The Hematologic System
Hematology is the branch of science that studies the form and structure of blood and blood-forming tissues. Two major components of blood are examined: plasma and formed elements (erythrocytes, or red blood cells [RBCs]; leukocytes, or white blood cells [WBCs]; and platelets, or thrombocytes).
Delivery of these formed elements throughout the body tissues is necessary for cellular metabolism, defense against injury and invading microorganisms, and acid-base balance. The formation and development of blood cells, which usually take place in the red bone marrow, are controlled by hormones (specifically erythropoietin) and feedback mechanisms that maintain an ideal number of cells.
The hematologic system is integrated with the lymphatic and immune systems; for a complete understanding of these systems see Chapters 7 and 13. The lymph nodes are part of the lymphatic system but also part of the hematopoietic (blood-forming) system and the lymphoid system, which consists of organs and tissues of the immune system (see Fig. 7-8).
Lymph fluid passes through these nodes, or valves, which are located in the lymph channels at 1-to 2-cm intervals. As the fluid passes through the nodes, it is purified of harmful bacteria and viruses. Networks of the lymphatic system are situated in several areas of the body and may be considered primary (thymus and bone marrow) or secondary (spleen, lymph nodes, tonsils, and Peyer’s patches of the small intestine).
All the lymphoid organs link the hematologic and immune systems in that they are sites of residence, proliferation, differentiation, or function of lymphocytes and mononuclear phagocytes (mononuclear phagocyte system: macrophage and monocyte cells capable of ingesting microorganisms and other antigens).
Lymphocytes are any of the nonphagocytic leukocytes (WBCs) found in the blood, lymph, and lymphoid tissues that make up the body’s immunologically competent cells. They are divided into two classes: B and T lymphocytes (see the section on leukocytosis in this chapter and Chapter 7). For example, in the hematologic system, the lymphocytes of the spleen produce approximately one third of the antibody available to the immune system.
SIGNS AND SYMPTOMS OF HEMATOLOGIC DISORDERS
Disruption of the hematologic system results in circulatory disorders as well as signs and symptoms noted in the hematologic tissues themselves. The circulatory disorders can be characterized by edema and congestion, infarction, thrombosis and embolism, lymphedema, bleeding and bruising, and hypotension and shock (Box 14-1).
Edema is the accumulation of excessive fluid within the interstitial tissues or within body cavities. Congestion is the accumulation of excessive blood within the blood vessels of an organ or tissue. The forms of lymphedema include cerebral edema, inflammatory edema, peripheral dependent edema, and pulmonary edema. Congestion may be localized, as with a venous thrombosis, or generalized, as with heart failure (e.g., congestive heart failure [CHF]), which results in congestion in the lungs, lower extremities, and abdominal viscera.
Infarction is a localized region of necrosis caused by reduction of arterial perfusion below a level required for cell viability. Such a situation occurs as a result of arterial obstruction due to atherosclerosis, arterial thrombosis, or embolism, when oxygen supply fails to meet the oxygen requirements of organs with end arteries, such as the gastrointestinal (GI) tract, the heart, and, less often, the kidneys and spleen. Cerebral cortical neurons (cerebral infarction) and myocardial cells (myocardial infarction) are most vulnerable to ischemia, although protective collateral blood flow develops in the heart through anastomoses.
A thrombus is a solid mass of clotted blood within an intact blood vessel or chamber of the heart. An embolus is a mass of solid, liquid, or gas that moves within a blood vessel to lodge at a site distant from its place of origin (see Fig. 12-28). Most emboli are thromboemboli. Thrombosis (development of a thrombus or clot) results from pathologic activation of the hemostatic mechanisms involving platelets, coagulation factors, and blood vessel walls. Endothelial injury, alteration in blood flow (stasis and turbulence), and hypercoagulability of the blood (e.g., protein abnormalities either primary or associated with cancers) promote thrombosis and thromboembolism.
Lymphedema, or chronic swelling of an area from accumulation of interstitial fluid (edema), occurs in hematolymphatic disorders secondary to obstruction of lymphatic vessels or lymph nodes. Obstruction may be of an inflammatory or mechanical nature from trauma, regional lymph node resection or irradiation, or extensive involvement of regional nodes by malignant disease.
Women who have been treated surgically for breast cancer with lymph node dissection, mastectomy, and/or radiation therapy are at double the risk of developing lymphedema of the arm and/or chest wall (see Chapter 13). When the obstruction that slows the lymph fluid exceeds the pumping capacity of the system, the fluid accumulates in the tissues in the extremity, causing edema in one or more limbs. This accumulation of fluid may become a source for bacterial growth, leading to infection, fibrosis, and possible loss of functional limb use.
Bleeding and bruising can occur from trauma of various types and are normal consequences of injury. However, when bleeding and bruising are elicited with minor trauma (e.g., brushing teeth) or bleeding continues longer than normal, there is more concern for a disorder of the blood. These symptoms are often a result of platelet abnormalities (function or quantity) such as idiopathic thrombocytopenic purpura, thrombotic thrombocytopenic purpura, or von Willebrand’s disease.
Purpura is a hemorrhagic condition that occurs when not enough normal platelets are available to plug damaged vessels or prevent leakage from even minor injury to normal capillaries. Purpura is characterized by movement of blood into the surrounding tissue (extravasation), under the skin, and through the mucous membranes, producing spontaneous ecchymoses (bruises) and petechiae (small, red patches) on the skin. When accompanied by a decrease in the circulating platelets, it is called thrombocytopenic purpura. In the acute form, bleeding can occur from any of the body orifices such as hematuria, nosebleed, vaginal bleeding, and bleeding gums.
Shock occurs when the circulatory system (heart as well as arteries) is unable to maintain adequate pressure in order to perfuse organs. Common clinical signs include tachycardia, tachypnea, cool extremities, decreased pulses, decreased urine output, and an altered mental status. Hypotension is typically present but may be initially absent. The end result is hypoxia to end-organ tissues, particularly the kidneys, brain, and heart.
Diagnosis as to the cause of shock should include an evaluation of the heart and the peripheral arteries (systemic vascular resistance). Myocardial infarction and heart failure are problems that make it difficult for the heart to pump an adequate amount of blood to the body. Decreased blood volume (hypovolemia) from hemorrhaging or severe volume depletion (e.g., nausea, vomiting, and diarrhea) also reduces the body’s ability to perfuse tissue.
Disorders that cause a decrease in the arterial pressure include sepsis (infection from any source), liver failure, severe pancreatitis, anaphylaxis, and thyrotoxicosis. The three most common classes of shock therefore are cardiogenic (heart related), hypovolemic, and causes related to reduced systemic vascular resistance, although many overlap (Table 14-1).

Lymphadenopathy is the abnormal enlargement of a lymph node(s). Lymph nodes filter lymph as it returns to the heart. Infectious organisms (e.g., Epstein-Barr virus [EBV] and tuberculosis) and autoimmune disorders (e.g., rheumatoid arthritis [RA] and systemic lupus erythematosus [SLE]) can cause an inflammatory expansion and enlargement of lymph nodes. Malignant diseases, such as lymphoma, chronic lymphocytic leukemia, and Hodgkin’s lymphoma, can also cause enlarged lymph nodes.
Lymph nodes are typically “rubbery” in feel, unattached to surrounding tissue (mobile), and small (usually less than 1 cm). Inflammatory nodes may be tender to the touch, warm, and enlarged but usually remain mobile and soft. While malignant nodes are often not tender or mobile, they are firm and enlarged. Most cases of lymphadenopathy are not malignancy related, although all instances of abnormal adenopathy should be investigated.
Enlargement of the spleen, or splenomegaly, is present in many hematologic diseases. The spleen is normally involved in removing old or deformed erythrocytes, producing antibodies, and removing antibody-laden bacteria or cells. When the spleen exceeds normal function in one of these areas, it becomes enlarged. For example, if a client has hereditary spherocytosis and forms abnormally shaped erythrocytes, the spleen attempts to remove all these cells, thereby increasing in size to accomplish this task.
Splenomegaly is often noted in people with infectious mononucleosis or malignancies such as Hodgkin’s lymphoma (where the spleen is infiltrated by disease). If the bone marrow is unable to produce cells (because of an infiltrative process), the spleen often assumes that role and becomes enlarged (extramedullary hematopoiesis).
AGING AND THE HEMATOPOIETIC SYSTEM
Although blood composition changes little with age, the percentage of the marrow space occupied by hematopoietic (blood-forming) tissue declines progressively. The percentage of bone marrow fat is equal to the person’s age, reaching a plateau at around age 50 years.
Other changes include decreased total serum iron, total iron-binding capacity, and intestinal iron absorption but with increased total body and bone marrow iron; increased fragility of plasma membranes; a rise in fibrinogen and increased platelet adhesiveness; red cell rigidity; and early activation of the coagulation system. Platelet morphology (form and structure) does not appear to change with age, but platelet count and function have been found to vary from normal to increased or decreased.
The cumulative effect of these changes appears in the form of disturbed blood flow in older subjects, leading to the development or aggravation of various circulatory disorders, especially hypertension, stroke, and diabetes. In addition, correlations found between hematologic changes and changes in behavioral patterns and some cognitive functions suggest that hematologic changes contribute to other changes associated with aging as well.5
Age-related changes in the peripheral blood include slightly decreased hemoglobin and hematocrit, although levels remain within the normal adult range. Low hemoglobin levels noted in aging adults can be caused by iron deficiency (usually via blood loss such as ulcer, telangiectasia, colon polyps, or cancer) or can be associated with a long-standing condition such as rheumatologic conditions often seen in a therapy practice (referred to as anemia of chronic disease). Vitamin B12, which is required to produce blood cells, and the subsequent development of anemia (resulting from a B12 deficiency) with its hematologic, neurologic, and GI manifestations, are discussed later in this chapter.
Aging is also associated with a decreased number of lymph nodes and diminished size of remaining nodes, decreased function of lymphocytes, and decline in cellular immunity owing to altered T-cell function (see the section on Effects of Aging on the Immune System in Chapter 7). The effect of aging on quantity, form, and structure of lymphocytes is not well documented.
BLOOD TRANSFUSIONS
Advances in treating hematologic/immunologic disorders through blood transfusions and bone marrow transplantation have provided new success in long-term treatment and a cure for some previously fatal disorders (see Chapter 21). Modern blood banking and transfusion medicine have developed techniques to administer only the blood component needed by the client, such as packed RBCs for anemia or cryoprecipitate for bleeding disorders.
Clients in a therapy setting who have undergone numerous surgical procedures (e.g., traumatic injuries) or elective orthopedic or cardiac procedures may also receive autologous blood transfusions (i.e., reinfusion of a person’s own blood) when significant blood loss may be a complication and a transfusion may be anticipated.
The development of recombinant human erythropoietin (rHuEpo, EPO, or Epogen), with its ability to stimulate erythropoiesis and elevate RBCs, has reduced the need for blood transfusion in a variety of clinical situations (e.g., chronic renal disease, hematologic malignancies, cancer-related anemia, and surgical procedures, especially joint arthroplasty and cardiac procedures).
Reaction to Blood and Blood Products
Because blood products are most often donated from another person, reactions may occur. The most common transfusion-related reaction is a febrile, nonhemolytic reaction (occurring in 0.5% to 1% of erythrocyte transfusions and 30% of platelet transfusions). The condition is characterized by an increase in temperature by more than 1° F during or soon after the transfusion. These reactions are a result of either donor leukocyte cytokines or alloantibodies of the recipient directed against the leukocytes of the donor.
Treatment includes stopping the transfusion, checking the blood for a direct hemolytic process (in the laboratory), and administering antipyretics or corticosteroids. Symptoms are usually transient, and the removal of donor leukocytes from the blood (leukocyte reduction) can reduce the risk of another similar reaction (Box 14-2).
Transfusion-Related Acute Lung Injury
Transfusion-related acute lung injury occurs in as many as 1 in every 2000 transfusions.108 This reaction may present with mild shortness of breath or appear clinically similar to adult respiratory distress syndrome. With appropriate respiratory intervention, most people recover without permanent pulmonary damage.154 Other complications may include transmission of disease (human immunodeficiency virus [HIV], hepatitis, cytomegalovirus), iron overload, air embolism, hypotension in clients taking angiotensin-converting enzyme inhibitors,115 and circulatory overload when blood is administered rapidly in large amounts.
Acute Hemolytic Transfusion Reaction
Less common (only 1 in every 25,000 transfusions) but more severe is the acute hemolytic transfusion reaction. This is due to ABO incompatibility: typically a mistake is made by giving the wrong blood to a person or blood is mislabeled. Symptoms begin soon after the transfusion is begun (see Box 14-2). Erythrocytes are destroyed intravascularly with resultant red plasma and red urine.
The mortality rate is high, ranging from 17% to 60%. The transfusion is immediately terminated and the client given cardiovascular support. Renal failure, disseminated intravascular coagulation, and severe hypotension may occur.
Delayed Hemolytic Transfusion
Delayed hemolytic transfusion reactions occur when the donated erythrocytes are quickly removed from the circulatory system because of an alloantibody. There are often asymptomatic reactions that are noted only because there was not a rise in the hemoglobin following the transfusion.
Allergic Reaction
If the client reacts to the donated plasma an allergic reaction may occur, with associated hives, rash, mucosal edema, wheezing, and other respiratory symptoms. These types of reactions are more typical with fresh frozen plasma and platelet transfusions and are seen in about 1% to 3% of all transfusions. Antihistamines and/or corticosteroids aid in treating the symptoms, and premedication prior to the next transfusion may help reduce or prevent subsequent reactions.
Anaphylaxis
True anaphylaxis is rare (approximately 1 in 20,000 to 50,000 transfusions) and may occur with or without allergic reactions. Symptoms include acute onset of hypotension and edema of the larynx with associated difficulty breathing. Nausea, abdominal pain, and diarrhea may accompany the reaction. This reaction can be severe and fatal and is associated with shock, respiratory failure, and vascular collapse. The earlier the symptoms occur, the more severe the reaction. Treatment consists of immediately discontinuing the transfusion, administering epinephrine and corticosteroids, and providing cardiovascular and respiratory support.
Septic Reactions
Rarely, septic reactions can occur secondary to bacterial contamination of blood products, principally platelets (they are not stored at cold temperatures). In March 2004, all blood banks began to routinely screen platelets for bacterial contamination with a subsequent reduction in septic reactions. Symptoms of such reactions include fever/chills; hypotension; headache; back, chest, and abdominal pain; and shortness of breath. Culture of the product and appropriate antibiotics and cardiovascular support are the mainstays of treatment.
