13 Bacterial pathogenicity
Key points
• Opportunist pathogens require a defect in host defence before they cause disease, whereas primary pathogens affect otherwise healthy individuals. The possession of virulence determinants generally differentiates pathogens from non-pathogens and, in turn, their number and potency separate opportunist from primary pathogens.
• Expression of virulence determinants is carefully regulated and may involve a form of chemical communication between bacteria known as quorum sensing.
• Adhesins are often involved the establishment of an infection. Bacterial adhesion may be mediated by fimbrial or non-fimbrial adhesins and generally involves interactions with host cell surface receptors or surface associated proteins such as fibronectin.
• Invasive pathogens gain entry and spread either by subverting host uptake mechanisms or by tissue disruption. Once established, pathogens must deploy a variety of mechanisms to avoid host defences.
• Multiplication within host tissues requires specific mechanisms to gain essential nutrients such as iron. Many pathogens make siderophores that compete with the host’s high-affinity iron transport and storage systems.
• Host damage may be direct, via the release of toxins, or indirect, via the effects of the host’s innate and adaptive immune responses (immunopathology).
• Endotoxins and exotoxins are recognized. Endotoxin is synonymous with the lipopolysaccharide or lipo-oligosaccharide of Gram-negative bacteria, and sufficient amounts elicit a cascade of responses leading to endotoxic shock.
• Exotoxins are proteins that cause damage or dysfunction by signalling at host cell membranes (type I), by damaging membranes (type II) or by entering target cells and directly altering function (type III).
Pathogenicity, or the capacity to cause disease, is a relatively rare quality among microbes. It requires the attributes of transmissibility or communicability from one host or reservoir to a fresh host, survival in the new host, infectivity or the ability to breach the new host’s defences, and virulence, a variable that is multifactorial and denotes the capacity of a pathogen to harm the host. Virulence (see Ch. 14) in the clinical sense is a manifestation of a complex parasite–host relationship in which the capacity of the organism to cause disease is considered in relation to the resistance of the host.
Types of bacterial pathogen
Bacterial pathogens can be classified into two broad groups, opportunists and primary pathogens. Both groups have a broad spectrum of virulence capabilities, hence their overlap. In addition, some pathogens are capable of infecting a wide range of host species; such pathogens are referred to as zoonoses, whereas other non-zoonotic pathogens are highly adapted to a single species or a closely related group of species. Both zoonotic and non-zoonotic pathogens can be either opportunist or primary pathogens and organisms which exist as harmless commensals in one species may be pathogens in a second species.
Opportunistic pathogens
These rarely cause disease in individuals with intact immunological and anatomical defences. Only when such defences are impaired or compromised, as a result of congenital or acquired disease, by the use of immunosuppressive therapy or surgical techniques, are these bacteria able to cause disease. Many opportunistic pathogens (e.g. coagulase-negative staphylococci) are part of the normal human flora, carried on the skin or mucosal surfaces where they cause no harm and may actually have a beneficial effect by preventing colonization by other potential pathogens. However, the introduction of these organisms into anatomical sites where they are not normally found, or removal of competing bacteria by the use of broad-spectrum antibiotics, may allow their localized multiplication and subsequent development of disease.
Primary pathogens
These bacteria are capable of establishing infection and causing disease in previously healthy individuals with intact immunological defences. However, they may more readily cause disease in individuals with impaired defences.
The above classification is applicable to the vast majority of pathogens. However, there are exceptions and variations within both categories of bacterial pathogens. Different strains of any bacterial species can vary in their genetic make-up and virulence. For example, the majority of Neisseria meningitidis strains are harmless commensals and are considered to be opportunistic bacteria; however, some hypervirulent clones of the organism can cause disease in the previously healthy individual. Conversely, people vary in their genetic make-up and susceptibility to invading bacteria, including meningococci.
Zoonoses and non-zoonotic pathogens
Some pathogens are found in a variety of animals and may be transferred to humans coming into contact with animals directly or indirectly. Familiar examples would include Escherichia coli O157, which are often found in association with cattle and other animals without causing apparent disease in these animals, but can cause gastrointestinal illness, as well as serious complications such as haemolytic uremic syndrome (HUS) when it infects man. By contrast, some pathogens are highly adapted to their host. For example, the pathogenic Nesseria species N. meningitidis and N. gonorrhea have only ever been isolated from human hosts and are not capable of infecting or initiating disease in other animals under normal circumstances.
Virulence determinants
Both opportunistic and primary pathogens possess virulence determinants or aggressins that facilitate pathogenesis. Possession of a single virulence determinant is rarely sufficient to allow the initiation of infection and production of pathology. Many bacteria possess several virulence determinants, all of which play some part at various stages of the disease process. In addition, not all strains of a particular bacterial species are equally pathogenic. For example, although six separate serotypes of encapsulated Haemophilus influenzae are recognized, serious infection is almost exclusively associated with isolates of serotype b. Moreover, even within serotype b isolates, 80% of serious infections are caused by six of more than 100 clonal types.
Different strains of a pathogenic species may cause distinct types of infection, each associated with possession of a particular complement of virulence determinants. Different strains of E. coli, for example, cause several distinct gastrointestinal diseases, urinary tract infections, septicaemia, meningitis and a range of other minor infections (see Ch. 26).
Expression and analysis of virulence determinants
Many pathogens produce an impressive armoury of virulence determinants in vitro. However, relatively early in the study of pathogenesis, it was appreciated that a knowledge of the behaviour of the pathogen in vivo is crucial to an understanding of virulence.
