Onset
The onset of leukemia varies from acute to insidious. In most instances the child displays remarkably few symptoms. For example, leukemia may be diagnosed when a minor infection, such as a cold, fails to completely disappear. The child continues to be pale, listless, irritable, febrile, and anorexic. Parents often suspect some underlying problem when they observe the weight loss, petechiae, bruising without cause, and continued complaints of bone and joint pain.
At other times leukemia is diagnosed after an extended history of signs and symptoms mimicking such conditions as rheumatoid arthritis or mononucleosis. In some cases the diagnosis of leukemia accompanies some totally unrelated event, such as a routine physical examination or injury.
The history not only yields valuable medical information regarding the subsequent course of the illness, but also bears heavily on the parents’ emotional reaction to the diagnosis. In most instances the diagnosis is an unexpected revelation of catastrophic proportion.
Staging and Prognostic Factors
The most important prognostic factors in determining long-term survival for children with ALL are the initial white blood cell count, the patient’s age at diagnosis, cytogenetics, the immunologic subtype, and the child’s sex (Table 36-3).
TABLE 36-3
FAVORABLE PROGNOSTIC FACTORS FOR ACUTE LYMPHOBLASTIC LEUKEMIA
| FACTOR | CRITERIA |
| Leukocyte count | <50,000/mm3 |
| Age | 2-10 yr |
| Immunologic subtype | CALLA-positive, early pre–B-cell |
| FAB morphology | L1 |
| Cytogenetics | Hyperdiploid (>50 chromosomes, DNA index >1.16); trisomies 4 and 10 and translocations t(12/21) (p21/q22) |
| Sex | Female |
| Leukemia cell burden | Minimal |
CALLA, Common acute lymphocytic leukemia antigen; DNA, deoxyribonucleic acid; FAB, French-American-British (classification system).
For children with AML, prognostic factors associated with a poorer prognosis include certain chromosome abnormalities (monosomy 7), a high white blood cell count (100,000/mm3), and AML developing after a myelodysplastic syndrome. The absence of Auer rods in the M1 subtype of AML has also been correlated with a low remission rate (Smith, Hasle, Cooper, 2011).
From the time of diagnosis, the nurse has some idea of the expected course the disease will follow. However, in some instances, because of the variety of cell types observed and the marked undifferentiation of immature cells, a definitive classification cannot be made or the diagnosis may be changed. Be aware of the importance of such events in counseling and supporting family members.
Diagnostic Evaluation
Leukemia is usually suspected from the history; physical manifestations; and a peripheral blood smear that contains immature forms of leukocytes, frequently in combination with low blood counts. Definitive diagnosis is based on bone marrow aspiration or biopsy. Typically the bone marrow shows a monotonous infiltrate of blast cells. Once the diagnosis is confirmed, an LP is performed to determine whether there is any CNS involvement, although a small number of children have CNS involvement and most are asymptomatic.
Therapeutic Management
Treatment of leukemia involves the use of IV and intrathecal chemotherapeutic agents. Cranial radiation is sometimes used for resistant CNS disease or testicular relapse. Typically leukemia treatment is divided into phases: (1) induction, which achieves a complete remission or clinical disappearance of leukemic cells; (2) intensification, or consolidation, therapy, which further decreases the total tumor burden; and (3) maintenance, which consists of further chemotherapy to ensure the disease stays in remission. Although the combination of drugs and possibility of irradiation may vary according to the institution, the patient’s prognostic or risk characteristics, and the type of leukemia being treated, the following general principles for each phase are consistently employed.
Remission Induction: Almost immediately after confirmation of the diagnosis, induction therapy is begun and lasts for 4 to 6 weeks (Margolin, Rabin, Steuber, et al, 2011). The principal drugs used for induction in ALL are the corticosteroids (prednisone or dexamethasone), vincristine, and l-asparaginase, with or without doxorubicin. Oral steroids are administered daily in divided doses to maintain consistently high blood levels. Vincristine is given by IV infusion once a week for a total of four to six doses, and l-asparaginase or doxorubicin is given at various schedules. A complete remission is determined by the absence of clinical signs or symptoms of the disease and the presence of less than 5% blast cells in the bone marrow. With AML the drug therapies differ from those used for lymphoblastic leukemia. The principal drugs used for induction therapy in AML are doxorubicin or daunomycin and cytosine arabinoside; various other drugs may be added.
Because many of the drugs also cause myelosuppression of normal blood elements, the period immediately after a remission can be critical. The body is defenseless against invading organisms (especially normal bacterial flora) and highly susceptible to spontaneous hemorrhage. Consequently, supportive therapy during this time is essential.
