49 Basic principles of antimicrobial chemotherapy
Overview
Chemotherapy is the term originally used to describe the use of drugs that are ‘selectively toxic’ to invading microorganisms while having minimal effects on the host. The term also embraces the use of drugs that target tumours and, in fact, has now come to be associated specifically with that branch of pharmacology. In this chapter, however, we intend the term to cover both usages, although, in the public mind at least, chemotherapy is usually associated with cytotoxic anticancer drugs that cause unwanted effects such as loss of hair, nausea and vomiting.
All living organisms are prey to infection. Humans, being no exception to this rule, are susceptible to diseases caused by viruses, bacteria, protozoa, fungi and helminths (collectively referred to as pathogens). The use of chemotherapeutic agents dates back to the work of Ehrlich and others and to the development of arsenical drugs such as salvarsan for the treatment of syphilis.1 The successful development of such agents during the past 80 years, particularly the ‘antibiotic revolution’, which began in the 1940s with the advent of penicillin, constitutes one of the most important therapeutic advances in the entire history of medicine.
Clearly, the feasibility of selective toxicity depends on the ability to exploit such biochemical differences as may exist between the infecting organism (or indeed cancer cells, our internal ‘invaders’) and the host. Chapter 6 outlined our own ‘host’ defences against infection, while the bulk of the chapters in this section of the book describe the drugs used to combat such infections. In this introductory chapter we consider, very broadly, the nature of these biochemical differences and outline the molecular targets of drug action.
Background
The term chemotherapy was coined by Ehrlich himself at the beginning of the 20th century to describe the use of synthetic chemicals to destroy infective agents. In recent years, the definition of the term has been broadened to include antibiotics—substances produced by some microorganisms (or by pharmaceutical chemists) that kill or inhibit the growth of other microorganisms. Here, we broaden it still further to include agents that kill or inhibit the growth of cancer cells.
Unhappily, our success in developing drugs to attack these invaders has been paralleled by their own success in counteracting the effects of the drugs, resulting in the emergence of drug resistance. And at present, the invaders—particularly some bacteria—seem close to getting the upper hand. This is a very important problem, and we will devote some space to the mechanisms of resistance and the means by which it is spread.
The Molecular Basis of Chemotherapy
Chemotherapeutic agents, then, are chemicals that are intended to be toxic to the pathogenic organism (or cancer cells) but innocuous to the host. It is important to remember that many microorganisms share our body spaces (e.g. the gut2) without causing disease (these are called commensals), although they may become pathogenic under adverse circumstances (i.e. if the host is immunocompromised).
Living organisms are classified as either prokaryotes, cells without nuclei (e.g. bacteria), or eukaryotes, cells with nuclei (e.g. protozoa, fungi, helminths). In a separate category are the viruses, which need to utilise the metabolic machinery of the host cell, and they thus present a particular kind of problem for chemotherapeutic attack. There remain those mysterious proteinaceous agents, the prions (see Ch. 39), which cause disease but resist all attempts at classification, and for which there is no known antidote at present.
In another category are cancer cells, which are clearly more similar to normal host cells than are any pathogenic invaders, and this makes the problem of implementing selective toxicity especially difficult. The principles of cancer chemotherapy are discussed in Chapter 55. Virtually all creatures, host and parasite alike, have the same basic DNA blueprint (an exception being the RNA viruses), so some biochemical processes are common to most, if not all, organisms. Finding agents that affect pathogens or cancers but not other human cells necessitates finding either qualitative or quantitative biochemical differences between them.
Bacteria cause most infectious diseases, and Figure 49.1 shows in simplified diagrammatic form the main structures and functions of a ‘generalised’ bacterial cell. Surrounding the cell is the cell wall, which characteristically contains peptidoglycan in all forms of bacteria except Mycoplasma. Peptidoglycan is unique to prokaryotic cells and has no counterpart in eukaryotes. Within the cell wall is the plasma membrane, which, like that of eukaryotic cells, consists of a phospholipid bilayer and proteins. It functions as a selectively permeable membrane with specific transport mechanisms for various nutrients. However, in bacteria the plasma membrane does not contain any sterols, and this may alter the penetration of some chemicals.
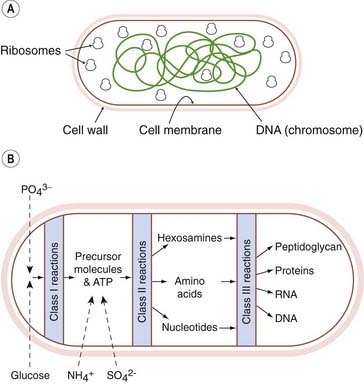
Fig. 49.1 Diagram of the structure and metabolism of a ‘typical’ bacterial cell.
[A] Schematic representation of a bacterial cell. [B] Flow diagram showing the synthesis of the main types of macromolecule of a bacterial cell. Class I reactions result in the synthesis of the precursor molecules necessary for class II reactions, which result in the synthesis of the constituent molecules; these are then assembled into macromolecules by class III reactions.
(Modified from Mandelstam J, McQuillen K, Dawes I (eds) 1982 Biochemistry of bacterial growth. Blackwell Scientific, Oxford.)
The function of the cell wall is to support the underlying plasma membrane, which is subject to an internal osmotic pressure of about 5 atmospheres in Gram-negative organisms, and about 20 atmospheres in Gram-positive organisms (see below). The plasma membrane and cell wall together comprise the bacterial envelope.
Bounded by the plasma membrane is the cytoplasm. As in eukaryotic cells, this contains soluble enzymes and other proteins, the ribosomes involved in protein synthesis, the small-molecule intermediates involved in metabolism as well as inorganic ions. The bacterial cell has no nucleus; instead, the genetic material, in the form of a single chromosome containing all the genetic information, lies in the cytoplasm with no surrounding nuclear membrane. In further contrast to eukaryotic cells, there are no mitochondria—cellular energy is generated by enzyme systems located in the plasma membrane.
Some bacteria have additional components such as a capsule and/or flagella, but the only additional structure with relevance for chemotherapy is the outer membrane outside the cell wall. The nature of this membrane enables bacteria to be classified according to whether they take up Gram’s stain (‘Gram-positive’) or not (‘Gram-negative’; for more details, see Ch. 50). In Gram-negative bacteria, this membrane may prevent penetration of antibacterial agents, and it also prevents easy access of lysozyme (a microbiocidal enzyme found in white blood cells, tears and other tissue fluids that breaks down peptidoglycan).
The biochemical reactions that are potential targets for antibacterial drugs are shown in Figure 49.1. There are three groups:
Other potential targets are the formed structures, for example the cell membrane, or in higher organisms (e.g. fungi and cancer cells) the microtubules or other specific tissues (e.g. muscle tissue in helminths). In considering these targets, emphasis will be placed on bacteria, but reference will also be made to protozoa, helminths, fungi, cancer cells and viruses. The classification that follows is clearly not rigid; a drug may affect more than one class of reactions or more than one subgroup of reactions within a class.