Animal models have been used to compare the virulence of naturally occurring variants differing in the expression of a particular determinant, and have provided much useful information. While some pathogens do not thrive or produce disease in typically available animal models, progress is being made in constructing better models by genetically manipulating the animals, for example, to express human receptors and other proteins important in human infection. Nevertheless, not all human clinical syndromes can be reproduced in animals, and extrapolation from animal studies to man can be misleading. Furthermore, the possibility that observed differences in virulence may be due to additional cryptic phenotypic or genotypic variations cannot always be excluded. Molecular techniques have been used to construct isogenic mutants of bacteria that differ only in the particular determinant of interest, and these constructs have allowed more detailed analysis of the role of such components in pathogenesis. More recently, comparative analysis of bacterial genome sequences have revealed the extent of genetic variation, as well as genetic mobility among bacterial strains.
Most studies of bacterial virulence determinants are by necessity performed in model systems in vitro. However, growth conditions in vitro differ significantly from those found in tissues, and as the expression of many virulence determinants is influenced by environmental factors, it is essential that such studies use cultural conditions that mimic as closely as possible those found in the host and that, where possible, confirmatory evidence is obtained that the phenomena observed actually occur during human infection.
Genetic studies have shown that expression of several different virulence determinants in a single bacterium is sometimes regulated in a coordinated fashion. Iron limitation, the situation encountered in host tissues, is one environmental stimulus that coordinately increases the production of many bacterial proteins, including virulence determinants such as haemolysin of E. coli and diphtheria toxin of Corynebacterium diphtheriae. In other bacteria, such as Staphylococcus aureus and Pseudomonas aeruginosa, some virulence determinants are expressed exclusively or maximally during the stationary phase of growth. Expression of these factors is associated with the production of inducer molecules or pheromones in the bacterial culture that accumulate as the bacteria grow until a threshold level is reached and gene expression is triggered – a process known as quorum sensing. The ability to regulate production of virulence determinants may save energy in situations in which expression is not required (e.g. in the environment) and quorum sensing may be important in establishing a sufficiently large population of bacteria in tissue to guarantee survival of the infecting organism. It is also clear that most organisms express some proteins only when in direct contact with host cells.
Molecular studies have also allowed mechanisms of transmission of virulence determinants to be investigated (see Ch. 6). Virulence determinants encoded by genomic DNA sequences, plasmids, bacteriophages and transposons have been reported. It is interesting that, in nature, these genetic elements can move between related organisms, transfer genes encoding virulence factors (e.g. toxins) horizontally, and transform the recipient bacteria to more adapted or more virulent pathogens. Apart from these genetic elements, there are other mechanisms by which some bacteria can exchange virulence genes. For example, neisseriae recognize and take up DNA fragments that contain specific sequences (uptake sequences) and incorporate them into their own genomes. In this way they can either vary the structure of an existing gene or, in the process, acquire a new set of genes. The genomes of many bacterial pathogens have been fully sequenced. The data reveal that several bacteria have acquired very large stretches of foreign DNA (often called pathogenicity islands) that contain virulence-related genes. This further demonstrates that the microbial population consists of a vibrant, kinetic and highly interactive community. In this community, bacteria evolve continuously and new pathogens, or old pathogens with newly acquired capabilities, may emerge as a result.
Establishment of infection
Potential pathogens may enter the body by various routes, including the respiratory, gastrointestinal, urinary or genital tract. Alternatively, they may enter tissues directly through insect bites, or by accidental or surgical trauma to the skin. Many opportunistic and several primary pathogens are carried as part of the normal human flora, which acts as a ready source of infection in the compromised host. For many primary pathogens, however, transmission to a new host and establishment of infection are more complex processes. Transmission of respiratory pathogens, such as Bordetella pertussis, may require direct contact with infectious material, as the organism cannot survive for any length of time in the environment. Sexually transmitted pathogens such as Neisseria gonorrhoeae and Treponema pallidum have evolved further along this route, and require direct person-to-person mucosal contact for transmission. Man is the only natural host for these pathogens, which die rapidly in the environment. The source of infection may be individuals with clinical disease or subclinically infected carriers, in whom symptoms may be absent or relatively mild either because the disease process is at an early stage or because of partial immunity to the pathogen.
In contrast, for many gastrointestinal pathogens such as Salmonella, Shigella and Campylobacter species, the primary source is environmental, and infection follows the ingestion of contaminated food or water. Many of these organisms also infect other animals, often without harmful effect, and these act as a reservoir of infection and source of environmental contamination (see Ch. 1).
Colonization
For many pathogenic bacteria, the initial interaction with host tissues occurs at a mucosal surface and colonization – the establishment of a stable population of bacteria in the host – normally requires adhesion to the mucosal cell surface. This allows the establishment of a focus of infection, which may remain localized or may subsequently spread to other tissues. Adhesion is necessary to avoid innate host defence mechanisms such as peristalsis in the gut and the flushing action of mucus, saliva and urine which removes non-adherent bacteria. For invasive bacteria, adhesion is an essential preliminary to penetration through tissues. Successful colonization also requires that bacteria are able to acquire essential nutrients, such as iron, for growth.
Adhesion
Adhesion involves surface interactions between specific receptors on the mammalian cell membrane (carbohydrates, proteins or glycolipids) and ligands (usually proteins or glycoproteins) on the bacterial surface. The presence or absence of specific receptors on mammalian cells contributes significantly to tissue specificity of infection. Non-specific surface properties of the bacterium, including surface charge and hydrophobicity, also contribute to the initial stages of the adhesion process. Several different mechanisms of bacterial adherence have evolved, all utilizing specialized cell surface organelles or macromolecules that help to overcome the natural forces of repulsion that exist between the pathogen and its target cell.