Transfusion as a source for hepatitis (B and C) has been reduced since the initiation of donor screening for the hepatitis antibody. People with hemophilia who received coagulation factor concentrates before 1984 have been at highest risk among transfusion recipients because of exposures to pooled blood products prepared from thousands of donors. The availability of nonhuman plasma factors has virtually eliminated the transmission of viruses among people with hemophilia.
The risk of HIV infection by transfusion is low overall, calculated at 1 in 1 million transfusions. The risk of HIV transmission by blood transfusion has been continually reduced through the elimination of high-risk individuals from blood donor pools and the use of more sensitive screening. Acquired immune deficiency syndrome (AIDS) has developed in a small percentage of people receiving transfusion of RBCs, platelets, or commercial coagulation factor concentrates. AIDS has also been reported in infants after neonatal exchange, but the majority of pediatric cases were associated with maternal transmission from mothers with HIV.
Bloodless Medicine
Bloodless medicine and surgery is the use of technologic and pharmaceutical techniques to minimize blood loss and avoid the use of allogenic blood transfusions. Bloodless medicine and surgery programs began over the last 20 years to meet the needs of Jehovah’s Witnesses, whose beliefs do not allow blood transfusions.
In recent years the number of bloodless medicine programs has increased due to a growing number of individuals seeking this type of treatment to avoid potential exposure to bloodborne pathogens or because of a family history of transfusion reactions.169
As bloodless programs have grown, researchers have found other advantages to avoiding blood transfusions. The length of time banked blood spends in storage can affect the hemoglobin molecule’s ability to release the oxygen it is carrying, potentially decreasing its oxygen-carrying capacity. Furthermore, cold storage of blood can negatively affect an RBC’s elasticity, potentially leading to its early destruction.140,169
Candidates for Bloodless Procedures
The first step in ensuring successful use of bloodless techniques is a thorough history, including a history of any personal or family history of bleeding abnormalities.70,140 Any history of bleeding abnormalities requires further evaluation.
Preoperative blood work is needed to detect anemia, since individuals with low preoperative hemoglobin levels are more likely to need a transfusion.71 Bloodless techniques can still be used if a low hemoglobin level is increased by giving recombinant human erythropoietin and iron.140,177
Bloodless Techniques
During surgical procedures, there are a variety of techniques that are used to avoid the need for a blood transfusion. Minimally invasive surgery can significantly reduce blood loss, as does meticulous surgical technique. Technologic advances, such as the gamma knife, harmonic scalpel, and argon beams, have also improved the surgeon’s ability to achieve hemostasis during procedures by coagulating vessels while causing less tissue damage.70
Another technique, acute normovolemic hemodilution, removing a quantity of a person’s blood and replacing it with intravenous crystalloid and colloid solution to maintain volume, can also be used.
With acute normovolemic hemodilution, fluid loss during a procedure is mainly the crystalloid/colloid solution, which limits loss of RBCs while preserving clotting factors. At the conclusion of the surgical procedure, the withdrawn blood is returned to the patient. Since this process is completed through a closed circuit, it is acceptable to most Jehovah’s Witnesses.170,177
Cell salvage techniques can be used in the intraoperative and postoperative phases to retranfuse lost blood. These techniques have been associated with infection and hemolysis but are believed to be safe as part of an overall blood loss management program.170
Postoperatively, steps should be taken to minimize blood loss by close observation for bleeding, with immediate steps being taken to regain hemostasis (halt bleeding). Additionally, postoperative phlebotomy should be kept to a minimum and the blood drawn using micro-sampling techniques.70,170 Use of recombinant human erythropoietin and iron should be continued as needed during the postoperative phase.
Finally, clinician acceptance of low hemoglobin levels is essential in bloodless medicine and surgery. Research has shown that the body can tolerate lower hemoglobin levels than would be thought acceptable without compromising oxygen delivery. Hemoglobin levels alone should not be used as the deciding factor for a blood transfusion. The individual’s condition and comorbidities should also be taken into account.169
The advances in bloodless medicine and surgery have led to use of these techniques in many surgical procedures, including the Whipple procedure, joint replacements, and coronary artery bypass.140
DISORDERS OF IRON ABSORPTION
Hemochromatosis is an autosomal recessive hereditary disorder characterized by excessive iron absorption by the small intestine. Most inherited forms of the disease are caused by abnormalities of the HFE gene located on chromosome 6. Although the exact mechanism of this gene is unknown, two hypotheses exist that may explain the pathology of the disease.
Pathogenesis
The crypt hypothesis suggests that the abnormal protein product of HFE is unable to interact with transferrin and leads to a decrease in absorption of iron by the intestinal crypt cells. This then triggers an inappropriate increase in iron absorbed by the intestinal villus cells.
The second hypothesis is based on a recently discovered peptide hormone produced by the liver, hepcidin, that appears to be the master regulator of iron homeostasis in human beings and other mammals. Hepcidin levels increase when iron plasma is high but is not produced when iron plasma is low. Hepcidin inhibits the release of iron by macrophages, but in its absence macrophages can release needed iron.
The product of HFE most likely plays a role in regulating the production of hepcidin; thus an abnormal gene product would alter iron metabolism, leading to increased absorption despite already high iron plasma levels.147
Most likely these two hypotheses are interrelated. Abnormalities of the hemochromatosis gene occur in 1 in every 200 people of Northern European descent, although not all people with the gene will develop the disease. Hemochromatosis is present at birth but remains asymptomatic until the development of iron overloading and onset of symptoms between ages 40 and 60 years (sometimes as early as age 30 years). The prevalence is equal among men and women, but men experience symptoms five to 10 times more often than do women (menstruation and pregnancy help to slow progression of the disorder).
Clinical Manifestations
The body typically absorbs iron at a rate equal to body requirements. But in hemochromatosis, there is an uncoupling between absorption and body needs. Excess iron is slowly deposited in cells, particularly in the liver, pancreas, heart, and, to a lesser extent, other endocrine glands (e.g., the pituitary gland).
Early signs and symptoms can include weakness, chronic fatigue, myalgias, joint pain, abdominal pain, hepatomegaly, elevated hemoglobin, and elevated liver enzymes. Continued iron overload leads to tissue damage. The liver is the most commonly affected organ, and clients may present with hepatomegaly without liver enzyme abnormalities. If the disease progresses without treatment, cirrhosis with liver failure may ensue.
Other complications of untreated hemochromatosis include diabetes mellitus, cardiac myopathy (with associated CHF) and arrhythmias, “bronzing” of the skin (from iron deposition in the dermis and increase of melanin), destructive arthritis, and impotence (men) or decreased libido (women) and sterility.
MEDICAL MANAGEMENT
Diagnosis can best be made through blood tests. The most sensitive test is the measurement of transferrin saturation. Levels higher than 60% in men and 50% in women are suggestive of the disease. If the client has a family history of the disease, genetic testing may be diagnostic. Ferritin levels greater than 1000 ng/ml (without evidence of inflammation) also suggest an iron overload state.
Definitive diagnosis requires a liver biopsy with measurement of iron stores; however, positive results from the genetic and blood tests may be sufficient for diagnosis. A liver biopsy is helpful when assessment of liver damage is required. In families where hemochromatosis has been previously diagnosed, all first-degree blood relatives should be genetically screened for hemochromatosis. Careful monitoring of affected family members can be done through blood tests. Monthly monitoring is required for those with known hemochromatosis.
TREATMENT.
Treatment should begin early in the disease process, when iron levels exceed normal values. Medical intervention consists of weekly to twice-weekly therapeutic phlebotomy. This is performed until iron stores are at normal levels, with ferritin levels less than 50 ng/ml (for some clients this may take 1 to 2 years), after which maintenance therapy is done as needed to maintain appropriate levels (about once every 3 months).
Chelating agents (i.e., agents that bind iron) may be given parenterally in cases where anemia or protein loss is severe. Phlebotomy, however, is less expensive and safer. Affected individuals are instructed to avoid ingesting alcohol since it increases the risk of developing cirrhosis nearly tenfold.
PROGNOSIS.
The prognosis is good, with normal life expectancy as long as the iron levels remain in the normal range and the disease has not caused organ damage. With treatment, liver function improves, cardiac failure may be reversed, skin color lightens, and about 40% of clients with diabetes mellitus have improved glycemic control. Cirrhosis does not improve with therapy and 30% go on to develop hepatocellular carcinoma. Hypogonadism and arthropathy typically do not improve with treatment, and joint symptoms may actually progress despite therapy.
DISORDERS OF ERYTHROCYTES
Definition
Anemia is a reduction in the oxygen-carrying capacity of the blood from an abnormality in the quantity or quality of erythrocytes (RBCs). The World Health Organization (WHO) has defined anemia in terms of the level of hemoglobin: less than 14 g/100 ml for men and less than 12 g/100 ml for women. Different ranges exist for men and women, infants and growing children, and different metabolic and physiologic states.
These normal values must be evaluated on an individual basis; normal levels may be inadequate if tissue oxygen delivery is impaired by pulmonary insufficiency, cardiac disorders, or an increase in hemoglobin oxygen affinity, whereas low levels may be appropriate if tissue oxygen requirements are decreased, as in the case of hypothyroidism.
Overview
Anemia is not a disease but rather a symptom of many other disorders, such as dietary deficiency (anemia due to folate or vitamin B12 deficiency); acute or chronic blood loss (iron deficiency); congenital defects of hemoglobin (sickle cell diseases); exposure to industrial poisons; diseases of the bone marrow; chronic inflammatory, infectious, or neoplastic disease; or any other disorder that upsets the balance between blood loss through bleeding or destruction of blood cells and production of blood cells.
Many types and causes of anemia exist; not all are discussed in this text. The most common anemias observed in a therapy setting fall into five broad disease-related categories: (1) iron deficiency associated with chronic GI blood loss secondary to NSAID use; (2) chronic diseases or inflammatory diseases, such as RA or SLE; (3) nutritional conditions (e.g., malabsorption syndrome leading to vitamin B12, folate, or iron deficiency; alcohol abuse leading to folate deficiency); (4) infectious diseases such as tuberculosis or AIDS; and (5) neoplastic disease (bone marrow failure). Anemia with neoplasia may be a complication of chemotherapy (e.g., with cisplatin, carboplatin, or taxol administration), as a consequence of radiation to the pelvis, or bone marrow infiltration.
Anemias are classified according to etiologic factors (Box 14-3) or morphology (form/structure) (Box 14-4). Descriptions of anemias based on erythrocyte morphology refer to the size and hemoglobin content of the RBC. In some anemias, variations occur in size (e.g., anisocytosis) or shape (e.g., poikilocytosis) of erythrocytes.
Etiologic Factors and Pathogenesis
Anemia results from (1) excessive blood loss, (2) increased destruction of erythrocytes, or (3) decreased production of erythrocytes. Anemia is the most common hematologic abnormality; only the anemias most commonly observed in rehabilitation or therapy settings are discussed here, using the three etiologic categories from Box 14-3 as a guideline.
The underlying pathogenesis can be multifactorial and depends on the condition causing the anemia. A number of physiologic compensatory responses to anemia occur, depending on the rapidity of onset and duration of anemia and the condition of the individual. In acute-onset anemia with severe loss of intravascular volume, peripheral vasoconstriction and central vasodilation occur to preserve blood flow to the vital organs.
If the anemia persists, small-vessel vasodilation will provide increased blood flow to ensure better tissue oxygenation. These vascular compensations result in decreased systemic vascular resistance, increased cardiac output, and tachycardia, resulting in a higher rate of delivery of oxygen-bearing erythrocytes to the tissues. Other compensatory mechanisms include an increase in plasma volume to maintain total blood volume and enhance tissue perfusion and stimulation of erythropoietin production to increase new erythrocyte production.
Excessive Blood Loss.: Excessive blood loss, such as occurs with GI bleeding in the client with a history of aspirin or NSAID use, is a cause of anemia seen in a therapy practice. Slow, chronic GI blood loss from medication or any GI disorder (e.g., peptic and duodenal ulcers, gastritis, GI cancers, hemorrhoids, diverticulosis, ulcerative colitis, and colon polyps) can result in iron-deficiency anemia.
Destruction of Erythrocytes.: Destruction of erythrocytes (hemolysis) can occur as a result of congenital RBC membrane abnormalities, lack of necessary enzymes needed for normal metabolism, autoimmune processes, or infection. All but the autoimmune processes are discussed elsewhere in this chapter.
Autoimmune hemolytic anemia (AIHA) is caused by an autoantibody that attaches to the RBC, leading to its destruction. The most common form of AIHA is warm antibody-mediated, an immunoglobulin (Ig) G autoantibody that binds to erythrocytes at body temperature. Macrophages are attracted to the attached autoantibody and release enzymes that begin to destroy the cell membrane. The resultant spherical cells are removed by the spleen.
Cold agglutinin disease is another autoimmune hemolytic process caused by IgM autoantibodies that bind to erythrocytes at temperatures less than 37° C and trigger complement fixation and clumping of erythrocytes. These complement-laden erythrocytes may be destroyed intravascularly or removed by the liver. Hemolytic anemia can be idiopathic or a result of collagen vascular diseases (e.g., SLE), lymphoproliferative diseases (e.g., chronic lymphocytic leukemia or lymphoma), or other malignancies. Medications such as dapsone, penicillin, quinidine, quinine, and methyldopa can also cause AIHA.
Decreased Production of Erythrocytes.: Anemias resulting in the underproduction of RBCs usually stem from either a lack of erythropoietin (as seen in kidney disease) or an inability of the bone marrow to respond to erythropoietin. Hyporesponsiveness of the bone marrow may be a result of a nutrient deficiency or a chronic disease such as RA, SLE, tuberculosis, or cancer.
Nutritional deficiency as a cause of anemia can occur at any age. Iron, vitamin B12, and folate are among the most important vitamins and minerals in the production of hemoglobin and the formation of erythrocytes. Iron is necessary for DNA synthesis, oxygen transport, and respiration. Iron deficiency can occur secondary to blood loss (RBCs are the principal site of iron storage), malabsorption, normal growth, and pregnancy. Menstruating women, pregnant women, growing children, lower socioeconomic groups, and older adults (as a result of economic constraints, lack of interest in food preparation, and poor dentition) are the most common groups to develop iron-deficiency anemia.
Vitamin B12 (cobalamin) is required for DNA synthesis. Deficiency of the vitamin may infrequently occur due to a lack in the diet (the body is very efficient at retaining cobalamin) but most often develops because of an absence of intrinsic factor (IF).
Normally, after cobalamin is ingested it combines with R binders in the stomach and then binds to IF in the small intestine. IF is produced by gastric parietal cells and is required for cobalamin absorption in the terminal small bowel. Without IF, cobalamin is not absorbed.
