Chapter 11 Haemopoietic and Lympho-Reticular Tissues
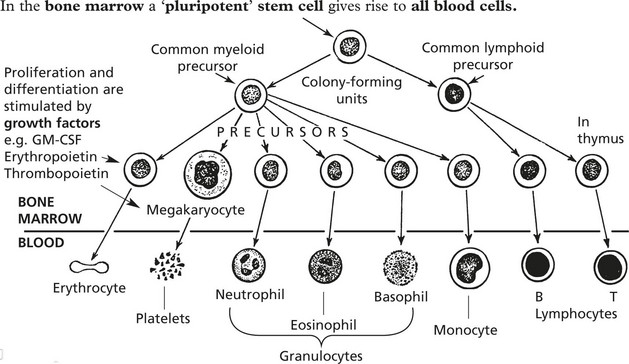
Haematology – Laboratory Tests
Investigation of blood diseases depends on examination of 1. peripheral blood and 2. bone marrow.
Blood count: this is normally done by sophisticated electronic machines.
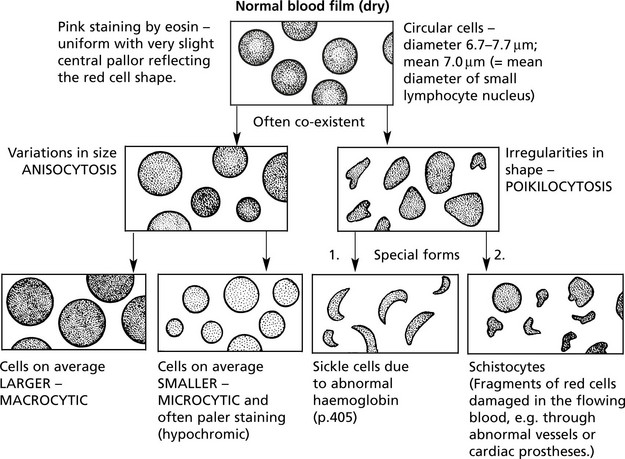
BLOOD FILM – stained by a Romanowski method.
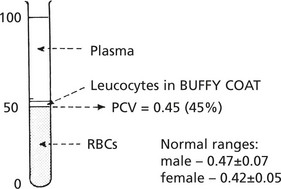
Another useful test is the measurement of the PACKED CELL VOLUME (PCV) or haematocrit.
This is obtained by centrifuging anticoagulated whole blood in a haematocrit tube.
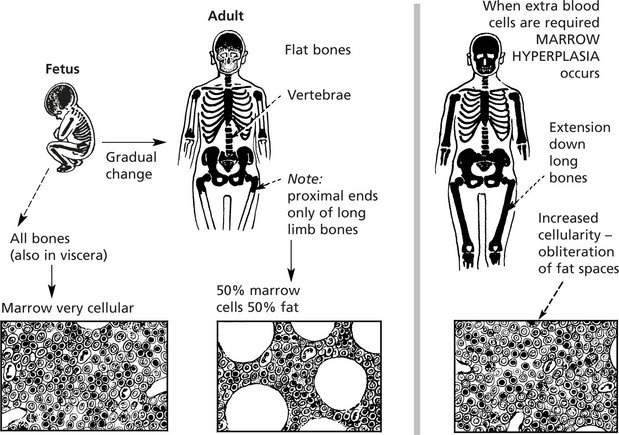
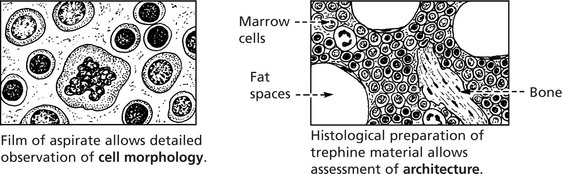
Examination of bone marrow is important in explaining abnormalities of the peripheral blood.
In the past, aspiration of marrow from the sternum was commonly performed. Now the posterior iliac crest is used – it is safer and allows marrow to be aspirated and a trephine biopsy to be taken.
Anaemia
The most important function of the red cell is the transport of oxygen bound to haemoglobin. The most common and important disorder associated with disease of the red cells is ANAEMIA, which is defined as a reduction below normal of the concentration of haemoglobin in the blood.
Anaemia in men – Hb < 13 g/dl: in women – < 11.5 g/dl.
Clinical Associations
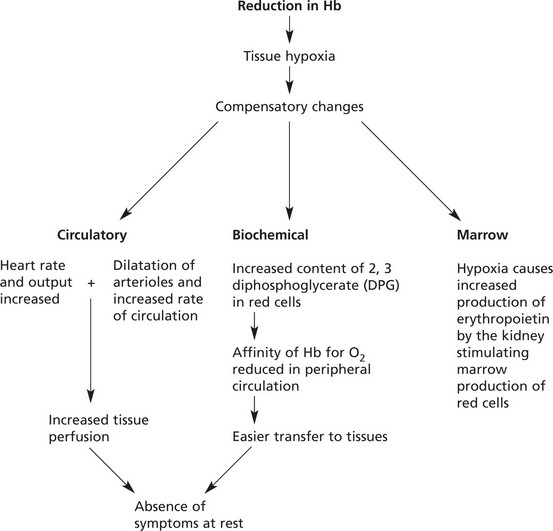
The effects of anaemia depend on its severity, rate of development and duration.
In slowly developing moderate anaemias, symptoms such as dyspnoea only appear on exertion, and even when the haemoglobin falls as low as 6–7 g/dl, clinical features may be slight.
Pathological Complications of Anaemia
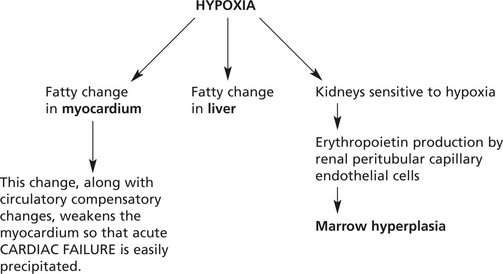
Note:Therefore blood transfusion used in the treatment of anaemia is given slowly and as packed cells to avoid fluid overload.
In very rapidly developing anaemias, the compensatory mechanisms cannot adjust adequately – the condition merges into shock.
Causes of Anaemia
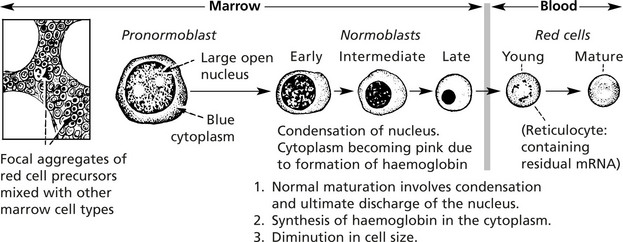
An understanding of the four main mechanisms by which anaemia develops depends on a knowledge of the life-cycle of red blood cells.
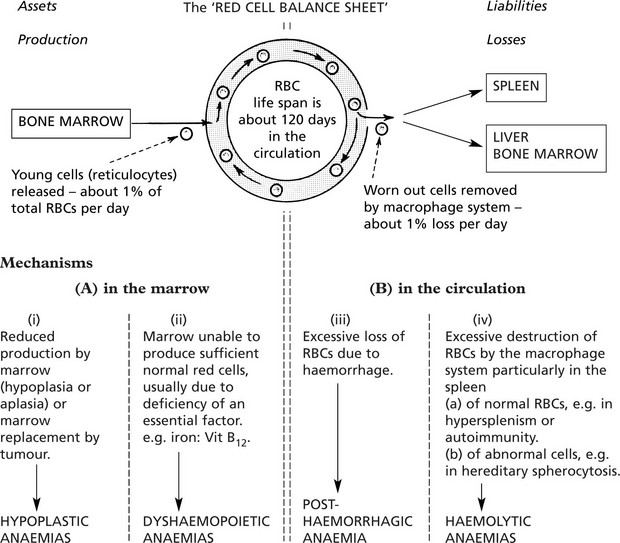
Hypoplastic and Aplastic Anaemias
These are rare conditions and, as the names imply, are due to marrow failure with diminished numbers or absence of haemopoietic cells. Usually all three marrow cell lines are affected, resulting in pancytopenia in the peripheral blood.
Marrow failure of this type is dealt with in detail on page 407.
Marrow failure due to extensive tumour infiltration or fibrosis also occurs.
The anaemias associated with miscellaneous chronic diseases (‘Secondary’ anaemias) are dealt with on page 408.
Dyshaemopoietic Anaemias
The usual cause of these anaemias is deficiency of an essential factor required for proper haemoglobin synthesis or erythroblast maturation and development. They are associated with a hypercellular marrow and are divided into two main groups:
(1) normoblastic and (2) megaloblastic, depending on the type of erythroblastic maturation in the marrow.
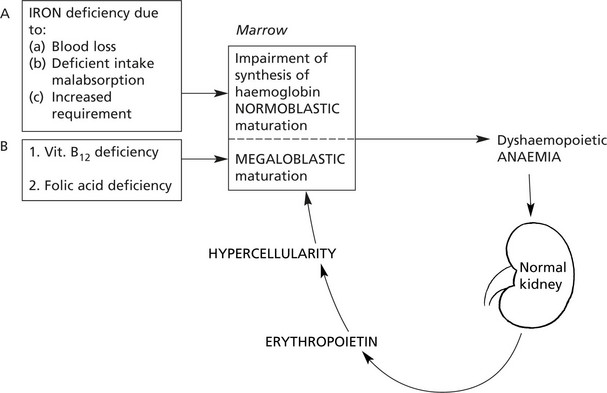
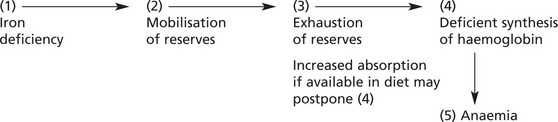
Iron Deficiency Anaemia
IRON DEFICIENCY ANAEMIA is the commonest anaemia on a world basis due to (a) poor nutrition, (b) intestinal parasites (esp. hookworm) causing bleeding and (c) multiple pregnancies.
In Western countries, in the adult male and post-menopausal women, iron deficiency anaemia is nearly always due to gastrointestinal blood loss from cancer, peptic ulceration, aspirin and non-steroidal ingestion, etc. Without IRON the haem component of the haemoglobin molecule cannot be synthesised.