Fimbrial adhesins
Electron microscopy of the surface of many Gram-negative and some Gram-positive bacteria reveals the presence of numerous thin, rigid, rod-like structures called fimbriae, or pili, that are easily distinguishable from the much thicker bacterial flagella (see p. 18). Fimbriae are involved in mediating attachment of some bacteria to mammalian cell surfaces. Different strains or species of bacteria may produce different types of fimbriae, which can be identified on the basis of antigenic composition, morphology and receptor specificity (Table 13.1). A broad division can be made between fimbriae, in which adherence in vitro is inhibited by D-mannose (mannose-sensitive fimbriae) and those unaffected by this treatment (mannose-resistant fimbriae).
Table 13.1 Examples of fimbriae produced by Gram-negative pathogens
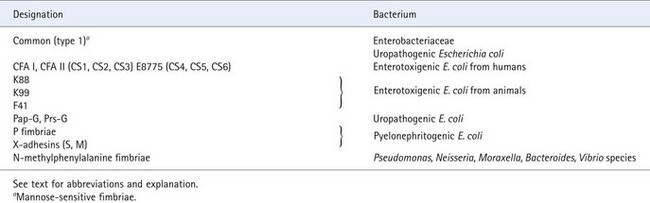
The antigenic composition of fimbriae can be complex. For instance, two fimbrial antigens called colonization factor antigen (CFA) I and II have been detected in enteropathogenic E. coli strains. CFA-II consists of three distinct fimbrial antigens designated as coli surface (CS) antigens 1, 2 and 3. Another E. coli strain, E8775, has been found to produce three other CS antigens, CS4, CS5 and CS6. Pyelonephritogenic E. coli isolates produce a group of adhesins called X-adhesins; two fimbrial types designated S and M on the basis of receptor specificity have been identified in this group. The genes encoding a second type of fimbrae, the Chaperone Usher (CU) fimbriae, also found in E. coli, are found as multiple homologous, and often cryptic, variants, potentially with specificities for different carbohydrate receptors.
The evolutionary significance of such heterogeneity may be that the ability of an individual bacterium to express several different types of fimbriae allows different target receptors to be used at different anatomical sites of the infected host. In vitro, production of fimbriae is influenced by cultural conditions such as incubation temperature and medium composition, which may switch off the production of fimbriae or induce a phase change from one fimbrial type to another.
For some fimbriae the association with infection is clear. Thus the K88 fimbrial antigen is clearly associated with the ability of E. coli K88 to cause diarrhoea in pigs; pigs lacking the appropriate intestinal receptors are spared the enterotoxigenic effects of E. coli strains of this type. In many other instances the association between production of fimbriae and infection remains putative at present. Production of fimbriae is controlled by either chromosomal or plasmid genes.
The structure of one of these fimbrial types – type 1 or common fimbriae – has been studied in detail. These consist of aggregates of a structural protein subunit called fimbrillin (or pilin) arranged in a regular helical array to produce a rigid rod-like structure 7 nm in diameter, with a central hole running along its length.
A highly conserved minor protein, located at both the tip and at intervals along the length of the fimbriae, mediates specific adhesion. Type 1 fimbriae bind specifically to D-mannose residues. Their role in vivo remains controversial; however, they may be involved in the pathogenesis of urinary tract infections.
Other Gram-negative bacteria, including those of the genera Pseudomonas, Neisseria, Bacteroides and Vibrio, produce fimbriae that share some homology, especially in the amino-terminal region of the fimbrillin subunits (the so-called N-methylphenylalanine fimbriae). These fimbriae have been shown to act as virulence determinants for Ps. aeruginosa and N. gonorrhoeae.
Non-fimbrial adhesins
Non-fimbrial adhesins include protein or polysaccharide structures that are surface exposed on the bacterial cell and/or secreted. Protein-based adhesins include the filamentous haemagglutinin of B. pertussis, a mannose-resistant haemagglutinin from Salmonella enterica serotype Typhimurium and a fibrillar haemagglutinin from Helicobacter pylori. Outer membrane proteins are involved in the adherence of many, if not most, pathogenic bacteria.
Exopolysaccharides present on the surface of some Gram-positive bacteria are also involved in adhesion. For example, Streptococcus mutans, which is involved in the pathogenesis of dental caries, synthesizes a homopolymer of glucose that anchors the bacterium to the tooth surface and contributes to the matrix of dental plaque. Actinomyces may adhere to other oral bacteria – a process called co-aggregation. Teichoic acid and surface proteins of coagulase-negative staphylococci mediate adherence of the bacterium to prosthetic devices and catheters, contributing to increasing numbers of hospital-acquired infections. Continued growth following attachment to these biomaterials may result in formation of a biofilm (see Ch. 4), which may hinder successful antibiotic treatment by restricting access of drugs to the bacterium.
Flagella act as adhesins in Vibrio cholerae and Campylobacter jejuni. Bacterial motility is also thought to be important in chemotaxis of these organisms and H. pylori towards intestinal cells, and in penetration of these bacteria through the mucous layer during colonization.
Binding to connective tissue proteins
Binding of pathogenic bacteria to a number of connective tissue proteins, including laminin, vitronectin and collagen, have been described. The best-studied of these host proteins is fibronectin, a complex multifunctional glycoprotein found in plasma and associated with mucosal cell surfaces, where it promotes numerous adhesion functions. Many pathogenic bacteria bind fibronectin at the bacterial surface, and for some organisms fibronectin has been shown to act as the cell surface receptor for bacterial adhesion. In Streptococcus pyogenes, lipoteichoic acid mediates attachment of the bacterium to the amino terminus of the fibronectin molecule. Attachment of S. aureus to cell surfaces also involves the amino terminus of fibronectin, but the bacterial ligand appears to be protein in this instance. T. pallidum also binds fibronectin. The significance of the interaction with fibronectin in the pathogenesis of syphilis and many other bacterial diseases needs further clarification.
