The Child with Hematologic or Immunologic Dysfunction

http://evolve.elsevier.com/wong/ncic

Administration of Medication, Ch. 27
Bone Marrow Aspiration and Biopsy, Ch. 27
Bone Marrow Transplantation, Ch. 36
Collection of Specimens, Ch. 27
Compliance, Ch. 27
Family-Centered Care of the Child with Chronic Illness or Disability, Ch. 22
Family-Centered Care of the Child During Illness and Hospitalization, Ch. 26
Family-Centered End-of-Life Care, Ch. 23
Hemolytic Disease of the Newborn, Ch. 9
Human Immunodeficiency Virus Encephalopathy, Ch. 37
Human Immunodeficiency Virus Infection and Acquired Immunodeficiency Syndrome, Ch. 20
Immunizations, Ch. 12
Infection Control, Ch. 27
Leukemias, Ch. 36
Lymphomas, Ch. 36
Pain Assessment; Pain Management, Ch. 7
Examination: Skin, Ch. 6
Preparation for Diagnostic and Therapeutic Procedures, Ch. 27
The Hematologic System and Its Function
Blood is composed of a fluid portion called plasma and a cellular portion known as the formed elements of the blood. The two components are approximately equal in volume. Plasma is about 90% water and 10% solutes. The principal solutes are albumin, electrolytes, and proteins. Among the proteins are clotting factors, globulins, circulating antibodies, and fibrinogen. The cellular elements sequentially develop into mature red blood cells (RBCs, erythrocytes), white blood cells (WBCs, leukocytes), and platelets (thrombocytes) (Fig. 35-1).
PATHOPHYSIOLOGY REVIEW
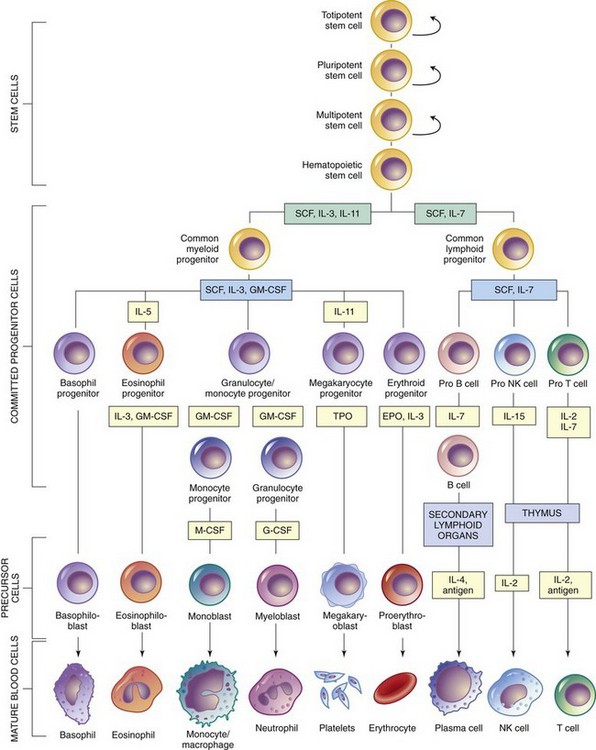
Fig. 35-1 Differentiation of hematopoietic cells. EPO, erythropoietin; G-CSF, granulocyte colony-stimulating factor; GM-CSF, granulocyte-macrophage colony-stimulating factor; IL, interleukin; M-CSF, macrophage colony-stimulating factor; NK, natural killer; SCF, stem cell factor; TPO, thrombopoietin. (From McCance KL, Huether SE: Pathophysiology: the biological basis for disease in adults and children, ed 6, St Louis, 2010, Mosby.)
The major hematopoietic (blood-forming) organs of the body are the red bone marrow (myeloid tissue) and the lymphatic system, which consists of lymph (fluid), lymphatic vessels, and lymphoid structures (the lymph nodes, spleen, thymus, and tonsils). Although the lymphatic system plays an important role in regulating blood cells, the lymph vessels and fluids do not produce cells. The lymph nodes regulate the manufacture of WBCs. The spleen and liver are the primary organs for hematopoiesis in the young fetus and for cell removal in postnatal life. Macrophages (formerly called reticular cells) are cells of mesodermal origin that are widely dispersed in the lining of the vascular and lymph channels. Macrophages form a network and are capable of phagocytosis (ingestion and digestion of foreign substances); formation of immune bodies; and differentiation into other cells, such as hemocytoblasts, myeloblasts, and lymphoblasts.
All of the formed elements of the blood, except to some extent the agranulocytes, are produced in myeloid tissue during postnatal life. During embryonic development the mesenchyme, spleen, liver, thymus, and yolk sac serve as additional sites of blood cell formation. In individuals with certain blood disorders these sites, particularly the spleen, can be stimulated to produce blood cells and constitute extramedullary hematopoiesis. In infants and young children all of the bone contains red marrow (so-called because of its color from the formation of erythrocytes), but as bone growth ceases near the end of adolescence, only the ribs, sternum, vertebrae, and pelvis continue to produce blood cells. The remainder of the bone marrow becomes yellow from deposition of fat. However, in conditions of increased demand for blood cells, the yellow marrow can revert to red marrow and become another hematopoietic source.
Although the progressive development of each blood cell is fairly well delineated, there is considerable controversy regarding the origin of the blood cell. One of the most widely held theories (monophyletic theory) is that each blood cell originates from a primordial (primitive) cell called a blast, or totipotential stem cell, which has the ability to self-replicate and transform into all the blood components.
The second-generation stem cell, called the pluripotent stem cell, is committed to produce erythroblast, myeloblast, monoblast, lymphoblast, or megakaryoblast (see Fig. 35-1). The blast cells sequentially develop into mature RBCs (erythrocytes), WBCs (leukocytes), platelets (thrombocytes), and other cells (such as mast cells and macrophages) (see Fig. 35-1).
Red Blood Cells (Erythrocytes)
The erythrocyte is formed from the hemocytoblast in the red bone marrow. As illustrated in Fig. 35-1, the pluripotential stem cell forms the proerythroblast. The initial cell of this series has a deep blue–staining (basophilic) cytoplasm and therefore is called a basophilic erythroblast. The chief change in the erythroblast is accumulation of hemoglobin in the cytoplasm. As the basophilic material decreases and the amount of hemoglobin increases, the cell comes to be called a polychromatic erythroblast, which describes its mixture of staining properties. At the same time that the nucleus decreases in size, the basophilic material disappears, so that the cell is uniformly stained by eosin dye—hence the name orthochromatic erythroblast, or normoblast. Finally, the normoblast completely loses its nucleus by a process of extrusion as it squeezes through the pores of the membrane into the capillary. Because of the loss of its nucleus, the cell caves in on both sides, which gives the mature erythrocyte its characteristic appearance as a biconcave disk. During each of these stages the different cells continue to undergo mitosis so that increasingly greater numbers of cells are produced. Because the mature RBC does not have a nucleus, it is unable to multiply.
The reticulocyte is the last stage of development before the mature erythrocyte. Reticulocytes are slightly larger than erythrocytes, and their presence indicates active RBC production (erythropoiesis). Ordinarily the total proportion of circulating reticulocytes is between 0.5% and 1.5%. The reticulocyte, or “retic,” count is a simple laboratory test frequently used to indirectly analyze hematopoiesis.
Regulation of Erythrocyte Production: The usual life span of the mature erythrocyte is 120 days. Apparently, as RBCs grow old, their membranes become fragile and eventually rupture. The contents of the cell fragment as they circulate through the blood vessels and are phagocytized by the macrophages in the spleen, liver, and bone marrow. The hemoglobin is broken down into the iron-containing pigment hemosiderin and the bile pigments biliverdin and bilirubin. Most of the iron is reused by the bone marrow for production of new RBCs or stored in the liver and other tissues for future use. The bile pigments are excreted by the liver in bile.
Normally there is a homeostatic balance between RBC production and destruction. This balance ensures adequate tissue oxygenation and a blood viscosity that allows the blood to flow freely through the vessels. The basic regulator of erythrocyte production is tissue oxygenation and renal production of erythropoietin (also called erythropoietic stimulating factor). In states of tissue hypoxia, the kidney releases erythropoietin into the bloodstream. As a result the bone marrow is stimulated to produce new RBCs. The major activity seems to be an increase in both maturation rate and mitosis at all stages of erythrocyte production, but primarily at the stem cell level.
During this rapid increase in RBC production, the circulating erythrocytes may not be totally mature. Consequently the number of reticulocytes may increase dramatically (as high as 30% or more of the total RBC count). Even normoblasts or nucleated RBCs may appear in the blood. A failure of this rise in erythrocyte and reticulocyte count to occur may indicate bone marrow failure.
Once tissue oxygenation is adequate, the production of erythropoietin ceases. Thus tissue oxygen requirements control both the stimulation and termination of erythrocyte production. Note that the basic regulatory mechanism is the ability of RBCs to transport oxygen to the tissues in response to their needs, not the circulating numbers of erythrocytes. Oxygen transport depends on both the number of circulating RBCs and the amount of normal hemoglobin in the cell. This explains why polycythemia (increase in the number of erythrocytes) occurs in conditions characterized by prolonged tissue hypoxia, such as cyanotic heart defects. (See Chapter 34.) If the circulating numbers of erythrocytes controlled erythropoietin release, this feedback mechanism would maintain erythrocyte production at a constant level (4.5 to 5.5 million/mm3 of blood) regardless of existing tissue hypoxia.
Functions of Erythrocytes: The major function of RBCs is to transport hemoglobin, which in turn carries oxygen to all cells of the body. However, erythrocytes have other significant functions: (1) they contain carbonic anhydrase, an enzyme that catalyzes the reaction between carbon dioxide and water, which allows large quantities of carbon dioxide to react with blood for transportation to the lungs; and (2) the hemoglobin, a protein, serves as an acid-base buffer, which, in combination with carbon dioxide, maintains the blood pH at a constant level.
Hemoglobin
Hemoglobin is a complex molecule composed of four globin chains. The type of hemoglobin in the cells depends on both the stage of life and the presence of any abnormalities in the genes that regulate the production of hemoglobin. Fetal hemoglobin, composed of two α and two γ chains, has a greater affinity for oxygen and is best suited to the fetal environment. During the latter part of pregnancy, the fetus begins developing adult hemoglobin (two α and two β chains). When a defect in hemoglobin synthesis is present (e.g., sickle cell disease [SCD] or thalassemia), fetal hemoglobin may be produced into adulthood. Research is currently underway to develop cell-free hemoglobin that can be used for oxygen and carbon dioxide transport. Hemoglobin values vary according to the child’s age. (See Appendix C.)
Several tests offer important information about hemoglobin. The hematocrit, which is approximately three times the concentration of hemoglobin (in grams per deciliter), indicates the percentage volume of circulating packed RBCs in the total blood. Under normal conditions, hemoglobin and the hematocrit are in a fixed relationship with each other and vary according to the child’s age and sex (Glader, 2007a; Richardson, 2007).
RBC indices are based on ratios of packed RBC volume, hemoglobin concentration, and RBC count, and they are a useful way of designating different types of anemias. Values for mean corpuscular volume (MCV) and mean corpuscular hemoglobin (MCH) do not stay constant during infancy and childhood (Glader, 2007b; Richardson, 2007). MCH concentration (MCHC) values, however, are more constant.
MCV is the average (mean) volume or size of a single RBC. The normal range for RBCs is given in Table 35-1.
TABLE 35-1
TESTS PERFORMED AS PART OF A COMPLETE BLOOD COUNT
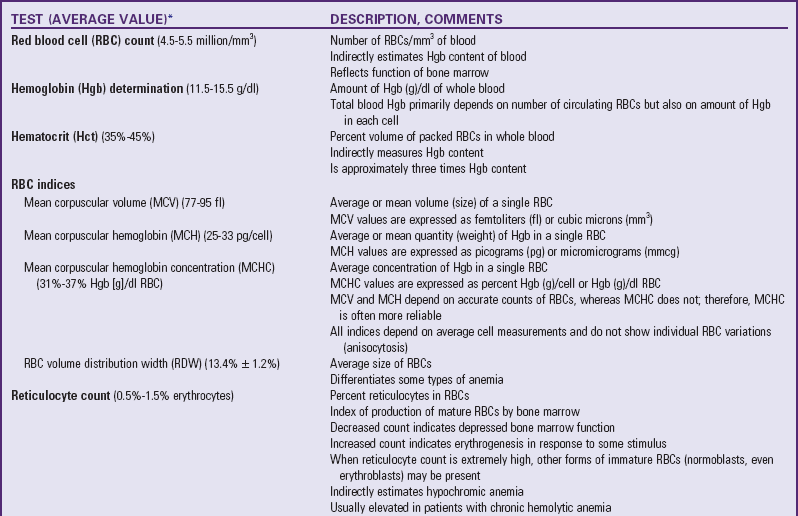

*See Appendix C for normal values according to ages.
MCH is the average weight of hemoglobin in each RBC (see Table 35-1). Normochromic cells are those with a normal hemoglobin content or normal MCH. Cells with below-normal MCH are termed hypochromic, and those with above-normal MCH are termed hyperchromic (McKenzie, 2004).
MCHC is the average concentration of hemoglobin in the RBC. MCHC is calculated from the amount of hemoglobin in 100 ml of RBCs rather than the amount of hemoglobin in whole blood. The normal MCHC value of 33 g/dl is reached at 6 months of age (Pesce, 2007).
Fig. 35-2 shows how RBC indices are used as indicators of different types of anemia.
