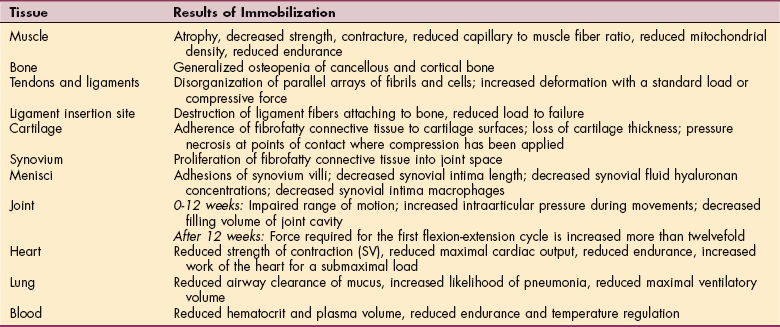
Arachidonic Acid Derivatives.: The synthesis of prostaglandins and leukotrienes begins with the cleavage (splitting) of arachidonic acid from membrane phospholipids by the action of the phospholipase (Fig. 6-13). Once this step is completed, either a cyclooxygenase (COX) enzyme or a lipoxygenase enzyme further metabolizes the arachidonic acid. The COX pathway leads to the production of several types of prostaglandins that modulate vasomotor tone and platelet aggregation (e.g., thromboxane is a strong platelet aggregator and vasoconstrictor, whereas prostacyclin [PGI2] is a strong platelet inhibitor and vaso- dilator). Clinically, prostaglandins are also important because they are mediators of the fever and pain responses associated with inflammation.24
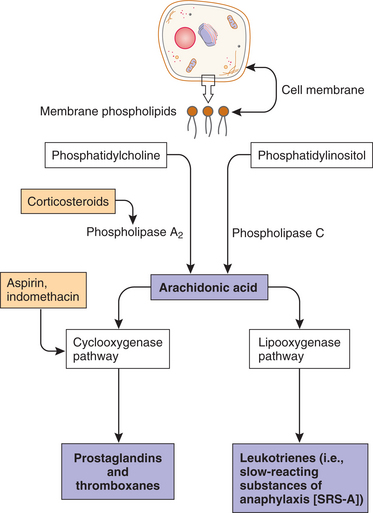
Figure 6-13 Production of prostaglandins and leukotrienes from damaged cell membranes. Note sites for pharmacologic (aspirin and prednisone) interventions. (Courtesy SH Tepper, PhD, PT, Winchester, VA, Shenandoah University.)
The lipoxygenase pathway leads to the production of leukotrienes. Leukotrienes occur naturally in leukocytes and produce allergic and inflammatory reactions similar to those of histamine. They are extremely potent mediators of immediate hypersensitivity reactions and inflammation, producing smooth muscle contraction, especially bronchoconstriction; increased vascular permeability; and migration of leukocytes to areas of inflammation. They are thought to play a role in the development of allergic and autoimmune disease such as asthma and rheumatoid arthritis. Certain leukotrienes (C4, D4, and E4) are collectively known as a slow-reacting substance of anaphylaxis (SRS-A), which is the name given when their potent bronchoconstrictor activity was discovered; they also cause leakage of fluid and proteins from the microvasculature.
The importance of the arachidonic acid metabolites in the inflammatory process is made evident by the excellent clinical response to treatment of acute and chronic inflammatory conditions with drugs that block the production of arachidonic acid (corticosteroids) or inhibit the enzyme and block the production of prostaglandins and cyclooxygenase (nonsteroidal antiinflammatory drugs [NSAIDs] such as aspirin or the newer COX-2 inhibitors). These antiinflammatory medications are commonly used for people with somatic pain or inflammatory conditions, especially rheumatoid arthritis.
Cytokines.: Leukocytes also produce polypeptide substances called cytokines (see Chapter 7) that have a wide range of inflammatory actions affecting either the cytokine-producing cells themselves (autocrine effects) or adjacent cells (paracrine effects). Cytokines also have a number of systemic “hormonal” inflammatory effects.
