48 Drug addiction, dependence and abuse
Overview
In this chapter we consider those drugs that are consumed because people choose to, and not because they are advised to by their doctor. Largely these drugs are taken because they are pleasurable (hedonic). A list of the more frequently used drugs is given in Table 48.1. It includes drugs that are also used for medicinal purposes (e.g. general anaesthetics, benzodiazepines, opioids and some psychostimulants), non-therapeutic drugs that are legal in many countries (e.g. nicotine and ethanol) and many other drugs that are widely used although their manufacture, sale and consumption have been declared illegal in most Western countries.
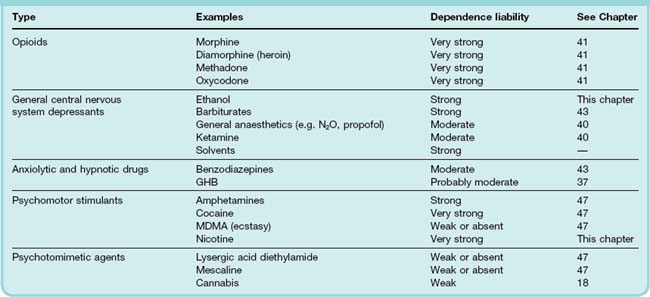
The reasons why the use of a particular drug is viewed as a problem to society—and hence may be considered ‘drug abuse’—are complex and largely outside the scope of this book. The drug and its pharmacological activity are only the starting point. For many, but not all, drugs of abuse, continued use leads to dependence. Here, we briefly review the classes of drug, the biological processes underlying drug dependence and describe in detail the pharmacology of two important drugs that have no place in therapeutics but are consumed in large amounts, namely nicotine and ethanol. Other drugs that are abused are described elsewhere in this book (see Table 48.1). ‘Lifestyle’ and ‘sport’ drugs are discussed in Chapter 58.
For further information on various aspects of drug abuse, see Winger et al. (2004), Karch (2006) and Koob & Le Moal (2006).
Drug Use and Abuse
A number of terms are used, sometimes interchangeably and sometimes incorrectly, to describe drug use and the consequences of administration of drugs. Terms that are best avoided are described in Table 48.2. Other, more useful terms are defined in the text below.
Table 48.2 Glossary of frequently used and ‘abused’ terms
| Addict | Person for whom the desire to experience a drug’s effects overrides any consideration for the serious physical, social or psychological problems that the drug may cause to the individual or others. Often used in non-scientific circles to convey criminal intent and so has fallen out of favour with those involved in treating people with drug problems |
| Drug misuse | Non-medicinal drug use (although some would not consider taking drugs to alter mood/induce hallucinations as ‘misuse’ or ‘abuse’) |
| Junkie | Pejorative term for someone who is dependent upon a drug |
| Narcotics | Originally used as a term to describe opioids as they induce sleep (narcosis). Subsequently this term has been used by non-scientists to describe a wide range of drugs of abuse (including cocaine which is a stimulant!) |
| Recreational drug use | Originally used to describe all drug abuse, it is now sometimes used to describe drug use in the bar/club/dance scene |
| Self-medication | Taking a drug of abuse to offset some underlying medical condition, e.g. pain, depression |
| Substance use | Some governments do not consider ethanol to be a drug, hence ‘substance use’ (or ‘substance abuse’) is used to include ethanol |
A vast and ever increasing array of drugs is used to alter mood and perception. These range from drugs that are also used as medicines, through non-medicinal synthetic drugs to herbal preparations (Table 48.1). The popularity of each varies between different societies across the world, and within societies popularity differs among different groups of individuals.1 Frequently, users will take more than one drug concomitantly or sequentially. Polydrug use is a very under-researched area both in regard to why it is done and how different drugs may interact, as well as in regard to the potential harm that may arise from such practices (e.g. ethanol alters cocaine metabolism resulting in the production of cocaethylene which is more potent than cocaine and has greater cardiovascular toxicity). Sequential use is often intended to reduce adverse effects when coming down off the first drug (e.g. use of benzodiazepines when coming down from stimulants).
At first sight, the drugs listed in Table 48.1 form an extremely heterogeneous pharmacological group; we can find little in common at the molecular and cellular level between say, morphine, cocaine and LSD. What links them is that people find their effects pleasurable (hedonic) and tend to want to repeat the experience. The drug experience may take the form of intense euphoria, mood elevation, hallucinations, stimulation, sedation or calming depending upon the specific drug taken.
Drug use involves effects on the brain that can be both acute and chronic (Fig. 48.1). The immediate, acute effect on mood is the reason the drug is taken. For some drugs (e.g. amphetamines, Ch. 47), this may be followed by a rebound negative or depressed phase. Persistent use of a drug may lead to compulsive drug use (addiction/dependence—a complex state that involves both psychological and physiological dependence) and to the development of tolerance. Psychological dependence can give rise to intense craving even when the user has been drug-free for months or years.
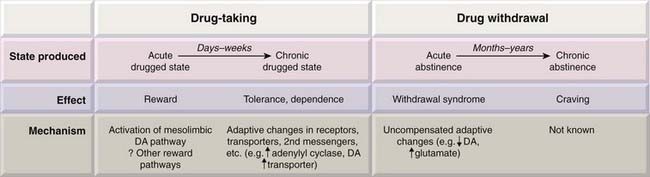
Fig. 48.1 Cellular and physiological mechanisms involved in drug dependence showing the relationship between the immediate and delayed effects of drug taking and drug withdrawal.
Drug Administration
For drugs that induce strong feelings of euphoria, there are two components to the experience: an initial rapid effect (the rush or buzz) and a more prolonged pleasurable effect (the high). The intensity of the initial effect is determined by how fast the drug enters the brain and activates its effector mechanism. For many casual drug users, ease of administration defines how the drug is taken (e.g. smoking, swallowing or snorting a drug is relatively easy). However, for other drug users chasing a more intense experience, the route of administration and the choice of individual drug become important. Intravenous injection or smoking results in faster absorption of a drug than when it is taken orally. Heroin (official name diamorphine), cocaine, amphetamines, tobacco and cannabis are all taken by one or other of these routes. Heroin is more popular as a drug of abuse than morphine. This is because it enters the brain more rapidly than morphine. However, heroin itself does not interact with opioid receptors but is rapidly deacetylated to 6-acetylmorphine and morphine, µ-receptor agonists (see Ch. 41).
Drug Harm
All drugs of abuse are harmful to a varying extent. Adverse effects can be the result of drug overdose (e.g. respiratory depression produced by opioids), of effects on tissues other than the brain (e.g. necrosis of the nasal septum resulting from chronic cocaine use), of the route of administration (e.g. HIV and other infections in drug users who share needles), of effects unrelated to the specific actions of the drug (e.g. carcinogenicity of tobacco smoke, severe bladder pain in regular ketamine users) or of use for illegal purposes (e.g. flunitrazepam or γ-hydroxybutyrate as date-rape drugs). Many major harms relate to the ability of some drugs to induce dependence (e.g. psychostimulants, opioids, ethanol and tobacco) or to reveal a susceptibility to psychotic illness in some individuals (e.g. amphetamines and cannabis).
An attempt to produce a rational scale of harm, based on assessment by an expert panel of physical risk, dependence liability and social cost was reported by Nutt et al. (2010), who have argued that such ratings should influence how governments police and punish people for supplying and using particular drugs. As expected, ethanol, heroin and cocaine were judged to be the most harmful, with cannabis, LSD and ecstasy (MDMA, see Ch. 47) much less so—an order that is not reflected in the classification of these drugs under UK law.2
Drug Dependence
Drug dependence describes the human condition in which drug taking becomes compulsive, taking precedence over other needs, often with serious adverse consequences. Dependence becomes a problem when:
Examples of the latter are the mental incapacity and liver damage caused by ethanol, the many diseases associated with smoking tobacco, the high risk of infection when injecting intravenously (especially HIV), the serious risk of overdosage with most opioids and the criminal behaviour resorted to when drug users need to finance their habit.
Dependence may involve a state of psychological as well as physical dependence. Family studies show clearly that susceptibility to dependence is an inherited characteristic, and many candidate genes have been reported, with a particular focus on genes involved in transmitter metabolism, receptors, etc. (see Mayer & Höllt, 2005). The general conclusion is that variants of many different genes each make a small contribution to the overall susceptibility of an individual to addiction—a familiar scenario that provides few pointers for therapeutic intervention. Polymorphisms in ethanol-metabolising genes (see later section on ethanol) are the best example of genes that directly affect the tendency to abuse a drug.
Drug-Induced Reward
The common feature of the various types of psychoactive drugs that are addictive is that all produce a rewarding experience (e.g. an elevation of mood or a feeling of euphoria or calmness).
In animal studies, where the state of mood cannot be inferred directly, reward is manifest as positive reinforcement, i.e. an increase in the probability of occurrence of any behaviour that is associated with the drug experience. In conditioned place preference studies, animals receive a drug or placebo and are then placed in different environments. Subsequently, when tested in a drug-free state, they will spend more time in the environment associated with a previous rewarding drug experience. Another way of determining if a drug is rewarding is to test whether or not animals will self-administer the drug by pressing a lever to obtain it. All dependence-producing drugs are self-administered by experimental animals. Hallucinogenic drugs are not, however, normally self-administered by animals, which may indicate that, unlike humans, they find the experience non-rewarding.
