50 Antibacterial drugs
Overview
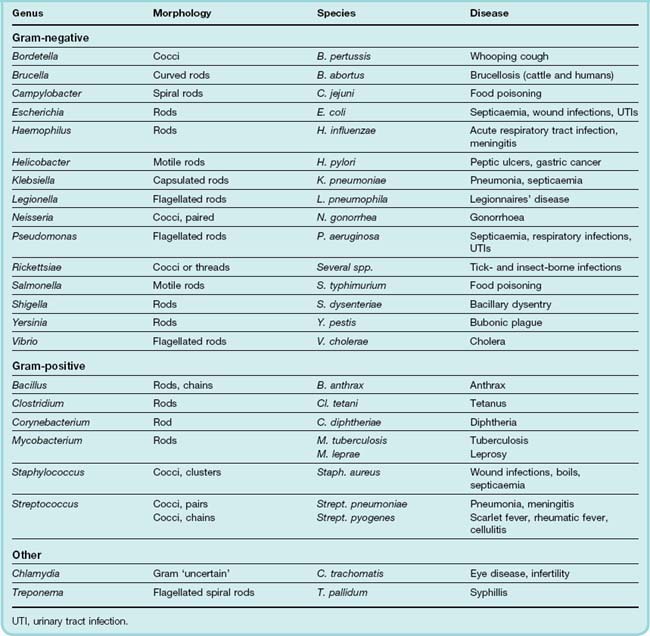
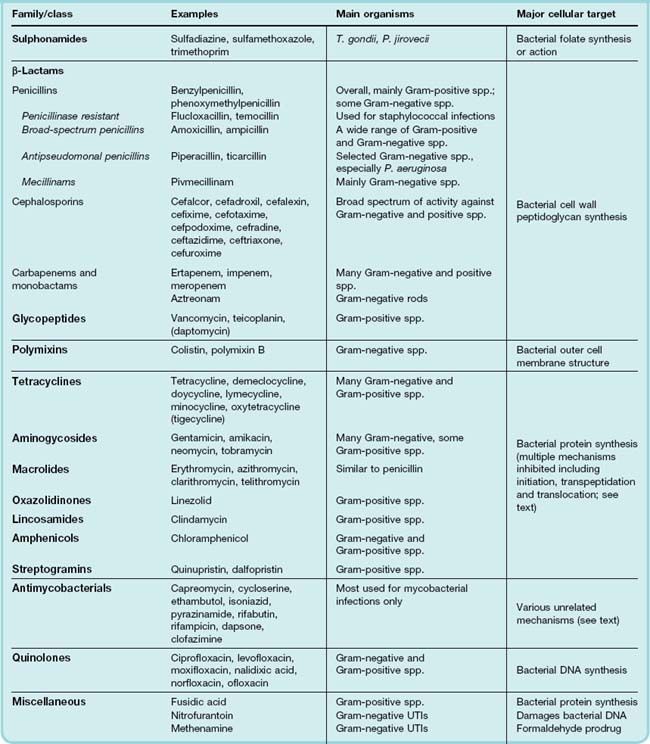
In this chapter, we continue to develop the ideas we introduced in the previous chapter. A detailed discussion of the bacteria of medical importance is beyond the scope of this book and the reader is referred to more specialist texts. However, information about some clinically significant pathogens is provided in Table 50.1 and an overview of the principal antibiotic ‘families’, together with their mechanisms of action, is given in Table 50.2. The major classes of antibacterial drugs are described, along with their mechanism of action, relevant pharmacokinetic properties and side effects.
Introduction
In 1928, Alexander Fleming, working at St Mary’s Hospital in London, observed that a culture plate on which staphylococci were being grown had become contaminated with a mould of the genus Penicillium, and that bacterial growth in the vicinity of the mould had been inhibited. He isolated the mould in pure culture and demonstrated that it produced an antibacterial substance, which he called penicillin. This substance was subsequently prepared in bulk, extracted and its antibacterial effects analysed by Florey, Chain and their colleagues at Oxford in 1940. They showed that it had powerful chemotherapeutic properties in infected mice, and that it was non-toxic, thus ushering in the ‘antibiotic era’. Seventy years later, the number of different types of antibiotics has grown 10-fold and the practice of medicine would be unthinkable without them.
Many organisms can be classified as being either Gram-positive or Gram-negative depending on whether or not they stain with Gram’s stain.1 This is not merely a taxonomic device, as it reflects several fundamental differences in (for example) the structure of their cell walls, and this in turn has implications for the action of antibiotics.
The cell wall of Gram-positive organisms is a relatively simple structure, 15–50 nm thick. It comprises about 50% peptidoglycan (see Ch. 49), 40–45% acidic polymer (which results in the cell surface being highly polar and carrying a negative charge) and 5–10% proteins and polysaccharides. The strongly polar polymer layer influences the penetration of ionised molecules and favours the penetration into the cell of positively charged compounds such as streptomycin.
The cell wall of Gram-negative organisms is much more complex. From the plasma membrane outwards, it consists of the following:
Difficulty in penetrating this complex outer layer is probably the reason why some antibiotics are less active against Gram-negative than Gram-positive bacteria. This is one reason for the extraordinary antibiotic resistance exhibited by Pseudomonas aeruginosa, a pathogen that can cause life-threatening infections in neutropenic patients and those with burns and wounds.
The cell wall lipopolysaccharide is also a major barrier to penetration. Antibiotics affected include benzylpenicillin (penicillin G), meticillin, the macrolides, rifampicin (rifampin), fusidic acid, vancomycin, bacitracin and novobiocin.
Antimicrobial Agents That Interfere With Folate Synthesis or Action
Sulfonamides
In a landmark discovery in the 1930s, Domagk demonstrated that it was possible for a drug to influence the course of a bacterial infection. The agent was prontosil, a dye that proved to be an inactive prodrug but which is metabolised in vivo to give the active product, sulfanilamide (Fig. 50.1). Many sulfonamides have been developed since, but their importance has declined in the face of increasing resistance. The only drugs still commonly used are sulfamethoxazole (usually in combination with trimethoprim as co-trimoxazole), sulfasalazine (poorly absorbed in the gastrointestinal tract, used to treat ulcerative colitis and Crohn’s disease; see Chs 26 and 29) and occasionally sufadiazine.
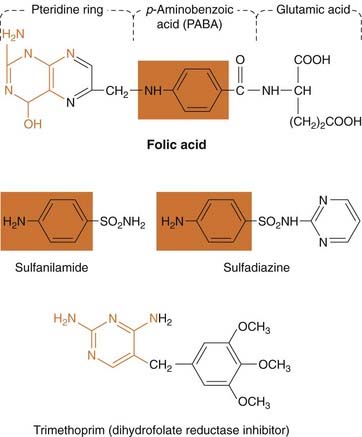
Fig. 50.1 Structures of two representative sulfonamides and trimethoprim.
The structures illustrate the relationship between the sulfonamides and the p-aminobenzoic acid moiety in folic acid (orange box), as well as the possible relationship between the antifolate drugs and the pteridine moiety (orange). Co-trimoxazole is a mixture of sulfamethoxazole and trimethoprim.
Mechanism of action
Sulfanilamide is a structural analogue of p-aminobenzoic acid (PABA; see Fig. 50.1), which is an essential precursor in the synthesis of folic acid, required for the synthesis of DNA and RNA in bacteria (see Ch. 49). Sulfonamides compete with PABA for the enzyme dihydropteroate synthetase, and the effect of the sulfonamide may be overcome by adding excess PABA. This is why some local anaesthetics, which are PABA esters (such as procaine; see Ch. 42), can antagonise the antibacterial effect of these agents.
