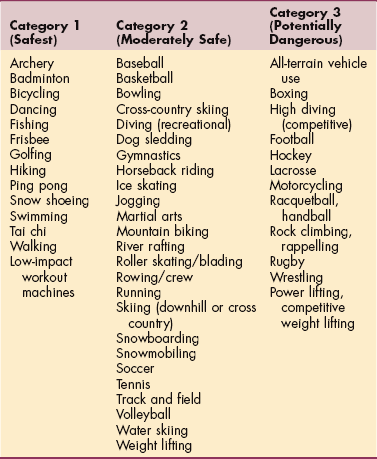
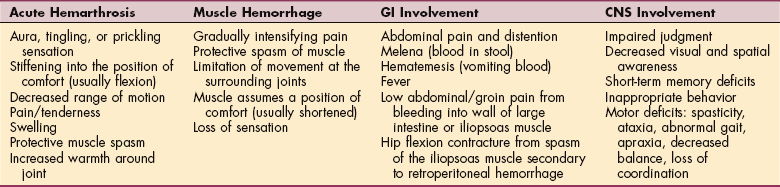
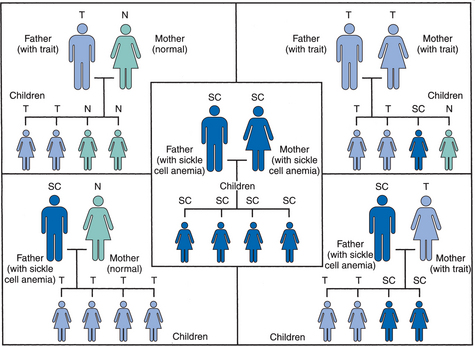
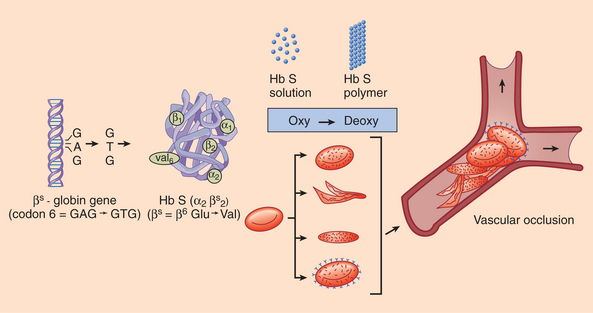
1. Abshire, T. An approach to target joint bleeding in hemophilia: prophylaxis for all or individualized treatment? J Pediatr. 2004;145(5):581–583.
2. ACOG committee opinion. Von Willebrand’s disease in gynecologic practice. Int J Gynaecol Obstet. 2002;76(3):336–337.
3. Adams, RJ, Brambilia, D. Discontinuing prophylactic transfusions used to prevent stroke in sickle cell disease. N Engl J Med. 2005;353(26):2769–2777.
4. Aderibigbe, A, Ologe, FE, Oyejola, BA. Hearing thresholds in sickle cell anemia patients: emerging new trends? J Natl Med Assoc. 2005;97(8):1135–1142.
5. Ajmani, RS, Rifkind, JM. Hemorheological changes during human aging. Gerontology. 1998;44(2):111–120.
6. Al-Majid, S. Cancer-induced fatigue and skeletal muscle wasting: the role of exercise. Biol Res Nurs. 2001;2:186–197.
7. Albrecht, K. Strength training guidelines for children, adolescents and adults with bleeding disorders. Hemaware. 2003;8(3):66–70.
8. American Academy of Pediatrics. Policy statement. Strength training by children and adolescents. Pediatrics. 2001;107(6):1470–1472.
9. American Cancer Society Acute lymphoblastic leukemia. Available at http://www.cancer.org Accessed February 10, 2007
10. American Cancer Society How many people get multiple myeloma? Available at http://www.cancer.org Accessed January 27 2007
11. Anderson, A, Holtzman, TS, Masley, J. Physical therapy in bleeding disorders. New York: National Hemophilia Foundation, 2000.
12. Ania, BJ, Suman, VJ, Sobell, JL, et al. Trends in the incidence of polycythemia vera among Olmsted County, Minnesota residents, 1935–1989. Am J Hematol. 1994;47:89.
13. Atkins, RC, Walters, MC. Haematopoietic cell transplantation in the treatment of sickle cell disease. Exp Opin Biol Ther. 2003;3(8):1215–1224.
14. Attal, M, Harousseau, J-L, Facon, T, et al. Single versus double autologous stem-cell transplantation for multiple myeloma. N Eng J Med. 2004;349:2495–2502.
15. Ballen, KK, Hasserjian, RP. Case 2-2005: a 39-year-old woman with headache, stiff neck, and photophobia. N Engl J Med. 2005;352(3):274–283.
16. Barlogie, B, Tricot, G, Anaissie, E, et al. Thalidomide and hematopoietic-cell transplantation for multiple myeloma. N Engl J Med. 2006;354:1021–1030.