Humans, of course, self-administer drugs without necessarily becoming addicted. To model the compulsive nature of addiction more accurately, extensions to the self-administration paradigm may be employed (see Deroche-Gamonet et al., 2004). Rats treated for a short time with ‘non-addictive’ doses of cocaine will self-administer the drug by bar pressing, but stop bar pressing when a signal is shown to indicate that the drug injector is disconnected, or if the drug injection is accompanied by punishment in the form of a foot shock. With more intense ‘addictive’ pretreatment, bar pressing persists at a high rate under these conditions. Models of this sort are considered more likely to replicate the situation of addiction in humans as a basis for testing therapeutic approaches, but in humans, drug dependence represents a stable change in brain function sustained by processes that are more complex and long lasting than the neurobiological changes so far studied in experimental animals.
Humans also have a choice as to whether or not they wish to experiment with and continue taking drugs—there may therefore be an element of risk taking when experimenting with drugs. In behavioural tests, some rats are observed to be much more impulsive than others (Dalley et al., 2007). These impulsive rats also show a higher rate of cocaine self-administration. Interestingly, the impulsive rats were also observed to have a lower level of expression of D2 and D3 dopamine receptors in the nucleus accumbens (see below for the importance of this brain region in drug use).
Reward pathways
 Virtually all dependence-producing drugs so far tested, including opioids, nicotine, amphetamines, ethanol and cocaine, activate the reward pathway—the mesolimbic dopaminergic pathway (see Ch. 38), that runs, via the medial forebrain bundle, from the ventral tegmental area (VTA) of the midbrain to the nucleus accumbens and limbic region (see Nestler, 2001). Even though for some of these drugs their primary sites of action may be elsewhere in the brain, they all increase the extracellular level of dopamine in the nucleus accumbens, as shown by microdialysis and other techniques (see Spanagel & Weiss, 1999). Opioids enhance the firing of VTA dopaminergic neurons by reducing the level of GABAergic inhibition (disinhibition) within the VTA, whereas amphetamine and cocaine act on dopaminergic nerve terminals in the nucleus accumbens to release dopamine or prevent its reuptake (see Ch. 14). Given that dopamine release in the nucleus accumbens is also enhanced by naturally rewarding stimuli, such as food, water, sex and nurturing, it would appear that drugs are simply activating, or overactivating, the body’s own pleasure system.
Virtually all dependence-producing drugs so far tested, including opioids, nicotine, amphetamines, ethanol and cocaine, activate the reward pathway—the mesolimbic dopaminergic pathway (see Ch. 38), that runs, via the medial forebrain bundle, from the ventral tegmental area (VTA) of the midbrain to the nucleus accumbens and limbic region (see Nestler, 2001). Even though for some of these drugs their primary sites of action may be elsewhere in the brain, they all increase the extracellular level of dopamine in the nucleus accumbens, as shown by microdialysis and other techniques (see Spanagel & Weiss, 1999). Opioids enhance the firing of VTA dopaminergic neurons by reducing the level of GABAergic inhibition (disinhibition) within the VTA, whereas amphetamine and cocaine act on dopaminergic nerve terminals in the nucleus accumbens to release dopamine or prevent its reuptake (see Ch. 14). Given that dopamine release in the nucleus accumbens is also enhanced by naturally rewarding stimuli, such as food, water, sex and nurturing, it would appear that drugs are simply activating, or overactivating, the body’s own pleasure system.
Chemical or surgical interruption of the VTA–accumbens dopaminergic pathway impairs drug-seeking behaviours in many experimental situations. Deletion of D2 receptors in a transgenic mouse strain eliminated the rewarding properties of morphine administration without eliminating other opioid effects, and it did not prevent the occurrence of physical withdrawal symptoms in morphine-dependent animals (Maldonado et al., 1997), suggesting that the dopaminergic pathway is responsible for the positive reward but not for the negative withdrawal effects. However, D2-receptor antagonists (antipsychotic drugs; see Ch. 45) have not been successful in treating addiction, and more recent evidence (see Heidbreder & Hagan, 2005) suggests that D3 receptors play an important role. The development of D3-receptor antagonists or partial agonists as treatments for drug abuse is awaited. Other mediators, particularly 5-hydroxytryptamine, glutamate and GABA, have also been implicated in the conditioning mechanisms that reinforce drug-seeking behaviour, and a variety of pharmacological strategies based on blocking these pathways are being explored (see Heidbreder & Hagan, 2005).
Psychological Dependence
Having experienced the rewarding effects of a drug, an individual may desire to repeat the experience. The memory of previous drug-induced experiences can be very intense and long lasting, giving rise to craving; it may drive an individual to take the drug again—referred to as relapse when someone is trying to come off a drug (see Weiss, 2005). Craving may be triggered by cues such as experiencing the environment that a person associates with previously taking the drug or the sight of drug administration paraphernalia (e.g. a crack pipe or syringe). Coupled with the direct rewarding effect of the drug, cessation of drug use may be associated with an aversive psychological effect from which the subject will attempt to escape by self-administering the drug.
The psychological factors in drug dependence are discussed in detail by Koob & Le Moal (2006) and summarised in Figure 48.2.
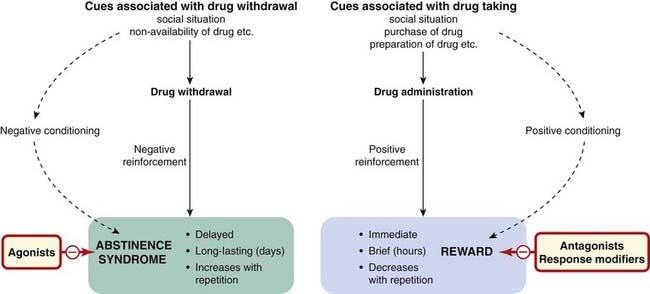
Physical Dependence
This condition is characterised by a withdrawal or abstinence syndrome whereby on cessation of drug administration or on administering an antagonist, adverse physiological effects are experienced over a period of days or weeks, the precise withdrawal responses being characteristic of the type of drug taken. Withdrawal responses can be observed in animals after chronic drug administration. The intensity of the withdrawal syndrome also varies between drugs of the same type (e.g. withdrawal from methadone is less intense but more prolonged than that from heroin, one of the reasons behind methadone maintenance treatment of heroin users). Pharmacological intervention can be used to reduce the intensity of the withdrawal (see Table 48.3). Several types of therapeutic drug, including antidepressant and antipsychotic agents, also produce withdrawal symptoms on cessation of administration but it is important to distinguish this type of commonly observed ‘rebound’ phenomenon from the physical dependence associated with drugs of abuse.
Table 48.3 Pharmacological approaches to treating drug dependence
| Mechanism | Example(s) |
|---|---|
| To alleviate withdrawal symptoms | Methadone (orally active) used short term to blunt opioid withdrawal Ibogaine (a naturally occurring psychoactive agent) used by some to reduce opioid withdrawal α2 Adrenoceptor agonists (e.g. clonidine, lofexidine) to diminish opioid, alcohol and nicotine withdrawal symptoms β Adrenoceptor antagonists (e.g. propranolol) to diminish excessive peripheral sympathetic activity Benzodiazepines, clomethiazole, topiramate and γ-hydroxybutyric acid (GHB) to blunt alcohol withdrawal |
| Long-term substitution | Methadone, buprenorphine or legal heroin to maintain opioid-dependent patients Nicotine patches or chewing gum Varenicline (α4β2 nicotinic receptor partial agonist) |
| Blocking response | Naltrexone to block opioid effects in drug-withdrawn patients Mecamylamine to block nicotine effects Immunisation against cocaine and nicotine to produce circulating antibody (still being developed) |
| Aversive therapies | Disulfiram to induce unpleasant response to ethanol |
| Reducing continued drug use (may act by reducing craving) | Bupropion (antidepressant with some nicotinic receptor antagonist activity) to reduce tobacco use Naltrexone to reduce ethanol use Clonidine (α2 adrenoceptor agonist) to reduce craving for nicotinea Acamprosate (NMDA receptor antagonist) to treat alcoholisma Topiramate and lamotrogine (antiepileptic agents) to treat alcoholism and cocaine usea γ-Hydroxybutyric acid (GHB) reported to reduce craving for alcohol and cocainea Baclofen reported to reduce opioid, alcohol and stimulant usea Ibogaine reported to reduce craving for stimulants and opioidsa |
Notes: Antidepressant, mood stabilising, anxiolytic and antipsychotic medications are useful when treating patients who, in addition to their drug use, also suffer from other mental disorders. The cannabinoid CB1-receptor antagonist rimonabant, in addition to its antiobesity effects, also reduces nicotine, ethanol, stimulant and opioid consumption. However, it also induces depression and its use has been discontinued.
a How effective these agents are at reducing the continued use of other drugs of abuse over and above the ones listed remains to be determined.
See Web links in the reference list for further information on treatments of drug dependence and Myrick & Anton (1998) for treatment of alcohol withdrawal.
Physical dependence is less important in sustaining drug-seeking behaviour than psychological dependence. A degree of physical dependence is common when patients receive opioid analgesics in hospital for several days, but this rarely leads to addiction. On the other hand, heroin users who are nursed through and recover fully from the physical abstinence syndrome are still extremely likely to revert to drug taking later. Therefore although physical dependence may influence the drive to retake a drug, it is not the major factor in long-term drug dependence.
