Chapter 18 Basic metabolic pathways and the origin of secondary metabolites
The biosynthesis of both primary and secondary metabolites is dependent on the highly organized structure of the plant and animal cell. Unlike animal cells, those of plants possess a rigid cell wall and are separated one from another by an intercellular structure, the middle lamella. Direct connection between adjacent cells is maintained by primary pit fields through which pass the plasmodesmata. Within the cell wall is the protoplast consisting of cytoplasm, nucleus and various organelles.
The light microscope shows the nucleus to contain various inclusions such as nucleoli, chromosomes (stainable during cell division) and the nuclear sap. The nucleus appears to be suspended within the cell by the cytoplasm, in which there may be large vacuoles with their own characteristic contents (crystals, aleurone grains, etc.). Other cytoplasmic inclusions are mitochondria, Golgi bodies, lysosomes and plastids (chloroplasts, chromoplasts, leucoplasts), but their structure is not resolvable, because of their small size. Electron microscopy shows a number of the subcellular organelles to have a highly organized fine structure suited to the many and varied biochemical processes which they perform.
Although, because of varied form and function, it is not possible to illustrate a ‘typical’ plant cell Fig. 18.1A shows diagrammatically the structures that might be expected in an unspecialized young root cell. Such a cell possesses a rigid wall, which immediately distinguishes it from an animal cell, but no chloroplasts and only small vacuoles are present. In a green plant cell (Fig. 18.1B) the same components are present but the large vacuole has oppressed the nucleus and cytoplasm towards the wall; green plastids often with starch granules are common.
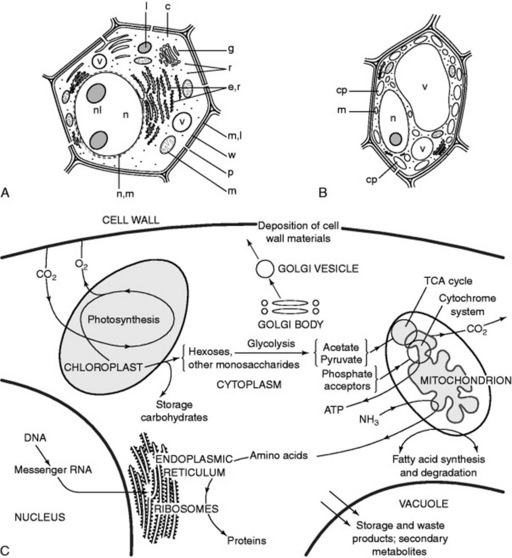
Fig. 18.1 The plant cell. A, diagrammatic representation of an undifferentiated cell: c, cytoplasm; e.r, endoplasmic reticulum; g, Golgi apparatus; l, lysosome; m, mitochondrion; m.l, middle lamella; n, nucleus; nl, nucleolus; n.m, nuclear membrane; p, pit in wall; r, ribosomes, w, primary cell wall; v, vacuole. B, green plant cell: cp, chloroplast; m, mitochondrion, n, nucleus; v, vacuole. C, Flow chart of some cell metabolites.
The organelles allow the creation of different chemical environments within one cell, and furthermore, by their structure they increase the area available for surface reactions which are all-important in biological systems. A description of the various organelles is given in the 14th edition of this book and Fig. 18.1C illustrates some aspects of their interdependence in the normal functioning of the cell. The molecular structures of these bodies have been extensively studied and details will be found in standard botanical texts.
Some basic metabolic pathways appear to be similar in both plants and animals, whereas others are more restricted in their occurrence. It is to the secondary plant products (i.e. those not necessarily involved in the essential metabolism of the cell) that the majority of vegetable drugs owe their therapeutic activity and so it is in these that pharmacognosists are particularly interested. However, as illustrated in Fig. 18.2, the production of these secondary metabolites is dependent on the fundamental metabolic cycles of the living tissue and so a brief indication of the latter will also be given; for fuller accounts of these, the student should consult a standard work on plant biochemistry.
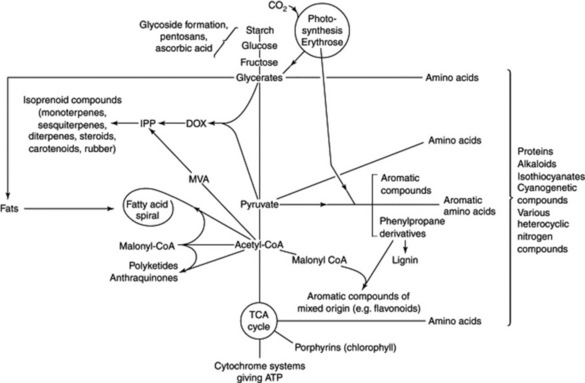
Fig. 18.2 Origins of some secondary metabolites in relation to the basic metabolic pathways of plants. DOX=deoxyxylulose; IPP=isopentenyl diphosphate; MVA=mevalonic acid.
ENZYMES
Many reactions occurring in the cell are enzyme-dependent, and before anything was known of the chemical nature of these substances it was recognized that they were organic catalysts produced by animal and vegetable cells. Their wide distribution and the delicacy of their operation has long been appreciated; they engineer reactions at normal temperatures and at pH values around neutral in a manner not possible in the laboratory.
An enzyme usually acts on one substance or class of substances, since it is specific for a particular atomic group or linkage. Specificity, however, varies; lipases are, in general, not highly specific, whereas fumarase acts only upon L-malate and fumarate, while D-malate is a competitive inhibitor of fumarase. Enzymes are also stereo- and regio- specific in their actions and, as it becomes possible to prepare more rare examples, organic chemists are becoming increasingly aware of enzyme potential for carrying out single-step transformations with complete stereochemical exactitude, an aspect important in the synthesis of many drugs. An enzyme will convert many thousand times its own weight, and the gradual diminution in activity which takes place is probably due to secondary reactions which bring about destruction of the enzyme.
The enzymology of the secondary metabolic pathways in plants has still been little investigated but progress is being made and here again cell cultures have proved useful in that they are often a better source for the isolation of enzymes than is the differentiated plant. In some cases, e.g. cell cultures of volatile oil-containing plants, little or no oil accumulates in the culture owing to the absence of storage receptacles but the relevant enzymes for terpenoid synthesis are still manufactured and preparations of them can be made.
New horizons for the study of the enzymology of secondary metabolism have now opened up as a result of advances in gene technology. In suitable instances, by cloning, the cDNA responsible for an enzyme’s synthesis can be expressed in another organism such as a bacterium and large amounts of enzyme prepared. By conventional methods only very small amounts of purified enzyme could be obtained from the original plant material. Of particular interest has been the isolation, characterization and cloning of the enzyme strictosidine synthase. This governs the key reaction for the commencement of the biosynthesis of the very many monoterpenoid indole alkaloids, namely the condensation of tryptamine and secologanin to give 3alpha;(S)-strictosidine (see Chapter 26); for a review (126 references) on this enzyme see T. M. Kutchan, Phytochemistry, 1993, 32, 493.
By means of relatively new technology, enzymes can be immobilized on a suitable carrier either in whole plant cells or as the isolated enzyme. In this way these biocatalysts can be repeatedly used in analytical and clinical chemistry, or to effect specific chemical transformations.
Like other catalysts, enzymes influence the rate of a reaction without changing the point of equilibrium. For example, lipase catalyses either the synthesis of glycerides from glycerol and fatty acids or the hydrolysis of glycerides, the final point of equilibrium being the same in either case. Similarly, β-glucosidase (prunase) has been used for both the synthesis and the hydrolysis of β-glucosides. In plants such reversible reactions may proceed in one direction or the other under different conditions, often resulting in daily and seasonal variations in the accumulation of metabolites.
Enzymes are colloidal in nature and consist of protein or contain protein as an essential part. They may be partly purified, and in some cases isolated, by the methods of protein chemistry (i.e. by fractional precipitation, dialysis and, more recently, gel and affinity chromatography, see Chapter 17). Most enzymes are soluble either in water or in dilute salt solutions and are precipitated by alcohol or acetone (acetone powders) and by high concentrations of salts. They are inactivated by heat, ultraviolet light and X-rays or by any treatment which brings about denaturation of proteins.