 While not necessarily clinically relevant, the action of a sulfonamide is to inhibit growth of the bacteria, not to kill them; that is to say, it is bacteriostatic rather than bactericidal. The action is vitiated in the presence of pus or products of tissue breakdown, because these contain thymidine and purines, which bacteria utilise directly, bypassing the requirement for folic acid. Resistance to the drugs, which is common, is plasmid mediated (see Ch. 49) and results from the synthesis of a bacterial enzyme insensitive to the drug.
While not necessarily clinically relevant, the action of a sulfonamide is to inhibit growth of the bacteria, not to kill them; that is to say, it is bacteriostatic rather than bactericidal. The action is vitiated in the presence of pus or products of tissue breakdown, because these contain thymidine and purines, which bacteria utilise directly, bypassing the requirement for folic acid. Resistance to the drugs, which is common, is plasmid mediated (see Ch. 49) and results from the synthesis of a bacterial enzyme insensitive to the drug.
Pharmacokinetic aspects
Most sulfonamides are given orally and, apart from sulfasalazine, are well absorbed and widely distributed in the body. There is a risk of sensitisation or allergic reactions when these drugs are given topically.
The drugs pass into inflammatory exudates and cross both placental and blood–brain barriers. They are metabolised mainly in the liver, the major product being an acetylated derivative that lacks antibacterial action.
Unwanted effects
Serious adverse effects necessitating cessation of therapy include hepatitis, hypersensitivity reactions (rashes including Stevens–Johnson syndrome and toxic epidermal necrolysis, fever, anaphylactoid reactions—see Ch. 57), bone marrow depression and acute renal failure due to interstitial nephritis or crystalluria. This last effect results from the precipitation of acetylated metabolites in the urine (Ch. 28). Cyanosis caused by methaemoglobinaemia may occur but is a lot less alarming than it looks. Mild to moderate side effects include nausea and vomiting, headache and mental depression.
Clinical uses of sulfonamides 
Trimethoprim
Mechanism of action
Trimethoprim is chemically related to the antimalarial drug pyrimethamine (Fig. 53.3), both being folate antagonists. Structurally (Fig. 50.1), it resembles the pteridine moiety of folate and the similarity is close enough to fool the bacterial dihydrofolate reductase, which is many times more sensitive to trimethoprim than the equivalent enzyme in humans.
Trimethoprim is active against most common bacterial pathogens as well as protozoa, and it too is bacteriostatic. It is sometimes given as a mixture with sulfamethoxazole as co-trimoxazole (Fig. 50.1). Because sulfonamides inhibit the same bacterial metabolic pathway, but upstream from dihydrofolate reductase, they can potentiate the action of trimethoprim (see Fig. 50.2). In the UK, its use is generally restricted to the treatment of Pneumocystis carinii (now known as P. jirovecii) pneumonia (a fungal infection), toxoplasmosis (a protozoan infection) as well as nocardiasis (a bacterial infection).
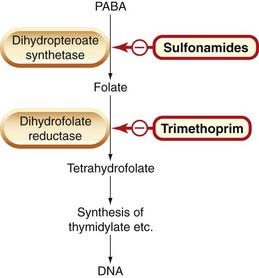
Fig. 50.2 The action of sulfonamides and trimethoprim on bacterial folate synthesis.
See Figure 25.2 for more detail of tetrahydrofolate synthesis, and Table 49.1 for comparisons of antifolate drugs. PABA, p-aminobenzoic acid.
Pharmacokinetic aspects
Trimethoprim is well absorbed orally, and widely distributed throughout the tissues and body fluids. It reaches high concentrations in the lungs and kidneys, and fairly high concentrations in the cerebrospinal fluid (CSF). When given with sulfamethoxazole, about half the dose of each is excreted within 24 h. Because trimethoprim is a weak base, its elimination by the kidney increases with decreasing urinary pH.
Unwanted effects
Folate deficiency, with resultant megaloblastic anaemia (see Ch. 25)—a toxic effect related to the pharmacological action of trimethoprim, can be prevented by giving folinic acid. Other unwanted effects include nausea, vomiting, blood disorders and rashes.
Antimicrobial agents that interfere with the synthesis or action of folate 
β-Lactam Antibiotics
Penicillins
The remarkable antibacterial effects of penicillin in humans were clearly demonstrated in 1941. A small amount of penicillin, extracted laboriously from crude cultures in the laboratories of the Dunn School of Pathology in Oxford, was given to a desperately ill policeman who had septicaemia with multiple abscesses. Although sulfonamides were available, they would have had no effect in the presence of pus. Intravenous injections of penicillin were given every 3 h. All the patient’s urine was collected, and each day the bulk of the excreted penicillin was extracted and reused. After 5 days, the patient’s condition was vastly improved, and there was obvious resolution of the abscesses. Furthermore, there seemed to be no toxic effects of the drug.2 Unfortunately, when the supply of penicillin was finally exhausted his condition gradually deteriorated and he died a month later.
While the penicillins are extremely effective antibiotics and are very widely used, they can be destroyed by bacterial amidases and β-lactamases (penicillinases; see Fig. 50.3). This forms the basis of one of the principal types of antibiotic resistance. Penicillins, often combined with other antibiotics, remain crucially important in antibacterial chemotherapy, and are the drugs of choice for many infections. A list of clinical uses is given in the clinical box.
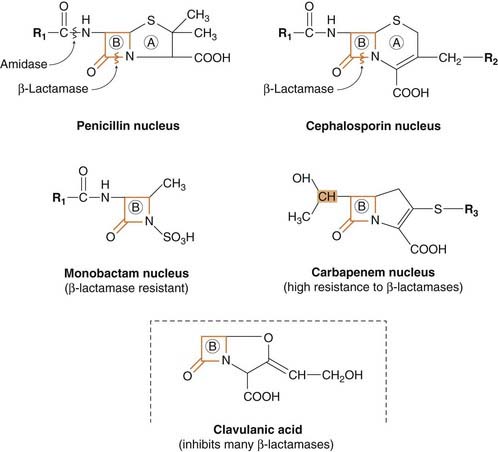
Fig. 50.3 Basic structures of four groups of β-lactam antibiotics and clavulanic acid.
The structures illustrate the β-lactam ring (marked B) and the sites of action of bacterial enzymes that inactivate these antibiotics (A, thiazolidine ring). Various substituents are added at R1, R2 and R3 to produce agents with different properties. In carbapenems, the stereochemical configuration of the part of the β-lactam ring shown shaded in orange here is different from the corresponding part of the penicillin and cephalosporin molecules; this is probably the basis of the β-lactamase resistance of the carbapenems. The β-lactam ring of clavulanic acid is thought to bind strongly to β-lactamase, meanwhile protecting other β-lactams from the enzyme.
Mechanisms of action
All β-lactam antibiotics interfere with the synthesis of the bacterial cell wall peptidoglycan (see Ch. 49, Fig. 49.3). After attachment to penicillin-binding proteins on bacteria (there may be seven or more types in different organisms), they inhibit the transpeptidation enzyme that crosslinks the peptide chains attached to the backbone of the peptidoglycan.
The final bactericidal event is the inactivation of an inhibitor of autolytic enzymes in the cell wall, leading to lysis of the bacterium. Some organisms, referred to as ‘tolerant’, have defective autolytic enzymes and are inhibited but not lysed in the presence of the drug. Resistance to penicillin may result from a number of different causes and is discussed in detail in Chapter 49.
Types of penicillin and their antimicrobial activity
The first penicillins were the naturally occurring benzylpenicillin (penicillin G) and its congeners, including phenoxymethylpenicillin (penicillin V). Benzylpenicillin is active against a wide range of organisms and is the drug of first choice for many infections (see clinical box). Its main drawbacks are poor absorption in the gastrointestinal tract (which means it must be given by injection) and its susceptibility to bacterial β-lactamases.