Pernicious anemia is an anemia due to a loss of IF. Antibodies against the membrane of gastric parietal cells cause an atrophy of these cells, resulting in a lack of IF production. Destruction of IF production sites may also occur with gastrectomy (see the section on Aging and the Gastrointestinal System in Chapter 16).
Other causes of vitamin B12 deficiency include bacterial overgrowth in the lumen of the intestine (competes for vitamin B12), surgical resection of the ileum (eliminates the site of vitamin B12 absorption), severe Crohn’s disease, and, more rarely, dietary deficiency (e.g., strict vegetarian diet) and tapeworm infection. Crohn’s disease can cause sufficient destruction of the ileum to retard vitamin B12 absorption.
Folic acid deficiency is a common cause of decreased production of erythrocytes. Folic acid deficiency has many causes, but it usually results from inadequate dietary intake, chronic alcoholism, malabsorption syndromes, anorexia, and consumption of overcooked food. In anemia due to folic acid deficiency associated with alcoholism, not only is the diet poor in folate, but alcohol inhibits the enzyme needed to absorb folate.
The common occurrence of folic acid deficiency during the growth spurts of childhood and adolescence and during the third trimester of pregnancy is explained by the increased demands for folate required for DNA synthesis in these circumstances. Pregnant women need six times the normal amount of folic acid to meet the needs of the developing fetus. Long-term use of anticonvulsants (e.g., primidone, diphenylhydantoin, phenobarbital), antimetabolites administered for cancer and leukemia, and certain oral contraceptives may interfere with folate absorption.
Anemia of chronic disease is very common in the therapy setting. It is characterized by a modest reduction in hemoglobin (9 to 11 g/dl), the presence of inflammation (secondary to a disease), and decreased responsiveness of the bone marrow to erythropoietin. Many diseases associated with inflammation have accompanying elevated levels of cytokines and interferons. The production of hepcidin, a protein synthesized by the liver, is induced by the presence of interferon 6 (IF-6); hepcidin both inhibits the absorption of iron from the gut and the release of iron from macrophages for bone marrow use. Clients with an underlying chronic illness usually do not need iron, and anemia of chronic disease does not respond to iron.
Bone marrow disorders constitute another source of anemia caused by decreased production of erythrocytes in a therapy practice. Aplastic anemia, marrow replacement with fibrotic tissue or tumor, acute leukemia, and infiltrative disease (e.g., lymphoma, myeloma, and carcinoma) fall into this etiologic category.
Anemias of radiation-induced bone marrow failure occur because the bone marrow stem cells are destroyed and mitosis (cell division) is inhibited, preventing the synthesis of RBCs. Antimetabolites used in cancer therapy also cause bone marrow failure by blocking the synthesis of purines or nucleic acids required for synthesis of DNA within the cell. Aplastic anemia may result from either damage to the stem cells or immune-mediated destruction of the stem cells.
Clinical Manifestations
Mild anemia often causes only minimal and usually vague symptoms such as fatigue until hemoglobin concentration and hematocrit fall below half of normal. As the anemia progresses, general signs and symptoms caused by the inability of anemic blood to supply the body tissues with enough oxygen may include weakness, dyspnea on exertion, easy fatigue, pallor or yellowing of skin (especially the palms of the hands, fingernails, mucosa, and conjunctiva), tachycardia, increased angina in preexisting coronary artery disease, leg ulcers (sickle cell), and, occasionally, koilonychia (Fig. 14-1).
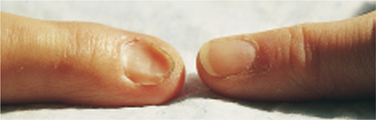
Figure 14-1 Normal nail (right) compared with nail referred to as koilonychia and sometimes called spoon-shaped nails or spoon nails (left). They are thin, depressed nails with lateral edges turned up and are concave from side to side. They may be idiopathic, congenital, or a hereditary trait and are occasionally due to iron-deficiency anemia. (Reprinted from Swartz MH: Textbook of physical diagnosis, ed 5, Philadelphia, 2006, Saunders.)
Pallor in dark-skinned people may be observed by the absence of the underlying red tones that normally give brown or black skin its luster. The brown-skinned individual demonstrates pallor with a more yellowish-brown color, and the black-skinned person appears ashen or gray.
Neuropsychiatric complications such as dementia, ataxia, psychosis, and peripheral neuropathies can develop in cases of B12 deficiency. These abnormalities are caused by lesions in the spinal column, the cerebrum, and peripheral nerves. The lack of cobalamin initially leads to demyelination of the nerves followed by axonal degeneration. Axonal death may result if cobalamin deficiency persists.
Reversal of symptoms may be possible if treatment is initiated before permanent damage to the nerves. The findings typically consist of a symmetric sensory neuropathy that begins in the feet and lower legs, although rarely it may involve the upper extremities, especially fine motor coordination of the hands. This upper extremity neuropathy may clinically manifest as problems with deteriorating handwriting.
Affected individuals may also describe moderate pain or paresthesias of the extremities, especially the feet. Individuals may interpret the neuropathy as difficulty with locomotion when, in fact, they are experiencing the loss of proprioception. The affected individual may need to hold on to the wall, countertops, or furniture at home due to difficulties maintaining balance. An associated positive Romberg’s sign may be present. Loss of motor function is a late manifestation of B12 deficiency.
Although a symmetrical neuropathy is the usual pattern, B12 deficiency occasionally presents as a unilateral neuropathy and/or bilateral but asymmetrical neuropathy. CNS manifestations range from mild cognitive changes to dementia to frank psychosis. Clients may present with personality changes and/or inappropriate behavior.
Complications depend on the specific type of anemia; severe anemia can cause heart failure and hypoxic damage to the liver and kidney with all the signs and symptoms associated with either of those conditions. Anemia in the presence of a coronary obstruction precipitates cardiac ischemia.
MEDICAL MANAGEMENT
Anemia in the early stages often goes unnoticed since symptoms may not be recognized until hemoglobin concentration is reduced to half of normal. Once symptoms become pronounced or noted on routine laboratory tests, the diagnosis is most often made by blood tests.
The RBC indexes indicate if the RBCs are normal (normocytic), larger than normal (macrocytic, as seen with B12 and folate deficiency), or smaller than normal (microcytic, as seen with thalassemias and iron deficiency). The peripheral smear may reveal structural characteristics, which give clues to the underlying cause of the anemia. For example, target cells (bull’s-eye erythrocytes) are often associated with thalassemia (Fig. 14-2) and microspherocytes can be seen in warm antibody-induced hemolysis, whereas sickled erythrocytes are noted with sickle cell disease (Fig. 14-3).
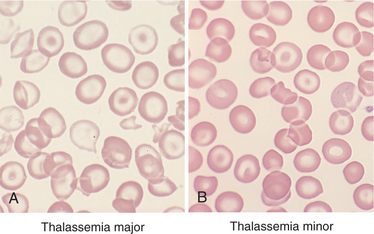
Figure 14-2 Thalassemia is a hemolytic hemoglobinopathy anemia characterized by microcytic, short-lived RBCs caused by deficient synthesis of Hb polypeptide chains. Classification of type depends on the chain involved (α-thalassemia, β-thalassemia). β-Thalassemia occurs in two forms: thalassemia major and thalassemia minor. Characteristic bull’s-eye or target cells are shown here in both forms. (Reprinted from Damjanov I, Linder J: Pathology: a color atlas, St Louis, 2000, Mosby.)
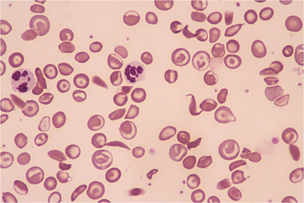
Figure 14-3 Target and sickle cells typical of sickle cell anemia (x200). (Reprinted from Goldman L: Cecil textbook of medicine, ed 22, Philadelphia, 2004, WB Saunders. Courtesy Jean Shafer.)
Personal and family history may point to congenital anemia, and a physical examination may elicit signs of primary hematologic diseases such as lymphadenopathy, hepatosplenomegaly, skin and mucosal changes, stool positive for blood, or bone tenderness.
Following these initial tests, more specific laboratory tests can be done to verify the diagnosis. These may include a complete blood cell count (CBC), an iron profile, serum ferritin, reticulocyte count, haptoglobin, B12 level, folate level, and lactate dehydrogenase.
TREATMENT.
Treatment of anemia is directed toward alleviating or controlling the causes, relieving the symptoms, and preventing complications. It is critical that the underlying cause of anemia is determined so that appropriate treatment can be given. For example, endoscopy to identify the source of GI blood loss for a client with a long-term history of NSAID use would indicate the need to stop taking the medication and prescribe the use of proton pump inhibitors (see Chapter 16).
Treating the underlying cause can include the replacement of deficient vitamins and minerals (e.g., vitamin B12, folate, or iron) or corticosteroids for warm-antibody AIHA. The anemia of cold agglutinin disease is typically mild, requiring only a warm environment, while bone marrow transplantation may be required for malignancies. Immunosuppressive therapy with antithymocyte globulin and cyclosporine is the initial treatment for clients with aplastic anemia, and individuals with kidney disease are given erythropoietin or darbepoetin.
PROGNOSIS.
The prognosis for anemia depends on the etiologic factors and potential treatment for the underlying cause. For example, the prognosis is good for anemia related to nutritional deficiency but poor for lymphoproliferative diseases. Likewise, treatment is aimed at correcting the underlying pathogenesis.
Untreated or misdiagnosed B12 deficiency can be progressive, resulting in irreversible neurologic damage. Anemia in the older adult (age 85 years or older) is associated with an increased risk of death. Although anemia was once considered a normal consequence of aging, it is now recognized as a sign of other disease in the older adult (e.g., hip fracture, RA, erosive gastritis, peptic ulcer, malnutrition, cirrhosis, ulcerative colitis) requiring further assessment.89
DISORDERS OF LEUKOCYTES
Alterations in blood leukocyte (WBC) concentration and in the relative proportions of the several leukocyte types are recognized as measures of the reaction of the body to infection, inflammation, tissue damage, or degeneration. In many instances, these alterations give useful indications of the nature of the pathologic process and may be seen in association not only with acute infections but also with many chronic ailments treated by the therapist.
Leukocytes may be classified in three main groups: granulocytes (basophils, eosinophils, neutrophils), monocytes, and lymphocytes. Granulocytes (granular leukocytes) contain lysing agents within their granules that are capable of digesting various foreign materials. The main type of granulocyte is the neutrophil, also called the polymorphonuclear leukocyte; these are usually not found in normal “healthy” tissue and are referred to as the first line of hematologic defense against invading pathogens.
Granulocytes are also involved in the pathophysiology of organ damage in ischemia/reperfusion, trauma, sepsis, or organ transplantation. Basophils and eosinophils are involved with allergic reactions and respond to parasitic and fungal infections.
Monocytes are the largest circulating blood cells and represent an immature cell until it leaves the blood and travels to the tissues. Once migrated, monocytes form macrophages when activated by foreign substances, such as bacteria. Monocytes/macrophages participate in inflammation by synthesizing numerous mediators and eliminating various pathogens.
Lymphocytes are further divided into B and T cells. B lymphocytes are responsible for the humoral portion of the immune system and are known to secrete antibodies that react with antigens and initiate complementmediated destruction or phagocytosis of foreign pathogens, particularly bacteria. T lymphocytes are in control of cell-mediated immunity and are able to recognize and destroy cells altered by viruses.
These cells are also responsible for coordinating the immune response through the release of lymphokines and inflammatory modulators, creating a cell-to-cell communication with B cells and monocytes. The exact role or function of leukocytes during inflammatory processes remains the subject of considerable investigation.
Leukocytosis
Leukocytosis, defined as an increase in the number of leukocytes in the blood, may occur as a result of a variety of causes (Box 14-5) and may also occur as a normal protective response to physiologic stressors such as strenuous exercise, emotional changes, temperature changes, anesthesia, surgery, pregnancy, some drugs, toxins, and hormones.
Leukocytosis develops within 1 or 2 hours after the onset of acute hemorrhage and is greater when the bleeding occurs internally (e.g., into the peritoneal cavity, pleural space, or joint cavity, or as a result of a skull fracture with associated intracranial bleed or subarachnoid hemorrhage) than when the bleeding is external.
Leukocytosis is a common finding in and characterizes many infectious diseases recognized by a count of more than 10,000 WBCs/mm3 (see Table 40-7). An elevated WBC count (greater than 50,000/mm3, with the majority of cells being neutrophils and neutrophil precursors) in response to a serious underlying process is referred to as a leukemoid reaction (see Box 14-5).
Leukocytosis frequently results from an increase in circulating neutrophils (neutrophilia), recruited in large numbers in the course of infections and in the presence of some rapidly growing neoplasms (e.g., leukemia, non–small cell lung cancer, renal cell carcinoma, and gastric carcinoma). The counts may be especially high in tumors with significant necrosis. Some tumors can also release hormone-like substances that cause leukocytosis.
Clinical Manifestations
Clinical signs and symptoms of leukocytosis are usually associated with symptoms of the conditions listed in Box 14-5 and may include fever, headache, shortness of breath, symptoms of localized or systemic infection, and symptoms of inflammation or trauma to tissue.
MEDICAL MANAGEMENT
DIAGNOSIS, TREATMENT, AND PROGNOSIS.
Major leukocyte functions are accomplished in the tissues so that the leukocytes in the blood are in transit from the site of production or storage to the tissues, even in normal people. Variations in the blood concentrations of each leukocyte type may be of brief duration and easily missed or may persist for days or weeks. Laboratory tests for detecting leukocyte abnormalities include total leukocyte count, leukocyte differential cell count (see Chapter 40), peripheral blood morphology, and bone marrow morphology.
Treatment is directed toward the underlying cause of the change in leukocytes and control of any infections. Prognosis depends on the etiology of the leukocytosis.
The importance of good handwashing (see Boxes 8-4 and 8-5) and hygiene practices cannot be overemphasized when treating immunocompromised clients. Some centers recommend that people with a WBC count of less than 1000/mm3 or a neutrophil count of less than 500/mm3 wear a protective mask. Therapists should ensure that these people are provided with equipment that has been disinfected according to standard precautions.
Leukopenia
Leukopenia, or reduction of the number of leukocytes in the blood below 5000/ml (see Chapter 40), can be caused by a variety of factors such as HIV (or other viral infection such as hepatitis), alcohol and nutritional deficiencies, drug-induced condition, and connective tissue disorders (e.g., SLE). It can occur in many forms of bone marrow failure, such as that following antineoplastic chemotherapy or radiation therapy or in overwhelming infections.