The activity of enzymes is markedly affected by the reaction of the medium and the presence of substances such as salts. It is well known, for example, that pepsin works only in an acid medium and trypsin in an alkaline one. In general, carbohydrases have pH optima of 3.8–7.5 and lipases optima of pH 5–8, while enzymes which act on bases all have optima more alkaline than pH 7.
The effect of heat on enzymes is of considerable importance in the drying of drugs. At low temperatures enzymic changes are not usually marked, although the proteolytic or protein-splitting enzymes in cod livers do bring about some hydrolysis at temperatures approaching zero. The optimum working temperatures of different enzymes vary, but they usually lie between 35 and 50 °C. At temperatures of about 60 °C destruction of the enzymes is usually fairly rapid, although considerable loss may take place below this temperature. When dry, enzymes show increased resistance to heat; thus, zymase, which in the presence of moisture is rapidly inactivated at 50 °C, will, when dry, resist a temperature of 85 °C.
Chemical nature
Following the isolation of urease in crystalline form by Sumner in 1926, many other enzymes have been prepared in a crystalline form and
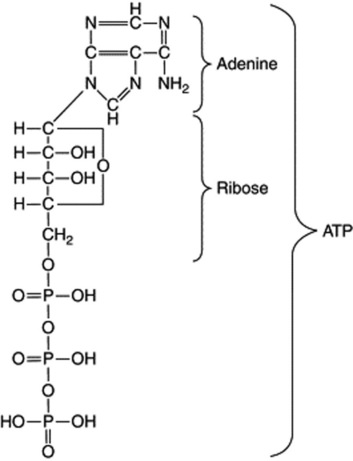
their protein nature has been established. Their molecular weights are high, but vary from about 9000 (hydrogenase) to about 1 000 000. In many cases the component amino acids are known and in some cases (e.g. ribonuclease) the amino acid sequence within the molecule has been determined. During the isolation of an enzyme, the purified protein (apoenzyme) may be inactive but regains its activity in the presence of an essential coenzyme or activator, which may be organic or inorganic.
Coenzymes
Some enzymes which require the presence of smaller organic molecules, called coenzymes, before they can function are of very common occurrence and participate in a large number of important biochemical reactions. One group of coenzymes consists of esters of phosphoric acid and various nucleosides. The adenosine and uridine phosphates contain one basic unit each (mononucleotides); they serve to transport energy in the form of high-energy phosphate bonds and this energy is made available for biochemical reactions in the presence of the appropriate enzyme by hydrolysis of the bond. Thus, the terminal phosphate bond of the adenosine triphosphate (ATP) on hydrolysis to adenosine diphosphate (ADP) affords 50 000 J mol−1.
Uridine triphosphate (UTP) is involved in the synthesis of sucrose via diphosphate glucose which is also associated with the formation of uronic acids and cellulose.
Nicotinamide-adenine dinucleotide (NAD) and nicotinamide-adenine dinucleotide phosphate (NADP) contain two basic units each and are termed dinucleotides. They function in oxidation–reduction systems with appropriate enzymes; the oxidized forms are written NAD+ and NADP+ and the reduced forms NADH and NADPH respectively.
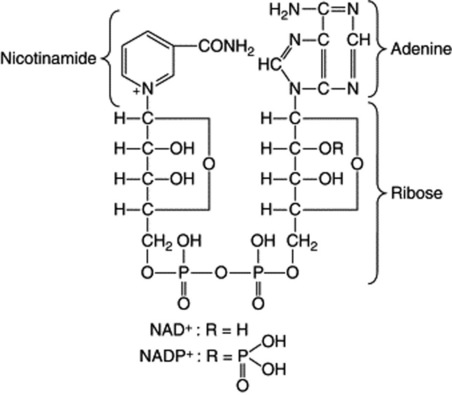
Another important coenzyme is coenzyme A (CoA), which contains the units adenosine-3,5-diphosphate, pantothenic acid-4-phosphate and thioethanolamine. It participates in the transfer of acetyl and acyl groups, acetyl-CoA (active acetate) having a central role in plant and animal metabolism.
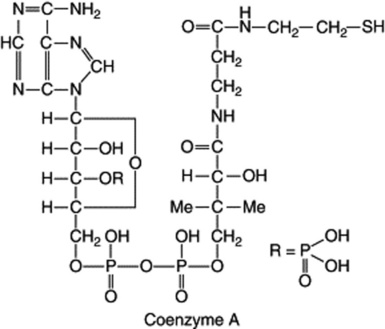
Riboflavine (Fig. 31.2) is a component of the two coenzymes flavin mononucleotide (FMN) and flavin adenine dinucleotide (FAD). They participate in the biological oxidation–reduction system, and FAD facilitates the transfer of H+ ions from NADH to the oxidized cytochrome system.
Other coenzymes are the decarboxylation coenzymes thiamine, biotin and pyridoxine (Fig. 31.2). Folic acid (Fig. 31.2) derivatives participate in enzymatic reactions which involve one-carbon fragment transfers.
A series of quinones e.g. plastoquinone and ubiquinone are widely distributed in plants, animals and microorganisms and function in biological electron transfer processes.
Classification of enzymes
As more enzymes are isolated, it becomes important to have a precise scheme of classification and nomenclature. Many long-established names such as pepsin, prunase, diastase and names for enzyme mixtures, such as emulsin and zymase, continue in use. A step towards uniform nomenclature was made when they were denoted by the name of the substrate and the termination ‘-ase’. Classes of enzymes were named similarly. Thus, the general term ‘esterase’ includes lipases, which hydrolyse fats; chlorophyllase hydrolyses chlorophyll; etc. The 1961 Report of the Commission on Enzymes*
* This report has a numbered classification for each enzyme. For example, the enzyme present in garlic, alliine-lyase, is numbered 4.4.1.4. The first number denotes the fourth of the six main groups already mentioned and the other numbers further subdivisions. Thus:
made the recommendation that the chemical reactions catalysed be generally adopted for classification and nomenclature. This, of course, presupposes that the exact chemical reaction is known; see also the 1984 International Union of Biochemistry publication (London, UK: Academic Press) Enzyme Nomenclature and any later reports. Enzymes are classified into six main groups (Table 18.1).
Table 18.1 Classification of some enzymes.
| Group | Trivial name | Systematic name |
|---|---|---|
| 1. Oxidoreductases | Glucose dehydrogenase | β-D-Glucose: NAD(P)-oxidoreductase |
| p-Diphenyl oxidase (lactase) | p-Diphenol: O2 oxidoreductase | |
| Peroxidase | Donor: H2O2 oxidoreductase | |
| Catalase | H2O2: H2O2 orthoreductase | |
| 2. Transferase | α-Glucan phosphorylase | α-1,4-Glucan: orthophosphate glucosyl-transferase |
| 3. Hydrolases | Lipase | Glycerol ester hydrolase |
| Chlorophyllase | Chlorophyll chlorophyllidohydrolyase | |
| Tannase | Tannin acyl-hydrolase | |
| α-Amylase | α-1,4-Glucan 4-glucano-hydrolyase | |
| Inulase | Inulin 1-fructanohydrolase | |
| 4. Lyases | Aldolyase | Ketose-l-phosphate aldehydelyase |
| Decarboxylase | L-Tryptophan-decarboxylase | |
| 5. Isomerases | Maleate isomerase | Maleate cis-trans-isomerase |
| 6. Ligases (Synthetases) | Asparagine synthetase | L-Aspartate: ammonia ligase (ADP) |
Oxidoreductases
As any oxidation implies a simultaneous reduction, the names ‘oxidase’ and ‘reductase’ may be applied to a single type of enzyme. Eleven different groups of oxidoreductases are recognized. Many oxidases (that is, enzymes that utilize molecular oxygen as acceptor) convert phenolic substances to quinones. They act on guaiacum resin to produce a blue colour which is used as a test for their detection. Oxidases include laccase (1.10.3.2), present in lac, and ascorbate oxidase (1.10.3.3), which is widely distributed in plants. Oxalate oxidase (1.2.3.4), a flavoprotein present in mosses and the leaves of higher plants, oxidizes oxalic acid into carbon dioxide and hydrogen peroxide. Peroxidases (1.11) are distinguished by the fact that they use hydrogen peroxide and not oxygen as the hydrogen acceptor. Catalase (1.11) must be regarded as an exception, since it catalyses the decomposition of hydrogen peroxide. An oxidoreductase of the morphine biosynthetic pathway (Chapter 26) has recently been purified and characterized; it catalyses the stereoselective reduction of salutaridine to 7(S)-salutaridinol using NADPH as cosubstrate.