Two important cytokines with overlapping functions are IL-1 and TNF. As many as fifteen ILs are now identified. Most ILs direct other cells to divide and differentiate, each interleukin acting on a particular group of cells that have receptors specific for that interleukin. TNF is thought to be capable of inducing most of the actions of IL-1 with the exception of activation of lymphocytes.
IL-1 has a number of local actions that promote the inflammatory reaction and a number of systemic actions that induce metabolic, hemodynamic, and hematologic alterations (Box 6-5). These alterations are discussed in some detail because of their importance in the clinical and laboratory diagnosis of inflammation. IL-1 causes fever by raising the production of prostaglandins in the hypothalamus and thereby resetting the threshold of temperature-sensitive neurons.
Fever in turn raises the systemic metabolism and increases the systemic consumption of oxygen by approximately 10% for each degree Celsius of body temperature elevation. As a result, a decrease in systemic vascular resistance occurs, thereby producing hypotension and an increase in cardiac output to increase the flow of blood and the delivery of oxygen to various organs. These hemodynamic changes are characteristic of severe systemic infections and a febrile condition.
IL-1 also causes characteristic changes in blood chemistry. Albumin and transferrin levels are decreased, while levels of coagulation factors, complement components, C-reactive protein, and serum amyloid A increase. These changes occur because IL-1 alters the rate of synthesis of these proteins by the liver. IL-1 also increases the number of neutrophils and decreases the number of lymphocytes in the circulation.
The Blood Coagulation, Fibrinolytic, and Complement Systems.: Plasma proteins produce chemical inflammatory mediators by the enzymatic activity of proteases on plasma proteins. Plasma proteases are enzymes that act as a catalyst in the breakdown of proteins. These plasma protein systems are the blood coagulation and fibrinolytic, kinin enzymatic, and complement systems.
All of these systems can become activated by contact with by-products of cell injury or foreign materials. Examples include contact with components of denuded vascular endothelial cells revealing their underlying basement membrane, which occurs with trauma to the vessel wall and contact with bacterial endotoxins. The key plasma protein in the activation sequence of these systems is clotting factor XII, also known as Hageman factor.
The blood coagulation system (Fig. 6-14) is formed in part by plasma proteins. The design is to bandage injuries with clots (coagulation), then disassemble (lyse) the clots when the job is done. The system protects against both hemorrhage and catastrophic clotting. To maintain homeostasis, these two processes must remain in balance.
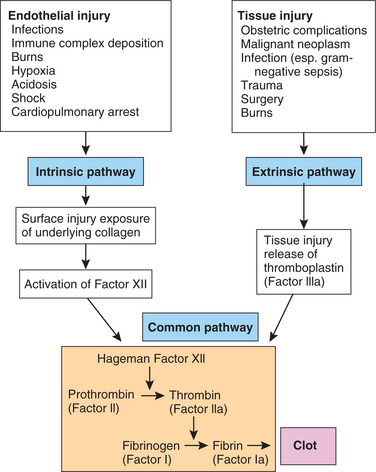
Figure 6-14 Clinical causes of the activation of a clotting cascade, intrinsic and extrinsic pathways of activation, and the mechanism by which both pathways lead to the formation of fibrin threads, or clot. In the chain reaction, inactive proenzymes (represented by Roman numerals) are converted into active enzymes (represented by Roman numerals followed by the letter “a.” The clotting cascade can follow two pathways: intrinsic and extrinsic. The intrinsic pathway is activated within the vascular compartment. The extrinsic pathway is activated outside the vascular compartment, when blood comes in contact with any tissue other than blood vessels. In the case of internal bleeding, both pathways are activated. (Courtesy SH Tepper, PhD, PT, Winchester, VA, Shenandoah University.)
Platelets circulating throughout the bloodstream are always ready to seal any damage to blood vessels with a hemostatic plug. When there is no need for the platelets, the smooth vascular walls prevent platelets from adhering and aggregating. At the same time, endothelial cells in the walls of the blood vessels make tissue plasminogen activator to prevent fibrin deposits from forming and for breaking down existing clots.