People with leukemia, lymphoma, myeloma, and Hodgkin’s lymphoma have serious underlying WBC abnormalities that contribute to the risk of infection associated with leukopenia. Unlike leukocytosis, leukopenia is never beneficial. As the leukocyte count decreases, the risk for various infections increases.
The risk of infection from leukopenia after bone marrow radiation has been reduced with continued improvements in medical treatment. The use of naturally occurring glycoproteins to help collect blood stem cells administered after chemotherapy reduces the duration of blood cell reduction and prevents the serious problems encountered in the past.
These glycoproteins are hematopoietic growth factors called colony-stimulating factors (CSFs) or, more specifically, granulocyte colony-stimulating factor (GCSF), or filgrastim (Neupogen). Growth factors move the stem cells from the bone marrow into the peripheral blood and can result in a temporary tenfold to hundredfold increase in the numbers of circulating stem cells at the time of bone marrow recovery. Filgrastim not only increases the number but also the function of granulocytes.
Clinical Manifestations
Leukopenia may be asymptomatic (and detected by routine tests) or associated with clinical signs and symptoms consistent with infection such as sore throat, cough, high fever, chills, sweating, ulcerations of mucous membranes (e.g., mouth, rectum, vagina), frequent or painful urination, or persistent infections.
MEDICAL MANAGEMENT
As with leukocytosis, diagnosis is by laboratory testing for leukocyte abnormalities. Treatment is directed toward elimination of the cause of the reduced leukocytes and control of any infections. Pharmacologic therapy includes the use of antibiotics, antifungal agents, and CSF drugs such as filgrastim (Neupogen). This drug markedly assists in decreasing the incidence of infection in people who have received bone marrow–depressing antineoplastic agents.
Basophilia
Basophils are a subtype of leukocytes involved in inflammatory and allergic reactions. Their granules contain heparin (an anticoagulant but without significant systemic effects), histamine (the cause of most of the systemic symptoms), chondroitin sulfate, platelet-activating factor, and other proteins.
The antibody IgE is the most common stimulator of basophilic degranulation. IgE may be secreted in response to the body’s detection of a foreign particle (such as insect venom or some drugs). The histamine and other proteins released by the basophil lead to a local inflammatory and allergic response. This response varies from person to person. Some clients may have reactions that are not modulated and develop anaphylaxis, asthma, urticaria (hives), and allergic rhinitis. Basophilia is primarily associated with myeloproliferative disorders, particularly chronic myeloid leukemia.
The remaining categories (e.g., basophilia/basopenia, eosinophilia/eosinopenia, neutrophilia/neutropenia) are all types of leukocytosis or leukopenia. The specific type is determined when the leukocyte differential (WBC count) determines the percentage of each type of granular and nongranular leukocyte.
Eosinophilia
Following the maturation and release of eosinophils from the bone marrow into the circulation, they soon migrate into tissue. These tissues are in areas that have contact with the external environment, such as the skin, GI tract, genital tract, and lungs. Eosinophils can be recruited to areas of inflammation by various antibodies and interleukins and stimulated to release chemokines, growth factors, cytokines, peroxidase, and other modulating proteins.
Chemokines are any of a group of low-molecular-weight cytokines (e.g., interleukins) identified on the basis of their ability to induce chemotaxis (cell movement; see Fig. 5-10) or chemokinesis (cell activity due to the presence of a chemical substance) in leukocytes in inflammation. Chemokines function as regulators of the immune system and may play roles in the circulatory system and CNS.
Eosinophils, however, are not central participants in defending the body against most infections but do play key roles in fighting parasitic infections, such as hookworm or strongyloidiasis. Eosinophils are also involved in allergic reactions such as asthma, cutaneous reactions, and other hypersensitivity states.
An elevation in the number of eosinophils in the blood, eosinophilia (eosinophil count greater than 500/μl) is seen most often in allergic reactions to drugs (aspirin, sulfonamides, penicillins), in hay fever, eczema, collagen vascular diseases (RA, eosinophilic fasciitis, periarteritis nodosa), and malignancies (Hodgkin’s lymphoma, mycosis fungoides, chronic myeloid leukemia, cancer of the stomach, pancreas, and lung).
Idiopathic hypereosinophilia syndrome (counts from 50,000 to 100,000/μl), eosinophilic leukemia, and Loeffler’s syndrome are uncommon illnesses are but associated with dramatic eosinophilia. Increased levels of eosinophils were also identified during outbreaks of eosinophilia myalgia syndrome, a connective tissue disease induced by the ingestion of contaminated L-tryptophan supplements, sometimes taken for insomnia or back pain.43
Neutrophilia
Granulocytes assist in initiating the inflammatory response, and they defend the body against infectious agents by phagocytosing bacteria and other infectious substances. Generally, the neutrophils (the most plentiful of the granulocytes) are the first phagocytic cells to reach an infected area, followed by monocytes; neutrophils and monocytes work together to phagocytose all foreign material present.
Granulocytosis (an excess of granulocytes in the blood) or neutrophilia (increased number of neutrophils in the blood) are terms used to describe the early stages of infection or inflammation. The capacity of corticosteroids or alcohol to diminish the accumulation of neutrophils in inflamed areas may be due to their ability to reduce cell adherence.
The many potential causes of neutrophilia include inflammation or tissue necrosis (e.g., after surgery from tissue damage, severe burns, myocardial infarction, pneumonitis, rheumatic fever, RA); acute infection (e.g., Staphylococcus, Streptococcus, Pneumococcus); drug-or chemical-induced causes (e.g., epinephrine, steroids, heparin, histamine); metabolic causes (e.g., acidosis associated with diabetes, gout, thyroid storm, eclampsia); and neoplasms of the liver, GI tract, or bone marrow. Physiologic neutrophilia may also occur as a result of exercise, extreme heat/cold, third-trimester pregnancy, and emotional distress.
Neutropenia
Neutropenia is the condition associated with a reduction in circulating neutrophils (less than 2500/μl). Causes are either acquired or congenital. Acquired neutropenias are typically a result of toxicity to neutrophil precursors in the bone marrow. This may be due to drugs (e.g., NSAIDS, sulfonamides, penicillins, anticonvulsants) or infectious agents (e.g., hepatitis B, cytomegalovirus, EBV, HIV). Other drugs can cause an autoimmunerelated peripheral destruction of neutrophils, leading to neutropenia.
Other causes of neutropenia include carcinoma of the lung, breast, prostate, and stomach and malignant hematopoietic disorders that can occupy enough of the bone marrow to cause global marrow failure with resultant pancytopenias (all cell lines are decreased in number). Congenital causes usually come to attention early in life and are much less common than acquired causes.
The longer an individual exists without neutrophils, the higher the risk for significant infection. Drug-induced neutropenia generally resolves in 10 to 12 days, while administration of GCSF may shorten the time to resolution.
Lymphocytosis/Lymphocytopenia
Lymphocytosis occurs most commonly in acute viral infections, especially those caused by EBV. Other causes include endocrine disorders (e.g., thyrotoxicosis, adrenal insufficiency) and malignancies (e.g., acute and chronic lymphocytic leukemia).
Lymphocytopenia may be acquired or congenital. Acquired lymphocytopenias can be attributed to abnormalities of lymphocyte production associated with neoplasms and immune deficiencies and destruction of lymphocytes by drugs, viruses, or radiation.
Other causes include corticosteroid therapy, severe systemic illnesses (e.g., military tuberculosis), SLE, sarcoid, or severe right-sided heart failure. For individuals with AIDS, lymphocytopenia can be a major problem, increasing their susceptibility to viral illnesses, malignancies, and fungal infections.
Monocytosis
Monocytosis, an increase in monocytes, is most often seen in chronic infections, such as tuberculosis and subacute endocarditis, and other inflammatory processes, such as SLE and RA. Monocytosis is present in more than 50% of people with collagen vascular disease (see Box 12-17).
Clients with sarcoid or other granulomatous processes may also have elevated monocytes. Monocytosis also exists as a normal physiologic response in newborns (first 2 weeks of life). Although not common, monocytes can go through a transformation, becoming leukemia, or an elevation of normal monocytes can be seen in malignancies such as Hodgkin’s and non-Hodgkin’s lymphoma. Monocytosis can be indicative of bone marrow recovery following a drug-induced loss of granulocytes.
NEOPLASTIC DISEASES OF THE BLOOD AND LYMPH SYSTEMS
Hematologic malignancies include diseases in any hematologic tissue (e.g., bone marrow, spleen, thymus) that arise from changes in stem cells or clonal (genetically identical cells) proliferation of abnormal cells. The primary hematologic disorders that result from stem cell abnormalities include the myeloproliferative disorders (e.g., polycythemia vera, essential thrombocythemia, chronic myeloid leukemia, and myelofibrosis with myeloid metaplasia) and acute myeloid leukemia. Myelofibrosis is the replacement of hematopoietic bone marrow with fibrous tissue such as fibroblasts and collagen.
Multiple myeloma and plasma cell diseases arise from clonal proliferation of abnormal plasma cells. Lymphoid malignancies are also a clonal proliferation of malignant cells and can be categorized according to the malignant cell type: B-cell, T-cell/natural killer cell, and Hodgkin’s lymphoma. For ease of discussion, the leukemias are presented together.
Bone Marrow and Stem Cell Transplantation
Bone marrow transplantation (BMT) is often a treatment choice for many of the neoplastic diseases of the blood and lymph systems. Both BMT and the more recent technique of stem cell transplantation are discussed in Chapter 21.
The Leukemias
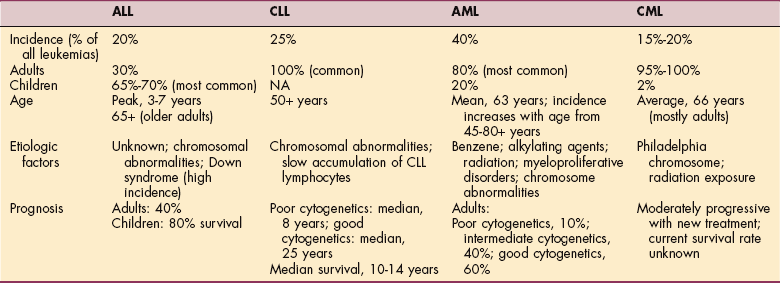
Leukemia is a malignant neoplasm of the blood-forming cells that replaces the normal bone marrow with a malignant clone (genetically identical cell) of lymphocytic or myelogenous cells. The disease may be acute or chronic based on its natural course; acute leukemias have a rapid clinical course, resulting in death in a few months without treatment, whereas chronic leukemias have a more prolonged course. The four major types of leukemia are acute or chronic lymphocytic and acute or chronic myeloid leukemia (Table 14-2).
When leukemia is classified according to its morphology (i.e., the predominant cell type and level of maturity), the following descriptors are used: lympho-, for leukemias involving the lymphoid or lymphatic system; myelo-, for leukemias of myeloid or bone marrow origin involving hematopoietic stem cells (see Fig. 21-6);-blastic, for leukemia involving large, immature (functionless) cells; and-cytic, for leukemia involving mature, smaller cells. If classified immunologically, T-cell/natural killer cell and B-cell leukemias are described.
Acute leukemia is an accumulation of neoplastic, immature lymphoid or myeloid cells in the bone marrow and peripheral blood. It is defined as more than 30% blasts in the bone marrow (the WHO classification accepts 20% as the definition of leukemia).
Chronic leukemia is a neoplastic accumulation of mature lymphoid or myeloid elements of the blood that usually progresses more slowly than an acute leukemic process and permits the production of greater numbers of more mature, functional cells. With rapid proliferation of leukemic cells, the bone marrow becomes overcrowded with abnormal cells, which then spill over into the peripheral circulation. Crowding of the bone marrow by leukemic cells inhibits normal blood cell production.
The three main symptoms that occur as a consequence of this infiltration and replacement process are (1) anemia and reduced tissue oxygenation from decreased erythrocytes, (2) infection from neutropenia as leukemic cells are functionally unable to defend the body against pathogens, and (3) bleeding tendencies from decreased platelet production (thrombocytopenia) (Fig. 14-4).
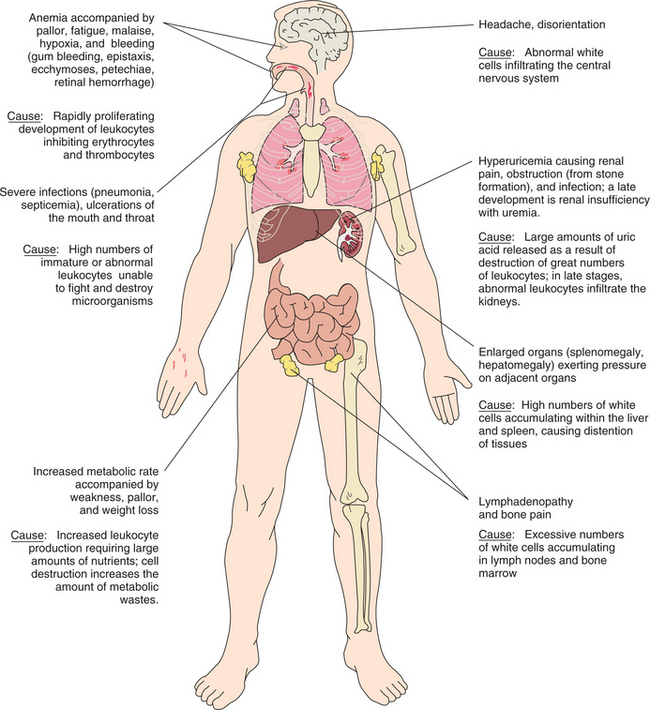
Figure 14-4 Pathologic basis for the clinical manifestations of leukemia. (Modified from Black JM, Matassarin-Jacobs E, editors: Luckmann and Sorensen’s medical-surgical nursing, ed 4, Philadelphia, 1993, W.B. Saunders.)
Leukemia is not limited to the bone marrow and peripheral blood. Abnormalities in the CNS or other organ systems can result from the infiltration and replacement of any tissue of the body with nonfunctional leukemic cells or metabolic complications related to leukemia.
Leukemia is a complex disease that requires careful identification of the subtype for appropriate treatment. Molecular probes can be used to establish a morphologic diagnosis of the type of leukemia, which then determines treatment and prognosis. These analyses are sufficiently sensitive to detect one leukemic cell among 100,000 or even in 1 million normal cells. Because of this extreme sensitivity, molecular markers have generally been used to determine the presence or absence of a few leukemic cells remaining after intensive therapy, so-called residual disease.