Drug dependence 
Tolerance
Tolerance (see Ch. 2) describes the decrease in pharmacological effect on repeated administration of a drug—it develops over time as does the state of dependence. It does not occur with all drugs of abuse.
Mechanisms of Dependence and Tolerance
Drug users report that visual cues—such as the sight of a crack pipe or of a syringe—can evoke intense memories of the drug experience and induce strong craving for the drug which may precipitate relapse. This suggests that associative learning may be a major factor in psychological dependence (Robbins et al., 2008). It has been suggested that drugs alter memory formation to enhance the recollection of previous drug experience. In this regard, it is of interest that several drugs produce changes in synaptic plasticity, a cellular correlate of memory formation (see Ch. 37). While cocaine, morphine, nicotine and ethanol enhance long-term potentiation (LTP) in the VTA by increasing the expression of AMPA receptors on the plasma membrane, cocaine also increases long-term depression (LTD) in the nucleus accumbens (Hyman et al., 2006).
It was for many years assumed that physical dependence and tolerance were produced by the same underlying adaptive mechanisms. This is now generally accepted to not be the case (see Bailey & Connor, 2005).
The mechanisms responsible for the withdrawal syndrome have been most fully characterised for opioid dependence but similar mechanisms may apply to cocaine and ethanol withdrawal. At the cellular level, withdrawal of opioids results in a rebound increase in cAMP production as a result of ‘superactivation’ of adenylyl cyclase as well as upregulation of the amount of this enzyme. This results in activation of protein kinase A (PKA), in an increase in adenosine as a consequence of the conversion of cAMP to adenosine and in activation of a transcription factor—cAMP response element binding protein (CREB). The rise in PKA activity increases the excitability of nerve terminals by phosphorylating neurotransmitter transporters thus increasing their ionic conductance (see Bagley et al., 2005) and in an increase in neurotransmitter release by a direct action on the secretory process (Williams et al., 2001). Withdrawal results in enhanced GABA release in various parts of the brain, probably through the mechanisms described above. The release of other neurotransmitters is also likely to be enhanced. On the other hand, the enhanced extracellular levels of adenosine, acting on presynaptic A1 receptors (see Ch. 16), acts to inhibit glutamate release at excitatory synapses, and thus counteracts the neuronal hyperexcitability that occurs during drug withdrawal, suggesting the possibility—not yet clinically proven—that adenosine agonists might prove useful in treating drug dependence. CREB, which is upregulated in the nucleus accumbens by prolonged administration of opioids or cocaine, plays a key role in regulating various components of cAMP signalling pathways, and transgenic animals lacking CREB show reduced withdrawal symptoms (see Chao & Nestler, 2004).
For drugs such as opioids that are agonists at specific receptors (see Ch. 41), cellular tolerance results from desensitisation of the receptor. On prolonged activation by an agonist, the µ opioid receptor (MOPr) is phosphorylated by various intracellular kinases—including G-protein-coupled receptor kinases (GRKs), protein kinase C (PKC), mitogen-activated protein kinase (MAPK) and Ca2+/calmodulin-dependent protein kinase II (CamKII)—which either directly desensitises the receptor or causes the binding to the receptor of other proteins, such as arrestins, that uncouple the receptor from its G-protein (see Bailey & Connor, 2005). In the intact animal, inhibition or knockout of these kinases reduces the level of tolerance. It has also been reported that blockade of neurokinin, calcitonin gene-related peptide (CGRP) and NMDA receptors reduces opioid tolerance in vivo. This may be because the activity of some of the kinases involved in MOPr desensitisation (e.g. PKC and CamKII) in neurons is enhanced when these other receptors are activated.
Clinical use of drugs in substance dependence 
Tobacco dependence
Alcohol dependence
Opioid dependence
Pharmacological Approaches to Treating Drug Addiction
From the discussion above, it will be clear that drug abuse involves many psychosocial and some genetic factors, as well as neuropharmacological mechanisms, so drug treatment is only one component of the therapeutic approaches that are used. The main pharmacological approaches (see O’Brien, 1997; Heidbreder & Hagan, 2005) are summarised in Table 48.3. For information on other approaches to the treatment of drug addiction, readers are advised to follow the Web link given at the end of this chapter to the National Institute on Drug Abuse (NIDA).
A new approach to the treatment of drug dependence, so far applied mainly to nicotine and cocaine, is the development of vaccines (see Bunce et al., 2003) consisting of the drug molecule complexed to a protein. Antibodies produced in response to injection of the complex also bind the free drug, thereby preventing it from reaching the brain. This strategy is effective in animal models involving self-administration, and clinical trials in humans are in progress.
Nicotine and Tobacco
Tobacco growing, chewing and smoking was indigenous throughout the American subcontinent and Australia at the time that European explorers first visited these places. Smoking spread through Europe during the 16th century, coming to England mainly as a result of its enthusiastic espousal by Raleigh at the court of Elizabeth I. James I strongly disapproved of both Raleigh and tobacco, and initiated the first antismoking campaign in the early 17th century with the support of the Royal College of Physicians. Parliament responded by imposing a substantial duty on tobacco, thereby setting up the dilemma (from which we show no sign of being able to escape) of giving the State an economic interest in the continuation of smoking at the same time that its official expert advisers were issuing emphatic warnings about its dangers.
Until the latter half of the 19th century, tobacco was smoked in pipes, and primarily by men. Cigarette manufacture began at the end of the 19th century, and now cigarettes account for 98% of tobacco consumption. Filter cigarettes (which give a somewhat lower delivery of tar and nicotine than standard cigarettes) and ‘low-tar’ cigarettes (which are also low in nicotine) constitute an increasing proportion of the total.3 Cigarette consumption across the globe continues to rise (Fig. 48.3).4 There are about 1.1 billion smokers in the world (18% of the population), and the number in developing countries is increasing rapidly. Six trillion (6 × 1012) cigarettes are sold each year, more than 900 cigarettes for every man, woman and child on the planet. In 2010, 12 million cigarettes per minute will be smoked around the world.
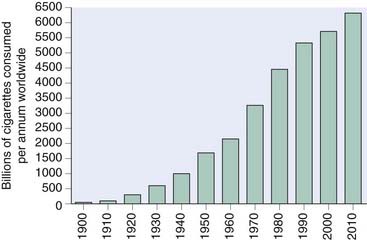
Fig. 48.3 Global cigarette consumption per annum 1900–2010.
(Data from http://www.tobaccoatlas.org/consumption.html.)
For reviews on nicotine and smoking, see Balfour & Fagerstrom (1996) and Benowitz (1996).
Tobacco smoking
Pharmacological Effects of Smoking
Nicotine5 is the main pharmacologically active substance in tobacco smoke. The acute effects of smoking can be mimicked by injection of nicotine and are blocked by mecamylamine, an antagonist at neuronal nicotinic acetylcholine receptors (nAChRs; see Ch. 13).
Effects on the central nervous system
The central effects of nicotine are complex and cannot be summed up overall simply in terms of stimulation or inhibition. At the cellular level, nicotine acts on nAChRs (see Ch. 38), which are widely expressed in the brain, particularly in the cortex and hippocampus, and are believed to play a role in cognitive function, as well as in the VTA, from which dopaminergic neurons project to the nucleus accumbens (the reward pathway, see above). nAChRs are ligand-gated cation channels located both pre- and postsynaptically, causing, respectively, enhanced transmitter release and neuronal excitation (see Wonnacott et al., 2005). Of the various subtypes of nAChR, the α4β2 and α7 subtypes (see Ch. 13) have received most attention, but other subtypes may also be involved in the rewarding effects of nicotine. As well as activating the receptors, nicotine also causes desensitisation, which may be an important component of its effects, because the effects of a dose of nicotine are diminished in animals after sustained exposure to the drug. Chronic nicotine administration leads to a substantial increase in the number of nAChRs (an effect opposite to that produced by sustained administration of most receptor agonists), which may represent an adaptive response to prolonged receptor desensitisation. It is likely that the overall effect of nicotine reflects a balance between activation of nAChRs, causing neuronal excitation, and desensitisation, causing synaptic block.
At the spinal level, nicotine inhibits spinal reflexes, causing skeletal muscle relaxation that can be measured by electromyography. This may be due to stimulation of the inhibitory Renshaw cells in the ventral horn of the spinal cord. The higher level functioning of the brain, as reflected in the subjective sense of alertness or by the electroencephalography (EEG) pattern, can be affected in either direction by nicotine, according to dose and circumstances. Smokers report that smoking wakes them up when they are drowsy and calms them down when they are tense, and EEG recordings broadly bear this out. It also seems that small doses of nicotine tend to cause arousal, whereas large doses do the reverse. Tests of motor and sensory performance (e.g. reaction time measurements or vigilance tests) in humans generally show improvement after smoking, and nicotine enhances learning in rats.
Some elaborate tests have been conducted to see, for example, whether the effect of nicotine on performance and aggression varies according to the amount of stress. Some tests border on nasty-mindedness, such as one in which subjects played a complicated logical game with a computer that initially played fair and then began to cheat randomly, causing stress and aggression in the subjects and a decline in their performance. Smoking, it was reported, did not reduce the anger but did reduce the decline in performance.