Changes in the blood: The red cells which show:

| Parameter | Low/High | Normal Range |
|---|---|---|
| MCV (mean cell volume) | LOW (< 80) | 80–92fl |
| MCH (mean cell haemoglobin) | LOW (< 27) | 27–32pg |
| MCHC (mean corpuscular haemoglobin concentration) | LOW (< 30) | 33 g/dl |
| Serum IRON | LOW | 10–30 mmol/l |
| Serum FERRITIN | LOW | 15–300 mg/l |
| Serum IRON BINDING CAPACITY | RAISED | 45–70 mmol/l |
Serum IRON SATURATION  |
LOW | 16–60% |
The reticulocyte count is NORMAL except following episodes of haemorrhage. Usually there are no changes in the leucocytes and platelets.
The bone marrow is hypercellular and contains small, poorly haemoglobinised normoblasts; iron stores are reduced.
Iron Metabolism
Iron is absorbed mainly in the duodenum and upper jejunum. Only small amounts are normally required to replace iron losses. Since the average diet contains more iron than is required, its absorption is controlled by the mucosal apoferritin mechanism.
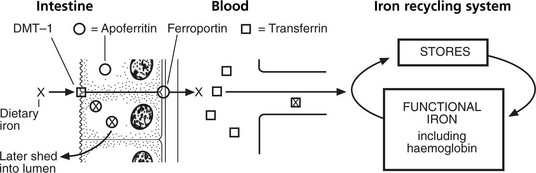
The iron (X) in the diet enters the mucosal cell through an apical uptake transporter (DMT-1) and having combined with apoferritin (O) is retained in the cell as ferritin (⊗).
Iron unbound by apoferritin passes through the cell through a basolateral transporter (ferroportin) and is transported in the blood to join the iron recycling system.
Note: New haemoglobin formed in the bone marrow contains 95% iron from recycling system, 5% from diet.
Note: This bound iron is subsequently shed along with the cell into the lumen.
The state of the iron stores controls the apoferritin (a form of intracellular transferrin) content of the intestinal mucosal cell by a feed-back mechanism.
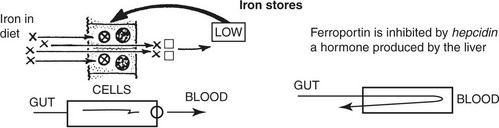
Acute Iron Overload
If a large dose of medicinal iron preparations is taken (particularly by children in error) the absorption and transport mechanisms are overwhelmed and free iron radicals exert very toxic effects.
The iron balance may be summarised as follows.
| INPUT (Adult male) |
BODY IRON Total 3–6g |
OUTPUT |
|---|---|---|
| Average: 1 mg/day derived from foods | (a) Functional iron in haemoglobin, myoglobin, enzyme systems, transferrin  70% at least 70% at least |
Average: 1 mg/day Skin desquamation and miscellaneous secretions |
| The average diet contains 10–20 mg iron, of which about 10% is absorbed. In the adult female, the average daily input is about 2 mg. | (b) Storage iron in liver, spleen, bone marrow as ferritin, haemosiderin  30% or less 30% or less |
Menstruation This extra loss of about 0.5 to 1 mg requires extra input in the female |
Anaemia results when this balance is upset in:
Usually anaemia develops slowly (except in cases of serious haemorrhage).
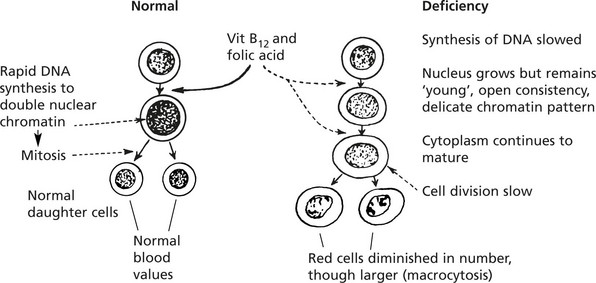
The Megaloblastic Anaemias
These dyshaemopoietic anaemias are almost always caused by deficiency of either vitamin B12 or folic acid which are intracellular co-enzymes, particularly important for the synthesis of DNA.
The effects of deficiency occur in most organs of the body but are prominent where cell turnover is rapid, e.g. in the marrow.

Pernicious Anaemia (PA)
This serious and severe anaemia was first described by the English physician Addison in the mid 19th century. At that time it was invariably fatal, but now is treatable. It is due to vitamin B12 deficiency and is always associated with achlorhydria and gastric mucosal atrophy, due to autoimmune gastritis.
Vitamin B12 metabolism and causes of deficiency:
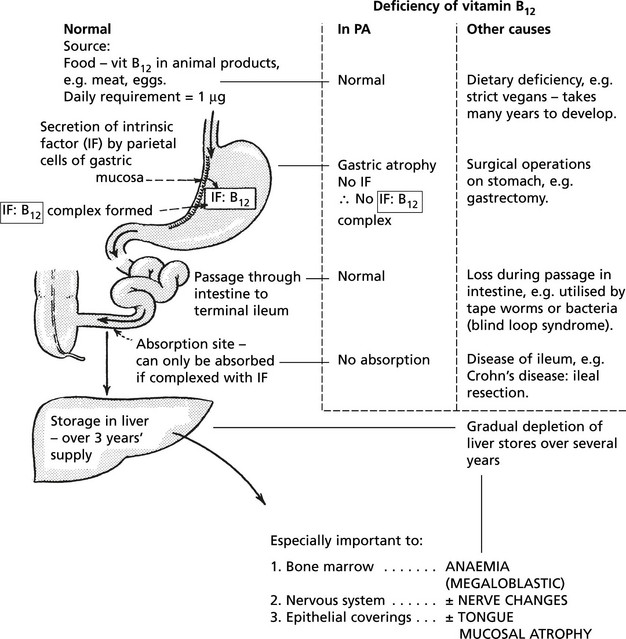
Released into blood stream bound to transcobalamin to act as important co-enzyme in several intracellular synthetic pathways but particularly of DNA.
Mechanism of Production of Gastritis
PA is an autoimmune disease. The gastric atrophy is caused by an immune reaction against parietal cell cytoplasmic constituents and specifically by antibodies to intrinsic factor (IF).
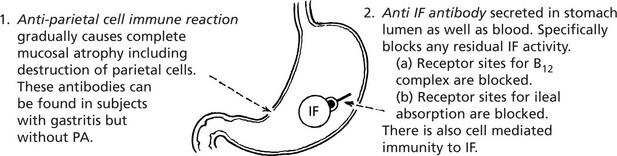
There is an increased familial incidence of PA and other organ specific autoimmune diseases, e.g. Hashimoto’s thyroiditis.
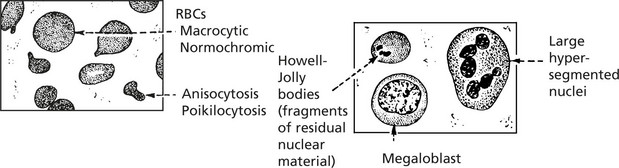
Blood Changes
There is a pancytopenia, i.e. reduction in RBCs, granulocytes and platelets but the red cells are larger (macrocytosis).
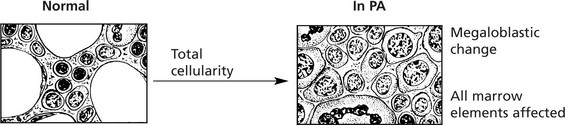
Marrow Changes
Hyperplasia – often complete cellularity in flat bones and extension down length of femur.
Folic Acid Deficiency
Folic Acid (Pteroyl-Glutamic Acid) Deficiency
| Normal metabolism | Deficiency |
|---|---|
| Source | Dietary |
| Polyglutamines in green vegetables, cereals, meat, fish and eggs (not milk) | Low intake of vegetables. Anorexia, alcoholism, poverty (elderly), infants (late weaning) |
| Minimum daily requirement | Increased requirements |
|---|---|
| 50 μg | Pregnancy (fetal growth) Infancy and childhood (rapid growth) |
| Body reserves 50–100 days | Haemolysis Malignancy |
| Absorption | Malabsorption |
|---|---|
As mono-glutamate in jejunum  |
Coeliac disease Surgical by-pass |
| Utilisation | Utilisation block |
|---|---|
| For DNA synthesis. Vit B12 is necessary for synthesis of the active tetrahydrofolate form (FH4) | Drugs e.g. methotrexate in cancer chemotherapy also anti-convulsants e.g. phenytoin. |
The Haemolytic Anaemias
Haemolytic Anaemias
In all haemolytic anaemias, there is a reduction in the life span of the red cells; due to an increased rate of red cell destruction – haemolysis.
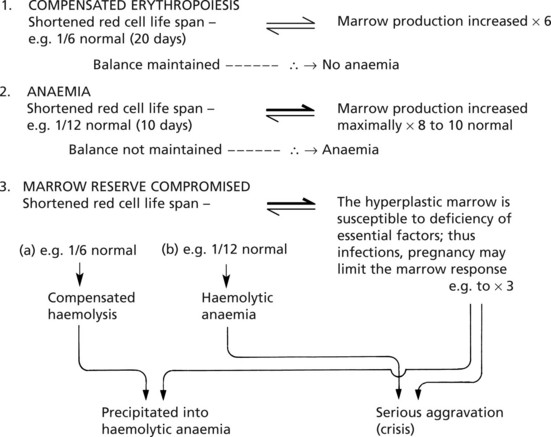
Functional reserve can compensate for a certain level of haemolysis but this fails when the degree of red cell loss is extreme or when the marrow function is compromised by other factors.
Effects of the Increased Degradation of Haemoglobin
In most haemolytic conditions, the red cells are removed and the haemoglobin degraded in the usual way, i.e. by phagocytosis by the macrophage system.
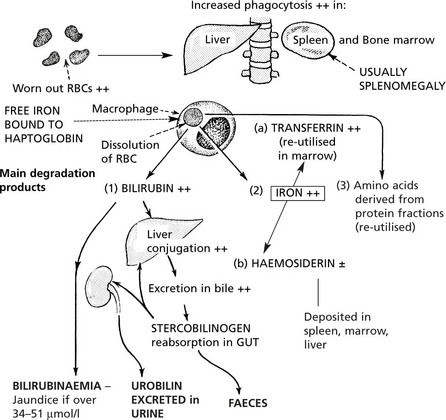