Over the last 25 years, death rates for leukemia have been falling significantly (57% decline) for children and more modestly for adults under 65 years. These declines in mortality reflect the advances made in the biologic and pathologic understanding of leukemia, technologic advances in medical care, and subsequent treatment that is more specifically targeted at the molecular level.
The aim of treatment is to bring about complete remission, or no evidence of the disease, with return to normal blood and marrow cells without relapse. For leukemia, a complete remission that lasts 5 years after treatment often indicates a cure. Future clinical and laboratory investigation will likely lead to the development of new, even more effective treatments specifically for different subsets of leukemia. The development of new chemotherapeutic and biologic agents combined with refined dose and schedule and stem cell transplantation has already contributed to the clinical success of treatment.
Acute Leukemia
Acute leukemia is a rapidly progressive malignant disease that results in the accumulation of immature, functionless cells called blast cells in the bone marrow and blood that block the development of normal cell development.
The two major forms of acute leukemia are acute lymphoblastic leukemia (ALL) and acute myelogenous leukemia (AML). Lymphocytic leukemia involves the lymphocytes and lymphoid organs, and myelogenous leukemia involves hematopoietic stem cells that differentiate into myeloid cells (monocytes, granulocytes, erythrocytes, and platelets).
Acute Myelogenous Leukemia
Incidence and Risk Factors.: AML is the most common leukemia in adults, constituting 80% of adult acute leukemias, while only 20% of AML patients are children. AML remains a rare disease, with about 12,000 cases per year.15
The incidence of AML increases with each decade of life, with the median age at onset of 63 years.114 People over the age of 70 years have a twelvefold increase of developing AML. Most cases of AML develop for unknown reasons, while some cases occur following treatment for another cancer (chemotherapy or radiation induced) or from a preexisting myelodysplastic syndrome.
Two types of chemotherapy treatment–related AML are described. The first typically occurs 5 to 7 years following exposure to an alkylating agent and is often heralded by a dysplastic phase. The other appears shortly after exposure to a topoisomerase II inhibitor (1 to 3 years) and lacks a dysplastic phase. Abnormalities in chromosome 7 are often seen in treatment-related AML and carry a worse prognosis than those cases that are idiopathic.163
Other risk factors for AML include previous radiation exposure and chemical/occupational exposure (e.g., benzene, herbicides, pesticides, cigarette smoking). Persons with uncommon genetic disorders, such as Down syndrome or Fanconi syndrome, also have a higher incidence of developing acute leukemia than the general population.
Pathogenesis.: AML is a heterogeneous group (not all the same) of neoplastic myeloid cells. Myeloid stem cells have the capability of differentiating into granulocytes, monocytes, erythrocytes, and platelets; neoplastic changes can occur along any line, resulting in many subtypes of AML.
Current techniques allow for cytogenetic analyses that can reveal specific chromosomal abnormalities where portions of a chromosome translocate (move) and fuse with another gene, creating a fusion gene. It is these abnormalities that lead to the development of leukemia, either allowing the cell to divide without regulation or failing to undergo programmed cell death (apoptosis).
Clinical Manifestations.: Initial clinical indications of AML are related to pancytopenia (reduction in all cell lines), reflecting leukemic cell replacement of bone marrow. Clients often have infections due to a lack of neutrophils or bleeding secondary to platelet deficiency (thrombocytopenia).
Spontaneous bleeding or bleeding with minor trauma often occurs in the skin and mucosal surfaces, manifested as gingival bleeding, epistaxis, mid-cycle menstrual bleeding, or heavy bleeding associated with menstruation (see Fig. 14-4). Petechiae (small, purplish spots caused by intradermal bleeding) are common clinical manifestations of thrombocytopenia particularly noted on the extremities after prolonged standing or minor trauma.
Fatigue, loss of energy, and shortness of breath with physical exertion are common due to anemia. Leukemia cells may infiltrate the skin (known as leukemia cutis), seen most often in the acute monocytic or myelomonocytic subtypes of AML. Leukemia cutis may present as multiple purplish papules or as a diffuse rash (Fig. 14-5).
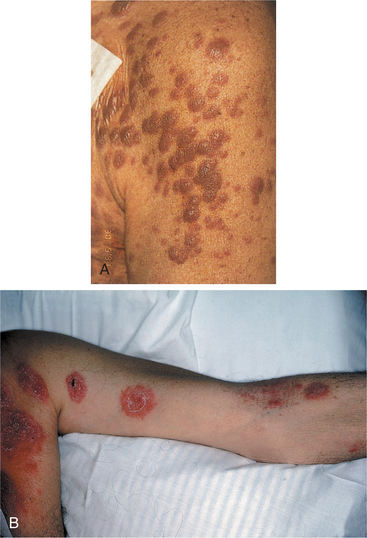
Figure 14-5 A, Leukemia cutis in an individual with monoblastic leukemia. B, Another example of leukemia cutis in the form of erythematous nodular tumors. (A, Reprinted from Hoffman R: Hematology: basic principles and practice, ed 4, Philadelphia, 2005, Churchill Livingstone. B, Reprinted from Noble J: Textbook of primary care medicine, ed 3, St Louis, 2001, Mosby.)
Modest splenomegaly is seen in 50% of clients with AML, whereas lymphadenopathy is uncommon. Discomfort in the bones, especially of the sternum, ribs, and tibia, caused by expanded leukemic marrow may occur. In the older adult, the disease can present insidiously with progressive weakness, pallor, a change in sense of well-being, and delirium.
CNS involvement is uncommon, occurring in only 1% to 2% of adults presenting with AML.30 These people present with symptoms similar to meningitis (e.g., headache, stiff neck, and fever). Some clients may develop cerebral bleeding or meningitis due to pancytopenia. In a small number of cases, AML may be more subtle, presenting at first with progressive fatigue and normal blood counts.
MEDICAL MANAGEMENT
Initial blood tests often reveal an elevated leukocyte count with an excessive amount of immature cells although low counts may be seen, especially in the elderly. Uncommonly initial counts may be normal. Diagnosis usually requires a bone marrow biopsy and aspiration in order to view bone marrow architecture and perform further tests.
Since most AML does not involve the CNS, lumbar punctures are not needed unless clinically indicated. Diagnosis involves several laboratory tests. First, immunophenotyping of blasts from peripheral blood or bone marrow is done in order to determine lineage of the cells (e.g., myeloid vs. lymphoid).
Treatment is determined by lineage, and AML treatment differs from ALL treatment. AML can be subdivided into various subtypes or classifications, and each subtype may receive varying initial therapy. The FAB system (French-American-British classification, first published in 1976) describes eight subdivisions classifying AML according to cell morphology and according to how the cells react to various stains.
The WHO has recently provided a revised classification of AML, retaining many aspects of the FAB system but adding cytogenetic information (chromosomal abnormalities) with associated prognosis (Table 14-3).90 Although the cytogenetic abnormalities do not determine initial therapy, they do guide postremission therapy and provide prognostic information.15
Table 14-3
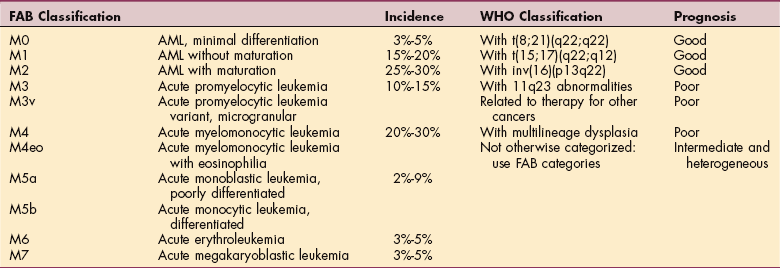
The FAB classification system, first published in 1976, describes eight subdivisions classifying AML according to cell morphology and how the cells react to various stains.
Modified from Jaffe ES, Harris NL, Stein H, editors: Pathology and genetics of tumours of haematopoietic and lymphoid tissues. Vol 3 of World Health Organization classification of tumours, Lyon, France, 2001, IARC Press.
The FAB system is clinically useful and widely accepted. The WHO classification is still undergoing changes as research provides more insight into the cytogenetic abnormalities of AML. Some recent abnormalities that have been identified include mutations of the FLT3 gene (found in about 30% of AML cases and associated with a poor prognosis) and NPM mutations (identified in 60% of AML cases with normal cytogenetics).
TREATMENT.
The diagnosis of acute leukemia is a medical emergency, especially if the WBC count is high (greater than 100,000/ml), placing the person at risk for cerebral hemorrhage caused by leukostasis (obstruction of and damage to blood vessels plugged with rigid, large blasts).
Treatment decisions are based on the subtype of AML. Most induction (initial) treatment protocols utilize aggressive combination chemotherapy in order to eradicate the neoplastic cells and restore normal hematopoiesis. Supportive care, including fluids, blood product replacement, and prompt treatment of infection with broad-spectrum antibiotics, is frequently needed during the 3-to 4-week hospitalization required for bone marrow recovery. Significant complications occur during this period, with a death rate of 5% to 10%.15
Induction chemotherapy is followed by consolidation chemotherapy, which is intended to maintain a complete remission. Consolidation treatment is administered in a cyclic fashion over 2 to 4 months.
The discovery of mutations and translocations that may be causing leukemia has led to the development of targeted therapeutic agents. One example is tretinoin. It is used against acute promyelocytic leukemia and targets the t(15,17) translocation. Experimental drugs are in clinical trials for persons with FLT3 mutations, and other agents will be designed in hopes of targeting the genetic abnormalities without affecting normal cells.
PROGNOSIS.
If left untreated, all leukemias are fatal. With induction treatment, approximately 80% of clients younger than 60 years achieve remission. The rate of remission decreases as age increases over 60 years.
According to the WHO classification, persons with AML with favorable cytogenetics (good prognosis) have a 60% 5-year survival rate, whereas those with a poor prognosis have only a 10% 5-year survival rate. Clients with an intermediate prognosis (the majority of AML cases) have a 40% 5-year survival rate.
Improvement in outcomes for people with a poor prognosis is a major goal of clinical research. Because this group rarely remains in remission after consolidation therapy, current protocols include allogeneic, unrelated-donor or cord-blood transplantation following induction (e.g., transplant done in first remission). Consolidation therapy is offered to those people with good prognostic cytogenetics. Autologous transplantation and consolidation therapy are the options for clients with normal cytogenetics but an intermediate risk for recurrence.
Acute Lymphoblastic Leukemia
Incidence and Risk Factors.: In contrast to AML, ALL is diagnosed more frequently in children. Of the 4000 cases of ALL identified each year, two thirds occur in children while only one third are diagnosed in adults.156 Yet of the 1500 annual deaths, almost two thirds occur in adults.
Like AML, most ALL cases develop for unknown reasons. A few risk factors have been identified including significant exposure to radiation and infection with HTLV-1 (human T-cell lymphoma/leukemia virus). Persons with genetic disorders such as Down syndrome also have an increased risk.
Questions have been raised as to whether electromagnetic fields, such as those generated in high-voltage power lines, increase the risk of developing ALL. Currently large studies are ongoing with initial data suggestive of no increased risk or a very slight increased risk. Answers should be available at the conclusion of these studies. Burkitt’s lymphoma can form a type of ALL that responds poorly to treatment.
Pathogenesis.: Abnormal cytogenetics or translocations and mutations are frequently seen in ALL cases. These genetic changes lead to abnormal cell growth and division or inability of the cells to decrease their growth and die (scheduled cell death or apoptosis). These various abnormalities are associated with poor or good prognosis. For instance, leukemic cells with more than 50 chromosomes (hyperdiploidy) and the translocation t(12;21) have a good prognosis. More than 50% of children with ALL have this defect, while only 10% of adults have it. Leukemic cells with less than 45 chromosomes are difficult to treat and carry a poor prognosis. Translocations t(4;11) (50% of infant cases) and t(9;22) (50% of adults over the age of 50 years) also have a poor prognosis).156
Clinical Manifestations.: ALL exhibits clinical signs and symptoms resulting from an abnormal bone marrow that is unable to engage in normal hematopoiesis. Fever and frequent infections indicate a lack of normal neutrophils, while easy bleeding and bruising are indicative of thrombocytopenia. Clients are often tired as a result of anemia.
ALL is more likely than AML to have leukemic cells spread to extramedullary sites. The CNS is frequently involved, causing headache, weakness, seizures, vomiting, difficulty with balance, and blurred vision.9 Testicles in males and ovaries in females commonly harbor leukemic cells and are difficult to reach with chemotherapy agents.
Bone and joint pain from leukemic infiltration or hemorrhage into a joint may be the initial symptoms (more common finding in children than adults). Involvement of the synovium may lead to symptoms suggestive of a rheumatic disease, especially in children.
Hepatosplenomegaly and lymphadenopathy (particularly in the mediastinum) are frequently encountered along with an enlarged thymus. If thymic swelling is significant, the client may exhibit difficulty breathing or upper extremity swelling from increased pressure on the bronchus or superior vena cava (superior vena cava syndrome), which requires immediate attention.
MEDICAL MANAGEMENT
The diagnosis of ALL is similar to that of AML (see diagnosis of AML above). The peripheral blood smear is examined, and additional special tests are performed using peripheral blood and bone marrow. A bone marrow biopsy and aspirate are required for many of these tests.
Bone marrow is used to perform flow cytometry/immunophenotyping to determine if the leukemic cells are myeloid or lymphoid in origin (rarely there are cells that express features of both). Cytogenetic studies are performed to aid in prognosis and assist in determining subsequent treatment. This is accomplished by culturing bone marrow cells or performing fluorescent in situ hybridization.
Since ALL commonly involves the CNS, a lumbar puncture is done to collect cerebral spinal fluid for analysis. As with AML, ALL is a heterogenous group of lymphoid leukemias with varying prognoses. The FAB classification of ALL includes the three subtypes L1, L2, and L3, while an expanded classification that relies on flow cytometry and cytogenetics establishes six subtypes: (1) early pre-B-cell ALL, (2) common ALL, (3) pre-B-cell ALL, (4) mature B-cell ALL (Burkitt’s lymphoma), (5) pre-T-cell ALL, and (6) mature T-cell ALL.
TREATMENT.
ALL treatment protocols vary depending on age, subtype of ALL, and genetic abnormalities associated with risk of recurrence. For example, mature B-cell ALL has a good prognosis and is treated with short-term intensive chemotherapy. Other subtypes require remission-induction therapy (initial intensive combination chemotherapy designed to induce a complete remission).