The molecular basis of antibacterial chemotherapy 
Biochemical Reactions as Potential Targets
Class I Reactions
Class I reactions are not promising targets for two reasons. First, bacterial and human cells use similar mechanisms to obtain energy from glucose (the Embden–Meyerhof pathway and the tricarboxylic acid cycle). Second, even if glucose oxidation is blocked, many other compounds (amino acids, lactate, etc.) can be utilised by bacteria as an alternative energy source.
Class II Reactions
Class II reactions are better targets because some pathways exist in pathogen, but not human, cells. For instance, human cells lack the ability, possessed by bacteria, to synthesise the so-called ‘essential’ amino acids as well as certain growth factors (termed vitamins in human physiology). Differences such as these represent potential targets. Another opportunity occurs when a pathway is identical in both bacteria and humans but exhibits a differential sensitivity to drugs. A prominent example is the folic acid pathway.
Folate
Folate biosynthesis is an example of a metabolic pathway found in bacteria but not in humans. Folate is required for DNA synthesis in both bacteria and in humans (see Chs 25 and 50). Humans cannot synthesise folate and must obtain it from the diet, and specific uptake mechanisms transport it into cells. By contrast, most species of bacteria, as well as the asexual forms of malarial protozoa, lack the necessary transport mechanisms and cannot make use of preformed folate but must synthesise their own de novo. This is a prime example of a difference that has proved to be extremely useful for chemotherapy. Sulfonamides contain the sulfanilamide moiety—a structural analogue of p-aminobenzoic acid (PABA), which is essential in the synthesis of folate (see Fig. 50.1). Sulfonamides compete with PABA for the enzyme involved in folate synthesis, and thus inhibit the metabolism of the bacteria. They are consequently bacteriostatic, not bactericidal3 (i.e. they suppress division of the cells but do not kill them), and are therefore only really effective in the presence of adequate host defences (see Chs 6 and 17).
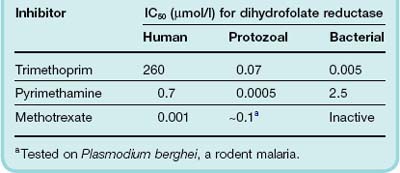
The utilisation of folate, in the form of tetrahydrofolate, as a co-factor in thymidylate synthesis is a good example of a pathway where human and bacterial enzymes exhibit a differential sensitivity to chemicals (Table 49.1; see Volpato & Pelletier, 2009). Although the pathway is virtually identical in microorganisms and humans, one of the key enzymes, dihydrofolate reductase, which reduces dihydrofolate to tetrahydrofolate (Fig. 50.2), is many times more sensitive to the folate antagonist trimethoprim in bacteria than in humans. In some malarial protozoa, this enzyme is somewhat less sensitive than the bacterial enzyme to trimethoprim but more sensitive to pyrimethamine and proguanil, which are used as antimalarial agents (Ch. 53). The relative IC50 values (the concentration causing 50% inhibition) for bacterial, malarial, protozoal and mammalian enzymes are given in Table 49.1. The human enzyme, by comparison, is very sensitive to the effect of the folate analogue methotrexate (Table 49.1) which is used to treat rheumatoid arthritis (Ch. 26) and cancer (Ch. 55). Methotrexate is inactive in bacteria because, being very similar in structure to folate, it requires active uptake by cells. Trimethoprim and pyrimethamine enter the cells by diffusion.
 The use of sequential blockade with a combination of two drugs that affect the same pathway at different points, for example sulfonamides and the folate antagonists, may be more successful than the use of either alone (e.g. in the treatment of Pneumocystis jirovecii pneumonia), and lower concentrations are effective when the two are used together. Thus, pyrimethamine and a sulfonamide (sulfadoxine) are used to treat falciparum malaria. An antibacterial formulation that contains both a sulfonamide and trimethoprim is co-trimoxazole; once widely used, this combination has become progressively less effective because of the development of sulfonamide resistance.
The use of sequential blockade with a combination of two drugs that affect the same pathway at different points, for example sulfonamides and the folate antagonists, may be more successful than the use of either alone (e.g. in the treatment of Pneumocystis jirovecii pneumonia), and lower concentrations are effective when the two are used together. Thus, pyrimethamine and a sulfonamide (sulfadoxine) are used to treat falciparum malaria. An antibacterial formulation that contains both a sulfonamide and trimethoprim is co-trimoxazole; once widely used, this combination has become progressively less effective because of the development of sulfonamide resistance.
Pyrimidine and purine analogues
Another example of a drug that interferes with a class II reaction is the pyrimidine analogue fluorouracil, which is used in cancer chemotherapy (Ch. 55). Fluorouracil is converted to a fraudulent nucleotide that interferes with thymidylate synthesis. Other cancer chemotherapy agents that give rise to fraudulent nucleotides are the purine analogues mercaptopurine and thioguanine. Flucytosine, an antifungal drug (Ch. 52), is deaminated to fluorouracil within fungal cells but to a much lesser extent in human cells, conferring a degree of selectivity.
Class III Reactions
As pathogen cells cannot take up their own unique macromolecules from the environment, class III reactions are particularly good targets for selective toxicity, and there are distinct differences between mammalian cells and parasitic cells in this respect.
The synthesis of peptidoglycan
The cell wall of bacteria contains peptidoglycan, a substance that does not occur in eukaryotes. It is the equivalent of a non-stretchable string bag enclosing the whole bacterium. In Gram-negative bacteria, this bag consists of a single thickness, but in Gram-positive bacteria there may be as many as 40 layers of peptidoglycan. Each layer consists of multiple backbones of amino sugars—alternating N-acetylglucosamine and N-acetylmuramic acid residues (Fig. 49.2)—the latter having short peptide side-chains that are crosslinked to form a polymeric lattice, which is strong enough to resist the high internal osmotic pressure and may constitute up to 10–15% of the dry weight of the cell. The cross-links differ in different species. In staphylococci, they consist of five glycine residues.
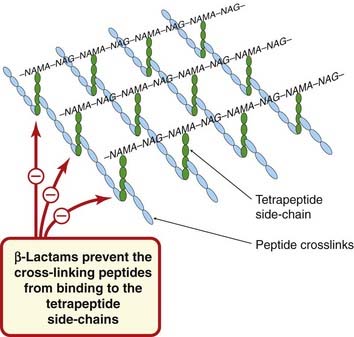
Fig. 49.2 Schematic diagram of a single layer of peptidoglycan from a bacterial cell (e.g. Staphylococcus aureus), showing the site of action of the β-lactam antibiotics.
In S. aureus, the peptide crosslinks consist of five glycine residues. Gram-positive bacteria have several layers of peptidoglycan. (NAG, N-acetylglucosamine; NAMA, N-acetylmuramic acid; more detail in Fig. 49.3.)
