8 Drug absorption and distribution
Overview
The physical processes of diffusion, penetration of membranes, binding to plasma protein and partition into fat and other tissues underlie the absorption and distribution of drugs. These processes are described, followed by more specific coverage of the process of drug absorption and related practical issue of routes of drug administration, and of the distribution of drugs into different bodily compartments. There is a short final section on special drug delivery systems designed to deliver drugs efficiently and selectively to their sites of action.
Introduction
Drug disposition is divided into four stages designated by the acronym ‘ADME’:
Absorption and distribution are considered here, together with routes of administration. Metabolism and excretion are covered in Chapter 9. We begin with a description of the physical processes that underlie drug disposition.
Physical Processes Underlying Drug Disposition
Drug molecules move around the body in two ways:
The chemical nature of a drug makes no difference to its transfer by bulk flow. The cardiovascular system provides a rapid long-distance distribution system. In contrast, diffusional characteristics differ markedly between different drugs. In particular, ability to cross hydrophobic diffusion barriers is strongly influenced by lipid solubility. Aqueous diffusion is part of the overall mechanism of drug transport, because it is this process that delivers drug molecules to and from the non-aqueous barriers. The rate of diffusion of a substance depends mainly on its molecular size, the diffusion coefficient for small molecules being inversely proportional to the square root of molecular weight. Consequently, while large molecules diffuse more slowly than small ones, the variation with molecular weight is modest. Many drugs fall within the molecular weight range 200–1000 Da, and variations in aqueous diffusion rate have only a small effect on their overall pharmacokinetic behaviour. For most purposes, we can regard the body as a series of interconnected well-stirred compartments within each of which the drug concentration is uniform. It is movement between compartments, generally involving penetration of non-aqueous diffusion barriers, that determines where, and for how long, a drug will be present in the body after it has been administered. The analysis of drug movements with the help of a simple compartmental model is discussed in Chapter 9.
The Movement of Drug Molecules Across Cell Barriers
Cell membranes form the barriers between aqueous compartments in the body. A single layer of membrane separates the intracellular from the extracellular compartments. An epithelial barrier, such as the gastrointestinal mucosa or renal tubule, consists of a layer of cells tightly connected to each other so that molecules must traverse at least two cell membranes (inner and outer) to pass from one side to the other. Vascular endothelium is more complicated, its anatomical disposition and permeability varying from one tissue to another. Gaps between endothelial cells are packed with a loose matrix of proteins that act as filters, retaining large molecules and letting smaller ones through. The cut-off of molecular size is not exact: water permeates rapidly whereas molecules of 80 000–100 000 Da permeate very slowly. In some organs, especially the central nervous system (CNS) and the placenta, there are tight junctions between the cells, and the endothelium is encased in an impermeable layer of periendothelial cells (pericytes). These features prevent potentially harmful molecules from leaking from the blood into these organs and have major pharmacokinetic consequences for drug distribution.1
In other organs (e.g. the liver and spleen), endothelium is discontinuous, allowing free passage between cells. In the liver, hepatocytes form the barrier between intra- and extravascular compartments and take on several endothelial cell functions. Fenestrated endothelium occurs in endocrine glands, facilitating transfer to the bloodstream of hormones or other molecules through pores in the endothelium. Formation of fenestrated endothelium (angiogenesis) is controlled by a specific endocrine gland-derived vascular endothelial growth factor (dubbed EG-VEGF). Endothelial cells lining postcapillary venules have specialised functions relating to leukocyte migration and inflammation: the sophistication of the intercellular junction can be appreciated from the observation that leukocyte migration can occur without any detectable leak of water or small ions (see Ch. 16).
There are four main ways by which small molecules cross cell membranes (Fig. 8.1):
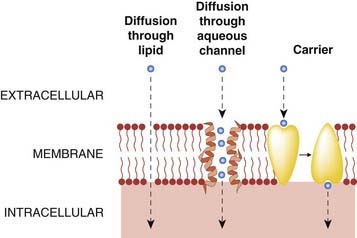
Fig. 8.1 Routes by which solutes can traverse cell membranes.
(Molecules can also cross cellular barriers by pinocytosis.)
Of these routes, diffusion through lipid and carrier-mediated transport are particularly important in relation to pharmacokinetic mechanisms. Diffusion through aquaporins (membrane glycoproteins that can be blocked by mercurial reagents such as para-chloromercurobenzene sulfonate) is probably important in the transfer of gases such as carbon dioxide, but the pores are too small in diameter (about 0.4 nm) to allow most drug molecules (which usually exceed 1 nm in diameter) to pass through. Consequently, drug distribution is not notably abnormal in patients with genetic diseases affecting aquaporins. Pinocytosis involves invagination of part of the cell membrane and the trapping within the cell of a small vesicle containing extracellular constituents. The vesicle contents can then be released within the cell, or extruded from its other side. This mechanism is important for the transport of some macromolecules (e.g. insulin, which crosses the blood–brain barrier by this process), but not for small molecules.
Diffusion through lipid and carrier-mediated transport will now be discussed in more detail.
Diffusion through Lipid
Non-polar molecules (in which electrons are uniformly distributed) dissolve freely in membrane lipids, and consequently diffuse readily across cell membranes. The number of molecules crossing the membrane per unit area in unit time is determined by the permeability coefficient, P, and the concentration difference across the membrane. Permeant molecules must be present within the membrane in sufficient numbers and must be mobile within the membrane if rapid permeation is to occur. Thus, two physicochemical factors contribute to P, namely solubility in the membrane (which can be expressed as a partition coefficient for the substance distributed between the membrane phase and the aqueous environment) and diffusivity, which is a measure of the mobility of molecules within the lipid and is expressed as a diffusion coefficient. The diffusion coefficient varies only slightly between different drugs, as noted above, so the most important variable is the partition coefficient (Fig. 8.2). Consequently, there is a close correlation between lipid solubility and the permeability of the cell membrane to different substances. For this reason, lipid solubility is one of the most important determinants of the pharmacokinetic characteristics of a drug, and many properties—such as rate of absorption from the gut, penetration into different tissues and the extent of renal elimination—can be predicted from knowledge of a drug’s lipid solubility.
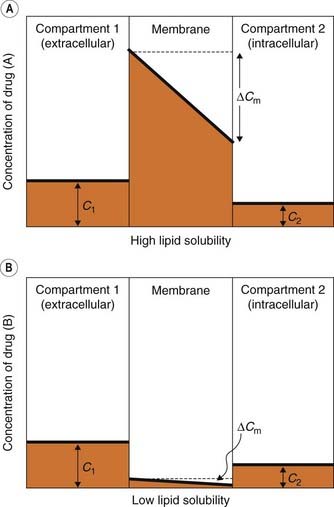
Fig. 8.2 The importance of lipid solubility in membrane permeation.