Semisynthetic penicillins, incorporating different side-chains attached to the penicillin nucleus (at R1 in Fig. 50.3), include β-lactamase-resistant penicillins (e.g. meticillin,3 flucloxacillin, temocillin) and broad-spectrum penicillins (e.g. ampicillin, amoxicillin). Extended-spectrum penicillins (e.g. ticarcillin, piperacillin) with antipseudomonal activity have gone some way to overcoming the problem of serious infections caused by P. aeruginosa. Amoxicillin and ticarcillin are sometimes given in combination with the β-lactamase inhibitor clavulanic acid (e.g. co-amoxiclav). Pivmecillinam is a prodrug of mecillinam, which also has a wide spectrum of action.
Pharmacokinetic aspects
Oral absorption of penicillins varies, depending on their stability in acid and their adsorption to foodstuffs in the gut. Penicillins can also be given by intravenous injection. Preparations for intramuscular injection are also available, including slow-release preparations such as benzathine penicillin (cf. long-lived preparations of insulin, Ch. 30). Benzathine penicillin may be useful in treating syphilis since Treponema pallidum is a very slowly dividing organism. Intrathecal administration (used historically to treat meningitis) is no longer used, as it can cause convulsions.4
The penicillins are widely distributed in body fluids, passing into joints; into pleural and pericardial cavities; into bile, saliva and milk; and across the placenta. Being lipid insoluble, they do not enter mammalian cells, and cross the blood–brain barrier only if the meninges are inflamed, when they reach therapeutically effective concentrations in the CSF.
Elimination of most penicillins occurs rapidly and is mainly renal, 90% being through tubular secretion. The relatively short plasma half-life is a potential problem in the clinical use of benzylpenicillin, although because penicillin works by preventing cell wall synthesis in dividing organisms, intermittent rather than continuous exposure to the drug can be an advantage.
Unwanted effects
Penicillins are relatively free from direct toxic effects (other than their proconvulsant effect when given intrathecally). The main unwanted effects are hypersensitivity reactions caused by the degradation products of penicillin, which combine with host protein and become antigenic. Skin rashes and fever are common; a delayed type of serum sickness occurs infrequently. Much more serious is acute anaphylactic shock which, although rare, may be fatal. When given orally, penicillins, particularly the broad-spectrum type, alter the bacterial flora in the gut. This can be associated with gastrointestinal disturbances and in some cases with suprainfection by other, penicillin-insensitive, microorganisms leading to problems such as pseudomembranous colitis (caused by C. difficile, see below).
Clinical uses of the penicillins
Cephalosporins and Cephamycins
Cephalosporins N and C, which are chemically related to penicillin, and cephalosporin P, a steroidal antibiotic that resembles fusidic acid (see below), were first isolated from Cephalosporium fungus. The cephamycins are β-lactam antibiotics produced by Streptomyces organisms, and they are closely related to the cephalosporins. They have the same mechanism of action as penicillins (see above).
Semisynthetic broad-spectrum cephalosporins have been produced by addition, to the cephalosporin C nucleus, of different side-chains at R1 and/or R2 (see Fig. 50.3). These agents are water soluble and relatively acid stable. They vary in susceptibility to β-lactamases. There are now a very large number of cephalosporins and cephamycins available for clinical use. Original members of the group such as cefradine, cefalexin and cefadroxil have largely been replaced with ‘second-generation’ drugs such as cefuroxime and cefaclor, or ‘third-generation’ drugs such as cefotaxime, ceftazidime, cefixime, cefpodoxime and ceftriaxone.
Resistance to this group of drugs has increased because of plasmid-encoded or chromosomal β-lactamase. Nearly all Gram-negative bacteria have a chromosomal gene coding for a β-lactamase that is more active in hydrolysing cephalosporins than penicillins, and in several organisms a single mutation can result in high-level constitutive production of this enzyme. Resistance also occurs when there is decreased penetration of the drug as a result of alterations to outer membrane proteins, or mutations of the binding-site proteins.
Pharmacokinetic aspects
Some cephalosporins may be given orally, but most are given parenterally, intramuscularly (which may be painful) or intravenously. After absorption, they are widely distributed in the body and some, such as cefotaxime, cefuroxime and ceftriaxone, cross the blood–brain barrier. Excretion is mostly via the kidney, largely by tubular secretion, but 40% of ceftriaxone is eliminated in the bile.
Unwanted effects
Hypersensitivity reactions, very similar to those seen with penicillin, may occur, and there may be some cross-sensitivity; about 10% of penicillin-sensitive individuals will have allergic reactions to cephalosporins. Nephrotoxicity has been reported (especially with cefradine), as has drug-induced alcohol intolerance. Diarrhoea is common and can be due to C. difficile.
Other β-Lactam Antibiotics
Carbapenems and monobactams (see Fig. 50.3) were developed to deal with β-lactamase-producing Gram-negative organisms resistant to penicillins.
Carbapenems
Imipenem, an example of a carbapenem, acts in the same way as the other β-lactams (see Fig. 50.3). It has a very broad spectrum of antimicrobial activity, being active against many aerobic and anaerobic Gram-positive and Gram-negative organisms. However, many of the ‘meticillin-resistant’ staphylococci are less susceptible, and resistant strains of P. aeruginosa have emerged during therapy. Imipenem was originally resistant to all β-lactamases, but some organisms now have chromosomal genes that code for imipenem-hydrolysing β-lactamases. It is sometimes given together with cilastatin, which inhibits its inactivation by renal enzymes. Meropenem is similar but is not metabolised by the kidney. Ertapenem has a broad spectrum of antibacterial actions but is licensed only for a limited range of indications. Most carbapenems are not orally active, and are used only in special situations.
Unwanted effects are generally similar to those seen with other β-lactams, nausea and vomiting being the most frequently seen. Neurotoxicity can occur with high plasma concentrations.
Monobactams
The main monobactam is aztreonam (see Fig. 50.3), which is resistant to most β-lactamases. It is given by injection and has a plasma half-life of 2 h. Aztreonam has an unusual spectrum of activity and is effective only against Gram-negative aerobic bacilli such as pseudomonads, Neisseria meningitidis and Haemophilus influenzae. It has no action against Gram-positive organisms or anaerobes.
Unwanted effects are, in general, similar to those of other β-lactam antibiotics, but this agent does not necessarily cross-react immunologically with penicillin and its products, and so does not usually cause allergic reactions in penicillin-sensitive individuals.
β-Lactam antibiotics
Penicillins
Cephalosporins and cephamycins
Glycopeptides
Vancomycin is a glycopeptide antibiotic, and teicoplanin is similar but longer lasting. Vancomycin acts by inhibiting cell wall synthesis (see Fig. 49.3). It is effective mainly against Gram-positive bacteria and has been used against MRSA. Vancomycin is not absorbed from the gut and is only given by the oral route for treatment of gastrointestinal infection with C. difficile. For parenteral use, it is given intravenously and has a plasma half-life of about 8 h.
The clinical use of vancomycin is limited mainly to pseudomembranous colitis (a clostridial infection sometimes associated with antibiotic therapy) and the treatment of some multiresistant staphylococcal infections. It is also valuable in severe staphylococcal infections in patients allergic to both penicillins and cephalosporins, and in some forms of endocarditis.
Unwanted effects include fever, rashes and local phlebitis at the site of injection. Ototoxicity and nephrotoxicity can occur, and hypersensitivity reactions are occasionally seen.
Daptomycin is a new lipopeptide antibacterial with a similar spectrum of actions to vancomycin. It is usually used, in combination with other drugs, for the treatment of MRSA.
Antimicrobial Agents Affecting Bacterial Protein Synthesis
Tetracyclines
Tetracyclines are broad-spectrum antibiotics. The group includes tetracycline, oxytetracycline, demeclocycline, lymecycline, doxycycline and minocycline. Tigelcycline is structurally related to the tetracycline family and has similar therapeutic and unwanted effects.
