The Hepatic, Pancreatic, and Biliary Systems
The liver has more than 500 separate functions such as the conversion and excretion of bilirubin (red bile pigment, which is an end-product of heme from hemoglobin in red blood cells [RBCs]). The liver is the sole source of albumin and other plasma proteins and also produces 500 to 1500 ml of bile each day. Other important functions of the liver include production of clotting factors and storage of vitamins. The liver and gut are the key organs in nutrient absorption and metabolism; nutrients bind to toxins in this pathway and aid in eliminating these toxins from the body.
The liver contributes to a functional immune system by reducing the amount of toxins that could impair the gut lining, which in turn helps prevent the entry of bacteria and viruses into the system. Bile acids, drugs, chemicals, and toxins undergo extensive enterohepatic circulation during the processes of metabolism. The liver also filters all of the blood from the gastrointestinal (GI) system and is therefore the primary organ for metastasis of intestinal cancer.
The pancreas is both an exocrine and an endocrine gland. Its primary function in digestion is exocrine secretion of digestive enzymes and pancreatic juices, transported through the pancreatic duct to the duodenum. Proteins, carbohydrates, and fats are broken down in the duodenum, aided by pancreatic and other secretions, which also help to neutralize the acidic substances passed from the stomach to the duodenum. The endocrine function involves the secretion of glucagon and insulin by islet of Langerhans cells for the regulation of carbohydrate metabolism. Pancreatic disease may result in a variety of clinical presentations, depending on whether the exocrine or endocrine function has been impaired.
The gallbladder, acting as a reservoir for bile, stores and concentrates the bile during fasting periods and then contracts to expel the bile into the duodenum in response to the arrival of food. Bile helps in alkalinizing the intestinal contents and plays a role in the emulsification, absorption, and digestion of fat. The signal for the gallbladder to contract comes from the release of cholecystokinin, a hormone released into the bloodstream from the wall of the duodenum and upper small intestine.
SIGNS AND SYMPTOMS OF HEPATIC DISEASE
Primary signs and symptoms of liver diseases vary and can include GI symptoms, edema/ascites, dark urine, light-colored or clay-colored feces, and right upper abdominal pain (Box 17-1). Impairment of the liver can result in hepatic failure when either the mass of liver cells is sufficiently diminished or their function is impaired as a result of cirrhosis, liver cancer, or infection and/or inflammation. Hepatic failure does not refer to one specific morphologic change but rather to a clinical syndrome that includes hepatic encephalopathy, renal failure (hepatorenal syndrome), endocrine changes, and jaundice.
Dark urine and light stools occur in association with jaundice (yellow pigmentation of skin, sclerae, and mucous membranes) (see the section on Jaundice in this chapter) when the serum bilirubin level increases from normal (0.1 to 1.0 mg/dl) to a value of 2 or 3 mg/dl. Any damage to the liver impairs bilirubin metabolism from the blood. Normally, bile converted from bilirubin causes brown coloration of the stool. Light-colored (almost white) stools and urine the color of tea or cola indicate an inability of the liver or biliary system to excrete bilirubin properly.
Skin changes associated with the hepatic system include jaundice, pallor, and orange or green skin. When bilirubin reaches levels of 2 to 3 mg/dl, the sclera of the eye takes on a yellow hue. When bilirubin level reaches 5 to 6 mg/dl, the skin becomes yellow. The changes described here in urine, stool, or skin color may be caused by hepatitis, gallbladder disease, pancreatic cancer blocking the bile duct, hepatotoxic medications, or cirrhosis. Other skin changes may include bruising, spider angiomas, and palmar erythema.
Spider angiomas (arterial spider, spider telangiectasis, or vascular spider) are branched dilations of the superficial capillaries, which may be vascular manifestations of increased estrogen levels (hyperestrogenism) (see Fig. 10-3). Spider angiomas and palmar erythema both occur in the presence of liver impairment as a result of increased estrogen levels normally metabolized by the liver. Palmar erythema (warm redness of the skin over the palms, also called liver palms) especially affects the hypothenar and thenar eminences and pulps of the finger. The soles of the feet may be similarly affected. The person may complain of throbbing, tingling palms.
Neurologic symptoms, such as confusion, sleep disturbances, muscle tremors, hyperreactive reflexes, and asterixis (see following discussion), may occur. When liver dysfunction results in increased serum ammonia and urea levels, peripheral nerve function can be impaired. Ammonia from the intestine (produced by protein breakdown) is normally transformed by the liver to urea, glutamine, and asparagine, which are then excreted by the renal system. When the liver does not metabolize and detoxify ammonia, ammonia is transported to the brain, where it reacts with glutamate (excitatory neurotransmitter) to produce glutamine. The reduction of brain glutamate impairs neurotransmission, leading to altered central nervous system (CNS) metabolism and function.
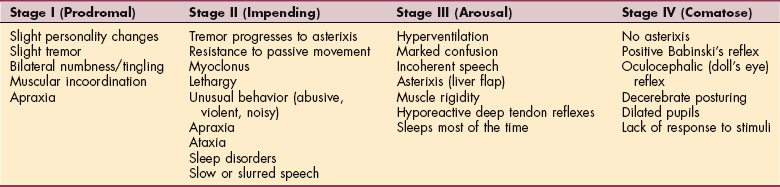
Asterixis and numbness or tingling (misinterpreted as carpal tunnel syndrome) can occur as a result of this ammonia abnormality, causing intrinsic nerve pathology. Asterixis, also called flapping tremors or liver flap, is a motor disturbance; specifically, it is the inability to maintain wrist extension with forward flexion of the upper extremities. A test for asterixis is asking the client to extend the wrist and hand with the rest of the arm supported on a firm surface or with the arms held out in front of the body. Observe for quick, irregular extensions and flexions of the wrist and fingers (Fig. 17-1). Altered neurotransmission, in the form of impaired inflow of joint and other afferent information to the brainstem reticular formation, causes the movement dysfunction.
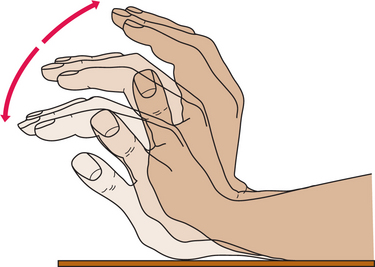
Figure 17-1 Flapping tremor. The flapping tremor elicited by attempted wrist extension while the forearm is fixed is the most common neurologic abnormality associated with liver failure. It can also be observed in uremia, respiratory failure, and severe heart failure. The tremor is absent at rest, decreased by intentional movement, and maximal on sustained posture. It is usually bilateral, although one side may be affected more than the other. (From Sherlock S, Dooley J: Diseases of the liver and biliary system, ed 9, Oxford, 1993, Blackwell Scientific Publications.) Blackwell Scientific Publications
Musculoskeletal locations of pain associated with the hepatic and biliary systems include thoracic pain between scapulae, right shoulder, right upper trapezius, right interscapular, or right subscapular areas. Sympathetic fibers from the biliary system are connected through the celiac and splanchnic (visceral) plexuses to the hepatic fibers in the region of the dorsal spine. These connections account for the intercostal and radiating interscapular pain that accompanies gallbladder disease. Although the innervation is bilateral, most of the biliary fibers reach the cord through the right splanchnic nerves, producing pain in the right shoulder.
Hepatic osteodystrophy, abnormal development of bone, can occur in all forms of cholestasis (bile flow suppression) and hepatocellular disease, especially in the alcoholic. Bone pain may occur especially in the presence of osteomalacia or more often, osteoporosis. Hepatic osteoporosis is secondary to osteoblastic dysfunction rather than to excessive bone resorption. The pathogenesis is probably complex, and factors include calcium malabsorption, alcohol, corticosteroid therapy, estrogen deficiency in the postmenopausal woman, and vitamin D deficiency with secondary hyperparathyroidism.
Vertebral wedging, vertebral crush fractures, and kyphosis can be severe; decalcification of the rib cage and pseudofractures occur frequently. Pseudofractures or Looser’s zones are narrow lines of radiolucency (areas of darkness on x-ray film) usually oriented perpendicular to the bone surface. This may represent a stress fracture that has been repaired by the laying down of inadequately mineralized osteoid, or these sites may occur as a result of mechanical erosion caused by arterial pulsations, as arteries frequently overlie sites of pseudofractures.
Osteoporosis associated with primary biliary cirrhosis and primary sclerosing cholangitis parallels the severity of liver disease rather than the duration. Painful osteoarthropathy may develop in the wrists and ankles as a nonspecific complication of chronic liver disease.
Portal hypertension, ascites, and hepatic encephalopathy are three other major complications of liver disease that are discussed in greater depth in this chapter as distinct clinical conditions.
AGING AND THE HEPATIC SYSTEM
The liver decreases in size and weight with advancing age, requiring more time to process substances, medications, and alcohol. Liver function test results (see Chapter 40), such as levels of aspartate transaminase (AST), alanine transaminase (ALT), γ-glutamyl transpeptidase (GGTP), alkaline phosphatase (ALP), and total serum bilirubin remain unchanged and within normal limits established for the adult. However, these tests often measure hepatic damage rather than overall function; abnormal values for these tests in older adults reflect disease rather than the effects of aging.
The decreased liver weight is accompanied by diminished blood flow. This combination of decreased liver mass and blood flow may account for some changes in drug elimination observed in the older adult. Many factors may influence drug metabolism in the older adult, including changes in body composition, decline in renal function, alterations in serum albumin levels (drugs such as sulfonamides and salicylates compete with bilirubin for binding sites on albumin, a transport plasma protein), individual variability, poor compliance, lack of understanding of drug therapy, presence of other systemic illnesses, and taking multiple drugs simultaneously (drug-drug interaction).
In the past, adjusting dosage downward based on hepatic function related to aging has not been considered necessary. However, emerging data on the use of pharmacologic agents in the older population suggest that the administration of many agents (especially chemotherapeutic agents) may be affected by physiologic changes occurring with age. Dose adjustments to compensate for pharmacokinetic and pharmacodynamic changes that occur in the older adult are being studied and adjustments for specific drugs presented.80
The liver itself becomes more fibrotic, but this change is not synonymous with pathologic cirrhotic changes. On autopsy, a buildup of brown pigment is seen, which is a result of accumulated unexcretable metabolic residue (end-stage metabolic products of lipids and proteins) acquired over a lifetime. No apparent physiologic significance has been identified with this pigment. Changes in liver oxidative status and function and the activity of the enzymatic antioxidant defense system, as well as the influence of aging on the susceptibility of the hepatic system to hepatotoxins, remain the focus of ongoing animal studies.118
Other studies are bringing to light the effects of physiologic age-related changes in the hepatic system such as the significance of growth hormone synthesis by the hepatic system responsible for maintaining various body tissues (e.g., bones, muscles, cardiovascular system, immune system, and CNS). Researchers are particularly interested in examining whether treatment with exogenous growth hormone can retard or reverse hepatic age-related changes in body structure and function.11
The pancreas undergoes structural changes, such as fibrosis, fatty acid deposits, and atrophy, but the pancreas has a large reserve capacity, and 90% of its function would have to be lost before any observable dysfunction occurs. Much remains unknown about the gallbladder in the aging process, but aging apparently has little effect on gallbladder size, contractility, or function. The gallbladder releases less bile into the liver, allowing more time for gallstones to develop. There is some evidence that moderate and vigorous physical activity enhances the function of the gallbladder as measured by reduced risk of gallbladder removal in physically active women.75
HEALING IN THE HEPATIC SYSTEM
In general, older organs may not adapt to injury as well as younger organs, so severe hepatic illnesses (e.g., severe hepatitis) may not be tolerated as well by the older person. Delayed or impaired tissue repair may require longer time for recovery of homeostasis. Chronic liver disease is associated with intrapulmonary shunting and reduced pulmonary vascular function. Vascular changes in the lungs have been well documented in people with chronic liver disease and reflect the importance of the liver in the control of vasoactive substances that regulate normal fluid balance, including lung fluids.28
Liver injury is followed by complete parenchymal regeneration, the formation of scars, or a combination of both. The outcome depends on the extent and chronicity of the insult. Chronic hepatic injury, such as chronic viral hepatitis or alcoholic liver injury, destroys the extracellular matrix framework. This type of destruction results in a combination of regenerated nodules separated by bands of fibrous connective tissue (fibrosis), which is termed cirrhosis.
Orthotopic liver transplantation has become an established therapy for end-stage liver disease (e.g., cirrhosis caused by alcohol abuse or hepatitis C [HCV]), acute liver failure, and primary biliary cirrhosis or primary sclerosing cholangitis, as well as for nonalcoholic cirrhosis and hepatic or biliary malignancy. Biliary atresia is the most common indication for pediatric liver transplantation. Theoretically, anyone with advanced, irreversible liver disease with certain mortality may be considered for a liver transplant provided the disease could be corrected by liver transplantation (see Table 21-5).
Current animal research is centered on identifying and harvesting specific stem cells from the bone marrow that under special conditions will convert into functioning liver tissue. In human research, a new procedure called hepatocyte transplantation is being pioneered. In this procedure, billions of donor liver cells are injected by intravenous infusion into the blood with the hope that the cells will correct life-threatening liver problems that would otherwise require a liver transplant. Other research efforts are working toward the development of bioartificial devices for liver support. An effective temporary liver support system could improve the chance of survival with or without a transplant as the final treatment (see Chapter 21).
LIVER
As a result of the extraordinary number of vital functions the liver performs, severe complications result when the liver has been damaged or is no longer functioning. Jaundice is a symptom that occurs with many types of diseases and disorders (both acute and chronic). End-stage complications occur most often because of cirrhosis and include portal hypertension, hepatic encephalopathy, ascites, and the hepatorenal syndrome. Any illness, toxin, or infection that leads to end-stage liver disease can display these complications.