Hydrolases
These include many different types, of which the following are some of pharmaceutical importance.
Lyases
Two important decarboxylases in the biosynthesis of monoterpenoid indole alkaloids and benzylisoquinoline alkaloids are L-tryptophan-decarboxylase (EC 4.1.1.27) and L-tyrosine/L-dopa-decarboxylase (EC 4.1.1.25), respectively.
Isomerases
Two enzymes important in pathways leading to medicinally important metabolites are isopentenyl diphosphate (IPP) isomerase (EC 5.3.3.2) amd chalcone isomerase (EC 5.5.1.6). The former is involved in the isomerization of IPP and dimethylallyl diphosphate (Fig. 18.18), an essential step in the formation of terpenoids, and the latter is a key enzyme catalysing the isomerization of chalcones to their corresponding flavanones (Fig. 18.11).
PHOTOSYNTHESIS
Photosynthesis, by which the carbon dioxide of the atmosphere is converted into sugars by the green plant, is one of the fundamental cycles on which life on Earth, as we know it, depends. Until 1940, when investigations involving isotopes were undertaken, the detailed mechanism of this ‘carbon reduction’ was unknown, although the basic overall reaction,
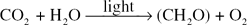
had been accepted for many years.
Photosynthesis occurs in the chloroplasts: green, disc-shaped organelles of the cytoplasm which are bounded by a definite membrane and which are autoreproductive. Separated from the rest of the cell, the chloroplasts can carry out the complete process of photosynthesis. Bodies with similar properties are found in cells of the red algae but these contain, in addition to chlorophyll as the principal pigment, other tetrapyrrole derivatives—the phycobilins.
The light microscope reveals no definite internal structure of the chloroplasts, but electron microscopy shows these bodies to have a highly organized structure in which the chlorophyll molecules are arranged within orderly structures (grana), each granum being connected with others by a network of fibres or membranes. According to one theory, the flat chlorophyll molecules themselves are orientated between layers of protein and lipid molecules so that the whole chloroplast can be looked upon as a battery containing several cells (the grana), each cell possessing layers of plates (the chlorophyll molecules).
Two fundamental processes which take place in photosynthesis, both of which require light, are the production of adenosine triphosphate (ATP) from adenosine diphosphate (ADP) and phosphate and the light-energized decomposition of water (the Hill reaction; named after the English biochemist Robert Hill, 1899–1991):
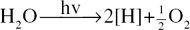
ATP is a coenzyme and the high energy of the terminal phosphate bond is available to the organism for the supply of the energy necessary for endergonic reactions. The Hill reaction produces free oxygen and hydrogen ions which bring about the conversion of the electron carrier, NADP, to its reduced form NADPH (see ‘Coenzymes’).
In this complicated process, two systems, Photosystem I and Photosystem II (also known as pigment systems I and II) are commonly referred to; they involve two chlorophyll complexes which absorb light at different wavelengths (above and below λ = 685 nm). Photosystem II produces ATP and Photosystem I supplies all the reduced NADP and some ATP. In these light reactions the chlorophyll molecule captures solar energy and electrons become excited and move to higher energy levels; on returning to the normal low-energy state, the electrons give up their excess energy, which is passed through a series of carriers (including in the case of Photosystem II plastoquinone and several cytochromes) to generate ATP. Photosystem I involves an electron acceptor and the subsequent reduction of ferredoxin in the production of NADPH. Reference to the current literature indicates that the nature and organization of the photosystems remains a very active research area. Students will have observed that an alcoholic solution of chlorophyll possesses, in sunlight, a red fluorescence—no carriers are available to utilize the captured energy and it is re-emitted as light. Hill first demonstrated in 1937 that isolated chloroplasts, when exposed to light, were capable of producing oxygen, provided that a suitable hydrogen acceptor was present. Work with isotopes has since proved that the oxygen liberated during photosynthesis is derived from water and not from carbon dioxide.
Following the light reactions, a series of dark reactions then utilize NADPH in the reduction of carbon dioxide to carbohydrate.
Current research suggests that terrestrial plants can be classified as C3, C4, intermediate C3–C4 and CAM plants in relation to photosynthesis.
C3 plants
The elucidation of the carbon reduction cycle (Fig. 18.3), largely by Calvin and his colleagues, was in large measure determined by methods dependent on exposing living plants (Chlorella) to 14C-labelled carbon dioxide for precise periods of time, some very short and amounting to a fraction of a second. The radioactive compounds produced were then isolated and identified. In this way a sequence for the formation of compounds was obtained. 3-Phosphoglyceric acid, a C3 compound, was the compound first formed in a labelled condition but it was only later in the investigation, after a number of 4-, 5-, 6- and 7-carbon systems had been isolated, that ribulose-1,5-diphosphate was shown to be the molecule with which carbon dioxide first reacts to give two molecules of phosphoglyceric acid. An unstable intermediate in this reaction is 2-carboxy-3-ketopentinol.
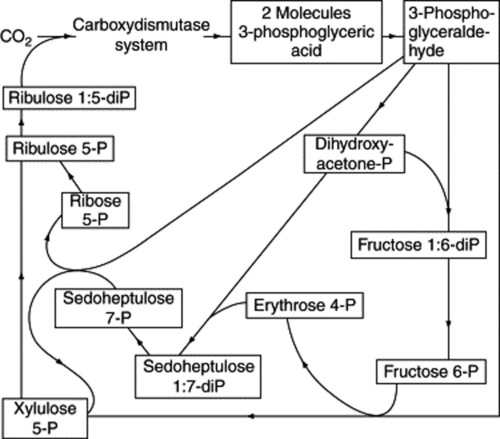
Fig. 18.3 The path of carbon in photosynthesis (after Calvin); the formulae of the sugars involved are given in Chapter 20.
C4 plants
Some plants which grow in semi-arid regions in a high light intensity possess an additional carbon-fixation system which, although less efficient in terms of energy utilization, is more effective in its use of carbon dioxide, so cutting down on photorespiration and loss of water. Such plants are known as C4 plants, because they synthesize, in the presence of light, oxaloacetic and other C4 acids. Carbon assimilation is based on a modified leaf anatomy and biochemistry. Its presence in plants appears haphazard and has been linked with the so-called Kranz syndrome of associated anatomical features. In the mesophyll cells pyruvate is converted via oxaloacetate to malate, utilizing carbon dioxide, and the malate, or in some cases aspartate, is transported to the vascular bundle cells, where it is oxidatively decarboxylated to pyruvate again, carbon dioxide and NADPH, which are used in the Calvin cycle. Pyruvate presumably returns to the mesophyll cells. Plants possessing this facility exhibit two types of photosynthetic cells which differ in their chloroplast type. Intermediate C3–C4 pathways are also known.
CAM plants
This term stands for ‘crassulacean acid metabolism’, so called because it was in the Crassulaceae family that the distinctive character of a build-up of malic acid during hours of darkness was first observed. Other large families, including the Liliaceae, Cactaceae and Euphorbiaceae, possess members exhibiting a similar biochemistry. As with C4 plants, this is an adaptation of the photosynthetic cycle of plants which can exist under drought conditions. When water is not available, respiratory carbon dioxide is recycled, under conditions of darkness, with the formation of malic acid as an intermediate. Carbon dioxide and water loss to the atmosphere are eliminated, a condition which would be fatal for normal C3 plants.