To build up this very large insoluble peptidoglycan layer on the outside of the cell membrane, the bacterial cell has the problem of how to transport the hydrophilic cytoplasmic ‘building blocks’ through the hydrophobic cell membrane structure. This is accomplished by linking them to a very large lipid carrier, containing 55 carbon atoms, which ‘tows’ them across the membrane. The process of peptidoglycan synthesis is outlined in Figure 49.3. First, N-acetylmuramic acid, attached to uridine diphosphate (UDP) and a pentapeptide, is transferred to the C55 lipid carrier in the membrane, with the release of uridine monophosphate. This is followed by a reaction with UDP–N-acetylglucosamine, resulting in the formation of a disaccharide pentapeptide complex attached to the carrier. This complex is the basic building block of the peptidoglycan. In Staphylococcus aureus, the five glycine residues are attached to the peptide chain at this stage. The building block is now transported out of the cell and added to the growing end of the peptidoglycan, the ‘acceptor’, with the release of the C55 lipid, which still has two phosphates attached. The lipid carrier then loses one phosphate group and thus becomes available for another cycle. Crosslinking between the peptide side-chains of the sugar residues in the peptidoglycan layer then occurs, the hydrolytic removal of the terminal alanine supplying the requisite energy.
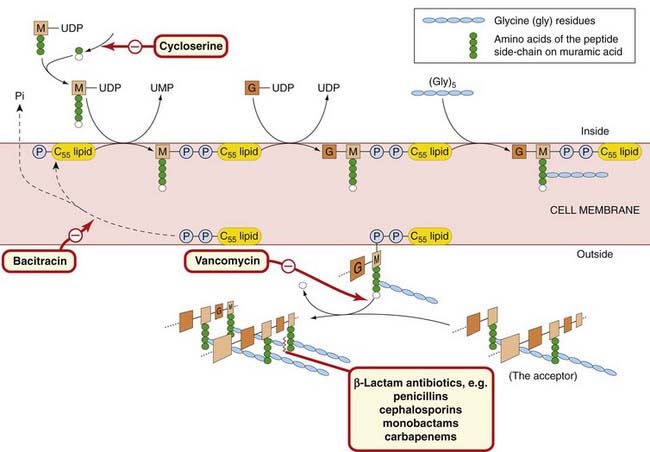
Fig. 49.3 Schematic diagram of the biosynthesis of peptidoglycan in a bacterial cell (e.g. Staphylococcus aureus), with the sites of action of various antibiotics.
The hydrophilic disaccharide–pentapeptide is transferred across the lipid cell membrane attached to a large lipid (C55 lipid) by a pyrophosphate bridge (–P–P–). On the outside, it is enzymically attached to the ‘acceptor’ (the growing peptidoglycan layer). The final reaction is a transpeptidation, in which the loose end of the (Gly) 5 chain is attached to a peptide side-chain of an M in the acceptor and during which the terminal amino acid (alanine) is lost. The lipid is regenerated by loss of a phosphate group (Pi) before functioning again as a carrier. G, N-acetylglucosamine; M, N-acetylmuramic acid; UDP, uridine diphosphate; UMP, uridine monophosphate.
This synthesis of peptidoglycan is a vulnerable step and can be blocked at several points by antibiotics (Fig. 49.3; Ch. 50). Cycloserine, which is a structural analogue of D-alanine, prevents the addition of the two terminal alanine residues to the initial tripeptide side-chain on N-acetylmuramic acid by competitive inhibition. Vancomycin inhibits the release of the building block unit from the carrier, thus preventing its addition to the growing end of the peptidoglycan. Bacitracin interferes with the regeneration of the lipid carrier by blocking its dephosphorylation. Penicillins, cephalosporins and other β-lactams inhibit the final transpeptidation by forming covalent bonds with penicillin-binding proteins that have transpeptidase and carboxypeptidase activities, thus preventing formation of the crosslinks.
Protein synthesis
Protein synthesis takes place in the ribosomes. Eukaryotic and prokaryotic ribosomes are different, and this provides the basis for the selective antimicrobial action of some antibiotics. The bacterial ribosome consists of a 50S subunit and a 30S subunit (Fig. 49.4), whereas in the mammalian ribosome the subunits are 60S and 40S. The other elements involved in peptide synthesis are messenger RNA (mRNA), which forms the template for protein synthesis, and transfer RNA (tRNA), which specifically transfers the individual amino acids to the ribosome. The ribosome has three binding sites for tRNA, termed the A, P and E sites.
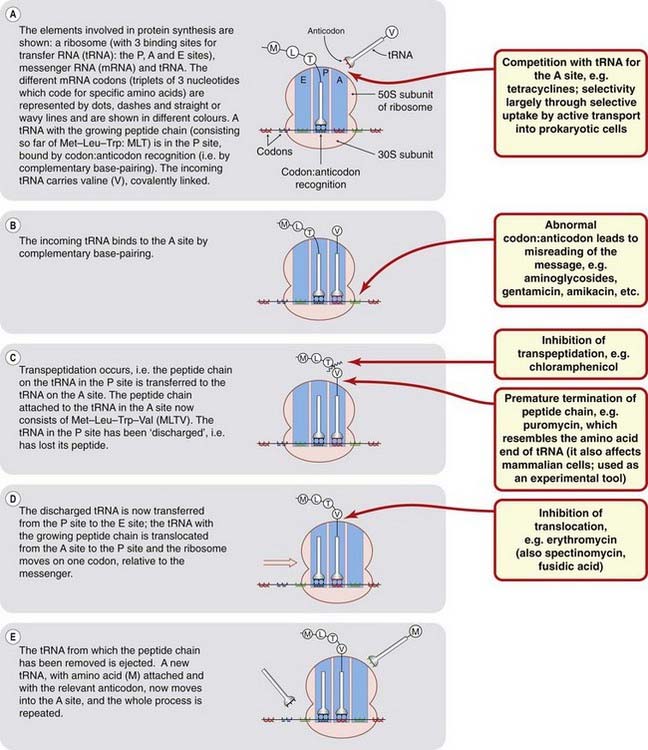
Fig. 49.4 Schematic diagram of bacterial protein synthesis, indicating the points at which antibiotics inhibit the process.
A simplified version of protein synthesis in bacteria is shown in Figure 49.4. To initiate translation, mRNA, transcribed from the DNA template (see below), is attached to the 30S subunit of the ribosome. The 50S subunit then binds to the 30S subunit to form a 70S subunit,4 which moves along the mRNA such that successive codons of the messenger pass along the ribosome from the A position to the P position. Antibiotics may affect protein synthesis at any one of these stages (Fig. 49.4; Ch. 50).
Nucleic acid synthesis
The nucleic acids of the cell are DNA and RNA. There are three types of RNA: mRNA, tRNA and ribosomal RNA (rRNA). The last of these is an integral part of the ribosome and is necessary for its assembly as well as for facilitating mRNA binding. The assembled ribosome also exhibits peptidyl transferase activity.