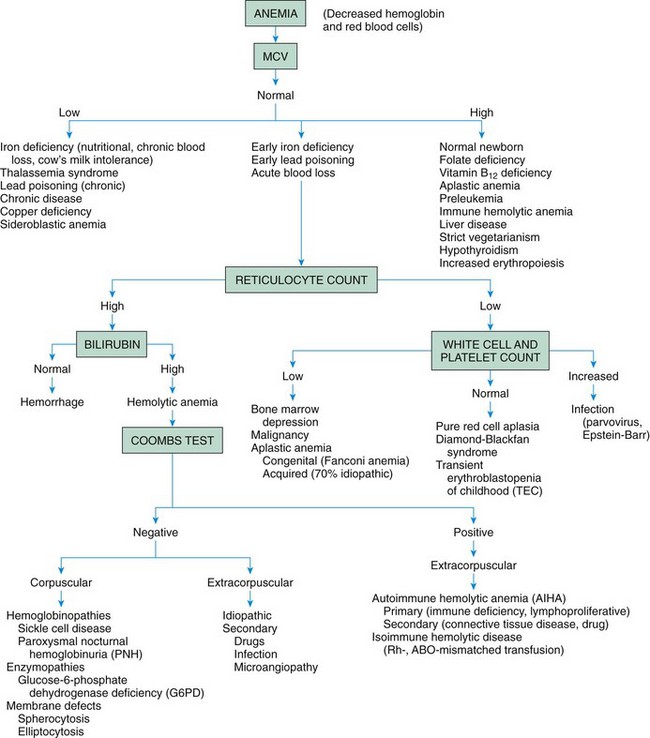
Fig. 35-2 Approach to the diagnosis of anemia by mean corpuscular volume (MCV) and reticulocyte count. (Modified from Lanzkowsky P: Manual of pediatric hematology and oncology, New York, 2005, Churchill Livingstone; Glader B: The anemias. In Kliegman RM, Jenson HB, Behrman RE, et al, editors: Nelson textbook of pediatrics, ed 18, Philadelphia, 2007, Saunders; and McKenzie SB: Introduction to anemia. In McKenzie SB, editor: Clinical laboratory hematology, Upper Saddle River, NJ, 2004, Pearson, Prentice Hall.)
White Blood Cells (Leukocytes)
The term leukocyte encompasses a number of cells with similar yet distinct functions. They are divided into two major classes—granulocytes and agranulocytes—based on the presence or absence, respectively, of granules within the cytoplasm of the cells.
Granulocytes: There are three types of granulocytes: neutrophils, basophils, and eosinophils. The name of each of these refers to the characteristic staining property of the granule during laboratory analysis. Neutrophils stain neutral to the dyes, whereas basophils stain purple to the basic methylene blue dye, and eosinophils take on a red color from acidic eosin dye. Because the nuclei of neutrophils have two or more lobules that are connected by fine chromatin strands, the term polymorphonuclear leukocytes (cells with many-formed nuclei), or simply polys, or segs (segmented or mature neutrophils) and bands (immature neutrophils with the nuclei connected) may be used collectively to refer to the neutrophils.
The granulocytes, like erythrocytes, are produced in the bone marrow. For this reason these cells are sometimes referred to as myelogenous leukocytes. These cells, in theory, originate from primitive stem cells, which develop into myeloblasts. As Fig. 35-1 illustrates, the genesis of neutrophils, basophils, and eosinophils is similar to the stages observed during erythrocyte production. The differentiation of myeloblasts into various mature WBCs is primarily a result of specialization within the cytoplasm and degeneration of the nucleus. Unlike erythrocytes, however, all WBCs are nucleated.
Accelerated production of immature granulocytes leads to increased numbers of bands in the peripheral circulation (referred to as a shift to the left in the complete blood count [CBC]), which is indicative of a bacterial infection. The absolute neutrophil count (ANC) reflects the body’s ability to handle bacterial infections. If the ANC is less than 500/mm3, a severe risk of infection is present.
Agranulocytes: The agranulocytes include two cell types: monocytes and lymphocytes. Characteristically these cells do not develop granules, and the nuclei are not lobulated. They originate in various lymphogenous organs, and for this reason are sometimes referred to as lymphogenous leukocytes. However, because stem cells and reticular cells are capable of differentiating into monocytes or lymphocytes, the origin of these cells is frequently designated as the lymphomyeloid complex, which includes bone marrow, lymph nodes, spleen, liver, thymus, subepithelial lymphoid tissue (tonsils, vermiform appendix, and intestinal lymphoid tissues), and connective tissues (mesenchymal cells of the reticuloendothelial system).
The monocytes follow the same sequence of development from the stem cell as the granulocytes (see Fig. 35-1). The monocytes in turn have the ability to exit the vessels and develop into macrophages, large cells that are highly effective phagocytes. Kupffer cells are macrophages located in the liver. Histiocytes are macrophages in the connective tissue. These names are remnants of the old reticular endothelial system designations.
Lymphocytopoiesis (lymphocyte formation) takes place anywhere in the lymphomyeloid complex. Lymphocytes develop from blast (stem) cells (see Fig. 35-1). The lymphocyte has the potential to develop into other cells, such as T cells or B cells (see p. 1410).
Regulation of Leukocyte Production: The exact life span of the leukocytes is not as clearly defined as that of the erythrocytes, since they exist in the circulation primarily for transport to extravascular areas, where they reside in reservoirs or where they are needed to resist infection. Therefore their survival rate is described in terms of three phases: (1) the hematopoietic phase, extending from the development of the blast cell to the delivery of the mature leukocyte into the circulation; (2) the intravascular phase, the period within the circulation; and (3) the extravascular phase, the time spent in the viscera or tissues.
Granulocytes have a half-life of 6 to 8 hours in the blood and, after entering the tissues, die over a period of 4 to 5 days. Agranulocytes live for an extended period because they remain in inflamed tissue areas longer than the granulocytes. Monocytes wander back and forth between the blood and tissues and are capable of becoming macrophages; their half-life in the blood is 8 to 10 hours, but their half-life in the tissue is 60 to 90 days.
The regulation of leukocytes is based on the body’s need for them. Tissue damage from bacterial or viral agents promotes leukocyte circulation and production. However, leukocytosis (increase in leukocytes) results from tissue destruction from almost any source, such as hemorrhage, neoplastic disease, toxicity, operative procedures, chemical and thermal injury, and tissue ischemia.
The leukocytes probably die as a result of their activity at the site of injury and are phagocytized by other newly formed WBCs. Effective control of the inflammatory process with subsequent tissue recovery most likely results in feedback to the bone marrow and causes lymphogenous organs to cease increased production of WBCs.
Functions of Leukocytes: Although all of the leukocytes play some role in the immune process, each of the WBCs has a specific role. Neutrophils and monocytes are effective phagocytes and as a result are primarily involved in inflammatory reactions. Neutrophilia (increased numbers of neutrophils) is most evident in an acute inflammation, whereas monocytosis (increased numbers of monocytes) is more evident in chronic conditions. The reason is that, as the affected area becomes acidic from tissue necrosis, neutrophils, which prefer a neutral environment, become less efficient, and monocytes, which become macrophages, become more powerful. These cells also increase in number during chronic inflammation. The other functions of lymphocytes in terms of the immune process are discussed on p. 1451.
The function of eosinophils is still not completely known. They seem to have parasiticidal properties because they can selectively destroy parasites. They may also function in the immediate type of allergic or anaphylactic hypersensitivity reaction, since eosinophilia (increased numbers of eosinophils) is well documented in such conditions. Eosinophils also are thought to release a substance called profibrinolysin, which, when activated to form fibrinolysin, digests fibrin and thereby helps dissolve a clot.
The function of basophils is also not completely understood, although basophilia (increased numbers of basophils) occurs during the healing phase of inflammation and during prolonged inflammation. Basophils in the blood exit the vessels and become mast cells in the tissue. They are responsible for histamine release, which results in increased permeability of the vessels to allow WBCs to exit the vessels at the site of injury.
Platelets
Platelets are actually small fragments of megakaryocytes. They are smaller than blood cells, do not possess a cellular structure, and consist of a clear substance containing granules. Platelets originate from part of the myelogenous group of WBCs (see Fig. 35-1). Platelets are formed when the megakaryocytic membrane invaginates, fuses within the cell to separate the cytoplasm, and then fragments.
Regulation of Platelet Production: The life span of platelets is estimated as 8 to 10 days. Apparently the body regulates platelet levels to maintain a fairly constant level (between 150,000 and 400,000/mm3). Platelet production is probably regulated by a hormone, thrombopoietin, but the source and mode of action of this substance are unknown. Old platelets are most likely removed by the liver and spleen.
Function of Platelets: The term thrombocyte means “clot” (thrombo) and “cell” (cyte) and accurately describes the main function of platelets. When there is a break in the continuity of a blood vessel, the platelets, which are normally flat and round or oval, come in contact with the wet vessel surface and dramatically change their shape to become swollen spheres with long, irregular projections called pseudopodia (false feet). As a result, the platelets begin to adhere to the wet endothelium and to each other. The first platelets at the site of injury release substances that attract other thrombocytes to the area. This causes a layering of platelets, which eventually forms a plug. This plug is large enough to partially or totally occlude the opening in the vessel wall but small enough to allow blood flow to continue unimpaired through the vessel.
In small vessel tears the platelet plug is sufficient to produce hemostasis, and additional blood coagulation is not necessary. When platelet counts are low, however, these numerous small ruptures, which occur continually in the body as a result of general functioning, are not repaired. Consequently, small hemorrhagic areas called petechiae form under the skin. They are similar in appearance to reddish freckles or tiny spider webs.
Platelets also influence hemostasis by releasing a substance called serotonin at the site of injury. Serotonin is a vasoconstrictor that produces vascular spasm to decrease the blood flow to the injured area.
Assessment of Hematologic Function
Several tests assess hematologic function, and additional procedures can identify the cause of the dysfunction. The following discussion is limited to a description of the most common and one of the most valuable tests, the CBC. Other procedures, such as those related to iron, coagulation, and immune status, are discussed throughout the chapter as appropriate.
The CBC consists of the following determinations: RBC count, WBC count, hematocrit, hemoglobin level, differential WBC count, RBC indices (MCV, MCH, and MCHC), and peripheral smear. Additional tests may be included, such as the reticulocyte count, RBC volume distribution width, and platelet count. Table 35-1 describes each of these. Most of the determinations can be performed on a small quantity of blood (micromethod), and values are computed automatically. The nurse should be familiar with the significance of the findings from the CBC (see Table 35-1) and should be aware of the normal values for age. (See Appendix C.)
The history and physical examination are essential to the identification of hematologic dysfunction, and the nurse is often the first person to suspect a problem based on information from these sources. Comments by the parent regarding the child’s lack of energy, food diary showing decreased sources of iron, frequent infections, and bleeding that is difficult to control offer clues to the more common disorders affecting the blood. A careful physical appraisal can reveal findings such as persistent fatigue, pallor, petechiae, or bruising that may indicate minor or serious hematologic conditions. Nurses need to be aware of the clinical manifestations of blood diseases to assist in recognizing symptoms and establishing a diagnosis.
Red Blood Cell Disorders
 Anemia is a reduction in RBCs mass and/or hemoglobin concentration compared with normal values for age (Brugnara, Oski, and Nathan, 2009; Glader, 2007a). The anemias are the most common hematologic disorders of infancy and childhood and are not diseases but manifestations of underlying pathologic processes (see Fig. 35-2).
Anemia is a reduction in RBCs mass and/or hemoglobin concentration compared with normal values for age (Brugnara, Oski, and Nathan, 2009; Glader, 2007a). The anemias are the most common hematologic disorders of infancy and childhood and are not diseases but manifestations of underlying pathologic processes (see Fig. 35-2).