Jaundice (Icterus)
Jaundice or icterus is not a disease but is rather a common symptom of many different diseases and disorders (Box 17-2). It is clinically characterized by yellow discoloration of the skin, sclerae, and mucous membranes. Jaundice occurs either as a result of an overproduction of bilirubin, defects in bilirubin metabolism (in uptake by the liver or conjugation), the presence of liver disease, or obstruction of bile flow.
In the normal breakdown of hemoglobin, the end product is bilirubin. In this metabolic process, the heme portion of hemoglobin is converted into biliverdin and then to bilirubin in the bone marrow and the spleen. Bilirubin is released into the bloodstream, where it binds to albumin, and is then taken up by hepatocytes to be conjugated with glucuronic acid. Once it is conjugated, it is then released into the bile. A small percentage of conjugated bile returns to the plasma and is excreted into the kidneys. In the terminal ileum and colon, conjugated bilirubin is deconjugated and excreted as colorless urobilinogen. Diseases that result in ineffective erythropoiesis (abnormal formation of erythrocytes) produce large amounts of bilirubin due to chronic hemolysis or destruction of cells.
Some diseases, such as Gilbert’s syndrome, have defects in the liver’s ability to conjugate bilirubin. Drugs, such as rifampin, may compete with bilirubin for uptake by the liver, decreasing the quantity of bilirubin the liver can process. Diseases, toxins, infections, and ischemia can cause generalized liver disease (acute and chronic), which reduces the capability of the liver to function normally and process bilirubin. Finally, bile ducts can be obstructed by diseases, tumors, and stones, leading to an elevation in bilirubin that has been conjugated.
As mentioned, jaundice is not clinically evident (particularly in the sclera of the eyes) until the plasma level reaches 3 mg/dl. Once the level reaches 5 to 6 mg/dl, the skin becomes a yellow color. Urine turns a darker color and stool is light in color. Signs and symptoms of liver disease may also be present.
Laboratory testing can aid in the specific diagnosis of jaundice. Bilirubin can be reported as conjugated or unconjugated, which provides a more accurate measurement than direct and indirect. Direct refers to the capability of the laboratory to directly measure conjugated bilirubin, while the indirect bilirubin refers to the unconjugated portion of bilirubin, which cannot be directly measured in the laboratory, and must be subtracted from the total bilirubin present in the blood.
Conjugated and direct are not always equivalent, particularly in disorders of bilirubin metabolism. In clients with jaundice, an elevation in the conjugated bilirubin is more common than unconjugated. Elevations in liver transaminases (AST and ALT) suggest that liver disease is involved. Many other tests are available for the specific diagnosis of jaundice, depending on the suspected process, and are included in the specific disease sections in this chapter.
Cirrhosis
Cirrhosis is the final common pathway of chronic, progressive inflammation of the liver. It is characterized pathologically by a progressive loss of normal tissue that is replaced with fibrosis and nodular regeneration. There are many diseases, medications, and toxins that can damage the liver and ultimately lead to cirrhosis, but the most common in the United States include alcohol abuse and HCV.
Overall, in the United States, cirrhosis is the twelfth leading cause of death, accounting for about 28,000 deaths a year.7 Cirrhosis of the liver occurs when inflammation (from disease or toxin) causes liver tissue damage and/or necrosis. With continued cycles of inflammation and healing, fibrous bands of connective tissue replace normal liver cells. These fibrous bands eventually constrict and partition the liver into irregular nodules. Once 80% to 90% of the liver is replaced with scar tissue, there is also significant loss of function, associated with decompensation of homeostasis.
The signs and symptoms of cirrhosis (Figs. 17-2 and 17-3) are multiple and varied, representing interference with major functions of the liver, and include processing dietary amino acids, carbohydrates, lipids, and vitamins; metabolizing cholesterol, hormones, vitamins, medications, and toxins; producing clotting factors and other plasma proteins; and storing glycogen.
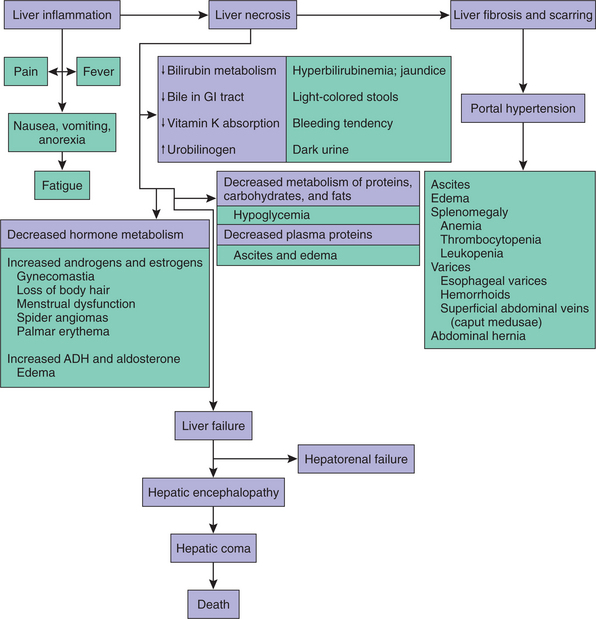
Figure 17-2 Pathologic basis (purple boxes) and resultant clinical manifestations (green boxes) associated with cirrhosis of the liver.
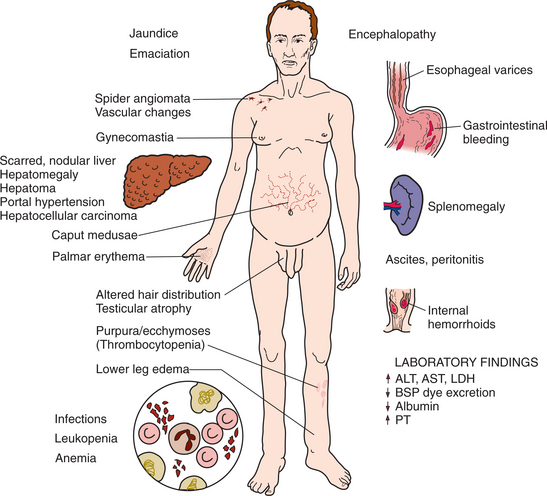
Figure 17-3 Liver cirrhosis. Clinical presentation and laboratory findings associated with liver cirrhosis. ALT, Alanine aminotransferase; AST, aspartate aminotransferase; LDH, lactate dehydrogenase; BSP, sulfobromophthalein; PT, prothrombin time. (From Black JM, Matassarin-Jacobs E, eds: Medical-surgical nursing: clinical management for continuity of care, ed 5, Philadelphia, 1997, WB Saunders.)
Clients with cirrhosis exhibit fatigue, weight loss, jaundice, coagulopathies, loss of ability to metabolize drugs, and hypoalbuminemia (the remaining serious complications are discussed later). History, physical examination, laboratory tests, and imaging tests aid in diagnosing the specific cause. Once cirrhosis has developed, it is usually not reversible, although each disease may have a specific therapy to reduce the risk of developing cirrhosis. Typically, complications are treated on an individual basis and transplantation provides the best therapy for long-term survival.
Portal Hypertension
Portal hypertension is defined as an increase in hepatic sinusoidal (sinuses in the liver where blood flows) pressure over 6 mm Hg. Portal refers to the area where blood vessels enter into the liver. Venous blood returning from the stomach, large and small intestine, pancreas, and spleen is transported via the portal vein to the liver (the splanchnic circulation). Most cases of portal hypertension are related to cirrhosis. Other causes include thrombus, tumor, infection, or may be idiopathic. With the development of cirrhosis, hyperdynamic vascular responses and mechanical factors prevent the flow of blood, resulting in increased pressure and resistance in the portal circulatory system (Fig. 17-4).
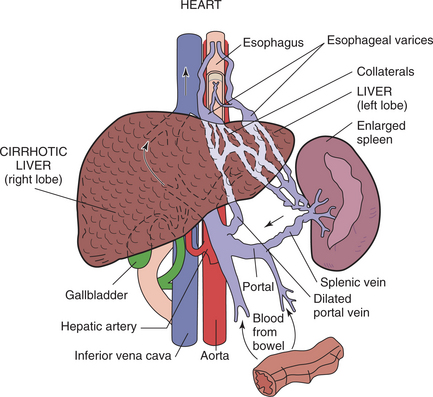
Figure 17-4 Portal hypertension. Normally, in the portal venous system (consisting of the portal veins, sinusoids, and hepatic veins), the portal veins carry blood from the GI tract, gallbladder, pancreas, and spleen to the liver. Veins collecting from these sites form the splenic vein and superior and inferior mesenteric veins, which in turn merge to create the portal vein. Portal hypertension occurs when portal venous pressure exceeds the pressure in the nonportal abdominal veins (e.g., inferior vena cava) by at least 6 mm Hg. As portal pressure rises, increased resistance to blood flow causes blood pooling in the spleen and the development of collateral channels formed in an effort to equalize pressures between these two venous systems. These collateral vessels or varices bypass the liver and cause large, tortuous veins, especially in the esophagus (esophageal varices).
A reduction in nitric oxide release from liver endothelial cells and production of endothelin-1 leads to intrahepatic vasoconstriction, while increased blood flow from the portal vein and splanchnic circulation further increases the pressure. Fibrosis, nodularity, and abnormal liver architecture combine to form mechanical barriers to blood flow and increase the resistance.
As a result of this increased portal pressure, blood that normally flows to the portal vein is reversed and blood begins to flow back to the stomach, esophagus, umbilicus, and rectum. This system is normally small with modest blood flow. However, in the cirrhotic state, blood flow increases in these vessels, causing dilation and expansion. These engorged vessels give rise to rectal varices, prominent vessels around the umbilicus (caput medusae), and gastroesophageal varices. The collateral veins of the stomach and esophagus are the most likely to bleed because of a lack of communicating vessels.
Gastroesophageal varices are one of the most serious complications of portal hypertension, occurring in 40% of people with cirrhosis. Endoscopy should be performed in all clients with cirrhosis to screen for varices. Follow-up endoscopy is scheduled, depending on the presence and severity of varices. Clinical manifestations of gastroesophageal bleeding include hematemesis or melena (or both). The blood is usually dark red in color. Over half of bleeds stop spontaneously and over 90% of bleeds can be controlled with therapy. But serious bleeding can quickly result in hypovolemia, shock, and death. Treatment is aimed at preventing bleeding by decreasing portal blood flow and intrahepatic pressure.
Nonselective β-blockers aid in preventing bleeding and rebleeding, whereas vasopressin, octreotide, and somatostatin are used to decrease splanchnic flow. Nitrates may relieve intrahepatic vasoconstriction. Endoscopic sclerotherapy can be used prophylactically or in clients who do not tolerate medications; variceal band ligation can be utilized in acute esophageal bleeding and to prevent rebleeding. Surgical shunts or more commonly, placement of a transjugular intrahepatic portacaval shunt (TIPS) can divert blood around the liver and away from the collateral systems.
Prognosis is poor for clients with repeated esophageal varices, and liver transplantation should be pursued.
Hepatic Encephalopathy
Hepatic encephalopathy or portosystemic encephalopathy refers to a potentially reversible, decreased level of consciousness in people with severe liver disease. This complication can occur with both acute and chronic liver disease. People with chronic end-stage liver disease often have an insidious onset; initially there are mild changes in ability to concentrate and complete complex tasks. As the hepatic encephalopathy progresses, mental status changes become more obvious and are classified in stages I to IV, depending on the severity of neurologic involvement (Table 17-1).
Stage I is characterized by impaired attention, depression, and some personality changes; neurologic signs include tremor and incoordination. Stage II displays drowsiness, sleep disorders, changes in behavior, and poor short-term memory; accompanying signs are asterixis (flapping tremor; see Fig. 17-1), ataxia, and slurred speech. The motor apraxia in stage II can be best observed by keeping a record of the client’s handwriting and drawings of simple shapes such as a circle, square, triangle, and rectangle. Progressive deterioration is apparent in these handwriting samples.
Confusion and somnolence are indicative of stage III encephalopathy associated with nystagmus, clonus, muscular rigidity, and hypoactive reflexes. Stage IV reflects severe encephalopathy; the person is in a comatose state and exhibits abnormal reflexes, such the oculocephalic (“doll’s eye”) reflex; decerebrate posturing may be present; pupils may be dilated; and there is no response to stimuli. There are characteristic electroencephalogram (EEG) findings at each stage.
The cause of hepatic encephalopathy has not been completely elucidated, although several key elements are known. First, laboratory animal models and clinical experience have shown that high-protein meals can lead to encephalopathy. It is felt that the liver is unable to process nitrogenous metabolites, leading to elevated levels of ammonia and other toxins (although these “other toxins” have not been well described).
The second element is the shunting of blood away from the hepatic portal system (because of cirrhosis) to the vena cava, which also worsens encephalopathy. Ammonia is one cause of encephalopathy that is fairly well documented but is certainly not the sole cause. Research is beginning to show how elevated ammonia and other metabolic abnormalities (including changes in neurotransmitters in the brain and circulating amino acid levels) combine to result in encephalopathy.
Ammonia is created by bacteria in the colon from the metabolism of protein and urea. Ammonia is absorbed into the portal blood system and is 5 to 10 times higher there than in the general circulatory system. The liver is typically able to metabolize ammonia, but with liver disease and shunting of blood away from the liver (particularly to the brain), ammonia levels rise. The kidneys are also a source of ammonia, which is increased in the face of diuretic use and hypokalemia. Muscles aid in ammonia removal but cirrhosis is often accompanied by muscle wasting. In the brain, ammonia appears to directly alter the function and signaling of nerve cells.