More specifically, when injury or bleeding occurs, a series of enzymes are activated sequentially to generate the enzyme thrombin, which converts the plasma protein fibrinogen to fibrin, the essential component of a blood clot. Fibrin forms a meshwork at bleeding sites to stop the bleeding and trap exudate, microorganisms, and foreign materials and keep this content contained in an area where eventually the greatest number of phagocytes will be found. This localizing effect prevents the spread of infection to other sites and begins the process of healing and tissue repair.
The fibrinolytic system (designed to dissolve these clots) is activated by the conversion of plasminogen to the enzyme plasmin (also known as fibrinolysin, which means “to loosen”). Plasmin splits or divides fibrin and lyses the blood clots. Both the coagulation and the fibrinolytic systems are activated in inflammation and function together in a system of checks and balances to preserve vascular function.
The products of fibrin degradation are chemotactic for leukocytes and increase vascular permeability. The kinin enzymatic system is also activated by Hageman factor and functions to produce bradykinin. Bradykinin is a mediator that causes dilatation and leakage of blood vessels and induces pain.
The complement system is composed of a group of plasma proteins that normally lie dormant in the blood, interstitial fluid, and mucosal surfaces. Then, through a series of enzymatic reactions, several plasma protein fragments (C3a, C3b, C5a, and C5b) are formed that are potent inflammatory mediators. These components are also active in immunologic processes. In the nomenclature used for the complement system, each complement component (C) is designated by a number (1 to 9). The individual subunits that make up each component are designated by a letter. For example, the first component of complement is designated C1. C1 is made up of three subunits that are designated C1q, C1r, and C1s. The protein fragments that are generated from the proteolytic degradation of complement components are also identified by a letter (a, b).
The complement system is activated by microorganisms or antigen-antibody complexes causing four events to occur that promote inflammation: (1) vasodilates the capillaries, which increases blood flow to the area, (2) facilitates the movement of leukocytes into the area by chemotaxis, (3) coats the surfaces of microbes to make them vulnerable to phagocytosis, and (4) formation of a membrane attack complex (MAC).
Complement activation can follow one of two pathways, the classic or the alternate pathway; each pathway produces the same active complement components. The products of the complement system bind to particles of foreign material, microorganisms, or other antigens, coating them to make them vulnerable to phagocytosis by leukocytes, a process called opsonization. Activation of the complement cascade by either pathway also results in the formation of the MAC. The MAC is inserted in cell membranes of the microorganism where it creates an opening (pore or channel) in the cell membrane, leading to influx of sodium and extracellular fluid, eventually leading to its lysis (Fig. 6-15). For example, in hemolytic anemia, MAC bores holes in the cell membrane of RBCs, causing their destruction.
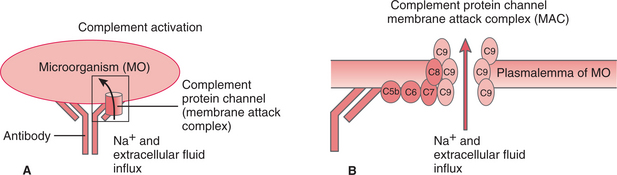
Figure 6-15 A, When an antibody attaches to an antigen (foreign protein) on a microorganism (MO), the antibody-antigen stimulates plasma-derived complement proteins to attach and form the membrane attack complex (MAC). B, This MAC forms a channel through the membrane of the invading cell and allows ions and extracellular fluid to enter, causing cytolysis (death of the microorganism). (Courtesy SH Tepper, PhD, PT, Winchester, VA, Shenandoah University.)
The plasma protease systems (blood coagulation, fibrinolytic, kinin enzymatic, and complement systems) are interconnected at several steps. This arrangement serves to amplify the stimulus for the inflammatory reaction as a balance mechanism. For example, the activation of the plasma protein Hageman factor can initiate both the coagulation (blood clotting) and the kinin systems (produces bradykinin causing dilation and vascular leakage).