17. Barnes, L. Non-Hodgkin’s lymphoma. Rehabil Oncol. 2000;18(2):14–16.
18. Beard, J, Tobin, B. Iron status and exercise. Am J Clin Nutr. 2000;72(Suppl 2):594S–597S.
19. Belhadj, K, Delfau-Larue, MH, Elgnaoui, T, et al. Efficiency of in vivo purging with rituximab prior to autologous peripheral blood progenitor cell transplantation in B-cell non-Hodgkin’s lymphoma, a single institution study. Ann Oncol. 2004;15(3):504–510.
20. Berenson, JR, et al. Zoledronic acid reduces skeletal-related events in patients with osteolytic metastases. Cancer. 2001;91(7):1191–1200.
21. Biagi, J, et al. Primary Hodgkin’s disease of the CNS in an immunocompetent patient: a case study and review of the literature. Neuro-oncol. 2000;2(4):239–243.
22. Bolton-Maggs, PH, Pasi, KJ. Haemophilia A and B. Lancet. 2003;361:1801–1809.
23. Braun-Moscovici, Y, et al. Methotrexate-treated arthritis and lymphoproliferative disease: coincidence only? Clin Rheumatol. 2001;20(1):80–82.
24. Bussel, J, Cines, D. Immune thrombocytopenia purpura, neonatal alloimmune thrombocytopenia, and post-transfusion purpura. In Hoffman R, Benz EJ, Shattil SJ, eds.: Hematology: basic principles and practice, ed 4, New York: Churchill Livingstone, 2005.
25. Bussel, J, Kuter, DJ, George, JN, et al. AMG 531, a thrombopoiesis-stimulating protein, for chronic ITP. N Engl J Med. 2006;355(16):1672–1681.
26. Buzzard, B, Beeton, K. Physiotherapy management of hemophilia. London: Blackwell Science, 2000.
26a. Cahalin, LP. Efficacy of diaphragmatic breathing in persons with chronic obstructive pulmonary disease: a review of the literature. J Cardiopulm Rehab. 2002;22(1):7–21.
27. Caley, B, Reidenbach, F. Growing older with hemophilia. Hemaware. 2003;8(4):59–63.
28. Campbell, PJ, Green, AR. The myeloproliferative disorders. N Engl J Med. 2006;355(23):2452–2466.
29. Carobbio, A, Finazzi, G, Guerini, V, et al. Leukocytosis is a risk factor for thrombosis in essential thrombocythemia: interaction with treatment, standard risk factors and Jak2 mutation status. Blood. 2006;109(6):2310–2313.
30. Castagnola, C, Nozza, A, Corso, A, et al. The value of combination therapy in adult myeloid leukemia with central nervous system involvement. Haematologica. 1997;82:577–580.
31. Centers for Disease Control and Prevention. White paper: recommendations for maintaining high quality care for person with bleeding and clotting disorders. Bethesda, MD: National Center for Infectious Diseases, 2000.
32. Chanan-Khan, A, Porter, CW. Immunomodulating drugs for chronic lymphocytic leukaemia. Lancet Oncol. 2006;7(6):480–488.
33. Chaperot, L, et al. Differentiation of antigen-presenting cells (dendritic cells and macrophages) for therapeutic application in patients with lymphoma. Leukemia. 2001;14(9):1667–1677.
33a. Chaves, PH. Association between mild anemia and executive function impairment in community-dwelling older women: the Women’s Health and Aging Study II. J Am Geriatr Soc. 2006;54(9):1429–1435.
33b. Chaves, PH. Looking at the relationship between hemoglobin concentration prevalent mobility difficulty in older women. Should the criteria currently used to define anemia in older people be reevaluated? J Am Geriatr Soc. 2002;50(7):1257–1264.
33c. Chaves, PH. What constitutes normal hemoglobin concentration in community-dwelling disabled older women? J Am Geriatr Soc. 2004;52(11):1811–1816.
34. Chiroazzi, N, Rai, KR, Ferrarini, M. Chronic lymphocytic leukemia. N Engl J Med. 2005;352(8):804–815.
35. Chorba, TL, Holman, RC, Clarke, MJ, et al. Effects of HIV infection on age and cause of death for persons with hemophilia A in the United States. Am J Hematol. 2001;66:229–240.
36. Coffino, M. Profile of Sharon Funk, 2001-2002 Physical Therapy Excellence Fellowship Recipient. Hemaware. 2001;6(6):29–32.
37. Coleman, EA. Feasibility of exercise during treatment for multiple myeloma. Cancer Nurs. 2003;26(5):410–419.
38. Cook, SK, Coleman, EA. Exercise decisions within the context of multiple myeloma, transplant, and fatigue. Cancer Nursing. 2004;27(2):108–118.
39. Criteria for the classification of monoclonal gammopathies, multiple myeloma and related disorders. A report of the International Myeloma Working Group. Br J Haematol. 2003;121:749–757.
40. Curt, GA. Impact of cancer-related fatigue on the lives of patients: new findings from the Fatigue Coalition. Oncologist. 2000;5(5):353–360.
41. Cvetkovic, RS, Perry, CM. Rituximab: a review of its use in non-Hodgkin’s lymphoma and chronic lymphocytic leukaemia. Drugs. 2006;66(6):791–820.