Nicotine and other nicotinic agonists such as epibatidine (Ch. 41) have significant analgesic activity.
Peripheral effects
The peripheral effects of small doses of nicotine result from stimulation of autonomic ganglia (see Ch. 13) and of peripheral sensory receptors, mainly in the heart and lungs. Stimulation of these receptors elicits various autonomic reflex responses, causing tachycardia, increased cardiac output and increased arterial pressure, reduction of gastrointestinal motility and sweating. When people smoke for the first time, they usually experience nausea and sometimes vomit, probably because of stimulation of sensory receptors in the stomach. All these effects decline with repeated dosage, although the central effects remain. Secretion of adrenaline and noradrenaline from the adrenal medulla contributes to the cardiovascular effects, and release of antidiuretic hormone from the posterior pituitary causes a decrease in urine flow.6 The plasma concentration of free fatty acids is increased, probably owing to sympathetic stimulation and adrenaline secretion.
Smokers weigh, on average, about 4 kg less than non-smokers, mainly because of reduced food intake; giving up smoking usually causes weight gain associated with increased food intake.
Pharmacokinetic Aspects
An average cigarette contains about 0.8 g of tobacco and 9–17 mg of nicotine, of which about 10% is normally absorbed by the smoker. This fraction varies greatly with the habits of the smoker and the type of cigarette.
Nicotine in cigarette smoke is rapidly absorbed from the lungs but poorly from the mouth and nasopharynx. Therefore, inhalation is required to give appreciable absorption of nicotine, each puff delivering a distinct bolus of drug to the CNS. Pipe or cigar smoke is less acidic than cigarette smoke, and the nicotine tends to be absorbed from the mouth and nasopharynx rather than the lungs. Absorption is considerably slower than from inhaled cigarette smoke, resulting in a later and longer-lasting peak in the plasma nicotine concentration (Fig. 48.4). An average cigarette, smoked over 10 min, causes the plasma nicotine concentration to rise to 15–30 ng/ml (100–200 nmol/l), falling to about half within 10 min and then more slowly over the next 1–2 h. The rapid decline results mainly from redistribution between the blood and other tissues; the slower decline is due to hepatic metabolism, mainly by oxidation to an inactive ketone metabolite, cotinine. This has a long plasma half-life, and measurement of plasma nicotine concentration provides a useful measure of smoking behaviour. A nicotine patch applied for 24 h causes the plasma concentration to rise to 75–150 nmol/l over 6 h and to remain fairly constant for about 20 h. Administration by nasal spray or chewing gum results in a time course intermediate between that of smoking and the nicotine patch.
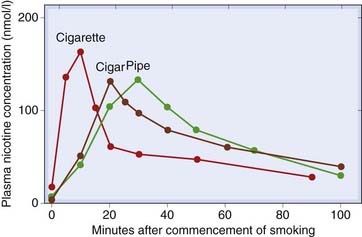
Tolerance and Dependence
As with other dependence-producing drugs, three separate processes—psychological dependence, physical dependence and tolerance—contribute to the overall state of dependence, in which taking the drug becomes compulsive.
The effects of nicotine associated with peripheral ganglionic stimulation show rapid tolerance, perhaps as a result of desensitisation of nAChRs by nicotine. With large doses of nicotine, this desensitisation produces a block of ganglionic transmission rather than stimulation (see Ch. 13). Tolerance to the central effects of nicotine (e.g. in the arousal response) is much less than in the periphery. The increase in the number of nAChRs in the brain produced by chronic nicotine administration in animals (see above) also occurs in heavy smokers. Because the cellular effects of nicotine are diminished, it is possible that the additional binding sites represent desensitised rather than functional receptors.
The addictiveness of smoking is due to the effects of nicotine combined with the ritual of smoking (see Le Foll & Goldberg, 2005). Rats choose to drink dilute nicotine solution in preference to water if given a choice, and in a situation in which lever pressing causes an injection of nicotine to be delivered—admittedly at high doses—they quickly learn to self-administer it. Similarly, monkeys who have been trained to smoke, by providing a reward in response to smoking behaviour, will continue to do so spontaneously (i.e. unrewarded) if the smoking medium contains nicotine, but not if nicotine-free tobacco is offered instead. Humans, however, are unlikely to become addicted to nicotine delivered from patches suggesting that other factors are also involved, such as the controlled pulsatile delivery associated with smoking.
Like other addictive drugs (see above), nicotine causes excitation of the mesolimbic reward pathway and increased dopamine release in the nucleus accumbens. Transgenic mice lacking the β2 subunit of the acetylcholine receptor lose the rewarding effect of nicotine and its dopamine-releasing effect, confirming the importance of the α4β2 nAChR subtype and mesolimbic dopamine release in the response to nicotine. In contrast to normal mice, the mutant mice could not be induced to self-administer nicotine, even though they did so with cocaine.
A physical withdrawal syndrome occurs in humans on cessation of smoking. Its main features are increased irritability, impaired performance of psychomotor tasks, aggressiveness and sleep disturbance. The withdrawal syndrome is much less severe than that produced by opioids, and it can be alleviated not only by nicotine but also by amphetamine, a finding consistent with the postulated role of dopamine in the reward pathway. The withdrawal syndrome lasts for 2–3 weeks, although the craving for cigarettes persists for much longer than this; relapses during attempts to give up cigarette smoking occur most commonly at a time when the physical withdrawal syndrome has long since subsided.
Pharmacology of nicotine
Harmful Effects of Smoking
The life expectancy of smokers is shorter than that of non-smokers. Smoking causes almost 90% of deaths from lung cancer, around 80% of deaths from bronchitis and emphysema, and around 17% of deaths from heart disease. About one-third of all cancer deaths can be attributed to smoking. Smoking is, by a large margin, the biggest preventable cause of death, responsible for about 1 in 10 adult deaths worldwide. Deaths from smoking are continuing to rise. In 1990, smoking was responsible for 10% (3 million out of 30 million) of deaths worldwide; by 2030, this is expected to increase to 17% (10 million out of 60 million), mainly due to the growth of smoking in Asia, Africa and Latin America (Peto et al., 1996).
The main health risks are as follows:
The agents probably responsible for the harmful effects are as follows:
Low-tar cigarettes give a lower yield of both tar and nicotine than standard cigarettes. However, it has been shown that smokers puff harder, inhale more and smoke more cigarettes when low-tar brands are substituted for standard brands. The end result may be a slightly reduced intake of tar and nicotine but an increase in carbon monoxide intake, with no net gain in terms of safety.
Other Effects of Smoking
Parkinson’s disease is approximately twice as common in non-smokers as in smokers. It is possible that this reflects a protective effect of nicotine, but it could be that common genetic or environmental factors underlie smoking behaviour and susceptibility to Parkinson’s disease. Ulcerative colitis appears to be a disease of non-smokers. Former smokers are at high risk for developing ulcerative colitis, while current smokers have the least risk. This tendency indicates that smoking cigarettes may prevent the onset of ulcerative colitis. In contrast, smoking tends to worsen the effects of Crohn’s disease. Earlier reports that Alzheimer’s disease is less common in smokers have not been confirmed; indeed there is evidence that smoking may increase the occurrence of Alzheimer’s disease in some genetic groups.
Effects of smoking
Pharmacological Approaches to Treating Nicotine Dependence
Most smokers would like to quit, but few succeed.7 The most successful smoking cure clinics, using a combination of psychological and pharmacological treatments, achieve a success rate of about 25%, measured as the percentage of patients still abstinent after 1 year. The two main pharmacological treatments (see George & O’Malley, 2004) are nicotine replacement therapy and bupropion (also used to treat depression; see Table 46.2). A nAChR partial agonist, varenicline has recently been introduced.
Nicotine replacement therapy is used mainly to assist smokers to quit by reducing craving and physical withdrawal symptoms. Because nicotine is relatively short acting and not well absorbed from the gastrointestinal tract, it is given either in the form of chewing gum, used several times daily, or as a transdermal patch that is replaced daily. These preparations cause various side effects, particularly nausea and gastrointestinal cramps, cough, insomnia and muscle pains. There is a risk that nicotine may cause coronary spasm in patients with heart disease. Transdermal patches may cause local irritation and itching. The conclusion of many double-blind trials of nicotine against placebo is that these preparations, combined with professional counselling and supportive therapy, roughly double the chances of successfully breaking the smoking habit, but the success rate measured as abstinence 1 year after ceasing treatment is still only about 25%. Nicotine on its own, without counselling and support, is no more effective than placebo, so its use as an over-the-counter smoking remedy has little justification. Although of limited value as an aid to abstinence, the long-term use of nicotine can significantly reduce cigarette consumption by smokers. In Sweden, the use of ‘smokeless tobacco’ is encouraged and the smoking-related death rate is much lower than elsewhere in Europe or North America.
The identification of the α4β2 nAChR subtype as the main nAChR subtype in the brain involved in the rewarding properties of tobacco smoking may allow selective agonists to be developed as nicotine substitutes with fewer side effects. Varenicline is a partial agonist at the α4β2 nicotinic receptor subtype and has differing levels of efficacy at other subtypes. Being a partial agonist it may provide a level of substitution while at the same time blocking the rewarding effect of smoking. It is effective in preventing relapse but there has been some concern that it may induce suicidal thoughts, suicide attempts, aggression and homicide. However, a recent large retrospective study (Gunnell et al., 2009) found no evidence of increased suicide or suicidal thoughts with varenicline, compared with other antismoking treatments.