From fructose, produced in the Calvin cycle, other important constituents, such as glucose, sucrose and starch, are derived: erythrose is a precursor in the synthesis of some aromatic compounds. Glucose-6-phosphate is among the early products formed by photosynthesis and it is an important intermediate in the oxidative pentose phosphate cycle and for conversion to glucose-1-phosphate in polysaccharide synthesis.
CARBOHYDRATE UTILIZATION
Storage carbohydrate such as the starch of plants or the glycogen of animals is made available for energy production by a process which involves conversion to pyruvate and then acetate, actually acetyl-coenzyme A, the latter then passing into the tricarboxylic acid cycle (Fig. 18.4). As a result of this, the energy-rich carbohydrate is eventually oxidized to carbon dioxide and water. During the process, the hydrogen atoms liberated are carried by coenzymes into the cytochrome system, in which energy is released in stages, with the possible formation of ATP from ADP and inorganic phosphate. Eventually the hydrogen combines with oxygen to form water.
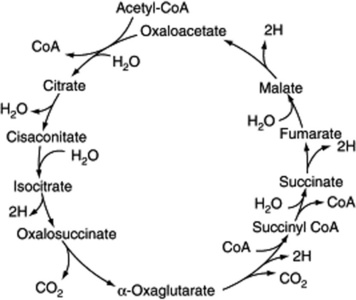
A number of pathways for the initial metabolism of glucose are known for various living tissues. One involves compounds which are found in the photosynthetic cycle, and it appears as a reversal of this cycle but the mechanism is apparently quite different. Another pathway is the Embden–Meyerhoff scheme of glycolysis (Fig. 18.5).
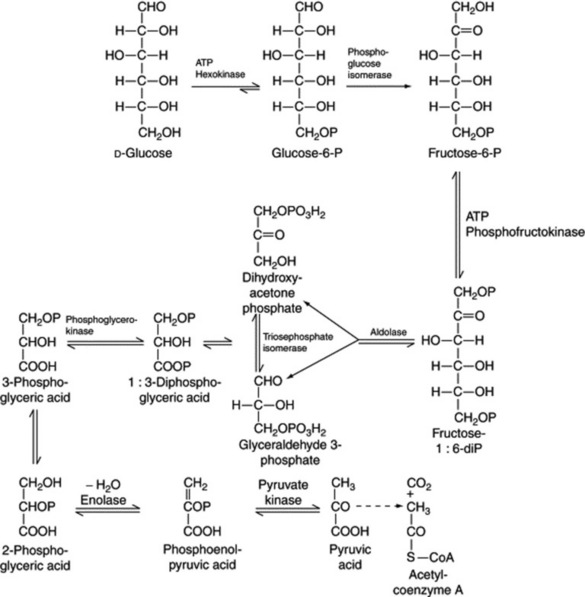
The kinetics and enzyme systems involved in the above pathways have been studied extensively. It will be noted that one molecule of glucose can give rise to two molecules of pyruvate, each of which isconverted to acetate, and one molecule of carbon dioxide. One turn of the TCA cycle represents the oxidation of one acetate to two molecules of carbon dioxide, giving rise to twelve molecules of ATP. The overall reaction for the metabolism of one molecule of glucose in terms of ADP and ATP is
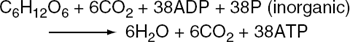
The above schemes, given in barest outline, are fundamental not only for the building up and breaking down of reserve foodstuffs, but also in that the intermediates are available for the biosynthesis of all other groups of compounds found in plants. The levels at which some of the important groups arise are indicated in Fig. 18.2.
GLYCOSIDES
Glycosides are formed in nature by the interaction of the nucleotide glycosides—for example, uridine diphosphate glucose (UDP-glucose)—with the alcoholic or phenolic group of a second compound. Such glycosides, sometimes called O-glycosides, are the most numerous ones found in nature. Other glycosides do, however, occur in which the linkage is through sulphur (S-glycosides), nitrogen (N-glycosides) or carbon (C-glycosides).
The formation and hydrolysis of an O-glycoside such as salicin may be represented:
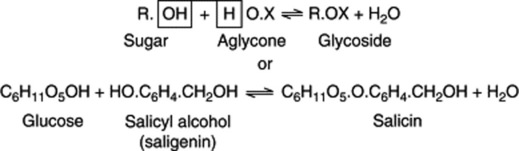
It will be noted that such reactions are reversible and in plants glycosides are both synthesized and hydrolysed under the influence of more or less specific enzymes. While glycosides do not themselves reduce Fehling’s solution, the simple sugars which they produce on hydrolysis will do so with precipitation of red cuprous oxide.
The sugars found in glycosides may be monosaccharides such as glucose, rhamnose and fucose or, more rarely, deoxysugars such as the cymarose found in cardiac glycosides. More than one molecule of such sugars may be attached to the aglycone either by separate linkages, which is rare, or, more commonly, as a di-, tri- or tetrasaccharide. Such complex glycosides are formed by the stepwise addition of sugars to the aglycone molecule.
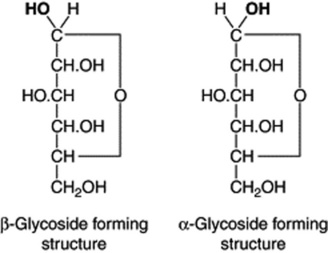
Since sugars exist in isomeric α- and β-forms, both types are theoretically possible. Practically all natural glycosides, however, are of the β-type, although the α-linkage is found in nature in some carbohydrates such as sucrose, glycogen and starch. In k-strophanthoside, a glycoside formed from the aglycone strophanthidin and strophanthotriose (cymarose + glucose + glucose) the outer glucose molecule has the α-linkage and the inner glucose the β-linkage. Isomeric glycosides may be prepared synthetically; for example, from glucose and methyl alcohol one obtains both the α- and β-methyl glucosides by introducing methyl groups into the OH groups printed in heavy type in the formulae of glucose below.
The term ‘glycoside’ is a very general one which embraces all the many and varied combinations of sugars and aglycones. More precise terms are available to describe particular classes. Some of these terms refer to the sugar part of the molecule, others to the aglycone, while others indicate some well-defined physical or pharmacological property. Thus, a glucoside is a glycoside having glucose as its sole sugar component; a pentoside yields a sugar such as arabinose; rhamnosides yield the methyl-pentose rhamnose; and rhamnoglucosides yield both rhamnose and glucose. Terms used for aglycones are generally self-explanatory (e.g. phenol, anthraquinone and sterol glycosides). The names ‘saponin’ (soap-like), ‘cyanogenetic’ (producing hydrocyanic acid) and ‘cardiac’ (having an action on the heart), although applied to these substances when little was known about them, are useful terms which do in fact bring together glycosides of similar chemical structure.
The older system of naming glycosides using the termination ‘-in’ (e.g. senegin, salicin, aloin, strophanthin) is too well established to be easily changed. It is not ideal, however, since many non-glycosidal substances (e.g. inulin and pectin) have the same termination. Modern workers more frequently use the termination ‘-oside’ (e.g. sennoside) and attempt to drop the older forms by writing sinigroside for sinigrin, salicoside for salicin and so on.
Although glycosides form a natural group in that they all contain a sugar unit, the aglycones are of such varied nature and complexity that glycosides vary very much in their physical and chemical properties and in their pharmacological action. In Part 5 they are classified according to that aglycone fragment with which they often occur in the plant.
FATS AND FATTY ACIDS
Fats, considered in more detail in Chapter 19, are triglycerides involving long-chain saturated or unsaturated acids and, as such, constitute an important food reserve for animals and plants (particularly in seeds). Related to the simple fats are the complex lipids, most of which are diesters of orthophosphoric acid; they have the status of fundamental cellular constituents.