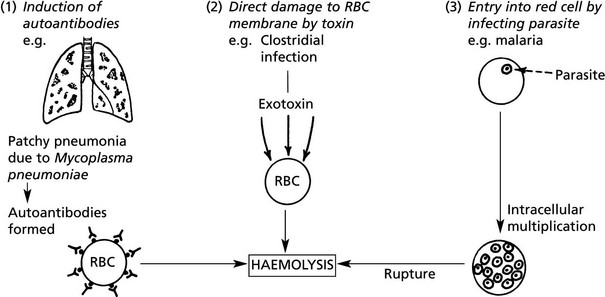
Extrinsic Haemolytic Anaemias
The causes of shortened red cell survival are divided into 2 main groups.
(1A) Autoimmune Haemolytic Anaemia
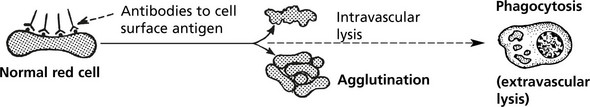
Auto-immune haemolytic anaemias are classified according to the temperature at which the reaction occurs and the presence of an underlying cause.
| warm (37 °C – usually IgG) | cold (< 30 °C – usually IgM) |
| Primary – so-called idiopathic haemolytic anaemia: usually in adults. | Idiopathic cold agglutination disease |
| Secondary – associated with | Mechanism 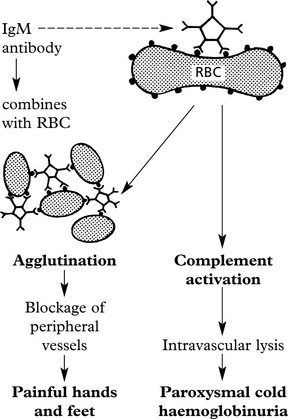 |
| Mechanism: Coating of RBC with IgG antibodies often against Rhesus ‘e’ antigens. 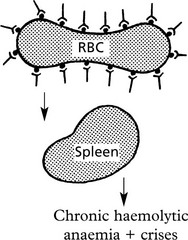
Destruction in spleen which is often enlarged. |
Incompatible Blood Transfusion/Haemolytic Disease of the Newborn
(1B) Destruction of RBCS is due to Iso-Antibodies
In these, the antibodies act against antigens which are derived from another individual of the same species.
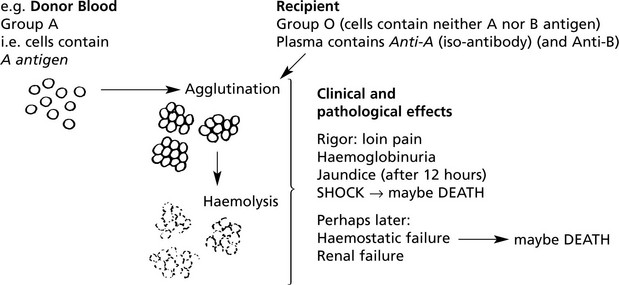
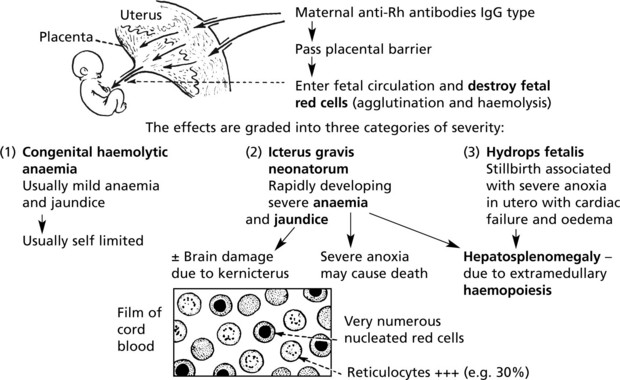
(1C) Haemolytic Disease of the Newborn (HDN)
This occurs in Rhesus positive fetuses conceived by Rh-negative mothers. The usual mechanism is as follows:
But during this pregnancy, iso-immunisation of the mother may occur.
Towards term and particularly during labour the placental barrier is breached and fetal RBCs enter the maternal circulation.
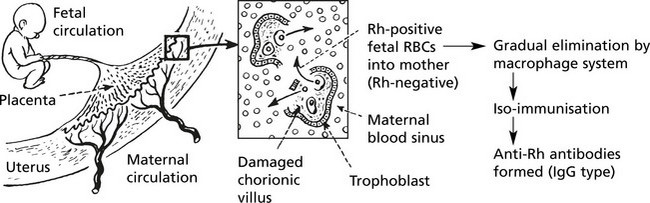
Haemolytic Disease of the Newborn
The basic mechanism is influenced by three important factors:

Subsequent pregnancies: All Rh-positive fetuses conceived by a mother who has acquired anti-Rh antibodies either during previous pregnancies or by blood transfusion (in this case the first fetus also) are at risk.

Extrinsic Haemolytic Anaemias
The results of damage to red cells within the circulation are:
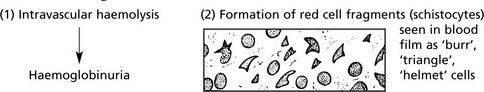
The main circumstances in which red cells are damaged are:
When the blood flow is subjected to undue turbulence or jet effect. An important example is prosthetic valves in the left side of the heart.


This condition is called microangiopathic haemolytic anaemia and occurs in many disease states involving small blood vessels e.g. malignant hypertension and/or intravascular coagulation.
The haemolysis is usually not severe enough to be of clinical significance, but haemoglobinuria may cause alarm.
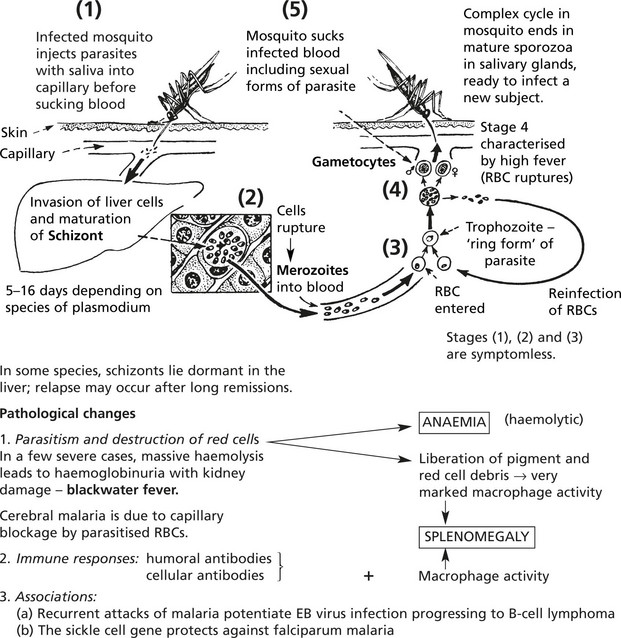
Extrinsic Haemolytic Anaemias – Malaria
Malaria is an endemic disease in many parts of Africa, Asia, Central and South America. Many millions of cases occur each year and the mortality is at least 1%. The disease is particularly severe in non-immune subjects from temperate climates; in endemic areas where the ‘herd’ immunity is high, a low grade chronic illness is common.
Female anopheline mosquitos act as intermediate hosts in the life cycle of the parasite, a protozoon (genusPlasmodium).
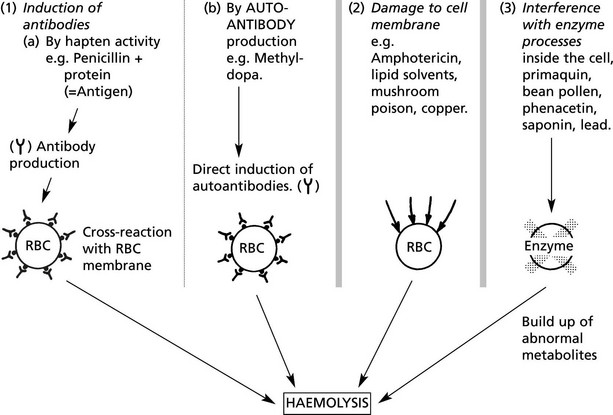
Intrinsic Haemolytic Anaemias
Intrinsic Defects – Usually Hereditary
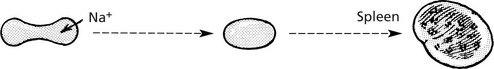
The primary defect is in the proteins which support the plasma membrane – SPECTRIN and ANKYRIN.
The cells assume a spherical shape due to instability of the plasma membrane.
Premature sequestration and destruction in splenic pulp. Splenomegaly is common.
Note: Splenectomy is usually ‘curative’ but the red cell defect remains.
The severity of the haemolysis is variable; many cases are compensated and not anaemic, but crises are common, e.g. if folate deficiency supervenes. Intercurrent infections may precipitate increased red cell destruction with jaundice or temporary bone marrow hypoplasia, e.g. parvovirus infection, with severe anaemia.

Usually hereditary. Glucose is the source of energy with which the red cell metabolism is maintained. Glycolysis is effected by two classic enzyme pathways.
The following simplified diagram outlines the pathways and indicates only the main enzymes and substrates.
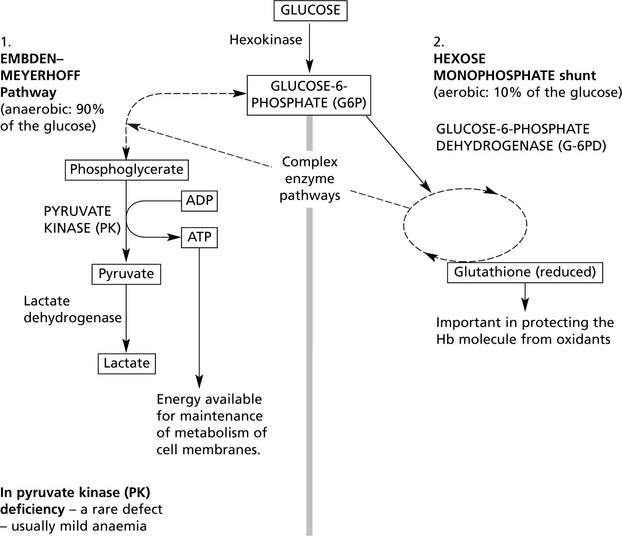
Over 400 different subtypes of G-6PD are described with a spectrum of activity. In G-6PD deficiency, a common defect especially in Africa, spontaneous anaemia is not common, but crises with anaemia and Heinz body formation occur under oxidant stress, e.g. chemicals and drugs; favism (bean products). The presence of the defect may seriously aggravate haemolysis due to other causes, e.g. haemolytic disease of the newborn.
G-6PD deficiency shows X-linked inheritance so that in the male the defect is fully expressed, while in the female heterozygote there are two populations of red cells in the blood (1) normal cells and (2) defective cells.
In both these defects and in the rare defects of other enzymes in the pathways which have been described the expression of the abnormal gene is very variable from case to case.
Disorders of Haemoglobin Synthesis
Normal Haemoglobin Molecule
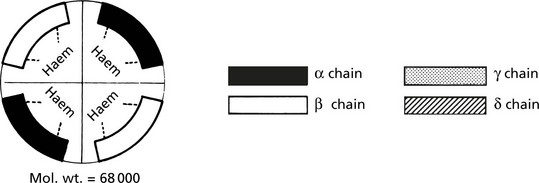
4 subunits each consisting of a haem core with a polypeptide chain attached. The haem core is constant while the polypeptide chains occur in pairs. 4 different polypeptide chains occur normally.
There are 3 types of normal haemoglobin.
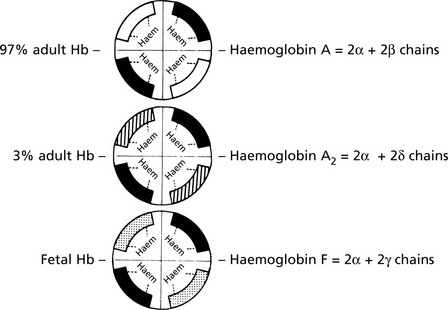
Fetal haemoglobin is replaced by adult haemoglobin during the first year (‘haemoglobin switching’). Note that the α chain occurs in all normal haemoglobins.
Two copies of the α chain genes (αα/αα) are present on chromosome 16; β, γ and δ genes are found in a complex on chromosome 11. A number of globin-like genes are also present but are omitted for simplicity.
In the thalassaemias and haemoglobinopathies there are abnormalities in the production and structure of haemoglobin chains due to mutation of these genes.
The Thalassaemias
These are inherited disorders in which synthesis of globin chains is diminished or absent.
They are common in the Mediterranean, Middle East and India.
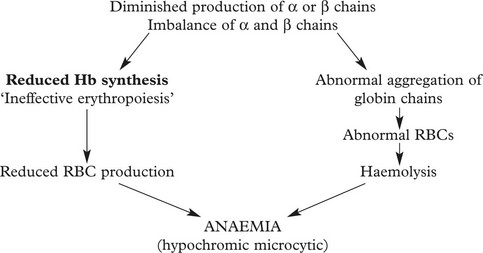
The severity of the disease depends on (i) the degree of globin chain abnormality and (ii) whether the patient is homozygous or heterozygous for the defect.
There are normally 4 α genes (αα/αα).
In the α-thalassaemias the genetic defects are illustrated as follows:
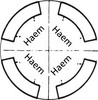
All 4 genes for the α chain are absent. Fetal Hb cannot be formed and the fetus dies in utero.
This leads to an excess of β chains which form tetramers called Hb-H.
There is a moderate microcytic hypochromic anaemia with splenomegaly.
β-Thalassaemia
β-thalassaemia major – two defective β genes.
The excess of α genes leads to severe anaemia with haemolysis. The marrow becomes hyperplastic and there is atrophy of bones. The main form of haemoglobin is HbF – which persists into adult life.
β-thalassaemia minor – one defective β gene.
The anaemia is mild. The red cells are microcytic and hypochromic and a mistaken diagnosis of iron deficiency anaemia can be made. These subjects act as carriers for β-thalassaemia major.
Sickle Cell Disease
This disorder is very common in Central Africa but also occurs in the Mediterranean, Middle East and India. It affects the black population of USA and the Caribbean.
It is due to a mutation in the β chain of haemoglobin (the amino-acid glutamic acid is substituted by valine) and is inherited as an autosomal recessive trait.
The effects are mild in heterozygotes (Hb AS) = sickle cell trait; they are severe in homozygotes (Hb SS) = sickle cell anaemia.
The effects are summarised as follows:
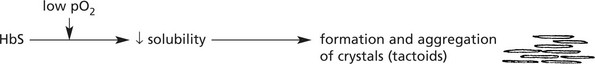
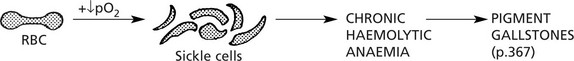
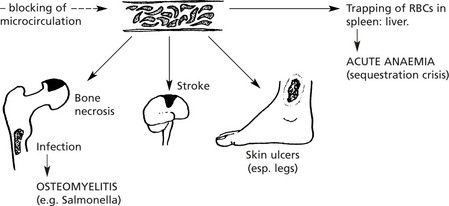
Acute sickle cell crises are often provoked by:
(1) infection, (2) cold, (3) low pO2 e.g. flying in unpressurised aircraft.
In addition, infection, especially by parvoviruses in childhood, may provoke an aplastic anaemia crisis.
Note: HbS confers resistance to malaria: in terms of evolution this confers a survival benefit to those carrying the HbS gene.
Anaemia of Chronic Disorders
This term describes the anaemias seen commonly in patients with chronic diseases
The anaemia is usually mild (Hb > 9 g/dl).
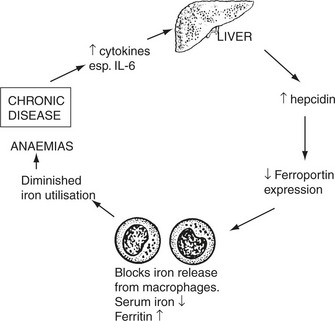
In addition, malignant tumour growth may cause anaemia by 2 more specific mechanisms.
Normocytic Normochromic Anaemias are also found in other systemic diseases.