[A] and [B] Figures show the concentration profile in a lipid membrane separating two aqueous compartments. A lipid-soluble drug (A) is subject to a much larger transmembrane concentration gradient (ΔCm) than a lipid-insoluble drug (B). It therefore diffuses more rapidly, even though the aqueous concentration gradient (C1–C2) is the same in both cases.
pH and ionisation
One important complicating factor in relation to membrane permeation is that many drugs are weak acids or bases, and therefore exist in both unionised and ionised form, the ratio of the two forms varying with pH. For a weak base, the ionisation reaction is:
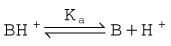
and the dissociation constant pKa is given by the Henderson–Hasselbalch equation
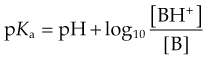
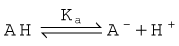
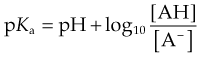
In either case, the ionised species, BH+ or A−, has very low lipid solubility and is virtually unable to permeate membranes except where a specific transport mechanism exists. The lipid solubility of the uncharged species, B or AH, depends on the chemical nature of the drug; for many drugs, the uncharged species is sufficiently lipid soluble to permit rapid membrane permeation, although there are exceptions (e.g. aminoglycoside antibiotics; see Ch. 50) where even the uncharged molecule is insufficiently lipid soluble to cross membranes appreciably. This is usually because of the occurrence of hydrogen-bonding groups (such as hydroxyl in sugar moieties in aminoglycosides) that render the uncharged molecule hydrophilic.
pH partition and ion trapping
Ionisation affects not only the rate at which drugs permeate membranes but also the steady-state distribution of drug molecules between aqueous compartments, if a pH difference exists between them. Figure 8.3 shows how a weak acid (e.g. aspirin, pKa 3.5) and a weak base (e.g. pethidine, pKa 8.6) would be distributed at equilibrium between three body compartments, namely plasma (pH 7.4), alkaline urine (pH 8) and gastric juice (pH 3). Within each compartment, the ratio of ionised to unionised drug is governed by the pKa of the drug and the pH of that compartment. It is assumed that the unionised species can cross the membrane, and therefore reaches an equal concentration in each compartment. The ionised species is assumed not to cross at all. The result is that, at equilibrium, the total (ionised + unionised) concentration of the drug will be different in the two compartments, with an acidic drug being concentrated in the compartment with high pH (‘ion trapping’), and vice versa. The concentration gradients produced by ion trapping can theoretically be very large if there is a large pH difference between compartments. Thus, aspirin would be concentrated more than four-fold with respect to plasma in an alkaline renal tubule, and about 6000-fold in plasma with respect to the acidic gastric contents. Such large gradients are not achieved in reality for two main reasons. First, the attribution of total impermeability to the charged species is not realistic, and even a small permeability will attenuate considerably the concentration difference that can be reached. Second, body compartments rarely approach equilibrium. Neither the gastric contents nor the renal tubular fluid stands still, and the resulting flux of drug molecules reduces the concentration gradients well below the theoretical equilibrium conditions. The pH partition mechanism nonetheless correctly explains some of the qualitative effects of pH changes in different body compartments on the pharmacokinetics of weakly acidic or basic drugs, particularly in relation to renal excretion and to penetration of the blood–brain barrier.
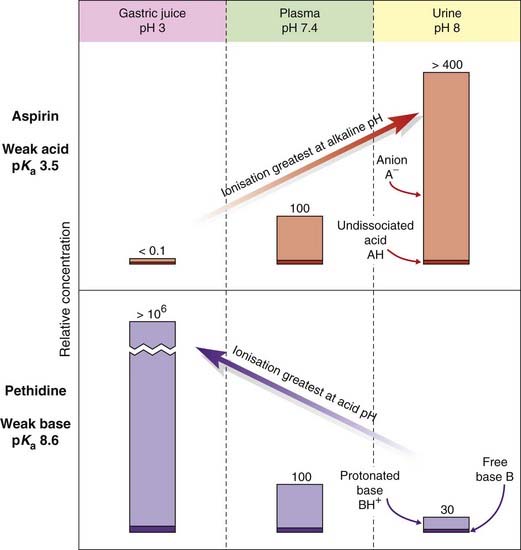
Fig. 8.3 Theoretical partition of a weak acid (aspirin) and a weak base (pethidine) between aqueous compartments (urine, plasma and gastric juice) according to the pH difference between them.
Numbers represent relative concentrations (total plasma concentration = 100). It is assumed that the uncharged species in each case can permeate the cellular barrier separating the compartments, and therefore reaches the same concentration in all three. Variations in the fractional ionisation as a function of pH give rise to the large total concentration differences with respect to plasma.
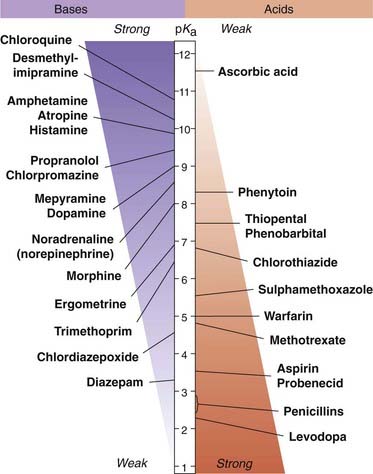
pH partition is not the main determinant of the site of absorption of drugs from the gastrointestinal tract. This is because the enormous absorptive surface area of the villi and microvilli in the ileum compared with the much smaller surface area in the stomach is of overriding importance. Thus, absorption of an acidic drug such as aspirin is promoted by drugs that accelerate gastric emptying (e.g. metoclopramide) and retarded by drugs that slow gastric emptying (e.g. propantheline), despite the fact that the acidic pH of the stomach contents favours absorption of weak acids. Values of pKa for some common drugs are shown in Figure 8.4.
There are several important consequences of pH partition:
Carrier-Mediated Transport
Many cell membranes possess specialised transport mechanisms that regulate entry and exit of physiologically important molecules, such as sugars, amino acids, neurotransmitters and metal ions. They are broadly divided into solute carrier (SLC) transporters and ATP-binding cassette (ABC) transporters. The former mediate passive movement of solutes down their electrochemical gradient, while the latter are active pumps fuelled by ATP. Over 300 human genes are believed to code these transporters, most of which act mainly on endogenous substrates, but some also transport foreign chemicals (‘xenobiotics’) including drugs (see Hediger et al., 2004). The role of such transporters in neurotransmitter function is discussed in Chapters 13, 14 and 36.
Organic cation transporters and organic anion transporters
Two structurally related SLC carriers of importance in drug distribution are the organic cation transporters (OCTs) and organic anion transporters (OATs). Generally, such transport systems involve a carrier molecule, i.e. a transmembrane protein that binds one or more molecules or ions, changes conformation and releases them on the other side of the membrane. Such systems may operate purely passively, without any energy source; in this case, they merely facilitate the process of transmembrane equilibration of a single transported species in the direction of its electrochemical gradient. The mechanism is called facilitated diffusion and the transporter is a ‘uniporter’. The OCTs (several families of SLC transporters) translocate dopamine, choline and various drugs including vecuronium, quinine and procainamide. They are uniporters and cause facilitated diffusion down the electrochemical gradient. OCT2 (transporter in proximal tubular cells in the kidney) concentrates drugs such as cisplatin (an important anticancer drug) in these cells, an explanation of its selective nephrotoxicity; related drugs (e.g. carboplatin, oxaliplatin) are not transported by OCT2 and are less nephrotoxic; competition with cimetididine for OCT2 offers possible protection against cisplatin nephrotoxicity (Fig. 8.5). Other SLCs are coupled to the electrochemical gradient of Na+ or other ions across the membrane, generated by ATP-dependent ion pumps (see Ch. 4); in this case, transport can occur against an electrochemical gradient. It may involve exchange of one molecule for another (‘antiport’) or transport of two molecules together in the same direction (’symport’). The OATs are responsible for the renal secretion of urate, prostaglandins, several vitamins and p-amino hippurate, and for drugs such as probenecid as well as many antibiotics, antiviral drugs, non-steroidal anti-inflammatory drugs and antineoplastic drugs among others. Uptake is driven by exchange with intracellular dicarboxylic acids (mainly α-ketoglutarate, partly derived from cellular metabolism and partly by co-transport with Na+ entering cells down its concentration gradient). Metabolic energy is provided by ATP for Na+/K+ exchange. Carrier-mediated transport, because it involves a binding step, shows the characteristic of saturation.