DNA is the template for the synthesis of both DNA and RNA. It exists in the cell as a double helix, each strand of which is a linear polymer of nucleotides. Each nucleotide consists of a base linked to a sugar (deoxyribose) and a phosphate. There are two purine bases, adenine (A) and guanine (G), and two pyrimidine bases, cytosine (C) and thymine (T). Single-strand DNA comprises alternating sugar and phosphate groups with the bases attached (Fig. 49.5). Specific hydrogen bonding between G and C and between A and T on each strand (i.e. complementary base pairing) is the basis of the double-stranded helical structure of DNA. The DNA helix is itself further coiled. In the test tube, the coil has 10 base pairs per turn. In vivo, the coil is unwound by about 1 turn in 20, forming a negative supercoil.
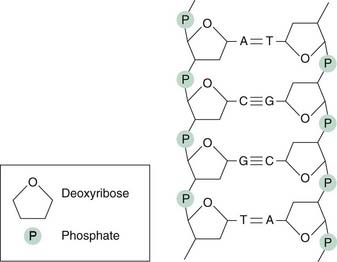
Each strand of DNA consists of a sugar–phosphate backbone with purine or pyrimidine bases attached. The purines are adenine (A) or guanine (G) and the pyrimidines are cytosine (C) or thymine (T). The sugar is deoxyribose. Complementarity between the two strands of DNA is maintained by hydrogen bonds (either two or three) between bases.
Initiation of DNA synthesis requires first the activity of a protein that causes separation of the strands. The replication process inserts a positive supercoil, which is relaxed by DNA gyrase (also called topoisomerase II; Fig. 49.6). During the synthesis of DNA, nucleotide units—each consisting of a base linked to a sugar and three phosphate groups—are added by base pairing with the complementary residues on the template. Condensation occurs by elimination of two phosphate groups, catalysed by DNA polymerase.
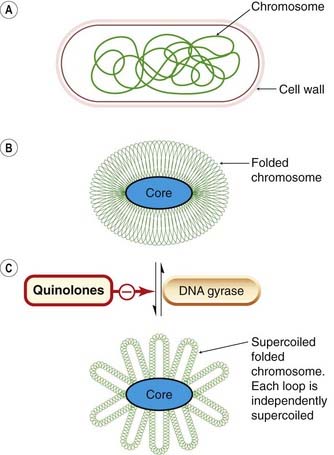
Fig. 49.6 Schematic diagram of the action of DNA gyrase: the site of action for quinolone antibacterials.
[A] Conventional diagram used to depict a bacterial cell and chromosome (e.g. Escherichia coli). Note that the E. coli chromosome is 1300 mm long and is contained in a cell envelope of 2 µm × 1 µm; this is approximately equivalent to a 50 m length of cotton folded into a matchbox. [B] Chromosome folded around RNA core, and then [C], supercoiled by DNA gyrase (topoisomerase II). Quinolone antibacterials interfere with the action of this enzyme.
(Modified from Smith J T 1985 In: Greenwood D, O’Grady F (eds) Scientific basis of antimicrobial therapy. Cambridge University Press, Cambridge, p. 69.)
RNA exists only in single-stranded form. The sugar moiety here is ribose, and the ribonucleotides contain the bases adenine, guanine, cytosine and uracil (U).
It is possible to interfere with nucleic acid synthesis in five different ways:
Inhibition of the synthesis of the nucleotides
This can be accomplished by an effect on the metabolic pathways that generate nucleotide precursors. Examples of agents that have such an effect have been described under class II reactions.
Alteration of the base-pairing properties of the template
Agents that intercalate in the DNA have this effect. Examples include acridines (proflavine and acriflavine), which are used topically as antiseptics. The acridines double the distance between adjacent base pairs and cause a frameshift mutation (Fig. 49.7), whereas some purine and pyrimidine analogues cause base mispairing.
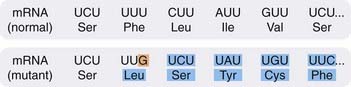
Fig. 49.7 An example of the effect on RNA and protein synthesis of a frameshift mutation in DNA.
A frameshift mutation is one that involves a deletion of a base or an insertion of an extra base. In the above example, an extra cytosine has been inserted in the DNA template, with the result that when mRNA is formed it has an additional guanine (G), as indicated in orange. The effect is to alter that codon and all the succeeding ones (shown in blue), so that a completely different protein is synthesised, as indicated by the different amino acids (Leu instead of Phe, Ser instead of Leu, etc.). A, adenine; C, cytosine; U, uracil.
Inhibition of either DNA or RNA polymerase
Dactinomycin (actinomycin D) binds to the guanine residues in DNA and blocks the movement of RNA polymerase, thus preventing transcription and inhibiting protein synthesis. The drug is used in cancer chemotherapy in humans (Ch. 55) and also as an experimental tool, but it is not useful as an antibacterial agent. Specific inhibitors of bacterial RNA polymerase that act by binding to this enzyme in prokaryotic but not in eukaryotic cells include rifamycin and rifampicin, which are particularly useful for treating tuberculosis (see Ch. 50). Aciclovir (an analogue of guanine) is phosphorylated in cells infected with herpes virus, the initial phosphorylation being by a virus-specific kinase to give the aciclovir trisphosphate, which has an inhibitory action on the DNA polymerase of the herpes virus (Ch. 51; Fig. 49.8).
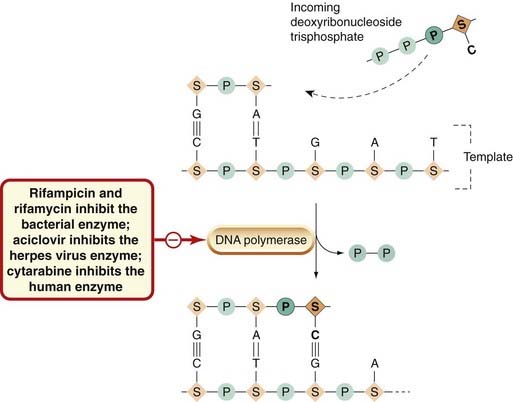
Fig. 49.8 Schematic diagram of DNA replication, showing some antibiotics that inhibit it by acting on DNA polymerase.
Nucleotides are added, one at a time, by base pairing to an exposed template strand, and are then covalently joined together in a reaction catalysed by DNA polymerase. The units that pair with the complementary residues in the template consist of a base linked to a sugar and three phosphate groups. Condensation occurs with the elimination of two phosphates. The elements added to the template are shown in darker colours and bold type. A, adenine; C, cytosine; G, guanine; P, phosphate; S, sugar; T, thymine.
RNA retroviruses have a reverse transcriptase (viral RNA-dependent DNA polymerase) that copies the viral RNA into DNA that integrates into the host cell genome as a provirus. Various agents (zidovudine, didanosine) are phosphorylated by cellular enzymes to the trisphosphate forms, which compete with the host cell precursors essential for the formation by the viral reverse transcriptase of proviral DNA.