Aplastic Anaemia
In this rare disorder there is:
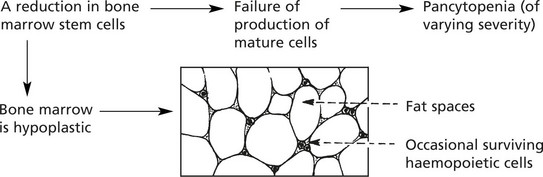
There are a number of different types
The Sideroblastic Anaemias
In these rare anaemias, failure of synthesis of the haem component of the haemoglobin molecule is indicated by the presence of a ring of iron granules around the normoblast nucleus.
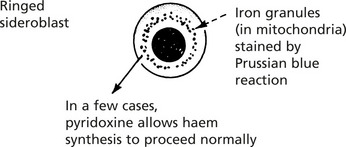
The condition arises secondarily in many of the chronic disease states mentioned on the previous page, or as a result of drug treatment or chemical poisoning (e.g. lead). Some cases are ‘primary’ – representing a form of myelodysplasia.
The anaemia is usually dimorphic; i.e. showing hypochromic and macrocytic features combined.
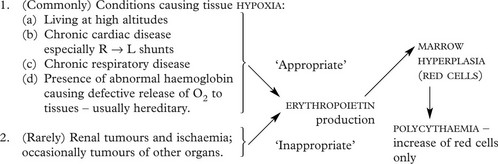
Polycythaemia
Polycythaemia
Polycythaemia (erythrocytosis) is defined by a haemoglobin level above the normal range: in true polycythaemia the red cell mass is increased and the haematocrit is always elevated.
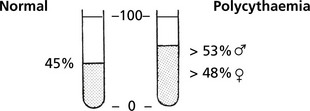
Note: Relative Polycythaemia is the result of fluid loss with a decrease in plasma volume and a normal red cell mass.
Secondary Polycythaemia
The red cell increase is the result of increased stimulation of marrow by erythropoietin. Two main groups of conditions are associated with this:
Neutrophil Granulocytes
Neutrophil Neutrophil Granulocyte (Polymorphonuclear Leucocyte)
The essential function of these cells is protection against microbial infection by phagocytosis and killing. They are produced alongside red blood cells, platelets and monocytes from a common stem cell in the marrow.
Marrow Production - Stained by a Romanowski Method
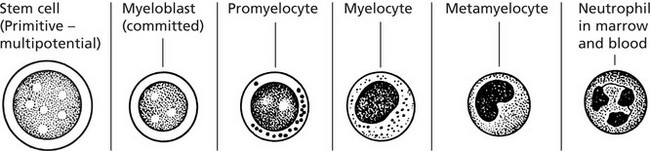
Note the nuclear changes progressing from large open nucleus and several nucleoli to the mature condensed multi-lobed state. The cytoplasm matures with the acquisition of specific granules.
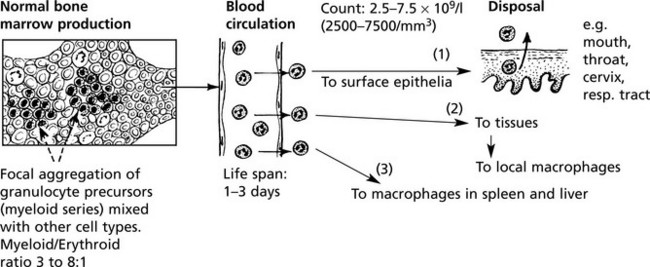
An increase in the number of circulating neutrophils reflects an increased activity in the tissues at the site of an acute inflammatory reaction
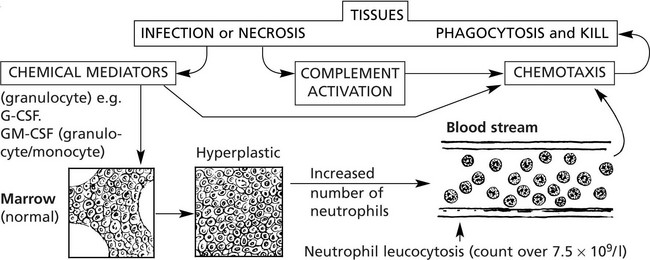
Disorders of Neutrophils – Agranulocytosis
Although a common finding in many bacterial infections, neutrophil leucocytosis does not occur in: (a) some acute bacterial infections, e.g. typhoid fever, brucellosis, (b) many chronic bacterial infection, e.g. TB, (c) most virus infections unless acute bacterial infection or necrosis occurs, (d) overwhelming infections with severe toxaemia.
In the absence (agranulocytosis) or diminished numbers of neutrophils – < 2.5 × 10/1 (2500/mm3) increased susceptibility to infection results.
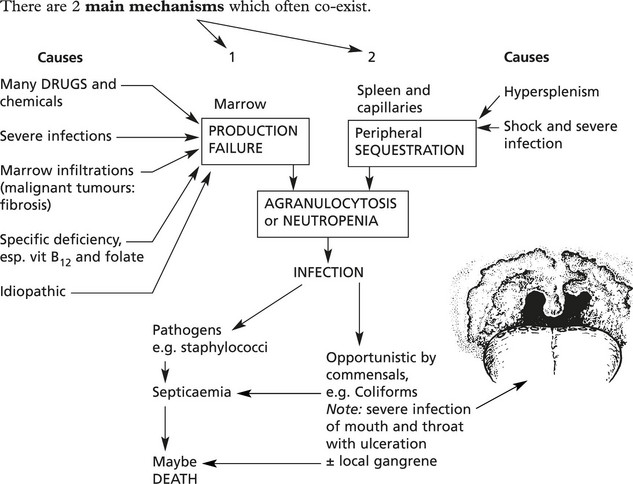
Disorders of Neutrophils
Disorders of Neutrophil Function
Divided into 3 groups: (1) chemotaxis, (2) microbial phagocytosis and (3) microbial kill.
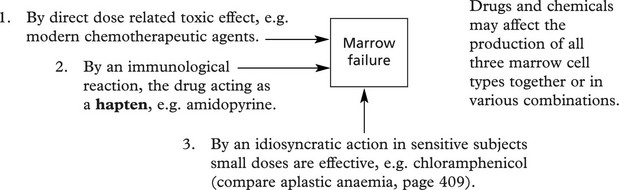
Chemotaxis and phagocytosis may be defective due to:
Microbial kill fails when there is an intracellular enzyme defect e.g. in chronic granulomatous disease (CGD) and the Chediak–Higashi syndrome where the neutrophils contain abnormal giant granules.
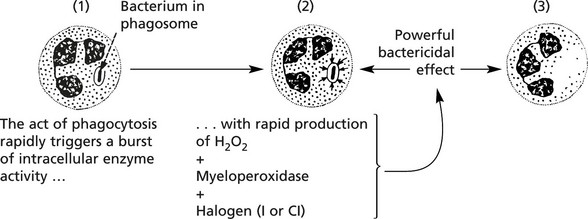
Chronic Granulomatous Disease (CGD)
It is a rare hereditary disorder in which the neutrophils have normal phagocytic function but are unable to kill certain bacteria. The disease is fully expressed only in males. It presents as multiple chronic abscesses and granulomas affecting skin, lungs, bones, spleen and liver.
The rapid production of H2O2 is defective (in the neutrophils and also in macrophages due to deficiency of NADPH-oxidase).