Mechanism of action
Following uptake into susceptible organisms by active transport, tetracyclines act by inhibiting protein synthesis (see Ch. 49, Fig. 49.4). The tetracyclines are regarded as bacteriostatic, not bactericidal.
Antibacterial spectrum
The spectrum of antimicrobial activity of the tetracyclines is very wide and includes Gram-positive and Gram-negative bacteria, Mycoplasma, Rickettsia, Chlamydia spp., spirochaetes and some protozoa (e.g. amoebae). Minocycline is also effective against N. meningitidis and has been used to eradicate this organism from the nasopharynx of carriers. However, widespread resistance to these agents has decreased their usefulness. Resistance is transmitted mainly by plasmids and, because the genes controlling resistance to tetracyclines are closely associated with genes for resistance to other antibiotics, organisms may develop resistance to many drugs simultaneously. The clinical use of the tetracyclines is given in the clinical box.
Pharmacokinetic aspects
The tetracyclines are generally given orally but can also be administered parenterally. Minocycline and doxycycline are virtually completely absorbed. The absorption of most other tetracyclines is irregular and incomplete but is improved in the absence of food. Because tetracyclines chelate metal ions (calcium, magnesium, iron, aluminium), forming non-absorbable complexes, absorption is decreased in the presence of milk, certain antacids and iron preparations.
Unwanted effects
The commonest unwanted effects are gastrointestinal disturbances caused initially by direct irritation and later by modification of the gut flora. Vitamin B complex deficiency can occur, as can suprainfection. Because they chelate Ca2+, tetracyclines are deposited in growing bones and teeth, causing staining and sometimes dental hypoplasia and bone deformities. They should therefore not be given to children, pregnant women or nursing mothers. Another hazard to pregnant women is hepatotoxicity. Phototoxicity (sensitisation to sunlight) has also been seen, particularly with demeclocycline. Minocycline can produce vestibular disturbances (dizziness and nausea). High doses of tetracyclines can decrease protein synthesis in host cells, an antianabolic effect that may result in renal damage. Long-term therapy can cause disturbances of the bone marrow.
Clinical uses of tetracyclines
Amphenicols
The principal agent is chloramphenicol which was originally isolated from cultures of Streptomyces. It inhibits bacterial protein synthesis by binding to the 50S ribosomal subunit (see Ch. 49, Fig. 49.4). The clinical uses of chloramphenicol are given in the box.
Antibacterial spectrum
Chloramphenicol has a wide spectrum of antimicrobial activity, including Gram-negative and Gram-positive organisms and rickettsiae. It is bacteriostatic for most organisms but kills H. influenzae. Resistance, caused by the production of chloramphenicol acetyltransferase, is plasmid mediated.
Pharmacokinetic aspects
Given orally, chloramphenicol is rapidly and completely absorbed and reaches its maximum concentration in the plasma within 2 h; it can also be given parenterally. The drug is widely distributed throughout the tissues and body fluids including the CSF. Its half-life is approximately 2 h. About 10% is excreted unchanged in the urine, and the remainder is inactivated in the liver.
Unwanted effects
The most important unwanted effect of chloramphenicol is severe, idiosyncratic depression of the bone marrow, resulting in pancytopenia (a decrease in all blood cell elements)—an effect that, although rare, can occur even with low doses in some individuals. Chloramphenicol must be used with great care in newborns, with monitoring of plasma concentrations, because inadequate inactivation and excretion of the drug (see Ch. 56) can result in the ‘grey baby syndrome’—vomiting, diarrhoea, flaccidity, low temperature and an ashen-grey colour—which carries 40% mortality. Hypersensitivity reactions can occur, as can gastrointestinal disturbances secondary to alteration of the intestinal microbial flora.
Aminoglycosides
The aminoglycosides are a group of antibiotics of complex chemical structure, resembling each other in antimicrobial activity, pharmacokinetic characteristics and toxicity. The main agents are gentamicin, streptomycin, amikacin, tobramycin and neomycin.
Mechanism of action
Aminoglycosides inhibit bacterial protein synthesis by blocking initiation (see Ch. 49). Their penetration through the cell membrane of the bacterium depends partly on oxygen-dependent active transport by a polyamine carrier system, and they have minimal action against anaerobic organisms. Chloramphenicol blocks this transport system. The effect of the aminoglycosides is bactericidal and is enhanced by agents that interfere with cell wall synthesis.
Resistance
Resistance to aminoglycosides is becoming a problem. It occurs through several different mechanisms, the most important being inactivation by microbial enzymes, of which nine or more are known. Amikacin was purposefully designed as a poor substrate for these enzymes, but some organisms have acquired enzymes that inactivate this agent as well. Resistance as a result of failure of penetration can be largely overcome by the concomitant use of penicillin and/or vancomycin.
Antibacterial spectrum
The aminoglycosides are effective against many aerobic Gram-negative and some Gram-positive organisms. They are most widely used against Gram-negative enteric organisms and in sepsis. They may be given together with a penicillin in streptococcocal infections and those caused by Listeria spp. and P. aeruginosa (see Table 50.1). Gentamicin is the aminoglycoside most commonly used, although tobramycin is the preferred member of this group for P. aeruginosa infections. Amikacin has the widest antimicrobial spectrum and can be effective in infections with organisms resistant to gentamicin and tobramycin.
Pharmacokinetic aspects
The aminoglycosides are polycations and therefore highly polar. They are not absorbed from the gastrointestinal tract and are usually given intramuscularly or intravenously. They cross the placenta but do not cross the blood–brain barrier, although high concentrations can be attained in joint and pleural fluids. The plasma half-life is 2–3 h. Elimination is virtually entirely by glomerular filtration in the kidney, 50–60% of a dose being excreted unchanged within 24 h. If renal function is impaired, accumulation occurs rapidly, with a resultant increase in those toxic effects (such as ototoxicity and nephrotoxicity; see below) that are dose related.
Unwanted effects
Serious, dose-related toxic effects, which may increase as treatment proceeds, can occur with the aminoglycosides, the main hazards being ototoxicity and nephrotoxicity.
The ototoxicity involves progressive damage to, and eventually destruction of, the sensory cells in the cochlea and vestibular organ of the ear. The result, usually irreversible, may manifest as vertigo, ataxia and loss of balance in the case of vestibular damage, and auditory disturbances or deafness in the case of cochlear damage. Any aminoglycoside may produce both types of effect, but streptomycin and gentamicin are more likely to interfere with vestibular function, whereas neomycin and amikacin mostly affect hearing. Ototoxicity is potentiated by the concomitant use of other ototoxic drugs (e.g. loop diuretics; Ch. 28) and susceptibility is genetically determined via mitochondrial DNA (see Ch. 11).
The nephrotoxicity consists of damage to the kidney tubules, and function recovers if the use of the drugs is stopped. Nephrotoxicity is more likely to occur in patients with pre-existing renal disease or in conditions in which urine volume is reduced, and concomitant use of other nephrotoxic agents (e.g. first-generation cephalosporins) increases the risk. As the elimination of these drugs is almost entirely renal, their nephrotoxic action can impair their own excretion, and a vicious cycle may develop. Plasma concentrations should be monitored regularly and the dose adjusted accordingly.
A rare but serious toxic reaction is paralysis caused by neuromuscular blockade. This is usually seen only if the agents are given concurrently with neuromuscular-blocking agents. It results from inhibition of the Ca2+ uptake necessary for the exocytotic release of acetylcholine (see Ch. 13).
Macrolides
The term macrolide relates to the structure—a many-membered lactone ring to which one or more deoxy sugars are attached. The main macrolide and related antibiotics are erythromycin, clarithromycin and azithromycin. Spiramycin and telithromycin are of minor utility.
Mechanism of action
The macrolides inhibit bacterial protein synthesis by an effect on translocation (Fig. 49.4). The drugs bind to the same 50S subunit of the bacterial ribosome as chloramphenicol and clindamycin, and any of these drugs may compete if given concurrently.