The kinin system can in turn activate the fibrinolytic system by producing plasmin (splits or divides fibrin and lyses blood clots). Plasmin then can activate the complement system and further amplify these protease loops by activating Hageman factor, once again starting the cycle (Fig. 6-16).
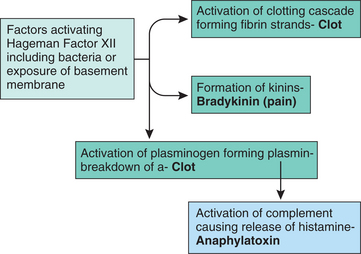
Figure 6-16 Clot formation. Revealed in this figure are the mechanisms for activating both the intrinsic and the extrinsic pathways for clot formation. Either of the above pathways leads to activation of the Hageman factor XII that results in the formation of a fibrin clot. (Courtesy SH Tepper, PhD, PT, Winchester, VA, Shenandoah University.)
PHAGOCYTOSIS.: One of the most important functions of the inflammatory reaction is to inactivate and remove the inflammatory stimulus and to begin the process of healing. The process of ingestion (phagocytosis) of microorganisms, other foreign substances, necrotic cells, and connective tissue constituents by specialized cells (phagocytes) is important in achieving this goal.
Although phagocytosis could be considered the next step in the process of acute inflammation (as a separate section after the section on Chemical Mediators of Inflammation), it is included here as part of the section on Chemical Mediators because the chemical mediators are what attract phagocytic cells to the area for removal of the dead tissue or microorganisms. After ingestion by phagocytic cells, microorganisms are killed or inactivated, and necrotic debris is removed to allow tissue healing to proceed.
The most important phagocytes involved in the inflammatory and healing reactions are neutrophils, monocytes, or when found in tissues of the body, macrophages. Macrophages have different names depending on their location (e.g., histiocytes in the skin, osteoclasts in bone, and microglial cells in the CNS).
The mechanism of phagocytosis is well understood. Phagocytosis is facilitated by the coating (opsonization) of particles to be ingested by immunoglobulin G (IgG) antibody or by the C3b component of complement. These opsonins bind to specific receptor sites located on the cell surface of neutrophils and macrophages. This receptor binding initiates a process of transmembrane signaling allowing calcium influx that activates cytoskeletal proteins within the cell. These cytoskeletal structures allow the movement of cell membranes that is necessary for phagocytosis.
The internalization of the opsonized particle begins by the enfolding of the cell surface membrane (Figs. 6-17 and 6-18). The membrane folds surround the particle to be ingested and seal it within a pouch that separates it from the cell surface and becomes an intracellular vacuole called the phagosome. The phagosomes fuse with lysosomes (containing digestive materials and bactericidal components) and acquire enzymes and other substances that allow the killing and degradation of microorganisms and other ingested materials. Many neutrophils (e.g., polymorphonuclear neutrophils [PMNs]) die in their battle with bacteria. Dead and dying leukocytes, mixed with tissue debris and lytic enzymes, form a viscous yellow fluid known as pus. Inflammations identified by their pus formations are called purulent or suppurative (see Table 6-4).
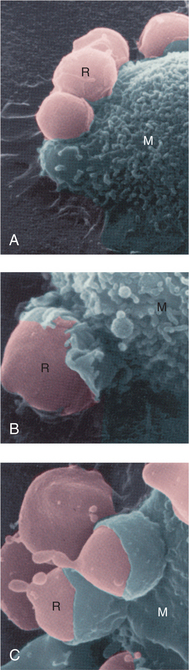
Figure 6-18 Phagocytosis. This series of scanning electron micrographs shows the progressive steps in phagocytosis of damaged red blood cells (RBCs) by a macrophage. A, RBCs (R) attach to the macrophage (M). B, Plasma membrane of the macrophage begins to enclose the RBC. C, The RBCs are almost totally ingested by the macrophage. (From Thibodeau GA, Patton KT: The human body in health and disease, ed 4, St. Louis, 2005, Mosby. Courtesy Emma Shelton.)