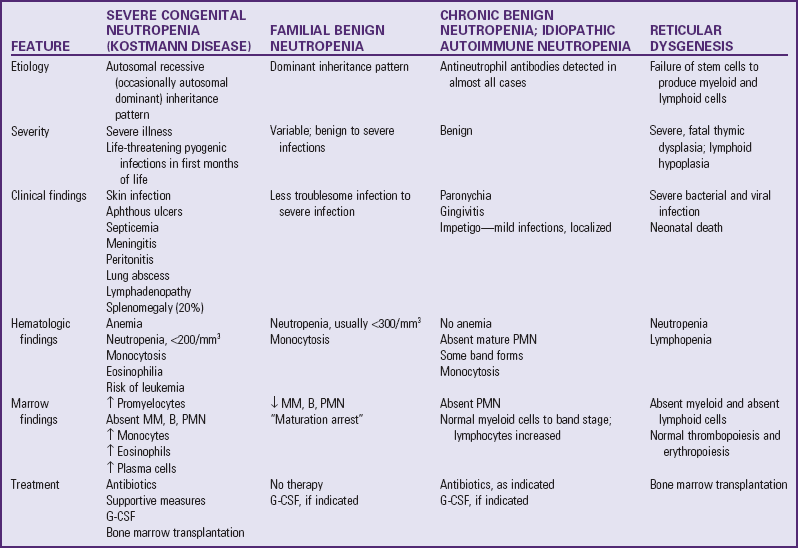
β-Thalassemia
Worldwide, thalassemia is a common genetic disorder affecting as many as 15 million people (Yalsh, 2009). The term thalassemia comes from the Greek word thalassa, meaning “sea,” and is applied to a variety of inherited blood disorders characterized by deficiencies in the rate of production of specific globin chains in hemoglobin. The name appropriately refers to people living near the Mediterranean Sea, namely, Italians, Greeks, and Syrians, or to their descendants. Evidence suggests that the high incidence of the disorder among these groups is a result of selective advantage of the trait in protecting against malaria, as is postulated for SCD. The disorder has a wide geographic distribution, however, probably as a result of genetic migration through intermarriages or possibly as a result of spontaneous mutation.
The thalassemias are classified according to the hemoglobin chain affected and the amount of the globin chain that is synthesized. The two major categories are α-thalassemia and β-thalassemia. Thalassemia is seen in various population groups, such as Asians, Africans, and inhabitants of the Mediterranean and Middle Eastern regions, and the majority of births of affected individuals occur in these groups (Cohen, Galanello, Rennell, et al, 2004; Cunningham, 2008).
β-Thalassemia is the most common of the thalassemias and occurs in four forms: two heterozygous forms, thalassemia minor (generally an asymptomatic silent carrier state) and thalassemia trait (which produces a mild microcytic anemia); thalassemia intermedia, which may involve either homozygous or heterozygous abnormalities and is manifested as splenomegaly and moderate to severe anemia; and a homozygous form, thalassemia major (also known as Cooley anemia), which results in a severe anemia that is not compatible with life without transfusion support.
Mode of Transmission
Thalassemia is an autosomal recessive disorder with varying expressivity. Both parents must be carriers to produce a child with β-thalassemia major. The typical mode of transmission is between parents who are heterozygous for thalassemia.
Pathophysiology and Clinical Manifestations
Normal postnatal HgbA is composed of two α and two β polypeptide chains. In β-thalassemia there is a partial or complete deficiency in the synthesis of the β chain of the hemoglobin molecule. Consequently there is a compensatory increase in the synthesis of α chain, and γ-chain production remains activated, which results in formation of defective hemoglobin. This unbalanced polypeptide unit is unstable; when it disintegrates, it damages the RBCs, which causes severe anemia. To compensate for the hemolytic process, an overabundance of erythrocytes is formed unless transfusion therapy suppresses the bone marrow. Excess iron from packed RBC transfusions and from the rapid destruction of defective cells is stored in various organs (hemosiderosis).