42. Dawson, TM, et al. Epstein-Barr virus, methotrexate, and lymphoma in patients with rheumatoid arthritis and primary Sjögren’s syndrome: case series. J Rheumatol. 2001;28(1):47–53.
43. D’Cruz, D. Autoimmune diseases associated with drugs, chemicals and environmental factors. Toxicol Lett. 2000:421–432. [112-113].
44. Dean, E. Oxygen transport deficits in systemic disease and implications for physical therapy. Phys Ther. 1997;77(2):187–202.
45. Demers, C, Derzko, C, David, M, et al. Gynaecological and obstetric management of women with inherited bleeding disorders. J Obstet Gynaecol Can. 2005;27(7):707–732.
46. Denis, CV, Kwack, K, Saffaripour, S, et al. Interleukin 11 significantly increases plasma von Willebrand factor and factor VIII in wild type and von Willebrand disease mouse models. Blood. 2001;97:465–472.
47. Deniz, K. Breast cancer in women after treatment for Hodgkin’s disease. Lancet Oncol. 2003;4(4):207–214.
48. Diamond, C, Taylor, TH, Im, T, et al. Highly active antiretroviral therapy is associated with improved survival among patients with AIDS-related primary central nervous system non-Hodgkin’s lymphoma. Curr HIVRes. 2006;4(3):375–378.
49. Diamond, PT. Severe anemia: implications for functional recovery during rehabilitation. Disabil Rehabil. 2000;22(12):574–576.
50. Diehl, V, Franklin, J, Pfreundschuh, M, et al. Standard and increased-dose BEACOPP chemotherapy compared with COPP-ABVD for advanced Hodgkin’s disease. N Engl J Med. 2003;348(24):2386–2395.
51. Diehl, V, Re, D, Josting, A. Hodgkin’s disease: clinical manifestations, staging, and therapy. In Hoffman R, Benz EJ, Shattil SJ, eds.: Hematology: basic principles and practice, ed 4, New York: Churchill Livingstone, 2005.
52. Diel, IJ, Solomayer, EF, Bastert, G. Bisphosphonates and the prevention of metastasis: first evidences from preclinical and clinical studies. Cancer. 2000;88(Suppl 12):3080–3088.
53. Dilley, A, et al. Von Willebrand disease and other inherited bleeding disorders in women with diagnosed menorrhagia. Obstet Gynecol. 2001;97(4):639.
54. Dores, GM, Metayer, C, Curtis, RE, et al. Second malignant neoplasms among long-term survivors of Hodgkin’s disease: a population-based evaluation over 25 years. J Clin Oncol. 2002;20:3484–3494.
55. Dunn, AL, Busch, MT, Wyly, JB. Arthroscopic synovectomy for hemophilic joint disease in a pediatric population. J Pediatr Orthop. 2004;24(4):414–426.
56. Dunn, AL, Busch, MT, Wyly, JB. Radionuclide synovectomy for hemophilic arthropathy: a comprehensive review of safety and efficacy and recommendation for a standardized treatment protocol. Thromb Haemost. 2002;87(3):383–393.
57. Durstine, JL, et al. Physical activity for the chronically ill and disabled. Sports Med. 2000;30(3):207–219.
58. Dyall, J, et al. Lentivirus-transduced human monocyte-derived dendritic cells efficiently stimulate antigen-specific cytotoxic T lymphocytes. Blood. 2001;97(1):114–121.
59. Dybjer, A, et al. Seropositive polyarthritis and skin manifestations in T-prolymphocytic leukemia. Leuk Lymphoma. 2000;37(3-4):437–440.
60. Ekstrand, BC, Lucas, JB, Horwitz, SM, et al. Rituximab in lymphocyte-predominant Hodgkin disease: results of a phase 2 trial. Blood. 2003;101(11):4285–4289.
61. Ewenstein, BM, Gomperts, ED, Pearson, S, et al. Inhibitor development in patients receiving recombinant factor VIII (recombinate rAHF/bioclate): a prospective pharmacovigilance study. Haemophilia. 2004;10(5):491–498.
62. Fallon, KE, Bishop, G. Changes in erythropoiesis assessed by reticulocyte parameters during ultralong distance running. Clin J Sport Med. 2002;12(3):172–179.
63. Fenig, E, et al. Pregnancy and radiation. Cancer Treat Rev. 2001;27(1):1–7.
64. Finazzi, G, Rambaldi, A, Guerini, V, et al. Risk of thrombosis in patients with essential thrombocythemia and polycythemia vera according to JAK2 V617F mutation status. Haematologica. 2007;92(1):135–136.
65. Fourney, DR. Percutaneous vertebroplasty and kyphoplasty for painful vertebral body fractures in cancer patients. J Neurosurg. 2003;98(1 Suppl):21–30.
66. Franchini, M, Veneri, D, Lippi, G. The use of recombinant activated factor VII in congenital and acquired von Willebrand disease. Blood Coagul Fibrinolysis. 2006;17(8):615–619.