About 2/3 of cases are X-linked (fully expressed in males): the remainder are recessive. The neutrophils are unable to reduce nitroblue tetrazolium (used as a diagnostic slide test).
Platelets and Coagulation
These small, non-nucleated discs, derived from the cytoplasm of the bone marrow megakaryocytes, are released into the blood stream by a budding process.
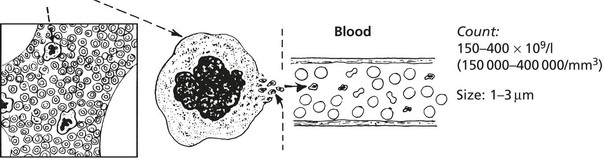
Platelets contain von Willebrand factor, adenosine diphosphate (ADP), vasoactive amines including serotonin (5HT) and histamine and phospholipid
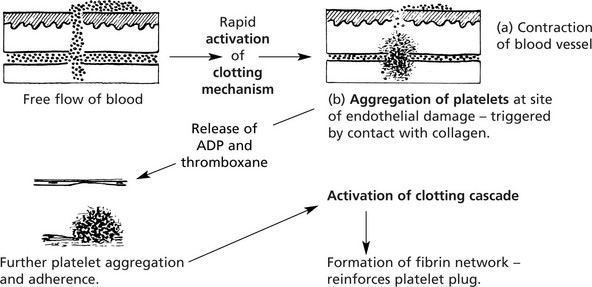

Disorders of Platelets
Disorders of the platelets include:
Thrombocytopenia
Although the normal platelet count ranges from 150–400 × 109/l, a risk of dangerous spontaneous haemorrhage is unusual unless the count falls below 30 × 109/l.
The causes of thrombocytopenia fall into two main groups:

PURPURA is the term used for disorders involving bleeding from capillaries, with the production of small petechial spots. Mild to moderate thrombocytopenia or defective platelet formation are the usual causes. Two examples are illustrative:
vWF is a large protein which potentiates the binding of platelets to subendothelial collagen and also acts as a carrier for coagulation factor VIII.
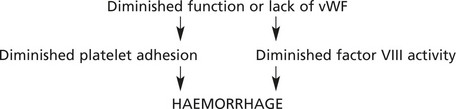
THROMBOCYTOSIS (increased platelet numbers) may occur in a mild form (with no clinical significance) reactive to a variety of disorders of chronic inflammation and cancer. Larger counts are seen in myeloproliferative disorders (e.g. essential thrombocytosis, chronic myeloid leukaemia) and are of serious clinical significance due to thrombosis.
The Coagulation Cascade
The coagulation of blood is the result of conversion of FIBRINOGEN to FIBRIN, but the chemical pathway leading to this final phase is a complicated cascade in which inert coagulation factors are serially activated. At each step augmentation takes place.
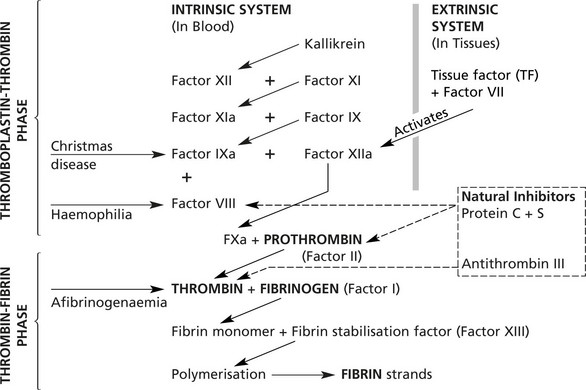
Note: ‘a’ denotes activated form
The system contains several feed-back loops (not illustrated) which can inhibit the cascade at various levels.
Anti-thrombin III and the protein C/protein S system are powerful inhibitors preventing uncontrollable coagulation.
Vitamin K is required for the production of factors II, VII, IX and X. calcium ions and phospholipids (mainly derived from platelets) are essential for many of the steps in thrombin production. Once thrombin is formed, it appears to stimulate several of the preceding reactions and also the polymerisation of fibrin.
Fibrinolytic System – This occurs following fibrin deposition as a protective mechanism against excess coagulation.

Inherited Defects of Coagulation
These are less common than acquired abnormalities, but are of considerable importance.
Haemophilia A
Haemophilia is due to deficiency of Factor VIII – an essential cofactor in the activation of Factor X in the intrinsic system.
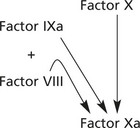
Factor VIII is encoded by a gene on the long arm of the X chromosome – haemophilia is inherited as an X-linked recessive, i.e. almost all patients are male. Their mothers are usually carriers, but about 30% of cases are due to new mutations. Around 1 in 10 000 is affected.
Clinical Features
The severity depends on the levels of Factor VIII.
| Level of Factor VIII | ||
|---|---|---|
| Severe: | Frequent spontaneous bleeding. | |
| <2% of normal | ||
| Moderate: | Bleeding after minor trauma – rarely spontaneous. | 2–10% |
| Mild: | Bleeding severe only after major trauma or surgery. | 10–50% |
Important sites of bleeding are:
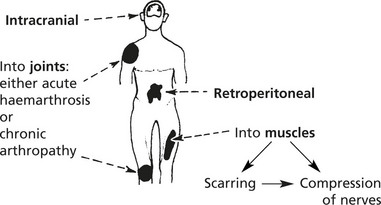
Note: In the past, the use of virus infected Factor VIII in treatment has caused HIV (AIDS) and Hepatitis C infection. Similar concerns apply to variant CJD (p.555).
Haemophilia B (Christmas Disease)
This is due to deficiency of Factor IX – also carried by a gene on the X chromosome. The clinical features are similar, but the prevalence is lower (1 in 30 000 males).
Rarely deficiencies of other coagulation factors are seen, e.g. fibrinogen, Factor XI and Factor V.
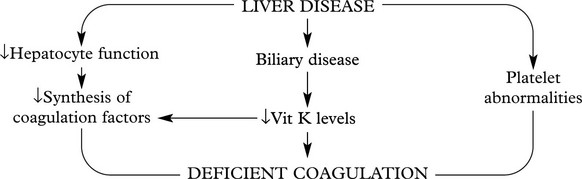
Acquired Defects of Coagulation
This large and heterogeneous group of conditions includes:
Vitamin K Deficiency
Vitamin K, a fat soluble vitamin, is essential for synthesis of Factors II, VII, IX and X in the liver. Deficiency leads to spontaneous bleeding, e.g. into skin and mucous membranes, and failure of blood clotting.
In adults, deficiency may be due to
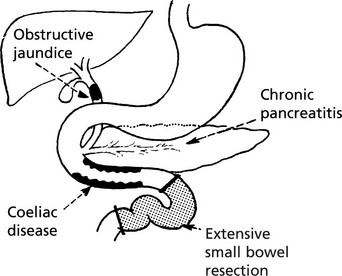
In neonates, vitamin K deficiency leads to bleeding – from GI tract and bruising. Breast milk contains little vitamin K – the disorder is uncommon in those receiving bottled milk. A severe deficiency may be seen in infants whose mothers took anticoagulants or anticonvulsants.
Disseminated Intravascular Coagulation
In this disorder the coagulation system is activated, but the consumption of platelets and clotting factors which follows leads to a paradoxical bleeding tendency.
The basic mechanism is as follows:
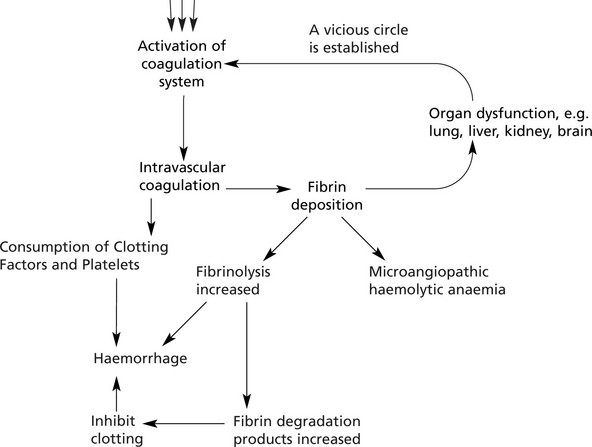
Patients frequently develop renal and hepatic failure and ‘shock lung’ [e.g. in the haemolytic uraemic syndrome (HUS) which is often associated with toxigenic E.coli infection – especially E.coli 0157 (see p.314)].
Thrombophilia – Thrombotic Disorders
The major types, causes and effects of thrombosis are discussed on pages 159–163.
Recently, a group of inherited disorders has been discovered where deficiency of natural anticoagulants leads to an increased risk of venous thrombosis. This may be deep venous thrombosis in the lower limbs or visceral, e.g mesenteric veins. There is also an increased risk of pulmonary embolism.
Antithrombin, a member of the SERPIN group of Serine protease inhibitors, is synthesised in the liver. It blocks the action of F IXa, F Xa, F XIa and thrombin. Deficiency is due to mutation of a gene on the long arm of chromosome 1 – the effects depend on the exact type of mutation. The prevalence is around 1 in 20 000.
Note: Heparin, the anticoagulant, acts by enhancing the action of antithrombin.
This is a Vitamin K dependent protein, synthesised in the liver. Together with protein S it blocks the action of Factor Va and Factor VIIIa. Around 1 in 15–30 000 is affected. In addition to deep thrombosis, patients also get superficial thrombophlebitis.
This protein is secreted by the liver, platelets and endothelial cells. Its function is to bind with protein C to inhibit Factors Va and VIIIa.
This is an inherited disorder where a mutation affects the Factor V gene. The protein is resistant to the effects of protein C – simulating protein C deficiency.
The possible role of minor deficiencies of these genetic factors in the more common forms of thrombosis is not yet established. The well known risk factors for thrombosis are listed below.
Acquired risk factors for thrombosis include:
| Arterial | Venous |
|---|---|
| ATHEROMA | IMMOBILITY |
| SMOKING | TRAUMA/SURGERY |
| HYPERTENSION | MALIGNANCY |
| MYELOPROLIFERATIVE DISORDERS | ORAL CONTRACEPTIVES |
| OBESITY |
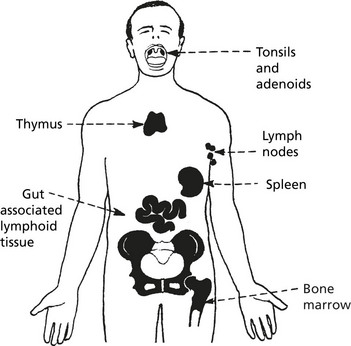
Lymphadenopathy
Lymph node enlargement may be localised or generalised. It may be due to:
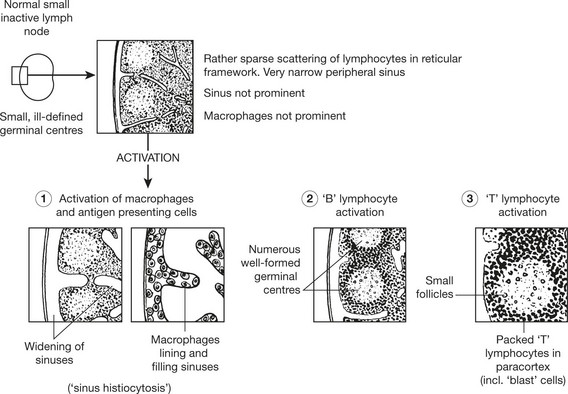
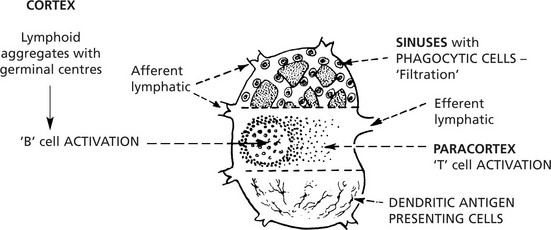
Lymphadenopathy – Infections
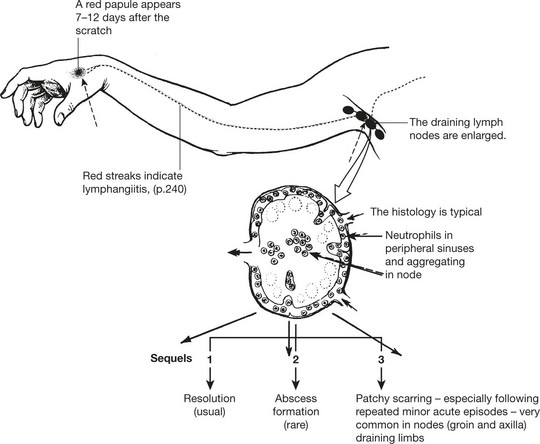
Cat Scratch Disease
This infection by a bacterium Bartonella henselae follows scratching by an infected cat. Similar histology is seen in lymphogranuloma venereum, caused by Klebsiella granulomatis. A small transient painless genital ulcer is followed by inguinal lymphadenopathy (figure). The diagnosis can be confirmed by the Frei test (injection of purified chlamydial antigen stimulates a delayed hypersensitivity reaction).
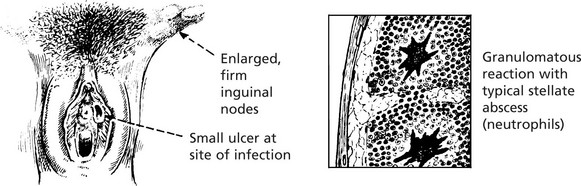
Lymphadenopathy
TUBERCULOSIS is associated with a granulomatous response (p.42), often with caseation; this is due to delayed type sensitivity.
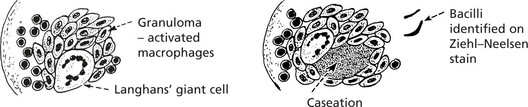
VIRAL LYMPHADENITIS – Many infections induce paracortical hyperplasia – with activation of T cells.
In GLANDULAR FEVER (Infectious mononucleosis), an infection by Epstein–Barr virus of young adults, the cervical lymph nodes are particularly involved.
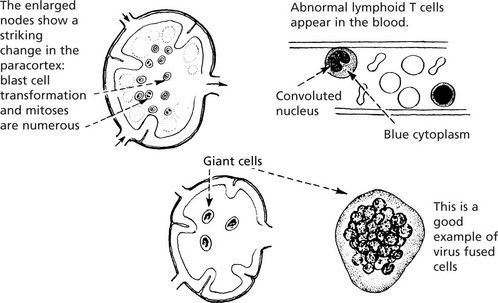
In MEASLES, the reactive nodes and lymphoid tissues generally contain unusual, multinucleate giant cells (Warthin–Finkeldey).
In TOXOPLASMOSIS, a protozoal disease (Toxoplasma gondii) which may cause serious damage to the fetus or neonate and present with lymph node enlargement in the adult, the histological appearances in the node may indicate the aetiology. The diagnosis is confirmed by serological tests.
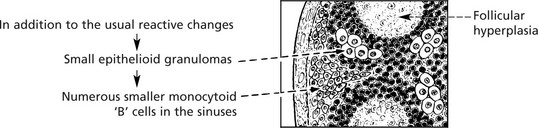
Lymphadenopathy – Non-Infective Causes
Foreign Material
Lymph nodes to which foreign particulate material drains often show marked enlargement due to accumulation of macrophages which have ingested the foreign material.
CHRONIC SKIN DISEASE (so-called dermatopathic lymphadenopathy) e.g. psoriasis, eczema and mycosis fungoides (p.435).
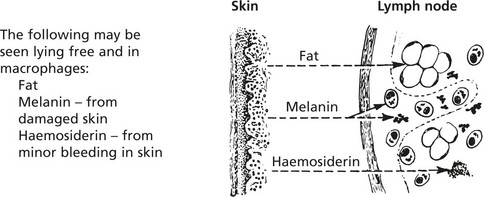
Sarcoidosis
In this disease of unknown aetiology non-caseating granulomas are seen.