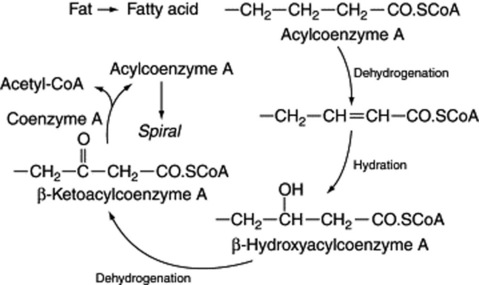
The fatty acids, on liberation from the fat, are available for the production of acetyl-CoA by the removal of C2 units. The elucidation of this spiral (Fig. 18.6) owes much to the work of Lynen and it was primarily investigated with animal tissues. In this β-oxidation sequence one turn of the spiral involves four reactions—two dehydrogenations, one hydration and a thiolysis liberating a two-carbon unit of acetyl-CoA. All these reactions can be shown by isolated enzyme studies to be reversible, and until about 1953 it was generally assumed that the biosynthetic route of fatty acids operated in the reverse direction, starting from acetyl-CoA. This, however, is not precisely so, as indicated below.
Biosynthesis of saturated fatty acids
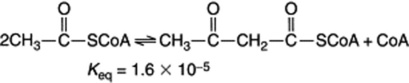
From the above it was generally assumed that the biosynthetic route for fatty acids operated in the reverse direction to the β-oxidative sequence for the degradation of these acids and started with acetyl-CoA. However, the unfavourable equilibrium of the initial thiolase reaction in such a pathway:
and the poor yield of long-chain fatty acids obtained with purified enzymes of the β-oxidation pathway together with other observations suggested the possibility that this was not necessarily the principal biosynthetic pathway. It was then shown, with animal tissues, that malonyl coenzyme A was necessary for the initial condensation with acetate. In fact, in palmitic acid (C16,) only one of the eight C2 units (i.e. the C15 + C16 carbons) is derived directly from acetyl-CoA; the other seven C2 units are attached by malonyl-CoA, which is formed by carboxylation of acetyl-CoA.
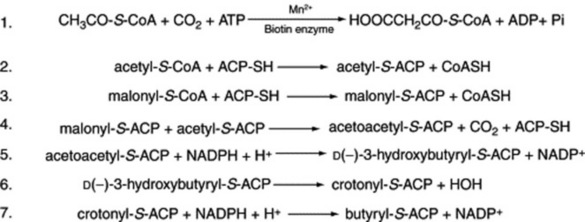
One feature of this anabolic pathway is the involvement of the acyl carrier protein (ACP) to produce fatty acyl thioesters of ACP. This conversion serves (1) to activate the relatively unreactive fatty acid and (2) to provide a carrier for the fatty acid acyl group. Thus, these ACP thioesters appear to be obligatory intermediates in fatty acid synthesis, whereas it is the fatty acid thioesters of coenzyme A which operate in the catabolic oxidation pathway. The synthesis is explained by the reactions shown in Fig. 18.7. Reactions 4–7 are then repeated, lengthening the fatty acid chain by two carbons (derived from malonyl-S-ACP) at a time. Eight enzymes have been identified as being involved in the synthesis of palmitoyl-ACP and stearoyl-ACP from acetyl-CoA.
In purified enzyme systems, when propionyl-CoA replaced acetyl- CoA, the product was a C17 acid and, similarly, butyryl-CoA gave primarily a C18 (stearic) acid. Thus, the formation of these C16, C17 and C18 acids may well depend on the availability of acetyl-, propionyl- and butyryl-CoA.
The system of biosynthesis of fatty acids in plants appears to operate in the same way as in animal tissues, but whereas in the latter the enzymes are located in the cytoplasm, in plants they are found in the mitochondria and chloroplasts. The mitochondria derived from the mesocarp of the avocado fruit have been used to demonstrate the incorporation of [14C]acetate into esterified long-chain fatty acids.
Biosynthesis of unsaturated fatty acids
As a number of unsaturated fatty acids have a particular significance in pharmaceutical products, a treatment of their formation is deferred until Chapter 19.
AROMATIC BIOSYNTHESIS
The shikimic acid pathway
This appears to be an important route from carbohydrate for the biosynthesis of the C6–C3 units (phenylpropane derivatives), of which phenylalanine and tyrosine are both examples. A scheme of biogenesis for these aromatic amino acids, as elucidated in various organisms, is given in Fig. 18.8; for higher plants, the presence of the enzyme system responsible for the synthesis of shikimic acid has been confirmed. An important branching point arises at chorismic acid; anthranilate synthase uses chorismic acid as a substrate to give anthranilic acid which is a precursor of tryptophan. The synthesis is controlled by the latter acting as a feedback inhibitor; chorismate mutase converts chorismic acid to prephenate, the precursor of phenylalanine and tyrosine, and a variety of control mechanisms appear to operate at the branching point. The opium alkaloids are synthesized via this pathway (Chapter 26) and two isoforms of chorismate mutase have been isolated and characterized from poppy seedlings (M. Benesova and R. Bode, Phytochemistry, 1992, 31, 2983).
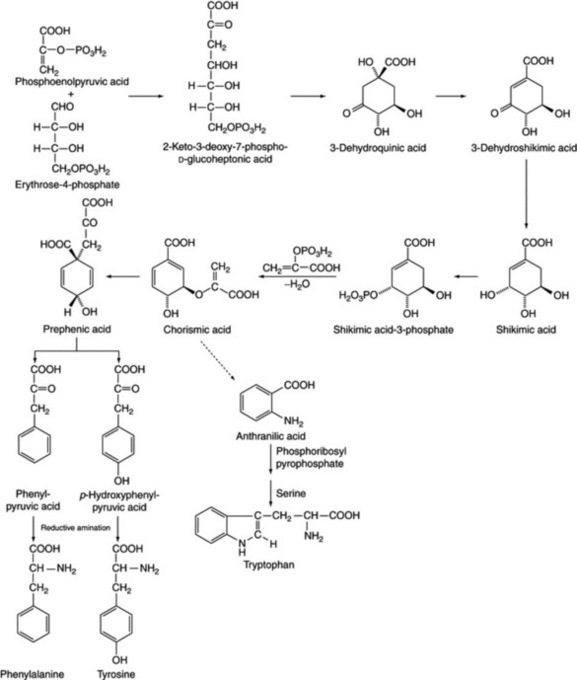
Although there are only small differences in the sequence of reactions for the shikimate pathway in bacteria, fungi and plants there are considerable differences in the molecular organization of the pathway. (For a review (123 refs), see J. Schmid and N. Amrhein, Phytochemistry, 1995, 39, 737.)
The shikimic acid pathway is also important in the genesis of the aromatic building blocks of lignin and in the formation of some tannins, vanillin and phenylpropane units of the flavones and coumarins (see Chapter 21). For a review see ‘Further reading’.
The acetate hypothesis
The central position of acetate in relation to the general metabolism of plants has already been indicated and it is possible to devise many possible routes by which acetate condensation could occur to give a variety of aromatic compounds. The general validity of the mechanism has been established by the use of labelled compounds but the detailed steps for many compounds remain to be established. Thus, the incorporation of [1-14C]acetate into 6-methylsalicylic acid by Penicillium griseofulvum takes place as below:
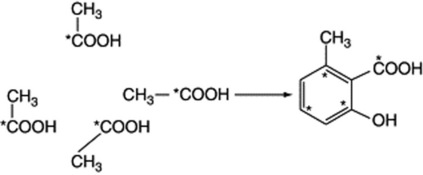
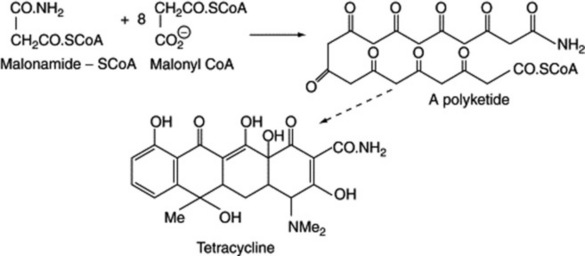
and the production of the mould anthraquinone metabolite endocrocin, from eight C2 units, is represented in Fig. 18.9. Decarboxylation of endocrocin affords emodin. The original chain lengthening, which can be represented as a condensation of acetate units, is the same as in fatty acid production (q.v. above) but does not require the reduction of =CO to =CH2. Thus, malonic acid plays a similar role in this aromatic synthesis to that in fatty acid formation and the chain is built up from the combination of malonyl units with a terminal acetyl (the starter) unit. Sometimes the starter unit is not acetate, as indicated with the formation of the tetracycline antibiotics from nine units; here malonamide-SCoA is the starter unit and by invoking standard biochemical reactions tetracycline is formed (Fig. 18.10). Higher plants also utilize the polyacetate–malonate pathway for the biosynthesis of emodin-type anthraquinones, such compounds all having substituents on both outer rings.