66a. Ganesh, R. Multiple myeloma of the cervical spine: treatment strategies for pain and spinal stability. J Neurosurg: Spine. 2006;5:140–145.
67. Gerard, L, Galicier, L, Boulanger, E, et al. Improved survival in HIV-related Hodgkin’s lymphoma since the introduction of highly active antiretroviral therapy. AIDS. 2003;17(1):81–87.
68. Gerber, L. Rehabilitation of the cancer patient. In: Rosenberg S, ed. Cancer: principles and practice of oncology. Philadelphia: J.B. Lippincott; 1997:2925–2956.
68a. Gilbert, MS, Radomisli, TE. Management of fixed flexion contracture of the elbow in haemophilia. Haemophilia. 1999;5(Suppl 1):39–42.
69. Gobbi, PG, Levis, A, Chisesi, T, et al. ABVD versus modified Stanford V versus MOPPEBVCAD with optional and limited radiotherapy in intermediate-and advanced-stage Hodgkin’s lymphoma: final results of a multicenter randomized trial by the Intergruppo Italiano Linfomi. J Clin Oncol. 2005;23(36):9198–9207.
70. Goodnought, LT, Shander, A, Spence, R. Bloodless medicine: clinical care without allogenic blood transfusion. Transfusion. 2003;43:668–676.
71. Goonaugh, LT, Shander, A, Spivak, JL. Detection, evaluation and management of anemia in the elective surgical patient. Anesth Analg. 2005;101:1858–1861.
72. Gorman, K. Sickle cell disease. Am J Nurs. 1999;99(3):1–14.
73. Greskovich, JF, Macklis, RM. Radiation therapy in pregnancy: risk calculation and risk minimization. Semin Oncol. 2000;27(6):633–645.
74. Guardino, AE, Rajapaksa, R, Ong, KH, et al. Production of myeloid dendritic cells (DC) pulsed with tumor-specific idiotype protein for vaccination of patients with multiple myeloma. Cytotherapy. 2006;8(3):277–289.
75. Gurney, B. Leg length discrepancy: a review. Gait & Posture. 2002;15(2):195–206.
76. Hardell, L, et al. Some aspects of the etiology of non-Hodgkin’s lymphoma. Environ Health Perspect. 1998;106(Suppl 2):679–681.
77. Hasenclever, D, Diehl, V. A prognostic score for advanced Hodgkin’s disease. International Prognostic Factors Project on Advanced Hodgkin’s Disease. N Engl J Med. 1998;339:1506–1514.
78. Herman-Hilker, S. Spring training: a model for sports and physical activity education. Hemaware. 2001;6(6):16–24.
78a. Hilliard, P. Haemophilia joint health score reliability study. Haemophilia. 2006;12:518–525.
79. Hockey, EG. Personal communication. Cleveland, Ohio: University Hospitals of Cleveland, 2001.
80. Hoffbrand, AV, Cohen, A, Hershko, C. Role of deferiprone in chelation therapy for transfusional iron overload. Blood. 2003;102:17–24.
81. Hogan, AM. An exploratory study of physiological correlates of neurodevelopmental delay in infants with sickle cell anaemia. Br J Haematol. 2006;132(1):99–107.
82. Hogan, AM, Kirkham, FJ, Prengler, M, et al. An exploratory study of physiological correlates of neurodevelopmental delay in infants with sickle cell anaemia. Br J Haematol. 2006;132(1):99–107.
83. Hollinger, FB, Kirtava, A, Oakley, M, et al. Blood safety monitoring among persons with bleeding disorder-United States, May 1998–June 2002. JAMA. 2003;289:541.
84. Hoppe, CC, Walters, MC. Bone marrow transplantation in sickle cell anemia. Curr Opin Oncol. 2001;13(2):85–90.
85. Horne, G, et al. Achieving pregnancy against the odds: successful implantation of frozen-thawed embryos generated by ICSI using spermatozoa banked prior to chemo/radiotherapy for Hodgkin’s disease and acute leukemia. Hum Reprod. 2001;16(1):107–109.
86. Huaux, JP, Geubel, A, Koch, MC, et al. The arthritis of hemochromatosis: a review of 25 cases with special reference to chondrocalcinosis, and a comparison with patients with primary hyperparathyroidism and controls. Clin Rheumatol. 1986;5:317.
87. Hunault, M, Harousseau, JL, Delain, M, et al. Better outcome of adult acute lymphoblastic leukemia after early genoidentical allogeneic bone marrow transplantation (BMT): a GOELAMS trial. Blood. 2004;104:3028–3037.
88. Iannone, R, Ohene-Frempong, K, Fuchs, EJ, et al. Bone marrow transplantation for sickle cell anemia: progress and prospects. Pediatr Blood Cancer. 2005;44(5):436–440.
89. Izaks, GJ, Westendorp, RG, Knook, DL. The definition of anemia in older persons. JAMA. 1999;281(18):1714–1717.
90. Jaffe, ES, Harris, NL, Stein, H, et al, eds. Pathology and genetics of tumours of haematopoietic and lymphoid tissues, vol 3 of World Health Organization classification of tumours. Lyons, France: IARC Press; 2001.