Clients with CNS involvement receive intrathecal chemotherapy periodically throughout their treatment, and those at high risk for CNS relapse may receive cranial radiation. Intensification therapy (consolidation) is administered following successful remission-induction treatment. This consists of a reinduction treatment (a readministration of induction agents) followed by cyclic use of other chemotherapy agents over a period of 2 years or longer, termed continuation therapy.
Selection of these agents is based on risk of recurrence, age, and subtype. Drugs, dosages, and scheduling can be individualized. For ALL subtypes at high risk, allogeneic BMT is offered. Adults frequently require transplantation and can achieve long-term survival rates of 45% to 75%.87
Ninety-eight percent of children and 85% of adults achieve a complete remission following remissioninduction therapy. With such favorable results, efforts are being made to reduce toxicities associated with aggressive combination chemotherapy, especially in children with standard risk of ALL with favorable cytogenetics.
A recent analysis has shown that children who survive cancer go on to develop significant long-term medical problems including other cancers, heart disease, cognitive dysfunction, joint replacement, and hearing/vision loss.137 Some of the strategies being implemented in protocols include a dose reduction of vincristine in infants, the omission of cranial irradiation in girls and young children (attempts are being made to completely omit irradiation for all people with ALL), and a dose reduction of methotrexate in clients with Down syndrome.
Another strategy is to provide targeted agents. Imatinib mesylate (Gleevec) is available for the treatment of cancers with the t(9;22) translocation. The use of targeted drugs reduces toxicity and alleviates the requirement of adjuvant agents.
PROGNOSIS.
Children with ALL do better than adults (the older the client, typically the worse the prognosis). The cure rate of children is approximately 80%, while adults average a 40% cure rate. Continuing research into the cytogenetics has provided significant prognostic information regarding various abnormalities.
T-cell ALL carries the best prognosis, while mature B-cell ALL is associated with the worst. ALL classified as L3 has a poor prognosis as well as those leukemic cells with the Philadelphia chromosome [t(9;22)] or t(4;11) translocation. Pre-B-cell ALL has an intermediate prognosis, while clients requiring 4 to 5 weeks to achieve complete remission with induction chemotherapy have a poor prognosis.
Chronic Leukemia
Chronic leukemia is a malignant disease of the bone marrow and blood that progresses slowly and permits numbers of more mature, functional cells to be made. Chronic leukemia has two major groups: chronic myeloid leukemia (CML) and chronic lymphocytic leukemia (CLL), each with several subtypes.
These are entirely different diseases and are presented separately. CML is also known as chronic myelogenous leukemia or chronic myelocytic leukemia. Other less-common forms include prolymphocytic leukemia (terminal transformation of CLL) and hairy cell leukemia (accounting for only 1% to 2% of adult leukemias).
Chronic Myeloid Leukemia
Incidence and Etiologic Factors.: CML is a neoplasm of the hematopoietic stem cell. The genetic abnormalities created in the stem cell are the result of acquired injury to the DNA and passed on to all related cell lines, resulting in increases in myeloid cells and clonal anomalies in erythroid cells and platelets. This leads to abnormal cells in peripheral blood and marked hyperplasia in the bone marrow.
CML accounts for 15% of all leukemias, with approximately 4600 cases diagnosed per year. This type of leukemia occurs mostly in adults, with only 2% developing in children. Although the exact etiologic factors are unknown, the incidence of CML is increased in people with severe radiation exposure. No chemical or other environmental risk factors are known to cause CML.
CML was the first form of leukemia with a known genetic predisposition (Philadelphia chromosome [Ph]; named after the city in which researchers first observed the chromosome). The specific genetic anomaly is a translocation that fuses the long arm of chromosome 22 to chromosome 9. Detached pieces of chromosome 9 adhere to the broken end of chromosome 22 and vice versa. This translocation occurs only in the stem cell and in the various blood cells derived from that stem cell. The chromosomes of the cells in other tissues are normal.
Pathogenesis.: CML originates in the hematopoietic stem cell (i.e., this cell has the ability to develop into any one of several blood cells; see Fig. 21-6) and involves overproduction of myeloid cells. The genetic defect detected in CML cells is called the Philadelphia chromosome. This translocation [t(9,22)] was the first consistent chromosomal anomaly identified in a cancer and is now detected in all cases of CML and known to be the cause of CML.
The abnormal chromosome develops from the accidental translocation and fusion of the BRC gene on chromosome 22 and the ABL gene on chromosome 9, creating a unique gene (BRC-ABL). The BRC-ABL gene encodes for an abnormal protein product that acts as a tyrosine kinase, resulting in a dysregulated proliferation signal.
Tyrosine kinase is an enzyme that is necessary for normal cell growth. In normal cells the enzyme turns on and off as it should, but in people with CML this enzyme appears to be in the permanent “switched on” state, eliminating the normal checks and balances on proliferation.
Unlike AML, CML permits the development of mature WBCs that generally can function normally. This important distinction from acute leukemia accounts for the less-severe early course of the disease.159
Clinical Manifestations.: Presenting signs and symptoms are often quite nonspecific. The most typical symptoms at presentation are fatigue, anorexia, and weight loss, although approximately 40% of affected individuals are asymptomatic. Sweats, malaise, and shortness of breath during physical activity are also reported.
Splenomegaly is present in 50% of all cases, with thrombocytosis being very common. Discomfort on the left side of the abdomen from the enlarged spleen is not uncommon.
The natural history of CML is a progression (over years) from a proliferative phase (chronic phase), into an accelerated phase (more symptoms but not acute leukemia), to an aggressive acute leukemia (blast crisis) that can be rapidly fatal within months.
MEDICAL MANAGEMENT
Early in the disease process, only vague symptoms or routine blood analysis may herald the disease. Peripheral blood counts and smears often demonstrate an abnormal leukocytosis, which leads to further testing. The peripheral blood smear will often reveal a wide range of cells in a variety of stages. Basophilia may also be seen. The appearance of both mature and immature cells in the peripheral blood is suggestive of CML but must be differentiated from other myeloproliferative disease.
As with other leukemias, laboratory findings, including blood and bone marrow testing, assist in establishing the diagnosis. The genetic abnormality in blood cells (Ph chromosome) is almost invariably present and may be detected in either the peripheral blood or bone marrow using fluorescent in situ hybridization, polymerase chain reaction (PCR) analysis, or karyotyping.
TREATMENT AND PROGNOSIS.
Recent research has significantly changed the treatment and prognosis of CML. In fact, because of the prolonged survival seen with current treatments, the long-term survival rate is unknown. Knowledge that the cause of the disease was an abnormal BCR-ABL gene led to the development of the drug imatinib mesylate (Gleevec), designed to target and inhibit tyrosine kinase (it blocks adenosine triphosphate from interacting with the abnormal protein product).
In studies comparing the previously used regimen of cytarabine and interferon, imatinib mesylate produced a 95% complete hematologic remission (blood and bone marrow look normal) compared to only 56% for the combination therapy. Imatinib mesylate is also better tolerated, with fewer adverse effects. After 18 months of treatment, 87% of clients were able to achieve a complete cytogenetic remission (cells lack the BRC-ABL gene) compared to only 35% on the combination therapy.135
Clients with evidence of the translocation in cells are at risk for relapse. Those without evidence of the abnormal clone had long-term CML-free survival. Investigations are still ongoing to determine the optimal length of treatment and long-term side effects. Some people will develop a relapse of CML that is resistant to imatinib mesylate. New drugs (e.g., nilotinib, dasatinib) have been and are being developed that can be used simultaneously with imatinib or in clinical situations where the leukemia is resistant to imatinib.195
For those clients with CML that is not responsive to these new treatments (or for individuals who are unable to tolerate imatinib), combination therapy can control the symptoms for a time. Allogeneic BMT continues to be an option that may provide a cure. This should be performed during the proliferative phase (chronic phase) within the first year of diagnosis. For those people who develop blast crisis, treatment is similar to acute leukemia treatment. If a remission is induced, these clients become candidates for BMT.
Chronic Lymphocytic Leukemia
Incidence and Etiologic and Risk Factors.: CLL is a common type of adult leukemia, accounting for 25% to 30% of all leukemias with almost 10,000 cases per year.91 The incidence of CLL increases with advancing age; 90% are older than 50 years, and men are affected more often than women. CLL may occur in as many as 3% of people over the age of 70 years.
The cause of CLL is also unknown but a few environment factors have been implicated, such as farming pesticides and the chemical warfare herbicide Agent Orange, although conclusive evidence is lacking. Some groups of people may have a genetic predisposition, including persons with a first-degree family member with CLL.204
Pathogenesis.: The cell type responsible for more than 95% of the cases of CLL is the B cell (T-cell CLL is uncommon). When a normal B cell is stimulated by an antigen, it enters a proliferative phase, creating clones able to fight infection. It is during this stage that a cell may develop a mutation and predispose it to become cancerous. A mutation allowing the cell to continue to proliferate places that cell at an advantage but the human host at a disadvantage. With continuous exposure to this antigen and the aid of stromal cells, cytokines, and chemokines, cells would be stimulated to proliferate and avoid apoptosis (programmed cell death), potentially leading to cancer.
CLL cells may develop from this type of cell since most initial CLL cells lack major cytogenetic abnormalities (translocations are uncommon). As the mutated B cell continues to divide, more mutations develop, creating a heterogenous group of abnormal cells. Different mutations appear to affect the course of the disease.
Mutations in the immunoglobulin-forming genes can proffer a prolonged, milder course of disease or a more aggressive, symptomatic illness. For example, clients with no or few V gene mutations or multiple CD38+ or ZAP-70+ mutations followed an aggressive, often fatal course, while cancerous clones with few CD38+ or ZAP-70+ mutations resulted in a more indolent course.34 ZAP-70+ is a protein normally found in T lymphocytes and is responsible for transferring signals inside the cell.
There are few unifying mutations except the deletion of 13q14.3, which is present in more than half of all people with CLL.123 The frequency of this mutation suggests that the genes in this region offer the clonal cells an advantage over other cells. Other mutations are noted to be in genes that control apoptosis or confer chemotherapy resistance, resulting in a poor prognosis.
Clinical Manifestations.: In the early stages of the disease, most clients remain asymptomatic or complain of vague, nonspecific symptoms such as fatigue or enlarged lymph nodes. Depending on the mutations present in the abnormal clone, clients may experience a prolonged, indolent course with few symptoms.
Those people with more aggressive CLL develop pancytopenia (with accompanying symptoms of infections, hemorrhage, and significant fatigue) and decreased immunoglobulin levels (also resulting in infections). With progression of the disease, clients may develop lymphadenopathy, splenomegaly, hepatomegaly, weight loss, bone pain, and bone marrow infiltration. Approximately 10% to 25% of clients develop the complication of AIHA,144 and immune thrombocytopenia occurs in 2% of cases.24
MEDICAL MANAGEMENT
Examination of the peripheral blood smear, CBC, and flow cytometry are performed to make a diagnosis. Bone marrow examination is not required but may be helpful. Staging is established according to the Rai and Binet systems.
The Rai system recognizes five stages (0 to IV) conferring a low (stage 0), intermediate (I, II), and high (III, IV) risk of progression. This staging determines which clients should receive treatment and when, but this staging system does not provide reliable prognostic information.
TREATMENT.
CLL has been difficult to treat and typically is without a cure. Better understanding of the pathogenesis of CLL is leading to improved treatments, but most initial treatment is based on clinical staging.
Low-risk clients require careful monitoring and frequent examinations with treatment initiated if symptoms worsen or if there is evidence of progressive leukemia. Intermediate-and high-risk clients are treated with chemotherapy (fludarabine and chlorambucil) accompanied by monoclonal antibodies. Alemtuzumab is the only Food and Drug Administration approved monoclonal antibody for the treatment of CLL, although rituximab is frequently used because of its better side-effect profile.
Immunomodulating agents (agents that change signals sent to the abnormal cells) are currently being studied and may prove to be beneficial as an adjuvant therapy.32 The use of autologous transplant for the treatment of CLL has increased recently but remains a successful option in a limited number of cases.
PROGNOSIS.
CLL continues to be a fatal disease with a significant impact on life expectancy, but in the last few years a trend toward an improvement in overall survival has taken place.
Prognostic information can be provided by analysis of mutations, which in the future may also better determine when therapy should be initiated (if an indolent course is most likely, therapy can be withheld until needed; an aggressive CLL may require therapy sooner). Since cytogenetic testing is not widely available, identification of the proteins ZAP-70+ or CD38+ may prove reliable surrogate markers for determining prognosis.34
Clients who present in Rai stage III to IV have an average life expectancy of 3 years, while those with less-aggressive leukemia may never require treatment (depending on the age at diagnosis). About 70% of people with CLL eventually require treatment.95
As mutations are discovered, they are being related to prognosis. As previously discussed, CLL, which displays ZAP-70+ or CD38+ mutations, has a poor prognosis, while mutations in the immunoglobulin heavy-chain variable region have a good prognosis (median survival of 25 years). Overall, CLL has a median life expectancy of 10 to 14 years.
Malignant Lymphomas
Lymphoma is a general term for cancers that develop in the lymphatic system. Lymphomas are divided into two groups: Hodgkin’s lymphoma (HL; also known as Hodgkin’s disease) and non-Hodgkin’s lymphoma (NHL). With the extensive progression in cytogenetic research that has occurred over the past 5 years, this distinction is beginning to obscure.
It is becoming more useful to categorize lymphomas according to their clinical behavior—indolent or aggressive—and their chromosome features. Currently HL is distinguished from other lymphomas by the presence of a characteristic type of cell known as the Reed-Sternberg cell. All other types of lymphoma are called NHL.
Hodgkin’s Lymphoma
Definition and Overview.: HL is lymphoid neoplasm with the primary histologic finding of giant ReedSternberg cells in the lymph nodes. These cells are part of the tissue macrophage system and have twin nuclei and nucleoli that give them the appearance of owl eyes (Fig. 14-6).
Figure 14-6 Reed-Sternberg cell. Named for Dorothy M. Reed, American pathologist (1874-1964) and Karl Sternberg, Austrian pathologist (1872-1935). This is one example of the large, abnormal, multinucleated reticuloendothelial cells in the lymphatic system found in HD. The number and proportion of Reed-Sternberg cells identified are the basis for the histopathologic classification of HD. (Reprinted from Kumar V, Cotran RS, Robbins SL: Basic pathology, ed 6, Philadelphia, 1997, WB Saunders. Courtesy Dr. Robert W. McKenna, University of Texas Southwestern Medical School, Dallas.)