Consequences of adhesion
In addition to preventing loss of the pathogen from the host, adhesion induces structural and functional changes in mucosal cells, and these may contribute to disease. For example, adherence of enteropathogenic E. coli (EPEC) to epithelial cells induces rearrangements of the cell cytoskeleton causing loss of microvilli and localized accumulation of actin, without subsequent invasion of the host cell. In contrast, adherence of H. pylori to gastric epithelial cells causes enhanced production of the pro-inflammatory chemokine interleukin (IL)-8, which contributes to gastric pathology. In both cases, these changes involve the induction of intracellular signalling pathways triggered after binding of the bacteria to specific receptors on the epithelial cell surface. Adhesion of bacteria to mammalian cells may also induce changes in bacterial gene expression.
Invasion
Once attached to a mucosal surface, some bacteria exert their pathogenic effects without penetrating the tissues of the host: toxins, other aggressins and induction of intracellular signalling pathways mediate tissue damage at local or distant sites. For a number of pathogenic bacteria, however, adherence to the mucosal surface represents but the first stage of the invasion of tissues. Examples of organisms that are able to invade and survive within host cells include mycobacteria and those of the genera Salmonella, Shigella, Escherichia, Yersinia, Legionella, Listeria, Campylobacter and Neisseria. Cell invasion confers the ability to avoid humoral host defence mechanisms and potentially provides a niche rich in nutrients and devoid of competition from other bacteria. However, the survival of bacteria in professional phagocytes, such as macrophages or polymorphonuclear leucocytes, depends on subverting intracellular killing mechanisms that would normally result in microbial destruction (see below). For some bacteria (e.g. Neisseria meningitidis), penetration through or between epithelial cells allows dissemination from the initial site of entry to other body sites.
Uptake into host cells
The initial phase of cellular invasion involves penetration of the mammalian cell membrane; rather than eliciting mechanisms for host cell penetration themselves, many intracellular pathogens induce phagocytic processes in the host cell to gain access.
Shigellae invade colonic mucosal cells but rarely penetrate deeper into the host tissues. Inside the cell, they are surrounded by a membrane-bound vesicle derived from the host cell. Soon after entry, this vesicle is lysed by the action of a plasmid-encoded haemolysin (haemolysis is just one way of detecting a membrane-damaging toxin), and the bacteria are released into the cell cytoplasm. Listeria monocytogenes produces a heat shock protein with a similar function, termed listeriolysin. Once free in the cell cytoplasm, shigellae multiply rapidly with subsequent inhibition of host cell protein synthesis. Several hours later the host cell dies, and bacteria spread to adjacent cells where the process of invasion is repeated.
Other bacteria remain within membrane-bound vesicles but modify these cellular compartments, preventing them from maturing and fusing with lysosomes. The pathogenic Neisseria, for example, elaborate a secreted toxin, immunoglobulin A protease, which in addition to cleaving immunoglobulin A during colonization of the mucosa (see below), is also capable of cleaving a key endocytic vesicle protein, LAMP1, which is responsible for acidification of the endosome. In this way the intracellular pathogen modifies the environment within the vesicle to favour its own survival. Most salmonellae proceed through the superficial layers of the gut and invade deeper tissues, in particular cells of the reticulo-endothelial system such as macrophages. Salmonellae also occupy a host-derived vesicle that does not lyse. In this case several vesicles coalesce to form large intracellular vacuoles. These vacuoles traverse the cytoplasm to reach the opposite side of the cell and initiate spread to adjacent cells and deeper tissues. Although invasion of host cells is essentially a bacterium-directed but a host-mediated process, the active participation of the bacterium and novel bacterial protein synthesis is needed.
Role of cell receptors
The availability of specific receptors defines the types of host cell that are involved. As a result, some pathogens can invade a wide range of cell types, whereas others have a more restricted invasive potential. Specific host receptors for some of the invasive pathogens have been identified. For example, Legionella pneumophila and Mycobacterium tuberculosis adhere to complement receptors on the surface of phagocytic cells. A specific receptor for Yersinia pseudotuberculosis belongs to a family of proteins termed integrins that form a network on the surface of host cells to which host proteins such as fibronectin can bind. Mimicry of the amino acid sequence (Arg-Gly-Asp) of fibronectin that mediates attachment to the integrins may represent a common mechanism of effecting intracellular entry. The ability to utilize integrins may not be restricted to intracellular bacteria. The filamentous haemagglutinin of B. pertussis may use the fibronectin integrin to mediate attachment in the respiratory tract.
A number of bacterial pathogens capable of crossing the blood–brain barrier, including N. meningitidis, Haemophilus influenza and Streptococcus pneumonia, as well as a number of neurotropic viruses and prions, have been shown to bind to the laminin receptor protein, suggesting that binding to this host receptor protein confers a neurotropism on these pathogens.
Survival and multiplication
To cause disease, most micro-organisms must survive on an epithelial surface, within a mucosal lumen or within host tissues and, at some stage multiply. Survival depends to a large extent on the organism’s ability to avoid, evade or resist host defences. Multiplication depends on acquiring all of the nutrients necessary for growth; the most extensively studied nutritional challenge to pathogens is iron acquisition.