Bupropion (Ch. 46) appears to be as effective as nicotine replacement therapy, even in non-depressed patients, and has fewer side effects. However, bupropion lowers the seizure threshold so should not be prescribed if there are other risk factors for seizures (including other drugs that lower seizure threshold). It is also contraindicated if there is a history of eating disorders or of bipolar mood disorder, and is used only with caution in patients with liver or renal disease. Because of these problems, nicotine remains the pharmacological treatment of choice in most cases.
Bupropion may act by increasing dopamine activity in the nucleus accumbens. It is a weak blocker of dopamine and noradrenaline uptake, but it is not clear that this accounts for its efficacy in treating nicotine dependence. It is usually given as a slow-release formulation.
Many other drugs have been tested clinically and shown to be useful in some cases. They include the following:
Ethanol
Judged on a molar basis, the consumption of ethanol far exceeds that of any other drug. The ethanol content of various drinks ranges from about 2.5% (weak beer) to about 55% (strong spirits), and the size of the normal measure is such that a single drink usually contains about 8–12 g (0.17–0.26 mol) of ethanol. Its low pharmacological potency is reflected in the range of plasma concentrations needed to produce pharmacological effects: minimal effects occur at about 10 mmol/l (46 mg/100 ml), and 10 times this concentration may be lethal. The average per capita ethanol consumption in the UK was 11.7 l/year (expressed as pure ethanol) in 2007, a figure that has doubled since 1970, the main changes having been a growing consumption of wine in preference to beer among adults and an increasing tendency for binge drinking, especially among young people.
For practical purposes, ethanol intake is often expressed in terms of units. One unit is equal to 8 g (10 ml) of ethanol, and is the amount contained in half a pint of normal strength beer, one measure of spirits or one small glass of wine. Based on the health risks described below, the current official recommendation is a maximum of 21 units/week for men and 14 units/week for women. It is estimated that in the UK, about 33% of men and 13% of women exceed these levels. The annual tax revenue from drink amounts to about £7 billion, whereas the health cost is estimated at £3 billion, and the social cost undoubtedly greater. Governments in most developed countries are attempting to curb alcohol consumption.
An excellent detailed review of all aspects of alcohol and alcoholism is provided by Spanagel (2009).
Pharmacological Effects of Ethanol
Effects on central nervous system neurons
The main effects of ethanol are on the central nervous system (CNS; see reviews by Charness et al., 1989; Spanagel, 2009), where its depressant actions resemble those of volatile anaesthetics (Ch. 40). At a cellular level, the effect of ethanol is depressant, although it increases neuronal activity—presumably by disinhibition—in some parts of the CNS, notably in the mesolimbic dopaminergic pathway that is involved in reward. The main acute cellular effects of ethanol that occur at concentrations (5–100 mM) relevant to alcohol consumption by humans are:
For reviews see Tabakoff & Hoffman (1996), Lovinger (1997) and Harris et al. (2008).
Ethanol enhances the action of GABA on GABAA receptors in a similar way to benzodiazepines (see Ch. 43). Its effect is, however, smaller and less consistent than that of benzodiazepines, and no clear effect on inhibitory synaptic transmission in the CNS has been demonstrated for ethanol. This may be because the effect of ethanol is seen only on some subtypes of GABAA receptor (see Ch. 37). Exactly which GABAA receptor subtypes are sensitive to ethanol is still unclear but those containing α6 and δ subunits appear to be important. Ethanol may also act presynaptically to enhance GABA release. The benzodiazepine inverse agonist flumazenil (see Ch. 43) reverses the central depressant actions of ethanol by a non-competitive interaction on the GABAA receptor. The use of flumazenil to reverse ethanol intoxication and treat dependence has not found favour for several reasons. Because flumazenil is an inverse agonist (see Ch. 2) at benzodiazepine receptors, it carries a risk of causing seizures, and it could cause an increase in ethanol consumption and thus increase long-term toxic manifestations.
Ethanol produces a consistent enhancement of glycine receptor function. This effect is likely to be due both to a direct interaction of ethanol with the α1 subunit of the glycine receptor and to indirect effects of ethanol mediated through PKC activation. Ethanol can also enhance glycine release from nerve terminals.
Ethanol reduces transmitter release in response to nerve terminal depolarisation by inhibiting the opening of voltage-sensitive calcium channels in neurons. It also reduces neuronal excitability by activating G-protein-activated inwardly rectifying K+ (GIRK) channels as well as potentiating calcium-activated potassium (BK) channel activity.
The excitatory effects of glutamate are inhibited by ethanol at concentrations that produce CNS depressant effects in vivo. NMDA receptor activation is inhibited at lower ethanol concentrations than are required to affect AMPA receptors (see Ch. 37). Other effects produced by ethanol include an enhancement of the excitatory effects produced by activation of nAChRs and 5-HT3 receptors. The relative importance of these various effects in the overall effects of ethanol on CNS function is not clear at present.
The depressant effects of ethanol on neuronal function resemble those of adenosine acting on A1 receptors (see Ch. 16). Ethanol in cell culture systems increases extracellular adenosine by inhibiting adenosine uptake, and there is some evidence that inhibition of the adenosine transporter may account for some of its CNS effects (Melendez & Kalivas, 2004).
Endogenous opioids also play a role in the CNS effects of ethanol, because both human and animal studies show that the opioid receptor antagonist naltrexone reduces the reward associated with ethanol.
Behavioural effects
The effects of acute ethanol intoxication in humans are well known and include slurred speech, motor incoordination, increased self-confidence and euphoria. The effect on mood varies among individuals, most becoming louder and more outgoing, but some becoming morose and withdrawn. At higher levels of intoxication, the mood tends to become highly labile, with euphoria and melancholy, aggression and submission, often occurring successively. The association between alcohol and violence is well documented.
Intellectual and motor performance and sensory discrimination show uniform impairment by ethanol, but subjects are generally unable to judge this for themselves. For example, bus drivers were asked to drive through a gap that they selected as the minimum for their bus to pass through; ethanol caused them not only to hit the barriers more often at any given gap setting, but also to set the gap to a narrower dimension, often narrower than the bus.
Much effort has gone into measuring the effect of ethanol on driving performance in real life, as opposed to artificial tests under experimental conditions. In an American study of city drivers, it was found that the probability of being involved in an accident was unaffected at blood ethanol concentrations up to 50 mg/100 ml (10.9 mmol/l); by 80 mg/100 ml (17.4 mmol/l), the probability was increased about four-fold, and by 150 mg/100 ml (32.6 mmol/l) about 25-fold. In the UK, driving with a blood ethanol concentration greater than 80 mg/100 ml is illegal.
The relationship between plasma ethanol concentration and effect is highly variable. A given concentration produces a larger effect when the concentration is rising than when it is steady or falling. A substantial degree of cellular tolerance develops in habitual drinkers, with the result that a higher plasma ethanol concentration is needed to produce a given effect. In one study, ‘gross intoxication’ (assessed by a battery of tests that measured speech, gait and so on) occurred in 30% of subjects between 50 and 100 mg/100 ml and in 90% of subjects with more than 150 mg/100 ml. Coma generally occurs at about 400 mg/100 ml, and death from respiratory failure is likely at levels exceeding 500 mg/100 ml.
Ethanol significantly enhances—sometimes to a dangerous extent—the CNS depressant effects of many other drugs, including benzodiazepines, antidepressants, antipsychotic drugs and opioids. Combined use of ethanol and cocaine leads to the formation of cocaethylene, a toxic metabolite of cocaine.
Neurotoxicity
In addition to the acute effects of ethanol on the nervous system, chronic administration also causes irreversible neurological damage (see Harper & Matsumoto, 2005). This may be due to ethanol itself, or to metabolites such as acetaldehyde or fatty acid esters. Binge drinking is thought to produce greater damage; probably due to the high brain concentrations of ethanol achieved and to repeated phases of withdrawal between binges. Heavy drinkers often exhibit convulsions and may develop irreversible dementia and motor impairment associated with thinning of the cerebral cortex (apparent as ventricular enlargement) detectable by brain-imaging techniques. Degeneration in the cerebellum and other specific brain regions can also occur, as well as peripheral neuropathy. Some of these changes are not due to ethanol itself but to accompanying thiamine deficiency, which is common in alcoholics.
Effects on other systems
The main acute cardiovascular effect of ethanol is to produce cutaneous vasodilatation, central in origin, which causes a warm feeling but actually increases heat loss.9 Paradoxically, there is a positive correlation between ethanol consumption and hypertension, possibly because ethanol withdrawal causes increased sympathetic activity. The beneficial effect of moderate drinking on cardiovascular function is discussed below.
Ethanol increases salivary and gastric secretion, perhaps a reason in some cultures for the popularity of a glass of sherry before dinner. This is partly a reflex effect produced by the taste and irritant action of ethanol. However, heavy consumption of spirits causes damage directly to the gastric mucosa, causing chronic gastritis. Both this and the increased acid secretion are factors in the high incidence of gastric bleeding in alcoholics. CNS depression predisposes to aspiration pneumonia and lung abscess formation. Acute pancreatitis may become chronic with pseudocyst formation (collections of fluid in the peritoneal sac), fat malabsorption and ultimately loss of B-cell function and insulin-dependent diabetes mellitus.