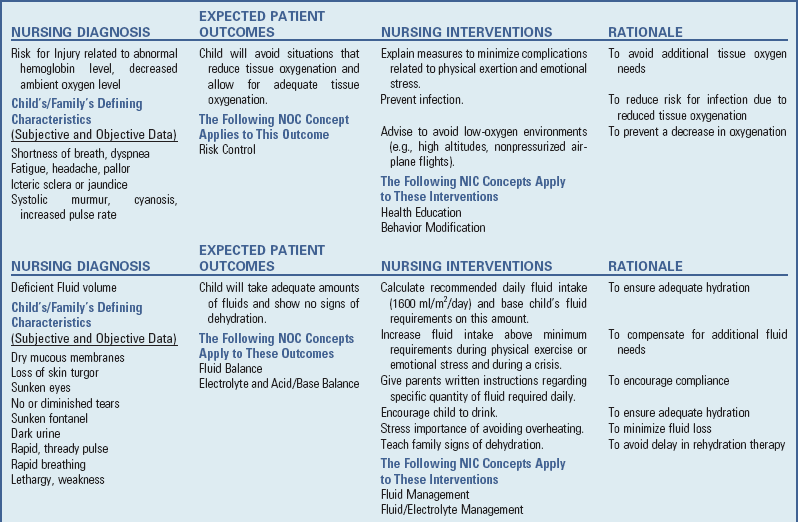
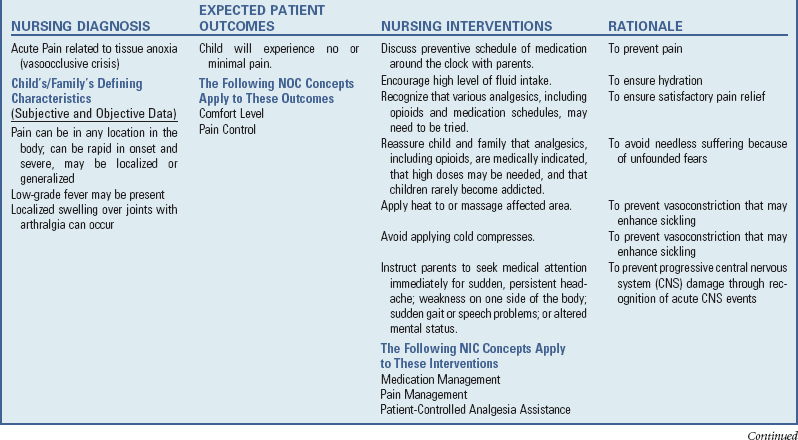
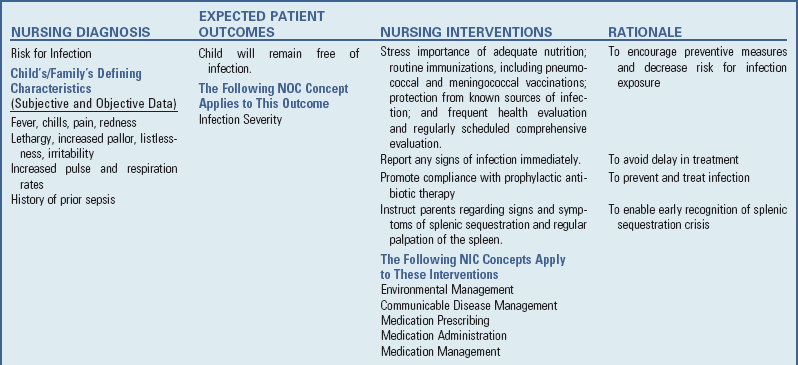
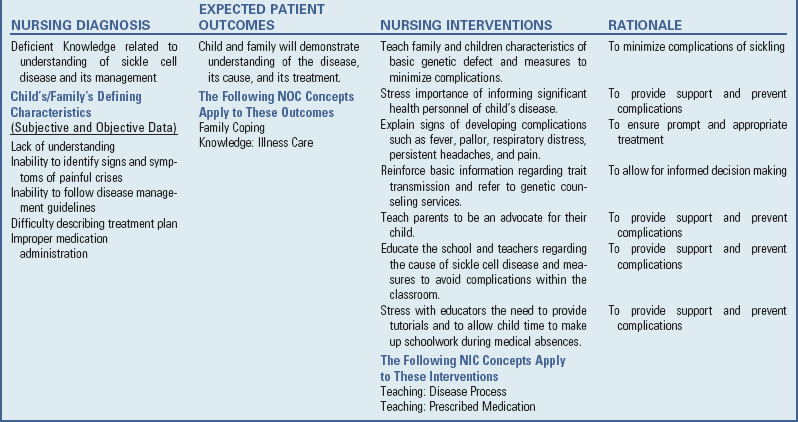
 Nursing Care Plan—The Child with Anemia
Nursing Care Plan—The Child with Anemia
Classification
The anemias can be classified using two basic approaches: (1) etiology as manifested by erythrocyte or hemoglobin depletion; and (2) morphology, the characteristic changes in RBC size, shape, and color (Box 35-1).
Although the morphologic classification is useful in the laboratory evaluation of anemia, the etiology provides direction for planning nursing care. For example, anemia with reduced hemoglobin concentration may be caused by a dietary depletion of iron, and the principal intervention is replenishing iron stores.
The main causes of anemia are (1) inadequate production of RBCs or RBC components, (2) increased destruction of RBCs, and (3) excessive loss of RBCs through hemorrhage. Each of these factors affects the amount of hemoglobin that is available to carry oxygen to the cells (see Box 35-1). Therefore the classification is based on the various conditions that can result from any of these physiologic changes.
Pathophysiology and Clinical Manifestations
The basic physiologic defect caused by anemia is a decrease in the oxygen-carrying capacity of blood and consequently a reduction in the amount of oxygen available to the cells. When the anemia has developed slowly, the child usually adapts to the declining hemoglobin level. Most children seem to have a remarkable ability to function well despite low levels of hemoglobin. Also, compensatory mechanisms such as a shift in the oxyhemoglobin dissociation curve may delay the development of any obvious signs (see p. 1193).
When the hemoglobin level falls sufficiently to produce clinical manifestations, the signs and symptoms (e.g., weakness, fatigue, and a waxy pallor in severe anemia) are due to tissue hypoxia (Box 35-2). Cyanosis, which results from an increased quantity of deoxygenated hemoglobin in arterial blood, is typically not evident. Anemia is caused by decreased levels of hemoglobin or RBCs, not inadequate oxygen saturation of existing hemoglobin.
Central nervous system manifestations include headache, dizziness, lightheadedness, irritability, slowed thought processes, decreased attention span, apathy, and depression. Growth retardation resulting from decreased cellular metabolism, and coexisting anorexia is a common finding in chronic severe anemia. It is frequently accompanied by delayed sexual maturation in the older child.
The effects of anemia on the circulatory system can be profound. A reduction in hemoglobin concentration that results in decreased oxygen-carrying capacity of the blood is associated with a compensatory increase in heart rate and cardiac output (see Box 35-2). Initially this greater cardiac output compensates for the lower oxygen-carrying capacity of the blood, since blood replenished with oxygen returns to the tissues at a faster than normal rate. The increased circulation and turbulence within the heart may produce a heart murmur. Because the cardiac workload increases during exercise, infection, or emotional stress, cardiac failure may occur.
Acute or chronic hemorrhage results in loss of plasma and all formed elements of the blood. After acute hemorrhage the body replaces plasma within 1 to 3 days, maintaining blood volume. However, this results in a low concentration of RBCs, which are gradually replaced within 3 to 4 weeks. During this period there is usually a normocytic normochromic anemia, provided that iron stores are sufficient for hemoglobin synthesis.
In chronic blood loss the actual number of RBCs may be normal because of continuous replacement. However, insufficient iron is available to form hemoglobin as quickly as it is lost. As a result, erythrocytes are usually microcytic and hypochromic.
Routine Screening: The Put Prevention into Practice program developed for the U.S. Public Health Service cites the following recommendations of major authorities (US Department of Health and Human Services, 2009):
American Academy of Family Physicians and U.S. Preventive Services Task Force—All children should be screened for anemia once during infancy.
American Academy of Pediatrics—Hemoglobin concentration or hematocrit should be measured once during infancy (between 9 and 12 months), early childhood (between 1 and 5 years), late childhood (between 5 and 12 years), and adolescence (between 14 and 20 years).
Canadian Task Force on the Periodic Health Examination—Hemoglobin concentration screening should be performed on children at high risk for iron deficiency anemia: preterm infants, infants born of a multiple pregnancy or to an iron deficient woman, and children in low socioeconomic groups.
Diagnostic Evaluation
In general, anemia may be suspected from findings on the history and physical examination, such as lack of energy, easy fatigability, and pallor. Unless the anemia is severe, however, the first clue to the disorder may be alterations in the CBC, such as decreased numbers of RBCs, and decreased hemoglobin and hematocrit levels. Although anemia is sometimes defined as a hemoglobin level below 10 or 11 g/dl, this arbitrary cutoff is inappropriate for all children, since hemoglobin levels normally vary with age. (See Appendix C.)
Various findings of the CBC are also significant, such as increased reticulocyte levels, which indicate the body’s response to an increased demand for RBCs. A peripheral smear may demonstrate significant changes in the shape of RBCs, such as sickled cells. Tests to measure the amount of hemoglobin in a single cell are helpful in determining the cause of the anemia (see Table 35-1 and p. 1412). Sometimes a bone marrow aspiration may be necessary to evaluate the body’s ability to produce normal cells. For example, in leukemia the bone marrow is hyperplastic (producing increased numbers of cells), whereas in aplastic anemia the bone marrow is hypoplastic (producing decreased numbers of cells) or aplastic (producing no cells).
Tests for hematologic function do not always reflect the immediate changes occurring in the blood. For example, in acute massive hemorrhage the hemoglobin and hematocrit values may not be reliable, since the plasma volume may not increase for several hours. Without the hemodilution caused by the reexpansion of the vascular space, the hemoglobin and hematocrit may be close to normal, and the RBC loss may not be apparent. Consequently, assessing the quantity of blood loss in a seriously ill child may be difficult. The estimated volume of blood loss must be analyzed in conjunction with the child’s total blood volume to determine the percentage of blood loss. Blood specimens obtained from central lines may more accurately reflect the patient’s status than specimens obtained from an extremity because of the vasoconstriction of the peripheral vasculature. Decreased blood pressure changes are a late sign because of the compensatory mechanisms.
Therapeutic Management
The objective of medical management is to reverse the anemia by treating the underlying cause. In nutritional anemias the specific deficiency is corrected. In blood loss from acute hemorrhage, RBC transfusion may be given. In patients with severe anemia, supportive medical care may include oxygen therapy, bed rest, and replacement of intravascular volume with intravenous (IV) fluids. In addition to these general measures, the nurse may implement more specific interventions depending on the cause. The next sections discuss these interventions.
Nursing Care Management
The physical examination yields valuable evidence regarding the severity of the anemia and some indication of its possible cause (see Fig. 35-2 and Box 35-2). In interviewing the family, the nurse stresses the following areas: (1) nutrition, especially if the child is lactose intolerant or has inadequate intake of iron; (2) past history of chronic, recurrent infection; (3) eating habits, particularly pica (consumption of nonnutritive substances such as dirt, starch, lead-based paint chips, paper); (4) bowel habits and presence of frank blood in stools or black, tarry stools as a result of chronic blood loss; and (5) familial history of hereditary diseases, such as SCD or thalassemia.
The nurse should also be aware of the importance of taking a thorough history to obtain pertinent information that may aid in identifying the cause of the anemia. For example, statements such as “The baby drinks lots of milk” or “My teenager is on a liquid or vegetarian diet” are clues to possible iron deficiency.
Prepare the Child and Family for Laboratory Tests: Several blood tests may be ordered sequentially. Therefore the child may undergo multiple finger or heel sticks or venipunctures. These invasive procedures need not be painful with the application of a topical anesthetic known as EMLA (a eutectic mixture of local anesthetics) or LMX (lidocaine) cream.
The nurse is responsible for preparing the child for the tests by (1) explaining the significance of each test, particularly why the tests are not all done at one time; (2) encouraging parents or another supportive person to be with the child during the procedure; and (3) allowing the child to try out with the equipment on a doll or participate in the actual procedure (e.g., by holding the Band-Aid).
Older children may appreciate the opportunity to observe the blood cells under a microscope or in photographs. This is especially important if a serious blood disorder, such as leukemia, is suspected, since it serves as a foundation for explaining the pathophysiology of the disorder.
Bone marrow aspiration is not a routine hematologic test but is essential for definitive diagnosis of the leukemias, lymphomas, and certain anemias. (Chapter 27 presents information on preparing the child.)
Decrease Tissue Oxygen Needs: Because the basic pathology in anemia is a decrease in the oxygen-carrying capacity of the RBCs, an important nursing responsibility is to minimize tissue oxygen needs by continual assessment of the child’s energy level. In most instances of anemia this is not necessary, but when it is, the nurse must implement several important interventions. These same interventions apply to any child with a nursing diagnosis of fatigue or activity intolerance.
 NURSING ALERT
NURSING ALERT
Signs of exertion include tachycardia, palpitations, tachypnea, dyspnea, hyperpnea, dizziness, lightheadedness, diaphoresis, and change in skin color. The child looks fatigued (sagging, limp posture; slow, strained movements; inability to tolerate additional activity; difficulty sucking in infants).
Assess the child’s level of tolerance for activities of daily living and play, and make adjustments to allow as much self-care as possible without undue exertion. During periods of rest the nurse measures vital signs and observes behavior to establish a baseline of nonexertion energy expenditure. During periods of activity the nurse repeats these measurements and observations to compare them with resting values.
Once a baseline of physical tolerance has been established, the nurse anticipates which activities will be physically taxing, such as dressing, feeding, or getting out of bed, and allows for conservation of energy by assisting the child as needed. Because dependency can be threatening, however, allow the child as much control in the environment as possible. For example, a child with severe anemia may be unable to walk to the bathroom but may be able to use a bedside commode or be transported in a wheelchair to the lavatory rather than having to use a bedpan. Scheduling activities throughout the day with planned rest periods in between maximizes the child’s energy potential without causing undue exertion. Anticipate and implement necessary safety measures (e.g., staying with the child when the child is out of bed and raising side rails when the child is in the bed to prevent falls).
Plan diversional activities that promote rest but prevent boredom and withdrawal. Because short attention span, irritability, and restlessness are common in anemia and increase stress demands on the body, plan appropriate activities such as:
• Listening to music, using a tape recorder
• Watching television or playing video games
• Reading or listening to stories or comics
• Continuing a favorite hobby, such as stamp collecting
Choosing the appropriate roommate, such as a child of similar age with a diagnosis that also requires restricted activity, is another helpful intervention.
If infants or young children are hospitalized, consider the importance of preventing separation from parents. Crying and fretfulness place greater stress demands on the body, which increase oxygen needs. Parents may need help in understanding the importance of their presence and the basis for their child’s mood changes.
Prevent Complications: Children with anemia are prone to infection because tissue hypoxia causes cellular dysfunction and the disturbed metabolic processes weaken the host’s defenses against foreign agents. Infection also worsens the anemia by increasing metabolic needs and, in instances of chronic infection, also interferes with erythropoiesis and shortens the survival time of RBCs. Take all of the usual precautions to prevent infection, such as practicing thorough hand washing, selecting an appropriate room in a noninfectious area, restricting the presence of visitors or hospital personnel with active infection, and maintaining adequate nutrition. The nurse also observes for signs of infection, particularly temperature elevation and leukocytosis. However, an elevated WBC count sometimes occurs in anemia without the presence of systemic or local infection.