Avoidance of host defence mechanisms
Colonization by bacterial pathogens, particularly of normally sterile areas of the body, results in the induction of specific and non-specific humoral and cell-mediated immune responses designed to eradicate the organism from the site of infection. Products of the organism that are not normally found within sterile tissues of the host may be chemotactic for phagocytic cells that are attracted to the site. Moreover, complement components may directly damage the bacterium and release peptides chemotactic for phagocytic cells. Other humoral antibacterial factors include lysozyme and the iron chelators transferrin and lactoferrin. Lysozyme is active primarily against Gram-positive bacteria but potentiates the activity of complement against Gram-negative organisms. Transferrin and lactoferrin chelate iron in body fluids, and reduce the amount of free iron to a level below that necessary for bacterial growth.
Pathogenic bacteria have evolved ways of avoiding or neutralizing these highly efficient clearance systems. As most of the interactions between the bacterium and the immune effectors involve the bacterial surface, resistance to these effects is related to the molecular architecture of the bacterial surface layers.
Capsules
Many bacterial pathogens need to avoid phagocytosis; production of an extracellular capsule is the most common mechanism by which this is achieved. Virtually all the pathogens associated with meningitis and pneumonia, including H. influenzae, N. meningitidis, E. coli and S. pneumoniae, have capsules, and non-capsulate variants usually exhibit much reduced virulence. Most capsules are polysaccharides composed of sugar monomers that vary among different bacteria. Polysaccharide capsules reduce the efficiency of host defences in a number of ways:
1. In the absence of specific antibody to the bacterium, the hydrophilic nature of the capsule may hinder uptake by phagocytes, a process that occurs more readily at hydrophobic surfaces. This may be overcome if the phagocyte is able to trap the bacterium against a surface, a process referred to as surface phagocytosis.
2. Capsules prevent efficient opsonization of the bacterium by complement or specific antibody, events that promote interaction with phagocytic cells. Capsules may either prevent complement deposition completely or cause complement to be deposited at a distance from the bacterial membrane where it is unable to damage the organism.
3. Capsules tend to be weakly immunogenic and may mask more immunogenic surface components and reduce interactions with both complement and antibody. In some cases, for example serogroup B N. meningitidis and serotype K1 E. coli, the capsular polysaccharide may mimic host polysaccharides moieties (e.g. brain sialic acid) and be seen as self antigen.
Streptococcal M protein
The M protein present on the surface of S. pyogenes is not a capsule but functions in a similar manner to prevent complement deposition at the bacterial surface. The M protein binds both fibrinogen and fibrin, and deposition of this material on the streptococcal surface hinders the access of complement activated by the alternative pathway.
Meningococcal factor H-binding protein
The complement system is a powerful weapon against bacterial pathogens, but can potentially damage host cells as well. The host must, therefore, protect itself against the damaging activities of complement. The serum glycoprotein factor H is a negative regulator of the complement system that protects host cells by binding to glycosamino glycans present on the surface of host but not to bacterial cells. It can downregulate the activity of complement and thus protect the host cell. N. meningitidis expresses a factor H-binding protein on its surface, which recruits this complement regulator, thus affording similar protection to the bacterial pathogen.
Resistance to killing by phagocytic cells
Some pathogens not only survive within macrophages and other phagocytes, but may actually multiply intracellularly. The normal sequence of events following phagocytosis involves fusion of the phagosome in which the bacterium is contained with lysosomal granules present in the cell cytoplasm. These granules contain enzymes and cationic peptides involved in oxygen-dependent and oxygen-independent bacterial killing mechanisms (see p. 117).
Different organisms use different strategies for survival (Table 13.2). M. tuberculosis is thought to resist intracellular killing by preventing phagosome–lysosome fusion; other bacteria are able to resist the action of such lysosomal components after fusion. Some organisms stimulate a normal respiratory burst but are intrinsically resistant to the effects of the potentially toxic oxygen radicals produced. Production of catalase by S. aureus and N. gonorrhoeae is thought to protect these organisms from such toxic products. The smooth lipopolysaccharide of many bacterial pathogens is also thought to contribute to their resistance to the effects of bactericidal cationic peptides present in the phagolysosome.
Table 13.2 Some strategies adopted by bacteria to avoid intracellular killing
| Species | Method |
|---|---|
| Mycobaeterium tuberculosis | Prevents phagosome–lysosome fusion |
| Salmonella serotype Typhi | Fails to stimulate oxygen-dependent killing |
| Staphylococcus aureus | Produces catalase to negate effect of toxic oxygen radicals |
| Pathogenic Neisseria species | Inhibits phagosome–lysosome acidification |
Antigenic variation
Variation in surface antigen composition during the course of infection provides a mechanism of avoidance of specific immune responses directed at those antigens. This strategy is most highly developed in blood-borne parasitic protozoa, such as trypanosomes, but is also exhibited by bacteria. Pathogenic Neisseria, for example, are capable of changing surface antigens using three highly efficient mechanisms. These are mutation of individual amino acids, phase variation (switching genes on and off) and horizontal exchange of DNA material. N. meningitidis can avoid the killing effect of antibodies against its major porin (PorA) by mutating amino acids and/or acquiring parts of or all of its porA gene from another meningococcal strain. The organism can switch off the expression of its capsule or its immunogenic proteins by shifts in the nucleotide sequence encoding them. The latter varies as a result of recombination or mutation during DNA replication.
Another mechanism of antigen variation in Neisseria is the genetic rearrangements demonstrated in the fimbriae. Usually only one complete fimbrillin gene is expressed, although there may be several incomplete ‘silent’ gene sequences present on the chromosome. Movement of parts of the incomplete gene sequences, either from within the genome of the expressing strain or from DNA released by a co-infecting strain to an expression locus, results in synthesis of a protein that may differ antigenically from the original.