Intensification, or Consolidation, Therapy: Intensification, or consolidation, therapy is used to further decrease the number of leukemic cells in the child’s body. Intensification therapy incorporates some of the following agents: l-asparaginase, high-dose methotrexate or intermediate-dose methotrexate with leucovorin rescue, vincristine, doxorubicin, steroids, cytarabine, intramuscular or oral methotrexate, and 6-mercaptopurine. The intensification phase consists of pulses of these agents given periodically during the first 6 months of treatment. The specific agents used for intensification therapy depend on the type of leukemia and the child’s risk factors.
Maintenance: The goal of maintenance therapy is to preserve remission and further reduce the number of leukemic cells. Combined drug regimens have been more successful in maintaining remissions and preventing drug resistance. A variety of agents are used during maintenance therapy, including a daily dose of oral 6-mercaptopurine, weekly doses of methotrexate, and intermittent pulses of steroids and vincristine, which are standard in most treatment regimens.
During maintenance therapy, weekly or monthly complete blood counts are taken to evaluate the marrow’s response to the drugs. If myelosuppression becomes severe (usually indicated by an ANC <1000/mm3), or if toxic side effects occur, therapy is temporarily stopped or the dose decreased.
Central Nervous System Prophylactic Therapy: Children with leukemia are at risk for invasion of the CNS by the leukemic cells. For this reason, all children receive CNS prophylactic therapy. Before the 1980s children with ALL received cranial-spinal irradiation. Because of the concern regarding late effects of cranial irradiation and secondary malignancies, this mode of therapy is now generally reserved for high-risk patients or those with resistant CNS disease. Depending on protocol, intrathecal methotrexate or triple intrathecal chemotherapy (consisting of methotrexate, cytarabine, and hydrocortisone) is used during induction, intensification, and maintenance therapy to prevent CNS disease.
Duration of therapy has been based on clinical experience comparing survival rates for various time intervals and is concerned with preventing deleterious effects of excessive treatment. Although the optimum time for discontinuing therapy is not known, current practice is to continue treatment for 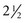 to 3 years. All children after cessation of therapy require regular medical evaluation for surveillance of relapse and long-term sequelae of treatment. Most relapses (16%) occur during the first year off therapy, about 2% to 3% of the relapses occur during each of the next 3 years, and very few relapses occur after 6 years (Margolin, Rabin, Steuber, et al, 2011).
to 3 years. All children after cessation of therapy require regular medical evaluation for surveillance of relapse and long-term sequelae of treatment. Most relapses (16%) occur during the first year off therapy, about 2% to 3% of the relapses occur during each of the next 3 years, and very few relapses occur after 6 years (Margolin, Rabin, Steuber, et al, 2011).
Reinduction After Relapse: For many children, additional therapy becomes necessary when a relapse occurs, as evidenced by the presence of leukemic cells within the bone marrow. Usually reinduction for ALL includes the use of prednisone and vincristine with a combination of other drugs not previously used. Although remissions may be achieved after more than one relapse, each relapse indicates an increasingly poor prognosis. However, more long-term second and subsequent remissions are occurring, and these may have better outlooks than previously thought.
A site that is resistant to chemotherapy and is responsible for leukemic relapse is the testes. A minority of males experience relapses during maintenance therapy or have occult disease after cessation of therapy. Treatment for testicular disease includes bilateral testicular irradiation, intensive systemic chemotherapy, and CNS prophylactic therapy (Landier, 2001).
Bone Marrow Transplantation: BMT has been used successfully in treating some children with ALL and AML. In general, BMT is not recommended for children with ALL during the first remission because of the excellent results possible with chemotherapy. The group with the best results has been those with ALL who received the graft during the second remission (Bollard, Krance, and Heslop, 2011). Because of the poorer prognosis in children with AML, transplantation may be considered during the first remission when a suitable donor is available (Bollard, Krance, and Heslop, 2011).
Prognosis: The majority of children with newly diagnosed leukemia who receive effective multiagent chemotherapy will survive. More than 80% of the children achieved long-term disease-free survival, and the majority of these children developed no obvious health problems from the leukemia or its treatment (Margolin, Rabin, Steuber, et al, 2011). Prognosis after transplantation varies with the timing of the procedure and the type of leukemia; reported ranges for long-term survival are between 25% and 50% (Bollard, Krance, and Heslop, 2011). However, because many of these children faced almost certain death without transplantation, even these low figures represent a major advance. Still, the use of BMT remains controversial.
Nursing Care Management
Nursing care of the child with leukemia is directly related to the regimen of therapy. Myelosuppression, drug toxicity, and leukemic infiltration cause secondary complications that necessitate supportive physical care. This discussion focuses on supportive interventions for the child with leukemia and the family. General aspects of care appropriate for the child with leukemia are discussed earlier under Nursing Care of the Child with Cancer.
Prepare the Family for Diagnostic and Therapeutic Procedures: From the time before diagnosis to cessation of therapy, children must undergo several tests, the most traumatic of which are bone marrow aspiration or biopsy and LP. Multiple finger sticks and venipunctures for blood analysis and drug infusion are common occurrences for several years after the diagnosis. Therefore the child needs an explanation of why each procedure is done and what can be expected. (See Preparation for Diagnostic and Therapeutic Procedures, Chapter 27.)