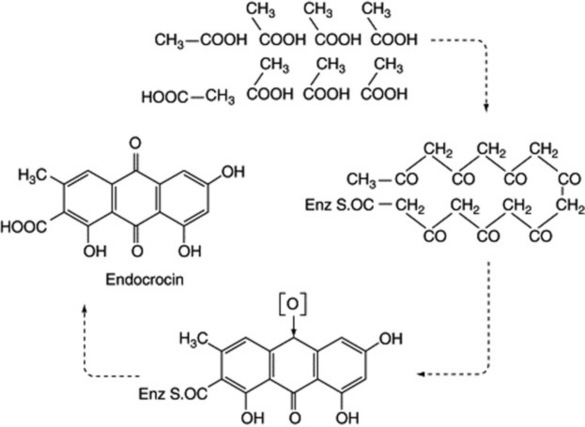
From the anthraquinones listed in Chapter 21, it will be seen that the structures of some anthraquinones are not fully explained by the acetate–malonate pathway and these are discussed below. However, there appear to be many exceptions to the rules in applying the general pathways to specific anthraquinones.
Compounds containing aromatic rings of different origin
The structures of anthraquinones such as alizarin, rubiadin, pseudopurpurin and morindadiol, pigments of the Rubiaceae and other families, cannot be readily explained on the acetate hypothesis.
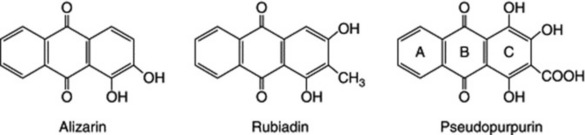
Circumstantial evidence that these compounds might be formed from naphthoquinones with the participation of mevalonic acid (a key precursor in the formation of isopentenyl units, q.v.) was provided by their co-occurrence in teak and other plants with naphthoquinones having an isopentenyl residue (e.g. formula below).
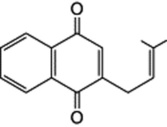
That mevalonate (itself derived from acetate) is involved in this formation has been shown by tracer experiments with Rubia tinctorum; ring C and carbon side-chain of pseudopurpurin and rubiadin are derived from mevalonate, and in the same plant shikimic acid has been shown to be incorporated into ring A of alizarin.
The aromatic rings of some compounds can be derived from both the shikimic acid and the acetic acid pathways. Thus, a phenylpropane formed by the former route may undergo chain lengthening by the addition of acetate units (via malonyl; see ‘Fatty Acids’, above) to give a polyketide and then, by ring closure, give a flavonoid derivative (Fig. 18.11).
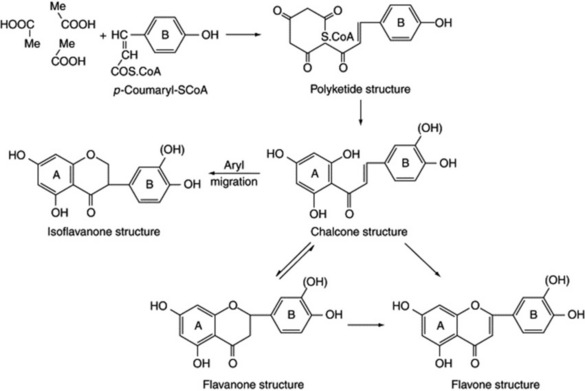
Isoflavones are formed in the same manner, with a rearrangement of the aryl-B ring in relation to the three carbons (Fig. 18.11). The flavonoids, in addition to their importance in plants as pigments, have interesting medicinal properties and are discussed in more detail in Chapter 21.
Rotenoids (insecticides of Derris and Lonchocarpus spp.) show a structural relationship to the isoflavones; see Chapter 40.
Stilbenes (q.v.) are also compounds containing aromatic units of different biogenetic origin.
AMINO ACIDS
Amino acids occur in plants both in the free state and as the basic units of proteins and other metabolites. They are compounds containing one or more amino groups and one or more carboxylic acid groups.Most of those found in nature are α-amino acids with an asymmetric carbon atom and the general formula R–CH(NH2)COOH. Some 20 different ones have been isolated from proteins, all having an L-configuration. Other amino acids occur in the free state and some having the D-configuration have been isolated from plants and microorganisms, where they may form antibiotic polypeptides.
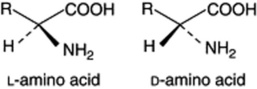
Many amino acids contain only carbon, hydrogen, oxygen and nitrogen, but other atoms may be present (e.g. sulphur in cystine, and iodine in thyroxin). As already mentioned, more than one amino group may be present (e.g. lysine, diaminocaproic acid) and more than one carboxylic acid group (e.g. aspartic or aminosuccinic acid). Some amino acids are aromatic such as phenylalanine, or heterocyclic such as proline (pyrrolidine nucleus), tryptophan (indole nucleus) and histidine (imidazole nucleus).
Amino acids are generally soluble in water but only slightly soluble in alcohol. A general test is to warm with ninhydrin, when, with the exception of proline, which gives a yellow, they give a pink, blue or violet colour. Amino acids do not respond to the biuret test (compare polypeptides and proteins). Certain amino acids are detected by more specific tests (e.g. histidine gives colour reactions with diazonium salts).
Amino acids found in proteins
These include α-alanine; arginine; asparagine (amide of aspartic acid), abundant in many plants, particularly etiolated seedlings; aspartic acid; aminosuccinic acid, involved in the biosynthesis of purines; cysteine, which contains sulphur; cystine or dicysteine (in hair and insulin); 3,5-di-iodotyrosine (in thyroid); glutamic acid (a component of the folic acid vitamins); glutamine (free in animals and plants, e.g. sugar-beet); glycine (aminoacetic acid); histidine; δ-hydroxylysine (in gelatin); hydroxyproline (in gelatin); leucine (α-aminocaproic acid); isoleucine; lysine; methionine (contains a sulphur atom); 3-monoiodotyrosine (in thyroid); phenylalanine; proline; serine (in phosphoproteins such as casein); threonine (in casein); thyroxin (the iodine-containing thyroid-hormone); 3,5,3′-triiodothyronine (in thyroid); tryptophan; tyrosine and valine.
Amino acids found free and not occurring in proteins
Following the pioneer research of Fowden, several hundred of these amino acids have been characterized and only a few (e.g. γ-aminobutyric acid, α-aminoadipic acid, pipecolic acid and δ-acetylornithine) are of wide occurrence. Seeds or fleshy organs of plants are the principal sites for the accumulation of these compounds where they may serve as a nitrogen reserve. Other examples in this class are: β-alanine (β-aminopropionic acid); citrulline (an intermediate in the cycle of urea synthesis); creatine; ergothioneine (a sulphur-containing constituent of some animal tissues and of ergot); and taurine (a component of bile acids). A number have teratogenic properties (see Chapter 39). 3-N-Oxalyl-L-2, 3-diaminopropanoic acid is a neurotoxin present in the seeds of Lathyrus sativus, the grass pea; it is the causal agent of human neurolathyrism, an irreversible paralysis of the lower limbs, which occurs in some drought prone areas of Ethiopia, India and Bangladesh (see Yu-Haey Kuo et al., Phytochemistry, 1994, 35, 911 for references and biosynthetic studies). Other non-protein amino acids provide plant protection from insects.