The borreliae that cause relapsing fever use a similar strategy to generate antigenic variation in their outer surface proteins. Other bacteria show strain-specific antigenic variability, for example group A streptococci produce up to 75 antigenically distinct serotypes of M protein.
The capacity for variation in surface antigens allows for longer survival of an individual organism in a host and means that antibody produced in response to infection by one strain of a pathogen may not protect against subsequent challenge with a different strain of that bacterium. This makes the variable antigens elusive targets for protective antibodies, and the development of vaccines based on inhibition of attachment or generation of opsonic or bactericidal antibodies is particularly difficult for these organisms.
Immunoglobulin A proteases
Several species of pathogenic bacteria that cause disease on mucosal surfaces produce a protease that specifically cleaves immunoglobulin A (IgA), the principal antibody type produced at these sites. These proteases are specific for human IgA isotype I. Nearly all of the pathogens causing meningitis possess an IgA protease and a polysaccharide capsule enabling them to persist on the mucosal surface and resist phagocytosis during the invasive phase of the disease.
Serum resistance
To survive in the bloodstream, bacteria must be able to resist lysis as a result of deposition of complement on the bacterial surface. In the Enterobacteriaceae, resistance is primarily due to the composition of the lipopolysaccharide present in the bacterial outer membrane. Smooth colonial variants that possess polysaccharide ‘O’ side chains are more resistant than rough colonial variants that lack such side chains (see below). The side chains sterically hinder deposition of complement components on the bacterial surface. Conversely, however, some O-chain polysaccharides activate complement by an alternative pathway leading to lysis of the bacterial cell. In N. meningitidis group B and E. coli K1, sialic acid capsules prevent efficient complement activation and, in N. gonorrhoeae, complement binds but forms an aberrant configuration in the bacterial outer membrane so that it is unable to effect lysis.
Iron acquisition
The concentration of free iron in body fluids and secretions is below that required for bacterial growth because it is bound (chelated) by high-affinity mammalian iron-binding proteins such as transferrin and lactoferrin. To multiply in body fluids or on mucous membranes bacterial pathogens have evolved efficient mechanisms for scavenging iron from mammalian iron-binding proteins. Bacteria such as E. coli, Klebsiella pneumoniae and some staphylococci produce extracellular iron chelators called siderophores for this purpose. Others, including Campylobacter jejuni and N. meningitides, do not produce siderophores themselves but are able to obtain iron from siderophores produced by other species or use host molecules such as noradrenalin, which has siderophore-like activity, as a source of iron. In an alternative strategy pathogens including N. meningitidis, Haemophilus parainfluenzae, H. influenzae type b, Staphylococcus epidermidis and S. aureus have specific receptors for transferrin and/or lactoferrin, on their surfaces, and are able to bind these proteins and remove the bound iron directly from these host proteins. Production of siderophores, their cell surface receptors, and receptors for transferrin, lactoferrin and other mammalian iron-binding proteins is iron-regulated and occurs mainly under conditions of iron restriction.
Two other mechanisms of iron acquisition from mammalian iron chelators have been described. Some Bacteroides species remove iron by proteolytic cleavage of the chelator. In L. monocytogenes, reduction of the Fe3+ ion to Fe2+ reduces the affinity for the chelator sufficiently for it to be removed by the bacterium.
Many bacteria express receptors for binding and/or internalizing other mammalian iron-containing molecules, such as haem, haemoglobin and haemoglobin–haemopexin complexes. These mammalian molecules are located intracellularly, where they may be available to intracellular pathogens; they may also be released by bacterial haemolysins that lyse the red cells.
Damage or dysfunction
In order for an infection to become apparent there must be sufficient host damage or dysfunction for the individual to become symptomatic. It is particularly important to appreciate that there are generally two components to this: the direct effect of the organism and the host response. In many cases a largely immune-mediated damage (immunopathology) can predominate. This may reflect an excessive innate response as in septic shock (see below), or the adaptive response as in tuberculosis (see Ch. 18). The most obvious means by which bacteria cause direct host damage or dysfunction is by the production of toxins.
Toxins
In many bacterial infections part or all the characteristic pathology of the disease is caused by toxins. Toxins may exert their pathogenic effects directly on a target cell or may interact with cells of the immune system resulting in the release of immunological mediators (cytokines) that cause pathophysiological effects (see Chs 8, 9 and 12). Such effects may not always lead to the death of the target cell but may selectively impair specific functions. Substances that have toxic physiological effects on target cells in vitro do not necessarily exert the same effects in vivo, but a number of toxins have been shown to be responsible for the typical clinical features of bacterial disease.
Two broad categories of toxin have been described: endotoxin, which is a component of the outer membrane of Gram-negative bacteria, and exotoxins, which are produced extracellularly by both Gram-negative and Gram-positive bacteria.
Endotoxin
Endotoxin, also called lipopolysaccharide or lipo-oligosaccharide, is a component of the outer membrane of Gram-negative bacteria and is released from the bacterial surface via outer membrane vesicles (blebs), which may be released from the bacterial cell surface, or by lysis and disintegration of the organism. Lipopolysaccharide is anchored into the bacterial outer membrane through a unique molecule termed lipid A (Fig. 13.1). Covalently linked to lipid A is an eight-carbon sugar, ketodeoxyoctonate, in turn linked to the chain of sugar molecules (saccharides) that form the highly variable O antigen structures of Gram-negative bacteria. On solid bacteriological media, bacteria carrying lipopolysaccharide with O antigen form smooth colonies with hydrophilic surfaces; in contrast those carrying lipopolysaccharide without the O antigen form rough colonies with hydrophobic surfaces.