The appearances clearly resemble tuberculosis, but the granulomas remain discrete and there is no caseation.
Sarcoid type granulomas are also seen in the lymph nodes of patients with other granulomatous diseases, e.g. Crohn’s disease and in lymph nodes draining tumours.
Spleen
The spleen is responsible for filtering the blood, phagocytosing debris and generating an immune response.
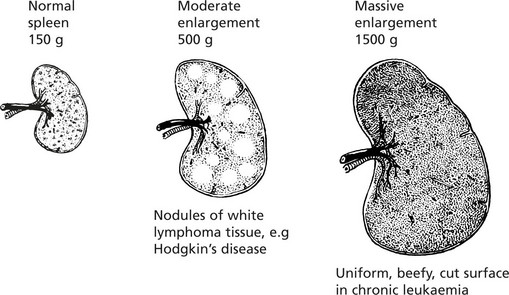
Normal average weight – 150 g.
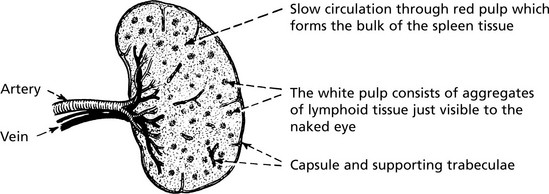
Microscopic Appearance
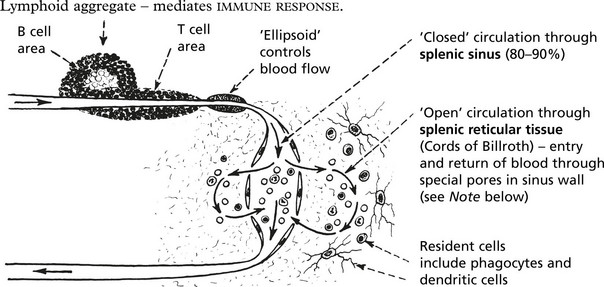
The red pulp is the site of filtration and phagocytosis of the following:
Note: The special pores allowing re-entry of RBCs to the circulation have a 2μm aperture: abnormal RBCs (e.g. spherocytes) cannot pass through and are trapped.
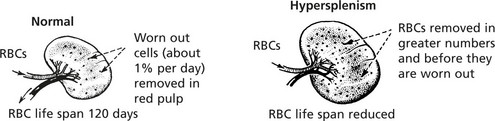
Splenomegaly
Enlargement of the spleen is an important and common clinical sign. Increase in the red pulp due to increased numbers of phagocytes and/or increased numbers of blood cells is the major component. In chronic infections, hyperplasia of the lymphoid tissue contributes.
Enlargement is associated with:
In acute systemic bacterial infections the spleen shows slight to moderate enlargement (200–400 g).
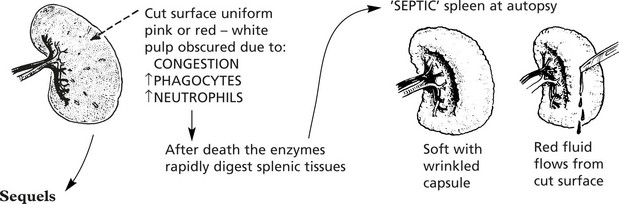
In non-pyogenic and chronic infections, there may be moderate enlargement.
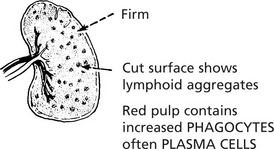
Congestive splenomegaly occurs in two main conditions:
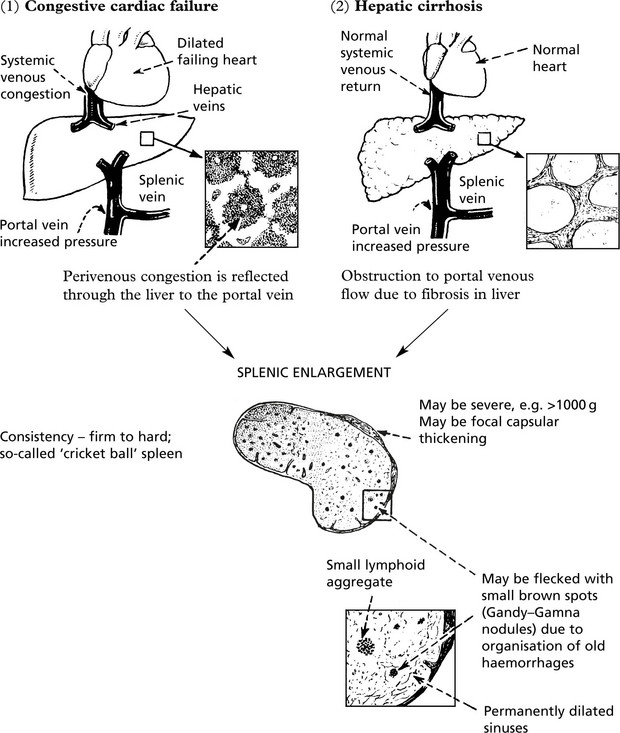
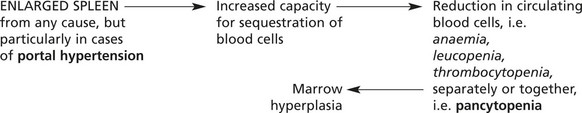
Splenomegaly associated with portal venous hypertension may have serious haematological effects (see Hypersplenism).
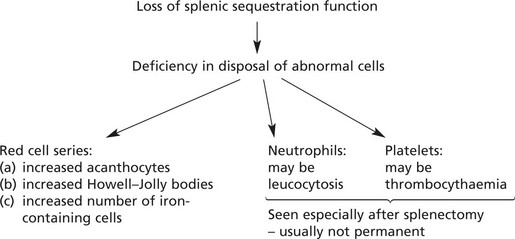
Splenic enlargement is associated with blood disorders in two main circumstances:
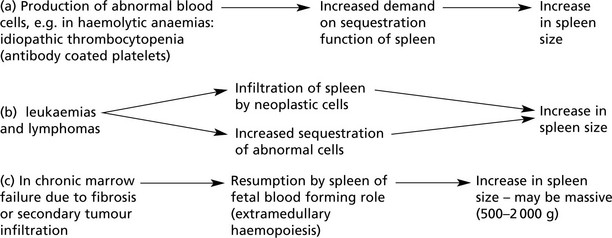
Sometimes these two processes combine to form a vicious circle.