Drawing multiple blood samples may present a problem with cumulative blood loss and necessitate blood replacement. This situation occurs most often in infants with severe anemia. To prevent this situation, blood may be withdrawn through a continuous IV line and replaced after the exact amount needed has been tested and discarded. As a precaution, keep a record of the volume of blood withdrawn. Using micromethods of testing whenever possible minimizes the amount of blood required for the test. The nurse needs to observe for cumulative effects of blood loss, particularly signs of shock and increased hypoxia, and to explain to parents the necessity for taking multiple blood samples and the reason for blood replacement.
The main complication of anemia is cardiac decompensation, which can result from excessive demands on the heart due to increased metabolic needs or cardiac overload. The nurse needs to observe for signs and symptoms of heart failure such as tachycardia, dyspnea, rales, moist respirations, cough, and sweating. Obviously, preventing heart failure by minimizing hypoxia and closely monitoring IV infusions is of first priority. Packed RBCs are usually administered to prevent circulatory hypervolemia. When blood transfusions are required in severe anemia to increase the hemoglobin level, follow all of the usual precautions for administering blood and observe for signs of transfusion reactions (Table 35-2).
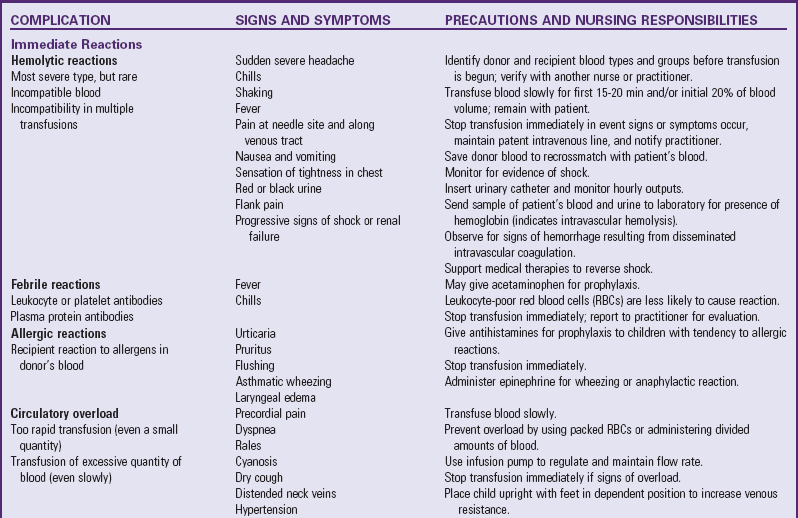
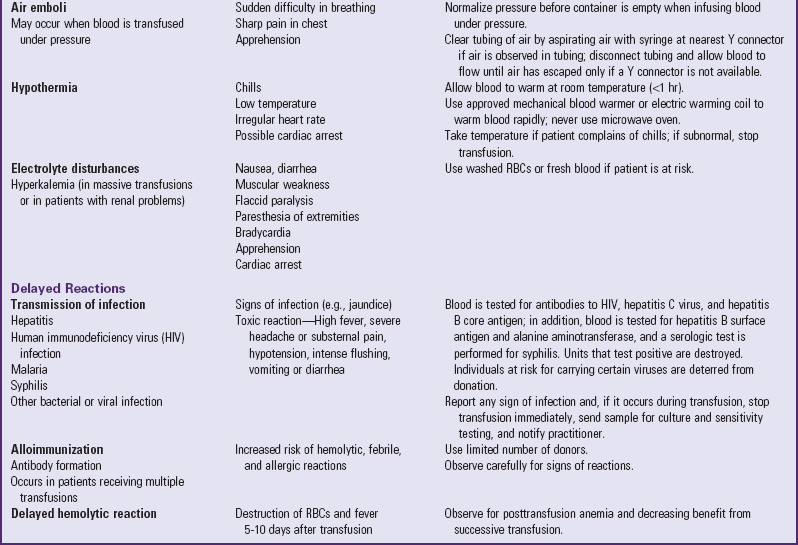
Blood Transfusion Therapy
Technologic advances in blood banking and transfusion medicine allow the administration of only the blood component needed by the child, such as packed RBCs in anemia or platelets for bleeding disorders (Table 35-3). Regardless of the blood component administered, the nurse must be aware of the possibility of transfusion reactions.
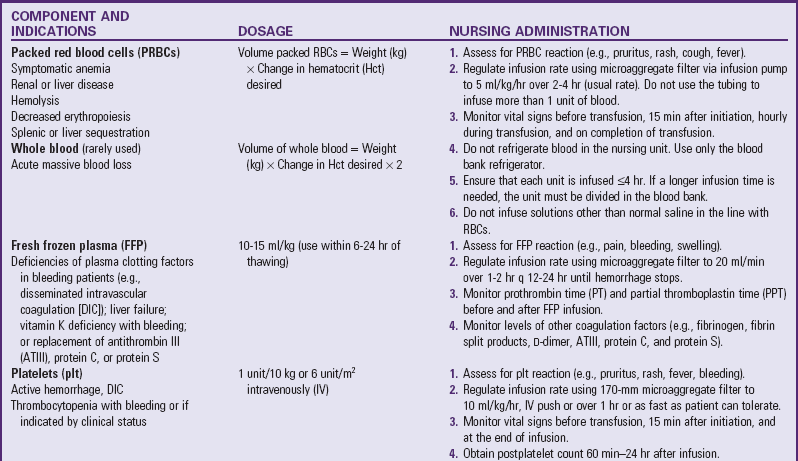

Although hemolytic reactions are rare, ABO incompatibility remains the most common cause of death from blood transfusion, and human error is usually responsible (e.g., administration of blood of the wrong type to the patient or mislabeling of a blood product) (Bell, 2007; Stainsby, Jones, Wells, et al, 2008). Blood is usually matched between the donor and recipient for blood group (A, B, AB, or O) and Rh factor (positive or negative). However, AB-type RBCs can be transfused into individuals with blood types A, B, and AB, and Rh-negative RBCs can be given to Rh-positive individuals. (See Chapter 9 for a discussion of blood groups and ABO and Rh incompatibility.)
When blood is mismatched, the A or B antiagglutinin is mixed with RBCs containing A or B agglutinogens, respectively, and agglutination (clumping) of the RBCs occurs. The agglutinins, which are bivalent, attach themselves to two different erythrocytes at the same time, causing the cells to clump together and clog small blood vessels. Over a few hours to days, the entrapped cells degenerate and hemolyze, liberating excessive quantities of hemoglobin into the circulation. The eventual hemolysis of large numbers of RBCs decreases the blood volume, which causes circulatory failure and shock. Treatment is aimed at replacing lost blood and using plasma volume expanders.
Acute kidney shutdown and eventual renal failure are the result of renal vasoconstriction caused by antigen-antibody complexes derived from the RBC surface. The greatly reduced blood flow leads to complete renal failure and death within 7 to 12 days. Treatment involves promoting diuresis with rapid administration of dilute IV fluids and diuretics such as furosemide and mannitol, and alkalinizing body fluids, which renders hemoglobin more soluble.
Another consequence of hemolysis is the release of large quantities of phospholipids, which are capable of stimulating disseminated intravascular coagulation (DIC) (see p. 1448). As a result, the plasma is depleted of the coagulation factors needed to prevent hemorrhage. Without treatment with heparin to prevent the coagulation and with blood components to initiate clotting, death from generalized hemorrhage can occur.
In addition to the nursing precautions and responsibilities outlined in Table 35-2, some other general guidelines that apply to all transfusions are:
• Take vital signs, including blood pressure, before administering blood to establish baseline data for intratransfusion and posttransfusion comparison; 15 minutes after initiation; hourly while blood is infusing; and on completion of the transfusion.
• Check the blood type and group of the recipient against the donor’s, regardless of the blood product used.
• Administer the first 50 ml of blood or initial 20% of volume (whichever is smaller) slowly and stay with the child.
• Administer with normal saline in a piggyback setup or have normal saline available.
• Administer blood through an appropriate filter to eliminate particles in the blood and prevent the precipitation of formed elements; gently shake the container frequently.
• Use blood within 30 minutes of its arrival from the blood bank. If it is not used, return it to the blood bank; do not store it in a regular unit refrigerator.
• Infuse a unit of blood (or the specified amount) within 4 hours. If the infusion will exceed this time, divide the blood into appropriate-size quantities by the blood bank, with the unused portion refrigerated under controlled conditions.
• If a reaction of any type is suspected, take vital signs, stop the transfusion, maintain a patent IV line with normal saline and new tubing, notify the practitioner, and do not restart the transfusion until the child’s condition has been medically evaluated.
Blood is usually administered to children by infusion pump; therefore the usual precautions and management related to pumps apply. When the blood infusion begins with a standard transfusion set, the filter chamber is filled to allow the total filter to be used. The drip chamber is partially filled with blood to permit counting of the drops. When the flow rate is adjusted, remember that blood administration sets do not use microdrops (60 drops/ml) but regular drops (usually 10 or 15 drops/ml). The nurse must consider this when calculating the flow rate.
Oxygen may be administered to provide optimum environmental conditions for hemoglobin saturation. Oxygen administration is of limited value, however, because each gram of hemoglobin is able to carry a limited amount of the gas. In addition, prolonged use of supplemental oxygen can decrease erythropoiesis. Therefore monitor the child closely for evidence of decreasing benefit from oxygen. One of the first signs of hypoxia is restlessness.
Anemia Caused by Nutritional Deficiencies
Anemia caused by an inadequate supply or loss of iron is the most prevalent nutritional disorder in the United States and the most preventable mineral disturbance. Without the intake of iron-fortified formula and cereals, term infants tend to develop iron deficiency, both with and without anemia, between 9 and 24 months of age (Andrews, Ullrich, and Fleming, 2009; Glader, 2007b). Adolescents are also at risk for iron deficiency because of their rapid growth rate, menses, poor eating habits, obesity, and strenuous activities (Andrews, Ullrich, and Fleming, 2009; Glader, 2007a; Nead, Halterman, Kaczorowski, et al, 2004). The decline in the prevalence of iron deficiency anemia in the United States over the past several decades may be due to the initiation of the U.S. Special Supplemental Food Program and the American Academy of Pediatrics’ promotion of formula for the first year of life (Altucher, Rasmussen, Barden, et al, 2005; Andrews, Ullrich, and Fleming, 2009). The promotion of iron supplement in the exclusively breast-fed infant, introduction of iron-fortified infant formula and cereal, weaning from the bottle by 1 year of age, limiting intake of cow’s milk to 16 to 24 oz/day, and delayed introduction of cow’s milk into the diet have all contributed to the decreased incidence of iron deficiency anemia in infants and young children (Andrews, Ullrich, and Fleming, 2009; Glader, 2007b; Richardson, 2007). However, iron deficiency still occurs in infants, children, adolescents, and child-bearing women of all races and ethnic groups (Andrews, Ullrich, and Fleming, 2009; Glader, 2007b; Richardson, 2007; White, 2005) and continues to be a significant health problem.
Critical Thinking Case Study—Iron Deficiency Anemia
Etiology
Iron deficiency anemia can be caused by any number of factors that decrease the supply of iron, impair its absorption, increase the body’s need for iron, or affect the synthesis of hemoglobin (Box 35-3). Although the clinical manifestations and diagnostic evaluation are similar regardless of the cause, the therapeutic and nursing care management depends on the specific reason for the iron deficiency. The following discussion is limited to iron deficiency anemia resulting from inadequate iron in the diet.
At birth the full-term infant has approximately a 0.5 g supply of iron, and an average of 0.8 mg of iron must be absorbed each day during the first 15 years of life (Glader, 2007b). During the last trimester of pregnancy, iron is transferred from the mother to the fetus at the rate of 4 mg/day. Most of the iron is stored in the circulating hemoglobin of the erythrocytes, and the remainder is deposited in the liver, spleen, and bone marrow. Maternally derived iron stores are adequate for the first 5 to 6 months in the full-term infant but for only about 2 to 3 months in premature infants or infants of multiple births. If dietary sources of iron are not supplied to meet the infant’s growth demands after depletion of fetal iron stores, iron deficiency anemia results. Physiologic anemia should not be confused with iron deficiency anemia resulting from nutritional causes.
Vegetarian diets, popular among teenage girls, have been associated with nutritional deficiencies. Some infants and toddlers who have been fed inappropriate vegetarian diets have had severe protein-energy malnutrition, as well as deficiencies of iron, vitamin B12, and vitamin D. Unrefined cereals contain substances that modify the absorption of minerals such as zinc, calcium, and iron. Individuals consuming strict vegetarian diets that include a large amount of unrefined cereals could be at a greater risk of rickets, vitamin B12 and folate deficiency, and iron deficiency anemia (Hubbard, 2004; Renda and Fischer, 2009).
Pathophysiology
Iron is required for the production of hemoglobin. One molecule of hemoglobin consists of protein (globin) combined with four molecules of a pigmented compound (heme). Each molecule of heme contains one atom of iron. When iron stores are deficient, the production of hemoglobin is reduced. Consequently, the main effect of iron deficiency is decreased hemoglobin level and reduced oxygen-carrying capacity of the blood.
Clinical Manifestations
The clinical manifestations are directly attributable to the reduction in the amount of oxygen available to the tissues and resemble those seen in any type of anemia. Usually the signs are insidious and obscure, and the severity is directly related to the duration of the dietary deficiency.