Although this malignancy originates in the lymphoid system and primarily involves the lymph nodes, it can spread to other sites such as the spleen, liver, bone marrow, and lungs. There are two subtypes of HL: classic HL (further divided into the categories of nodular sclerosing HL, mixed-cellularity HL (MCHL), lymphocyte-rich classic HL, and lymphocyte-depleted HL) and nodular lymphocyte-predominant HL (LPHL). LPHL is uncommon and represents only 4% to 5% of HL cases.
Incidence and Risk Factors.: Classic HL can occur in both children and adults but peaks at two different ages: between the ages of 25 and 30 years and after the age of 55 years. Children younger than 5 years rarely develop this disease, while only 10% of HL cases occur in children 16 years old and younger. LPHL typically has only one peak incidence around the fourth decade. Approximately 7800 cases of HL are diagnosed in the United States each year (typically men more than women).91
Although the exact cause of HL remains under investigation, certain risk factors have been identified (Table 14-4). One factor that has been related to HL is previous infection with EBV. The DNA of this virus has been found in the Reed-Sternberg cells of about 50% of clients with classic HL (about 90% in developing countries).
Table 14-4
Risk Factors for Malignant Lymphomas
| NHL | HL |
| Age (increased risk with increasing age) | Familial |
| Gender (males more than females) | |
| Environmental Contaminants | |
| Herbicides and pesticides (?) | |
| Benzene (?) | |
| Polychlorinated biphenyls (PCBs) (?) | |
| Radiation | |
| Viral Infection | |
| EBV, mononucleosis virus | EBV, mononucleosis virus |
| Human T-lymphotrophic virus type I (HTLV-1) | |
| HIV | |
| Congenital Immunodeficiency Syndromes | |
| Hepatitis C (?) | |
| Immunocompromise/immunodeficiency | |
| Chronic disease or illness; autoimmune diseases | Chronic disease or illness; autoimmune diseases |
| Immunosuppressants | Immunosuppressants |
| Cancer treatment with alkylating or cytotoxic agents | Cancer treatment with alkylating or cytotoxic agents |
| Inherited immune deficiencies (e.g., collagen vascular disease) | SLE |
| HIV | HIV |
| AIDS | AIDS |
| Helicobacter pylori bacteria (gastric lymphoma) | Ulcerative colitis |
| Methotrexate | Drug abuse |
| Obesity (women) | Obesity (men) |
Pathogenesis.: HL is a B-cell–type malignancy with clonal expansion of a malignant B cell. The reason for the transformation from a normal B cell to a malignant cell is still under investigation, but significant progress has been made. Recent evidence suggests that an infection or inflammation may be involved. As just discussed, genes from EBV are found in half of all HL cases in the industrialized world.
Products from these genes (LMP1, LMP2, and EBNA1) have the ability to mimic transmembrane receptors and activate the transcription factor NF-κB (which is normally inhibited). NF-κB, through its function of regulating dozens of genes within the cell, plays a key role in the proliferation and survival of the malignant clones. The products of these genes include cytokines and chemokines.
Many cytokines, particularly interleukins, are involved in producing an environment where the Reed-Sternberg cells thrive. For example, some interleukins attract inflammatory cells (eosinophils, monocytes, and mast cells), which aid in the survival of the cell. In the laboratory, if these inflammatory cells are not surrounding the Reed-Sternberg cells, they do not survive.
Other interleukins expressed by the Reed-Sternberg cells inhibit the activation of T cells, which normally destroy abnormal cells. This inhibition creates an area of local immunosuppression and allows the Reed-Sternberg cell to evade detection. Some interleukins act as growth factors, encouraging proliferation, metastasis, and angiogenesis. Another protein induced by NF-κB, termed c-FLIP, is incorporated into a complex that signals cell death but is not functional in that capacity, thereby evading apoptosis and making the cell immortal.99
Reed-Sternberg cells also express receptors for receiving signals from inflammatory cells, creating “cross-talking” between the malignant cells and surrounding inflammatory cells, which may contribute to the ability of the Reed-Sternberg cells to metastasize.
Clinical Manifestations.: See Fig. 14-7.
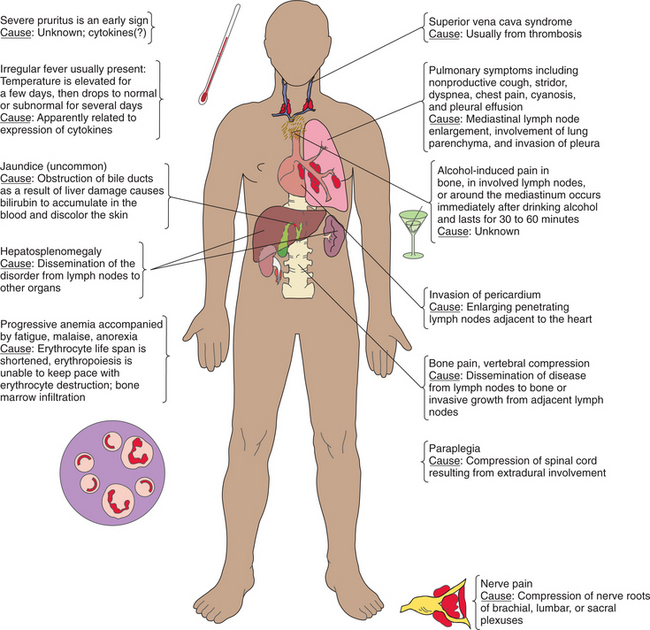
Classic HL and LPHL present with different clinical manifestations and progression of disease. Because of these distinctions, these two subgroups are discussed separately.
Classic Hodgkin’s Lymphoma.: Classic HL begins in a group of lymph nodes and spreads contiguously to other lymph node chains. The cervical, axillary, and para-aortic lymph nodes and mediastinum are the most common initial locations for involvement (Fig. 14-8). These lymph nodes are typically nontender and firm.51

Figure 14-8 Enlarged cervical lymph node associated with HD. (Reprinted from del Regato J, Spjut HJ, Cox JD: Cancer: diagnosis, treatment, and prognosis, ed 6, St Louis, 1985, Mosby–Year Book.)
Nodular sclerosing HL typically presents with supradiaphragmatic lymph node involvement, while MCHL often exhibits smaller involved lymph nodes in a subdiaphragmatic location or involves organs. Clients who have disease below the diaphragm, MCHL, or “B” symptoms are more likely to develop splenic involvement. Splenic involvement is seen in 30% to 40% of people with HL, but detection is often difficult. An enlarged spleen does not necessary indicate involvement, while a normal size spleen does not rule out involvement. HL in the liver is rare and seen only with splenic involvement.51
Bone marrow involvement occurs in less than 10% of newly diagnosed cases. If lymph nodes become large and bulky, they can lead to further symptoms, such as tracheal or bronchial compression (with accompanying shortness of breath) or obstruction of the GI tract. Lymph nodes may grow and enlarge and finally perforate the lymph node capsule, continuing to grow and invade into adjacent tissue or organs. This can occur in the lung, pericardium, pleura, chest wall, gut, or bone.51
Effusions (collections of fluid) may also develop in the lung, heart, or abdominal cavity. Tumor can spread not only from lymph node to adjacent lymph node, but via the bloodstream to lung, liver, bone marrow, and bone. Involvement in these areas is often indicative of extensive disease. As bone marrow is replaced, infections, anemia, and thrombocytopenia result.51
Primary involvement of the CNS is rare, and dissemination of disease to the CNS is uncommon.21 Occasionally spinal cord involvement may occur in the dorsal and lumbar regions, and compression of nerve roots of the brachial, lumbar, or sacral plexus can cause nerve root pain. Epidural involvement (also uncommon) causes back and neck pain with hyperreflexia. Extremity involvement is characterized by pain, nerve irritation, and obliteration of the pulse.
A significant number of people (25%) present with “B” symptoms. The fever associated with HL is intermittent and occurs with drenching night sweats. Clients may also complain of fatigue, pruritus, and pain associated with drinking alcohol.
Lymphocyte-Predominant Hodgkin’s Lymphoma.: LPHL typically presents with one-node involvement rather than groups of involved lymph nodes. This occurs in peripheral lymph nodes such as the cervical, axillary, or inguinal lymph node chains. Unlike classic HL, LPHL does not follow an orderly pattern of spread, but can be found in lymph nodes distant from the original node of disease. LPHL infrequently involves the bone marrow, spleen, or thymus. “B” symptoms rarely occur. LPHL has an indolent clinical course with long disease-free intervals. Relapse is common but responds well to treatment.148
Pregnancy and Hodgkin’s Lymphoma.: Since the mean age at diagnosis of HL is 32 years, it is not uncommon for women to develop HL while pregnant. Diagnostic staging can be accomplished safely with magnetic resonance imaging (MRI) because it does not use ionizing radiation133; it has no adverse impact on the natural course of HL; and HL has no effect on the course of gestation, delivery, or the incidence of prematurity or spontaneous abortions. The risk of metastatic involvement of the fetus by HL is negligible.62
The management of HL during pregnancy must be individualized. Many women have been successfully treated while pregnant without adverse effects on the fetus.63 In cases of disease onset early in pregnancy, the recommendation may be made to consider a therapeutic abortion. Women presenting in later pregnancy are often able to have therapy delayed until after delivery or can undergo modified or standard combination chemotherapy and radiation therapy.62
Antiretroviral treatment and prophylaxis for opportunistic infection may also be administered for HIVpositive women.102 With the increased use of ABVD therapy (Adriamycin [doxorubicin], bleomycin, vinblastine, and dacarbazine) and reduced reliance on radiation therapy, most women do not have to receive radiation. However, with appropriate shielding, the estimated fetal dose of radiation can be reduced by 50% or more in most cases if required.73 In nonpregnant women, to further reduce any risk, it is advisable to delay pregnancy for 12 months after completion of radiation therapy.63
Long-term semen banking is available for men whose future fertility may be compromised by suppression of spermatogenesis secondary to administration of chemotherapy or radiotherapy treatment. Banking of a single ejaculate before chemotherapy or radiotherapy treatment may preserve potential fertility without compromising the oncology treatment.85
Hodgkin’s Lymphoma in AIDS.: Although HL does not occur as frequently in HIV-positive clients as does NHL, people with HIV are still at increased risk of developing HL (about eight-to tenfold increase). When it occurs, the histology is usually MCHL associated with aggressive, disseminated disease and systemic symptoms. Since the introduction of highly active antiretroviral therapy (HAART), the incidence of HL in clients with HIV has not changed significantly, but there has been an improvement in mortality rate, particularly due to the HAART therapy and reduced AIDS-related deaths.67
MEDICAL MANAGEMENT
Diagnosis depends on identification of Reed-Sternberg cells in lymph node tissue or at other sites that exhibit the characteristic markers of CD15+ and CD30+. This can often be achieved by excisional biopsy (needle biopsy usually does not provide enough cells for diagnosis since only 2% to 3% of the cells in a lymph node are malignant) of involved tissue (usually an accessible lymph node). Biopsied tissue is examined under the microscope for Reed-Sternberg cells and tested for markers using immunohistochemistry.
Once the diagnosis is made, staging of the disease is accomplished through a complete physical examination; blood tests (sedimentation rate); and computed tomographic (CT) scan of the chest, abdomen, and pelvis (Fig. 14-9). Often a bone marrow biopsy or aspirate is needed to determine the extent of the disease. Because systemic chemotherapy is utilized, extensive staging is no longer required, including exploratory laparotomy or splenectomy (which can be more dangerous than helpful).
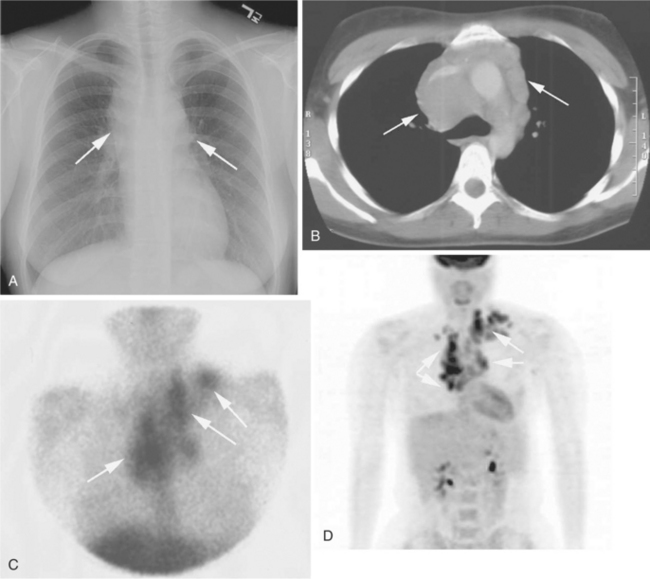
Figure 14-9 HD as seen on chest radiograph (A); CT scan of the chest (B); gallium scan of the head, neck, and chest (C); and PET scan (D). The arrows indicate sites of diseases. Note that PET and CT scans provide more detailed information compared with chest radiograph and gallium scan. (Reprinted from Goldman L: Cecil textbook of medicine, ed 22, Philadelphia, 2004, WB Saunders.)
The stage of the neoplasm depends on the number of nodes involved, the location of the nodes, the presence of “B” symptoms (fever, weight loss, night sweats), the sedimentation rate, and size of nodes. Staging is by the Ann Arbor system or the modified version called the Cotswolds classification, denoting stages I to IV (see Table 14-6), which helps determine treatment.
Table 14-6
Pettersson Classification of Hemophilic Arthropathy
This classification is a joint scoring system based on radiographic findings used to classify and monitor joint changes and damage.
*Possible total joint score is 0-13 points.
Reprinted from Anderson A, Holtzman TS, Masley J: Physical therapy in bleeding disorders, New York, 2000, National Hemophilia Foundation.
Three prognostic groups have also been proposed: early favorable, early unfavorable, and advanced disease. Advanced disease is further divided according to the presence of seven risk factors: advanced disease plus zero to three risk factors is staged as advanced disease with favorable prognosis, and advanced disease with greater than three risk factors is termed advanced disease with unfavorable prognosis.77
Some factors denoting a worse prognosis include a low serum albumin, low hemoglobin, age over 45 years, and male gender (despite stage). Other factors that carry a poorer prognosis include “B” symptoms, stage IV disease, too high or too low lymphocyte count, or high WBC count. These factors are often considered in subsequent treatment options.
TREATMENT.
Cure of HL is the primary treatment goal through the use of chemotherapy and, in some cases, both chemotherapy and radiation. The specific treatment is guided by the stage of the disease at diagnosis. The combination chemotherapy of choice is ABVD. Seventy-five percent of clients are cured with this treatment (with or without radiation).