Antimicrobial spectrum
The antimicrobial spectrum of erythromycin is very similar to that of penicillin, and it is a safe and effective alternative for penicillin-sensitive patients. Erythromycin is effective against Gram-positive bacteria and spirochaetes but not against most Gram-negative organisms, exceptions being N. gonorrhoeae and, to a lesser extent, H. influenzae. Mycoplasma pneumoniae, Legionella spp. and some chlamydial organisms are also susceptible (see Table 50.1). Resistance can occur and results from a plasmid-controlled alteration of the binding site for erythromycin on the bacterial ribosome (Fig. 49.4).
Azithromycin is less active against Gram-positive bacteria than erythromycin but is considerably more effective against H. influenzae and may be more active against Legionella. It has excellent action against Toxoplasma gondii, killing the cysts. Clarithromycin is as active, and its metabolite is twice as active, against H. influenzae as erythromycin. It is also effective against Mycobacterium avium-intracellulare (which can infect immunologically compromised individuals and elderly patients with chronic lung disease), and it may also be useful in leprosy and against Helicobacter pylori (see Ch. 29). Both these macrolides are also effective in Lyme disease.
Pharmacokinetic aspects
The macrolides are administered orally. Erythromycin can also be given parenterally, although intravenous injections can be followed by local thrombophlebitis. All three diffuse readily into most tissues but do not cross the blood–brain barrier, and there is poor penetration into synovial fluid. The plasma half-life of erythromycin is about 90 min; that of clarithromycin is three times longer, and that of azithromycin 8–16 times longer. Macrolides enter and indeed are concentrated within phagocytes—azithromycin concentrations in phagocyte lysosomes can be 40 times higher than in the blood—and they can enhance intracellular phagocyte killing of bacteria.
Erythromycin is partly inactivated in the liver; azithromycin is more resistant to inactivation, and clarithromycin is converted to an active metabolite. Their inhibition of the P450 cytochrome system can affect the bioavailability of other drugs leading to clinically important interactions, for example with theophylline (see Ch. 56). The major route of elimination is in the bile.
Unwanted effects
Gastrointestinal disturbances are common and unpleasant but not serious. With erythromycin, the following have also been reported: hypersensitivity reactions such as rashes and fever, transient hearing disturbances and, rarely, following treatment for longer than 2 weeks, cholestatic jaundice. Opportunistic infections of the gastrointestinal tract or vagina can occur.
Antimicrobial agents affecting bacterial protein synthesis
Antimicrobial Agents Affecting Topoisomerase
Quinolones
The quinolones include the broad-spectrum agents ciprofloxacin, levofloxacin, ofloxacin, norfloxacin and moxifloxacin as well as a narrow-spectrum drug used in urinary tract infections—nalidixic acid. These agents inhibit topoisomerase II (a bacterial DNA gyrase), the enzyme that produces a negative supercoil in DNA and thus permits transcription or replication (see Fig. 50.4).
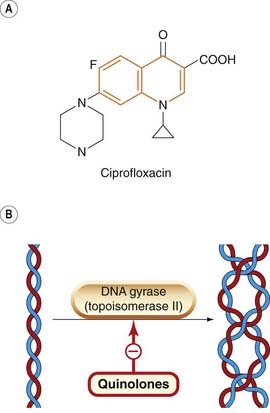
Fig. 50.4 A simplified diagram of the mechanism of action of the fluoroquinolones.
[A] An example of a quinolone (the quinolone moiety is shown in orange). [B] Schematic diagram of (left) the double helix and (right) the double helix in supercoiled form (see also Fig. 49.6). In essence, the DNA gyrase unwinds the RNA-induced positive supercoil (not shown) and introduces a negative supercoil.
Antibacterial spectrum and clinical use
The fluoroquinolone ciprofloxacin is the most commonly used and typical of the group. It is a broad-spectrum antibiotic effective against both Gram-positive and Gram-negative organisms, and also against the Enterobacteriaceae (the enteric Gram-negative bacilli), including many organisms resistant to penicillins, cephalosporins and aminoglycosides, and against H. influenzae, penicillinase-producing N. gonorrhoeae, Campylobacter spp. and pseudomonads. Of the Gram-positive organisms, streptococci and pneumococci are only weakly inhibited, and there is a high incidence of staphylococcal resistance. Ciprofloxacin should be avoided in MRSA infections. Clinically, the fluoroquinolones are best reserved for infections with facultative and aerobic Gram-negative bacilli and cocci.5 Resistant strains of S. aureus and P. aeruginosa have emerged. Further details of the clinical use of the fluoroquinolones are given in the box.
Pharmacokinetic aspects
Fluoroquinolones are well absorbed orally. The drugs accumulate in several tissues, particularly in the kidney, prostate and lung. All quinolones are concentrated in phagocytes. Most fail to cross the blood–brain barrier, but ofloxacin does so. Aluminium and magnesium antacids interfere with the absorption of the quinolones. Elimination of ciprofloxacin and norfloxacin is partly by hepatic metabolism by P450 enzymes (which they can inhibit, giving rise to interactions with other drugs; see below) and partly by renal excretion. Ofloxacin is excreted in the urine.
Unwanted effects
In hospitals, infection with C. difficile may prove hazardous (see below) but otherwise unwanted effects are infrequent, usually mild and reversible. The most frequent manifestations are gastrointestinal disorders and skin rashes. Arthropathy has been reported in young individuals. Central nervous system symptoms—headache and dizziness—have occurred, as have, less frequently, convulsions associated with central nervous system pathology or concurrent use of theophylline or a non-steroidal anti-inflammatory drug.
There is a clinically important interaction between ciprofloxacin and theophylline (through inhibition of P450 enzymes), which can lead to theophylline toxicity in asthmatics treated with the fluoroquinolones. The topic is discussed further in Chapter 27.
Antimicrobial agents affecting DNA topoisomerase II
Miscellaneous and Less Common Antibacterial Agents
Metronidazole
Metronidazole was introduced as an antiprotozoal agent (see Ch. 53), but it is also active against anaerobic bacteria such as Bacteroides, Clostridia spp. and some streptococci. It is effective in the therapy of pseudomembranous colitis (see below), and is important in the treatment of serious anaerobic infections (e.g. sepsis secondary to bowel disease). It has a disulfiram-like action (see Ch. 48), so patients must avoid alcohol while taking metronidazole.
Streptogramins
Quinupristin and dalfopristin are cyclic peptides, which inhibit bacterial protein synthesis by binding to the 50S subunit of the bacterial ribosome. Dalfopristin changes the structure of the ribosome so as to promote the binding of quinupristin. Individually, they exhibit only very modest bacteriostatic activity, but combined together as an intravenous injection they are active against many Gram-positive bacteria.
The combination is used to treat serious infections, usually where no other antibacterial is suitable. For example, the combination is effective against MRSA and is also active against vancomycin-resistant Enterococcus faecium.
Both drugs undergo extensive first-pass hepatic metabolism and must therefore be given as an intravenous infusion. The half-life of each compound is 1–2 h.
Unwanted effects include inflammation and pain at the infusion site, arthralgia, myalgia and nausea, vomiting and diarrhoea. To date, resistance to quinupristin and dalfopristine does not seem to be a major problem.
Clindamycin
The lincosamide, clindamycin, is active against Gram-positive cocci, including many penicillin-resistant staphylococci and many anaerobic bacteria such as Bacteroides spp. It acts in the same way as macrolides and chloramphenicol (Fig. 49.4). In addition to its use in infections caused by Bacteroides organisms, it is used to treat staphylococcal infections of bones and joints. It is also given topically, as eye drops, for staphylococcal conjunctivitis.