Cytarabine (cytosine arabinoside) is used in cancer chemotherapy (Ch. 55). Its trisphosphate derivative is a potent inhibitor of DNA polymerase in mammalian cells. Foscarnet inhibits viral RNA polymerase by attaching to the pyrophosphate-binding site.
Inhibition of DNA gyrase
Figure 49.6 is a simplified scheme showing the action of DNA gyrase. The fluoroquinolones (cinoxacin, ciprofloxacin, nalidixic acid and norfloxacin) act by inhibiting DNA gyrase, and these chemotherapeutic agents are used particularly in infections with Gram-negative organisms (Ch. 50). These drugs are selective for the bacterial enzyme because it is structurally different from the mammalian enzyme. Some anticancer agents, for example doxorubicin, act on the mammalian topoisomerase II.
Direct effects on DNA itself
Alkylating agents form covalent bonds with bases in DNA and prevent replication. Compounds with this action are used only in cancer chemotherapy and include nitrogen mustard derivatives and nitrosoureas (Ch. 55). Mitomycin also binds covalently to DNA. No antibacterial agents work by these mechanisms. Bleomycin, an anticancer drug, causes fragmentation of the DNA strands following free radical formation (Ch. 55).
Biochemical reactions as potential targets for chemotherapy
The Formed Structures of the Cell as Potential Targets
The Membrane
The plasma membrane of bacterial cells is similar to that in mammalian cells in that it consists of a phospholipid bilayer in which proteins are embedded, but it can be more easily disrupted in certain bacteria and fungi.
Polymixins are cationic peptide antibiotics, containing both hydrophilic and lipophilic groups, which have a selective effect on bacterial cell membranes. They act as detergents, disrupting the phospholipid components of the membrane structure, thus killing the cell.
Unlike mammalian and bacterial cells, fungal cell membranes comprise large amounts of ergosterol. This facilitates the attachment of polyene antibiotics (e.g. nystatin and amphotericin; Ch. 52), which act as ionophores and cause leakage of cations.
Azoles such as itraconazole kill fungal cells by inhibiting ergosterol synthesis, thereby disrupting the function of membrane-associated enzymes. The azoles also affect Gram-positive bacteria, their selectivity being associated with the presence of high levels of free fatty acids in the membrane of susceptible organisms (Ch. 52).
Intracellular Organelles
Microtubules and/or microfilaments
The benzimidazoles (e.g. albendazole) exert their anthelminthic action by binding selectively to parasite tubulin and preventing microtubule formation (Ch. 54). The vinca alkaloids vinblastine and vincristine are anticancer agents that disrupt the functioning of microtubules during cell division (Ch. 55).
Food vacuoles
The erythrocytic form of the malaria plasmodium feeds on host haemoglobin, which is digested by proteases in the parasite food vacuole, the final product, haem, being detoxified by polymerisation. Chloroquine exerts its antimalarial action by inhibiting plasmodial haem polymerase (Ch. 53).
Muscle Fibres
Some anthelminthic drugs have a selective action on helminth muscle cells (Ch. 54). Piperazine acts as an agonist on parasite-specific chloride channels gated by GABA in nematode muscle, hyperpolarising the muscle fibre membrane and paralysing the worm; avermectins increase Cl− permeability in helminth muscle—possibly by a similar mechanism. Pyrantel (now seldom used) and levamisole are agonists at nematode acetylcholine nicotinic receptors on muscle, causing contraction followed by paralysis (Ch. 54).
Resistance to Antibacterial Drugs
Since the 1940s, the development of effective and safe drugs to deal with bacterial and other infections has revolutionised medical treatment, and the morbidity and mortality associated with these diseases has been dramatically reduced. Unfortunately, the development of effective antibacterial drugs has been accompanied by the emergence of drug-resistant organisms. This is not unexpected, because the short generation time of many bacterial species affords ample opportunity for evolutionary adaptation. The phenomenon of resistance imposes serious constraints on the options available for the medical treatment of many bacterial infections. Resistance to chemotherapeutic agents can also develop in protozoa, in multicellular parasites (see, for example, Martin & Robertson, 2000; St Georgiev, 2000) and in populations of malignant cells (discussed in Ch. 55). Here, however, we will confine our discussion mainly to the mechanisms of resistance in bacteria.
Antibiotic resistance in bacteria spreads in three ways:
Understanding the mechanisms involved in antibiotic resistance is crucial for the sensible clinical use of existing medicines and in the design of new antibacterial drugs. One byproduct of the studies of resistance in bacteria was the development of plasmid-based techniques for DNA cloning, leading to the use of bacteria to produce recombinant proteins for therapeutic use (see Ch. 59).
Genetic Determinants of Antibiotic Resistance
Chromosomal Determinants: Mutations
The spontaneous mutation rate in bacterial populations for any particular gene is very low, and the probability is that approximately only 1 cell in 10 million will, on division, give rise to a daughter cell containing a mutation in that gene. However, as there are likely to be very many more cells than this over the course of an infection, the probability of a mutation causing a change from drug sensitivity to drug resistance can be quite high with some species of bacteria and with some drugs. Fortunately, the presence of a few mutants is not generally sufficient to produce resistance: despite the selective advantage that the resistant mutants possess, the drastic reduction of the population by the antibiotic usually enables the host’s natural defences (see Ch. 6) to prevail. However, the outcome may not be quite so happy if the primary infection is caused by a drug-resistant strain.
Resistance resulting from chromosomal mutation is important in some instances, notably infections with meticillin-resistant S. aureus (MRSA; see below) and in tuberculosis, but apart from these examples this type of resistance is of limited clinical relevance, possibly because the mutants often have reduced pathogenicity.
Gene Amplification
Recently it has been discovered that gene duplication and amplification are important mechanisms for resistance in some organisms (Sandegren & Andersson, 2009). According to this idea, treatment with antibiotics can induce an increased number of copies for pre-existing resistance genes such as antibiotic-destroying enzymes and efflux pumps.
Extrachromosomal Determinants: Plasmids
In addition to the chromosome itself, many species of bacteria contain extrachromosomal genetic elements called plasmids that exist free in the cytoplasm. These are also genetic elements that can replicate independently. Structurally, they are closed loops of DNA that may comprise a single gene or as many as 500 or even more. Only a few plasmid copies may exist in the cell but often multiple copies are present, and there may also be more than one type of plasmid in each bacterial cell. Plasmids that carry genes for resistance to antibiotics (r genes) are referred to as R plasmids. Much of the drug resistance encountered in clinical medicine is plasmid determined. It is not known how these genes arose.
The whole process can occur with frightening speed. S. aureus, for example, is a past master of the art of antibiotic resistance. Having become completely resistant to penicillin through plasmid-mediated mechanisms, this organism, within only 1–2 years, was able to adapt to its replacement, meticillin (de Lancastre et al., 2007).