91. Jemal, A. Cancer statistics 2007. CA Cancer J Clin. 2007;57(1):43–66.
92. Joyner, LC. Leg length discrepancy and target joints. Hemaware. 2003;8(3):51–52.
93. Joyner, LC. Night splints. An option for treating chronic muscle tightness. Hemaware. 2003;8(3):53–55.
94. Karavatas, SG. Physical therapy management of patients with multiple myeloma: musculoskeletal considerations. Rehab Oncol. 2006;24(3):11–16.
95. Keating, MJ, O’Brien, S, Lerner, S, et al. Long-term follow-up of patients with chronic lymphocytic leukemia (CLL) receiving fludarabine regimens as initial therapy. Blood. 1998;92:1165–1171.
96. Keeney, S, Cumming, AM. The molecular biology of von Willebrand disease. Clin Lab Haematol. 2001;23:209–230.
97. Kempton, CL, Soucie, JM, Abshire, TC. Incidence of inhibitors in a cohort of 838 males with hemophilia A previously treated with factor VIII concentrates. J Thromb Haemost. 2006;4(12):2576–2581.
98. Kern, M, Blanchette, V, Stain, AM, et al. Clinical and cost implications of target joints in Canadian boys with severe hemophilia A. J Pediatr. 2004;145:628–634.
99. Khan, G. Epstein-Barr virus, cytokines, and inflammation: a cocktail for the pathogenesis of Hodgkin’s lymphoma? Exp Hematol. 2006;34(4):399–406.
100. Kinney, TR, et al. Safety of hydroxyurea in children with sickle cell anemia: results of the HUG KIDS Study, a phase I/II trial. Blood. 1999;94(5):1550–1554.
101. Kinney, TR, Sleeper, LA, Wang, WC, et al. Silent cerebral infarcts in sickle cell anemia: a risk factor analysis. Pediatrics. 1999;103:640–645.
102. Klepfish, A, et al. Advanced Hodgkin’s disease in a pregnant HIV seropositive woman: favorable mother and baby outcome following combined anticancer and antiretroviral therapy. Am J Hematol. 2000;63(1):57–58.
103. Klinge, J, Auberger, K, Auerswald, G, et al. Prevalence and outcome of intracranial haemorrhage in haemophiliacs-a survey of the paediatric group of the German Society of Thrombosis and Haemostasis (GTH). Eur J Pediatr. 1999;158(Suppl 3):S162–S165.
104. Kulkarni, R, Ponder, KP, James, AH, et al. Unresolved issues in diagnosis and management of inherited bleeding disorders in the perinatal period: a white paper of the Perinatal Task Force of the Medical and Scientific Advisory Council of the National Hemophilia Foundation, USA. Haemophilia. 2006;12(3):205–211.
105. Kyle, RA, Therneau, TM, Rajkumar, SV, et al. A long-term study of prognosis in monoclonal gammopathy of undetermined significance. N Engl J Med. 2002;346:564–569.
106. Lal, A, Fung, EB, Pakbaz, Z, et al. Bone mineral density in children with sickle cell anemia. Pediatr Blood Cancer. 2006;47(7):901–906.
107. Landolfi, R, Marchioli, R, Kutti, J, et al. Efficacy and safety of low-dose aspirin in polycythemia vera. N Engl J Med. 2004;350:114–124.
108. Lenahan, SE, et al. Transfusion-related acute lung injury secondary to biologically active mediators. Arch Pathol Lab Med. 2001;125(4):523–526.
109. Lethagen, S, Widell, A, Berntorp, E, et al. Clinical spectrum of hepatitis C-related liver disease and response to treatment with interferon and ribavirin in haemophilia or von Willebrand disease. Br J Haematol. 2001;113:87–93.
110. Levine, AM. Acquired immunodeficiency syndrome-related lymphoma: clinical aspects. Semin Oncol. 2000;27(4):442–453.
111. Lim, ST, Levine, AM. Recent advances in acquired immunodeficiency syndrome (AIDS)-related lymphoma. Cancer J Clin. 2005;55:229–241.
112. Little, RF, et al. HIV-associated non-Hodgkin lymphoma: incidence, presentation, and prognosis. JAMA. 2001;285(14):1880–1885.
113. Lorigan, P, Radford, J, Howell, A, et al. Lung cancer after treatment for Hodgkin’s lymphoma: a systematic review. Lancet Oncol. 2005;6(10):773–779.
114. Lowenberg, B, Downing, JR, Burnett, R. Acute myeloid leukemia. N Engl J Med. 1999;341:1051–1062.
114a. Lundin, B. Compatible scales for progressive and additive MRI assessments of haemophilic arthropathy. Haemophilia. 2005;11(12):109–115.
115. Mair, B, Leparc, GF. Hypotensive reactions associated with platelet transfusions and angiotensin-converting enzyme inhibitors. Vox Sang. 1998;74(1):27–30.
116. Manches, O, Plumas, J, Lui, G, et al. Anti-Gal-mediated targeting of human B lymphoma cells to antigen-presenting cells: a potential method for immunotherapy using autologous tumor cells. Haematologica. 2005;90(5):625–634.