Although infants with iron deficiency anemia tend to be underweight, many are overweight because of excessive milk ingestion (known as milk baby). These children become anemic for two reasons: (1) milk, a poor source of iron, is given almost to the exclusion of solid foods; and (2) increased fecal loss of blood occurs in 50% of iron deficient infants fed cow’s milk. This asymptomatic loss of hemoglobin causes iron deficiency (Glader, 2007b; Richardson, 2007). Although chubby, these infants are pale (sometimes porcelain-like), usually demonstrate poor muscle development, and are prone to infection.
Although the mechanism is unknown, iron deficiency anemia enhances the leakage of plasma proteins, which causes edema; retarded growth; and decreased serum concentration of the proteins albumin, gamma globulin, and transferrin (a protein that binds iron and transports it through the plasma). Other manifestations of iron deficiency include irritability, tachycardia, fatigue, glossitis, angular stomatitis, and koilonychia (concave or “spoon” fingernails). The association between iron deficiency anemia and impaired neurocognitive function (attention span, alertness, and learning) in both infants and adolescents has been well established, but the mechanism by which iron deficiency anemia impairs neurologic function is unknown (Akman, Cebecci, Okur, et al, 2004; Andrews, Ullrich, and Fleming, 2009; Glader, 2007b).
Diagnostic Evaluation
Laboratory tests that measure or describe hemoglobin, the morphologic changes in the RBC, and iron concentration are usually performed (see Table 35-1). The RBC count may be normal, borderline, or moderately reduced in the child with iron deficiency anemia. Typically the nearly normal number of erythrocytes is strikingly out of proportion to the low hemoglobin concentration. RBCs are typically small (microcytic), so that MCV is decreased (see Box 35-1). For infants 1 year of age, an MCV below 70 femtoliters (fl) is considered diagnostic. In children from 1 to 10 years of age, an MCV value of 70 fl plus the child’s age in years is a quick calculation of the lower limit of normal.
The reticulocyte count is usually normal or slightly reduced because of decreased stores of iron (see Table 35-1). However, in severe anemia, when tissue hypoxia elicits an erythropoietic response, the reticulocyte count may be elevated to 3% or 4%. The level of erythrocyte protoporphyrin, the immediate precursor of heme, becomes elevated in RBCs whenever heme synthesis is disturbed.
In terms of differential diagnosis, a stool analysis for occult blood (guaiac test) is commonly performed to confirm or rule out the possibility of chronic fecal blood loss, especially from milk intolerance or structural anomalies such as diverticulitis.
Iron Studies: In addition to tests that indirectly indicate the level of iron by revealing the effects of iron deficiency on the RBCs, several other tests are usually performed that more directly measure the amount of circulating iron. The serum iron concentration (SIC) is the amount of circulating iron and is normally about 70 mcg/dl in infants and slightly higher in older children. Lower limits of SIC vary not only with age but also with the time of day; they are highest in the morning, when the test should be performed.
The total iron-binding capacity (TIBC) is the amount of transferrin (iron-binding globulin), which is necessary for the transport of iron in the bloodstream. When combined with transferrin, the iron is loosely bound to the globulin molecule so that it can be released easily to tissue cells anywhere in the body. In iron deficiency anemia TIBC is elevated above the normal range of 350 mcg/dl (6 months to 2 years) or 450 mcg/dl (children >2 years and adults). The elevated TIBC represents the body’s compensatory mechanism to absorb more iron from exogenous sources than normally during states of deficiency. Transferrin saturation is calculated by dividing the SIC by the TIBC and multiplying the result by 100 to express the value as a percentage. A transferrin saturation of 10% suggests anemia.
These biochemical test (ferritin, TIBC, SIC) are acute phase reactants, which means in an inflammatory setting a positive acute phase reactant may overestimate iron stores (Andrews, Ullrich, and Fleming, 2009; Glader, 2007b). Other tests not affected by inflammation that are available in some clinical laboratories are the serum transferring receptor (elevated in iron deficiency) and reticulocyte hemoglobin content, which has been used to accurately assess iron status in adults (Andrews, Ullrich, and Fleming, 2009; Glader, 2007b).
Therapeutic Management
Prevention is the primary goal and is achieved through optimum nutrition and appropriate iron supplementation. In infants, the following guidelines are recommended to prevent iron deficiency (American Academy of Pediatrics, 1999; Glader, 2007a)
• Use only breast milk or iron-fortified formula (containing 7 to 12 mg/L for full-term infants and 15 mg/L for preterm infants of iron) for the first 12 months.
• Iron supplementation of 1 mg/kg/day should be provided by 4 to 6 months of age in full-term infants and 2 mg/kg/day by 2 months of age in preterm infants.
• Administer iron drops at a dosage of 2 to 3 mg/kg/day to a maximum of 15 mg/day of elemental iron to breast-fed preterm infants after 2 months of age, and give iron-fortified infant cereal when solid foods are introduced.
• Limit the amount of formula to no more than 1 L/day to encourage intake of iron-rich solid foods.
Once the diagnosis of iron deficiency anemia is made, therapeutic management focuses on increasing the amount of supplemental iron the child receives. This usually occurs through dietary counseling and the administration of oral iron supplements. In formula-fed infants the most convenient and best sources of supplemental iron are iron-fortified commercial formula and iron-fortified infant cereal (Glader, 2007b; Mabry-Hernandez, 2009). Iron-fortified formula provides a relatively constant and predictable amount of iron and is not associated with an increased incidence of gastrointestinal symptoms, such as colic, diarrhea, or constipation. Infants younger than 12 months of age should not be given fresh cow’s milk to decrease the possibility of gastrointestinal blood loss from intolerance to the milk protein.
Addition of iron-rich foods to the diet may not provide sufficient supplemental quantities of the mineral. Oral supplements of ferrous iron are given because this form is more readily absorbed than ferric iron and results in higher hemoglobin levels. Ingested iron is absorbed largely from the duodenum, and absorption is facilitated by an acid environment. Children normally absorb an average of 10% to 20% of the iron in oral supplements, but during periods of iron deficiency they absorb an additional 5% to 10%. Oral iron supplementation is prescribed as 3 to 6 mg of elemental iron per kilogram per day. Lower dosages of iron are associated with fewer side effects. Ideally the daily dose of iron should be given in two or three divided doses between meals. Side effects of oral iron therapy include nausea, gastric irritation, diarrhea or constipation, and anorexia, but these occur infrequently, especially in infants. If the iron produces vomiting and diarrhea, it should be administered with meals and in gradually increasing doses.
The response to oral iron therapy is reflected in a peak increase in the reticulocyte count by the fifth to the tenth day of administration. Following the reticulocyte rise, the hemoglobin and hematocrit levels and RBC count increase. The hemoglobin level rises as much as 0.5 g/dl/24 hr; therefore a substantial increase should occur by the end of 1 month (Glader, 2007b; Richardson, 2007).
If the hemoglobin level is very low or if the level fails to rise after 1 month of oral therapy, it is important to assess whether the iron is being administered correctly. Parenteral iron administration is painful; expensive; and occasionally associated with regional lymphadenopathy, transient arthralgias, or serious allergic reaction (Andrews, Ullrich, and Fleming, 2009; Glader, 2007b). Therefore parenteral iron administration is reserved for children who have iron malabsorption or chronic hemoglobinuria. The Z-track method of intramuscular injection must be used to minimize staining of the skin, and careful observation is required because of the risk of anaphylaxis. Transfusions are indicated for the most severe anemia and in cases of serious infection, cardiac dysfunction, or surgical emergency when anesthesia is required. Packed RBCs (2 to 3 ml/kg), not whole blood, are used to minimize the chance of circulatory overload. Supplemental oxygen is administered when tissue hypoxia is severe.
Next to iron deficiency, folate deficiency is the most common micronutrient deficiency (Watkins, Whitehead, and Rosenblatt, 2009). Causes of folate deficiency include inadequate diet, overcooking of vegetables with loss of folates, and malabsorption. The deficiency is treated with adequate prepared and intake of folate-enrich foods and/or 1 mg of folate daily (Watkins, Whitehead, and Rosenblatt, 2009). As macrocytic anemias, both folate and vitamin B12 deficiencies result in defective ribonucleic acid (RNA) and deoxyribonucleic acid (DNA) synthesis (Kaferle and Strzoda, 2009).
Vitamin B12 deficiency commonly develops when the gastric mucosa fails to secrete sufficient intrinsic factor, which is essential for absorption of vitamin B12. Deprived of vitamin B12, the bone marrow produces fewer but macrocytic RBCs. The erythrocytes are usually immature and, because of their extremely fragile cell membranes, are rapidly destroyed during circulation. Treatment initially involves the administration of 100 mcg or higher dose of vitamin B12 parenteral therapy for several days, followed by injections of 500 or 1000 mcg of vitamin B12 every 1 to 2 months. Researchers compared oral and parenteral vitamin B12 therapy and found that they yielded comparable benefits (Bolarman, Kadikoylu, Yukselen, et al, 2003; Butler, Vidal-Alaball, Cannings-John, et al, 2006).
Prognosis: Prognosis for a child with iron deficiency anemia, folate deficiency, or vitamin B12 deficiency is very good. However, there is evidence that if the anemia is severe and longstanding, then diminished cognitive function, behavioral changes, delayed infant growth and development, decreased exercise tolerance, and impaired immune function may develop (Andrews, Ullrich, and Fleming, 2009; Richardson, 2007).
Nursing Care Management
A primary nursing objective is to prevent nutritional anemia through family education. Nurses need to be aware of recommendations regarding iron supplementation during infancy and appropriate sources of dietary iron. The nurse should encourage parents to limit the quantity of milk, to use iron-fortified infant formulas, and to introduce solid foods. This may be difficult when parents believe milk is best for the infant and equate the resultant weight gain with “healthiness.” Although milk is an excellent food, it is deficient in iron, vitamin C, zinc, and fluoride. Sources of each of these nutrients and the role they play in preventing deficiencies need to be discussed with the family, especially the person responsible for feeding the infant. For example, the mother may have less decision-making power regarding feeding than the grandmother who cares for the child.
Also stress that overweight is not synonymous with good health. If the infant has obvious signs of anemia, such as pallor, listlessness, frequent infections, and muscular weakness, point these out as evidence of suboptimum health. In some instances it is helpful to chart the hemoglobin or hematocrit values to visually impress on parents the change in iron levels. Often, increased blood values correspond to improved physical status and reinforce the benefit of dietary or oral iron supplementation.
Instructing parents regarding proper administration of oral iron supplements is an essential nursing responsibility. Several factors, such as stomach acidity, affect the absorption of iron (see Box 13-1).
 DRUG ALERT
DRUG ALERT
Iron Supplements
Ideally iron supplements are administered in two divided doses between meals, when the presence of free hydrochloric acid is greatest, and are accompanied by a citrus fruit or juice, which helps reduce iron to its most soluble state.
An adequate dosage of oral iron turns the stools a tarry green or black color. The nurse advises parents of this normally expected change and inquires about its occurrence on follow-up visits. Absence of the greenish black stool may be a clue to poor compliance. If compliance is an issue, make every effort to institute strategies to improve adherence to the medication regimen, such as administering the drug once a day at the most convenient time. (See Compliance, Chapter 27.)
NURSING ALERT
Because iron ingested in excessive quantities is toxic, even fatal, parents should keep no more than a 1-month supply in the home and store it safely away from the reach of children.
Oral iron supplements are available in liquid or tablet form. Liquid preparations may temporarily stain the teeth. If possible, take the medication through a straw or give it through a syringe or medicine dropper placed toward the back of the mouth. Brushing the teeth after administration of the drug lessens the discoloration.
Counseling families whose children are anemic is often a difficult and challenging task. Meal planning must be based on the family’s budget, cultural pattern, and food preferences (see Cultural Competence box). Often this requires more than a brief discussion with the mother or usual caregiver about foods high in iron (see Table 13-2). For teaching to be effective, the nurse may need to offer recipes, assist in planning a shopping list, and investigate food prices for economy. Because the physical effects of anemia are insidious, parents may not consider their child ill and consequently may view the medication and diet changes as unnecessary. Stressing the physical and behavioral improvements and what effect the improved diet will have on all family members may encourage parents to adhere to the treatment plan.
 CULTURAL COMPETENCE
CULTURAL COMPETENCE
Tea Drinking and Nonheme Iron Absorption
In cultures in which tea is drunk as a common beverage, administer iron with some other liquid because the tannins in tea form an insoluble complex with nonheme iron that is from foods other than meat. In addition, the phytates in legumes and maize and the phenolic compounds in herbal teas may adversely affect the uptake of iron (Diaz, Rosado, Allen, et al, 2003). There is clear evidence to show that tea drinking limits the absorption of nonheme iron whereas ascorbic acid enhances iron absorption (Nelson and Poulter, 2004; Thankachan, Walczyk, Muthayya, et al, 2008).
Diet education of teenagers is difficult, especially because teenage girls are particularly prone to following weight-reduction diets. Emphasizing the effect of anemia on appearance (pallor) and energy level (difficulty maintaining popular activities) may be useful. (See Table 13-2 and Mineral Disturbances, Chapter 13, for sources of iron-rich foods.)
Anemias Caused by Increased Destruction of Red Blood Cells
Excessive destruction or hemolysis of erythrocytes can occur from a defect in the RBC (intracorpuscular defect) that shortens the life span of the cell so that production cannot keep pace with destruction. In sickle cell anemia and thalassemia, erythrocyte life spans are decreased because of a hemoglobin defect, whereas in spherocytosis erythrocyte life span is decreased due to a defective red cell membrane. Extracorpuscular factors are those conditions that cause hemolysis in otherwise normal RBCs. A classic example is blood group incompatibility, such as hemolytic disease of the newborn or incompatibility secondary to mismatched blood transfusion. Damage to a normal red cell may be caused by toxic drugs, burns, poisonings (such as from lead), infections (such as malaria), and splenic sequestration (hypersplenism).