The Transfer of Resistance Genes between Genetic Elements within the Bacterium
Transposons
Some stretches of DNA are readily transferred (transposed) from one plasmid to another and also from plasmid to chromosome or vice versa. This is because integration of these segments of DNA, which are called transposons, into the acceptor DNA can occur independently of the normal mechanism of homologous genetic recombination. Unlike plasmids, transposons are not able to replicate independently, although some may replicate during the process of integration (Fig. 49.9), resulting in a copy in both the donor and the acceptor DNA molecules. Transposons may carry one or more resistance genes (see below) and can ‘hitch-hike’ on a plasmid to a new species of bacterium. Even if the plasmid is unable to replicate in the new host, the transposon may integrate into the new host’s chromosome or into its indigenous plasmids. This probably accounts for the widespread distribution of certain of the resistance genes on different R plasmids and among unrelated bacteria.
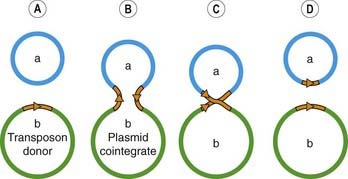
Fig. 49.9 An example of the transfer and replication of a transposon (which may carry genes coding for resistance to antibiotics).
[A] Two plasmids, a and b, with plasmid b containing a transposon (shown in brown). [B] An enzyme encoded by the transposon cuts DNA of both donor plasmid and target plasmid a to form a cointegrate. During this process, the transposon replicates. [C] An enzyme encoded by the transposon resolves the cointegrate. [D] Both plasmids now contain the transposon DNA.
Gene cassettes and integrons
Plasmids and transposons do not complete the tally of mechanisms that natural selection has provided to confound the hopes of the microbiologist/chemotherapist. Resistance—in fact, multidrug resistance—can also be spread by another mobile element, the gene cassette, which consists of a resistance gene attached to a small recognition site. Several cassettes may be packaged together in a multicassette array, which can, in turn, be integrated into a larger mobile DNA unit termed an integron. The integron (which may be located on a transposon) contains a gene for an enzyme, integrase (recombinase), which inserts the cassette(s) at unique sites on the integron. This system—transposon/integron/multiresistance cassette array—allows particularly rapid and efficient transfer of multidrug resistance between genetic elements both within and between bacteria.
The Transfer of Resistance Genes between Bacteria
The transfer of resistance genes between bacteria of the same and indeed of different species is of fundamental importance in the spread of antibiotic resistance. The most important mechanism in this context is conjugation. Other gene transfer mechanisms, transduction and transformation, are of little importance in spreading resistance genes.
Conjugation
Conjugation involves cell-to-cell contact during which chromosomal or extrachromosomal DNA is transferred from one bacterium to another, and is the main mechanism for the spread of resistance. The ability to conjugate is encoded in conjugative plasmids; these are plasmids that contain transfer genes that, in coliform bacteria, code for the production by the host bacterium of proteinaceous surface tubules, termed sex pili, which connect the two cells. The conjugative plasmid then passes across from one bacterial cell to another (generally of the same species). Many Gram-negative and some Gram-positive bacteria can conjugate. Some promiscuous plasmids can cross the species barrier, accepting one host as readily as another. Many R plasmids are conjugative. Non-conjugative plasmids, if they co-exist in a ‘donor’ cell with conjugative plasmids, can hitch-hike from one bacterium to the other with the conjugative plasmids. The transfer of resistance by conjugation is significant in populations of bacteria that are normally found at high densities, as in the gut.
Transduction
Transduction is a process by which plasmid DNA is enclosed in a bacterial virus (or phage) and transferred to another bacterium of the same species. It is a relatively ineffective means of transfer of genetic material but is clinically important in the transmission of resistance genes between strains of staphylococci and of streptococci.
Transformation
A few species of bacteria can, under natural conditions, undergo transformation by taking up DNA from the environment and incorporating it into the genome by normal homologous recombination. Transformation is probably not of importance clinically.
Resistance to antibiotics
Biochemical Mechanisms of Resistance to Antibiotics
The Production of an Enzyme That Inactivates the Drug
Inactivation of β-lactam antibiotics
The most important example of resistance caused by inactivation is that of the β-lactam antibiotics. The enzymes concerned are β-lactamases, which cleave the β-lactam ring of penicillins and cephalosporins (see Ch. 50). Cross-resistance between the two classes of antibiotic is not complete, because some β-lactamases have a preference for penicillins and some for cephalosporins.
Staphylococci are the principal bacterial species producing β-lactamase, and the genes coding for the enzymes are on plasmids that can be transferred by transduction. In staphylococci, the enzyme is inducible (i.e. its synthesis is not expressed in the absence of the drug) and minute, subinhibitory, concentrations of antibiotics de-repress the gene and result in a 50- to 80-fold increase in expression. The enzyme passes through the bacterial envelope and inactivates antibiotic molecules in the surrounding medium. The grave clinical problem posed by resistant staphylococci secreting β-lactamase was tackled by developing semisynthetic penicillins (such as meticillin) and new β-lactam antibiotics (the monobactams and carbapenems), and cephalosporins (such as cephamandole), that are less susceptible to inactivation. The growing problem of MRSA is discussed below.
Gram-negative organisms can also produce β-lactamases, and this is a significant factor in their resistance to the semisynthetic broad-spectrum β-lactam antibiotics. In these organisms, the enzymes may be coded by either chromosomal or plasmid genes. In the former case, the enzymes may be inducible, but in the latter they are produced constitutively. When this occurs, the enzyme does not inactivate the drug in the surrounding medium but instead remains attached to the cell wall, preventing access of the drug to membrane-associated target sites. Many of these β-lactamases are encoded by transposons, some of which may also carry resistance determinants to several other antibiotics.
Inactivation of chloramphenicol
Chloramphenicol is inactivated by chloramphenicol acetyltransferase, an enzyme produced by resistant strains of both Gram-positive and Gram-negative organisms, the resistance gene being plasmid borne. In Gram-negative bacteria, the enzyme is produced constitutively, resulting in levels of resistance five-fold higher than in Gram-positive bacteria, in which the enzyme is inducible.
Inactivation of aminoglycosides
Aminoglycosides are inactivated by phosphorylation, adenylation or acetylation, and the requisite enzymes are found in both Gram-negative and Gram-positive organisms. The resistance genes are carried on plasmids, and several are found on transposons.
Many other examples of this kind are given by Wright (2005).
Alteration of Drug-Sensitive or Drug-Binding Site
The aminoglycoside-binding site on the 30S subunit of the ribosome may be altered by chromosomal mutation. A plasmid-mediated alteration of the binding site protein on the 50S subunit also underlies resistance to erythromycin, and decreased binding of fluoroquinolones because of a point mutation in DNA gyrase A has recently been described. An altered DNA-dependent RNA polymerase determined by a chromosomal mutation is reported to be the basis for rifampicin resistance.