Ammonia also appears to affect glutamate and γ-aminobutyric acid (GABA) signaling. The GABA receptor complex is the principal nerve network, which inhibits the CNS. When GABA or benzodiazepines bind to this complex, there is an inhibition in the functioning of the brain. The liver contains high concentrations of GABA, and dysfunction of the liver may contribute to higher levels of GABA.
Ammonia is known to combine with α-ketoglutarate in the CNS to form glutamate, which is in turn used to produce GABA. Ammonia production is therefore tied to glutamate and GABA production. Although serum ammonia levels are used to monitor therapy, the level does not correspond well with the severity of encephalopathy, reinforcing the fact that other mechanisms are involved in the development of encephalopathy.
The development of hepatic encephalopathy warrants a careful evaluation and correction of the cause. Serious and common causes include bleeding, infection (particularly spontaneous bacterial peritonitis), hypovolemia, or electrolyte abnormalities (hypokalemia). Other common factors that may precipitate or severely aggravate hepatic encephalopathy include constipation, diuretics, increased dietary protein, and CNS-depressant drugs, such as alcohol, benzodiazepines (e.g., Librium, Valium, Dalmane, and Tranxene), and opiates.
Because protein can precipitate or worsen encephalopathy, many of the symptoms can be improved by eliminating or reducing sources of protein (i.e., stopping any internal bleeding and restricting dietary protein to 60 g/day). Health care providers must also be aware of any subtle changes in mental status since the ability to drive is often impaired. In one study, clients with cirrhosis but no evidence of hepatic encephalopathy underwent psychomotor testing; 60% were found unfit to drive and 25% displayed questionable driving skills.147 Driving must be assessed on a case-by-case basis.
Lactulose (or Lactitol) is a first-line pharmacologic therapy that decreases nitrogenous compounds from being absorbed from the gut and increases transit time through the intestine (the goal is 2 to 4 bowel movements a day). Ammonia-lowering therapy in suspected encephalopathy cases can be beneficial even when the ammonia level is normal since the production is tied to other toxins.
Neomycin is an antibiotic that decreases the bacterial count in the intestine but carries nephrotoxicities and ototoxicities as a result of systemic absorption. The new medication rifaximin (a nonabsorbable antibiotic) has been shown in preliminary studies to be effective in hepatic encephalopathy.93 Flumazenil is a benzodiazepine-receptor antagonist that may block the GABA receptor complex, reversing inhibition. It has been shown to improve hepatic encephalopathy in the short term but has no effect on survival.3
Reversal of hepatic encephalopathy is typically successful when a source is identified, corrected, and treated appropriately. However, without intervention, mortality is high, as the person’s condition progresses into coma. Similar to most complications of end-stage liver disease, liver transplantation provides the best long-term treatment.
Ascites
Ascites is the abnormal accumulation of fluid within the peritoneal cavity. Ascites is most often caused by cirrhosis (85% of cases), but other diseases associated with ascites include heart failure, abdominal malignancies, nephrotic syndrome, infection, and malnutrition.
The mechanism for the accumulation of fluid in the case of cirrhosis is principally a result of portal hypertension. High pressure in the vessels attempting to pass blood through the cirrhotic liver leads to vasodilatation of the splanchnic vessels (vessels to the gut or viscera), which in turn decreases the filling of the vessels going to the kidney. The renin-angiotensin-aldosterone system is activated, resulting in sodium and water retention. However, because of the high pressure in the liver and splanchnic vessels, excessive lymph is produced that leaks into the tissues and eventually into the abdominal cavity.10
Ascites becomes clinically detectable when more than 500 ml has accumulated, causing weight gain, abdominal distention, increased abdominal girth, and eventually, peripheral edema (Fig. 17-5). Dyspnea with increased respiratory rate occurs when the fluid displaces the diaphragm.
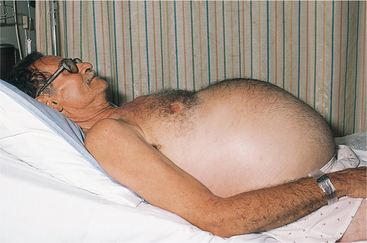
Figure 17-5 Ascites in an individual with cirrhosis. Distended abdomen, dilated upper abdominal veins, and inverted umbilicus are classic manifestations. Peripheral edema associated with developing ascites may be observed first by the therapist. (From Swartz MH: Textbook of physical diagnosis, ed 5, Philadelphia, 2006, WB Saunders.)
Diagnosis of ascites is usually based on clinical manifestations in the presence of liver disease. Paracentesis is used as the initial test in people with new-onset ascites to determine the cause. Fluid is sent to the laboratory for chemical and microscopic evaluation. Abdominal ultrasonography can aid in locating pockets of ascitic fluid that may be loculated (formed or divided into small cavities or compartments).
In people with established cirrhosis, paracentesis can be diagnostic and therapeutic. Large volume paracentesis with administration of albumin is the treatment of choice for tense ascites (i.e., when a person is no longer able to breathe or eat comfortably), followed by the use of diuretics to reduce reaccumulation of fluid.
Treatment of mild-to-moderate ascites includes sodium restriction accompanied by diuretic use.140 Fluid restriction is appropriate when the serum sodium decreases to less than 120 to 125 mEq/L. True refractory ascites (i.e., meaning not responsive to therapy) is uncommon and can be treated with serial large-volume paracentesis or TIPS. This alleviates pressure in the portal area but may induce hepatic encephalopathy caused by bypassing the liver (see Hepatic Encephalopathy section).
Prophylactic antibiotics may be used in some clients with a history of spontaneous bacterial peritonitis (SBP) and are often administered to people with a GI bleed.172 The development of refractory ascites is associated with a poor prognosis, with a 12-month survival of 25%. Liver transplantation provides the best treatment option but is not always readily available.
Complications include hepatorenal syndrome (discussed later) and SBP. The microbial source of SBP (infection of the ascitic fluid) is the gut, where organisms (typically Escherichia coli, streptococci [mostly pneumococci], and Klebsiella) are translocated into lymph nodes and then into the ascitic fluid. Bacterial peritonitis is symptomatic in 87% of cases (fever, chills, abdominal pain, mental status changes, and tenderness), but symptoms can often be subtle. Without diagnosis by paracentesis and antibiotic treatment, the infection can be fatal.
Hepatorenal Syndrome
Hepatorenal syndrome is characterized by renal dysfunction in people with portal hypertension and advanced liver failure, but with normal renal tubular function (i.e., the kidney tubular cells function). About 7% to 15% of people with end-stage cirrhosis will develop this syndrome, which portends a poor prognosis. This syndrome is hypothesized to occur as a result of portal hypertension in which increased vascular pressure from liver disease causes vasodilation of the blood vessels in the splanchnic circulation (the body’s attempt to reduce this pressure). This vasodilation leads to underfilling of the arteries and a low blood pressure elsewhere in the body, which particularly affects the blood pressure in the kidney (which is very sensitive to blood pressure). Because the blood pressure the kidney “sees” is low, there is an activation of the renin-angiotensin-aldosterone system in an attempt to constrict the blood vessels. This leads to constriction of the vessels in the limbs and to the brain, as well as the kidneys. The total effect is that the vasoconstrictors have a greater effect than the vasodilators, and kidney dysfunction develops.46
Criteria defining the diagnosis of hepatorenal syndrome were established by the International Ascites Club and include the presence of advanced liver disease with portal hypertension; serum creatinine of greater than 1.5 mg/dl; urine protein of less than 500 mg/dl; and most importantly, the absence of other causes for kidney involvement.
Common illnesses found in people with cirrhosis that can cause renal insufficiency include infection (particularly spontaneous bacterial peritonitis), shock, medications, bleeding, and fluid losses. Renal obstruction should be “ruled out” by ultrasonography. With the presence of these features, along with the failure to improve after diuretics are removed and 1 to 1.5 liters of saline given, suggests the diagnosis of hepatorenal syndrome.
Hepatorenal syndrome is classified into two types. Type 1 is rapid both in onset and progression to renal failure and carries a poor short-term prognosis. Type 2 is more insidious in onset with progression over months; ascites is often the key feature of this type. Because of the intense vasoconstriction, treatment centers around the use of vasodilators and albumin, which aid in increasing blood flow to the kidneys (such as vasopressin analogues or proportional variant-adrenergic agonists). Transjugular intrahepatic portosystemic shunts (TIPS) may also be of benefit.9
Optimal treatment consists of liver transplantation, which provides a 5-year posttransplant survival rate of 70%. Although many people will improve with medical treatment, liver transplantation should be pursued because of poor long-term prognosis. Hemodialysis may be required to bridge treatment until a transplant is available.
Hepatitis
Hepatitis is an acute or chronic inflammation of the liver caused by a virus, a chemical, a drug reaction, or alcohol abuse. Classifications of hepatitis discussed in this text are listed in Box 17-2. Six different identifiable hepatitis viruses (A, B, C, D, E, and G) are responsible for more than 95% of all viral-induced cases worldwide. The letter F was not skipped; it represents the fulminant form of hepatitis, the generic term for any rapidly progressing form of liver inflammation resulting in hepatic encephalopathy within a few weeks of developing infection. Fulminant hepatitis is not strictly viral induced but can develop as a result of any form of hepatitis.
Other viral causes of hepatitis include Epstein-Barr virus (mononucleosis), herpes simplex virus types I and II, varicella-zoster virus, measles, or cytomegalovirus (CMV). Hepatitis from any cause produces very similar symptoms and usually requires a careful client history to establish the diagnosis.
People with mild-to-moderate acute hepatitis rarely require hospitalization. The emphasis is on preventing the spread of infectious agents and avoiding further liver damage when the underlying cause is drug-induced or toxic hepatitis. Persons with fulminant hepatitis (which has a severe, sudden intensity and is sometimes fatal) require special management because of the rapid progression of the disease and the potential need for urgent liver transplantation.
Chronic Hepatitis
Chronic hepatitis comprises several diseases that are grouped together because they have common clinical manifestations and are all marked by chronic necroinflammatory injury that can lead insidiously to cirrhosis and end-stage liver disease. The disease is defined as chronic with evidence of ongoing injury for 6 months or more.
Previously, chronic hepatitis was classified as either chronic persistent hepatitis (CPH) or chronic active hepatitis (CAH), but recent advances in understanding of the causes and natural history of this type of liver injury have led to the elimination of these terms. Now chronic hepatitis is described in diagnostic terms that include the etiology, degree of active inflammation and injury (i.e., grade: mild, moderate, severe, or I, II, III), and the degree of scarring or how advanced the process is (i.e., stage: I, II, or III; IV represents cirrhosis). Stages of disease are usually irreversible.
Chronic hepatitis has multiple causes, including viruses, medications, metabolic abnormalities, and autoimmune disorders. Despite extensive testing, some cases cannot be attributed to any known cause and are probably the result of as yet unidentified viruses. Hepatitis B (HBV), with or without hepatitis D (HDV); HCV; and hepatitis G (HGV) can progress to chronic hepatitis.
Most people with chronic hepatitis are asymptomatic, and when symptoms occur, these are nonspecific and mild, with fatigue, malaise, loss of appetite, polyarthralgias, and intermittent right upper quadrant discomfort. Some people report sleep disturbances or difficulty in concentrating. Symptoms of advanced disease or an acute exacerbation include nausea, poor appetite, weight loss, muscle weakness, itching, dark urine, and jaundice. Once cirrhosis is present, weakness, weight loss, abdominal swelling, edema, easy bruising, muscle wasting and weakness, gastrointestinal bleeding, and hepatic encephalopathy with mental confusion may arise.
The diagnosis of chronic viral hepatitis is based on serologic testing. The strict definition of chronic hepatitis (from any cause) is based on histologic features of hepatocellular necrosis and chronic inflammatory cell infiltration in the liver, but the diagnosis can usually be made from clinical features and blood test results alone.
Liver biopsy is important to assess the severity of underlying liver disease (grade and stage) and to determine the need for antiviral treatment. The treatment for chronic viral hepatitis has improved substantially in the last decade and depends on the underlying cause and grade and stage of disease. With the advances currently being made in the fields of antiviral and immunomodulatory therapeutics, it is anticipated that the considerable progress made in treating these diseases over the past decade will continue in the future.
The prognosis in chronic hepatitis is variable depending on the development of cirrhosis and other complications such as hepatocellular carcinoma (HCC). Male gender, moderate-to-severe alcohol consumption, and other coexistent liver disorders are the factors that increase the rate of progression to cirrhosis; HCV is the major risk factor of development of liver cancer. The 5-year survival rate for compensated cirrhosis is greater than 90%, but the prognosis and survival rate for decompensation (characterized by development of variceal bleeding, ascites, and hepatic encephalopathy) are extremely poor. Progression of chronic hepatitis to decompensated cirrhosis is an indication for liver transplantation.
Fulminant Hepatitis
Fulminant hepatitis is the generic term for any rapidly progressing form of liver inflammation that results in hepatic encephalopathy (confusion, stupor, and coma) within a few weeks of developing infection. This type of hepatitis is rare, occurring in less than 1% of persons with acute viral hepatitis, but can be fatal. The most common causes are acetaminophen hepatotoxicity (20% to 25% of all cases of fulminant hepatitis),79 idiosyncratic drug reaction, hepatitis A (HAV) and HBV, and hepatic ischemia. Encephalopathy may progress to cerebral edema, which is the most common cause of death. In addition to liver failure, numerous complications can occur, including infection, hypoglycemia, coagulation defects, lactic acidosis, GI hemorrhage, electrolyte disturbances, and renal insufficiency.