Ethanol produces a variety of endocrine effects. In particular, it increases the output of adrenal steroid hormones by stimulating the anterior pituitary gland to secrete adrenocorticotrophic hormone. However, the increase in plasma hydrocortisone usually seen in alcoholics (producing a ‘pseudo-Cushing’s syndrome’; Ch. 32) is due partly to inhibition by ethanol of hydrocortisone metabolism in the liver.
Diuresis is a familiar effect of ethanol. It is caused by inhibition of antidiuretic hormone secretion, and tolerance develops rapidly, so that the diuresis is not sustained. There is a similar inhibition of oxytocin secretion, which can delay parturition. Attempts have been made to use this effect in premature labour, but the dose needed is large enough to cause obvious drunkenness in the mother. If the baby is born prematurely despite the ethanol, it too may be intoxicated at birth, sufficiently for respiration to be depressed. The procedure evidently has serious disadvantages.
Acute toxic effects on muscle are exacerbated by seizures and prolonged immobility; severe myositis (‘rhabdomyolysis’) with myoglobinuria can cause acute renal failure. Chronic toxicity affects particularly cardiac striated muscle giving rise to alcoholic cardiomyopathy and chronic heart failure.
Chronic ethanol consumption may also result in immunosuppression, leading to increased incidence of infections such as pneumonia (immunisation with pneumococcal vaccine is important in chronic alcoholics); and increased cancer risk, particularly of the mouth, larynx and oesophagus.
Male alcoholics are often impotent and show signs of feminisation. This is associated with impaired testicular steroid synthesis, but induction of hepatic microsomal enzymes by ethanol, and hence an increased rate of testosterone inactivation, also contributes.
Effects of ethanol on the liver
Together with brain damage, liver damage is the most common serious long-term consequence of excessive ethanol consumption (see Lieber, 1995). Increased fat accumulation (fatty liver) progresses to hepatitis (i.e. inflammation of the liver) and eventually to irreversible hepatic necrosis and fibrosis. Cirrhosis is an end stage with extensive fibrosis and foci of regenerating hepatocytes that are not correctly ‘plumbed in’ to the blood and biliary systems. Diversion of portal blood flow around the cirrhotic liver often causes oesophageal varices to develop, which can bleed suddenly and catastrophically. Increased fat accumulation in the liver occurs, in rats or in humans, after a single large dose of ethanol. The mechanism is complex, the main factors being:
With chronic ethanol consumption, many other factors contribute to the liver damage. One is malnutrition, for alcoholic individuals may satisfy much of their calorie requirement from ethanol itself. Three hundred grams of ethanol (equivalent to one bottle of whisky) provides about 2000 kcal but, unlike a normal diet, it provides no vitamins, amino acids or fatty acids. Thiamine deficiency is an important factor in causing chronic neurological damage (see above). The hepatic changes occurring in alcoholics are partly due to chronic malnutrition but mainly to the cellular toxicity of ethanol, which promotes inflammatory changes in the liver.
The overall incidence of chronic liver disease is a function of cumulative ethanol consumption over many years. Therefore, overall consumption, expressed as g/kg of body weight per day multiplied by years of drinking, provides an accurate predictor of the incidence of cirrhosis. An increase in the plasma concentration of the liver enzyme γ-glutamyl transpeptidase (a marker of CYP induction) often raises the suspicion of alcohol-related liver damage, although not specific to ethanol.
Effects on lipid metabolism, platelet function and atherosclerosis
Moderate drinking reduces mortality associated with coronary heart disease, the maximum effect—about 30% reduction of mortality overall—being achieved at a level of 2–3 units/day (see Groenbaek et al., 1994). The effect is much more pronounced (> 50% reduction) in men with high plasma concentrations of low-density-lipoprotein cholesterol (see Ch. 23).10 Most evidence suggests that ethanol, rather than any specific beverage, such as red wine, is the essential factor.
Two mechanisms have been proposed. The first involves the effect of ethanol on the plasma lipoproteins that are the carrier molecules for cholesterol and other lipids in the bloodstream (see Ch. 23). Epidemiological studies, as well as studies on volunteers, have shown that ethanol, in daily doses too small to produce obvious CNS effects, can over the course of a few weeks increase plasma high-density-lipoprotein concentration, thus exerting a protective effect against atheroma formation.
Ethanol may also protect against ischaemic heart disease by inhibiting platelet aggregation. This effect occurs at ethanol concentrations in the range achieved by normal drinking in humans (10–20 mmol/l) and probably results from inhibition of arachidonic acid formation from phospholipid. In humans, the magnitude of the effect depends critically on dietary fat intake, and it is not yet clear how important it is clinically.
The effect of ethanol on fetal development
The adverse effect of ethanol consumption during pregnancy on fetal development was demonstrated in the early 1970s, when the term fetal alcohol syndrome (FAS) was coined.
The features of full FAS include:
A lesser degree of impairment, termed alcohol-related neurodevelopmental disorder (ARND), results in behavioural problems, and cognitive and motor deficits, often associated with reduced brain size. Full FAS occurs in about 3 per 1000 live births and affects about 30% of children born to alcoholic mothers. It is rare with mothers who drink less than about 5 units/day, and most common in binge drinkers who sporadically consume much larger amounts, resulting in high peak levels of ethanol. ARND is about three times as common. Although there is no clearly defined safe threshold, there is no evidence that amounts less than about 2 units/day are harmful. There is no critical period during pregnancy when ethanol consumption is likely to lead to FAS, although one study suggests that FAS incidence correlates most strongly with ethanol consumption very early in pregnancy, even before pregnancy is recognised, implying that not only pregnant women, but also women who are likely to become pregnant, must be advised not to drink heavily. Experiments on rats and mice suggest that the effect on facial development may be produced very early in pregnancy (up to 4 weeks in humans), while the effect on brain development is produced rather later (up to 10 weeks).
Effects of ethanol
Pharmacokinetic Aspects
Metabolism of ethanol
Ethanol is rapidly absorbed, an appreciable amount being absorbed from the stomach. A substantial fraction is cleared by first-pass hepatic metabolism. Hepatic metabolism of ethanol shows saturation kinetics (see Chs 9 and 10) at quite low ethanol concentrations, so the fraction of ethanol removed decreases as the concentration reaching the liver increases. Thus, if ethanol absorption is rapid and portal vein concentration is high, most of the ethanol escapes into the systemic circulation, whereas with slow absorption more is removed by first-pass metabolism. This is one reason why drinking ethanol on an empty stomach produces a much greater pharmacological effect. Ethanol is quickly distributed throughout the body water, the rate of its redistribution depending mainly on the blood flow to individual tissues, as with volatile anaesthetics (see Ch. 40).
Ethanol is about 90% metabolised, 5–10% being excreted unchanged in expired air and in urine. This fraction is not pharmacokinetically significant but provides the basis for estimating blood ethanol concentration from measurements on breath or urine. The ratio of ethanol concentrations in blood and alveolar air, measured at the end of deep expiration, is relatively constant, 80 mg/100 ml of ethanol in blood producing 35 µg/100 ml in expired air, this being the basis of the breathalyser test. The concentration in urine is more variable and provides a less accurate measure of blood concentration.
Ethanol metabolism occurs almost entirely in the liver, and mainly by a pathway involving successive oxidations, first to acetaldehyde and then to acetic acid (Fig. 48.5). Since ethanol is often consumed in large quantities (compared with most drugs), 1–2 mol daily being by no means unusual, it constitutes a substantial load on the hepatic oxidative systems. The oxidation of 2 mol of ethanol consumes about 1.5 kg of the co-factor nicotinamide adenine dinucleotide (NAD+). Availability of NAD+ limits the rate of ethanol oxidation to about 8 g/h in a normal adult, independently of ethanol concentration (Fig. 48.6), causing the process to show saturating kinetics (Ch. 10). It also leads to competition between the ethanol and other metabolic substrates for the available NAD+ supplies, which may be a factor in ethanol-induced liver damage (see Ch. 57). The intermediate metabolite, acetaldehyde, is a reactive and toxic compound, and this may also contribute to the hepatotoxicity. A small degree of esterification of ethanol with various fatty acids also occurs in the tissues, and these esters may also contribute to long-term toxicity.
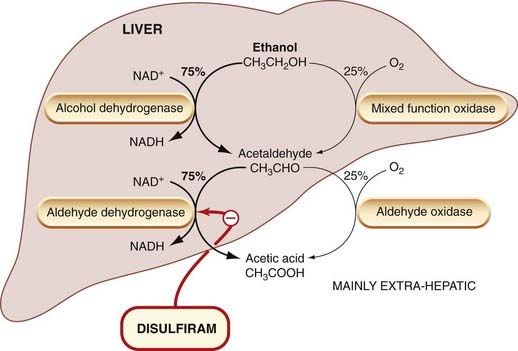
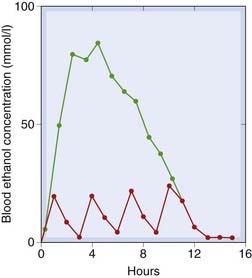
Fig. 48.6 Zero-order kinetics of ethanol elimination in rats.