Splenic enlargement may be a feature of Langerhans cell histiocytosis.
The diseases associated with this type of enlargement are rare.
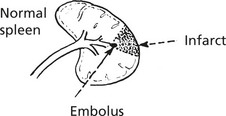
Diseases of the Spleen – Miscellaneous
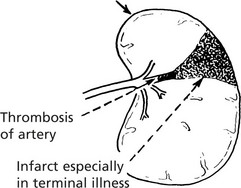
Thymus
This ‘primary’ lymphoid organ is concerned with the development and maturation of T lymphocytes which are then distributed to the lymphoid tissues and to the circulating pool.
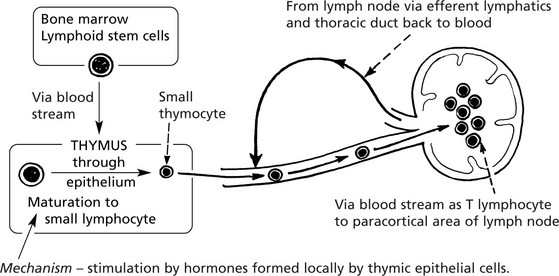
This activity is maximal in the fetal and childhood stages. Involution is rapid after puberty.
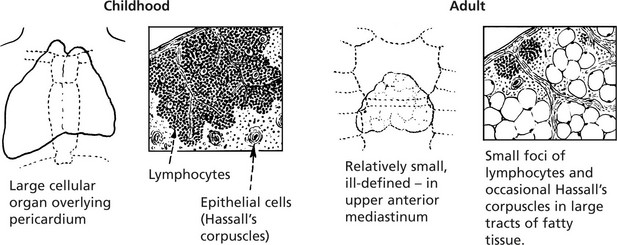
Congenital Thymic Aplasia or Hypoplasia
The resulting T cell deficiency is associated with disordered cell-mediated immune response.
Thymic Hyperplasia
This is associated with a number of conditions:
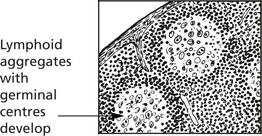
Thymic Tumours
Thymomas are rare epithelial tumours: with an admixture of lymphoid cells: they rarely metastasise. A small number are carcinomas. They may have auto-immune associations similar to thymic hyperplasia.
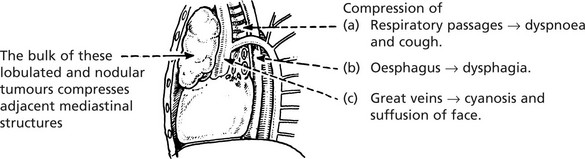
The Thymus in Myasthenia Gravis (MG)
MG is a disease of voluntary muscles in which weakness is the main feature. The association with the thymus is striking in that 80% of all patients with MG have either thymic hyperplasia or thymoma.
The detailed pathological changes in MG and the mechanisms by which the thymus is linked with the disease are described on pages 109, pages 614.
Neoplastic Lymphadenopathy
Lymph node enlargement may be due to
Secondary Tumour Invasion
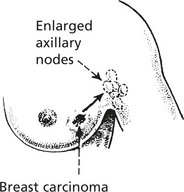
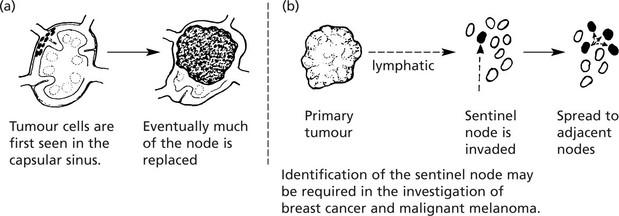
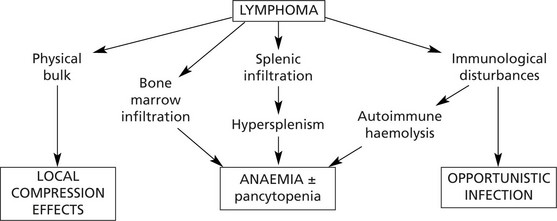
Lymphomas
Lymphomas are malignant tumours derived from lymphoid cells. They may arise within lymph nodes or at other sites (extra-nodal lymphomas).
Classically, they are divided into 2 main groups.
In recent years, modern immunocytochemical, molecular biological and cytogenetic techniques have been applied to the study of lymphomas. The traditional division between Hodgkin’s and non-Hodgkin’s lymphoma has been challenged but remains of major clinical importance.
Non-Hodgkin’s Lymphomas
These tumours mainly affect middle aged to elderly patients.
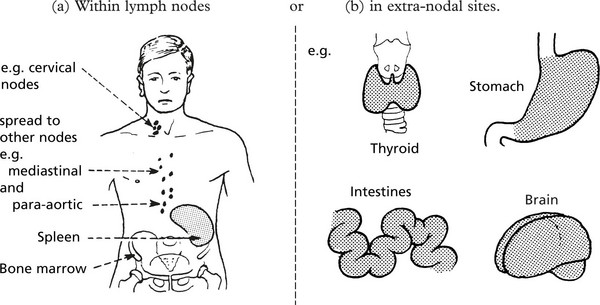
Degree of Malignancy
The natural history tends to separate lymphomas into two main groups which have links with the morphological appearances.
Pathological Complications
Classification
This is a confusing area. Traditional classifications relied on morphology alone. The World Health Organisation Classification (2008) which includes immunohistochemistry and genetic data is now generally accepted. A simplified version follows:
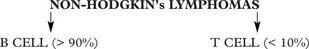
| a) High Grade Tumours Diffuse large B cell lymphoma. Burkitt’s lymphoma. |
Anaplastic large cell lymphomas (some do not express T cell markers). |
| b) Low Grade Tumours Small lymphocytic lymphoma (equivalent to B-CELL, p.442). Lymphoplasmacytic lymphoma. Mantle cell lymphoma. Follicular lymphoma. Extranodal marginal zone B-cell lymphoma Myeloma/Plasmacytoma. |
T cell prolymphocytic leukaemia Peripheral T cell lymphoma. Adult T cell leukaemia. Angioimmunoblastic T cell lymphoma. Enteropathy-type T cell lymphoma. Mycosis fungoides (skin). |
According to this classification each type is considered separately for treatment purposes.
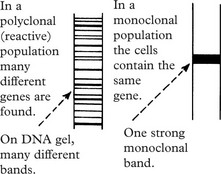
The classification relies on cell surface markers which distinguish the B and T cells and their subsets. B and T cell monoclonality can be detected by gene rearrangements, and some types of lymphoma have characteristic translocations.
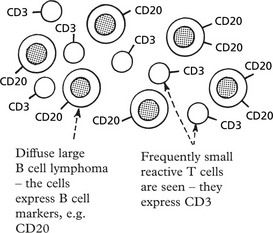
Molecular Genetic Analysis
In B cell differentiation different immunoglobulin genes are found by gene rearrangements, while in T cell differentiation a similar process occurs with T cell antigen receptors (p.92).
Follicular Lymphoma
This is one of the commonest forms – a ‘B’ cell lymphoma.
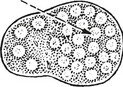
The neoplastic follicles are the malignant equivalent of normal germinal centres.
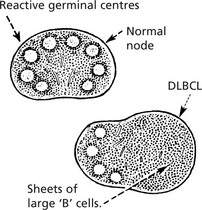
The tumour cells express the anti-apoptosis gene bcl–2 which immortalises them. There is often a translocation t (14;18) which joins the bcl–2 gene to the immunoglobulin heavy chain gene.
The lymphoma is slowly progressive but often eventually fatal. Sometimes it transforms into a diffuse large ‘B’ cell lymphoma (DLBCL).
Burkitt’s Lymphoma
This is a high grade lymphoma found mainly in Equatorial Africa, New Guinea and other malaria endemic areas. Similar tumours are seen in AIDS patients. The relationship with Epstein–Barr virus has already been discussed (p.81) – as has the translocation t (8;14) which activates the c-myc oncogene (p.152). The histological appearance is typical.

Endemic (African type) Burkitt’s lymphoma typically presents with lesions in the jaw, or in the abdomen, e.g. ovary, liver, gastrointestinal tract. The less common cases in the West typically involve lymph node, marrow and gut.

The disease may respond dramatically to aggressive treatment.
Plasma Cell Tumours
Solitary Plasmacytoma
A very small number of plasma cell tumours are solitary at presentation, e.g. in a long bone, the nasopharynx, a lymph node, alimentary tract. In 50% of cases, multiple myeloma occurs within 10 years.
Multiple Myeloma (Myelomatosis)

The disease has an incidence of 10/100 000 per annum with an age peak in the 7th decade.
The median survival, even with treatment, is 3 years.
Pathological effects are considered under two main headings:
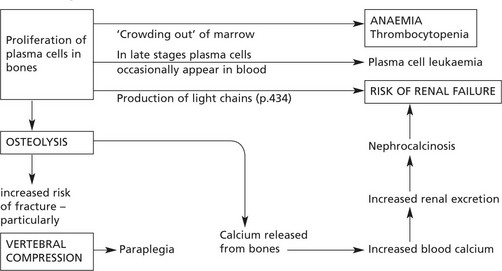
Myeloma, being a monoclonal tumour, will produce a single Ig. The common heavy chains are G (60%) and A (20%). In Waldenström’s macroglobulinaemia the immunoglobulin is always IgM.
Light chains of κ type are encountered more frequently than λ.
The presence of a single type of Ig is reflected in the electrophoretic pattern
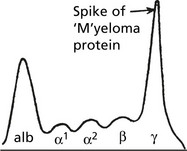
Release of Light Chains
In many myelomas, some of the Ig molecules are incompletely formed, and unattached light chains are released with important effects. Because of their low molecular weight the light chains:
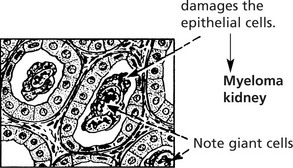
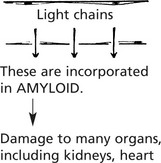
Infections, often of opportunistic type, and particularly pneumonias are common because the immune response is deficient since the high levels of ‘M’ protein are non-functioning.
Non-Hodgkin’s Lymphoma – T Cell
Mycosis Fungoides
This is a primary T cell (CD4) lymphoma of the skin occurring usually in middle age. It presents as a scaly, red macule progressing to skin plaques and then nodules.
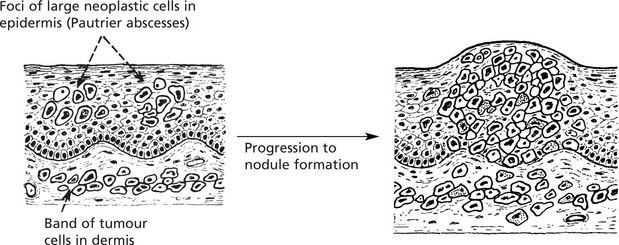
After many years the lymphoma may become generalised or exfoliate into the blood (Sézary syndrome).
Anaplastic Large Cell Lymphoma
The large malignant T cells (usually expressing surface marker CD30) often spread within the sinuses of the node and may mimic the cells in Hodgkin’s disease.