Diagnosis is made in the presence of a combination of hepatic encephalopathy, acute liver disease (elevated serum bilirubin and transaminase levels), and liver failure. Treatment is supportive because the underlying etiologic factors of liver failure are rarely treatable short of liver transplantation.
Prognosis is determined in part by the cause of the condition. If the prognosis is deemed poor and no contraindications to transplantation are present, the person should be immediately considered for transplantation. Short-term prognosis without liver transplantation is very poor, and the mortality rate is high (80%). Despite the poor prognosis, complete recovery can occur as a result of liver cell regeneration with recovery of liver function. The development of artificial liver support devices has not been shown to improve outcome nor are they widely available.
Viral Hepatitis21
Overview.: Each of the recognized hepatitis viruses belongs to a different virus family, and each has a unique epidemiology. Characteristics of these strains of viruses are presented in Table 17-2. The identification of the specific virus is made difficult by the fact that a long incubation period often occurs between acquisition of the infection and development of the first symptoms. The incubation period for HAV is 15 to 50 days, 1 to 6 months for HBV, and 1 week to 6 months for HCV. Not all causative agents have been identified, and because hepatitis can be easily spread before symptoms appear, morbidity is high in terms of loss of time from school or work. More than half and possibly as many as 90% of all cases go unreported because symptoms are mild or even subclinical.
Table 17-2
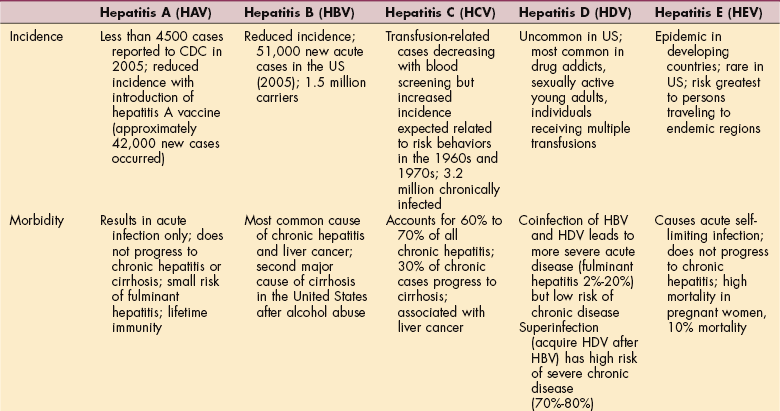
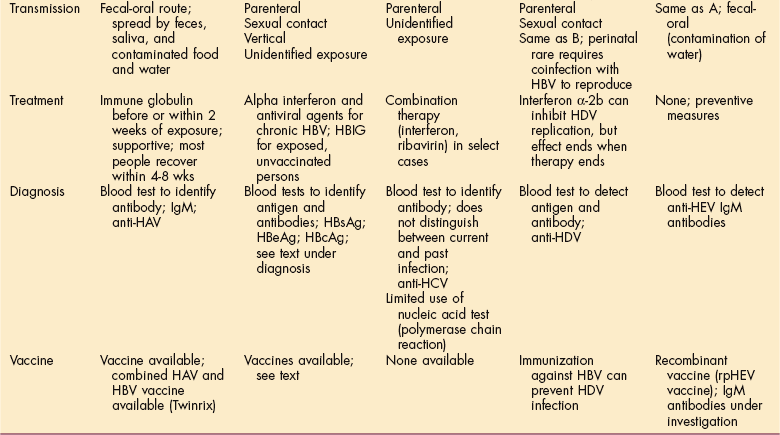
HBsAg, Hepatitis B surface antigen; HBeAg, hepatitis B “e” antigen; HBcAg, hepatitis B core antigen; IgM, immunoglobulin M. From Centers for Disease Control and Prevention (CDC): National Center for Infectious Diseases (online): http://www.cdcod/diseases/hepatitis.2007.
Hepatitis A virus (HAV), formerly known as infectious hepatitis, is transmitted by the oral-fecal route. The oral-fecal route of transmission is primarily from poor or improper handwashing and personal hygiene, particularly after using the bathroom and then handling food for public consumption. This route of transmission also may occur through the shared used of oral utensils such as straws, silverware, and toothbrushes.
Major outbreaks of HAV occur when people consume contaminated water or food. HAV most commonly affects children, men who have sex with men (MSM), and people who live or travel in underdeveloped countries (Table 17-3). HAV is rarely transmitted through transfused blood, and little placental transmission occurs, although the antibody is often detected in infants of infected mothers.
Table 17-3
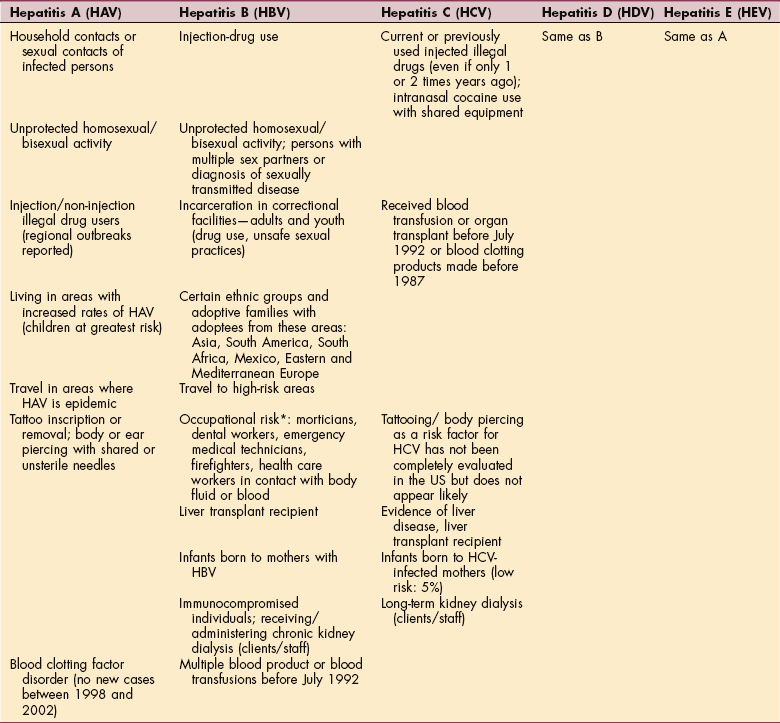
*HBV can also survive in dried blood at least 1 week.
From Centers for Disease Control and Prevention (CDC): National Centers for Infectious Diseases (online): http://www.cdc.gov/ncidod/diseases/hepatitis.2007.
HAV is highly contagious, with the peak time of viral excretion and contamination occurring during the 2-week period before the onset of jaundice. Thus the greatest danger of infection is during the incubation period, when a person is unaware that the virus is present. The illness can last from 4 to 8 weeks; it generally lasts longer and is more severe in persons older than 50 years or in people with chronic, underlying liver disease.42
Hepatitis B virus (HBV) is transmitted percutaneously (i.e., puncture of the skin) or through mucosal contact. HBV is highly infectious: 100 times more infectious than human immunodeficiency virus (HIV) and 10 times more infectious than HCV. Because HBV can be transmitted through heterosexual or homosexual intercourse, it is considered a sexually transmitted disease. The average incubation period is 90 days (with a range of 60 to 150 days) with symptoms occurring around 60 days.94
Hepatitis C virus (HCV), formerly posttransfusion non-A, non-B hepatitis associated with blood transfusion, is now most commonly associated with injection-drug use. As with HAV, the period of infectivity begins before the onset of symptoms, and the person may become a lifetime carrier of this virus. Clinically, HCV is very similar to HBV and often is asymptomatic; the acute HCV infection is usually mild. Chronic HCV varies greatly in its course and outcome from asymptomatic with normal liver function to mild degree of liver injury and overall good prognosis to severe symptomatic HCV with complications of cirrhosis and end-stage liver disease.
Hepatitis D virus (HDV), or delta virus, is a defective single-stranded RNA that presents as a coinfection or superinfection of HBV. This virus requires hepatitis B surface antigen (HBsAg) for its replication, so only individuals with HBV are at risk for hepatitis D. Risk factors and transmission mode are the same as for HBV; parenteral drug users have a high incidence of HDV. The symptoms of HDV are similar to those of HBV except that clients are more likely to have fulminant hepatitis and to develop chronic active hepatitis and cirrhosis.
Hepatitis E virus (HEV), previously known as enteric non-A, non-B hepatitis, is transmitted by contaminated water via the oral-fecal route and clinically resembles HAV. It is believed to be nonfatal, although it has been clearly associated with liver damage. A 20% to 25% mortality rate exists in pregnant women from fulminant hepatitis.2 This virus tends to occur in poor socioeconomic conditions, primarily occurs in developing countries (contaminated waste water and sewage), and is rare in the United States. No specific treatment is available for HEV, but ensuring clean drinking water remains the best preventive strategy.
Hepatitis G virus (HGV) is most prevalent in African countries. It is parenterally transmitted, although vertical and sexual transmissions are well documented. Parenteral refers to transmission in some other mode than via the alimentary canal (enteric system) such as by subcutaneous, intramuscular, intraorbital, intracapsular, intraspinal, intrasternal, or intravenous injection. It may cause acute or chronic infection, but little information about this viral cause of hepatitis is known.146 HGV has been identified as the causative agent of approximately 20% of posttransfusion hepatitis cases and approximately 15% of community-acquired hepatitis cases that are not caused by HAV. Although present in liver tissue, pathogenicity to the liver has not been proved.
Incidence and Risk Factors.: Each year, approximately 500,000 Americans are infected with some form of hepatitis virus; annually about 15,000 persons die from its complications. HAV is the predominant type of hepatitis, causing 40% to 60% of acute viral hepatitis cases, although the number of affected people in the United States has been on the decline, with about 4500 acute cases reported in 2005 (probably 42,000 new cases occurred when taking into account asymptomatic and unreported cases).167
Since HAV is transmitted via the fecal-oral route, one risk factor for acquiring the virus is working at a daycare center; children who attend daycare centers are also at higher risk. Because of the HAV vaccine the infection rate in this population has improved; this may change the prevalence of the disease, with more cases being reported in adults (adults display a more severe clinical course than children).33 Another risk factor for HAV is visiting or living in an underdeveloped country where the rate is high.
The incidence of HBV has also declined, with only 5400 acute cases reported in 2005 and a total of about 51,000 new cases (if asymptomatic and reported cases are taken into account).167 Prevalence of HBV infection has significantly decreased in most ethnic populations except blacks, who continue to demonstrate an elevated prevalence of three times that of other racial/ethnic populations.94 Prevalence varies most according to risk factors for acquisition, so for parenterally transmitted hepatitis (B, C, D, and G), the highest rates are among persons with direct percutaneous blood exposures such as injection-drug users.
Common risk factors for HBV include sexual relations, injection-drug use, sharing needles, needlesticks, and perinatal (vertical) transmission from mother to child. Injection-drug use and intimate contact with another person with HBV are the two most frequent sources of HBV in the United States. Transfusion-related HBV is rare, since the initiation of donor screening for HBV and the HBV vaccine have improved the incidence in dialysis clients and workers. Only 670 new cases of HCV were reported in 2005; including asymptomatic cases, the actual number of new cases was probably close to 20,000 cases. Men were more likely to contract HCV than women, but the incidence was fairly equal among various ethnic groups/populations. HCV deceased 58% among the age group of 20 to 39 years, where it had been the highest.
In the past, HCV infection was commonly acquired through blood transfusion. Currently, because of donor screening, the incidence of HCV from transfusion is uncommon. The number of people with chronic HCV is significant: about 3.2 to 4 million people are chronically infected with HCV. Most of these cases were acquired during the 1970s and 1980s when the rates were the highest.
Risk factors for HCV are similar to those for HBV. In 2005, injection-drug use was the most common risk factor and having multiple sex partners second (although transmitting HCV by sexual contact is inefficient). Currently, 68% of the new cases of HCV occur among injection-drug users sharing needles. Up to one third of individuals with a bleeding disorder are coinfected with HIV and persons with a bleeding disorder who were infected with HIV also contracted HCV (before screening for HCV was made available). Vertical transmission is not common in HCV. See Table 17-3 for other risk factors.
The number of cases of people who developed HBV/HCV infection from dialysis or work-related exposure has become low. People undergoing chronic hemodialysis are at risk for HBV/HCV infection because the process of hemodialysis requires vascular access for prolonged periods. In an environment where multiple clients receive dialysis concurrently, repeated opportunities exist for person-to-person transmission of infectious agents, directly or indirectly via contaminated devices, equipment and supplies, environmental surfaces, or hands of personnel. Furthermore, individuals receiving hemodialysis are immunosuppressed, which increases their susceptibility to infection, and they require frequent hospitalizations and surgery, which increases their opportunities for exposure to nosocomial infections.106
HBV is relatively stable in the environment and remains viable for at least 7 days on environmental surfaces at room temperature. HBsAg has been detected in dialysis centers on clamps, scissors, dialysis machine control knobs, and doorknobs. Thus blood-contaminated surfaces that are not routinely cleaned and disinfected represent a reservoir for HBV transmission. Dialysis staff members can transfer virus to clients from contaminated surfaces by their hands or gloves or through use of contaminated equipment and supplies.106
HEV is most common in developing countries. On the basis of serological tests, an estimated one-third of the world’s population has been infected with HEV. In India, the lifetime prevalence is more than 60%.153
Pathogenesis.: The viruses associated with hepatitis are not typically cytopathic (destroy cells), yet the body’s reaction to the virus often creates significant inflammation; the intensity of the disease depends on the degree of immune response.18,135 Initially, cytokines (interferons) and natural killer cells are employed to remove virus from the body. Later, antigen-specific T cells (maturated in the lymph tissue) enter the liver to aid in the removal of virus; antibodies prevent spread of virus and provide immunity against further infection. In adults with intact immunity, this response is able to clear the virus. However, in infants, young children, and the immunosuppressed, the immune system is unable to mount an adequate defense and the virus continues to replicate and reside in the liver, leading to a chronic state. In these people, there is a weak response with few antigen-specific T cells.