Hereditary Spherocytosis
Hereditary spherocytosis (HS), a common hemolytic disorder, is caused by a defect in the proteins that form the RBC membrane (Grace and Lux, 2009; Segel, 2007). It occurs in most ethnic groups, but is primarily prevalent in persons of northern European heritage with a reported incidence of 1 in 5000 (Grace and Lux, 2009; Segel, 2007).
The condition is transmitted in an autosomal dominant pattern. However, 25% of cases are thought to represent new mutations or inheritance through an autosomal recessive mode or autosomal dominant mode with reduced penetrance (Segel, 2007; Shah and Vega, 2004). The affected cells have a smaller surface area relative to their volume than normal RBCs, so that they become inflexible spheres known as spherocytes. The inflexibility of these cells makes it difficult for them to circulate through the spleen and leads to their early destruction.
Clinical manifestations vary widely and include anemia (usually mild), splenomegaly (usually modest and does not correlate with the severity of disease), and jaundice (most often scleral icterus). HS frequently manifests in the first 24 hours of the newborn’s life as severe hyperbilirubinemia. Folic acid supplementation should be given to these children to prevent deficiency due to the rapid cell turnover. Laboratory findings include hemoglobin level between 7 and 10 g/dl, reticulocyte count of 3% to 15% (inversely correlated with hemoglobin level), and an increase in osmotic fragility.
Aplastic crisis, which results in a sudden cessation of RBC production by the bone marrow, is a serious complication. Hemoglobin and hematocrit values drop rapidly, which results in severe anemia. Transfusion support may be needed, and close monitoring of the child’s cardiovascular status is necessary.
Splenectomy, a treatment for HS, is generally reserved for children older than 5 years of age with symptomatic anemia. The splenectomy corrects the hemolysis but not the RBC defect. Occasionally splenectomy is performed in children younger than 5 years of age who are severely anemic and are showing signs of failure to thrive. Children who are scheduled to undergo a splenectomy should be evaluated for the presence of gallstones before surgery. If gallstones are present, a cholecystectomy is performed at the time of splenectomy.
Because of the risk of life-threatening bacterial infection after splenectomy, these children are immunized with the pneumococcal, meningococcal, and Haemophilus influenzae type b vaccines before surgery and receive prophylactic penicillin for several years after splenectomy. Instruct parents in the importance of seeking immediate medical attention if their child develops a fever of 38.3° C (101° F) or higher as a common sign of infection or postsplenectomy sepsis (Grace and Lux, 2009; Lanzkowsky, 2005; Shah and Vega, 2004). Given the lifelong increased risk of severe infection and thromboembolic complications in splenectomized children, partial splenectomy has been investigated for selected patients with HS with the goal of decreasing hemolysis while maintaining splenic phagocytic function (Grace and Lux, 2009; Segel, 2007).
Sickle Cell Anemia
Sickle cell anemia (SCA) is one of a group of diseases collectively termed hemoglobinopathies in which normal adult hemoglobin (hemoglobin A [HgbA]) is partly or completely replaced by abnormal sickle hemoglobin (HgbS). SCD refers to a group of hereditary disorders, all of which are related to the presence of HgbS. Although the name SCD is sometimes used to refer to SCA, this usage is incorrect. The correct terms for SCA are HgbSS disease and homozygous sickle cell disease. In the United States the most common forms of SCD are:
SCA—The homozygous form of the disease (HgbSS), in which valine, an amino acid, is substituted for glutamic acid at the sixth position of the β chain.
Sickle cell C disease—A heterozygous variant of SCD (HgbSC), characterized by the presence of both HgbS and hemoglobin C (HgbC), in which lysine is substituted for glutamic acid at the sixth position of the β chain.
Sickle thalassemia disease—A combination of sickle cell trait and β-thalassemia trait. In the β+ (beta plus) form some normal adult hemoglobin can still be produced. In the β0 (beta zero) form there is no ability to produce normal adult hemoglobin.
Of the SCDs, SCA is the most common form in African-Americans in the United States, followed by sickle cell C disease and sickle β-thalassemia. Numerous other sickle syndromes exist in which HgbS is paired with rare mutant globins.
SCD is one of the most common genetic diseases worldwide, affecting approximately 90,000 Americans; those of other nationalities, such as Africans, Hispanics, Italians, Greeks, Iranians, and Turks; and individuals of Arab, Caribbean, and Asian Indian descent. The incidence of the disease varies in different geographic locations. Among African-Americans, the incidence of sickle cell trait is about 8%, whereas among inhabitants of West Africa the frequency of sickle cell trait is reportedly as high as 40%. The high incidence of sickle cell trait in these individuals is believed to be a selective protection against death from malaria caused by endemic Plasmodium falciparum infection (Driscoll, 2007). The physiologic basis for the influence of malaria on the sickle gene (so-called balanced polymorphism) is not well understood (Heeney and Dover, 2009).
Mode of Transmission
The gene that determines the production of HgbS is situated on an autosome. When both parents have sickle cell trait, there is a 25% chance with each pregnancy of producing an offspring with SCA. In the United States it is estimated that 1 in 12 African-Americans carries the trait; therefore the risk of two African-American parents having a child with the disease is 0.7%. Other forms of SCD occur through the union of two individuals who carry the heterozygous form of hemoglobin variants.
Basic Defect
The basic defect responsible for the sickling of erythrocytes is contained in the globin fraction of hemoglobin, which is composed of 574 amino acids. Under conditions of dehydration, acidosis, hypoxia, and temperature elevation, the relatively insoluble HgbS changes its molecular structure to form long, slender crystals. These filamentous crystals cause distortion of the cell membrane, so that the cell changes from a pliable disk to a crescent- or sickle-shaped RBC. The filamentous forms are associated with much greater viscosity than those with the normal holly leaf structure of HgbA.
In most instances the sickling response is reversible under conditions of adequate oxygenation and hydration. During this time the RBCs are indistinguishable from normal erythrocytes on peripheral examination. RBCs with HgbS can sickle and unsickle under appropriate conditions. After repeated cycles of sickling and unsickling, the RBCs become irreversibly sickled.
Although the defect is inherited, the sickling phenomenon is usually not apparent until later in infancy because of the presence of fetal hemoglobin (HgbF). HgbF is composed of two α and two γ polypeptide chains. At 32 weeks of gestation, the production of β and δ chains begins. These combine with α chains to form the major adult hemoglobins: HgbA (two α and two β chains) and HgbA2 (two α and two δ chains). The newborn with SCD is generally asymptomatic because of the protective effect of HgbF (60% to 80%), but this rapidly decreases during the first year, so that the child is at risk for sickle cell–related complications (Heeney and Dover, 2009; Driscoll, 2007).
Sickle Cell Trait: Persons with sickle cell trait have the same basic defect, but only about 35% to 45% of the total hemoglobin is HgbS. The remainder is HgbA. Normally these individuals are asymptomatic. Although complications are rare, they have been described in individuals with sickle cell trait. Nonpainful gross hematuria is the major complication, seen primarily in the teenage and adult years. Under conditions of extreme or prolonged deoxygenation, such as riding in a nonpressurized aircraft or undergoing military training, splenic sequestration with profound anemia can occur, resulting in death.
Pathophysiology and Clinical Manifestations
The clinical manifestations of SCA are primarily the result of (1) obstruction caused by the sickled RBCs’ adhesion to vascular endothelium accompanied by inflammatory process and (2) increased RBC destruction. The entanglement and enmeshing of rigid sickle-shaped cells block the microcirculation, causing vasoocclusion (Fig. 35-3). The resultant absence of blood flow to adjacent tissues causes local hypoxia, which leads to tissue ischemia and infarction (cellular death) (Box 35-4). The sickle cell adhesion to the vascular endothelium actually sets in motion the abnormal adhesion-inflammation–increased adhesion of the sickling cycle (Redding-Lallinger and Knoll, 2006). Most of the complications seen in SCA can be traced to this cycle and its impact on various organs of the body (Fig. 35-4).
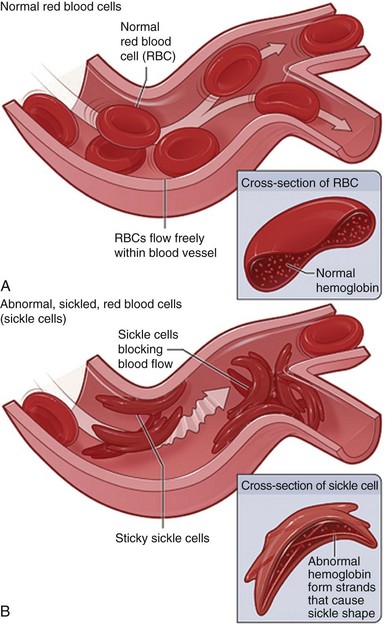
Fig. 35-3 A, Normal red blood cells (RBCs) flowing freely in a blood vessel. The inset image shows a cross-section of a normal red blood cell with normal hemoglobin. B, Abnormal, sickled red blood cells clumping and blocking blood flow in a blood vessel. (Other cells also may play a role in this clumping process.) The inset image shows a cross-section of a sickle cell with abnormal hemoglobin. (From National Heart, Lung, and Blood Institute: What is sickle cell anemia? Bethesda Md, August 2008, National Institutes of Health.)
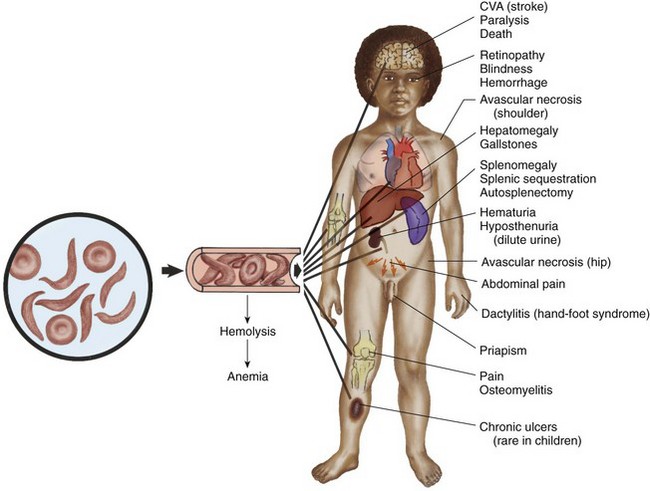
Fig. 35-4 Effects of sickled red blood cells on circulation with related complications. CVA, Cerebrovascular accident.
Initially the spleen may become enlarged from congestion and engorgement with sickled cells. This repeated insult to the splenic sinuses results in infarction. The functioning cells are gradually replaced by fibrotic tissue until, by the age of 5 years, the spleen has decreased in size and has been totally replaced by a fibrous mass (functional asplenia). Without the spleen to filter bacteria and to promote the release of large numbers of phagocytic cells, these individuals are highly susceptible to infection.
The liver is also altered in form and function. Liver failure and necrosis are the result of severe impairment of hepatic blood flow from anemia and capillary obstruction. Moderate hepatomegaly is common by age 1 and usually persists throughout childhood and early adulthood. The rapid destruction of RBCs often results in the development of pigmented gallstones. Obstruction of the common bile duct by gallstones is uncommon; therefore cholecystectomy is generally not recommended for asymptomatic patients. If recurrent episodes of right upper abdominal pain occur, cholecystectomy may be indicated.
Kidney abnormalities are probably the result of the same cycle of congestion of glomerular capillaries and tubular arterioles with sickle cells and hemosiderin, tissue necrosis, and eventual scarring. The principal results of kidney ischemia are hematuria, inability to concentrate urine, enuresis, and occasionally nephrotic syndrome.
Bone changes include hyperplasia and congestion of the bone marrow, which result in osteoporosis, widening of the medullary spaces, and thinning of the cortices. As a result of the weakening of bone, especially in the lumbar and thoracic regions, skeletal deformities, particularly lordosis and kyphosis, may occur. Because of chronic hypoxia, the bone becomes susceptible to osteomyelitis, frequently from Salmonella organisms. Aseptic necrosis of the femoral head from chronic ischemia is an occasional problem.
Changes in the central nervous system are primarily vascular and result from the same cyclic reaction of occlusion, ischemia, and infarction. Stroke, or cerebrovascular accident, is a major complication that occurs in approximately 11% of children with SCD by the age of 20 years and can result in permanent paralysis or death (DeBaun and Vichinsky, 2007; Driscoll, 2007). Any number of neurologic symptoms can indicate a minor cerebral insult, such as headache, aphasia, weakness, convulsions, visual disturbances, or unilateral hemiplegia. Loss of vision is usually the result of progressive retinopathy and retinal detachment. Cognitive impairment from SCA without any overt signs of neurologic injury is known as silent cerebral infarct (Driscoll, 2007; Buchanan, DeBaun, Quinn, et al, 2004). Silent cerebral infarct occurs in 22% of children with SCA and is defined as an abnormality on magnetic resonance imaging (Driscoll, 2007; Steen, Fineberg-Buchner, Hankins, et al, 2005; Buchanan, DeBaun, Quinn, et al, 2004).
Heart problems are mainly attributable to the stress of chronic anemia, which can eventually result in decompensation and failure. Cardiomegaly is visualized on chest radiographic examination, and a systolic flow murmur is frequently present as a consequence of the anemia. An echocardiogram shows cardiomegaly, septal hypertrophy, and impaired contractility (Heeney and Dover, 2009; Lanzkowsky, 2005).