The onset of clinical manifestations in thalassemia major may be insidious and not recognized until late infancy or early toddlerhood (Box 35-5). The clinical effects of thalassemia major are primarily attributable to (1) defective synthesis of HgbA, (2) structurally impaired RBCs, and (3) the shortened life span of the erythrocyte. The major consequences of thalassemia are caused by the pathologic condition, resultant chronic hypoxia, and iron overload from the supportive treatment of multiple blood supplements (Fig. 35-5 and Box 35-5).

Fig. 35-5 A young girl with β-thalassemia demonstrating mild frontal bossing of the right forehead and mild maxillary prominence. (Courtesy James DeLeon, Texas Children’s Hospital, Houston.)
Anemia results from the body’s inability to maintain a level of erythropoiesis commensurate with hemolysis. The bone marrow compensates by producing large numbers of immature cells, such as normoblasts and erythroblasts; large cells that are extremely thin and form bizarre shapes; and target cells, which have abnormal staining properties. As a result of the excessive production of abnormal RBCs, their life span is severely shortened.
Aplastic crises after infection, folic acid deficiencies from the demands of bone marrow hyperplasia, and progressive hemolysis from repeated blood transfusions all worsen anemia. The spleen becomes greatly enlarged as a result of extramedullary hematopoiesis, rapid destruction of the defective erythrocytes, and, rarely, progressive fibrosis from hemochromatosis. Splenomegaly may progress until the organ’s very size interferes with the function of other abdominal organs and respiratory expansion.
With progressive anemia, signs of chronic hypoxia—namely, headache, irritability, precordial and bone pain, decreased exercise tolerance, listlessness, and anorexia—may develop. Another common symptom in these children is frequent epistaxis, although the exact reason is unknown. Hyperuricemia and gout from rapid cellular catabolism also occur.
Hemosiderosis refers to excess iron storage in various tissues of the body, especially the spleen, liver, lymph glands, heart, and pancreas, but without associated tissue injury. Hemochromatosis refers to excess iron storage that results in cellular damage. It is not known how iron storage causes tissue destruction. Chronic hypoxia is believed to be an important contributing factor.
In thalassemia, excess of hemosiderin, the iron-containing pigment from the breakdown of hemoglobin, results from decreased hemoglobin synthesis and increased hemolysis of transfused erythrocytes. Decreased production of hemoglobin results in an excess supply of available iron. In addition, the body probably responds to the anemia by increasing the rate of gastrointestinal absorption of dietary iron, since ineffective erythropoiesis is a potent controlling factor in exogenous iron use. However, the primary source of additional iron is from the hemolysis of supplemental erythrocytes and the rapid destruction of defective RBCs. With the prophylactic use of deferoxamine and/or oral chelators (deferiprone and deferasirox) to minimize excess iron storage, the characteristic changes in body structures from hemochromatosis have been greatly reduced.
Retarded growth and, especially, delayed sexual maturation are common findings. There is evidence that both may also be caused by pituitary failure, although the exact reasons for this are unclear, but the impaired growth is probably also related to hemochromatosis. It is possible that the endocrine glands are extremely sensitive to iron toxicity and that even small amounts of deposited iron can produce organ dysfunction. Children with severe disease usually exhibit significant growth retardation. The development of secondary sexual characteristics is delayed or absent in many adolescents (Cunningham, Sankaran, Nathan, et al, 2009).
Diagnostic Evaluation
Hematologic studies reveal characteristic changes in the RBCs (e.g., microcytosis, hypochromia, anisocytosis, poikilocytosis, target cells, and basophilic stippling of various stages). Low hemoglobin and hematocrit levels often occur in severe anemia, although they are typically less pronounced than the reduction in the RBC count because of the proliferation of immature erythrocytes.
Hemoglobin electrophoresis confirms the diagnosis and is helpful in distinguishing the type and severity of the thalassemia because it analyzes the quantity and kind of hemoglobin variants found in the blood. In β-thalassemia, levels of HgbF and HgbA2 (a type of normal adult hemoglobin) are elevated because neither depends on β chain polypeptides for synthesis.
