F., Mull R., Karbwang J. Population pharmacokinetics and therapeutic response of CGP 56697 (artemether + benflumetol) in malaria patients. Br. J. Clin. Pharmacol.. 1998;46:553-561. (Deals with the pharmacokinetics of this increasingly important combination therapy)
Fidock D.A., Rosenthal P.J., Croft S.L., et al. Antimalarial drug discovery: efficacy models for compound screening. Nat. Rev. Drug. Discov.. 2004;3:509-520. (Useful review dealing with mechanisms of antimalarial drug action and new concepts for screening future candidates)
Foley M., Tilley L. Quinoline antimalarials: mechanisms of action and resistance. Int. J. Parasitol.. 1997;27:231-240. (Good, short review; useful diagrams)
Greenwood B.M., Fidock D.A., Kyle D.E., et al. Malaria: progress, perils, and prospects for eradication. J. Clin. Invest.. 2008;118:1266-1276. (Good overview of the disease, its current and future treatment)
Lanteri C.A., Johnson J.D., Waters N.C. Recent advances in malaria drug discovery. Recent. Pat. Antiinfect. Drug Discov.. 2007;2:95-114. (This comprehensive review focuses mainly upon chemical leads but also has a good section on drug targets and ways of optimising existing therapies)
Newton P., White N. Malaria: new developments in treatment and prevention. Annu. Rev. Med.. 1999;50:179-192. (Excellent review of drug treatment and management of malaria)
O’Brien C. Beating the malaria parasite at its own game. Lancet. 1997;350:192. (Clear, succinct coverage of mechanisms of action and resistance of current antimalarials and potential new drugs; useful diagram)
Odeh M. The role of tumour necrosis factor-alpha in the pathogenesis of complicated falciparum malaria. Cytokine. 2001;14:11-18.
Shanks G.D., Kain K.C., Keystone J.S. Malaria chemoprophylaxis in the age of drug resistance. II. Drugs that may be available in the future. Clin. Infect. Dis.. 2001;33:381-385. (A useful look ahead to new drugs)
Haque R., Huston C.D., Hughes M., et al. Amebiasis. N. Engl. J. Med.. 2003;348:1565-1573. (Good review; concentrates on the pathogenesis of the disease but has a useful table of drugs and their side effects)
Stanley S.L. Pathophysiology of amoebiasis. Trends. Parasitol.. 2001;17:280-285. (A good account of the human disease that incorporates some results from animal models also)
Stanley S.L. Amoebiasis. Lancet. 2003;361:1025-1034. (Comprehensive and easy-to-read account of this disease, covering all aspects from diagnosis to treatment. Excellent)
Aksoy S., Gibson W.C., Lehane M.J. Interactions between tsetse and trypanosomes with implications for the control of trypanosomiasis. Adv. Parasitol.. 2003;53:1-83. (A very substantial and comprehensive article covering the biology of the tsetse fly, which also discusses alternative methods from controlling the insect population. Less good on drug therapy, but if you are interested in the biology of the insect vector of trypanosomiasis, then this is for you)
Burchmore R.J., Ogbunude P.O., Enanga B., Barrett M.P. Chemotherapy of human African trypanosomiasis. Curr. Pharm. Des.. 2002;8:256-267. (Very good concise article; nice discussion of future therapeutic possibilities)
Burri C., Brun R. Eflornithine for the treatment of human African trypanosomiasis. Parasitol. Res.. 2003;90(Suppl. 1):S49-S52. (The title is self-explanatory)
Denise H., Barrett M.P. Uptake and mode of action of drugs used against sleeping sickness. Biochem. Pharmacol.. 2001;61:1-5. (Good coverage of drug therapy)
Gehrig S., Efferth T. Development of drug resistance in Trypanosoma brucei rhodesiense and Trypanosoma brucei gambiense. Treatment of human African trypanosomiasis with natural products (Review). Int. J. Mol. Med.. 2008;22:411-419. (Good overview of drug therapy including sections on mechanisms of drug resistance)
Keiser J., Stich A., Burri C. New drugs for the treatment of human African trypanosomiasis: research and development. Trends. Parasitol.. 2001;17:42-49. (Excellent review on an increasingly threatening disease)
Kennedy P.G. The continuing problem of human African trypanosomiasis (sleeping sickness). Ann. Neurol.. 2008;64:116-126. (Reviews the disease and also the problems with current drug therapies)
Legros D., Ollivier G., Gastellu-Etchegorry M., et al. Treatment of human African trypanosomiasis—present situation and needs for research and development. Lancet Infect. Dis.. 2002;2:437-440.