With the formation of sickled erythrocytes, mechanical fragility increases, which decreases the life span of the RBC. Hemolysis occurs both during intravascular circulation and as a result of stagnation of sickled cells in the congested spleen (see Fig. 35-3). Although the body attempts to compensate through stimulated erythropoietic activity, as evidenced by a hyperplastic bone marrow, the rate of destruction exceeds the rate of production. A normocytic normochromic anemia results. With increased hemolysis, hemosiderosis (increased storage of iron) is present in the liver, spleen, bone marrow, kidneys, and lymph nodes (see Box 35-4).
Other Signs and Symptoms: In addition to the effects of sickling on various organ structures, the child with SCA may have a variety of complaints such as exercise intolerance, anorexia, jaundiced sclera, and gallstones. Chronic leg ulcers are common in adolescents and adults and are thought to be a result of decreased circulation due to vasoocclusion and tissue ischemia. Other generalized effects include retardation of growth in both height and weight, delayed sexual maturation, and decreased fertility. When the child reaches adulthood, full sexual development and adult height are usually achieved.
Sickle Cell Crises: The clinical manifestations of SCA vary markedly in severity and frequency. The most acute symptoms of the disease occur during periods of exacerbation called crises. There are several types of episodic crisis: vasoocclusive, acute splenic sequestration, aplastic, hyperhemolytic, stroke, chest syndrome, and infection related. The crises may occur individually or concomitantly with one or more other crises.
Vasoocclusive crisis (VOC), preferably called a painful episode or event, is the most common type of non–life-threatening crisis. It is characterized by ischemia causing mild to severe pain that may last from minutes to days. A child experiencing a VOC alone may have localized or generalized pain, acute abdominal pain from visceral hypoxia or gallstones, priapism (an unwanted painful penile erection), and arthralgia. The pain is often migratory with the presence of a low-grade fever.
VOCs can result in a variety of skeletal problems. One of the more frequent is hand-and-foot syndrome (dactylitis), which occurs primarily in young children ages 6 months to 2 years. It is caused by infarction of short tubular bones and is characterized by pain and swelling of the soft tissue over the hands and feet. It usually resolves spontaneously within a couple of days to weeks. Localized swelling over joints with arthralgia can occur from erythrostasis with sickle cells.
Sequestration crisis is caused by the pooling of large quantities of blood, usually in the spleen and infrequently in the liver, which causes a decrease in blood volume and ultimately shock. The splenic crisis may be acute or chronic. The chronic manifestation is termed hypersplenism. The acute form occurs most commonly in children between 2 months and 5 years of age and may result in death from profound anemia and cardiovascular collapse. Splenic sequestration has occurred in older children and adolescents with sickle cell C disease or sickle β-thalassemia.
Aplastic crisis is diminished RBC production, usually triggered by infection with a virus (especially the human parvovirus) or other organism. When it is superimposed on the rapid destruction of RBCs, a profound anemia results. Packed RBC transfusion is occasionally required in children exhibiting signs and symptoms of congestive heart failure.
Megaloblastic anemia is attributed to an excessive nutritional need for folic acid and/or vitamin B12 during periods of pronounced erythropoiesis. Because infection is not always antecedent to aplastic or hypoplastic crises, it is possible that folic acid deficiency is a causative agent.
Hyperhemolytic crisis is an accelerated rate of RBC destruction characterized by anemia, jaundice, and reticulocytosis. This complication frequently suggests other coexisting conditions, such as viral illness; transfusion reactions to alloantibodies; or glucose-6-phosphate dehydrogenase (G6PD) deficiency, which is also common in African-Americans.
A cerebrovascular accident is a sudden and severe complication, often with no related illnesses. Sickled cells block the major blood vessels in the brain, which results in cerebral infarction causing variable degrees of neurologic impairment. Repeat strokes causing progressively greater brain damage occur in approximately 70% of children who have already experienced one stroke and do not receive monthly transfusions (Heeney and Dover, 2009). The mortality rate for untreated children is approximately 20% (Heeney and Dover, 2009).
Another serious complication is acute chest syndrome, which is clinically similar to pneumonia. It is the presence of a new pulmonary infiltrate associated with chest pain, fever, cough, tachypnea, wheezing, and hypoxia (Driscoll, 2007; Heeney and Dover, 2009). Researchers believe that a VOC or infection results in sickling in the small blood vessels of the lungs, with ensuing occlusion, stasis, and anemia. Repeated episodes of chest syndrome may cause restrictive lung disease and pulmonary hypertension.
Overwhelming infection, especially with Streptococcus pneumoniae and H. influenzae type b as a result of defective splenic function, is the major cause of death in children with SCD under the age of 5 years. Repeated insults on the splenic sinuses by sickled cells result in impaired filtration and function, which allows the development of septicemia and possibly subsequent death.
Diagnostic Evaluation
Although SCA has been reported during the neonatal period and early part of infancy, it may not be recognized until the toddler or preschool period, during a crisis precipitated by an acute upper respiratory tract or gastrointestinal infection. However, early diagnosis (before 3 months of age) facilitates initiation of appropriate interventions to minimize complications. Several specific tests detect abnormal hemoglobin in the homozygous or heterozygous form of the disease.
Examination of a stained blood smear may reveal a few sickled RBCs. Because the erythrocyte assumes its normal discoid shape under adequate oxygenation, however, no sickled cells may be present even in the homozygous form of the disease. Whenever sickle cells are found, diagnostic test results are usually positive for SCA, not sickle cell trait.
For screening purposes the sickle turbidity test (Sickledex) is performed on anticoagulated blood, which is mixed with a special solution. Because HgbS is normally much less soluble than HgbA or HgbF (as well as other variants), when it is combined with this solution, it forms a cloudy or turbid mixture. All other forms of hemoglobin result in a clear suspension. This test is a reliable screening method that uses blood from a finger or heel stick and yields accurate results in 3 minutes. If the test result is positive, however, hemoglobin electrophoresis is necessary to distinguish between those children with the trait and those with the disease.
In hemoglobin electrophoresis, the blood is specially prepared and separated into various hemoglobins by high-voltage electrophoresis. The resulting pattern of the separated peptides as it appears on paper is referred to as fingerprinting of the protein. This test is accurate, rapid, and specific for detecting the homozygous and heterozygous forms of the disease, as well as the percentages of the various hemoglobins.
Screening of Newborns: Screening for SCD in the newborn period can identify children with hemoglobinopathies. Screening of newborns for SCD is performed in the majority of states, the District of Columbia, Puerto Rico, and the Virgin Islands (Driscoll, 2007; Pack-Mabien and Haynes, 2009). It provides early identification of these children before complications develop. Early diagnosis facilitates parental education regarding the importance of immunizations, penicillin prophylaxis, detection of splenomegaly, and the need to report fever and increasing pallor, all of which may be lifesaving (DeBaun and Vichinsky, 2007; Heeney and Dover, 2009).
The death rate from splenic sequestration and septicemia has decreased. Teach parents to palpate the child’s spleen and seek medical attention at the first sign of complications (DeBaun and Vichinsky, 2007; Heeney and Dover, 2009; Pack-Mabien and Haynes, 2009). Penicillin prophylaxis is started by 2 months of age, and parents are instructed to seek medical attention if their child develops a fever of 38.3° C (101° F) or higher (see Evidence-Based Practice box).
Therapeutic Management
The aims of therapy are (1) to prevent the sickling phenomenon, which is responsible for the pathologic sequelae; and (2) to treat the medical emergency of sickle cell crisis. The successful achievement of these aims depends on prompt nursing interventions and medical therapies, child and family preventive measures, and innovative treatment interventions.
Three general forms of treatment are available: supportive-symptomatic, specific, and curative. Hydroxyurea is the only effective drug approved by the U.S. Food and Drug Administration (FDA) to reduce the incidence of recurrent severe painful episodes and acute chest syndrome by increasing the concentration of HgbF and ultimately to reduce complications (DeBaun and Vichinsky, 2007; Pack-Mabien and Haynes, 2009). Recent research is investigating the use of hydroxyurea to prevent stroke (Gulbis, Haberman, DuFour, et al, 2005; Heeney and Ware, 2008). Hematopoietic stem cell transplantation (HSCT) is the only potential for cure of SCD with a high risk of neurologic complications (DeBaun and Vichinsky, 2007; Driscoll, 2007).
Medical management of a crisis is directed at supportive, symptomatic, and specific treatments. The main objectives are to provide (1) bed rest to minimize energy expenditure and to improve oxygen utilization; (2) hydration through oral and IV therapy; (3) electrolyte replacement, since hypoxia results in metabolic acidosis, which also promotes sickling; (4) analgesia for severe pain from vasoocclusion (see pp. 1432-1433); (5) blood replacement to treat anemia and to reduce the viscosity of the sickled blood; and (6) antibiotic therapy to treat any existing infection.
Administration of pneumococcal, H. influenzae type b, and meningococcal vaccines is recommended for these children because of their susceptibility to infection from functional asplenia. (See Immunizations, Chapter 12.) Oral penicillin prophylaxis is recommended by 2 months of age to reduce the chance of pneumococcal sepsis (see Evidence-Based Practice box, p. 1430). The nurse assumes an important role in helping the family comply with a medication regimen and seek medical attention immediately when the child has a fever of 38.3° C (101° F) or higher, an increase in spleen size, or severe pallor (Akinyanju, Otaigbe, and Ibidapo, 2005; Driscoll, 2007; Pack-Mabien and Haynes, 2009).
Oxygen therapy is of little therapeutic value unless the patient is hypoxic (Heeney and Dover, 2009). Oxygen administration is usually not effective in reversing sickling or reducing pain because the oxygen is not able to reach the enmeshed sickled RBCs through the clogged vessels (Chiocca, 1996; Perkins, 2001). In addition, prolonged administration of oxygen can depress bone marrow activity, which further aggravates the anemia (Khoury and Grimsley, 1995).
The use of blood transfusions is another important component of care. An RBC transfusion is used in aplastic, hyperhemolytic, and splenic sequestration crises; in stroke prevention; and before general surgery. Exchange transfusion is a successful, rapid method of reducing the number of circulating sickle cells and therefore slowing down the vicious circle of hypoxia, tissue ischemia, and injury. It is used in acute chest syndrome and after a stroke to prevent recurrence and further tissue damage. Routine transfusions to maintain the hemoglobin value between 9 and 10 g/dl in children with central nervous system disease can minimize the chances of further neurologic problems. In the event of major surgery, exchange or partial exchange transfusions may be given preoperatively to prevent anoxia and suppress the formation of new sickle cells. A simple packed RBC transfusion to raise the hemoglobin value to 10 g/dl, as well as maintenance hydration preoperatively and postoperatively, is sufficient to prevent sickling complications secondary to anesthesia (DeBaun and Vichinsky, 2007; Heeney and Dover, 2009; Vichinsky, Haberkern, Neumayr, et al, 1995). However, sickle cell patients with a higher surgical risk, which may include a history of pulmonary disease, previous acute chest syndrome, stroke, or multiple hospitalizations, should undergo exchange transfusion or multiple simple transfusions to reduce the chance of SCD complications by decreasing the level of HgbS to less than 30%.
Multiple transfusions carry the risk of transmission of viral infection, hyperviscosity, transfusion reactions, alloimmunization, and hemosiderosis (Hankins, Jeng, Harris, et al, 2005; Heeney and Dover, 2009). To reduce iron overload, chelation therapy may be started (see p. 1438). Historically, deferoxamine (Desferal), as an effective parenteral chelator, was administered at 30 to 40 mg/kg over 8 to 10 hours for 5 or 6 nights per week. Deferoxamine is now used either alone or in combination with an oral chelator. Deferasirox (approved by the FDA in 2004) and deferiprone (not been approved by the FDA, but approved in more than 40 other countries) are two new oral iron chelators used worldwide alone or in combination with deferoxamine (Cunningham, Sankaran, Nathan, et al, 2009).
The appropriate time for beginning treatment is controversial, but chelation is often initiated when the ferritin level is higher than 1000 ng/ml or after a year or more of three or four weekly transfusions. Because the ferritin level is also an acute phase reactant, it is recognized as an ineffective or inaccurate test to determine iron overload (DeBaun and Vichinsky, 2007; Olivieri, 1999). In contrast, testing of a liver biopsy specimen allows an accurate and direct measurement of iron concentration but is not without risk of hemorrhage, infection, and discomfort (Cunningham, Sankaran, Nathan, et al, 2009; Olivieri, 1999). Emerging noninvasive alternatives for liver biopsy based on the magnetic properties of iron are known as magnetic susceptometry and superconducting quantum interference devices. Both devices provide an accurate measurement of hepatic iron (Brown, Subramony, May, et al, 2009; DeBaun and Vichinsky, 2007). However, neither of these specialized devices is readily available to the majority of centers.
Children with HgbSS disease and HgbS-β0-thalassemia have the highest risk of stroke and should be monitored with transcranial Doppler ultrasonography (TCD) (Driscoll, 2007; Heeney and Dover, 2009). TCD is a cost-effective, noninvasive ultrasound technique that screens for stroke risk in children who have SCD. TCD is performed yearly on children with SCD from 2 to 16 years of age and measures the intracranial vascular flow within the large cerebral arteries (Bulas, 2005; DeBaun and Vichinsky, 2007; Platt, 2006). The recommended treatment for children with confirmed abnormal TCD findings is chronic transfusion therapy (DeBaun and Vichinsky, 2007; Armstrong-Wells, Grimes, Sidney, et al, 2009). Multiple transfusions carry the risk of transmission of viral infection, hyperviscosity, transfusion reactions, alloimmunization, and hemosiderosis, which must be discussed with the parents or legal guardian before the initiation of a transfusion program.