Unwanted effects consist mainly of gastrointestinal disturbances, and a potentially lethal condition, pseudomembranous colitis, may develop. This is an acute inflammation of the colon caused by a necrotising toxin produced by a clindamycin-resistant organism, Clostridium difficile, which may form part of the normal faecal flora.6 Metronidazole (see above) is usually effective in the treatment of this condition; vancomycin, given orally, is an alternative.
Oxazolidinones
Hailed as the ‘first truly new class of antibacterial agents to reach the marketplace in several decades’ (Zurenko et al., 2001), the oxazolidinones inhibit bacterial protein synthesis by a novel mechanism: inhibition of N-formylmethionyl-tRNA binding to the 70S ribosome. Linezolid is the first member of this new antibiotic family to be introduced. It is active against a wide variety of Gram-positive bacteria and is particularly useful for the treatment of drug-resistant bacteria such as MRSA, penicillin-resistant Streptococcus pneumoniae and vancomycin-resistant enterococci. The drug is also effective against some anaerobes, such as Clostridium difficile. Most common Gram-negative organisms are not susceptible to the drug. Linezolid can be used to treat pneumonia, septicaemia, and skin and soft tissue infections. Its use is restricted to serious bacterial infections where other antibiotics have failed, and there have so far been few reports of linezolid resistance.
Unwanted effects include thrombocytopenia, diarrhoea, nausea and, rarely, rash and dizziness. Linezolid is a non-selective inhibitor of monoamine oxidase, and appropriate precautions need to be observed (see Ch. 46).
Fusidic Acid
Fusidic acid is a narrow-spectrum steroid antibiotic active mainly against Gram-positive bacteria. It acts by inhibiting bacterial protein synthesis (Fig. 49.4). As the sodium salt, the drug is well absorbed from the gut and is distributed widely in the tissues. Some is excreted in the bile and some metabolised. It is used in combination with other antistaphylococcal agents in staphylococcal sepsis, and topically for staphylococcal infections (e.g. as eye drops).
Unwanted effects such as gastrointestinal disturbances are fairly common. Skin eruptions and jaundice can occur. Resistance occurs if it is used systemically as a single agent.
Nitrofurantoin
Nitrofurantoin is a synthetic compound active against a range of Gram-positive and Gram-negative organisms. The development of resistance in susceptible organisms is rare, and there is no cross-resistance. Its mechanism of action is not known. It is given orally and is rapidly and totally absorbed from the gastrointestinal tract and just as rapidly excreted by the kidney. Its use is confined to the treatment of urinary tract infections.
Unwanted effects such as gastrointestinal disturbances are relatively common, and hypersensitivity reactions involving the skin and the bone marrow (e.g. leukopenia) can occur. Hepatotoxicity and peripheral neuropathy have also been reported.
Methanamine has a similar clinical utility to nitrofurantoin and shares several of its unwanted effects. It exerts its effects by conversion (in acidic urine) to formaldehyde.
Polymixins
The polymixin antibiotics in use are polymixin B and colistin (polymixin E). They have cationic detergent properties and exert their antibacterial action by disrupting the outer cell membrane (Ch. 49). They have a selective, rapidly bactericidal action on Gram-negative bacilli, especially pseudomonads and coliform organisms. They are not absorbed from the gastrointestinal tract. Clinical use of these drugs is limited by their toxicity and is confined largely to gut sterilisation and topical treatment of ear, eye or skin infections caused by susceptible organisms.
Unwanted effects may be serious and include neurotoxicity and nephrotoxicity.
Miscellaneous antibacterial agents
Antimycobacterial Agents
The main mycobacterial infections in humans are tuberculosis and leprosy, chronic infections caused by Mycobacterium tuberculosis and M. leprae, respectively. A particular problem with both these organisms is that they can survive inside macrophages after phagocytosis, unless these cells are ‘activated’ by cytokines produced by T-helper (Th)1 lymphocytes (see Ch. 17).
Drugs Used To Treat Tuberculosis
For centuries, tuberculosis was a major killer disease, but the introduction of streptomycin in the late 1940s followed by isoniazid and, in the 1960s, of rifampicin and ethambutol revolutionised therapy, and tuberculosis came to be regarded as an easily treatable condition. Regrettably, this is so no longer; strains with increased virulence or exhibiting multidrug resistance are now common (Bloom & Small, 1998). Tuberculosis is again a major threat; the World Health Organization estimates that one-third of the world’s population (2 billion people) are currently infected with the bacillus, 10% of whom will develop the disease at some point in their lifetime. Infection rates are falling very slowly, but in 2008 there were over 9 million new cases and the disease killed about 1.8 million people. Poverty-stricken countries in Africa and Asia bear the brunt of the disease, partly because of an ominous synergy between mycobacteria (e.g. M. tuberculosis, M. avium-intercellulare) and HIV. About one-third of HIV-associated deaths are caused by tuberculosis. The disease is out of control in many countries, and it is now the world’s leading cause of death from a single agent.
Our counterattack is led by the first-line drugs isoniazid, rifampicin, rifabutin, ethambutol and pyrazinamide. Some second-line drugs available are capreomycin, cycloserine, streptomycin (rarely used now in the UK), clarithromycin and ciprofloxacin. These are used to treat infections likely to be resistant to first-line drugs, or when the first-line agents have to be abandoned because of unwanted reactions.
To decrease the probability of the emergence of resistant organisms, compound drug therapy is a frequent strategy. This commonly involves:
Isoniazid
The antibacterial activity of isoniazid is limited to mycobacteria. It halts the growth of resting organisms (i.e. is bacteriostatic) but can kill dividing bacteria. It passes freely into mammalian cells and is thus effective against intracellular organisms. Isoniazid is a prodrug that must be activated by bacterial enzymes before it can exert its inhibitory activity on the synthesis of mycolic acids, important constituents of the cell wall peculiar to mycobacteria. Resistance to the drug, caused by reduced penetration into the bacterium, may be encountered, but cross-resistance with other tuberculostatic drugs does not occur.
Isoniazid is readily absorbed from the gastrointestinal tract and is widely distributed throughout the tissues and body fluids, including the CSF. An important point is that it penetrates well into ‘caseous’ tuberculous lesions (i.e. necrotic lesions with a cheese-like consistency). Metabolism, which involves acetylation, depends on genetic factors that determine whether a person is a slow or rapid acetylator of the drug (see Chs 11 and 56), with slow inactivators enjoying a better therapeutic response. The half-life in slow inactivators is 3 h and in rapid inactivators, 1 h. Isoniazid is excreted in the urine partly as unchanged drug and partly in the acetylated or otherwise inactivated form.
Unwanted effects depend on the dosage and occur in about 5% of individuals, the commonest being allergic skin eruptions. A variety of other adverse reactions have been reported, including fever, hepatotoxicity, haematological changes, arthritic symptoms and vasculitis. Adverse effects involving the central or peripheral nervous systems are largely consequences of pyridoxine deficiency and are common in malnourished patients unless prevented by administration of this substance. Isoniazid may cause haemolytic anaemia in individuals with glucose 6-phosphate dehydrogenase deficiency, and it decreases the metabolism of the antiepileptic agents phenytoin, ethosuximide and carbamazepine, resulting in an increase in the plasma concentration and toxicity of these drugs (Ch. 57).
Rifampicin
Rifampicin acts by binding to, and inhibiting, DNA-dependent RNA polymerase in prokaryotic but not in eukaryotic cells (Ch. 49). It is one of the most active antituberculosis agents known, and is also effective against leprosy (see below) and most Gram-positive bacteria as well as many Gram-negative species. It enters phagocytic cells and can therefore kill intracellular microorganisms including the tubercle bacillus. Resistance can develop rapidly in a one-step process and is thought to be caused by chemical modification of microbial DNA-dependent RNA polymerase, resulting from a chromosomal mutation (see Ch. 49).