Persons classified as having early favorable disease (stage IA or IIA) receive either four courses of ABVD or two courses of ABVD plus involved-field radiation treatment. Early unfavorable disease (stage IA, IB, IIA, IIB, a large mass in the chest, or other unfavorable characteristics) is typically treated with four courses of ABVD plus involved-field radiation treatment.
Advanced disease (stages III and IV) with favorable prognosis treatment includes six courses of ABVD. Advanced disease with unfavorable prognosis can be treated with aggressive combination chemotherapy using BEACOPP (bleomycin, etoposide, Adriamycin [doxorubicin], cyclophosphamide, vincristine, procarbazine, prednisone) or Stanford V (with or without radiation). Other therapies are available for advanced disease; all have significant toxicities and varying effectiveness.50,69
As therapy progresses, some oncologists are utilizing positron emission tomography (PET) scans to determine the effect of treatment. For example, if a large mass in the chest of a child shrinks significantly with chemotherapy, radiation can be avoided. PET and gallium scans (a nuclear medicine scan) are also used to determine if there is any residual disease upon completion of therapy.
For people with relapsed HL after combination chemotherapy or for those clients with disease that did not completely respond to treatment (10% to 15%), current data support the use of high-dose chemotherapy with autologous stem cell or peripheral blood stem cell transplantation. Biologic treatments such as monoclonal antibody–based therapies (e.g., rituximab) are still being investigated,60 while new efforts are being made to develop drugs that could inhibit genes or protein products that contribute to the pathogenesis of HL.
Current treatments are being evaluated for long-term toxicity since the survival rate has improved. ABVD is associated with a long-term risk of less than 1% for developing acute leukemia, while radiation (mantle and para-aortic fields) has a 1% per year lifetime risk of developing a solid tumor in smokers and 0.5% in nonsmokers. The most common solid tumors noted include lung and breast cancers.47,113
Those who survive HL have a 13% risk of developing a solid tumor at 15 years that increases to 22% at 25 years of survival (this increase does not appear to plateau).54 Clients who received radiation to the chest also have a higher risk of fatal myocardial infarction (presumed to be caused by damage to the intimal lining of the coronary arteries).
ABVD has not been shown to cause infertility in men or women, as do the older alkylating agents previously used. Because of the increased long-term complications, the use of radiation has been limited to only areas in field and lower doses are being used, particularly in children. The combination therapy of ABVD appears to be a better long-term choice for treatment.54
PROGNOSIS.
HL is now considered one of the most curable forms of cancer, and death rates have decreased 60% since the early 1970s. Ninety-three percent of clients are alive at 1 year following treatment. The 5-year survival rate is about 85%, while the 10-year rate is 78%. At 15 years and beyond, the relative rate is 68%. The cause of death in the majority of cases prior to 15 years is recurrent disease. After 15 years, most die of other causes, including neoplasms derived from HL therapy.
Non-Hodgkin’s Lymphomas
Overview and Incidence.: NHL comprises a large group (about 30 specific types described) of lymphoid malignancies that present as solid tumors arising from cells of the lymphatic system. Over 67,000 people develop NHL per year, making it the fifth most common cancer in the United States.
The incidence rate has doubled since 1970, in part due to the increase in HIV-related disease, but the reasons for the remaining cases are unknown. Most of the increase has been noted in women, but NHL is still more common in men. Ninety-five percent of NHL occurs in adults and only 5% develop in children; yet the types of NHL in adults are different than those seen in children. The average age of onset in adults is between 60 and 70 years.
The lymph nodes are usually involved first, and any extranodal lymphoid tissue, particularly the spleen, thymus, and GI tract, may also be involved. The bone marrow is commonly infiltrated by lymphoma cells, but this is rarely the primary site of a lymphoma.
Lymphomas are classified according to the Revised European/American Lymphoma/WHO system, which relies on the histochemical, genetic, and cytologic features. Lymphomas are classified as either B cell or T cell. B-cell lymphomas are more common than T-cell lymphomas.
The clinical course for each of the NHLs, even subtypes, is variable. The most common lymphoma is diffuse large B-cell lymphoma (DLBCL), which comprises 33% of all NHLs. It is an aggressive, fast-growing tumor. Studies have demonstrated three subtypes with different response rates and prognosis.
Burkitt’s lymphoma is a highly aggressive B-cell tumor requiring intensive treatment; only 1% to 2% of lymphomas are classified as Burkitt’s. Follicular lymphoma is slow growing and clinically indolent (slow growing, painless, continually relapsing). Follicular lymphomas constitute about 14% of lymphomas. Follicular lymphoma, although indolent, may transform to a more rapidly progressive form, DLBCL.
Mantle cell lymphoma makes up only 2% of lymphomas but is typically widespread at diagnosis. Although these cells grow at an intermediate rate, the prognosis is poor. Clinical staging of NHL is according to the Ann Arbor system, ranging from stage I to stage IV (Box 14-6). Compared with HL, NHLs are more likely to present in an extranodal site, and the progression of the NHL does not follow the orderly anatomic progression from one lymph node to the next. Stage I and II NHLs are uncommon because the disease is much more likely to be disseminated at the time of diagnosis.
Etiologic and Risk Factors.: Studies in the 1990s linked NHL to two widespread environmental contaminants: exposure to benzene, which originates from cigarette smoke, gasoline, automobile emissions, and industrial pollution, and polychlorinated biphenyls (PCBs) found throughout the food chain (highest in meats, dairy products, and fish).76
One large study was unable to confirm the connection of benzene to lymphoma.200 Of benzene exposure that occurs in the environment, 70% is derived from vehicle exhaust emissions. The increase of environmental benzene has closely paralleled the rise in frequency of hematologic malignancies.136
In people with HIV, the risk of developing NHL is significantly elevated compared with noninfected people. Other predisposing risk factors for lymphoma are listed in Table 14-4.146
A wide variety of primary and secondary immunodeficiencies have been associated with an increased incidence of lymphomas. This phenomenon may reflect a decrease in the host’s surveillance mechanism against transformed cells or be from prolonged exposure to oncogenic agents, such as EBV, as a consequence of failure to mount an adequate immune response. The presence of Helicobacter pylori (bacteria) in the stomach lining is associated with the development of gastric lymphoma, but this comprises a very small proportion of cases. Low-dose methotrexate therapy used for classic and juvenile RA carries an increased risk of lymphoproliferative disease.23,42
Pathogenesis.: Although the exact cause of NHL is unknown, studies using techniques of molecular biology have provided some clues to the pathogenesis. The malignant lymphomas develop from the malignant transformation of a single lymphocyte that is arrested at a specific stage of B-or T-lymphoid cell differentiation and begins to multiply, eventually crowding out healthy cells and creating tumors, which enlarge lymph nodes.
Because immunosuppressed people have a greater incidence of the disease, an immune mechanism is suspected. Unlike in HL, T-cell function is minimally affected (30%), but B-cell abnormalities are more common in NHL (70%). In children, virtually all malignant lymphomas are high-grade, aggressive neoplasms.
Clinical Manifestations.: The NHLs are variable in clinical presentation and course, varying from indolent disease to rapidly progressive disease. Lymphadenopathy is the first symptom of NHL, with painless enlargement of isolated or generalized lymph nodes of the cervical, axillary, supraclavicular, inguinal, and femoral (pelvic) chains. This development may occur slowly and progressively or rapidly depending on lymphoma type.
Extranodal sites of involvement may include the nasopharynx, GI tract, bone (accompanied by bone pain), thyroid, testes, and soft tissue. Abdominal lymphoma may cause abdominal pain and fullness, GI obstruction or bleeding, ascites, back pain, and leg swelling.
Lymph node enlargement in the chest can lead to compression of the trachea or bronchus, causing shortness of breath and coughing. Development of the superior vena cava (SVC) syndrome can occur secondary to compression of the SVC by enlarged nodes; this causes edema of the upper extremities and face. SVC syndrome is life threatening and requires immediate attention.
NHL presenting as polyarthritis has been reported, and Sjögren’s syndrome is associated with malignant lymphomas.59,191 Constitutional symptoms include fever, night sweats, pallor, fatigue, and weight loss; when present, these systemic B symptoms typically predict a poor prognosis.
Primary CNS lymphoma is an NHL restricted to the nervous system. Presenting symptoms may include headache, confusion, seizures, extremity weakness/numbness, personality changes, difficulty speaking, and lethargy. Prior to the spread of HIV, this type of lymphoma was rare.
HIV and NHL.: NHL is more common in clients with HIV than is HL and is an AIDS-defining illness. Typically lymphomas that occur in clients with HIV are aggressive, fast-growing tumors. The two major subtypes of lymphomas are CNS and systemic lymphomas (with or without CNS involvement).
Two rare lymphomas seen more frequently in people with HIV are primary effusion lymphoma and plasmablastic lymphoma of the oral cavity, but the most common types are DLBCL and Burkitt’s lymphoma. Tumor is frequently diffusely spread at the time of diagnosis, with extranodal involvement common.
As discussed above, many illnesses that are accompanied by a reduced immune system demonstrate an increased incidence of NHL. Prior to aggressive HIV therapy (i.e., HAART), lymphomas in persons with HIV were associated with a very poor prognosis.
Currently the use of HAART has significantly reduced the risk of developing NHL and also improved tolerance for chemotherapy once diagnosed with NHL. This reduction is based on higher CD4 counts and improving the immune system. It appears that if HAART therapy is not effective, people with AIDS still have the same increased risk of developing NHL.111 The improved immune status derived from HAART has increased treatment options for HIV-related lymphoma. Clients are now treated with the intent to cure, receiving chemotherapy, immune modulators, and BMT.
MEDICAL MANAGEMENT
Accurate diagnosis is important because of the other clinical conditions that can mimic malignant lymphomas (e.g., infection, tuberculosis, SLE, lung and bone cancer). Molecular genetic techniques that take advantage of the clonal nature of this malignancy are now being applied to better characterize and diagnose the lymphomas. However, at the present time a biopsy is still required to confirm the underlying cause of persistent enlargement of lymph nodes present on clinical examination.
CT scans of the chest, abdomen, and pelvis are helpful in staging, while MRI is used to image the brain and spinal cord. Bone marrow may be examined for staging and peripheral blood may be tested, but blood abnormalities are not present until the disease is in an advanced stage. If clinical symptoms warrant, a lumbar puncture for spinal fluid may be performed. Immunohistochemistry, flow cytometry, or cytogenetic testing is often done to distinguish one type of NHL from another.
The gallium scan (Ga scan, scintigraphy) using radiotracer (gallium-67) uptake is 85% to 90% accurate to predict residual disease after chemotherapy and is able to differentiate between active tumor tissue and fibrosis (uptake only occurs in viable lymphoma tissue, not in fibrotic or necrotic tissue). PET imaging is becoming more widespread and can be performed to aid in the initial diagnosis and help ascertain if a lymph node is malignant or benign. PET is also used following chemotherapy (frequently along with CT) to determine if the lymphoma is reduced in size and the treatment is effective.
TREATMENT.
Treatment varies for NHL depending on the type. In general, fast-growing tumors can be cured but require aggressive treatment. Slow-growing tumors often cannot be cured, but the clinical course is chronic and therapy is often reserved until symptoms develop, such as for follicular lymphoma. Localized disease (stage I or II) may be treated with radiation, whereas disseminated disease requires radiation and chemotherapy.
The most common chemotherapy combination is CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone). Another combination omits the doxorubicin (because of its effects on the heart) and is called CVP. Other agents include chlorambucil, fludarabine, and etoposide. Since many risk factors for NHL are associated with a reduced immune system, immune modulators, such as interferon and monoclonal antibodies, have been employed to combat NHL.
Combining the monoclonal antibody rituximab (Rituxan) with chemotherapy (CHOP) has produced high rates of response and is the treatment of choice for many NHLs, including DLBCL.41 Clinical studies suggest that the immune modulator rituximab may alter the sensitivity of B-cell lymphoma to chemotherapy as well as induce apoptosis and cause the lysis of B cells.
BMT may be used for clients who relapse or do not completely respond to treatment (which often occurs with aggressive lymphomas). Combined with intensive chemotherapy, BMT (autologous or allogeneic) can be curative. In 2002, 4300 transplants were performed. One fourth of these were allogeneic, while the remainder was autologous. Nonmyeloablative (i.e., the doses of chemotherapy are not high enough to ablate the bone marrow) transplants can be performed for older clients who normally cannot tolerate high-dose chemotherapy, but graft-versus-host complications are problematic.
For some lymphomas, chemotherapy becomes palliative because of an inability to overcome drug resistance within the lymphoma cells; attempts at overcoming specific drug resistance mechanisms have had limited success. Other strategies involve the use of antigen-presenting cells for taking up, processing, and presenting tumor protein in a vaccine strategy. This may provide a new immune mode to eradicate lymphoma, particularly tumor cells that persist following therapy.33,116
Radioimmunotherapy, radioactive labeling of a monoclonal antibody, is also under investigation to provide targeted therapy and provide tumor-free grafts for transplant.19,197
The optimal management of women with NHL who are pregnant requires special considerations because of the poor prognosis without treatment. Treatment during the first trimester is associated with significant risk to the developing fetus and should be avoided. Treatment during the second or third trimester should include standard chemotherapy despite the potential risk to the developing fetus.153
PROGNOSIS.
Good prognostic features include age under 60 years, limited disease at diagnosis (stage I or II), lack of extranodal disease, and a normal lactate dehydrogenase (LDH) level. Individuals with NHL survive for long periods when involvement is only regional. The presence of diffuse disease reduces survival time.
The indolent lymphomas are usually systemic and widespread and cure cannot be achieved, whereas intermediate-and fast-growing lymphomas are more likely to present as treatable and even curable localized disorders but require aggressive therapy.
The prognosis for people with high-grade lymphomas depends on their response to treatment. More specifically, DLBCL can be cured in 40% to 50% of clients with therapy, follicular lymphoma has a 5-year survival of 60% to 70% (although it is eventually fatal), 20% of clients with mantle cell are alive at 5 years, and Burkitt’s lymphoma has a 50% 5-year survival with intensive therapy. In general, the 5-year survival rate for NHL is 63% and the 10-year survival rate is 49%.
Traditionally high-grade NHL associated with AIDS was associated with an extremely poor prognosis. But since the advent of antiretroviral therapy for HIV and a multidisciplinary approach to complex AIDS cases involving malignancy, return to functional health has become possible for many individuals; survival rates approach those seen in persons without HIV.48,111,112
Prognostic indicators for decreased survival in HIV NHL include age greater than 35 years, history of injection drug use, CD4 cell count less than 100/100 ml, a history of AIDS before the diagnosis of lymphoma, stage III or IV disease, and/or elevated LDH levels.110