In HCV, the virus is able to bypass the immune system in most cases. Current theories include T-cell exhaustion, T-cell dysfunction, viral escape mutations, or rapid T-cell deletion in the liver.114 Antibodies are not produced soon after infection with HCV and in some cases do not develop at all.
Clinical Manifestations.: Most cases of acute viral hepatitis are asymptomatic and never reported (for HAV, HBV, and HCV). Up to 70% of children under the age of 6 years remain asymptomatic with HAV infection.42 Infants, children under the age of 5, and immunosuppressed adults typically have no symptoms associated with acute HBV infection, while most cases of HCV infection are subclinical and not reported.
Classic symptoms of acute hepatitis are often the same, regardless of the responsible virus. Most individuals present with malaise, fatigue, mild fever, nausea, vomiting, anorexia, right upper quadrant discomfort, and occasionally diarrhea. Jaundice, dark urine, and clay-colored stools may also be observed, particularly with acute HAV, HDV, HEV, and frequently in HBV (30% of cases).
Acute HCV usually does not present with jaundice. In over 95% of adults with normal immunity, infection with HBV is self-limited; but in 5% of adults, 30% of children (under the age of 5), 90% of infants, and those with immunodeficiencies the infection becomes chronic. Most people who acquire HCV become chronic carriers of the disease (60% to 85%).
Some people may develop extrahepatic manifestations (more frequent in HCV than in HBV) such as essential mixed cryoglobulinemia, porphyria cutanea tarda, lichen planus, rheumatoid arthritis, Hodgkin’s lymphoma, and diabetes mellitus. Rheumatologic and skin manifestations are the most common. Individuals with acute HBV may also demonstrate extrahepatic symptoms including rash, arthralgias, and arthritis.
MEDICAL MANAGEMENT
Prevention takes place at three levels: primary, secondary, and tertiary. Primary prevention involves primary immunization (HAV, HBV, and HEV), education regarding food preparation and proper handwashing, avoiding needle punctures by contaminated needles (or other similar infective material), and practicing protective sex or avoiding sexual contact during the period of HBsAg positivity.
Secondary prevention involves passive immunization following exposure to HAV or HBV, travel precautions when visiting areas where hepatitis is endemic (e.g., avoid drinking unbottled water or beverages served with ice; avoid eating foods rinsed in contaminated water, such as fruits and vegetables; and avoid eating shellfish).
Tertiary prevention involves education to those infected about preventing possible infectivity to others and self-care during active infection (e.g., avoid strenuous activity and ingestion of hepatotoxins, such as alcohol and acetaminophen; some advocate alternative treatment such as herbs, acupuncture, and dietary measures).
Hepatitis A.
Preventive measures include HAV vaccine, standard precautions, and immune globulin (IG), which is a preparation of antibodies against HAV. IG can be used before infection and provides passive immunity for 3 to 5 months or can be given to household and intimate contacts of people with HAV. When given after exposure, IG is 80% to 90% effective in preventing HAV infection. The sooner IG is given after exposure, the more efficacious it is.42 People who have received the HAV vaccine at least 1 month before exposure do not require IG.
HAV vaccine is recommended (before exposure) for persons 1 year of age and older (as a standard childhood vaccination), adults wishing to obtain immunity, or anyone who is at risk for infection (e.g., persons traveling to areas with intermediate to high rates of endemic HAV, persons living in communities with high endemic rates or periodic outbreaks of HAV infection, men who have sex with men or who are bisexual and their partners of either gender, and clients with chronic liver disease). HAV vaccine confers 97% to 100% protection in children and 94% to 100% in adults.
Hepatitis B.
Hepatitis B is a preventable disease achieved by administering a HBV vaccine. Currently, two vaccines are available as single-antigen vaccines: Recombivax HB and Engerix-B. Three are available as combination vaccines: Twinrix (for HAV and HBV in adults), Comvax (for HBV and Haemophilus influenzae type B in children), and Pediarix (for HBV, diphtheria and tetanus toxoids, pertussis, and poliovirus in children). The vaccine is given in 3 doses over a period of 6 months.
Life-long protective immunity develops in more than 90% of healthy adults. In the unvaccinated person, hepatitis B IG (HBIG) is used for postexposure prophylaxis (PEP) and should be administered soon after the exposure, followed by the HBV series at 0, 1, and 6 months. Effectiveness of PEP decreases with time between exposure and treatment. PEP should be given in less than 7 days from exposure for needlesticks and less than 14 for sexual exposure. Immunity to HBV also confers immunity to HDV; therefore HBV vaccination is recommended for preexposure immunization prophylaxis for HDV.
Once individuals begin to engage in behaviors associated with high-risk groups, they may become infected before vaccine can be given. A major obstacle in eliminating HBV is identifying persons and vaccinating them before they become infected. For this reason, in the United States, it is now recommended that all infants, health care workers, and persons in the high-risk category for HBV receive the HBV vaccine.
According to the Occupational Safety and Health Administration (OSHA) Bloodborne Pathogen Standard (1991), HBV vaccination must be offered to all employees within 10 days of employment. Records related to this vaccination must be maintained. Employees who decline vaccination must sign a standardized declination form.
Children younger than 5 years of age, if infected, have a high risk of becoming HBV carriers. Testing to identify pregnant women who are HBsAg positive and providing their infants with immunoprophylaxis effectively prevents HBV transmission during the perinatal period. The HBsAg is a core protein antigen of the HBV present in the nuclei of infected cells. Presence of HBsAg in the blood usually indicates the individual is infectious. HBc antibodies appear during the acute infection but do not protect against reinfection.
The World Health Organization (WHO) has recommended that all countries integrate HBV vaccination into their national immunization programs, and much progress has been made toward the goal to control, eliminate, and eradicate hepatitis in the coming generations.163
Hepatitis C.
Currently, no vaccine is available to prevent HCV because of the rapidity of HCV mutations in adaptation to the environment72 and no immunoglobulin (Ig) is effective in treating exposure. The only means of preventing new cases of HCV are to screen the blood supply, encourage health care professionals to take precautions when handling blood and body fluids, and educate people about high-risk behaviors (particularly injection-drug users and those involved with multiple sex partners).
Hepatitis E.
A new recombinant protein vaccine (rpHEV) has been developed and shown effective in preventing HEV in high-risk populations.153
DIAGNOSIS.
In addition to the history and clinical examination, serologic and molecular testing help provide an accurate diagnosis. Serology is the standard for diagnosis of viral hepatitis (see Table 17-2). People with a positive IgM HAV have acute disease, whereas IgG HAV is present at the onset of disease and remains in the blood for life. IgG HAV signifies previous exposure, whereas IgM HAV signifies only acute infection.
Serology relating to HBV changes as the disease progresses or is contained. HBV has an acute phase, a convalescent phase, and a chronic phase (Table 17-4). Early in HBV infection, the serologic marker HBsAg is positive, but the antibodies are negative until an immune response has been mounted by the body. Acute infection is demonstrated by a positive HBsAg and the presence of antibodies total anti-HBc (antibody to the hepatitis B core antigen) and IgM anti-HBc (IgM is an acute antibody). Resolving infection has eliminated the HBsAg, and antibodies remain present (anti-HBsAg, or the antibody to the surface antigen, may also be present). A serologic pattern positive for anti-HBc and anti-HBs demonstrates past infection and existing immunity.
Table 17-4
Interpretation of Serologic Results of Hepatitis B Testing

HBsAg, Hepatitis B surface antigen; anti-HBc, antibody to hepatitis B core antigen; anti-HBs, antibody to hepatitis B surface antigen, HBeAg, hepatitis B e antigen.
Chronic infection is marked by the presence of HBsAg and anti-HBc. Vaccinated people display a positive anti-HBsAg. HBeAg appears early in the acute infection and continued presence in the chronic state suggests increased infectivity, greater number of virus replication, and the need for antiviral therapy.
HBV deoxyribonucleic acid (DNA) can be detected in persons with chronic infection by polymerase chain reaction (PCR) techniques89; this information is useful in treatment and prognosis. None of the serodiagnostic tests are able to determine the current extent of liver damage; a liver biopsy provides information about the severity of disease and establishes grading and degree of fibrosis but is only necessary in the case of chronic hepatitis.
Testing for HCV includes an initial screening for anti-HCV, which, if positive, is followed by a more specific serologic test (such as RIBA) or a nucleic acid test (NAT). The best method is the detection of HCV ribonucleic acid (RNA) in the serum. Liver biopsy is not required but is often helpful in determining severity and making treatment decisions.
Transaminases (enzymes normally present in hepatocytes) are released from the acutely damaged hepatocytes, and serum transaminase levels rise, often to levels exceeding normal levels by twentyfold. An elevated serum bilirubin level is usually found. Liver function tests (see Table 40-5) can indicate other viral liver diseases, drug toxicity, or alcoholic hepatitis. Researchers are working to develop improved molecular techniques for increased diagnostic precision for all the hepatitis viruses.104
TREATMENT.
Treatment options have expanded and prevention methods are available for two of the six viruses although no cure is available for the viruses that are responsible for chronic liver disease (HBV, HDV, and HGV). The development of new antiviral agents that inhibit steps in the viral replication process is under investigation.110 A potential cure for HCV has been announced (see further discussion later). Any hepatic irritants, such as alcohol, medications, or chemicals (e.g., occupational exposure to carbon tetrachloride), must be avoided in all types of hepatitis.
Hepatitis A.
Treatment for HAV is primarily symptomatic and supportive. Good sanitation and hygiene practices should be followed. Hospitalization may be required for excessive vomiting and subsequent fluid and electrolyte imbalance.
Hepatitis B.
Treatment for acute HBV is symptomatic. Only clients with chronic disease receive treatment, which depends on the severity of the disease. People with normal liver enzyme values, an HBV DNA level of less than 1021 copies/ml, and findings on biopsy that are normal are considered “inactive carriers.” Their prognosis is good and compares favorably with people who are not infected with HBV. People with abnormal liver enzymes (elevated AST), HBV DNA levels greater than 1021 copies/ml, and architecture on liver biopsy consistent with active disease require treatment. Pegylated interferon and lamivudine, adefovir, and entecavir (nucleoside analogues) are current therapies available. Interferon is expensive and has many side effects, but therapy duration is limited and maintenance therapy is not required.
Nucleoside analogues can be given orally, cost less than interferon, and have few side effects but require continued treatment to maintain response. The newest of these drugs for chronic HBV infection, telbivudine (Tyzeka) is an oral medication that reduces the viral load by inhibiting HBV reverse transcriptase and HBV replication. It is indicated for adults with evidence of viral replication and either persistent elevations in serum aminotransferases or histologically active HBV. Combination therapy is frequently used. Since people with cirrhosis and HBV are at high risk for the development of HCC, ultrasonography of the liver every 6 to 12 months is advisable.
Hepatitis C.
Since the majority of complications occur in clients who develop cirrhosis from HCV, treatment is provided to those at risk for developing cirrhosis. This includes people with a history of alcohol abuse, people who contracted HCV at a young age, or those with active disease by liver biopsy. Because people with genotypes 2 and 3 (subtypes of HCV) have a high likelihood of responding to therapy, a liver biopsy is often not required before therapy.
Researchers reported at the 38th annual Digestive Disease Week conference in Washington, D.C., that HCV is curable and should be treated (with peginterferon alfa-2a alone or with ribavirin) even if the person has no symptoms.149 A study using combination therapy consisting of pegylated interferon and ribavirin (Virazole, a nucleoside analogue) for 3 to 12 months (depending on genotype and response) provided a sustained clearance of HCV in about 55% of clients.32
The treatment involves a new “pegylated” form of the commonly used HCV drug interferon. By attaching polyethylene glycol (PEG) molecules to the interferon, its half-life is lengthened, allowing it to stay in the body much longer. The new interferon formulation is given as a weekly injection rather than three times a week. PEG interferon has been reported to reverse infection-related liver damage previously believed impossible. The study showed reductions in liver fibrosis, or scar tissue, in three out of four people given the new treatment. Half of those with cirrhosis also showed improvement.150 Many potential side effects and contraindications exist for the use of interferon that prevents this treatment from being applied to all individuals with HCV infection.
Surveillance by ultrasonography every 6 to 12 months is recommended to detect HCC. Administration of HAV and HBV vaccine is recommended for anyone with chronic HCV because of the potential for increased severity of acute hepatitis superimposed on existing liver disease.154
PROGNOSIS.
Prognosis varies with each type. A substantial proportion of HBV morbidity and mortality that occurs in the health care setting can be prevented by vaccinating health care workers against HBV. In addition, health care workers must practice infection control measures (see Appendix A). Other prophylactic strategies are based on avoidance of high-risk behavior and use of IG.
Hepatitis A.
HAV is almost always a self-limited disease and rarely leads to fulminant hepatitis requiring transplantation (about 0.3% to 0.6% of cases). It does not lead to chronic hepatitis or cirrhosis. Most people recover fully and become immune to HAV.
Hepatitis B.
In adults with normal immune status, most (94% to 98%) recover completely from newly acquired HBV infections, eliminating virus from the blood and producing neutralizing antibody that creates immunity from future infection. In immunosuppressed persons (including hemodialysis clients), infants, and young children, most newly acquired HBV infections result in chronic infection.