Therapeutic Management
The objective of supportive therapy is to maintain sufficient hemoglobin levels to prevent bone marrow expansion and bony deformities and to provide sufficient RBCs to support growth and normal physical activity. Transfusions are the foundation of medical management, with a goal of maintaining the hemoglobin level above 9.5 g/dl, an aim that may require transfusions as often as every 3 weeks. The advantages of this therapy include (1) improved physical and psychologic well-being because of the ability to participate in normal activities, (2) decreased cardiomegaly and hepatosplenomegaly, (3) fewer bone changes, (4) normal or near-normal growth and development until puberty, and (5) fewer infections.
One of the potential complications of frequent blood transfusions is iron overload (hemosiderosis). Because the body has no effective means of eliminating the excess iron, the mineral is deposited in body tissues. To minimize the development of hemosiderosis and hemochromatosis, deferoxamine, an iron-chelating agent, is given with oral supplements of vitamin C. Vitamin C should be given only to patients who are ascorbate depleted and only while deferoxamine is being administered. Administration of vitamin C significantly augments iron excretion in response to deferoxamine, particularly in patients with vitamin C deficiency (Cunningham, Sankaran, Nathan, et al, 2009). As postulated, vitamin C may delay the conversion of ferritin to hemosiderin, which allows more iron to remain in chelatable form.
Deferoxamine is given intravenously or subcutaneously at home via a portable infusion pump over a period of 8 to 10 hours (usually during sleep) for 5 to 7 days a week. Significant liver fibrosis, cardiac dysfunction and growth impairment may be prevented if chelation therapy is adequate during childhood (Cunningham, Sankaran, Nathan, et al, 2009). Therefore adherence to an intensive schedule is required for substantial chelation therapy. The availability of oral chelators (deferiprone and deferasirox) is a major advance in the care of patient undergoing long-term transfusion therapy. Deferasirox was approved by the FDA in 2004 for the treatment of patients 2 years and older with chronic iron overload secondary to recurrent blood transfusions (Cunningham, Sankaran, Nathan, et al, 2009; Raphael, Bernhardt, Mahoney, et al, 2009). A daily dose of 20 to 30 mg/kg of deferasirox is generally well tolerated, with mild gastrointestinal events and rash as common toxicities (Cohen, Glimm, and Porter, 2008). Deferiprone is currently available only on a compassionate-use basis in the United States (Neufeld, 2008).
 DRUG ALERT
DRUG ALERT
Chelation
New strategies for chelation, such as combination parenteral and oral chelation therapy and organ-targeted chelation, may soon have a considerable impact on the quality of life of patients with thalassemia (Cohen, Galanello, Rennell, et al, 2004; Naithani, Chandra, and Sharma, 2005; Cunningham, Sankaran, Nathan, et al, 2009).
New methods to assess cardiac and liver iron overload using magnetic resonance imaging have recently been developed (Cohen, Galanello, Rennell, et al, 2004; Hankins and Aygun, 2009).
In some children with severe splenomegaly who require repeated transfusions, a splenectomy may be necessary to decrease the disabling effects of abdominal pressure and to increase the life span of supplemental RBCs. Over time the spleen may accelerate the rate of RBC destruction and therefore increase transfusion requirements. After a splenectomy children generally require fewer transfusions, although the basic defect in hemoglobin synthesis remains unaffected. A major postsplenectomy complication is severe and overwhelming infection. Therefore these children are often on prophylactic antibiotics with close medical supervision for many years and should receive the pneumococcal and meningococcal vaccines in addition to regularly scheduled immunizations. (See Immunizations, Chapter 12.)