117. Manno, CS, Pierce, GF, Arruda, VR, et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat Med. 2006;12(3):342–347.
118. Mannucci, PM. Treatment of von Willebrand’s disease. N Engl J Med. 2004;351(7):683–694.
119. Marchese, VG, Chiarello, LA. Relationships between specific measures of body function, activity, and participation in children with acute lymphoblastic leukemia. Rehab Oncol. 2004;22(2):5–9.
120. May, C, et al. Therapeutic haemoglobin synthesis in beta-thalassaemic mice expressing lentivirus-encoded human beta-globin. Nature. 2000;406(6791):82–86.
121. McNeil, J. Keeping fit during the college years. Hemaware. 2003;8(3):61–65.
122. Medical Research Council Haemostasis & thrombosis database resource sites-factor VIII database. Available at http://europium.csc.mrc.ac.uk Accessed January 26, 2007.
123. Migliazza, A, et al. Nucleotide sequence, transcription map, and mutation analysis of the 13q14 chromosomal region deleted in B-cell chronic lymphocytic leukemia. Blood. 2001;97(70):2098–2104.
124. Moore, BD. Neurocognitive outcomes in survivors of childhood cancer. J Pediatr Psychol. 2005;30:51–63.
125. Morbidity and Mortality Weekly Report. Update: newborn screening for sickle cell disease. MMWR. 2000;49(32):729–731.
126. Morcos, AC. Exercise heals. Hemaware. 2003;8(3):74–78.
127. Moylan, CA, Muir, AJ. Treatment of hepatitis C in special populations. Clin Liver Dis. 2005;9(4):567–577.
128. National Comprehensive Cancer Network Clinical practice guidelines inn oncology: cancer-related fatigue. V.1.2006. Available at http://www.nccn.org Accessed January 29, 2007.
129. National Comprehensive Cancer Network Multiple myeloma. V.2.2007. Available at http://www.nccn.org Accessed January 29, 2007.
130. National Hemophilia Foundation. Hemophilia, sports, and exercise. New York: National Hemophilia Foundation, 1996.
131. National Hemophilia Foundation. Hepatitis updates. Hemaware. 2000.
132. Nelson, J. Hemophilia and exercise. PT Magazine. 1994;2:60–66.
133. Nicklas, AH, Baker, ME. Imaging strategies in the pregnant cancer patient. Semin Oncol. 2000;27(6):623–632.
134. Norian, JM. Total knee arthroplasty in hemophilic arthropathy. J Bone Joint Surg (Am). 2002;84-A(7):1138–1141.
135. O’Brien, SG, Guilhot, F, Larson, RA, et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med. 2003;348(11):994–1004.
136. O’Connor, SR, Farmer, PB, Lauder, I. Benzene and non-Hodgkin’s lymphoma. J Pathol. 1999;189(4):448–453.
137. Oeffinger, KC, Mertens, AC, Sklar, CA, et al. Chronic health conditions in adult survivors of childhood cancer. N Engl J Med. 2006;355(15):1572–1582.
138. Ogundipe, O, et al. Sickle cell disease and nitrous oxide-induced neuropathy. Clin Lab Haematol. 1999;21(6):409–412.
139. Oyono-Enguelle, S, et al. Cardiorespiratory and metabolic responses to exercise in HbSC sickle cell patients. Med Sci Sports Exerc. 2000;32(4):725–731.
140. Ozawa, S, Shander, A, Ochani, TD. A practical approach to achieving bloodless surgery. AORN J. 2001;74(1):34–45.
141. Painter, P, et al. Low-functioning hemodialysis patients improve with exercise training. Am J Kidney Dis. 2000;36(3):600–608.
142. Palli, D, et al. Hodgkin’s disease risk is increased in patients with ulcerative colitis. Gastroenterol. 2000;119(3):647–653.
143. Panepinto, JA, et al. Universal versus targeted screening of infants for sickle cell disease: a cost-effectiveness analysis. J Pediatr. 2000;136(2):201–208.
144. Patel, DV, Rai, KR. Chronic lymphocytic leukemia. In Hoffman R, Benz EJ, Shattil SJ, eds.: Hematology: basic principles and practice, ed 4, New York: Churchill Livingstone, 2005.
145. Pellett, R. Sickle cell disease. Rehabil Oncol. 2000;18(2):10–13.
146. Persson, B, Fredrickson, M. Some risk factors for non-Hodgkin’s lymphoma. Int J Occup Med Environ Health. 1999;12(2):135–142.
147. Petrangelo, A. Hereditary hemochromatosis-a new look at an old disease. N Engl J Med. 2004;350(23):2383–2397.
148. Pileri, SA, Falini, B, Stein, H. Pathobiology of Hodgkin’s lymphoma. In Hoffman R, Benz EJ, Shattil SJ, eds.: Hematology: basic principles and practice, ed 4, New York: Churchill Livingstone, 2005.