Although the consequences of acute HBV can be severe (1% die from acute hepatitis), most of the serious sequelae associated with the disease occur in persons in whom chronic infection develops. Approximately 15% of people who acquire the disease as adults and 25% of people who acquired HBV as a child die prematurely from cirrhosis or liver cancer.94
Despite reinfection rates, orthotopic liver transplantation for cirrhosis caused by HBV provides the best treatment. Continued treatment of the virus with the administration of HBIG (intravenously or intramuscularly) used in combination with lamivudine has been found to be efficacious, making liver transplantation outcomes successful.
Hepatitis C.
Acute HCV infection only leads to death in 1.3% of cases. The majority of disease sequelae occurs from chronic disease, particularly cirrhosis. Approximately 20% to 30% of all cases of HCV progress to cirrhosis. Between 1% and 5% of people with HCV-related cirrhosis develop HCC each year.31
HCV-related cirrhosis is the most common reason for liver transplantation and between 8000 and 12,000 people die each year from HCV-related disease.4 The number of annual deaths is most likely to triple as people with chronic disease develop end-stage liver disease.8
The progression of HCV is accelerated if a person is also coinfected with the acquired immunodeficiency syndrome (AIDS) virus, causing cirrhosis more quickly and twice as often.156 HIV/HCV coinfection among individuals with bleeding disorders results in mortality; more than 5000 people have died, and the numbers continue to climb.111
Hepatitis D.
HDV by itself appears to be relatively harmless, but it has a high morbidity and mortality rapidly leading to hepatic failure and cirrhosis when it accompanies HBV.
Hepatitis E.
HEV is typically a self-limited acute hepatitis, usually lasting 1 to 4 weeks. Severity of symptoms increases with age. It does not progress to chronic disease. When it occurs in epidemics, it can cause substantial rates of death and complications, especially in pregnant women. The overall fatality rate is estimated to be 3%; pregnant women who develop the infection have the highest risk of acute hepatic failure.153
Drug-Related Hepatotoxicity
Injury to the liver can be caused by many drugs or toxins. More than 600 medicinal agents, chemicals, and herbal remedies are recognized as producing hepatic injury.79 For example, herbal products, such as chaparral, comfrey, germander, Jin Bu Huan, mistletoe, nutmeg, ragwort, sassafras, senna, or tansy, can cause liver-related complications (ranging from acute hepatitis and jaundice to cirrhosis and death from liver failure) in anyone with an underlying liver disease.
Although OTC and prescription medications are often thought to be the only agents to cause liver injury, complementary or alternative medications, such as chaparral, germander, pennyroyal oil, Jin Bu Huan, and Sho-saiko-to, are also known to be hepatotoxic.159
Chemicals, such as carbon tetrachloride, trichloroethylene, derivatives of benzene and toluene, vinyl chloride, and organic pesticides, can also lead to liver injury. Carbon tetrachloride was the most common industrial chemical considered an occupational inhalation poison until it was banned in most countries. Ingestion of poisonous mushrooms, including Amanita phalloides and related species (rare in the United States but more common in Europe), can lead to fulminant liver failure.
Although uncommon, drugs are currently the most common cause of acute liver failure.117 A recent study of the WHO database demonstrated acetaminophen, troglitazone, valproate, stavudine, and halothane to be the five drugs most frequently associated with hepatotoxicity and death (particularly acetaminophen).16,70
The incidence of hepatotoxicity as a result of these noninfectious agents is difficult to determine since many cases may be subclinical (i.e., mild symptoms) or misdiagnosed, leading to underreporting. One French study demonstrated a rate of 14 cases/100,000 people in the general population; 12% required hospitalization, and 6% died as a result of acute liver failure.148 Hepatotoxicity has also been the principal reason for removing several new drugs from the market.49
Drugs or chemicals typically cause either predictable (or dose-related) or unpredictable (or idiosyncratic) liver injury (Box 17-3). Drugs that cause predictable liver damage are dose-related (i.e., a specific toxic dose most likely will cause damage) and result in injury soon after ingestion. Acetaminophen is the most common example of predictable drug-related liver disease. Unpredictable reactions occur without warning, are unrelated to dose, and may occur days to months after ingestion.
Etiologic Risk Factors
A host of factors may enhance susceptibility to drug-induced liver disease, including age (adults more so than children), gender (women have a higher risk than men), obesity, malnutrition, pregnancy, concurrent medication use, history of drug reactions, and genetic variability (probably the most important factor). Preexisting liver diseases and comorbidities appear to alter the rate of recovery rather than affect the risk of developing hepatotoxicity.141 Alcohol use and fasting may increase the likelihood of acetaminophen-related hepatotoxicity.171
Pathogenesis
Drugs and toxins can result in liver injury via numerous mechanisms.62 Some of these mechanisms include programmed cell death (apoptosis) as a result of tumor necrosis factor (TNF); binding of drug to cellular proteins, which causes an immunologic response, leading to cell death137; inhibiting metabolism of drugs; and mitochondrial dysfunction with formation and accumulation of reactive oxidative species.123
The pattern of injury is often drug dependent. Patterns of liver injury include hepatocellular, cholestatic, mixed, hypersensitivity (or immunologic), and mitochondrial injury. Hepatocellular injury is defined by inflammation of the hepatocytes, breakup of the hepatic lobule (comprised of the central vein, the portal vein, hepatic artery, bile duct, and hepatic cords), and hepatocellular necrosis.
Isoniazid is an example of a drug that can cause this type of injury. Cholestatic injury is demonstrated by mild bile duct injury with pooling of bile in the hepatocytes. Amoxicillin-clavulanic acid is one drug that can cause this type of injury. Mixed patterns exhibit both hepatocellular and cholestatic patterns. Hypersensitivity reactions often show an eosinophilic infiltration of the portal triad; phenytoin can lead to this type of response. Mitochondrial injury is demonstrated by small droplets of fat (or microvesicular steatosis) within the hepatocyte. Valproic acid has been known to cause this type of cellular change.
Clinical Manifestations
The manifestations of drug-related liver disease can range from mild symptoms to fulminant liver failure. Vague symptoms, including fatigue, nausea, and right upper quadrant pain or discomfort, may be the first indications of hepatotoxicity. Other symptoms associated with liver injury are jaundice, pruritus, and dark urine. Fever and rash are often present with a hypersensitivity reaction. As discussed, drugs often cause a specific pattern of injury. The most readily identifiable patterns are hepatocellular and cholestatic.
Hepatocellular liver disease frequently presents with abdominal discomfort/pain, fatigue, and jaundice; the clinical course is acute and can be severe. Cholestatic liver disease is manifested clinically by jaundice and pruritus. In contrast to the hepatocellular pattern, cholestatic disease is often less acutely serious, but healing may be prolonged and chronic, taking weeks to months.
MEDICAL MANAGEMENT
Since drug-induced hepatotoxicity is uncommon, people who present with symptoms consistent with liver disease should have a complete evaluation to avoid missing more common causes. This includes laboratory testing for viral hepatitis, autoimmune liver diseases, Wilson’s disease, hemochromatosis, and α1-antitrypsin deficiency. Ultrasonography, computed tomography (CT) scanning, magnetic resonance imaging (MRI), or endoscopic retrograde cholangiopancreatography (ERCP) are useful to identify biliary obstruction. A history of heavy alcohol consumption may be consistent with alcohol-related hepatitis.
A review of medications taken, remembering to question exposures to chemicals, herbals, and OTC medications, may point toward drug-related liver disease. Although many drugs have more immediate effects, unpredictable injury may not exhibit symptoms for months.
Tests used to detect liver injury include ALT, bilirubin, and ALP. ALT values that are at least three times normal are suggestive of liver injury, whereas ALP values of two times normal are considered abnormal. A bilirubin more than twice the upper limit of normal and associated with an abnormal ALT or ALP suggests disease. An elevation in the ALT value, with minimal changes in ALP, is consistent with hepatocellular damage, whereas a predominantly elevated ALP is associated with cholestatic disease.
Significant elevations in these laboratory values are not predictive of prognosis since the liver has the prodigious ability to heal. Function is often a better indicator of prognosis. Several laboratory tests indicate how well the liver is functioning. Prothrombin/international normalized ratio (INR) and albumin demonstrate the liver’s ability to synthesize proteins, and total bilirubin and conjugated bilirubin reflect the liver’s ability to move bilirubin from the blood into bile.112
The liver also has the ability to adapt to some medications. This ability is demonstrated when the introduction of a drug leads to transient elevations in liver enzyme tests, but there is no progression of injury. In these cases the drug does not have to be stopped. For example, isoniazid often causes a transient, minor increase in liver enzymes but is permanently stopped in only 1 in 1000 affected clients.115
TREATMENT AND PROGNOSIS.
Treatment for drug-related hepatotoxicity most often consists of removal of the causative agent and providing supportive care. Two exceptions include the usage of N-acetylcysteine soon after toxic ingestion of acetaminophen and intravenous carnitine for valproate-induced hepatotoxicity. Reexposure or rechallenge of the offending agent should be avoided, particularly if the drug has an unpredictable liver toxicity and the hepatotoxicity was immune-related. Reexposure could lead to an even more severe and serious reaction.
Liver injury accompanied by increases in liver enzyme levels may worsen for days to weeks before improvement. In some severe cases, improvement of liver enzyme levels suggests liver failure rather than healing of injury caused by severe necrosis and loss of hepatocytes. In this setting, laboratory values indicative of liver function (such as prothrombin time/INR and albumin) and symptoms (such as encephalopathy) are better indicators of hepatic function.
Chronic liver disease can also develop; up to 5% to 6% of drug-induced liver disease can become chronic (principally with a cholestatic pattern of injury). Methyldopa, minocycline, and nitrofurantoin are drugs that have been associated with this problem. A poor prognosis is seen in clients who develop jaundice, impaired liver function, and encephalopathy within 26 weeks of the onset of symptoms.124 Prognosis is the most serious and poor for those people with hepatocellular damage accompanied by jaundice, with a mortality of 10% to 50% (known as Hy’s law). This type of injury is more likely than others to require liver transplantation.15
Autoimmune Hepatitis
Autoimmune hepatitis is a chronic progressive, inflammatory disorder of the liver of unknown cause. It occurs in adults and children and is characterized by the presence of abnormal liver histology, autoantibodies, elevated levels of serum immunoglobulins, and frequent association with other autoimmune diseases. Autoimmune hepatitis is one of the three major autoimmune liver diseases, along with primary biliary cirrhosis and primary sclerosing cholangitis. Variant forms of autoimmune hepatitis have been termed overlap syndromes because they share features of several liver diseases.
The International Autoimmune Hepatitis Group has classified two types of autoimmune hepatitis: type 1 and type 2.5 Both forms are more common among women than men and have similar clinical and serum biochemical features. Type 2 is rare and most often seen in childhood and in young adulthood; it is associated with more severe and advanced disease at presentation. Autoimmune hepatitis is seen worldwide and in various ethnic groups, although type 2 is uncommon in North America.
Etiologic Factors and Pathogenesis
The cause and pathogenesis of autoimmune hepatitis are not known. The disease appears to occur among genetically predisposed individuals on exposure to as-yet unidentified environmental agents, triggering a cascade of T cell–mediated events directed at liver antigens.67
Purported triggering agents include viruses (such as measles, hepatitis, CMV, and Epstein-Barr viruses) and drugs (such as methyldopa, nitrofurantoin, diclofenac, interferon, pemoline, minocycline, and atorvastatin). It is uncertain if drugs actually trigger autoimmune hepatitis or merely cause a drug-mediated hepatitis with features similar to autoimmune hepatitis. Most people with autoimmune hepatitis, however, have no identifiable trigger. Factors that predispose an individual to autoimmune hepatitis are not certain, but specific HLA genes have been linked to the development of autoimmune hepatitis and probably play a major role.
Type 1 is associated with human leukocyte antigen (HLA)-DR3 and HLA-DR4. Studies have shown that HLA-DR3–associated disease correlates to earlier onset, particularly in girls and young women, and more severe disease; whereas HLA-DR4 is associated with milder disease (although extrahepatic findings are more common) and better response to treatment.67 HLA-DR2 may be protective.35
Autoreactive T cells and their proinflammatory response appear to cause the liver destruction seen in autoimmune hepatitis. Research continues to identify the antigens that are targeted by T cells, but one possibility is the asialoglycoprotein receptor, which is a liver membrane protein found in hepatocytes.
Continued T-cell response with chronic liver damage may ensue because of a failure of CD4+CD25+ regulatory T cells (T-regs) to regulate the response of CD8+ and CD4+CD25 cells (autoreactive T cells). Normally, T-regs control the adaptive immune response of CD8+ and CD4+CD25-T cells by suppressing the proliferation and function of these T cells, which allows for self-tolerance maintenance. In autoimmune hepatitis, the low number and defective function of T-regs leads to a change in cytokine production, proliferation, and apoptosis of autoreactive T cells.87
Antibodies are also produced and vary, depending on the type of autoimmune hepatitis. Antinuclear antibodies (ANA), smooth-muscle antibodies, atypical perinuclear antineutrophilic cytoplasmic antibodies (pANCA), and antiactin antibodies are seen with type 1 autoimmune hepatitis. Type 2 is associated with liver-kidney microsome 1 (LKM-1) and liver cytosol 1 (ALC-1) antibodies. Although present in the serum, there is little evidence to support the theory that these autoantibodies cause the disease.67 Yet measurement of these antibodies can be helpful in the diagnosis of autoimmune hepatitis.