Depending on the child’s age, one way of beginning diagnostic preparation is to explain the tests, procedures, and treatment plan.* Using a drawing or letting the child look at a drop of blood under a microscope not only teaches, but also fosters trust between the nurse and the child. It also allows the nurse to assess the child’s level of understanding. An error many health professionals make is to overestimate children’s knowledge about their bodies. For example, a bone marrow aspiration makes sense only when it is clarified that the center of a bone is hollow and contains the cells that later become “working” blood cells or leukemic cells.
Provide Continued Emotional Support: Nursing care of the child with leukemia is based on typical problems the family confronts during the treatment phases. It is not unusual for a child who discontinues therapy after 2 or 3 years and maintains a permanent remission to experience many side effects. Therefore the nurse’s role is one of continual support, guidance, clarification, and judgment. Parents need to know how to recognize symptoms that demand medical attention. Although some of the reactions discussed are expected, parents should still report them to their practitioner. Warning parents of their possible occurrence beforehand also allows parents to prepare. At the same time, it reassures them that these reactions are not caused by a return of leukemic cells.
The nurse must also use judgment in recognizing which side effects are normal reactions and which indicate toxicity. Frequently it is the office or clinic nurse who screens such telephone calls and gives advice, when appropriate. Usually nausea and vomiting are not indications for drug cessation. However, severe vomiting may require immediate intervention to prevent dehydration. Signs of infection, mucosal ulceration, hemorrhagic cystitis, peripheral neuropathy, and constipation require medical evaluation.
Another aspect of continued emotional support involves prognosis. Leukemia is not invariably fatal, but present statistics must be correctly interpreted. Although more than 95% of children with ALL achieve an initial remission and almost 80% of them live 5 years or longer, remember that these are average estimates and apply to those children treated with the most successful protocols since diagnosis (Shusterman and Meadows, 2000). For the low-risk child the chances may be better, but for the high-risk child they may be significantly poorer. Of those who do survive after discontinuing therapy, a portion will relapse. At present only the passage of time is positive confirmation that the child is “cured” of the disease.
The nurse must be familiar with these statistics to interpret them correctly to parents. At the same time, the nurse must realize that a realistic understanding of the chances for survival requires an adjustment period. For example, it is not unusual for parents to interpret the “95% remission” as the probability for a cure. When a relapse occurs, parents may for the first time be able to “hear” the facts.
Statistics are numbers. Sometimes they bring hope, and at other times they bring despair. Although they are important in terms of research, better treatment, and identification of high- or low-risk populations, they present a general picture of what to expect. The nurse who is working with family members must individualize the numbers to relate to the people. An understanding of each member’s emotional needs, as well as competent care of physical ones, is essential to the positive, growth-promoting support of the family. Chapter 23 discusses comprehensive emotional support for the family through all phases of the illness.
 Tumors of the CNS account for about 20% of all childhood cancers and have an annual incidence of 2.4 per 100,000 children under 15 years of age. About 60% of the tumors are infratentorial (below the tentorium cerebelli), which means that they occur in the posterior part of the brain, primarily in the cerebellum or brainstem. This anatomic distribution accounts for the frequency of symptoms resulting from increased ICP. The other tumors are supratentorial or lie within the midbrain structures.
Tumors of the CNS account for about 20% of all childhood cancers and have an annual incidence of 2.4 per 100,000 children under 15 years of age. About 60% of the tumors are infratentorial (below the tentorium cerebelli), which means that they occur in the posterior part of the brain, primarily in the cerebellum or brainstem. This anatomic distribution accounts for the frequency of symptoms resulting from increased ICP. The other tumors are supratentorial or lie within the midbrain structures. 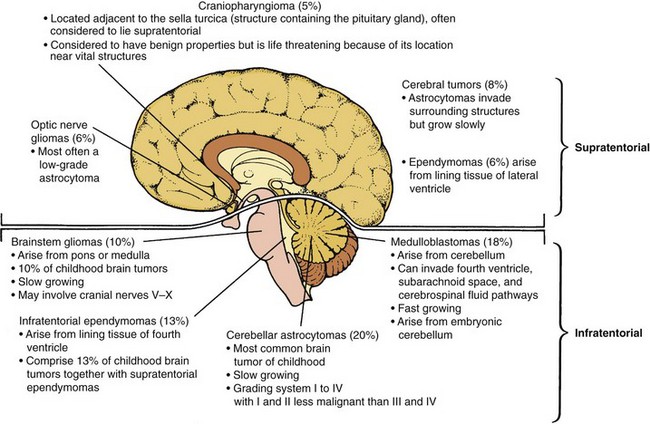
 Case Study—Brain Tumor
Case Study—Brain Tumor

 NURSING ALERT
NURSING ALERT