Rifampicin is given orally and is widely distributed in the tissues and body fluids (including CSF), giving an orange tinge to saliva, sputum, tears and sweat. It is excreted partly in the urine and partly in the bile, some of it undergoing enterohepatic cycling. The metabolite retains antibacterial activity but is less well absorbed from the gastrointestinal tract. The half-life is 1–5 h, becoming shorter during treatment because of induction of hepatic microsomal enzymes.
Unwanted effects are relatively infrequent. The commonest are skin eruptions, fever and gastrointestinal disturbances. Liver damage with jaundice has been reported and has proved fatal in a very small proportion of patients, and liver function should be assessed before treatment is started. Rifampicin causes induction of hepatic metabolising enzymes (Ch. 10), resulting in an increase in the degradation of warfarin, glucocorticoids, narcotic analgesics, oral antidiabetic drugs, dapsone and oestrogens, the last effect leading to failure of oral contraceptives.
Ethambutol
Ethambutol has no effect on organisms other than mycobacteria. It is taken up by the bacteria and exerts a bacteriostatic effect after a period of 24 h, although the mechanism by which this occurs is unknown. Resistance emerges rapidly if the drug is used alone. Ethambutol is given orally and is well absorbed. It can reach therapeutic concentrations in the CSF in tuberculous meningitis. In the blood, it is taken up by erythrocytes and slowly released. Ethambutol is partly metabolised and is excreted in the urine.
Unwanted effects are uncommon, the most important being optic neuritis, which is dose related and is more likely to occur if renal function is decreased. It results in visual disturbances manifesting initially as red–green colour blindness progressing to a decreased visual acuity. Colour vision should be monitored during prolonged treatment.
Pyrazinamide
Pyrazinamide is inactive at neutral pH but tuberculostatic at acid pH. It is effective against the intracellular organisms in macrophages because, after phagocytosis, the organisms are contained in phagolysosomes where the pH is low. Resistance develops rather readily, but cross-resistance with isoniazid does not occur. The drug is well absorbed after oral administration and is widely distributed, penetrating well into the meninges. It is excreted through the kidney, mainly by glomerular filtration.
Unwanted effects include gout, which is associated with high concentrations of plasma urates. Gastrointestinal upsets, malaise and fever have also been reported. Serious hepatic damage due to high doses was once a problem but is less likely with lower dose/shorter course regimens now used; nevertheless, liver function should be assessed before treatment.
Capreomycin
Capreomycin is a peptide antibiotic given by intramuscular injection. Unwanted effects include kidney damage and injury to the auditory nerve, with consequent deafness and ataxia. The drug should not be given at the same time as streptomycin or other drugs that may cause deafness.
Cycloserine
Cycloserine is a broad-spectrum antibiotic that inhibits the growth of many bacteria, including coliforms and mycobacteria. It is water soluble and destroyed at acid pH. It acts by competitively inhibiting bacterial cell wall synthesis. It does this by preventing the formation of D-alanine and the D-Ala-D-Ala dipeptide that is added to the initial tripeptide side-chain on N-acetylmuramic acid, i.e. it prevents completion of the major building block of peptidoglycan (see Fig. 49.3). It is absorbed orally and distributed throughout the tissues and body fluids, including CSF. Most of the drug is eliminated in active form in the urine, but approximately 35% is metabolised.
Cycloserine has unwanted effects mainly on the central nervous system. A wide variety of disturbances may occur, ranging from headache and irritability to depression, convulsions and psychotic states. Its use is limited to tuberculosis that is resistant to other drugs.
Antituberculosis drugs
First-line drugs
Second-line drugs
Drugs Used To Treat Leprosy
Leprosy is one of the most ancient diseases known to mankind and has been mentioned in texts dating back to 600 BC. It is a chronic disfiguring illness with a long latency, and historically sufferers have been ostracised and forced to live apart from their communities although, in fact, the disease is not particularly contagious. Once viewed as incurable, the introduction in the 1940s of dapsone, and subsequently rifampicin (see above) and clofazimine in the 1960s, completely changed our perspective on leprosy. It is now generally curable, and the global figures show that the prevalence rates for the disease have dropped by 90% since 1985 and there has been a 20% annual decrease in the number of new cases detected since 2001. The disease has been eliminated from 113 out of 122 countries where it was considered to be a major health problem. In 2009, some 200 000 new cases were reported. The bulk of these (75%) are in the Indian subcontinent, Brazil or Mozambique.
Multidrug treatment regimens (which fortunately seem to defy drug resistance) initiated by the World Health Organization in 1982 are now the mainstay of treatment. Paucibacillary leprosy, leprosy characterised by one to five numb patches, is mainly tuberculoid7 in type and is treated for 6 months with dapsone and rifampicin. Multibacillary leprosy, characterised by more than five numb skin patches, is mainly lepromatous in type and is treated for at least 2 years with rifampicin, dapsone and clofazimine. The effect of therapy with minocycline or the fluoroquinolones is being investigated.
Dapsone
Dapsone is chemically related to the sulfonamides and, because its action is antagonised by PABA, probably acts through inhibition of bacterial folate synthesis. Resistance to the drug has steadily increased since its introduction and treatment in combination with other drugs is now recommended.
Dapsone is given orally; it is well absorbed and widely distributed through the body water and in all tissues. The plasma half-life is 24–48 h, but some dapsone persists in certain tissues (liver, kidney and, to some extent, skin and muscle) for much longer periods. There is enterohepatic recycling of the drug, but some is acetylated and excreted in the urine. Dapsone is also used to treat dermatitis herpetiformis, a chronic blistering skin condition associated with coeliac disease.
Unwanted effects occur fairly frequently and include haemolysis of red cells (usually not severe enough to lead to frank anaemia), methaemoglobinaemia, anorexia, nausea and vomiting, fever, allergic dermatitis and neuropathy. Lepra reactions (an exacerbation of lepromatous lesions) can occur, and a potentially fatal syndrome resembling infectious mononucleosis has occasionally been seen.
Clofazimine
Clofazimine is a dye of complex structure. Its mechanism of action against leprosy bacilli may involve an action on DNA. It also has anti-inflammatory activity and is useful in patients in whom dapsone causes inflammatory side effects.
Clofazimine is given orally and accumulates in the body, being sequestered in the mononuclear phagocyte system. The plasma half-life may be as long as 8 weeks. The antileprotic effect is delayed and is usually not evident for 6–7 weeks.
Unwanted effects may be related to the fact that clofazimine is a dye. The skin and urine can develop a reddish colour and the lesions a blue–black discoloration. Dose-related nausea, giddiness, headache and gastrointestinal disturbances can also occur.
Possible New Antibacterial Drugs
In contrast to the rapid discoveries and developments that characterised the ‘heroic’ years of antibiotic research spanning approximately 1950–1980, and which produced virtually all our existing drugs, the flow has since dried up, with only two totally novel antibiotics introduced during this period (Jagusztyn-Krynicka & Wysznska, 2008). However, the problem of resistance is now acute and a major worry. Novel antibiotic candidates continue to be discovered in plants (Limsuwan et al., 2009) and bacteria (Sit & Vederas, 2008) as well as through traditional medicinal chemistry. In addition, researchers in the front line of this important field are pressing all the latest conceptual technologies into the fray: bioinformatics, utilising information derived from pathogen genome sequencing, is one such approach (Bansal, 2008). The hunt for, and targeting of, bacterial virulence factors is showing some promise (Escaich, 2008). New types of screening procedures have been devised (Falconer & Brown, 2009) which would reveal novel types of targets, and sophisticated pharmacodynamic profiling brought to bear on the problem (Lister, 2006). The world awaits developments with bated breath.
References and Further Reading
Allington D.R., Rivey M.P. Quinupristine/dalfopristin: a therapeutic review. Clin. Ther.. 2001;23:24-44.