In children with recurrent life-threatening splenic sequestration, splenectomy may be a lifesaving measure. However, the spleen usually atrophies on its own through progressive fibrotic changes (functional asplenia) by 6 years of age in children with SCA. Surgical splenectomy or autosplenectomy has several benefits because the spleen is the major site of sickling, sequestration, and destruction of RBCs.
Prognosis: The prognosis varies, but most patients live into their fifth decade. The greatest risk is usually in children younger than 5 years of age, and the majority of deaths in these children are caused by overwhelming infection. As the child grows older, however, the crises usually become less severe and less frequent, although death in early adulthood is not uncommon. Consequently SCA is a chronic illness with a potentially terminal outcome. Physical and sexual maturation are delayed in adolescents with SCA. Although adults achieve normal height, weight, and sexual function, the delay may present problems to the adolescent (Heeney and Dover, 2009; Redding-Lallinger, and Knoll, 2006). In some young people chronic pain becomes a significant problem. HSCT offers a curative approach for some children with SCD with an event-free survival of 95% (Driscoll, 2007; Haining, Duncan, and Lehmann, 2009).
Children and adolescents younger than 16 years of age who have severe complications (stroke, recurrent acute chest syndrome, or refractory pain) and have a human leukocyte antigen (HLA)–matched donor available are the best candidates for transplantation (DeBaun and Vichinsky, 2007; Haining, Duncan, and Lehmann, 2009). Eventually, improved survival with other HSCT modalities, such as umbilical cord blood transplantation, haploidentical transplants, and nonmyeloablative conditioning regimens, may augment sibling donor protocols and widen the availability of HSCT as a potential cure to SCD patients (Driscoll, 2007).
Nursing Care Management
Many nurses are involved in SCA screening programs to identify persons with the abnormal hemoglobin so that therapy can be implemented for homozygotes and genetic counseling provided for heterozygotes. The nurse should seek medical attention immediately for young children who exhibit any of the signs previously described and whose families belong to a racial or geographic group know to be at risk for SCD.
Nursing Care Plan—The Child with Sickle Cell Disease
Assessment of the child in sickle cell crisis includes all areas and systems that can be affected by circulatory obstruction, including vital signs; neurologic signs; vision; hearing; and the respiratory, gastrointestinal, renal, and musculoskeletal systems. It is also important to identify the location and intensity of pain.
Minimize Tissue Deoxygenation: Anything that increases cellular metabolism also results in tissue hypoxia. For the child, minimization of tissue deoxygenation includes (1) taking frequent rest breaks during physical activities; (2) avoiding contact sports if the spleen is enlarged, because rupture will cause massive internal hemorrhage; (3) avoiding environments with low oxygen concentration, such as high altitudes or nonpressurized airplane flights; and (4) avoiding known sources of infection. If the child has even a mild infection, the parents must seek medical attention at once.
Promote Hydration: The nurse emphasizes the importance of adequate hydration to prevent sickling and delay the vasoocclusion and hypoxia-ischemia cycle. The nurse calculates the child’s fluid requirements (approximately 1600 ml/m2/day), which is the minimum daily fluid intake. The nurse also assesses the child’s usual fluid consumption to evaluate its adequacy and makes adjustments based on this knowledge. It is not sufficient to advise parents to “force fluids” or “encourage drinking.” They need specific instructions on how many glasses or bottles of fluid are required. Many foods are also a source of fluid, particularly soups, popsicles, yogurt, ice cream, sherbet, gelatin, and puddings.
Encourage children to drink by giving them a special cup, thermos, or water bottle with a straw from which to drink throughout the day. The nurse advises parents to take advantage of times of thirst, such as on awakening or after playing; to serve frequent small portions; and to leave the cup within easy reach for self-service. Flavored ice pops and crushed ice “slurpies” are sources of fluid commonly accepted by children.
Because the kidneys’ ability to concentrate urine is impaired, the child is especially prone to dehydration. Dilute urine or urine of low specific gravity is no longer a valid sign of adequate hydration. Parents should observe for other indications of fluid loss, such as dry mucous membranes, dry diapers, weight loss, and a sunken fontanel in infants. In addition, without the ability to conserve water by concentrating urine, the child is prone to dehydration from environmental factors, particularly overheating. The nurse alerts parents to the need for the child to wear proper indoor and outdoor clothing and avoid excessive exposure to the sun.
Increased fluid intake combined with impaired kidney function result in the problem of enuresis. Parents who are unaware of this fact frequently employ the usual measures to discourage bed-wetting, such as limiting fluids at night, and may resort to punishment and shaming to force bladder control. The nurse discusses this problem with the parents, stressing that the child’s ability to concentrate urine is impaired. Reminding the child to urinate frequently during the day is helpful, and waking the child during the night may prove beneficial if the child’s sleep patterns are not disturbed. Parents who are toilet training their toddlers should be aware of the more frequent pattern of urination and increased difficulty in learning control. Enuresis is treated as a complication of the disease to alleviate parental pressure on the child and to prevent any fluid restriction.
Minimize Crises: Because infection is the major cause of death due to the body’s inability to resist infection, the nurse stresses to parents the importance of adequate nutrition, frequent medical supervision, proper hand washing, and isolation from known sources of infection. Keep in mind that children also need to live a normal life. Overprotection can be as devastating emotionally as an infection is physically. Parents need to be aware of the need to seek prompt medical care at the first sign of any infection.
Teach the family the signs and symptoms of crises and advise them to seek medical attention immediately when any are present. Teaching parents spleen palpation for earlier detection of splenic sequestration can reduce mortality from this serious complication.
Promote Supportive Therapies: The success of many of the medical therapies relies heavily on nursing implementation. Management of pain is an especially difficult problem and often involves experimenting with various analgesics, including opioids, and various schedules before relief is achieved. Unfortunately, these children tend to be undermedicated, which results in “clock watching” and demands for additional doses sooner than might be expected. Often this incorrectly raises suspicion of drug addiction, when in fact the problem is one of inadequate pain control (see Family-Centered Care box). In choosing and scheduling analgesics, the goal is prevention of pain.
 FAMILY-CENTERED CARE
FAMILY-CENTERED CARE
Fear of Addiction
Although the pain during a sickle cell crisis is usually severe and opioids are needed, many families fear that their child will become addicted to the narcotic. Unfortunately, misinformed health professionals may foster this unfounded fear, which results in needless suffering. Extremely few children who receive opioids for severe pain become behaviorally addicted to the drug (Gribbons, Zahr, and Opas, 1995; Howard and Davies, 2007). Families and older children, especially adolescents, need to be reassured that opioids are medically indicated, high doses may be needed, and children rarely become addicted.
Pain is the most common and debilitating symptom experienced by patients with SCD (Driscoll, 2007; Shaiova and Wallenstein, 2004) (see Fig. 35-4). Beyer (2000) found that 15 of 21 children with SCD who were receiving IV pain medication for a painful crisis continued to report moderate to severe pain. Recommendations for continuous adjustment of analgesics were emphasized in this study.
The chronic nature of this pain can greatly affect the child’s development. A multidisciplinary approach is best for its management. When mild to moderate pain is reported, acetaminophen (Tylenol) or ibuprofen is initially used. If acetaminophen or ibuprofen alone is not effective, codeine can be added. The dosages of the drugs are titrated (adjusted) to a therapeutic level. Opioids such as immediate- and sustained-release morphine, oxycodone, hydromorphone, and methadone are administered parenterally or orally for severe pain and are given around the clock. Patient-controlled analgesia (PCA) has been used successfully for sickle cell–related pain. PCA reinforces the patient’s role and responsibility in managing the pain and provides flexibility, since pain may vary in severity over time. If PCA devices are not available or delayed in set-up, a recent study reported intranasal diamorphine was safely combined with oral morphine to manage initial sickle cell pain in most pediatric cases until commencement of IV opiates (Telfer, Lahoz, Ali, et al, 2009).
DRUG ALERT
Meperidine
Meperidine (pethidine [Demerol]) is not recommended. Normeperidine, a metabolite of meperidine, is a central nervous system stimulant that produces anxiety, tremors, myoclonus, and generalized seizures when it accumulates with repetitive dosing. Patients with SCD are particularly at risk for normeperidine-induced seizures (Howard and Davies, 2007; National Institutes of Health, 2002).
Medication given by mouth can be as effective as IV medication when equianalgesic dosages are prescribed.
The nurse should combine any pain management program with psychologic support to help the child deal with the depression, anxiety, and fear that accompany the disease. This includes regular visits with the child to discuss his or her concerns during the hospitalization and positive reinforcement of adaptive coping skills, such as successful methods of dealing with the pain and compliance with treatment prescriptions. To reduce the negative connotation associated with the term crisis, it is best to say “pain episode.”
Frequently heat to the affected area is soothing. Cold compresses are not applied to the area because doing so enhances vasoconstriction and occlusion. Bed rest is usually well tolerated during a crisis, although the actual rest obtained depends a great deal on pain alleviation and the use of organized schedules of nursing care. Although the objective of bed rest is to minimize oxygen consumption, some activity, particularly passive range-of-motion exercises, is beneficial to promote circulation. Usually the best course is to let children determine their activity tolerance.
If blood transfusions or exchange transfusions are given, the nurse has the responsibility of observing for signs of transfusion reaction (see Table 35-2). Because hypervolemia from too rapid transfusion can increase the workload of the heart, the nurse also must be alert to signs of cardiac failure.
In splenic sequestration, gently measure the size of the spleen, since increasing splenomegaly is an ominous sign. A decrease in the size of the spleen denotes response to therapy. The nurse also closely monitors vital signs and blood pressure to detect impending shock. Anemia is typically not a presenting complication in VOCs but is a critical problem in other types of crisis. The nurse monitors for evidence of increasing anemia and institutes appropriate nursing intervention.
Oxygen administration is not beneficial in vasoocclusive episodes unless hypoxemia is present (Heeney and Dover, 2009). It does not reverse sickled RBCs, and if used in a nonhypoxic patient, it decreases erythropoiesis (Vichinsky and Styles, 1996). Because prolonged oxygen therapy can aggravate the anemia, report any signs of lack of therapeutic benefit, such as restlessness, increased pallor, and continued pain.
Record intake, especially of IV fluids, and output. The child’s weight should be taken on admission, since it serves as a baseline for evaluating hydration. Because diuresis can result in electrolyte loss, the nurse observes for signs of hypokalemia and should be familiar with normal serum electrolyte values to report changes. Nurses also need to be aware of the signs of chest syndrome and stroke, both potentially fatal complications (see Nursing Alert, p. 1434).
Decrease Surgical Risks: The main surgical risk is hypoxia from anesthesia. However, emotional stress, the demands of wound healing, and infection potentially increase the sickling phenomenon, both in children with the disease and in those with the trait. The primary nursing objectives are to minimize each of these threats preoperatively and postoperatively by keeping the child well hydrated, preparing the child psychologically, and preventing infection.
Encourage Screening and Genetic Counseling: Screening is recommended during the neonatal period, since early diagnosis allows earlier, more prevention-oriented treatment, such as prophylactic antibiotic therapy and parent education about potential complications. The advantages of trait identification lie in selective reproduction of offspring not afflicted with HgbS. Alternate methods of childbearing include artificial insemination, adoption, and abortion of affected fetuses. However, for some these alternatives are unacceptable.
To be effective, combine screening with genetic counseling and long-term follow-up. The nurse can be instrumental in such programs by conducting parent education sessions, providing follow-up for the family in the home, disseminating correct information about the disease and trait to the community, and rendering support to parents of children newly diagnosed with the trait or the disease. A primary consideration in genetic counseling is informing parents of the 25% chance with each pregnancy of having a child with the disease when both parents carry the trait. (See Chapter 5.)
Prenatal diagnosis is possible through amniocentesis or fetoscopy and fetal blood sampling during the sixteenth week of gestation. Analysis of amniotic cells for a DNA fragment associated with the gene responsible for sickled β-globulin chain synthesis can be performed as early as the twelfth week with chorionic sampling. In the event the fetus is affected, the decision regarding termination of the pregnancy should be left to the couple.
Explain the Disease: Because SCA may be recognized when the child is a toddler, most of the nurse’s counseling is directed at the parents. The nurse explains to parents the basic effect of tissue hypoxia on RBCs and the effect of sickling on the circulation (see Fig. 35-3). Taking time to establish a sound basis of understanding regarding why certain measures are beneficial to the child encourages parents to practice them.*†
The nurse advises the parents to inform all treating practitioners of the child’s condition. The use of a medical identification bracelet is another way of ensuring awareness of the disease. Some people view such identification as “negative labeling.” The nurse can stress the benefits of displaying this information, especially in emergencies when anesthesia may be required.
Support the Family: Families need the opportunity to discuss their feelings regarding transmitting a potentially fatal, chronic illness to their child. Some parents are able to cope with this fact; some feel great guilt and remorse for giving their child the disease, whereas others regret not knowing that they carried the trait. For many parents decision making regarding subsequent pregnancies is fraught with doubt and ambivalence.
Because of the widely publicized prognosis for children with SCA, many parents express their fear of death. The prognosis varies; with early diagnosis and treatment these children are living longer. The predictors of a severe course of SCA are a low hemoglobin level (approximately 7 g/dl), dactylitis or painful episode, and elevated WBC count exhibited before the age of 24 months (DeBaun and Vichinsky, 2007; Miller, Sleeper, Pegelow, et al, 2000). The nurse should care for the family as for any family with a child who has a chronic and life-threatening illness, and give some consideration to the siblings’ reactions, the stress on the marital relationship, and the childrearing attitudes displayed toward the child (see Nursing Care Plan). (See Chapters 22 and 23.)
 NURSING CARE PLAN
NURSING CARE PLAN