Rats were given ethanol orally (104 mmol/kg) either as a single dose or as four divided doses. The single dose results in a much higher and more sustained blood ethanol concentration than the same quantity given as divided doses. Note that, after the single dose, ethanol concentration declines linearly, the rate of decline being similar after a small or large dose, because of the saturation phenomenon.
(From Kalant H et al. 1975 Biochem Pharmacol 24: 431.)
Alcohol dehydrogenase is a soluble cytoplasmic enzyme, confined mainly to liver cells, which oxidises ethanol at the same time as reducing NAD+ to NADH (Fig. 48.5). Ethanol metabolism causes the ratio of NAD+ to NADH to fall, and this has other metabolic consequences (e.g. increased lactate and slowing down of the Krebs cycle). The limitation on ethanol metabolism imposed by the limited rate of NAD+ regeneration has led to attempts to find a ‘sobering up’ agent that works by regenerating NAD+ from NADH. One such agent is fructose, which is reduced by an NADH-requiring enzyme. In large doses, it causes a measurable increase in the rate of ethanol metabolism, but not enough to have a useful effect on the rate of return to sobriety.
Normally, only a small amount of ethanol is metabolised by the microsomal mixed function oxidase system (see Ch. 9), but induction of this system occurs in alcoholics. Ethanol can affect the metabolism of other drugs that are metabolised by the mixed function oxidase system (e.g. phenobarbitone, warfarin and steroids), with an initial inhibitory effect produced by competition, followed by enhancement due to enzyme induction.
Nearly all the acetaldehyde produced is converted to acetate in the liver by aldehyde dehydrogenase (Fig. 48.5). Normally, only a little acetaldehyde escapes from the liver, giving a blood acetaldehyde concentration of 20–50 µmol/l after an intoxicating dose of ethanol in humans. The circulating acetaldehyde usually has little or no effect, but the concentration may become much larger under certain circumstances and produce toxic effects. This occurs if aldehyde dehydrogenase is inhibited by drugs such as disulfiram. In the presence of disulfiram, which produces no marked effect when given alone, ethanol consumption is followed by a severe reaction comprising flushing, tachycardia, hyperventilation, and considerable panic and distress, which is due to excessive acetaldehyde accumulation in the bloodstream. This reaction is extremely unpleasant but not harmful, and disulfiram can be used as aversion therapy to discourage people from taking ethanol. Some other drugs (e.g. metronidazole; see Ch. 50) produce similar reactions to ethanol. Interestingly, a Chinese herbal medicine, used traditionally to cure alcoholics, contains daidzin, a specific inhibitor of aldehyde dehydrogenase. In hamsters (which spontaneously consume alcohol in amounts that would defeat even the hardest two-legged drinker, while remaining, as far as one can tell in a hamster, completely sober), daidzin markedly inhibits alcohol consumption.
Genetic factors
In 50% of Asian people, an inactive genetic variant of one of the aldehyde dehydrogenase isoforms (ALDH-2) is expressed; these individuals experience a disulfiram-like reaction after alcohol, and the incidence of alcoholism in this group is extremely low (see Tanaka et al., 1997; Tyndale, 2003).
Metabolism and toxicity of methanol and ethylene glycol
Methanol is metabolised in the same way as ethanol but produces formaldehyde instead of acetaldehyde from the first oxidation step. Formaldehyde is more reactive than acetaldehyde and reacts rapidly with proteins, causing the inactivation of enzymes involved in the tricarboxylic acid cycle. It is converted to another toxic metabolite, formic acid. This, unlike acetic acid, cannot be utilised in the tricarboxylic acid cycle and is liable to cause tissue damage. Conversion of alcohols to aldehydes occurs not only in the liver but also in the retina, catalysed by the dehydrogenase responsible for retinol–retinal conversion. Formation of formaldehyde in the retina accounts for one of the main toxic effects of methanol, namely blindness, which can occur after ingestion of as little as 10 g. Formic acid production and derangement of the tricarboxylic acid cycle also produce severe acidosis.
Methanol is used as an industrial solvent and also to adulterate industrial ethanol in order to make it unfit to drink. Methanol poisoning is quite common, and used to be treated by administration of large doses of ethanol, which acts to retard methanol metabolism by competition for alcohol dehydrogenase. Fomepizole inhibits alcohol dehydrogenase and is now preferred if available. Such treatment may be in conjunction with haemodialysis to remove unchanged methanol, which has a small volume of distribution.
Poisoning with ethylene glycol, used in automobile antifreeze and brake fluid, is a medical emergency. It is rapidly absorbed from the gut and metabolised to glycolate and then more slowly to oxalate. Glycolate interferes with metabolic processes and produces metabolic acidosis. It affects the brain, heart and kidneys. Treatment is with alkali such as sodium bicarbonate to reverse the acidosis, pyridoxine and thiamine to promote conversion to non-toxic metabolites and haemodialysis.
Metabolism of ethanol
Tolerance and Dependence
Tolerance to the effects of ethanol can be demonstrated in both humans and experimental animals, to the extent of a two- to three-fold reduction in potency occurring over 1–3 weeks of continuing ethanol administration. A small component of this is due to the more rapid elimination of ethanol. The major component is cellular tolerance, which accounts for a roughly two-fold decrease in potency and which can be observed in vitro (e.g. by measuring the inhibitory effect of ethanol on transmitter release from synaptosomes) as well as in vivo. The mechanism of this tolerance is not known for certain (see Little, 1991). Ethanol tolerance is associated with tolerance to many anaesthetic agents, and alcoholics are often difficult to anaesthetise.
Chronic ethanol administration produces various changes in CNS neurons, which tend to oppose the acute cellular effects that it produces (see above). There is a small reduction in the density of GABAA receptors, and a proliferation of voltage-gated calcium channels and NMDA receptors.
A well-defined physical abstinence syndrome develops in response to ethanol withdrawal. As with most other dependence-producing drugs, this is probably important as a short-term factor in sustaining the drug habit, but other (mainly psychological) factors are more important in the longer term (see above). The physical abstinence syndrome usually subsides in a few days, but the craving for ethanol and the tendency to relapse last for very much longer.
The physical abstinence syndrome in humans, in severe form, develops after about 8 h. In the first stage, the main symptoms are tremor, nausea, sweating, fever and sometimes hallucinations. These last for about 24 h. This phase may be followed by seizures (‘rum fits’). Over the next few days, the condition of ‘delirium tremens’ develops, in which the patient becomes confused, agitated and often aggressive, and may suffer much more severe hallucinations. A similar syndrome of central and autonomic hyperactivity can be produced in experimental animals by ethanol withdrawal. Treatment of this medical emergency is by sedation with large doses of a benzodiazepine such as chlordiazepoxide (Ch. 43) together with large doses of thiamine.
Pharmacological Approaches to Treating Alcohol Dependence
Alcohol dependence (‘alcoholism’) is common (4–5% of the population) and, as with smoking, difficult to treat effectively. The main pharmacological approaches (see Garbutt, 2009; Table 48.3) are the following:
References and Further Reading
Bagley E.E., Gerke M.B., Vaughan C.W., et al. GABA transporter currents activated by protein kinase A excite midbrain neurons during opioid withdrawal. Neuron. 2005;45:433-445.
Bailey C.P., Connor M. Opioids: cellular mechanisms of tolerance and physical dependence. Curr. Opin. Pharmacol.. 2005;5:60-68.
Bunce C.J., Loudon P.T., Akers C., et al. Development of vaccines to help treat drug dependence. Curr. Opin. Mol. Ther.. 2003;5:58-63.
Chao J., Nestler E.J. Molecular neurobiology of addiction. Annu. Rev. Med.. 2004;55:113-132. (Useful review article by leading scientists in addiction research)
Dalley J.W., Fryer T.D., Brichard L., et al. Nucleus accumbens D2/3 receptors predict trait impulsivity and cocaine reinforcement. Science. 2007;315:1267-1270. (An exciting first description of the role of dopamine receptors and impulsivity in drug self-administration)
Deroche-Gamonet V., Belin D., Piazza P.V. Evidence for addiction-like behaviour in the rat. Science. 2004;305:1014-1017. (See also commentary by T.E. Robinson in the same issue, pp. 951–953, Experimental strategy for distinguishing between self-administration and addiction-like behaviour in rats)
Heidbreder C.A., Hagan J.J. Novel pharmacological approaches for the treatment of drug addiction and craving. Curr. Opin. Pharmacol.. 2005;5:107-118. (Describes the numerous theoretical strategies, based mainly on monoamine pharmacology, for treating addiction)
Hyman S.E., Malenka R.C., Nestler E.J. Neural mechanisms of addiction: the role of reward-related learning and memory. Annu. Rev. Neurosci.. 2006;29:565-598. (Extensive review on how drugs of abuse can alter memory and learning processes)
Karch S.B., editor. Drug abuse handbook, second ed, Boca Raton: CRC Press, 2006.
Koob G.F., Le Moal M. Neurobiology of addiction. London: Academic Press; 2006. (A very extensive book covering many aspects of addiction from the neuroscientist’s perspective)
Maldonado R., Saiardi A., Valverde O., et al. Absence of opiate rewarding effects in mice lacking dopamine D2 receptors. Nature. 1997;388:586-589. (Use of transgenic animals to demonstrate role of dopamine receptors in reward properties of opiates)
Mayer P., Höllt V. Genetic disposition to addictive disorders—current knowledge and future perspectives. Curr. Opin. Pharmacol.. 2005;5:4-8. (Describes the current inconclusive understanding of the genetic basis of addiction)
Measham F., Moore K. Repertoires of distinction. Exploring patterns of weekend polydrug use within local leisure scenes across the English night time economy. Criminol. Crim. Justice. 2009;9:437-464.