Prognosis: Most children treated with blood transfusions and early chelation therapy survive well into adulthood (Cunningham, Sankaran, Nathan, et al, 2009). The most common causes of death are heart disease, postsplenectomy sepsis, and multiorgan failure secondary to hemochromatosis (Cunningham, Sankaran, Nathan, et al, 2009). A curative treatment for some children is HSCT. (See Chapter 36.) Children younger than 16 years of age who undergo allogeneic HSCT have a high rate of complication-free survival; approximately 80% of these children are cured (Lucarelli and Gaziev, 2008). An experimental approach for correction of thalassemia through the introduction of new genetic material into pluripotent stem cells is ongoing but continues to have shortcomings (Cunningham, Sankaran, Nathan, et al, 2009; Ye, Chang, Lin, et al, 2009).
Nursing Care Management
The objectives of nursing care are to (1) promote compliance with transfusion and chelation therapy, (2) assist the child in coping with the anxiety-provoking treatments and the effects of the illness, (3) foster the child’s and family’s adjustment to a chronic illness, and (4) observe for complications of multiple blood transfusions. Basic to each of these goals is explaining to parents and older children the defect responsible for the disorder, its effect on RBCs, and the potential effects of untreated hemosiderosis (such as delayed growth and maturation and heart disease). Because this condition is prevalent among families of Mediterranean descent, the nurse also inquires about the family’s previous knowledge about thalassemia. All families with a child with thalassemia should be tested for the trait and referred for genetic counseling.
Support the Family: As with any chronic illness, the family’s needs must be met for optimum adjustment to the stresses imposed by the disorder. (See Chapter 22.) Sources of information for the family are the Cooley’s Anemia Foundation* and the Thalassemia Action Group.* Genetic counseling for the parents and fertile offspring is mandatory, and both prenatal diagnosis using amniocentesis or fetal blood sampling and screening for thalassemia trait are available. There has been a marked decline in the number of new cases of thalassemia worldwide. This may be a result of education and testing of parents.
Assist in Coping with the Effects of the Disorder: Body image alterations, decreased growth, and sexual immaturity are frequently difficult adjustment problems for older children. These children feel different from their peers, and the delayed sexual development is a major issue for the maturing adolescent with an improved life expectancy. Adolescents need an opportunity to express their thoughts and feelings about these complex issues. They can learn grooming measures that make them appear more sexually mature, such as wearing up-to-date clothing, adopting new hairstyles, and wearing well-applied makeup. Children with the characteristic bone changes may benefit from surgery or use of orthodontic appliances to improve facial structure.
With frequent transfusion therapy there is less restriction on physical activity because of severe anemia, and the nurse should encourage these children to pursue activities that they are able to tolerate. The frequency of treatment, however, can interfere with a normal lifestyle. To minimize disruptions and improve cooperation, the nurse can help arrange for blood transfusions and medical supervision at times that interfere least with the child’s regular activities, especially school.
 Understanding the role that factor deficiencies play in promoting bleeding tendencies requires a review of the normal coagulation process of the blood. Although the coagulation process is complex, clotting depends on three main elements: vascular events, platelets, and clotting factors.
Understanding the role that factor deficiencies play in promoting bleeding tendencies requires a review of the normal coagulation process of the blood. Although the coagulation process is complex, clotting depends on three main elements: vascular events, platelets, and clotting factors. Critical Thinking Exercise—Bleeding
Critical Thinking Exercise—Bleeding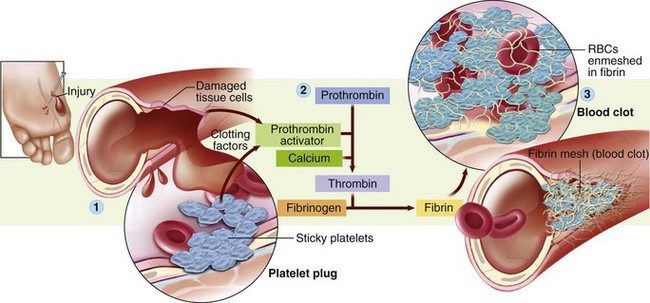
 NURSING ALERT
NURSING ALERT EMERGENCY TREATMENT
EMERGENCY TREATMENT
 years.
years.