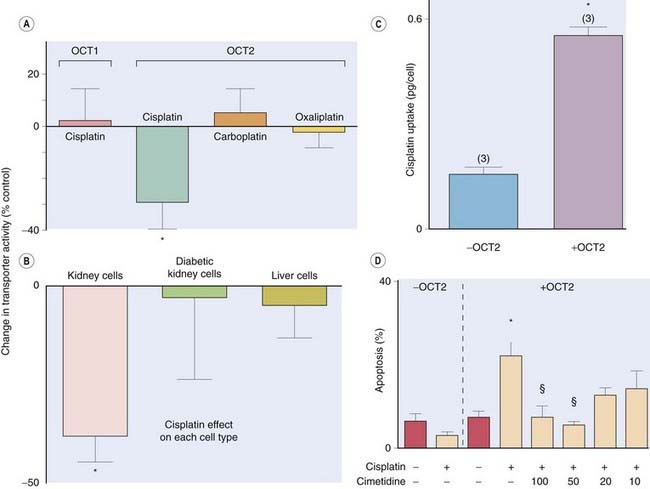
Fig. 8.5 Human organic cation transporter 2 (OCT2) mediates cisplatin nephrotoxicity.
OCT2 is expressed in kidney whereas OCT1 is expressed in liver. Cisplatin (100 µmol/l) influences the activity of OCT2 but not of OCT1, each expressed in a cultured cell line [A], whereas the less nephrotoxic drugs carboplatin and oxaliplatin do not. Cisplatin similarly influences OCT2 activity in fresh human kidney tubule cells but not in fresh hepatocytes or kidney cells from diabetic patients who are less susceptible to ciplatin nephrotoxicity [B]. Cisplatin accumulates in cells that express OCT2 [C] and causes cell death [D]. Cimetidine competes with cisplatin for OCT2 and concentration dependently protects against cisplatin-induced apoptosis [D]—cimetidine concentrations are in µmol/l.
(Data redrawn from Ciarimboli G et al. 2005 Am J Pathol 167: 1477–1484.)
Carriers of this type are ubiquitous, and many pharmacological effects are the result of interference with them. Thus nerve terminals have transport mechanisms for accumulating specific neurotransmitters, and there are many examples of drugs that act by inhibiting these transport mechanisms (see Chs 13, 14 and 36). From a general pharmacokinetic point of view, however, the main sites where SLCs, including OCTs and OATs, are expressed and carrier-mediated drug transport is important are:
P-glycoprotein transporters
P-glycoproteins (P-gp; P for ‘permeability’), which belong to the ABC transporter superfamily, are the second important class of transporters, and responsible for multidrug resistance in cancer cells. They are present in renal tubular brush border membranes, in bile canaliculi, in astrocyte foot processes in brain microvessels, and in the gastrointestinal tract. They play an important part in absorption, distribution and elimination of many drugs, and are often co-located with SLC drug carriers, so that a drug that has been concentrated by, for example, an OAT transporter in the basolateral membrane of a renal tubular cell may then be pumped out of the cell by a P-gp in the luminal membrane.
Polymorphic variation in the genes coding SLCs and P-gp contributes to individual genetic variation in responsiveness to different drugs. OCT1 transports several drugs, including metformin (used to treat diabetes; see Ch. 30), into hepatocytes (in contrast to OCT2 which is active in renal proximal tubular cells, see above). Metformin acts partly through intracellular effects within hepatocytes. Single nucleotoide polymorphisms (SNPs; Ch. 56) that impair the function of OCT1 influence the effectiveness of metformin (Fig. 8.6). This is but one example of many genetic influences on drug effectiveness or toxicity via altered activity of carriers that influence drug disposition. Furthermore, induction or competitive inhibition of transport can occur in the presence of a second ligand that binds the carrier, so there is a potential for drug interaction (see Fig. 8.5 and Ch. 56). The characteristics of transport systems are discussed later, when patterns of distribution and elimination in the body as a whole are considered more fully.
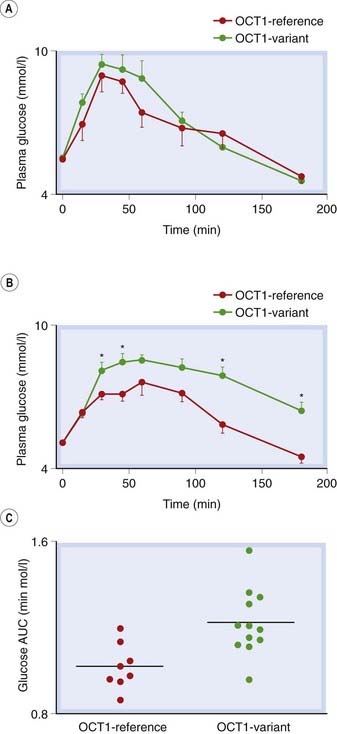
Fig. 8.6 Genetic variants of organic cation transporter 1 (OCT1) are associated with different responses to metformin in healthy humans.
[A] An oral glucose tolerance test (OGTT) gave similar plasma glucose responses in control subjects with only reference OCT1 alleles versus subjects with at least one reduced function OCT1 allele. [B] In contrast, after metformin treatment the OGTT response was less in the same reference subjects than in those with reduced function OCT1 alleles. [C] Glucose exposure estimated by area under the glucose time curves (AUC) was significantly lower in subjects with only reference OCT1 alleles, P = 0.004.
(Data redrawn from Yan Shu et al. 2007 J Clin Invest 117: 1422–1431.)
In addition to the processes so far described, which govern the transport of drug molecules across the barriers between different aqueous compartments, two additional factors have a major influence on drug distribution and elimination. These are:
Movement of drugs across cellular barriers 
Binding of Drugs to Plasma Proteins
At therapeutic concentrations in plasma, many drugs exist mainly in bound form. The fraction of drug that is free in aqueous solution can be less than 1%, the remainder being associated with plasma protein. It is the unbound drug that is pharmacologically active. Such seemingly small differences in protein binding (e.g. 99.5 versus 99.0%) can have large effects on free drug concentration and drug effect. Such differences are common between human plasma and plasma from species used in preclinical drug testing, and must be taken into account when estimating a suitable dose for ‘first time in human’ studies. The most important plasma protein in relation to drug binding is albumin, which binds many acidic drugs (e.g. warfarin, non-steroidal anti-inflammatory drugs, sulfonamides) and a smaller number of basic drugs (e.g. tricyclic antidepressants and chlorpromazine). Other plasma proteins, including β-globulin and an acid glycoprotein that increases in inflammatory disease, have also been implicated in the binding of certain basic drugs, such as quinine.
The amount of a drug that is bound to protein depends on three factors:
As a first approximation, the binding reaction can be regarded as a simple association of the drug molecules with a finite population of binding sites, exactly analogous to drug–receptor binding (see Ch. 2):

The usual concentration of albumin in plasma is about 0.6 mmol/l (4 g/100 ml). With two sites per albumin molecule, the drug-binding capacity of plasma albumin would therefore be about 1.2 mmol/l. For most drugs, the total plasma concentration required for a clinical effect is much less than 1.2 mmol/l, so with usual therapeutic doses the binding sites are far from saturated, and the concentration bound [DS] varies nearly in direct proportion to the free concentration [D]. Under these conditions, the fraction bound, [DS]/([D] + [DS]), is independent of the drug concentration. However, some drugs, for example tolbutamide (Ch. 30), work at plasma concentrations at which the binding to protein is approaching saturation (i.e. on the flat part of the binding curve). This means that adding more drug to the plasma increases its free concentration disproportionately. Doubling the dose of such a drug can therefore more than double the free (pharmacologically active) concentration. This is illustrated in Figure 8.7.
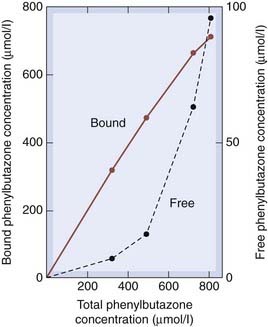
Fig. 8.7 Binding of phenylbutazone to plasma albumin.
The graph shows the disproportionate increase in free concentration as the total concentration increases, owing to the binding sites approaching saturation.