Ball P. Future of the quinolones. Semin. Resp. Infect.. 2001;16:215-224. (Good overview of this class of drugs)
Blondeau J.M. Expanded activity and utility of the new fluoroquinolones: a review. Clin. Ther.. 1999;21:3-15. (Good overview)
Blumer J.L. Meropenem: evaluation of a new generation carbapenem. Int. J. Antimicrob. Agents. 1997;8:73-92.
Bryskier A. Ketolides—telithromycin, an example of a new class of antibacterial agents. Clin. Microbiol. Infect.. 2000;6:661-669.
Duran J.M., Amsden G.W. Azithromycin: indications for the future? Expert. Opin. Pharmacother.. 2000;1:489-505.
Greenwood D., editor. Antimicrobial chemotherapy, third ed, Oxford: Oxford University Press, 1995. (Good all-round textbook)
Lowy F.D. Staphylococcus aureus infections N. Engl. J. Med. 339:1998:520-541 (Basis of Staph. aureus pathogenesis of infection, resistance; extensive references; impressive diagrams)
Moellering R.C. Principles of anti-infective therapy. In: Mandell G.L., Douglas R.G., Bennett J.E., editors. Principles and practice of infectious disease. New York: John Wiley, 1985.
Perry C.M., Jarvis B. Linezolid: a review of its use in the management of serious gram-positive infections. Drugs. 2001;61:525-551.
Quagliarello V.J., Scheld W.M. Treatment of bacterial meningitis. N. Engl. J. Med.. 1997;336:708-716.
Raoult D., Drancourt M. Antimicrobial therapy of rickettsial diseases. Antimicrob. Agents. Chemother.. 1994;35:2457-2462.
Sato K., Hoshino K., Mitsuhashi S. Mode of action of the new quinolones: the inhibitory action on DNA gyrase. Prog. Drug Res.. 1992;38:121-132.
Shimada J., Hori S. Adverse effects of fluoroquinolones. Prog. Drug Res.. 1992;38:133-143.
Stojiljkovic I., Evavold B.D., Kumar V. Antimicrobial properties of porphyrins. Expert. Opin. Investig. Drugs. 2001;10:309-320.
Tillotson G.S. Quinolones: structure–activity relationships and future predictions. J. Med. Microbiol.. 1996;44:320-324.
Zurenko G.E., Gibson J.K., Shinabarger D.L., et al. Oxazolidinones: a new class of antibacterials. Curr. Opin. Pharmacol.. 2001;1:470-476. (Easy-to-assimilate review that discusses this relatively new group of antibacterials)
Resistance (see also reading list in Ch. 49)
Bax R., Mullan N., Verhoef J. The millennium bugs—the need for and development of new antibacterials. Int. J. Antimicrob. Agents. 2000;16:51-59. (Good review that includes an account of the development of ‘resistance’ and a round-up of potential new drugs)
Courvalin P. Evasion of antibiotic action by bacteria. J. Antimicrob. Chemother.. 1996;37:855-869. (Covers developments in the understanding of the genetics and biochemical mechanisms of resistance)
Gold H.S., Moellering R.C. Antimicrobial drug resistance. N. Engl. J. Med.. 1996;335:1445-1453. (Excellent well-referenced review; covers mechanisms of resistance of important organisms to the main drugs; has useful table of therapeutic and preventive strategies, culled from the literature)
Heym B., Honoré N., Truffot-Pernot C., et al. Implications of multidrug resistance for the future of short-course chemotherapy of tuberculosis: a molecular study. Lancet. 1994;344:293-298.
Iseman M.D. Treatment of multidrug-resistant tuberculosis. N. Engl. J. Med.. 1993;329:784-791.
Livermore D.M. Antibiotic resistance in staphylococci. J. Antimicrob. Agents. 2000;16:S3-S10. (Overview of problems of bacterial resistance)
Michel M., Gutman L. Methicillin-resistant Staphylococcus aureus and vancomycin-resistant enterococci: therapeutic realities and possibilities. Lancet. 1997;349:1901-1906. (Excellent review article; good diagrams)
Nicas T.I., Zeckel M.L., Braun D.K. Beyond vancomycin: new therapies to meet the challenge of glycopeptide resistance. Trends Microbiol.. 1997;5:240-249.
Woodford N., Johnson A.P., Morrison D., Speller D.C. Current perspectives on glycopeptide resistance. Clin. Microbiol. Rev.. 1995;8:585-615. (Comprehensive review)
Bacterial evasion of host defences and miscellaneous
Bloom B.R., Small P.M. The evolving relation between humans and Mycobacterium tuberculosis. Lancet. 1998;338:677-678. (Editorial comment)
Loferer H. Mining bacterial genomes for antimicrobial targets. Mol. Med. Today. 2000;6:470-474. (An interesting article focusing on the way in which a better understanding of the bacterial genome may lead to new drugs)
New approaches to antibacterial drug discovery
These papers have been provided for those who want to learn more about the work underway to develop novel antibacterials (These papers have been provided for those who want to learn more about the work underway to develop novel antibacterials. Some are quite technical in nature)
Bansal A.K. Role of bioinformatics in the development of new antibacterial therapy. Expert. Rev. Anti. Infect. Ther.. 2008;6:51-65.
Escaich S. Antivirulence as a new antibacterial approach for chemotherapy. Curr. Opin. Chem. Biol.. 2008;12:400-408.
Falconer S.B., Brown E.D. New screens and targets in antibacterial drug discovery. Curr. Opin. Microbiol.. 2009;12:497-504.
Jagusztyn-Krynicka E.K., Wyszynska A. The decline of antibiotic era—new approaches for antibacterial drug discovery. Pol. J. Microbiol.. 2008;57:91-98.
Limsuwan S., Trip E.N., Kouwen T.R., et al. Rhodomyrtone: a new candidate as natural antibacterial drug from Rhodomyrtus tomentosa. Phytomedicine. 2009;16:645-651.
Lister P.D. The role of pharmacodynamic research in the assessment and development of new antibacterial drugs. Biochem. Pharmacol.. 2006;71:1057-1065.
O’Neill A.J. New antibacterial agents for treating infections caused by multi-drug resistant Gram-negative bacteria. Expert. Opin. Investig. Drugs. 2008;17:297-302.
Sit C.S., Vederas J.C. Approaches to the discovery of new antibacterial agents based on bacteriocins. Biochem. Cell Biol.. 2008;86:116-123.
http://www.who.int (Once again, the World Health Organization Web site is a mine of information about the demographics and treatment of infectious diseases. The sections on leprosy and tuberculosis are especially worthwhile studying. The site includes photographs, maps and much statistical information, as well as information on drug resistance. Highly recommended)
1Named after the Danish physician who devised the technique.
2Although this was the first evidence of the dramatic antibacterial effect of penicillin when given systemically in humans, topical penicillin had actually been used with success in five patients with eye infections 10 years previously by Paine, a graduate of St Mary’s who had obtained some penicillin mould from Fleming.
3Meticillin (previous name: methicillin) was the first β-lactamase-resistant penicillin; it is now not used clinically because it was particularly associated with interstitial nephritis but is remembered in the acronym ‘MRSA’—meticillin-resistant Staphylococcus aureus.
4Indeed, penicillins applied topically to the cortex are used to induce convulsions in an experimental setting.
5When ciprofloxacin was introduced, clinical pharmacologists and microbiologists sensibly suggested that it should be reserved for organisms already resistant to other drugs so as to prevent emergence of resistance. However, by 1989 it was already estimated that it was prescribed for 1 in 44 of Americans, so it would seem that the horse had not only left the stable but had bolted into the blue!
6This may also occur with some penicillins and cephalosporins.
7The basis of the difference between tuberculoid and lepromatous disease appears to be that the T cells from patients with the former vigorously produce interferon-γ, which enables macrophages to kill intracellular microbes, whereas in the latter case the immune response is dominated by interleukin-4, which blocks the action of interferon-γ (see Ch. 17).