149. Platt, OS. Preventing stroke in sickle cell anemia. N Engl J Med. 2005;353(26):2743–2745.
150. Platt, OS. The acute chest syndrome of sickle cell disease. N Engl J Med. 2000;342(25):1904–1907.
151. Platt, OS, Brambilla, DJ, Rosse, WF, et al. The Cooperative Study of Sickle Cell Disease: mortality in sickle cell disease: life expectancy and risk factors for early death. N Engl J Med. 1994;330:1639.
152. Plug, I, Van Der Bom, JG, Peters, M, et al. Mortality and causes of death in patients with hemophilia. J Thromb Haemost. 2006;4(3):510–516.
153. Pohlman, B, Macklis, RM. Lymphoma and pregnancy. Semin Oncol. 2000;27(6):657–666.
154. Popovsky, MA. Transfusion-related acute lung injury. Curr Opin Hematol. 2000;7(6):402–407.
155. Poulson, B, Gudas, SA. Multiple myeloma. Rehab Oncol. 2003;21(3):8–10.
156. Pui, C-H, Evans, WE. Treatment of acute lymphoblastic leukemia. N Engl J Med. 2006;354(2):166–178.
157. Richardson, PG, Sonneveld, P, Schuster, MW, et al. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N Engl J Med. 2005;352(24):2487–2498.
158. Riedel, DA, Pottern, LM. The epidemiology of multiple myeloma. Hematol Oncol Clin North Am. 1992;6:225.
159. Rizzo, A. CML: lessons learned when the therapist becomes the patient. Rehab Oncol. 2003;21(3):4–6.
160. Rodeghiero, F, Castaman, G, Dini, E. Epidemiological investigation of the prevalence of von Willebrand’s disease. Blood. 1987;69:454–459.
161. Rodon, P, et al. Multiple myeloma in elderly patients: presenting features and outcome. Eur J Haematol. 2001;66(1):11–17.
162. Rodruiquez-Merchan, EC. Therapeutic options in the management of articular contractures in haemophiliacs. Haemophilia. 1999;5(Suppl 1):5–9.
163. Rund, D, Ben-Yehuda, D. Therapy-related leukemia and myelodysplasia: evolving concepts of pathogenesis and treatment. Hematology. 2004;9(3):179–187.
164. Rund, D, Rachmilewitz, E. ?-thalassemia. N Engl J Med. 2005;353(11):1135–1146.
165. Sawka, MN, et al. Blood volume: importance and adaptations to exercise training, environmental stresses, and trauma/sickness. Med Sci Sports Exerc. 2000;32(2):332–348.
166. Schafer, AI. Thrombocytosis. N Engl J Med. 2004;350:1211–1219.
167. Schatz, J, McClellan, CB. Sickle cell disease as a neurodevelopmental disorder. Ment Retard Dev Disabil Res Rev. 2006;12(3):200–207.
168. Schwarz, HP, Schlokat, U, Mitterer, A, et al. Recombinant von Willebrand factor-insight into structure and function through infusion studies in animals with severe von Willebrand disease. Semin Thromb Hemost. 2002;28(2):215–226.
169. Shander, A. The safety and efficacy of bloodless cardiac surgery. Semin Cardiothorac Vasc Anesth. 2005;9(1):53–63.
170. Shander, A, Goodnough, LT. Objectives and limitations of bloodless medical care. Curr Opin Hematol. 2006;13:462–470.
171. Sharathkumar, A, Lillicrap, D, Blanchette, VS, et al. Intensive exposure to factor VIII is a risk factor for inhibitor development in mild hemophilia A. J Thromb Haemost. 2004;1(6):1228–1236.
172. Sharpe, K, Ashenden, MJ, Schumacher, YO. A third generation approach to detect erythropoietin abuse in athletes. Haematologica. 2006;91(3):356–363.
173. Shaskey, DJ, Green, GA. Sports hematology. Sports Med. 2000;29(1):27–38.
174. Silva, M, Luck, JV, Jr. Long-term results of primary total knee replacement in patients with hemophilia. J Bone Joint Surg (Am). 2005;87(1):85–91.
175. Sokolova, Y. Acute shoulder pain and swelling in a 68-year-old man. J Musc Med. 2000;17(11):699–700.
176. Soucie, JM, et al. Mortality among males with hemophilia: relations with source of medical care. The Hemophilia Surveillance System Project Investigators. Blood. 2000;96(2):437–442.
176a. Soucie, JM, Cianfrini, C, Janco, RL, et al. Joint range-of-motion limitations among young males with hemophilia: prevalence and risk factors. Blood. 2004;103(7):2467–2473.
177. Spahn, DR, Casutt, M. Eliminating blood transfusions. N Aspects Perspect Anesth. 2000;93(1):242–255.
178. Steen, RG, Miles, MA, Helton, KJ, et al. Cognitive impairment in children with hemoglobin SS sickle cell disease: relationship to MR imaging findings and hematocrit. AJNR Am J Neuroradiol. 2003;24(3):382–389.
179. Steinberg, MH, Barton, F, Castro, O, et al. Effect of hydroxyurea on mortality and morbidity in adult sickle cell anemia: risks and benefits up to 9 years of treatment. JAMA. 2003;289:1645–1651.