(Data from Brodie B, Hogben C A M 1957 J Pharm Pharmacol 9: 345.)
Binding sites on plasma albumin bind many different drugs, so competition can occur between them. If two drugs (A and B) compete in this way, administration of drug B can reduce the protein binding, and hence increase the free plasma concentration, of drug A. To do this, drug B needs to occupy an appreciable fraction of the binding sites. Few therapeutic drugs affect the binding of other drugs because they occupy, at therapeutic plasma concentrations, only a tiny fraction of the available sites. Sulfonamides (Ch. 50) are an exception, because they occupy about 50% of the binding sites at therapeutic concentrations and so can cause harmful effects by displacing other drugs or, in premature babies, bilirubin (Ch. 56). Much has been made of binding interactions of this kind as a source of untoward drug interactions in clinical medicine, but this type of competition is less important than was once thought (see Ch. 56).
Partition into Body Fat and Other Tissues
Fat represents a large, non-polar compartment. In practice, this is important for only a few drugs, mainly because the effective fat:water partition coefficient is relatively low for most drugs. Morphine, for example, although quite lipid soluble enough to cross the blood–brain barrier, has a lipid:water partition coefficient of only 0.4, so sequestration of the drug by body fat is of little importance. Thiopental, by comparison (fat:water partition coefficient approximately 10), accumulates substantially in body fat. This has important consequences that limit its usefulness as an intravenous anaesthetic to short-term initiation (‘induction’) of anaesthesia (Ch. 40).
The second factor that limits the accumulation of drugs in body fat is its low blood supply—less than 2% of the cardiac output. Consequently, drugs are delivered to body fat rather slowly, and the theoretical equilibrium distribution between fat and body water is approached slowly. For practical purposes, therefore, partition into body fat when drugs are given acutely is important only for a few highly lipid-soluble drugs (e.g. general anaesthetics; Ch. 40). When lipid-soluble drugs are given chronically, however, accumulation in body fat is often significant (e.g. benzodiazepines; Ch. 43). Some dugs and environmental contaminants, if ingested intermittently, accumulate slowly but progressively in body fat.
Body fat is not the only tissue in which drugs can accumulate. Chloroquine—an antimalarial drug (Ch. 53)—has a high affinity for melanin and is taken up by the retina, which is rich in melanin granules, accounting for its ocular toxicity. Tetracyclines (Ch. 50) accumulate slowly in bones and teeth, because they have a high affinity for calcium, and should not be used in children for this reason. Very high concentrations of amiodarone (an antidysrhythmic drug; Ch. 21) accumulate in liver and lung during chronic use, causing hepatitis and interstitial pulmonary fibrosis.
Binding of drugs to plasma proteins
Drug Absorption and Routes of Administration
The main routes of drug administration and elimination are shown schematically in Figure 8.8. Absorption is defined as the passage of a drug from its site of administration into the plasma. It is important for all routes of administration except intravenous injection, where it is complete by definition. There are instances, such as topical administration of a steroid cream to skin or inhalation of a bronchodilator aerosol to treat asthma (Ch. 27), where absorption as just defined is not required for the drug to act, but in most cases the drug must enter plasma before reaching its site of action.
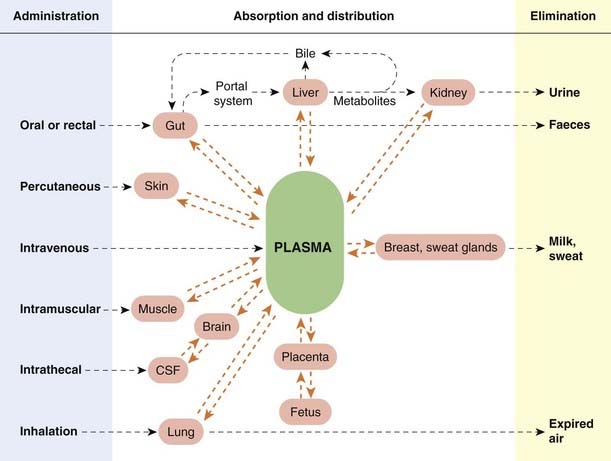
The main routes of administration are:
Oral Administration
Most drugs are taken by mouth and swallowed. Little absorption occurs until the drug enters the small intestine.
Drug Absorption from the Intestine
For most drugs the mechanism of absorption is the same as for other epithelial barriers, namely passive transfer at a rate determined by the ionisation and lipid solubility of the drug molecules. Figure 8.9 shows the absorption of various weak acids and bases as a function of pKa. As expected, strong bases of pKa 10 or higher are poorly absorbed, as are strong acids of pKa less than 3, because they are fully ionised. The arrow poison curare used by South American Indians contains quaternary ammonium compounds that block neuromuscular transmission (Ch. 13). These strong bases are poorly absorbed from the gastrointestinal tract, so the meat from animals killed in this way was safe to eat.
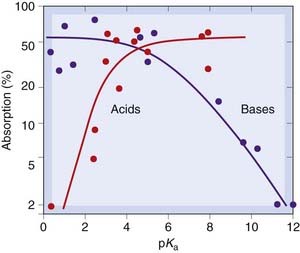
Fig. 8.9 Absorption of drugs from the intestine, as a function of pKa, for acids and bases.
Weak acids and bases are well absorbed; strong acids and bases are poorly absorbed.
(Redrawn from Schanker L S et al. 1957 J Pharmacol 120: 528.)
In a few instances, intestinal drug absorption depends on carrier-mediated transport rather than simple lipid diffusion. Examples include levodopa, used in treating Parkinson’s disease (see Ch. 39), which is taken up by the carrier that normally transports phenylalanine, and fluorouracil (Ch. 55), a cytotoxic drug that is transported by the system that carries natural pyrimidines (thymine and uracil). Iron is absorbed via specific carriers in the epithelial cell membranes of jejunal mucosa, and calcium is absorbed by means of a vitamin D-dependent carrier system.
Factors Affecting Gastrointestinal Absorption
Typically, about 75% of a drug given orally is absorbed in 1–3 h, but numerous factors alter this, some physiological and some to do with the formulation of the drug. The main factors are:
Gastrointestinal motility has a large effect. Many disorders (e.g. migraine, diabetic neuropathy) cause gastric stasis and slow drug absorption. Drug treatment can also affect motility, either reducing (e.g. drugs that block muscarinic receptors; see Ch. 13) or increasing it (e.g. metoclopramide, an antiemetic used in migraine to facilitate absorption of analgesic). Excessively rapid movement of gut contents (e.g. in some forms of diarrhoea) can impair absorption. Several drugs (e.g. propranolol) reach a higher plasma concentration if they are taken after a meal, probably because food increases splanchnic blood flow. Conversely, splanchnic blood flow is greatly reduced by hypovolaemia or heart failure, with a resultant reduction of drug absorption.
Particle size and formulation have major effects on absorption. In 1971, patients in a New York hospital were found to require unusually large maintenance doses of digoxin (Ch. 21). In a study on normal volunteers, it was found that standard digoxin tablets from different manufacturers resulted in grossly different plasma concentrations (Fig. 8.10), even though the digoxin content of the tablets was the same, because of differences in particle size. Because digoxin is rather poorly absorbed, small differences in the pharmaceutical formulation can make a large difference to the extent of absorption.
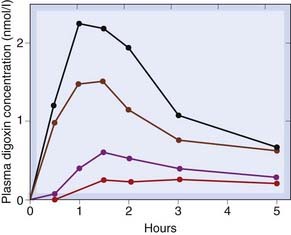
Fig. 8.10 Variation in oral absorption among different formulations of digoxin.
The four curves show the mean plasma concentrations attained for the four preparations, each of which was given on separate occasions to four subjects. The large variation has caused the formulation of digoxin tablets to be standardised since this study was published.