In addition to acquiring resistance to β-lactams susceptible to β-lactamase, some strains of S. aureus have even become resistant to some antibiotics that are not significantly inactivated by β-lactamase (e.g. meticillin), because they express an additional β-lactam-binding protein coded for by a mutated chromosomal gene.
See Lambert (2005) for other examples of this type of action.
Decreased Drug Accumulation in the Bacterium
An important example of decreased drug accumulation is the plasmid-mediated resistance to tetracyclines encountered in both Gram-positive and Gram-negative bacteria. In this case, resistance genes in the plasmid code for inducible proteins in the bacterial membrane, which promote energy-dependent efflux of the tetracyclines, and hence resistance. This type of resistance is common and has greatly reduced the therapeutic value of the tetracyclines in human and veterinary medicine. Resistance of S. aureus to erythromycin and the other macrolides, and to fluoroquinolones, is also brought about by energy-dependent efflux. Inhibitors of such pumps may be useful adjuncts to antibiotics (Van Bambeke et al., 2006).
There is also recent evidence of plasmid-determined inhibition of porin synthesis, which could affect those hydrophilic antibiotics that enter the bacterium through these water-filled channels in the outer membrane. Altered permeability as a result of chromosomal mutations involving the polysaccharide components of the outer membrane of Gram-negative organisms may confer enhanced resistance to ampicillin. Mutations affecting envelope components have been reported to affect the accumulation of aminoglycosides, β-lactams, chloramphenicol, peptide antibiotics and tetracycline.
The Development of a Pathway That Bypasses the Reaction Inhibited By the Antibiotic
Resistance to trimethoprim is the result of plasmid-directed synthesis of a dihydrofolate reductase with low or zero affinity for trimethoprim. It is transferred by transduction and may be spread by transposons.
Sulfonamide resistance in many bacteria is plasmid mediated and results from the production of a form of dihydropteroate synthetase with a low affinity for sulfonamides but no change in affinity for PABA. Bacteria causing serious infections have been found to carry plasmids with resistance genes to both sulfonamides and trimethoprim.
Biochemical mechanisms of resistance to antibiotics
Current Status of Antibiotic Resistance in Bacteria
The most disturbing development of resistance has been in staphylococci, one of the commonest causes of hospital bloodstream infections, many strains of which are now resistant to almost all currently available antibiotics (de Lencastre et al., 2007). In addition to resistance to some β-lactams through production of β-lactamase and the production of an additional β-lactam-binding protein that also renders them resistant to meticillin, S. aureus may also manifest resistance to other antibiotics as follows:
Infections with MRSA have become a major problem, particularly in hospitals, where they can spread rapidly among elderly and/or seriously ill patients, and patients with burns or wounds. Until recently, the glycopeptide vancomycin was the antibiotic of last resort against MRSA but, ominously, strains of MRSA showing decreased susceptibility to this drug were isolated from hospitalised patients in the USA and Japan in 19975 and, more recently, in the community. MRSA infections are rising; Bax et al. (2000) report prevalence in US hospitals as rising from 11–13% in 1985/6 to 26% in 1998.
The fact that vancomycin resistance seems to have developed spontaneously could have major clinical implications—and not only for nosocomial (those contracted in hospital) MRSA infections. It had been thought that antibiotic-resistant bacteria were dangerous only to seriously ill, hospitalised patients, in that the genetic burden of multiple resistance genes would lead to reduced virulence. Distressingly, however, there is now evidence that the spectrum and frequency of disease produced by meticillin-susceptible and meticillin-resistant staphylococci are similar.
In the past few years, enterococci have been rapidly developing resistance to many chemotherapeutic agents and have emerged as the second most common nosocomial pathogen. Non-pathogenic enterococci are ubiquitous in the intestine, have intrinsic resistance to many antibacterial drugs, and can readily become resistant to other agents by taking up plasmids and transposons carrying the relevant resistance genes. Such resistance is easily transferred to invading pathogenic enterococci.
Enterococci, already multiresistant, have recently developed resistance to vancomycin. This is apparently achieved by substitution of D-Ala-D-Ala with D-Ala-D-lactate in the peptide chain attached to N-acetylglucosamine-N-acetylmuramic acid (G-M) during the first steps of peptidoglycan synthesis (see Fig. 49.3; Ch. 50). This is becoming a major problem in hospitalised patients, and in the USA vancomycin resisitance has increased from 0.5% to 18% in less than a decade (Bax et al., 2000). A particular concern is the possibility of transfer of vancomycin resistance from enterococci to staphylococci, because they can co-exist in the same patient.
Many other pathogens are developing or have developed resistance to commonly used drugs. This list includes Pseudomonas aeruginosa, Streptococcus pyogenes, Streptococcus pneumoniae, Neisseria meningitidis, N. gonorrhoeae, Haemophilius influenzae and H. ducreyi, as well as Mycobacterium, Campylobacter and Bacteroides species. Some strains of M. tuberculosis are now able to evade every antibiotic in the clinician’s armamentarium, and tuberculosis, once easily treatable, is now reported to be causing more deaths worldwide than malaria and AIDS together. Fortunately, some glycopeptide and other antibiotics (e.g. teicoplanin, daptomycin and linezolide see Ch. 50) that are used to treat resistant Gram-positive strains have largely maintained their potency. Even so, there is a danger of resistance arising if they are wrongly utilised.
Prescribers and consumers must also bear a responsibility for the burgeoning problem of resistance. Indiscriminate use of antibiotics in human and veterinary medicine, and their use in animal foodstuffs, has undoubtedly encouraged the growth of resistant strains. Some governmental and regulatory bodies (e.g. the European Union) have devised political and social measures to curb such excesses, and these have been at least partly successful.
The issue around declining antibiotic efficacy is, however, not solely to do with bacterial countermeasures. There has been a declining interest in the pharmaceutical industry in researching novel antibiotics. Historically, the area has been one of the mainstays of the industry, but most of the drugs available today are the result of incremental changes in the structures of a relatively small number of basic molecular structures, such as the β-lactam nucleus. By common consent, the days when it was possible to discover new and effective drugs in this way are long gone.
Hubris has also played a part. In 1967, the US Surgeon General effectively announced that infectious diseases had been vanquished, and that the researchers should turn their attention to chronic diseases instead. As a result, many pharmaceutical companies scaled down their efforts in the area, and only in the past few years has activity been resumed as the pressing need for novel compounds has been recognised (Barrett & Barrett, 2003).
However, nature has endowed microorganisms with fiendishly effective adaptive mechanisms for outwitting our best therapeutic strategies, and so far several have been effortlessly keeping pace with our attempts to eradicate them. This challenging situation has been reviewed in depth by Shlaes (2003) and Barrett & Barrett (2003).