Non-protein amino acids, considered oddities of plant biosynthesis 40 years ago, now constitute a group receiving increasing attention because of their possible physiological and ecological significance.
Biosynthesis of amino acids
As amino acids are also the precursors of some secondary metabolites, their biosyntheses will be considered below. They arise at various levels of the glycolytic and TCA systems.
Nitrogen appears to enter the metabolism of the organism by reductive amination of α-keto acids; pyruvic, oxalacetic and α-ketoglutaric acids give alanine, aspartic acid and glutamic acid, respectively (Fig. 18.12).
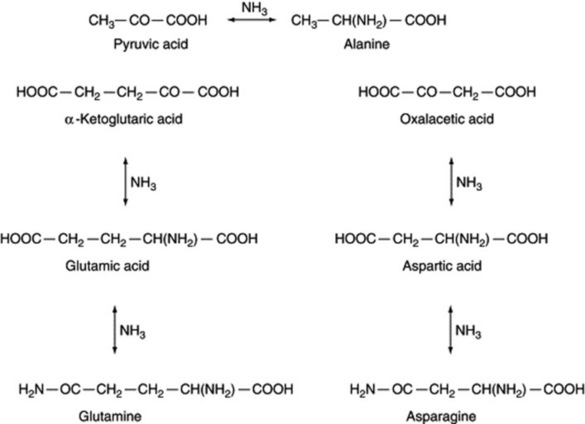
By transamination reactions with other appropriate acids, alanine, aspartic acid and glutamic acid serve as α-amino donors in the formation of other amino acids. The general transamination reaction may be written:
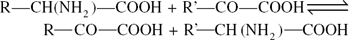
Glutamic acid, in particular, appears to be a central product in amino-acid metabolism and glutamic acid dehydrogenase has been reported in a number of plant tissues. The enzyme functions in conjunction with NAD:
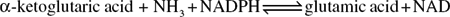
That the nitrogen of ammonia first appears in the dicarboxylic amino acids and is later transferred to other nitrogen compounds was demonstrated in some of the earliest plant biochemistry experiments (around 1940), which utilized 15N.
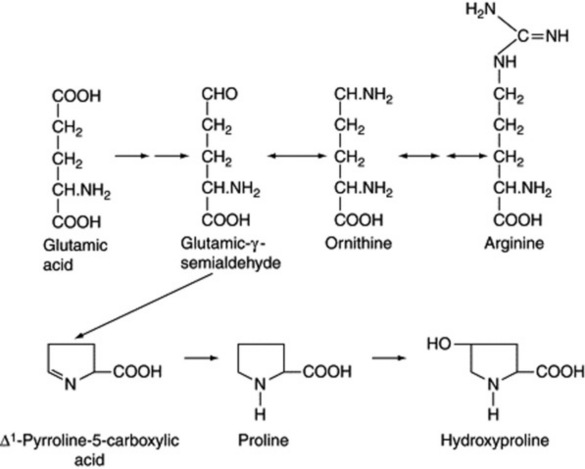
Proline, hydroxyproline, ornithine and arginine
These amino acids are of importance in the secondary metabolism of some plants in that they are precursors of a number of alkaloids. They are metabolically connected to glutamic acid (Fig. 18.13) and their formation in plant cells is complex in that the reactions are strictly compartmentalized. The enzymes involved have been characterized, and for the formation of ornithine it is the N-acetyl derivatives which are involved. Arginine appears to be synthesized from ornithine in all organisms via the reactions of the urea cycle. (For a review giving the role of the mitochondria, cytoplasm and plastids in these reactions, see P. D. Shargool et al., Phytochemistry, 1988, 27, 1571.)
Serine and glycine
Together with cysteine and cystine, these amino acids arise at the triose level of metabolism. Preparations from rat liver use the pathway indicated in Fig. 18.14 for the formation of serine.
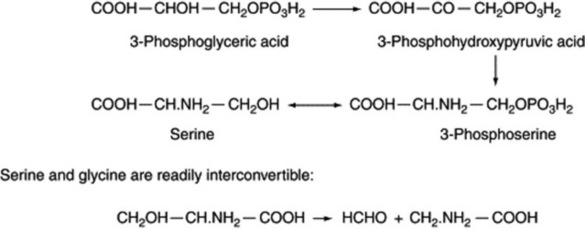
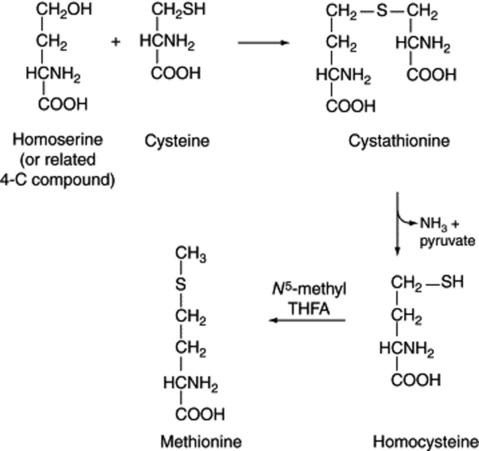
In animal tissues it has been shown that tetrahydrofolic acid (THFA) is responsible for the removal of the hydroxymethyl group of serine to form hydroxymethyltetrafolic acid. As this compound serves as a source of formate and methyl groups in many reactions, the β-carbon of serine may be their original source; this applies to the formation of methionine (Fig. 18.15), itself an important methyl donor in plant biochemistry.
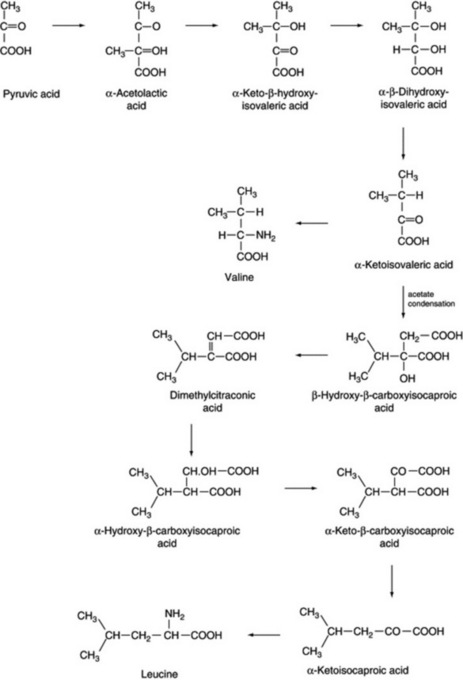
Alanine, valine and leucine
Studies with microorganisms and yeasts have shown these amino acids to be derived from pyruvate. There is evidence that α-ketoisovaleric acid is aminated to form valine and that it can also condense with acetate to form an intermediate which on decarboxylation and amination affords leucine (Fig. 18.16).
Isoleucine
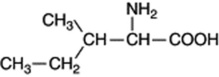
is formed by a similar series of reactions to valine but commencing with α-aceto-α-hydroxypropionic acid instead of α-acetolactic acid.
Lysine
Lysine, H2N–(CH2)4–CH(NH2)–COOH, is derived, in plants, from aspartate involving a pathway utilizing 2,3-dihydropicolinic acid and diaminopimelic acid. It is the precursor of some alkaloids of Nicotiana, Lupinus and Punica.
For a review of the biosynthesis and metabolism of aspartate-derived amino acids (lysine, threonine, methionine, S-adenosyl methionine) see R. A. Azevedo et al., Phytochemistry, 1997, 46, 395.