(From Lindenbaum J et al. 1971 N Engl J Med 285: 1344.)
Therapeutic drugs are formulated pharmaceutically to produce desired absorption characteristics. Capsules may be designed to remain intact for some hours after ingestion in order to delay absorption, or tablets may have a resistant coating to give the same effect. In some cases, a mixture of slow- and fast-release particles is included in a capsule to produce rapid but sustained absorption. More elaborate pharmaceutical systems include modified-release preparations that permit less frequent dosing. Such preparations not only increase the dose interval but also reduce adverse effects related to high peak plasma concentrations following administration of a conventional formulation. Osmotically driven ‘minipumps’ can be implanted experimentally, and some oral extended-release preparations that are used clinically use the same principle, the tablet containing an osmotically active core and being bound by an impermeable membrane with a precisely engineered pore to allow drug to exit in solution, delivering drug at an approximately constant rate into the bowel lumen. Such preparations may, however, cause problems related to high local concentrations of drug in the intestine (an osmotically released preparation of the anti-inflammatory drug indometacin, Ch. 26, had to be withdrawn because it caused small bowel perforation), and are sensitive to variations in small bowel transit time that occur during ageing and with disease.
Physicochemical factors (including some drug interactions; Ch. 56) affect drug absorption. Tetracycline binds strongly to Ca2+, and calcium-rich foods (especially milk) prevent its absorption (Ch. 50). Bile acid-binding resins such as colestyramine (used to treat diarrhoea caused by bile acids) bind several drugs, for example warfarin (Ch. 24) and thyroxine (Ch. 33).
When drugs are administered by mouth, the intention is usually that they should be absorbed and cause a systemic effect, but there are exceptions. Vancomycin is very poorly absorbed, and is administered orally to eradicate toxin-forming Clostridium difficile from the gut lumen in patients with pseudomembranous colitis (an adverse effect of broad-spectrum antibiotics caused by appearance of this organism in the bowel). Mesalazine is a formulation of 5-aminosalicylic acid in a pH-dependent acrylic coat that degrades in the terminal ileum and proximal colon, and is used to treat inflammatory bowel disease affecting this part of the gut. Olsalazine is a prodrug (see below) consisting of a dimer of two molecules of 5-aminosalicylic acid that is cleaved by colonic bacteria in the distal bowel and is used to treat patients with distal colitis.
Bioavailability and bioequivalence
To get from the lumen of the small intestine into the systemic circulation, a drug must not only penetrate the intestinal mucosa, it must also run the gauntlet of enzymes that may inactivate it in gut wall and liver, referred to as ‘presystemic’ or ‘first-pass’ metabolism or clearance. The term bioavailability is used to indicate the fraction (F) of an orally administered dose that reaches the systemic circulation as intact drug, taking into account both absorption and local metabolic degradation. F is measured by determining the plasma drug concentration versus time curves in a group of subjects following oral and (on a separate occasion) intravenous administration (the fraction absorbed following an intravenous dose is 1 by definition). The areas under the plasma concentration time curves (AUC) are used to estimate F as AUCoral/AUCintravenous. Bioavailability is not a characteristic solely of the drug preparation: variations in enzyme activity of gut wall or liver, in gastric pH or intestinal motility all affect it. Because of this, one cannot speak strictly of the bioavailability of a particular preparation, but only of that preparation in a given individual on a particular occasion, and F determined in a group of healthy volunteer subjects may differ substantially from the value determined in patients with diseases of gastrointestinal or circulatory systems.
Bioavailability relates only to the total proportion of the drug that reaches the systemic circulation and neglects the rate of absorption. If a drug is completely absorbed in 30 min, it will reach a much higher peak plasma concentration (and have a more dramatic effect) than if it were absorbed more slowly. Regulatory authorities—which have to make decisions about the licensing of products that are ‘generic equivalents’ of patented products—require evidence of ‘bioequivalence’ based on the maximum concentration achieved (Cmax) and time between dosing and Cmax (tmax) as well as AUC(0–∞). For most drugs, each of these parameters (AUC(0–∞), Cmax, tmax) must lie between 80% and 125% of the lead product for the new generic product to be accepted as bioequivalent.
Sublingual Administration
Absorption directly from the oral cavity is sometimes useful (provided the drug does not taste too horrible) when a rapid response is required, particularly when the drug is either unstable at gastric pH or rapidly metabolised by the liver. Glyceryl trinitrate and buprenorphine are examples of drugs that are often given sublingually (Chs 21 and 41, respectively). Drugs absorbed from the mouth pass directly into the systemic circulation without entering the portal system, and so escape first-pass metabolism by enzymes in the gut wall and liver.
Rectal Administration
Rectal administration is used for drugs that are required either to produce a local effect (e.g. anti-inflammatory drugs for use in ulcerative colitis) or to produce systemic effects. Absorption following rectal administration is often unreliable, but this route can be useful in patients who are vomiting or are unable to take medication by mouth (e.g. postoperatively). It is used to administer diazepam to children who are in status epilepticus (Ch. 44), in whom it is difficult to establish intravenous access.
Application to Epithelial Surfaces
Cutaneous Administration
Cutaneous administration is used when a local effect on the skin is required (e.g. topically applied steroids). Appreciable absorption may nonetheless occur and lead to systemic effects.
Most drugs are absorbed very poorly through unbroken skin. However, a number of organophosphate insecticides (see Ch. 13), which need to penetrate an insect’s cuticle in order to work, are absorbed through skin, and accidental poisoning occurs in farm workers.
 A case is recounted of a 35-year-old florist in 1932. ‘While engaged in doing a light electrical repair job at a work bench he sat down in a chair on the seat of which some “Nico-Fume liquid” (a 40% solution of free nicotine) had been spilled. He felt the solution wet through his clothes to the skin over the left buttock, an area about the size of the palm of his hand. He thought nothing further of it and continued at his work for about 15 minutes, when he was suddenly seized with nausea and faintness … and found himself in a drenching sweat. On the way to hospital he lost consciousness.’ He survived, just, and then 4 days later: ‘On discharge from the hospital he was given the same clothes that he had worn when he was brought in. The clothes had been kept in a paper bag and were still damp where they had been wet with the nicotine solution.’ The sequel was predictable. He survived again but felt thereafter ‘unable to enter a greenhouse where nicotine was being sprayed’. Transdermal dosage forms of nicotine are now used to reduce the withdrawal symptoms that accompany stopping smoking (Ch. 48).
A case is recounted of a 35-year-old florist in 1932. ‘While engaged in doing a light electrical repair job at a work bench he sat down in a chair on the seat of which some “Nico-Fume liquid” (a 40% solution of free nicotine) had been spilled. He felt the solution wet through his clothes to the skin over the left buttock, an area about the size of the palm of his hand. He thought nothing further of it and continued at his work for about 15 minutes, when he was suddenly seized with nausea and faintness … and found himself in a drenching sweat. On the way to hospital he lost consciousness.’ He survived, just, and then 4 days later: ‘On discharge from the hospital he was given the same clothes that he had worn when he was brought in. The clothes had been kept in a paper bag and were still damp where they had been wet with the nicotine solution.’ The sequel was predictable. He survived again but felt thereafter ‘unable to enter a greenhouse where nicotine was being sprayed’. Transdermal dosage forms of nicotine are now used to reduce the withdrawal symptoms that accompany stopping smoking (Ch. 48).
Transdermal dosage forms, in which the drug is incorporated in a stick-on patch applied to the skin, are used increasingly, and several drugs—for example oestrogen and testosterone for hormone replacement (Ch. 34)—are available in this form. Such patches produce a steady rate of drug delivery and avoid presystemic metabolism. Fentanyl is available in a patch to treat intermittent breakthrough pain (Ch. 41). However, the method is suitable only for lipid-soluble drugs and is relatively expensive.