Clinical Manifestations
Autoimmune hepatitis is represented by a wide spectrum of clinical manifestations. While most clients present with nonspecific, mild, or chronic liver symptoms, up to 40% present with acute symptoms of hepatitis, including fulminant hepatitis with cirrhosis and liver failure. Autoimmune hepatitis is usually progressive and chronic, although a minority of cases are characterized by a fluctuating course.
The most common presenting symptoms are fatigue (85%), jaundice (46%), anorexia (30%), myalgias (30%), and diarrhea (28%). Other symptoms include abdominal pain, arthralgias (particularly small joints), and malaise. Weight loss is uncommon and should prompt an evaluation for another disorder.
The presence of another autoimmune disorder, such as thyroiditis, ulcerative colitis, type 1 diabetes, rheumatoid arthritis, and celiac disease,1 is seen in one-third of affected individuals. Physical examination may be normal, but 78% of clients present with hepatomegaly. Individuals with fulminant hepatitis often have profound jaundice.
Complications of autoimmune hepatitis are similar to other chronic liver diseases, particularly the development of cirrhosis. Although occurring less frequently than hepatitis associated with a virus, another serious complication is the development of HCC.
MEDICAL MANAGEMENT
The diagnosis of autoimmune hepatitis can be difficult since no one test is diagnostic for the disease. The diagnosis is based on a synthesis of clinical, biochemical, and histologic information. Laboratory findings consistent with autoimmune hepatitis include elevated aminotransferase values, hypergammaglobulinemia, elevated bilirubin and ALP values, and the presence of autoantibodies. At presentation aminotransferase values are typically between 150 U/L and 500 U/L, but people with severe disease at presentation can have significantly elevated aminotransferase values in the thousands.
Over 80% of affected people have hyperbilirubinemia, although this is typically mild with values less than 3 mg/dl. Serum ALP values are usually elevated, but values twice the upper limit of normal are likely indicative of another disease. Serum globulins are elevated up to three times normal, particularly gamma globulin and IgG.
As discussed, autoantibodies are produced and can be helpful in making the diagnosis of autoimmune hepatitis, but 10% of cases do not exhibit autoantibody production. The most common autoantibodies seen in type 1 are ANA and smooth muscle. Titers of at least 1: 80 in adults are considered a positive value.67 Anti-LKM-1 and anti-ALC-1 antibodies are more specific to type 2.
The magnitude of the elevations of aminotransferase and gamma globulin does not necessarily correlate with the extent of liver damage, making liver biopsy important. Although histologic appearance of autoimmune hepatitis is similar to chronic hepatitis (can also be seen in other liver diseases), there are characteristic findings of autoimmune hepatitis that include a mononuclear-cell infiltrate of the interface around the portal triad, particularly with plasma cells. Fibrosis is another common finding.
TREATMENT.
The mainstay of therapy is immunosuppression. Over 80% of clients achieve remission with prednisone or prednisone plus azathioprine. Serum aminotransferases and gamma globulin levels can be used to monitor response to treatment. Therapy can be withdrawn once in remission, although relapse is high, occurring in 50% to 85% of affected people, particularly during the first 6 months.
Therapy for relapsed disease is the same as that used for the initial treatment and can successfully induce remission of the disease. Individuals who experience repeated relapses tend to have a poorer prognosis.105 Maintenance therapy with azathioprine is often needed and life-long maintenance therapy is frequently used in persons with type 2 disease or cirrhosis at diagnosis.
Liver transplantation may be required for clients who develop end-stage liver disease because of intolerance to therapy (and have progression of the disease) or who have progression of the disease while receiving adequate therapy. Five-year survival rates after liver transplantation are 80% to 90%, and the 10-year survival rate is approximately 75%.37 Autoimmune hepatitis reoccurs in about 40% of clients who undergo liver transplantation,133 although this is often mild and related to the immunosuppressive medications used after transplantation.
PROGNOSIS.
Prognosis depends on the stage of disease at the time of diagnosis and initiation of therapy, but 10-year survival rates (meaning not requiring liver transplantation or causing death) surpass 90%, whereas 20-year survival rates are probably less than 80%. People that respond to treatment often have lifespans similar to healthy persons.27 Individuals who present with cirrhosis have 20-year survival rates of only 40%.
Alcohol-Related Liver Disease
Overview, Incidence, and Risk Factors
Although over two-thirds of Americans drink alcohol, only a minority develop problems leading to chronic alcohol abuse and severe liver disease. Yet alcohol problems result in significant morbidity and mortality; over 40% of deaths from cirrhosis are alcohol-related and 30% of HCC cases are a result of alcohol-related liver disease.
More men than women acquire liver disease, but women develop the disease after a shorter exposure to alcohol and while consuming lower quantities of alcohol as compared to men. In men 6 to 8 alcohol-containing beverages daily (60 to 80 g/day) over a 5-year period can lead to liver disease, while only 3 to 4 drinks/day are needed to cause the same effect in women.
Women are more vulnerable to the effects of alcohol than men. Women produce substantially less of the gastric enzyme alcohol dehydrogenase, which breaks down ethanol in the stomach. As a result, women absorb 75% more alcohol into the bloodstream. Other effects and gender differences are discussed further in Chapter 2.
About 90% of heavy drinkers develop fat accumulation in the liver (the first sign of alcohol abuse), yet although many persons are heavy drinkers for long periods of time, only a minority progress to develop alcoholic hepatitis and alcoholic cirrhosis. Research suggests that genetics may play an important role in preventing liver damage despite chronic alcohol exposure.
Alcohol-related liver disease encompasses alcoholic hepatitis and alcoholic cirrhosis. Alcoholic hepatitis occurs in only 10% to 35% of heavy drinkers and is the precursor for the development of alcoholic cirrhosis. People with alcoholic hepatitis are nine times more likely than people with fatty liver infiltration to develop cirrhosis.81
Some cofactors that may predispose to cirrhosis include coexisting HCV, smoking, and obesity. Although much progress has been made in understanding potential causes, many questions remain such as determining which factors lead to severe liver damage and the mechanisms that protect other heavy drinkers.
Pathogenesis
The initial physiologic change observed in the liver with alcohol exposure is the accumulation of fat, which is reversible with abstinence. In some people, with continued exposure to alcohol, there is a progression of damage to the liver consisting of inflammation, necrosis of individual cells, and early fibrosis, termed alcoholic hepatitis. With continued heavy drinking, micronodular fibrosis (small bands of fibers) can develop and eventually progress to large bands of fibrosis, creating large nodules of fibrotic liver tissue (macronodular cirrhosis). Once cirrhosis has developed, HCC may result.
Research has demonstrated that heavy quantities of alcohol can have significant detrimental effects. Many mechanisms have been described that may be responsible for liver injury caused by alcohol consumption. Damage most likely requires several of these processes to be present, and genetic factors probably play a role in prevention or progression of liver injury. A few of these mechanisms include the toxic buildup of acetaldehyde, inflammatory reaction of immune cells, overproduction of reactive oxidative species (ROS) and reduction of antioxidants, mitochondrial dysfunction, and abnormal metabolism of methionine and S-adenosylmethionine (SAM).
Acetaldehyde is an intermediate product in the metabolism of alcohol. Typically, alcohol is converted to acetaldehyde by the enzymes alcohol dehydrogenase and cytochrome P450 2E1 (CYP2E1); acetaldehyde is in turn metabolized to acetate by aldehyde dehydrogenase. When the metabolism process is overwhelmed by alcohol, acetaldehyde can build up. Acetaldehyde is a reactive molecule and is able to modify proteins. These altered proteins are then not only unable to function appropriately in the hepatocyte but may also elicit an immune response, bringing inflammatory leukocytes into the liver and contributing to cellular injury.
Alcohol can also alter the balance between prooxidants and antioxidants, causing oxidative stress and liver injury. When the enzyme CYP2E1 is activated, it leaks electrons. These electrons create reactive species (such as superoxide anions), which then react with proteins, lipids, and DNA, or deplete antioxidants, leading to the inability of the hepatocyte to function. Reactive molecules can also signal other pathways, such as activating transcription factors, to produce TNF. Hepatocytes can be very sensitive to the damaging effects of TNF as a result of the abnormally increased level of acetaldehyde.86
Another factor contributing to oxidative stress is alcohol-related mitochondrial dysfunction. Mitochondria utilize oxygen and produce ROS but are protected from oxidative stress by glutathione (which is transported from the cytosol). In the presence of alcohol, hepatic mitochondria produce increased amounts of superoxide molecules, but the transport of glutathione into the mitochondria does not function, leaving the mitochondria to the damaging effects of ROS.
Alcohol also interrupts the important metabolism of methionine to SAM. The enzyme responsible for this conversion is methionine adenosyltransferase (MAT), which is depressed by alcohol.96 SAM is a necessary molecule, donating methyl groups in many enzymatic reactions involving DNA, RNA, phospholipids, and proteins. SAM is the precursor of glutathione and may be hepatoprotective; the depletion of SAM can result in abnormal functioning of hepatocytes and sensitization of hepatocytes to destructive cytokines.
Clinical Manifestations
The initial histologic change noted in heavy drinkers is fatty liver infiltrate. This is often asymptomatic and detected only on laboratory evaluation. Even clients with alcoholic hepatitis and/or cirrhosis may be asymptomatic. Others may present with nausea, vomiting, abdominal pain, jaundice, anorexia, fever, and weight loss.
The most common sign in people with fatty liver or alcoholic hepatitis is hepatomegaly; 60% of all people with alcohol-related liver disease exhibit jaundice and ascites (typically in clients with severe disease). Splenomegaly is more common as the disease worsens and hepatic encephalopathy can exist in varying degrees, ranging from mild cognitive impairment to coma. Clients with alcoholic hepatitis or cirrhosis often display spider angiomata, liver tenderness, and edema.97
MEDICAL MANAGEMENT
The diagnosis of alcoholic hepatitis is made using the appropriate history of heavy drinking, consistent laboratory values, and no evidence of other diseases that could cause liver injury. Because alcohol can worsen preexisting diseases, such as viral hepatitis, the diagnosis of alcohol-related liver disease should not be made without a thorough evaluation.
A history of heavy drinking may not be present because of client denial. Only 50% of clients who abuse alcohol are identified by their physician. Elevated levels of aminotransferases (AST and ALT) are the most common abnormalities seen in alcohol-related liver disease. The AST is rarely over 300 to 500 U/L, while the ALT is typically only slightly elevated, giving an AST/ALT ratio of greater than 2. Serum ALP and bilirubin can range from normal to 1000 U/L and 20 to 40 mg/dl, respectively.
Often, despite advanced disease, laboratory levels may be only mildly elevated. Alcohol-related liver disease is usually accompanied by malnutrition and is evident in the low albumin levels and elevated prothrombin time (PT). Erythrocyte count is often low (anemia) and thrombocytopenia may be present; the white blood cell (WBC) count frequently is elevated. Because the diagnosis of alcohol-related liver disease can competently be made using history, physical, and laboratory information, liver biopsy is rarely needed.
TREATMENT AND PROGNOSIS.
Treatment of alcoholic hepatitis centers on cessation of alcohol, nutritional support and education, and prevention of the complications of end-stage disease. Corticosteroids can be used for severe cases of alcoholic hepatitis, whereas SAM, an antioxidant, may reduce mortality and decrease the need for liver transplantation.95
Anti-TNF-α agents, such as etanercept, may be beneficial99; however, only preliminary data are available from pilot studies and these medications are not used routinely. Alcoholic hepatitis carries a poor prognosis if the person continues to drink alcohol. Frequently, the disease will stabilize if the person stops drinking.
Clients who develop cirrhosis (but are asymptomatic) and are able to abstain from alcohol have a prognosis of 80% at 5 years. Liver transplantation is offered to clients who have end-stage liver disease and are able to stop drinking, typically for at least a period of 6 months. This often allows for sufficient improvement to the point of not requiring a liver transplant. Outcomes of transplantation for alcohol-related liver disease are similar to other groups requiring transplantation,136 with a 7-year survival of 60%.
Prognosis depends on the severity of disease, other coexisting illnesses (i.e., HCV), nutritional status, the client’s ability to abstain from alcohol, and the presence of end-stage liver disease. The prognosis and severity of disease can be determined using the discriminant function (DF) equation; the more recent model for end-stage disease (MELD) predicts survival and is often used in allocating livers for transplantation.38 People with a DF score greater than 32 have a 50% mortality within 30 days.
Primary Biliary Cirrhosis
Primary biliary cirrhosis (PBC) is a chronic, progressive liver disorder with uncertain cause. Similar to autoimmune hepatitis, PBC has an autoimmune basis, but unlike autoimmune hepatitis, the areas of destruction involve the small bile ducts. The incidence of PBC has been stable over time although the prevalence has been increasing, which is most likely a result of earlier diagnosis and prolonged survival. PBC occurs most frequently in women (80% to 90% of cases) between the ages of 40 to 60 years.
PBC is a slowly progressive (irreversible), chronic liver disease that causes inflammatory destruction of the small intrahepatic bile ducts, decreased bile secretion, cirrhosis, and ultimately, liver failure. An autoimmune attack against the bile duct is probably an important pathogenetic element, but the precipitating event and contribution of genetic and environmental factors are uncertain. The disease is associated with other autoimmune disorders, including scleroderma, Sjögren’s syndrome, thyroiditis, pernicious anemia, and renal tubular acidosis. Although it is most common in whites from North America and Europe, cases have occurred in all races.