180. Stephensen, D. Rehabilitation of patients with haemophilia after orthopedic surgery. Haemophilia. 2005;11(Suppl 1):26–29.
181. Stokes, M, Young, A. The contribution of reflex inhibition to arthrogenous muscle weakness. Clin Sci. 1984;67:714.
182. Strong, A, Karavatas, SG. Recommended exercise protocol to decrease cancer-related fatigue and muscle wasting in patients with multiple myeloma: an evidence-based systematic review. Top Geriatr Rehabil. 2006;22:172–186.
183. Sullivan, KJ, et al. Nitric oxide successfully used to treat acute chest syndrome of sickle cell disease in a young adolescent. Crit Care Med. 1999;27(11):2563–2568.
184. Sultan, SM, Ioannou, Y, Isenberg, DA. Is there an association of malignancy with systemic lupus erythematosus? An analysis of 276 patients under long-term review. Rheumatol (Oxford). 2000;39(10):1147–1152.
185. Takahashi, R, et al. A newly developed biphosphonate, YM529, is a potent apoptosis inducer of human myeloma cells. Leuk Res. 2001;25(1):77–83.
186. ten Cate, H. Pathophysiology of disseminated intravascular coagulation in sepsis. Crit Care Med. 2000;28(9 Suppl):S9–S11.
187. Thomas, VN. The role of cognitive-behavioral therapy in the management of pain in patients with sickle cell disease. J Adv Nurs. 1998;27:1002–1009.
188. Vacek, MM, Ma, H, Gemignani, F, et al. High-level expression of hemoglobin A in human thalassemic erythroid progenitor cells following lentiviral vector delivery of an antisense snRNA. Blood. 2003;101:104–111.
189. Vichinsky, EP. Pulmonary hypertension in sickle cell disease. N Engl J Med. 2004;350:857–859.
190. Vichinsky, EP, et al. The perioperative complication rate of orthopedic surgery in sickle cell disease: report of the National Sickle Cell Surgery Study Group. Am J Hematol. 1999;62(3):129–138.
191. von Kempis, J, et al. Intravascular lymphoma presenting as symmetric polyarthritis. Arthritis Rheum. 1998;41(6):1126–1130.
192. Wagstaff, AJ, Keating, GM. Anagrelide: a review of its use in the management of essential thrombocythaemia. Drugs. 2006;66:111–131.
193. Walters, MC. Impact of bone marrow transplantation for symptomatic cell disease: an interim report. Blood. 2000;95(6):1918–1924.
194. Walters, MC. Stem cell therapy for sickle cell disease: transplantation and gene therapy. Hematol Am Soc Hematol Educ Prog. 2005:66–73.
195. Walz, C, Sattler, M. Novel targeted therapies to overcome imatinib mesylate resistance in chronic myeloid leukemia (CML). Crit Rev Oncol Hematol. 2006;57(2):145–164.
196. Wang, A, Frick, N. HIV/AIDS updates: exercise may be even more beneficial than you think. Hemaware. 2000.
197. White, CA. Radioimmunotherapy in non-Hodgkin’s lymphoma: focus on 90Y-ibritumomab tiuxetan (Zevalin). J Exp Ther Oncol. 2004;4(4):305–316.
198. Wirthwein, DP, et al. Death due to microvascular occlusion in sickle-cell trait following physical exertion. J Forensic Sci. 2001;46(2):399–401.
199. Wolk, A, et al. A prospective study of obesity and cancer risk. Cancer Causes Control. 2001;12(1):13–21.
200. Wong, O, Raabe, GK. Non-Hodgkin’s lymphoma and exposure to benzene in a multinational cohort of more than 308,000 petroleum workers, 1937 to 1996. J Occup Environ Med. 2000;42(5):554–568.
201. World Health Organization Fifty-ninth World Health Assembly 2006. Sickle cell anaemia: report by the secretariat Available at http://www.who.int/gb/ebwha/pdf_files/WHA59/A59_9-en.pdf Accessed Jan. 25, 2007.
202. Wright, MJ. Proficiency of balance in children and youth who have had acute lymphoblastic leukemia. Phys Ther. 2005;85(8):782–790.
203. Yeh, HS, Berenson, JR. Treatment for myeloma bone disease. Clin Cancer Res. 2006;12(20 Pt 2):6279s–6284s.
204. Yuille, MR, et al. Familial chronic lymphocytic leukemia: a survey and review of published studies. Br J Haematol. 2000;109(4):794–799.
205. Zavadsky, AJ. Platelet disorders and their implications on physical therapy intervention. Rehab Onol. 2001;19(3):11–13.
206. Zeerleder, S, Hack, E, Wuillemin, WA. Disseminated intravascular coagulation in sepsis. Chest. 2005;128:2864–2875.
207. Zhan, F, Huang, Y, Colla, S, et al. The molecular classification of multiple myeloma. Blood. 2006;108(6):2020–2028.
208. Zoller, H, Vogel, W. Iron supplementation in athletes-first do no harm. Nutrition. 2004;20(7–8):615–619.