Nasal Sprays
Some peptide hormone analogues, for example of antidiuretic hormone (Ch. 32) and of gonadotrophin-releasing hormone (see Ch. 34), are given as nasal sprays, as is calcitonin (Ch. 35). Absorption is believed to take place through mucosa overlying nasal-associated lymphoid tissue. This is similar to mucosa overlying Peyer’s patches in the small intestine, which is also unusually permeable.
Eye Drops
Many drugs are applied as eye drops, relying on absorption through the epithelium of the conjunctival sac to produce their effects. Desirable local effects within the eye can be achieved without causing systemic side effects; for example, dorzolamide is a carbonic anhydrase inhibitor that is given as eye drops to lower ocular pressure in patients with glaucoma. It achieves this without affecting the kidney (see Ch. 28), thus avoiding the acidosis that is caused by oral administration of acetazolamide. Some systemic absorption from the eye occurs, however, and can result in unwanted effects (e.g. bronchospasm in asthmatic patients using timolol eye drops for glaucoma).
Administration by Inhalation
Inhalation is the route used for volatile and gaseous anaesthetics (see Ch. 40), the lung serving as the route of both administration and elimination. The rapid exchange resulting from the large surface area and blood flow makes it possible to achieve rapid adjustments of plasma concentration. The pharmacokinetic behaviour of inhalation anaesthetics is discussed more fully in Chapter 40.
Drugs used for their effects on the lung are also given by inhalation, usually as an aerosol. Glucocorticoids (e.g. beclometasone dipropionate) and bronchodilators (e.g. salbutamol; Ch. 27) are given in this way to achieve high local concentrations in the lung while minimising systemic side effects. However, drugs given by inhalation in this way are usually partly absorbed into the circulation, and systemic side effects (e.g. tremor following salbutamol) can occur. Chemical modification of a drug may minimise such absorption. For example, ipratropium, a muscarinic receptor antagonist (Chs 13 and 27), is a quaternary ammonium ion analogue of atropine. It is used as an inhaled bronchodilator because its poor absorption minimises systemic adverse effects.
Administration by Injection
Intravenous injection is the fastest and most certain route of drug administration. Bolus injection rapidly produces a high concentration of drug, first in the right heart and lungs and then in the systemic circulation. The peak concentration reaching the tissues depends critically on the rate of injection. Administration by steady intravenous infusion avoids the uncertainties of absorption from other sites, while avoiding high peak plasma concentrations caused by bolus injection.
Subcutaneous or intramuscular injection of drugs usually produces a faster effect than oral administration, but the rate of absorption depends greatly on the site of injection and on local blood flow. The rate-limiting factors in absorption from the injection site are:
Absorption from a site of injection (sometimes but not always desirable, see below) is increased by increased blood flow. Hyaluronidase (an enzyme that breaks down the intercellular matrix, thereby increasing diffusion) also increases drug absorption from the site of injection. Conversely, absorption is reduced in patients with circulatory failure (’shock’) in whom tissue perfusion is reduced (Ch. 22).
Methods for Delaying Absorption
It may be desirable to delay absorption, either to produce a local effect or to prolong systemic action. For example, addition of adrenaline (epinephrine) to a local anaesthetic reduces absorption of the anaesthetic into the general circulation, usefully prolonging the anaesthetic effect (Ch. 42). Formulation of insulin with protamine or zinc produces a long-acting form (see Ch. 30). Procaine penicillin (Ch. 50) is a poorly soluble salt of penicillin; when injected as an aqueous suspension, it is slowly absorbed and exerts a prolonged action. Esterification of steroid hormones (e.g. medroxyprogesterone acetate, testosterone propionate; Ch. 34) and antipsychotic drugs (e.g. fluphenazine decanoate; Ch. 45) increases their solubility in oil and slows their rate of absorption when they are injected in an oily solution.
Another method used to achieve slow and continuous absorption of certain steroid hormones (e.g. estradiol; Ch. 34) is the subcutaneous implantation of solid pellets. The rate of absorption is proportional to the surface area of the implant.
Intrathecal Injection
Injection of a drug into the subarachnoid space via a lumbar puncture needle is used for some specialised purposes. Methotrexate (Ch. 55) is administered in this way in the treatment of certain childhood leukaemias to prevent relapse in the CNS. Regional anaesthesia can be produced by intrathecal administration of a local anaesthetic such as bupivacaine (see Ch. 42); opioid analgesics can also be used in this way (Ch. 41). Baclofen (a GABA analogue; Ch. 37) is used to treat disabling muscle spasms. It has been administered intrathecally to minimise its adverse effects. Some antibiotics (e.g. aminoglycosides) cross the blood–brain barrier very slowly, and in rare clinical situations where they are essential (e.g. nervous system infections with bacteria resistant to other antibiotics) can be given intrathecally or directly into the cerebral ventricles via a reservoir.
Intravitreal Injection
Ranibizumab (monoclonal antibody fragment that binds to vascular endothelial growth factor; Ch. 22) is given by intravitreal injection by ophthalmologists treating patients with wet age-related macular degeneration.
Drug absorption and bioavailability
Distribution of Drugs in the Body
Body Fluid Compartments
Body water is distributed into four main compartments, as shown in Figure 8.11. The total body water as a percentage of body weight varies from 50% to 70%, being rather less in women than in men.
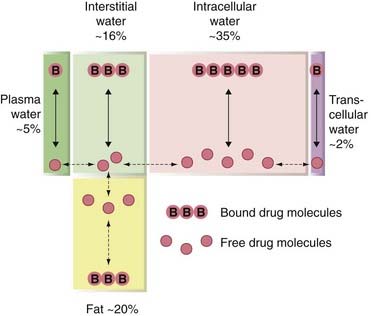
Fig. 8.11 The main body fluid compartments, expressed as a percentage of body weight.
Drug molecules exist in bound or free form in each compartment, but only the free drug is able to move between the compartments.
Extracellular fluid comprises the blood plasma (about 4.5% of body weight), interstitial fluid (16%) and lymph (1.2%). Intracellular fluid (30–40%) is the sum of the fluid contents of all cells in the body. Transcellular fluid (2.5%) includes the cerebrospinal, intraocular, peritoneal, pleural and synovial fluids, and digestive secretions. The fetus may also be regarded as a special type of transcellular compartment. Within each of these aqueous compartments, drug molecules usually exist both in free solution and in bound form; furthermore, drugs that are weak acids or bases will exist as an equilibrium mixture of the charged and uncharged forms, the position of the equilibrium depending on the pH.
The equilibrium pattern of distribution between the various compartments will therefore depend on:
To enter the transcellular compartments from the extracellular compartment, a drug must cross a cellular barrier, a particularly important example in the context of pharmacokinetics being the blood–brain barrier.
The Blood–Brain Barrier
The concept of the blood–brain barrier was introduced by Paul Ehrlich to explain his observation that intravenously injected dye stained most tissues yet the brain remained unstained. The barrier consists of a continuous layer of endothelial cells joined by tight junctions and surrounded by pericytes. The brain is consequently inaccessible to many drugs with a lipid solubility that is insufficient to allow penetration of the blood–brain barrier. However, inflammation can disrupt the integrity of the blood–brain barrier, allowing normally impermeant substances to enter the brain (Fig. 8.12); consequently, penicillin (Ch. 50) can be given intravenously (rather than intrathecally) to treat bacterial meningitis (which is accompanied by intense inflammation).
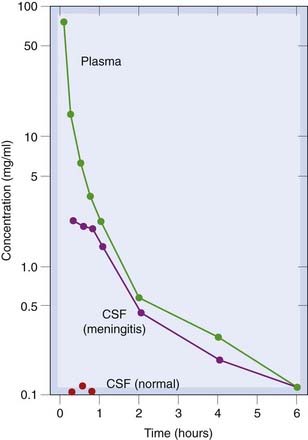
Fig. 8.12 Plasma and cerebrospinal fluid concentrations of an antibiotic (thienamycin) following an intravenous dose (25 mg/kg).