Nestler E.J. Molecular basis of long-term plasticity underlying addiction. Nat. Rev. Neurosci.. 2001;2:119-128. (Good review article focusing on long-term changes in gene expression associated with drug dependence)
Nestler E.J. Molecular mechanisms of drug addiction. Neuropharmacology. 2004;47(Suppl. 1):24-32.
Nutt D., King L.A., Phillips L.D. Drug harms in the UK: a multicriteria decision analysis. Lancet. 2010;376:558-565.
O’Brien C.P. A range of research-based pharmacotherapies for addiction. Science. 1997;278:66-70. (Useful overview of pharmacological approaches to treatment)
Robbins T.W., Ersche K.D., Everitt B.J. Drug addiction and the memory systems of the brain. Ann. N. Y. Acad. Sci.. 2008;1141:1-21. (Review of how different forms of memory play important roles in drug dependence)
Spanagel R., Weiss F. The dopamine hypothesis of reward: past and current research. Trends Neurosci.. 1999;22:521-527. (Summarises evidence for activation of mesolimbic dopamine pathways as a factor in drug dependence)
Weiss F. Neurobiology of craving, conditioned reward and relapse. Curr. Opin. Pharmacol.. 2005;5:9-19. (Review of recent studies on the neurobiology of addiction, focusing mainly on animal models)
Williams J.T., Christie M.J., Manzoni O. Cellular and synaptic adaptations mediating opioid dependence. Physiol. Rev.. 2001;81:299-343.
Winger G., Woods J.H., Hofmann F.G. A handbook on drug and alcohol abuse, fourth ed. New York: Oxford University Press; 2004. (Short and informative textbook on biomedical aspects)
Balfour D.J.K., Fagerstrom K.O. Pharmacology of nicotine and its therapeutic use in smoking cessation and neurodegenerative disorders. Pharmacol. Ther.. 1996;72:51-81. (Review of the pharmacology of nicotine and its usefulness as replacement therapy)
Benowitz N.L. Pharmacology of nicotine: addiction and therapeutics. Annu. Rev. Pharmacol.. 1996;36:597-613. (General review article including information on potential therapeutic uses of nicotine other than reduction of smoking)
George T.P., O’Malley S.S. Current pharmacological treatments for nicotine dependence. Trends Pharmacol. Sci.. 2004;25:42-48.
Gunnell D., Irvine D., Wise L., Davies C., Martin R.M. Varenicline and suicidal behaviour: a cohort study based on data from the General Practice Research Database. BMJ. 2009;339:b3805.
Hung R.J., McKay J.D., Gaborieau V., et al. A susceptibility locus for lung cancer maps to nicotinic acetylcholine receptor subunit genes on 15q25. Nature. 2008;452:633-637. (Original paper showing a genetic link between cancer and single nucleotide polymorphisms in the nicotinic receptor)
Le Foll B., Goldberg S.R. Control of the reinforcing effects of nicotine by associated environmental stimuli in animals and humans. Trends Pharmacol. Sci.. 2005;26:287-293.
Peto R., Lopez A.D., Boreham J., et al. Mortality from smoking worldwide. Br. Med. Bull.. 1996;52:12-21.
Wonnacott S., Sidhpura N., Balfour D.J.K. Nicotine: from molecular mechanisms to behaviour. Curr. Opin. Pharmacol.. 2005;5:53-59. (Useful review on the acute and long-term CNS effects of nicotine)
Charness M.E., Simon R.P., Greenberg D.A. Ethanol and the nervous system. N. Engl. J. Med.. 1989;321:442-454.
Garbutt J.C. The state of pharmacotherapy for the treatment of alcohol dependence. J. Subst. Abuse Treat.. 2009;36(Suppl.):S15-S21. (Reviews current drugs and potential new approaches)
Groenbaek M., Deis A., Sørensen T.I., et al. Influence of sex, age, body mass index and smoking on alcohol intake and mortality. BMJ. 1994;308:302-306. (Large-scale Danish study showing reduced coronary mortality at moderate levels of drinking, with increase at high levels)
Harper C., Matsumoto I. Ethanol and brain damage. Curr. Opin. Pharmacol.. 2005;5:73-78. (Describes deleterious effects of long-term alcohol abuse on brain function)
Harris R.A., Trudell J.R., Mihic S.J. Ethanol’s molecular targets. Sci. Signal.. 2008. 1, re7. (Short review of potential molecular actions of alcohol relevant to its effects on the brain)
Lieber C.S. Medical disorders of alcoholism. N. Engl. J. Med.. 1995;333:1058-1065. (Review focusing on ethanol-induced liver damage in relation to ethanol metabolism)
Little H.J. Mechanisms that may underlie the behavioural effects of ethanol. Prog. Neurobiol.. 1991;36:171-194.
Lovinger D.M. Alcohols and neurotransmitter gated ion channels: past, present and future. Naunyn-Schmiedebergs Arch. Pharmacol.. 1997;356:267-282. (Review article arguing that alcohol effects depend on interaction with synaptic ion channels)
Melendez R.I., Kalivas P.W. Last call for adenosine transporters. Nat. Neurosci.. 2004;7:795-796. (Commentary on a study supporting a role for adenosine in the CNS effects of ethanol)
Myrick H., Anton R.F. Treatment of alcohol withdrawal. Alcohol Health Res W. 22:1998:38-43 Available online at http://pubs.niaaa.nih.gov/publications/arh22-1/38-43.pdf.. http://pubs.niaaa.nih.gov/publications/arh22-1/38-43.pdf
Spanagel R. Alcoholism: a systems approach from molecular physiology to addictive behaviour. Physiol. Rev.. 2009;89:649-705. (Comprehensive review article, very useful for reference)
Tabakoff B., Hoffman P.L. Alcohol addiction: an enigma among us. Neuron. 1996;16:909-912. (Review of alcohol actions at the cellular and molecular level—ignore the silly title)
Tanaka F., Shiratori Y., Yokusuka O., et al. Polymorphism of alcohol-metabolizing genes affects drinking behaviour and alcoholic liver disease in Japanese men. Alcohol. Clin. Exp. Res.. 1997;21:596-601. (Describes polymorphism of aldehyde and alcohol dehydrogenases, and their effect on drinking behaviour)
Tyndale R.F. Genetics of alcohol and tobacco use in humans. Ann. Med.. 2003;35:94-121. (Detailed review of the many genetic factors implicated in alcohol and nicotine consumption habits)
http://www.ash.org.uk/ (ASH, an antismoking organisation)
http://www.drugscope.org.uk/ (DrugScope, an independent organisation providing advice on various aspects of drug abuse)
http://www.nida.nih.gov/ (National Institute on Drug Abuse [NIDA], US government organisation providing information to scientists and the general public on various aspects of drug abuse)
http://www.drugabuse.gov/PODAT/PODATIndex.html (Provides access to the NIDA publication Principles of Drug Addiction Treatment: A Research Based Guide, second ed.)
http://www.ias.org.uk/resources/factsheets/factsheets.html (An excellent range of factsheets relating to all aspects of alcohol consumption and its consequences from the Institute of Alcohol Studies [UK])
1A recent survey in one UK city showed that among Friday-night clubbers the choice of drug was associated with the type of music the clubs played (Measham & Moore, 2009).
2In determining society’s attitude towards drugs, the media play an influential role. In the UK, deaths following consumption of ecstasy (around 60 per year) are often widely reported in the press and on television but deaths due to heroin overdose (much more prevalent at around 700 per year) are largely ignored unless the victim is famous.
3Smokers, however, adapt by smoking more low-tar cigarettes and inhaling more deeply so as to maintain their nicotine consumption.
4In contrast to the global picture, in the UK consumption has dropped by over 50% from its peak in the 1970s, the main factors being increased price, adverse publicity, restrictions on advertising, the compulsory publication of health warnings and, most recently, a ban on smoking in public places. Still, however, around 9.4 million adults (just over 20% of the adult population) in the UK smoke, with little difference between men and women. About 10% of children aged 10–15 are regular smokers.
5From the plant Nicotiana, named after Jean Nicot, French ambassador to Portugal, who presented seeds to the French king in 1560, having been persuaded by natives of South America of the medical value of smoking tobacco leaves. Smoking was believed to protect against illness, particularly the plague.
6This may explain why, in years gone by, men smoked cigars while chatting over drinks after dinner.
7Freud tried unsuccessfully to give up cigars for 45 years before dying of cancer of the mouth at the age of 83.
8It also reduces postmenopausal flushing, which may represent a physiological oestrogen withdrawal response.
9The image of a large St Bernard dog carrying a small keg of brandy around its neck to revive avalanche victims is an apocryphal one created by the English painter, Edwin Landseer, who in 1820 produced a painting called ‘Alpine Mastiffs Reanimating a Distressed Traveller’. With their keen sense of smell, such dogs were useful in searching for people buried in the snow, but taking a tot of brandy would only have enhanced the victim’s heat loss.
10This beneficial effect of moderate drinking outweighs the risk of adverse effects (e.g. accidents, cancers, liver damage) only in men over 45 and women over 55.