Multidrug resistance
Many pathogenic bacteria have developed resistance to the commonly used antibiotics. Examples include the following:
References and Further Reading
Amyes S.G.B. Magic bullets, lost horizons: the rise and fall of antibiotics. London: Taylor & Francis; 2001. (Thought-provoking book by a bacteriologist with wide experience in bacterial resistance and genetics; he opines that unless the problem of antibiotic resistance is solved in the next 5 years, ‘we are going to slip further into the abyss of uncontrollable infection’)
Knodler L.A., Celli J., Finlay B.B. Pathogenic trickery: deception of host cell processes. Mol. Cell. Biol.. 2001;2:578-588. (Discusses bacterial ploys to subvert or block normal host cellular processes: mimicking the ligands for host cell receptors or signalling pathways. Useful list of examples)
Martin R.J., Robertson A.P. Electrophysiological investigation of anthelmintic resistance. Parasitology. 2000;120(Suppl.):S87-S94. (Uses patch clamp technique to study ion channels in resistant and non-resistant nematode tissue)
Recchia G.D., Hall R.M. Gene cassettes: a new class of mobile element. Microbiology. 1995;141:3015-3027. (Detailed coverage of this unusual mechanism)
Shlaes D.M. The abandonment of antibacterials: why and wherefore? Curr. Opin. Pharmacol.. 2003;3:470-473. (A good review that explains the reasons underlying the resistance problem and the regulatory and other hurdles that must be overcome before new antibacterials appear on to the market; almost apocalyptic in tone)
Tan Y.T., Tillett D.J., McKay I.A. Molecular strategies for overcoming antibiotic resistance in bacteria. Mol. Med. Today. 2000;6:309-314. (Succinct article reviewing strategies to exploit advances in molecular biology to develop new antibiotics that overcome resistance; useful glossary of relevant terms)
Volpato J.P., Pelletier J.N. Mutational ‘hot-spots’ in mammalian, bacterial and protozoal dihydrofolate reductases associated with antifolate resistance: sequence and structural comparison. Drug. Resist. Updat.. 2009;12:28-41.
Zasloff M. Antimicrobial peptides of multicellular organisms. Nature. 2002;415:389-395. (Thought-provoking article about the potent broad-spectrum antimicrobial peptides possessed by both animals and plants, which are used to fend off a wide range of microbes; it is suggested that exploiting these might be one answer to the problem of antibiotic resistance)
Barrett C.T., Barrett J.F. Antibacterials: are the new entries enough to deal with the emerging resistance problem? Curr. Opin. Biotechnol.. 2003;14:621-626. (Good general review with some compelling examples and a round-up of new drug candidates)
Bax R., Mullan N., Verhoef J. The millennium bugs—the need for and development of new antibacterials. Int. J. Antimicrob. Agents. 2000;16:51-59. (Excellent review of the problem of resistance and some of the new agents in the pipeline)
Courvalin P., Trieu-Cuot P. Minimizing potential resistance: the molecular view. Clin. Infect. Dis.. 2001;33:S138-S146. (Reviews the potential contribution of molecular biology to preventing the spread of resistant bacteria)
de Lencastre H., Oliveira D., Tomasz A. Antibiotic resistant Staphylococcus aureus: a paradigm of adaptive power. Curr. Opin. Microbiol.. 2007;10:428-435. (A little specialised but worth reading. It details the extraordinary ability of this organism to survive attack by virtually all the drugs in our antibiotic arsenal)
Hawkey P.M. The origins and molecular basis of antibiotic resistance. Br. Med. J.. 1998;7159:657-659. (Succinct overview of resistance; useful, simple diagrams; this is one of 12 papers on resistance in this issue of the journal)
Jones M.E., Peters E., Weersink A.M., et al. Widespread occurrence of integrons causing multiple antibiotic resistance in bacteria. Lancet. 1997;349:1742-1743.
Lambert P.A. Bacterial resistance to antibiotics: modified target sites. Adv. Drug Deliv. Rev.. 2005;57:1471-1485. (Excellent review dealing with this important topic. Numerous examples drawn from studies with many different bacterial species)
Levy S.B. The challenge of antibiotic resistance. Sci. Am.. 1998;March:32-39. (Simple, clear review by an expert in the field; excellent diagrams)
Michel M., Gutman L. Methicillin-resistant Staphylococcus aureus and vancomycin-resistant enterococci: therapeutic realities and possibilities. Lancet. 1997;349:1901-1906. (Good review article; useful diagram; suggests schemes for medical management of infections caused by resistant organisms)
Noble W.C., Virani Z., Cree R.G. Co-transfer of vancomycin and other resistance genes from Enterococcus faecalis NCTC 12201 to Staphylococcus aureus. FEMS Microbiol. Lett.. 1992;72:195-198.
St Georgiev V. Membrane transporters and antifungal drug resistance. Curr. Drug Targets. 2000;1:184-261. (Discusses various aspects of multidrug resistance in disease-causing fungi in the context of targeted drug development)
Sandegren L., Andersson D.I. Bacterial gene amplification: implications for the evolution of antibiotic resistance. Nat. Rev. Microbiol.. 2009;7:578-588.
Van Bambeke F., Pages J.M., Lee V.J. Inhibitors of bacterial efflux pumps as adjuvants in antibiotic treatments and diagnostic tools for detection of resistance by efflux. Recent. Pat. Antiinfect. Drug Discov.. 2006;1:157-175.
van Belkum A. Molecular epidemiology of methicillin-resistant Staphylococcus aureus strains: state of affairs and tomorrow’s possibilities. Microb. Drug Resist.. 2000;6:173-187.
Walsh C. Molecular mechanisms that confer antibacterial drug resistance. Nature. 2000;406:775-781. (Excellent review outlining the mechanisms of action of antibiotics and the resistance ploys of bacteria; very good diagrams)
Woodford N. Biological counterstrike: antibiotic resistance mechanisms of Gram-positive cocci. Clin. Microbiol. Infect.. 2005;3:2-21. (A useful reference that classifies antibiotic resistance as one of the major public health concerns of the 21st century and discusses drug treatment for resistant strains)
Wright G.D. Bacterial resistance to antibiotics: enzymatic degradation and modification. Adv. Drug Deliv. Rev.. 2005;57:1451-1470. (This comprehensive review gives details of the many pathways that have evolved in bacteria to destroy antibiotics. A little complex but fascinating reading)
1Mercury-containing compounds were also once used for treating syphilis. ‘One night with Venus, a lifetime with Mercury’ was a saying of that time.
2Humans harbour about 2 kg of bacteria in the gut, comprising a large ‘forgotten organ’ in the body with important metabolic functions.
3Whether a drug is bactericidal rather than bacteriostatic is determined according to a strict technical criterion, but in practice it can be difficult to differentiate the two actions during therapy.
4You query whether 30S + 50S = 70S? Yes it does, because we are talking about Svedberg units, which measure sedimentation rate, not mass.
5Noble et al. (1992) have reported transfer of vancomycin resistance from enterococci to staphylococci. If this occurred in a clinical environment, it would be disastrous. Some microbiologists have suggested that Noble and his team should be autoclaved.