In normal rabbits, no drug reaches the cerebrospinal fluid (CSF), but in animals with experimental Escherichia coli meningitis the concentration of drug in CSF approaches that in the plasma.
(From Patamasucon & McCracken 1973 Antimicrob Agents Chemother 3: 270.)
Furthermore, in some parts of the CNS, including the chemoreceptor trigger zone, the barrier is leaky. This enables domperidone, an antiemetic dopamine receptor antagonist (Ch. 29 & 39) that does not penetrate the blood–brain barrier but does access the chemoreceptor trigger zone, to be used to prevent the nausea caused by dopamine agonists such as apomorphine when these are used to treat advanced Parkinson’s disease. This is achieved without loss of efficacy, because dopamine receptors in the basal ganglia are accessible only to drugs that have traversed the blood–brain barrier.
Methylnaltrexone bromide is a peripherally acting µ-opioid receptor antagonist used in treating opioid-induced constipation in patients requiring opioids as part of palliative care. It has limited gastrointestinal absorption and does not cross the blood–brain barrier, so does not block the desired CNS opioid effects. Several peptides, including bradykinin and enkephalins, increase blood–brain barrier permeability. There is interest in exploiting this to improve penetration of chemotherapy during treatment of brain tumours. In addition, extreme stress renders the blood–brain barrier permeable to drugs such as pyridostigmine (Ch. 13), which normally act peripherally.2
Volume of Distribution
The apparent volume of distribution, Vd, (see Ch. 10) is defined as the volume of fluid required to contain the total amount, Q, of drug in the body at the same concentration as that present in the plasma, Cp:
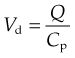
Values of Vd have been measured for many drugs (see Table 8.1).3 It is important to avoid identifying a given range of Vd too closely with a particular anatomical compartment. For example, insulin has a measured Vd similar to the volume of plasma water but exerts its effects on muscle, fat and liver via receptors that are exposed to interstitial fluid but not to plasma (Ch. 30).
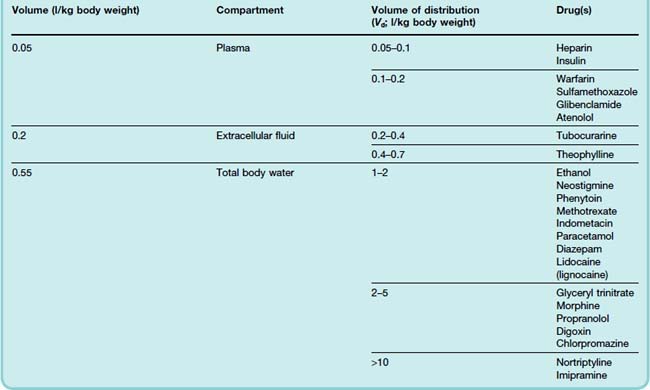
Drugs Confined to the Plasma Compartment
The plasma volume is about 0.05 l/kg body weight. A few drugs, such as heparin (Ch. 24), are confined to plasma because the molecule is too large to cross the capillary wall easily. More often, retention of a drug in the plasma following a single dose reflects strong binding to plasma protein. It is, nevertheless, the free drug in the interstitial fluid that exerts a pharmacological effect. Following repeated dosing, equilibration occurs and measured Vd increases. Some dyes, such as Evans blue, bind so strongly to plasma albumin that its Vd is used experimentally to measure plasma volume.
Drugs Distributed in the Extracellular Compartment
The total extracellular volume is about 0.2 l/kg, and this is the approximate Vd for many polar compounds, such as vecuronium (Ch. 13), gentamicin and carbenicillin (Ch. 50). These drugs cannot easily enter cells because of their low lipid solubility, and they do not traverse the blood–brain or placental barriers freely.
Distribution throughout the Body Water
Total body water represents about 0.55 l/kg. This approximates the distribution of relatively lipid-soluble drugs that readily cross cell membranes, such as phenytoin (Ch. 44) and ethanol (Ch. 48). Binding of drug outside the plasma compartment, or partitioning into body fat, increases Vd beyond total body water. Consequently, there are many drugs with Vd greater than the total body volume, such as morphine (Ch. 41), tricyclic antidepressants (Ch. 46) and haloperidol (Ch. 45). Such drugs are not efficiently removed from the body by haemodialysis, which is therefore unhelpful in managing overdose with such agents.
Drug distribution
Special Drug Delivery Systems
Several approaches are used or in development to improve drug delivery and localise the drug to the target tissue. They include:
Biologically Erodible Nanoparticles
Microspheres of biologically erodible polymers (see Varde & Pack, 2004) can be engineered to adhere to mucosal epithelium in the gut. Such particles can be loaded with drugs, including high-molecular-weight substances, as a means of improving absorption, which occurs both through mucosal absorptive epithelium and also through epithelium overlying Peyer’s patches. This approach has yet to be used clinically, but microspheres made from polyanhydride co-polymers of fumaric and sebacic acids by a technique known as phase inversion nanoencapsulation have been used to produce systemic absorption of insulin and of plasmid DNA following oral administration in rats, potentially enabling gene therapy (Ch. 59) to be administered orally. Various polymer nanoparticles, that can be loaded with drug molecules and targeted to specific tissues, are in development for many therapeutic applications (see Singh & Lillard, 2008), particularly as a means of delivering cytotoxic drugs specifically to cancer cells (see Ch. 55).
Prodrugs
Prodrugs are inactive precursors that are metabolised to active metabolites; they are described in Chapter 9. Some of the examples in clinical use confer no obvious benefits and have been found to be prodrugs only retrospectively, not having been designed with this in mind. However, some do have advantages. For example, the cytotoxic drug cyclophosphamide (see Ch. 55) becomes active only after it has been metabolised in the liver; it can therefore be taken orally without causing serious damage to the gastrointestinal epithelium. Levodopa is absorbed from the gastrointestinal tract and crosses the blood–brain barrier via an amino acid transport mechanism before conversion to active dopamine in nerve terminals in the basal ganglia (Ch. 39). Zidovudine is phosphorylated to its active triphosphate metabolite only in cells containing the appropriate reverse transcriptase, hence conferring selective toxicity towards cells infected with HIV (Ch. 51). Valaciclovir and famciclovir are each ester prodrugs, respectively of aciclovir and of penciclovir. Their bioavailability is greater than that of aciclovir and penciclovir, which are themselves prodrugs that are converted into active metabolites in virally infected cells (Ch. 51).
Other problems could theoretically be overcome by the use of suitable prodrugs; for example, instability of drugs at gastric pH, direct gastric irritation (aspirin was synthesised in the 19th century in a deliberate attempt to produce a prodrug of salicylic acid that would be tolerable when taken by mouth), failure of drug to cross the blood–brain barrier and so on. Progress with this approach remains slow, however, and the optimistic prodrug designer ‘will have to bear in mind that an organism’s normal reaction to a foreign substance is to burn it up for food’.
Antibody–Drug Conjugates
One of the aims of cancer chemotherapy is to improve the selectivity of cytotoxic drugs (see Ch. 55). One interesting possibility is to attach the drug to an antibody directed against a tumour-specific antigen, which will bind selectively to tumour cells.
Packaging in Liposomes
Liposomes are minute vesicles produced by sonication of an aqueous suspension of phospholipids. They can be filled with non-lipid-soluble drugs, which are retained until the liposome is disrupted. Liposomes are taken up by reticuloendothelial cells, especially in the liver. They are also concentrated in malignant tumours, and there is a possibility of achieving selective delivery of drugs in this way. Amphotericin, an antifungal drug used to treat systemic mycoses (Ch. 52), is available in a liposomal formulation that is less nephrotoxic and better tolerated than the conventional form, albeit considerably more expensive. In the future, it may be possible to direct drugs or genes selectively to a specific target by incorporating antibody molecules into liposomal membrane surfaces.