Myler P.J. Searching the Tritryp genomes for drug targets. Adv. Exp. Med. Biol.. 2008;625:133-140. (A review of the use of bioinformatics to search the published sequences of some protozoan genomes to identify novel drug targets)
Berman J. Current treatment approaches to leishmaniasis. Curr. Opin. Infect. Dis.. 2003;16:397-401. (Good general review that includes some data on new clinical trials)
Handman E., Bullen D.V.R. Interaction of Leishmania with the host macrophage. Trends. Parasitol.. 2002;18:332-334. (Very good article describing how this parasite colonises macrophages and evades intracellular killing; easy to read)
Jayanarayan K.G., Dey C.S. Microtubules: dynamics, drug interaction and drug resistance in Leishmania. J. Clin. Pharm. Ther.. 2002;27:313-320. (Deals with the action on parasite microtubules of antileishmanial drugs—very specialised)
Kumari S., Kumar A., Samant M., et al. Discovery of novel vaccine candidates and drug targets against visceral leishmaniasis using proteomics and transcriptomics. Curr. Drug. Targets.. 2008;9:938-947. (Reviews the use of sophisticated bioinformatic tools to facilitate the development of new vaccines)
Mishra J., Saxena A., Singh S. Chemotherapy of leishmaniasis: past, present and future. Curr. Med. Chem.. 2007;14:1153-1169. (Self-explanatory title!)
Sacks D., Toben-Trauth N. The immunology of susceptibility and resistance to Leishmania major in mice Nat. Rev. Immunol. 2:2002:845-858 (A lengthy article that explores the host response to Leishmania parasite infection using mouse models of the disease; fascinating and authoritative—but only attempt it if your immunology is up to scratch!)
Verma N.K., Dey C.S. Possible mechanism of miltefosine-mediated death of Leishmania donovani. Antimicrob. Agents Chemother.. 2004;48:3010-3015.
Warren E., George S., You J., Kazanjian P. Advances in the treatment and prophylaxis of Pneumocystis carinii pneumonia. Pharmacotherapy. 1997;17:900-916.
http://malaria.who.int/ (The WHO home page dealing with the global malaria programme contains links to all the major information on the site dealing with malaria—a terrific starting point for further investigation)
http://www.oneworldhealth.org (The Web page of the visionary ‘non-profit pharmaceutical company’, with details of their current programmes dealing with global health issues)
http://www.mmv.org/ (The Web page of the Medicines for Malaria Venture, the first private–public partnership established to bring together funding and expertise from a number of sources to tackle malaria)
1The disease was once considered to arise from marshy land, hence the Latin name ‘mal aria’, meaning bad or poisonous air.
2WHO reported 87 cases of ‘airport malaria’ in 12 countries between 1969 and 1999. ‘Weekend malaria’, which occurs when city dwellers in Africa spend weekends in the countryside, is becoming more of a problem.
3Hence the invention of palatable drinks containing the drug, including, of course, the famous ‘tonic’ drunk together with gin and other beverages.
4Having been used for thousands of years in China as a herbal extract for treating ‘fevers’, the active compound artemsinin was isolated by Chinese chemists in 1972. This was ignored in the West for more than 10 years, until the WHO recognised its importance, and in 2002 placed it on the WHO ‘essential drugs’ list for malaria treatment. The herbs are noted for their extreme bitterness, and their name derives from Artemisia, wife and sister of the fourth century king of Halicarnassus; her sorrow on his death led her to mix his ashes with whatever she drank to make it bitter.