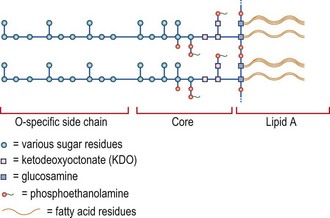
Fig. 13.1 Diagrammatic representation of the structure of bacterial lipopolysaccharide.
After Rietschel ET, Galanos C, Lüderlitz O 1975 Structure, endotoxicity and immunogenicity of the lipid A component of bacterial lipopolysaccharide. In: Schlessinger D (ed.) Microbiology, pp. 307–314. American Society for Microbiology, Washington, DC.
The term endotoxin was originally introduced to describe the component of Gram-negative bacteria responsible for the pathophysiology of endotoxic shock, a syndrome with a high mortality rate, particularly in immunocompromised or otherwise debilitated individuals. Endotoxin activates complement via the alternative pathway, but most of the biological activity of the molecule is attributable to lipid A. Both endotoxin and lipid A are potent activators of macrophages, resulting in the induction of a range of cytokines involved in the regulation of immune and inflammatory responses (see Ch. 9). Although endotoxin from Gram-negative organisms remains central to our understanding of septic shock, other structural and secreted components of bacteria that interact with pattern recognition receptors (see Ch. 9) can contribute to the pathogenesis of this clinical syndrome. Thus the multi-system effects, often involving complement, blood clotting factor and kinin activation together with extensive cytokine release, should not be seen as exclusive to Gram-negative infection.
Exotoxins
Exotoxins, in contrast to endotoxin, are diffusible proteins secreted into the external medium by the pathogen. Most pathogens secrete various protein molecules that facilitate adhesion to, or invasion of, the host. Many others cause damage to host cells. The damage may be physiological, for example cholera toxin promotes electrolyte (and fluid) excretion from enterocytes without killing the cells, or pathological, where the toxin (e.g. diphtheria toxin) inhibits protein synthesis and induces cell death. Exotoxins vary in their molecular structure, biological function, mechanism of secretion and immunological properties. The list of bacterial exotoxins is vast and increasing; however, they are often classified by their mode of action on animal cells:
• Type I (membrane acting) toxins bind surface receptors and stimulate transmembrane signals, and include the super-antigenic toxins.
• Type II (membrane damaging) toxins directly affect membranes, forming pores or disrupting lipid bilayers.
• Type III (intracellular effector) toxins translocate an active enzymatic component into the cell and modify an intracellular target molecule.
Examples of exotoxins and their effects on target cells are shown in Table 13.3. Bacteria secrete toxins and other proteins using a number of distinct mechanisms that are understood to varying extents. Some of these systems are listed in Gram-negative bacteria as types I–VI, although this number does not reflect the total number of pathways that are known to mediate secretion of proteins across the bacterial envelope. Figure 13.2 shows the basic components of the types I and V pathways which are relatively well characterized. In type I, at least three proteins get together to form a channel through which large molecules (such as haemolysin of E. coli) are exported. In type V, however, a single precursor protein that consists of three domains is secreted sequentially across the inner and outer membranes. Although these latter proteins are called autotransporters, it is now known that both secretion steps require the activity of a number of additional cellular proteins. A typical example of an autotransporter is the IgA1 protease of Neisseria spp.
Table 13.3 Some effects of bacterial exotoxins
| Toxic effect | Examples |
|---|---|
| Lethal action | |
| Effect on neuromuscular junction | Clostridium botulinum toxin A |
| Effect on voluntary muscle | Tetanus toxin |
| Damage to heart, lungs, kidneys, etc. | Diphtheria toxin |
| Pyrogenic effect | |
| Increase in body temperature and polyclonal T cell activation | Super-antigenic exotoxins of Staphylococcus aureus and Streptococcus pyogenes (e.g. staphylococcal toxic shock syndrome toxin 1) |
| Action on gastrointestinal tract | |
| Secretion of water and electrolytes | Cholera and Escherichia coli enterotoxins |
| Pseudomembranous colitis | Clostridium difficile toxins A and B |
| Bacillary dysentery | Shigella toxin (Shiga toxin) |
| Vomiting | Staphylococcus aureus enterotoxins A–E |
| Action on skin | |
| Necrosis | Clostridial toxins; staphylococcal α-toxin |
| Erythema | Diphtheria toxin; streptococcal erythrogenic toxin |
| Permeability of skin capillaries | Cholera enterotoxin; E. coli heat-labile toxin |
| Nikolsky signa | Staphylococcus aureus epidermolytic toxin |
| Cytolytic effects | |
| Lysis of blood cells | Staphylococcus aureus α-, β- and δ-lysins; leucocidin Streptolysin O and S Clostridium perfringens α and θ toxins |
| Inhibition of metabolic activity | |
| Protein synthesis | Diphtheria toxin; shiga toxin |
a Separation of epidermis from dermis.
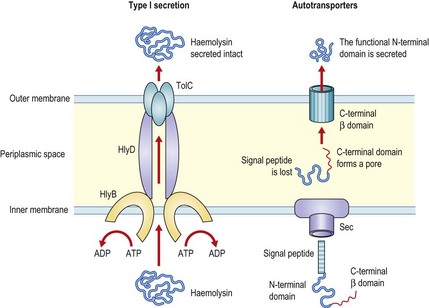
Fig. 13.2 Diagrammatic representation of type I and type V (autotransporter) secretion of exotoxins across the bacterial cell membrane. ADP, adenosine diphosphate; ATP, adenosine triphosphate; Hly, haemolysin; Tol, special receptor.