Coated Implantable Devices
Impregnated coatings have been developed that permit localised drug delivery from implants. Examples include hormonal delivery to the endometrium from intrauterine devices, and delivery of antithrombotic and antiproliferative agents (drugs or radiopharmaceuticals) to the coronary arteries from stents (devices inserted via a catheter after a diseased coronary artery has been dilated with a balloon). Stents reduce the occurrence of re-stenosis, but this can still occur at the margin of the device. Coating stents with drugs such as sirolimus (a potent immunosuppressant; see Ch. 26) embedded in a surface polymer prevents this important clinical problem.
References and Further Reading
Drug distribution (including blood–brain barrier)
Bauer B., Hartz A.M.S., Fricker G., Miller D.S. Modulation of P-glycoprotein transport function at the blood–brain barrier. Exp. Biol. Med.. 2005;230:118-127. (Reviews mechanisms by which P-glycoprotein activity can be modulated, including direct inhibition by specific competitors, and functional and transcriptional modulation)
Ciarimboli G. Organic cation transporters. Xenobiotica. 2008;38:936-971. (Discusses species- and tissue-specific distribution of different OCT isoforms and polymorphisms in OCTs as a source of variation in drug response)
de Boer A.G., van der Sandt I.C.J., Gaillard P.J. The role of drug transporters at the blood–brain barrier. Ann. Rev. Pharmacol. Toxicol. 2003;43:629-656. (Reviews the role of carrier- and receptor-mediated transport systems in the blood–brain barrier; these include P-glycoprotein, multidrug-resistance proteins 17, nucleoside transporters, organic anion transporters and large amino acid transporters, the transferrin-1 and -2 receptors, and the scavenger receptors SB-AI and SB-BI)
Eraly S.A., Bush K.T., Sampogna R.V., et al. The molecular pharmacology of organic anion transporters: from DNA to FDA? Mol. Pharmacol.. 2004;65:479-487. (Reviews aspects of the molecular biology and pharmacology of the organic anion transporters, and discusses their structural biology, paired genomic organisation, developmental regulation, toxicology and pharmacogenetics)
Hediger M.A., Romero M.F., Peng J.-B., et al. The ABCs of solute carriers: physiological, pathological and therapeutic implications of human membrane transport proteins. Pflug. Arch.. 2004;447:465-468.
Koepsell H. Polyspecific organic cation transporters: their functions and interactions with drugs. Trends Pharmacol. Sci.. 2004;25:375-381. (Reviews organic cation transporters [OCT]1–3, which are expressed in gut, liver, kidney, heart, placenta, lung and brain, and facilitate diffusion of structurally diverse organic cations including monoamine neurotransmitters and many drugs; studies in knockout mice implicate OCT1 in the hepatic uptake and biliary excretion of cationic drugs, and OCT1 and 2 in renal proximal tubules participate in secreting cationic drugs into urine)
McNamara P.J., Abbassi M. Neonatal exposure to drugs in breast milk. Pharm. Res.. 2004;21:555-566. (Review)
Neff M.W., Robertson K.R., Wong A.K., et al. Breed distribution and history of canine mdr1-1 delta, a pharmacogenetic mutation that marks the emergence of breeds from the collie lineage. Proc. Natl. Acad. Sci. USA. 2004;101:11725-11730. (The breed distribution and frequency of mdr1-1 delta have applications in veterinary medicine, whereas the allele’s history recounts the emergence of formally recognised breeds from an admixed population of working sheepdogs)
Petzinger E., Geyer J. Drug transporters in pharmacokinetics. Naunyn-Schmiederberg’s Arch. Pharmacol.. 2006;372:465-475. (Emphasises the interplay between drug metabolism and drug transport, especially in liver)
Ritter C.A., Jedlitschky G., Schwabedissen H.M.Z., et al. Cellular export of drugs and signaling molecules by the ATP-binding cassette transporters MRP4 (ABCC4) and MRP5 (ABCC5). Drug Metab. Rev.. 2005;37:253-278. (Members of the multidrug resistance-associated protein [MRP] subfamily of ATP-binding cassette transporters, MRP4 and 5 are organic anion transporters; they transport nucleotides and nucleotide analogues, and also cyclic nucleotides, so are implicated in signal transduction. MRP4 also transports conjugated steroids, prostaglandins and glutathione)
Sasaki M., Suzuki H., Aoki J., et al. Prediction of in vivo biliary clearance from the in vitro transcellular transport of organic anions across a double-transfected Madin–Darby canine kidney II monolayer expressing both rat organic anion transporting polypeptide 4 and multidrug resistance associated protein 2. Mol. Pharmacol.. 2004;66:450-459. (Double-transfected Madin–Darby canine kidney cell monolayer may be useful in analysing hepatic transport of organic anions and in predicting in vivo biliary clearance)
Cornford E.M., Cornford M.E. New systems for delivery of drugs to the brain in neurological disease. Lancet Neurol.. 2002;1:306-315. (Reviews augmentation of pinocytosis to deliver drugs to the brain. Macromolecules can be conjugated to peptidomimetic ligands that bind peptide receptors, and are then internalised and transported in small vesicles across the cytoplasmic brain–capillary barrier. Such conjugates can remain effective in animal models of neurological disease)
Mahato R.I., Narang A.S., Thoma L., Miller D.D. Emerging trends in oral delivery of peptide and protein drugs. Crit. Rev. Ther. Drug Carrier Syst.. 2003;20:153-214. (Various strategies currently under investigation include amino acid backbone modifications; formulation approaches; chemical conjugation of hydrophobic or targeting ligand; and use of enzyme inhibitors, mucoadhesive polymers and absorption enhancers)
Mizuno N., Niwa T., Yotsumoto Y., Sugiyama Y. Impact of drug transporter studies on drug discovery and development. Pharmacol. Rev.. 2003;55:425-461. (Reviews drug transport in intestine, liver, kidney and brain, and its roles in absorption, distribution and excretion)
Singh R., Lillard J.W. Nanoparticle-based targeted drug delivery. Exp. Mol. Pathol.. 2008;86:215-223.
Taguchi A., Sharma N., Saleem R.M. Selective postoperative inhibition of gastrointestinal opioid receptors. N. Engl. J. Med.. 2001;345:935-940. (Speeds recovery of bowel function and shortens hospitalisation: notionally ‘poor’ absorption is used to advantage by providing a selective action on the gut)
Varde N.K., Pack D.W. Microspheres for controlled release drug delivery. Exp. Opin. Biol. Ther.. 2004;4:35-51. (Describes methods of microparticle fabrication and factors controlling the release rates of encapsulated drugs; recent advances for delivery of single-shot vaccines, plasmid DNA and therapeutic proteins are discussed)
1This is illustrated by strain and species differences. For example, collie dogs lack the multidrug resistance gene (mdr1) and a P-glycoprotein that contributes importantly to the blood–brain barrier, with consequences for veterinary medicine because ivermectin (an anthelminthic drug, Ch. 54,) is consequently severely neurotoxic in the many breeds with collie ancestry (see Neff et al., 2004).
2This has been invoked to explain the central symptoms of cholinesterase inhibition experienced by some soldiers during the Gulf War. These soldiers may have been exposed to cholinesterase inhibitors (developed as chemical weapons and also, somewhat bizarrely, used externally during the conflict to prevent insect infestation) in the context of the stress of warfare.
3The experimental measurement of Vd is complicated by the fact that Q does not stay constant (because of metabolism and excretion of the drug) during the time that it takes for it to be distributed among the various body compartments that contribute to the overall Vd. It therefore has to be calculated indirectly from a series of measurements of plasma concentrations as a function of time (see Fig. 10.1).