Although three-quarters of cases are of T cell type, the remainder are ‘null’ cell type, i.e. they express neither T nor B cell markers, causing further diagnostic difficulties. The cells express ALK-1 which resolves these difficulties.
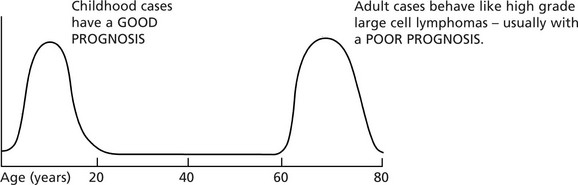
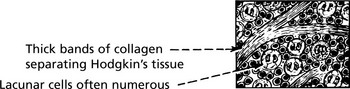
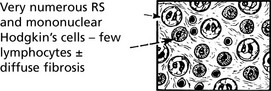
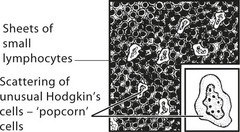
Hodgkin’s Disease
Incidence
This disease accounts for 20% of lymphomas. It may occur at any age but there are 2 peaks of incidence.
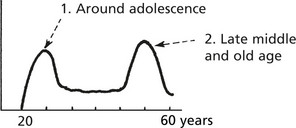
Presentation
Most patients present with painless enlargement of one or more lymph node groups – cervical, axillary, mediastinal. One quarter complain of systemic ‘B’ symptoms (see Staging) – fever, night sweats, weight loss, itch. The risk of infections is increased by immunosuppression.
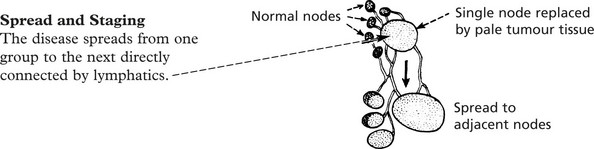
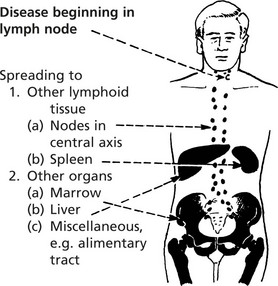
| Stage I | Disease involving single node or group of nodes |
| Stage II | Disease in more than one site – all lesions either below or above the diaphragm |
| Stage III | Disease on both sides of diaphragm |
| Stage IV | Widespread involvement of extralymphoid sites ± lymph node involvement |
The suffix (E) to the numeral indicates Extranodal disease, (A) denotes absence of systemic symptoms, (B) presence of these (see above).
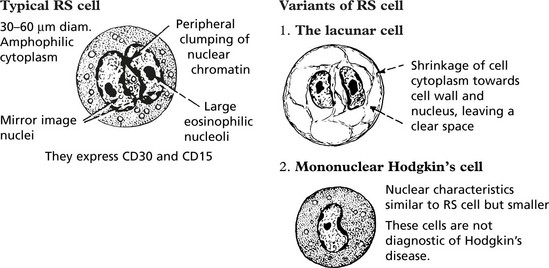
Hodgkin’s disease is diagnosed on the basis of distinctive large tumour cells known as Reed–Sternberg cells, which are now known to be ‘B’ cells of germinal centre origin.

Leukaemias
Leukaemias are primary malignant tumours of haemopoietic cells.
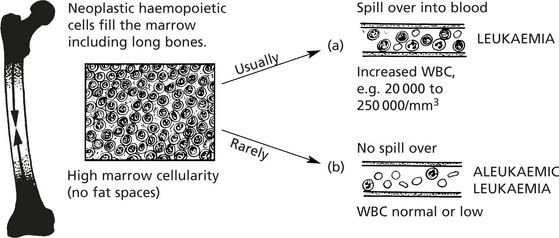
Spread Of Leukaemia
Leukaemic cells circulate in blood and/or lymph and can therefore spread anywhere in the body. Nodular deposits are, however, uncommon.
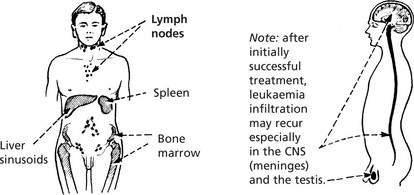
Classification
Leukaemias are classified according to the rate of progression and the lineage of the tumour cells. The 4 main forms are:
| MYELOID (granulocyte: monocyte series) | LYMPHOID (B and T lymphocyte series) | |
|---|---|---|
| ACUTE LEUKAEMIA – Rapid progression – Numerous primitive ‘blast’ cells. |
(i) ACUTE MYELOBLASTIC LEUKAEMIA |
(iii) ACUTE LYMPHOBLASTIC LEUKAEMIA |
| CHRONIC LEUKAEMIA – Slow progression – Cells almost mature. |
(ii) CHRONIC MYELOID LEUKAEMIA |
(iv) CHRONIC LYMPHOCYTIC LEUKAEMIA |
Rare leukaemias affect red cells, megakaryocytes and plasma cells.
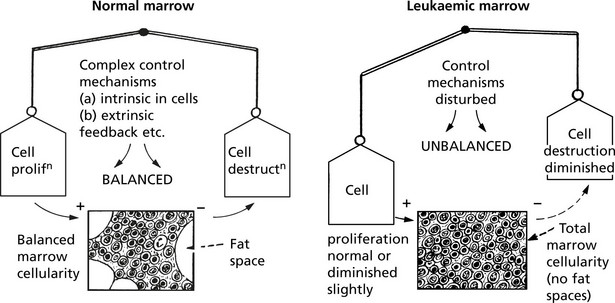
Acute Myeloblastic Leukaemia
This is the commonest acute leukaemia of adults – 80% of patients are over 60 years old. Young adults and children may also be affected.
It may arise de novo or in association with chronic myeloid leukaemia, myeloproliferative disorders, myelodysplasia. The onset and progression to marrow failure is rapid, clinically presenting as anaemia, haemorrhage or serious infection.
The WHO classification describes four broad categories:

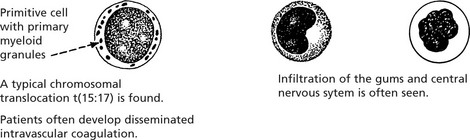
Chronic Myeloid Leukaemia (CML)
This form of leukaemia arises from malignant transformation of a primitive stem cell – but with the production of differentiated cells – particularly neutrophils.
This is a disease of middle age. There are 3 phases:
Rarely patients develop myelofibrosis with marrow failure.
Philadelphia (Ph) Chromosome
In over 95% of cases of CML there is a specific cytogenetic change t(9;22)(q34;q11) with the formation of the ‘Ph chromosome’.
The change involves a translocation of the c-abl gene (an oncogene) from chromosome 9 to chromosome 22 where hybridisation with the bcr gene occurs. A tyrosine kinase inhibitor (IMATINIB) which targets this pathway has been introduced.
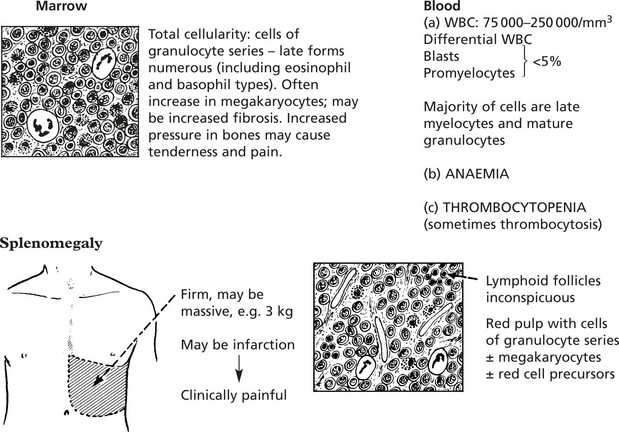
Chronic Lymphocytic Leukaemia
This is a common form of leukaemia (25% of all cases) and particularly affects middle aged and elderly patients. It is a monoclonal proliferation of small lymphocytes and is best regarded as the leukaemic form of lymphocytic lymphoma.
Most cases are of B cell type – less than 5% are of T cell lineage.
There is typically lymph node enlargement and splenomegaly.
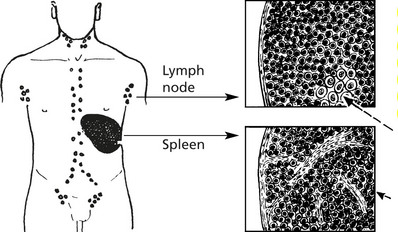
The lymph node and splenic architecture is obscured by closely packed small lymphocytes.
Mitotic activity is minimal, but very small foci of larger active cells (proliferation centres) are the source of new neoplastic cells.
Germinal centres destroyed by infiltrating lymphocytes
In some cases the lymphoid enlargement precedes the leukaemic phase.
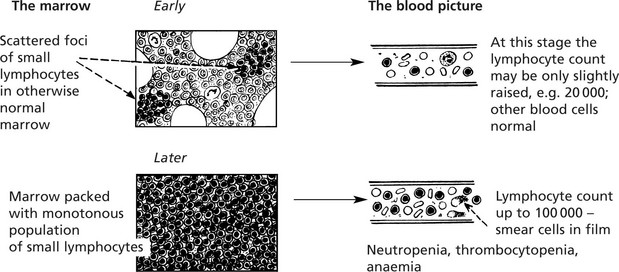
In addition to anaemia these patients often suffer recurrent infections due to defective immunity.
In a minority of cases (approximately 5%) transformation to a high grade ‘B’ cell lymphoma results (Richter syndrome).
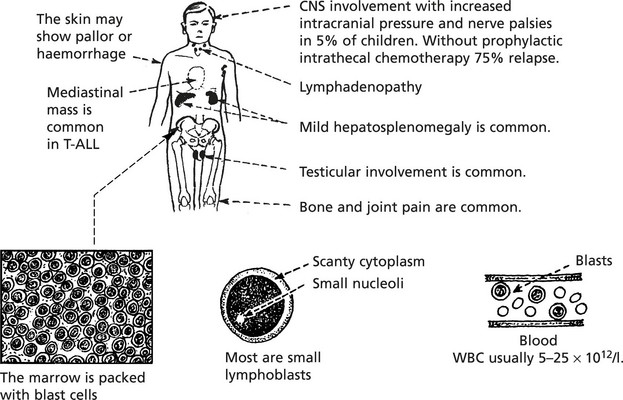
Acute Lymphoblastic Leukaemia (ALL)
This term describes a group of leukaemias of lymphocytic precursors. ALL is the commonest form of childhood leukaemia but is also seen in adults.
Like AML, immunological and genotypic classification is increasingly important. Broadly, they are classified as follows:
Myeloproliferative Disorders
In this group of diseases, neoplastic transformation of a haemopoietic precursor cell may lead to excess production of erythrocyte, leucocyte or platelet precursors. In many cases more than one element is affected. Transformation to acute leukaemia can occur in all forms. This can be summarised as follows:
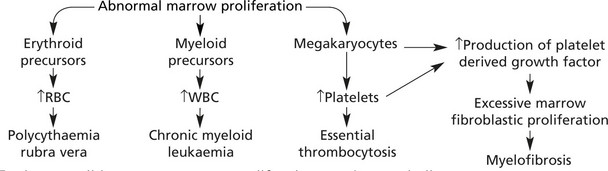
In these conditions excess marrow proliferation may be seen in liver, spleen and other organs – extramedullary haemopoiesis (EHM).
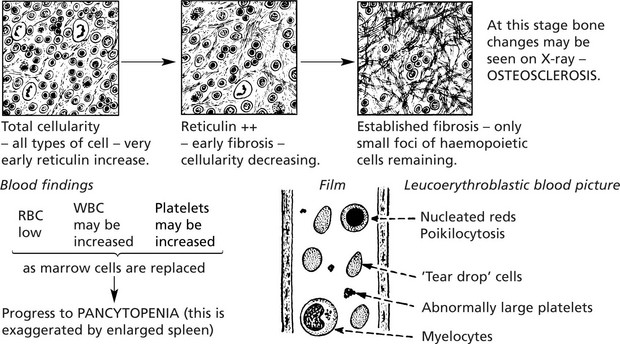
Myelofibrosis
When haemopoietic cellular proliferation is overshadowed by progressive fibrosis of the marrow, splenomegaly (often of massive proportions) due to extramedullary haemopoiesis is often found.
Marrow: ordinary marrow puncture usually results in a ‘dry tap’, therefore trephine or cutting needle biopsy is required
2. Myelodysplastic Syndromes (MDS)
These are primary disorders of stem cells associated with several different chromosomal abnormalities. They lead to ineffective haemopoiesis of varying types, with the appearance in the peripheral blood of abnormal cells. A significant number progress to leukaemia.