Enterotoxins cause symptoms of gastrointestinal disease, including diarrhoea, dysentery and vomiting. In some cases the disease is caused by ingestion of preformed toxin in food, but in most cases colonization of the intestine is required before toxin is made.
Cholera toxin and heat-labile toxins of enterotoxigenic E. coli (ETEC) do not induce inflammatory changes in the intestinal mucosa, but perturb the processes that regulate ion and water exchange across the intestinal epithelium (see Chs 26 and 30). In contrast, the enterotoxins of Clostridium difficile, C. perfringens type A and Bacillus cereus cause structural damage to epithelial cells, resulting in inflammation. Another gastrointestinal pathogen, enteropathogenic E. coli (EPEC), mediates damage by a process that involves secretion of a protein, Tir, directly across both bacterial membranes and the host cell membrane in a single step. Tir is subsequently inserted into the host cell membrane where it acts as a receptor for a bacterial adhesin, intimin. Binding of intimin to Tir results in phosphorylation of the latter protein; this in turn leads to a signalling cascade and cytoskeletal rearrangements within the host cell.
B. pertussis, the causative agent of whooping cough, produces various extracellular products, including a tracheal cytotoxin that inhibits the beating of cilia on tracheal epithelial cells, pertussis toxin, which exhibits several systemic effects, and an adenylate cyclase that interferes with phagocyte function.
Another group of toxins causes damage to subepithelial tissues following penetration and multiplication of the pathogen at the site of infection. Many of these toxins also inhibit or interfere with components of the host immune system. Membrane-damaging toxins such as staphylococcal α and β toxins, streptolysin O and streptolysin S, and C. perfringens α and θ toxins inhibit leucocyte chemotaxis at subcytolytic concentrations, but cause necrosis and tissue damage at higher concentrations.
Systemic effects of toxins
Some toxins cause damage to internal organs following absorption from the focus of infection. Included in this category are the toxins causing diphtheria, tetanus and botulism, and those associated with streptococcal scarlet fever, staphylococcal toxic shock syndrome and haemolytic uremic syndrome associated with Shiga toxin-producing enteropathogens. The diphtheria toxin, the gene for which is bacteriophage-encoded, inhibits protein synthesis in mammalian cells. Tetanus toxin, in contrast, exerts its effect by preventing the release of inhibitory neurotransmitters whose function is to prevent overstimulation of motor neurones in the central nervous system, resulting in the convulsive muscle spasm characteristic of tetanus. Diphtheria and tetanus toxins represent the sole determinant of disease and are neutralized by specific antitoxin antibody. As a result, vaccination with toxoids (formalin-inactivated toxins) derived from these toxins is highly effective (see Ch. 70).
Botulism results from the ingestion of preformed toxin produced by Clostridium botulinum in food contaminated with this bacterium, and is not a true infectious disease. The toxic activity is due to a family of serologically distinct polypeptide neurotoxins that prevent release of acetylcholine at neuromuscular junctions, resulting in the symptoms of flaccid paralysis. These toxins have been used clinically in treating squints and muscle spasm.
Other toxins cause disseminated multi-system organ damage. Such pathology is seen in staphylococcal toxic shock syndrome caused by certain strains of S. aureus that produce a toxin designated toxic shock syndrome toxin 1 (TSST-1). This toxin belongs to a group of functionally related proteins collectively referred to as superantigens, which include the staphylococcal enterotoxins, staphylococcal exfoliative toxin and streptococcal pyrogenic exotoxin A. These molecules are potent T cell mitogens whose reactivity with lymphocytes induces cytokine release; they may initiate tissue damage by mechanisms similar to those postulated to account for Gram-negative endotoxic shock (see p. 165).
Other extracellular aggressins
Many bacteria secrete a range of enzymes that may be involved in the pathogenic processes.
Proteus spp. and some other bacteria that cause urinary tract infections produce ureases that break down urea in the urine, and the release of ammonia may contribute to the pathology. The urease produced by the gastric and duodenal pathogen H. pylori is similarly implicated in the virulence of the organism. L. pneumophila produces a metalloprotease thought to contribute to the characteristic pathology seen in legionella pneumonia.
Many other degradative enzymes, including mucinases, phospholipases, elastases collagenases and hyaluronidases, are produced by pathogenic bacteria. Many non-pathogenic bacteria and opportunistic pathogens also produce such enzymes; in most cases their role in pathogenesis requires further clarification.
An understanding of the basic mechanisms of pathogenesis is important for the design of new or improved vaccines and appropriate therapies. Such knowledge is also invaluable in the analysis of ‘new’ bacterial pathogens that are recognized from time to time. However, for some bacterial diseases, for example syphilis, such approaches have still not defined the mechanisms of pathogenesis or the virulence determinants involved, and new strategies employed by such successful pathogens may yet be discovered.
Alouf JE, Freer JH. Bacterial Protein Toxins, ed 2. London: Academic Press; 1999.
Hacker J, Hessemann J. Molecular Infection Biology: Interaction Between Microorganisms and Cells. Hoboken, NJ: John Wiley, 2000.
Lamont R, ed. Bacterial Invasion of Host Cells (Advances in Molecular and Cellular Microbiology). Cambridge University Press, 2004.
Locht C, Simonet M. Bacterial Pathogenesis: Molecular and Cellular Mechamisms. Caister Academic Press, 2012.
Mims C, Nash A, Stephen J. Mims’ Pathogenesis of Infectious Disease, ed 5. London: Academic Press; 2000.
Salyers AA, Whitt DD. Bacterial Pathogenesis: A Molecular Approach, ed 3. Washington: ASM Press; 2011.
Williams P, Ketley J, Salmond G. Bacterial Pathogenesis, Vol 28. London: Academic Press. 1998.