Etiologic Factors and Pathogenesis
The underlying cause of primary biliary cirrhosis has a basis in aberrant autoimmunity. People affected with PBC express antimitochondrial antibodies (90% to 95% of cases). These are specifically targeted toward large enzyme complexes associated with oxidative phosphorylation (pyruvate dehydrogenase complex-E2 [PDC-E2]).45
Although these enzymes occur throughout the body, only the epithelial cells of the small bile ducts in the liver are affected. This may be related to how a bile duct epithelial cell processes glutathione once the bile duct cell undergoes apoptosis.61 Autoreactive T cells then respond to these antibodies, causing inflammation. As the ducts are destroyed, toxic substances build up in the liver, intensifying the damage. Chronic inflammation leads to fibrosis, cirrhosis, and eventually, without treatment, liver failure.
Research has also been investigating factors that lead to the production of antimitochondrial antibodies. Several hypotheses are centered on molecular mimicry.61 Evidence suggests that antibodies made against bacteria have similarities to the PDC-E2 in mitochondrial cells, thus producing antibodies that target bile duct cells. Some of the bacteria proposed to play a role in this process are E. coli, Novosphingobium aromaticivorans, lactobacilli, and Chlamydia.61 Other possible environmental factors include chemicals, such as halogenated hydrocarbons found in pesticides and detergents, but more research is needed to verify this association.19,76
Clinical Manifestations
Most people with PBC are asymptomatic (50% to 60%) at diagnosis; the remainder may present with symptoms ranging from minor annoyances to advanced disease. The most common presenting symptom is fatigue, which is noted in over 20% of affected clients at diagnosis. With progression of the disease, fatigue becomes more significant, occurring in about 80% of people,43 and may be disabling.128
Pruritus is frequently present (20% to 70%), particularly in the perineal area and the palmar and plantar surfaces of the hands and feet, although it can be diffuse. Pruritus often worsens at night or when the environment is warm. Contact with fibers, such as wool, can also aggravate the pruritus. Less commonly noted is the presence of right upper quadrant pain (about 10%).
Other symptoms of the disease include hyperlipidemia, osteopenia, and the presence of other autoimmune diseases (i.e., Sjögren’s and scleroderma).78,168 In the later stages of the disease, clients may exhibit portal hypertension, malabsorption, fat-soluble vitamin deficiencies, and steatorrhea (the result of impairment of excretion of bile into the intestine). Complications from advanced liver disease include ascites, bleeding from esophageal varices, and hepatic encephalopathy.
The physical examination is typically normal in asymptomatic people, but skin manifestations often occur as the disease progresses. Pruritus frequently leads to excoriations of the skin, and spider nevi, thickening of the skin, and increased skin pigmentation are often detected with advancement. Xanthelasmas are seen in 5% to 10% of persons with the disease (Fig. 17-6). Hepatomegaly is noted in 70% of clients, yet splenomegaly is uncommon at presentation, often developing only near the end stages of PBC. Jaundice is observed later in the illness, often months to years after diagnosis. Muscle wasting, edema, and ascites often herald liver failure.
Figure 17-6 Xanthelasma. Multiple, soft yellow plaques involving the eyelid (lower and upper). Lipid-laden foam cells seen in the dermis tend to cluster around blood vessels. Lipid deposits can also be seen along the extensor surfaces of the body (not shown) such as the heels, elbows, and dorsum of the hands. (A, From Yanoff M: Ophthalmology, ed 2, St. Louis, 2004, Mosby; B, from Rakel RE: Textbook of family medicine, ed 7, Philadelphia, 2007, WB Saunders.)
MEDICAL MANAGEMENT
The diagnosis of PBC is based on a triad of criteria: presence and elevation of antimitochondrial antibodies (titer 1: 40 or more), elevated levels of serum liver enzymes (particularly serum ALP and γ- glutamyltransferase) for more than 6 months, and liver biopsy histology consistent with PBC. A probable diagnosis of PBC can be made with only the presence of antimitochondrial antibodies and elevated liver enzymes, but a definite diagnosis requires a liver biopsy. A liver biopsy may be beneficial for several reasons. First, the stage of the disease can be defined since the magnitude of the elevation of antimitochondrial titers is not indicative of the severity of the disease. Second, 5% to 10% of clients with PBC do not manifest antimitochondrial antibodies. Third, liver biopsy gives a baseline with which to compare response to treatment.
The histology of PBC is divided into four successive stages referred to as stages 1 to 4, as seen on evaluation of the liver biopsy. As the liver is affected asymmetrically, a biopsy may not give a complete picture of the disease process. If more than one stage is observed on biopsy, the stage assigned is the highest of those seen. The hallmark of PBC is asymmetric inflammation and destruction of the bile duct within the portal triads. In stage 1 the damage is confined to the portal triad. Stage 2 is characterized by a reduction in the number of bile ducts and an inflammatory involvement of the surrounding parenchyma. Stage 3 disease demonstrates fibrosis, which connects the affected portal triads, whereas stage 4 manifests frank cirrhosis with regenerative nodules and accompanying liver failure.
The clinical course of the disease is variable and early diagnosis is important in order to initiate therapeutic measures before the development of advanced disease.
TREATMENT.
Most people with PBC are now treated with ursodeoxycholic acid (UDCA), or ursodiol, a bile acid taken in capsules. It is currently the only drug approved by the Food and Drug Administration (FDA) for the treatment of PBC. Twenty-five to thirty percent of clients will have a complete response with UDCA,77 which can reduce serum liver enzymes, cholesterol, and IgM; it also reduces pruritus.121
UDCA has also been shown to reduce the risk of death or need of liver transplantation after 4 years of therapy and can be effective for up to 10 years.126,129 It appears to be safe and has few adverse effects. If used in clients with early-stage disease, it may delay the development of hepatic fibrosis and esophageal varices, with survival rates slightly lower than that seen in an age-sex–matched control population.24,25,127
UDCA has little effect in clients with advanced disease, and better options are still needed for this subgroup. Clients who have a complete response to UDCA require indefinite therapy. Progression, however, occurs in many people, requiring additional medical treatment or liver transplantation. Colchicine and methotrexate are two drugs that are often utilized when UDCA is no longer efficacious; other drugs are under investigation.134
Liver transplantation is considered for clients with end-stage liver disease (i.e., hepatorenal syndrome, diuretic-resistant edema, bleeding esophageal varices, and hepatic encephalopathy), unacceptable quality of life caused by intractable symptoms, or anticipated death in less than 1 year.91
Many laboratory values and factors are followed to aid in determining the appropriate time for liver transplantation such as the bilirubin, AST/ALT ratio, serum albumin, prothrombin time, and presence of complications from portal hypertension. Clients who receive a liver transplant often have difficulty weaning from immunosuppressants, and evidence of recurrent disease appears in 15% of clients at 3 years and 30% at 10 years.113
Complications of PBC, such as fatigue, pruritus, osteoporosis, fat-soluble vitamin deficiencies, portal hypertension, and hyperlipidemia, also receive treatment. Fatigue is difficult to treat and can be debilitating. One preliminary study showed some improvement in daytime drowsiness with the use of modafinil in people who were able to tolerate the drug.60
Cholestyramine and rifampin are the principal drugs used to relieve pruritus, although side effects may not be tolerable. PBC-associated osteoporosis occurs in up to one-third of clients with PBC, but severe bone disease (i.e., with multiple fractures) is uncommon except in more advanced disease. Treatment with alendronate may increase bone mineral density, but long-term studies are needed to demonstrate efficacy.54
Liver transplantation leads to an initial worsening of osteopenia, but bone mineral density typically returns to baseline after 1 year and can subsequently continue to improve. Because of a lack of bile acid secretion, fat-soluble vitamins (i.e., vitamins D, A, and K) are not absorbed in the intestine. This typically occurs in the later stages of the disease and clients can be given supplements as needed.
Many clients with PBC have significantly elevated serum lipids, yet they appear not to be at increased risk for cardiovascular complications, usually making treatment with a cholesterol-lowering agent unnecessary.88 People with PBC may have early esophageal bleeding, which cause portal hypertension leading to later esophageal bleeds.
Treatment consists of placement of a distal splenorenal shunt by endoscopic rubber-band ligation. If this is not successful, a transjugular intrahepatic portosystemic stent-shunt is placed. Survival is not significantly altered by treatment and many people are stable for years without requiring liver transplant.
PROGNOSIS.
Clients who are diagnosed with stage 1 or 2 disease typically have a better response to treatment. In one study, people with stage 1 or 2 disease who were treated with UDCA for a mean average of 8 years demonstrated a survival rate similar to healthy controls. However, in this same study, those people with stage 3 or 4 disease were at significantly higher risk of requiring a liver transplant.25
Liver transplantation offers persons with liver failure improved survival. The survival rate at 1 year is 92%, and the 5-year rate is 85%. Clients with an elevated bilirubin level above 10 mg/dl have an average life expectancy of 2 years without liver transplantation. Death is usually a result of hepatic failure or complications of portal hypertension associated with cirrhosis.
Intrahepatic Cholestasis of Pregnancy
Intrahepatic cholestasis (suppression of bile flow) of pregnancy is characterized by pruritus and cholestatic jaundice; it usually occurs in the last trimester of each pregnancy and promptly resolves after delivery. Most women present with pruritus, since clinical jaundice is evident in only a minority of women. Of the women who develop pregnancy-induced intrahepatic cholestasis, up to 50% have other family members who have experienced jaundice during pregnancy or after the use of oral contraceptives. Women who have experienced cholestatic jaundice of pregnancy should avoid contraceptives because of a high risk of disease recurrence.
Cholestasis of pregnancy is most likely related to the inhibitory effect estrogen has on bile formation in susceptible women. Maternal health is unaffected by this condition, but the effects on the unborn child can be serious, including fetal distress, stillbirth, prematurity, and an increased risk of intracranial hemorrhage during delivery. UDCA can be used to relieve symptoms and is well tolerated by both mother and fetus. No significant effects are apparent on the liver of the mother, although the risk of developing gallstones is increased.
Vascular Disease of the Liver
Congestive heart failure is the major cause of liver congestion, especially in the Western world, where ischemic heart disease is so prevalent. Backward congestion of the liver and the decreased perfusion of the liver secondary to decline in cardiac output result in abnormal liver function tests. Hepatic encephalopathy may contribute to the altered mentation seen with severe congestive heart failure. Decreased cerebral perfusion, hypoxemia, and electrolyte imbalance are usually the major contributors to the confused state that can be seen in people with severe congestive heart failure.
Portal vein obstruction can be the result of thrombosis, infection, constriction, or invasion. Thrombosis of the portal system is typically caused by a hypercoagulable state or secondary to inflammation, cirrhosis, trauma, or cancer. Inflammatory diseases that affect thrombosis of the portal vein include pancreatitis and inflammatory bowel disease. HCC and pancreatic cancer are the two most common malignancies to cause thrombosis; they also lead to constriction and invasion of the vein.
Ischemic hepatitis or hypoxic hepatitis results when tissue of the liver does not receive adequate oxygen. This may occur in any disease process that reduces blood flow (cardiac failure), reduces oxygen supply (respiratory failure), or increases oxygen requirements (sepsis).
The Budd-Chiari syndrome is an uncommon disease manifested by a thrombosis in and on the hepatic veins where the veins open into the inferior vena cava. Symptoms include pain, ascites, and impaired liver function. Hypercoagulable states, cancer, and infections are the most common causes.
Liver Neoplasms
Hepatic neoplasms can be divided into three groups: benign neoplasms, primary malignant neoplasms, and metastatic malignant neoplasms. Cancer arising from the liver itself is called primary; liver cancer that has spread from somewhere else is labeled secondary or metastatic.
Primary liver tumors may arise from hepatocytes, connective tissue, blood vessels, or bile ducts and are either benign or malignant (Table 17-5). Primary malignant cancer is almost always found in a cirrhotic liver and is considered a late complication of cirrhosis. Benign and malignant neoplasms can also occur in women taking oral contraceptives. A few rare tumors arise from the bile ducts within the liver and are associated with certain hormonal drugs and cancers. Cholangiocarcinomas are discussed later in this chapter.
Table 17-5
Classification of Primary Liver Neoplasms
| Origin | Benign | Malignant |
| Hepatocytes | Adenoma | Hepatocellular carcinoma |
| Connective tissue | Fibroma | Sarcoma |
| Blood vessels | Hemangioma | Hemangioendothelioma |
| Bile ducts | Cholangioma | Cholangiocarcinoma |
Benign Liver Neoplasms
Cavernous Hemangioma.: About 7% of autopsied livers contain hemangiomas, making this lesion the most common benign liver tumor. It is of unknown etiology and occurs in all age groups, more commonly among women. The pathology is similar to that of hemangiomas anywhere in the body; it is a blood-filled mass of variable size and can be located anywhere in the liver. Multiple lesions are observed in about 10% of cases. Areas of thrombosis are common, and older lesions may begin to calcify. The end stage is a fibrous scar.
Most hepatic hemangiomas are asymptomatic until they become large enough to cause a sense of fullness or upper abdominal pain. Hepatomegaly or an abdominal mass is the most common physical finding. About 10% of clients with clinically detectable lesions are febrile. Hepatic hemangiomas are often discovered coincidentally on CT scan, by MRI, or during laparotomy; otherwise the diagnosis is made by contrast-enhanced serial CT, MRI (particularly if small), or single-photon emission CT (SPECT). Needle aspiration or biopsy is avoided because of the risk of hemorrhage.
Treatment is not usually recommended because most hepatic hemangiomas have a benign course with negligible risk of malignancy and minimal chance of spontaneous hemorrhage. Surgical resection may be performed if the hemangioma is consistently symptomatic, producing pain or fever, or if the tumor is large enough for traumatic rupture to be considered a risk (e.g., a palpable lesion in an athlete). Other methods, such as radiofrequency ablation, arterial embolization, and systemic glucocorticoids, have had limited success.