PEPTIDES AND PROTEINS
The term ‘peptide’ includes a wide range of compounds varying from low to very high molecular weights and showing marked differences in physical, chemical and pharmacological properties. The lowest members are derived from only two molecules of amino acid, but higher members have many amino-acid units and form either peptides, simple proteins (albumins, globulins, prolamines, glutalins, etc.) or more complex proteins, conjugated proteins, in which other groupings form part of the molecule—for example, carbohydrate in mucoproteins, the very complex chlorophyll molecule in the protein of chloroplasts, phosphorus-containing proteins such as casein, nucleoproteins, in which proteins are combined with nucleic acid, and the lipoproteins of the cytoplasm, in which protein is combined with lipids. Among such substances with relatively low molecular weight are some antibiotics which have a cyclic polypeptide structure (e.g. gramicidin, bacitracin and polymyxin); peptide hormones such as oxytocin and vasopressin from the posterior pituitary gland; and glutathione, which is found in nearly all living cells.
All these more or less complex compounds have two or more molecules of amino acid united by a peptide linkage which results from the elimination of water, an OH coming from one amino acid and an H from the other.
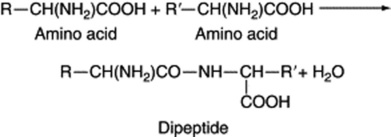
A dipeptide of the Sapindaceous plant Blighia sapida has hypoglycaemic properties and although more complex, penicillin also has a dipeptide structure. Tripeptides have three amino-acid components and polypeptides from ten upwards. Peptides are usually defined as protein-like substances having molecular weights below 10 000. In typical proteins the molecular weight is higher, ranging from about 30 000 to 50 000 in the relatively simple prolamines and glutelins and reaching very high values, sometimes several million, in the complex proteins such as those in sheep’s wool.
Protein synthesis takes place in association with the ribosomes, which are small bodies found in the cytoplasm and particularly in the endoplasmic reticulum area (see Fig. 18.1). The amino acids are brought to the ribosomes associated with a transfer-RNA molecule and by the action of the ribosomes, using a sequence dictated by a particular messenger-RNA molecule, are linked to form the peptide chains of the particular protein. Although not directly relevant to most pharmacognostical studies, the story of the nucleic acids and their vital role in the control of cell metabolism is a fascinating one which it is suggested students study from a standard work on biochemistry.
ISOPRENOID COMPOUNDS
Studies on the pyrogenic decomposition of rubber led workers in the latter half of the nineteenth century to believe that isoprene could be regarded as a fundamental building block for this material. As a result of the extensive pioneering investigations into plant terpene structures, Ruzicka published in 1953 his ‘biogenetic isoprene rule’, which indicated that the apposition of isoprenoid units could be used to explain not only the formation of rubber and the monoterpenes, but also many other natural products, including some, such as sterols and triterpenes, with complex constitutions. The value of the rule lay in its broad unifying concept, which allowed the postulation of a rational sequence of events which might occur in the biogenesis of these otherwise unrelated compounds. Examples of various structures to which the rule can be applied are indicated in Fig. 18.17.
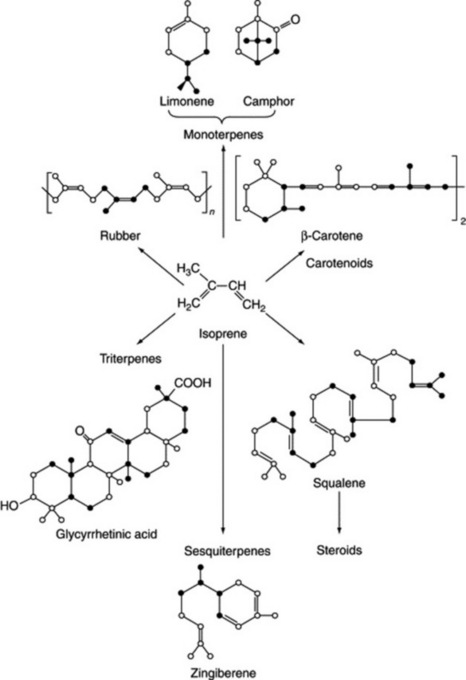
The task set biochemists was to investigate the validity of the rule, and the work on this subject constitutes a brilliant example of modern biochemistry; however, as will be seen below, this chapter of research is still unfinished. By 1951 it had been established that acetic acid was intimately involved in the synthesis of cholesterol, squalene, yeast sterols and rubber. The use of methyl- and carboxyl-labelled acetic acid with animal tissues indicated that the methyl and carboxyl carbons alternated in the skeleton of cholesterol or squalene and that the lateral carbon atoms all arose from the methyl group of acetic acid.The discovery, in 1950, of acetyl-coenzyme A, the so-called ‘active acetate’, gave further support to the role of acetate in biosynthetic processes.
The mevalonic acid pathway
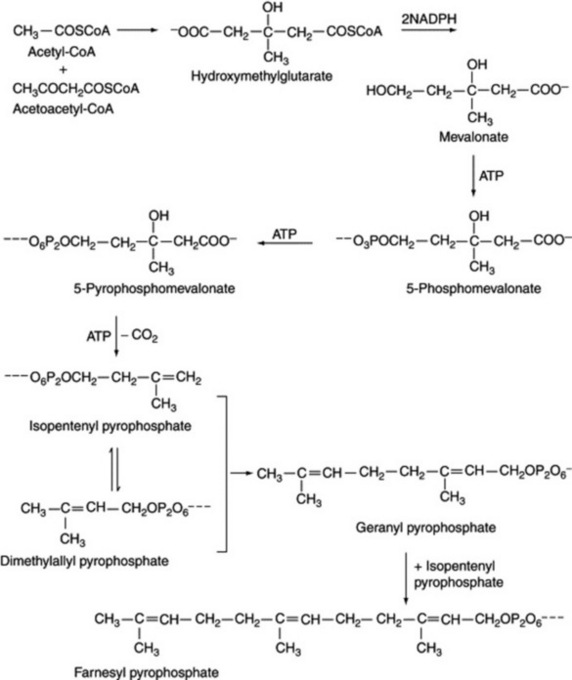
The next major advance in the elucidation of the isoprenoid biosynthetic route was the discovery in 1956 of mevalonic acid and the demonstration of its incorporation, by living tissues, into those compounds to which the isoprene rule applied. Mevalonic acid (3,5-dihydroxy-3-methylvaleric acid) is a C6 acid and, as such, is not the ‘active isoprene’ unit which forms the basic building block of the isoprenoid compounds. During the next four years, by research involving the use of tracer techniques, inhibitor studies, cell-free extracts, partition chromatography and ionophoresis as well as synthetic organic chemistry, it was established that the C5 compound for which biochemists had been seeking so long was isopentenyl pyrophosphate; it is derived from mevalonic acid pyrophosphate by decarboxylation and dehydration. Isoprenoid synthesis then proceeds by the condensation of isopentenyl pyrophosphate with the isomeric dimethylallyl pyrophosphate to yield geranyl pyrophosphate. Further C5 units are added by the addition of more isopentenyl pyrophosphate. These preliminary stages in the biosynthesis of isoprenoid compounds are shown in Fig. 18.18.
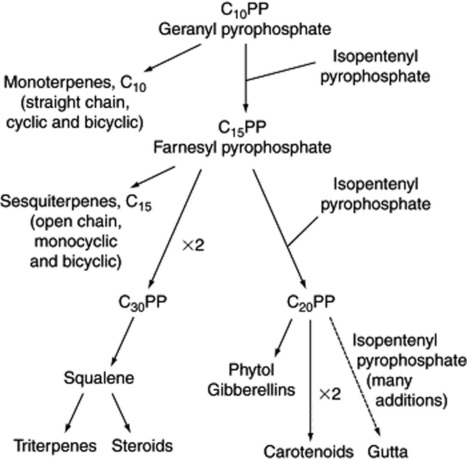
From geranyl and farnesyl pyrophosphates various structures can be built up (see Fig. 18.19).
Studies, particularly by Cornforth and Popják, involving the use of stereospecifically 3H- and 14C-labelled mevalonic acid, have demonstrated the stereochemical mechanism of the initial stages of isoprenoid formation. Only the (R)-form of mevalonic acid gives rise to the terpenoids, the (S)-form appearing to be metabolically inactive. In the formation of isopentenyl pyrophosphate, the elimination is trans and the elimination of the proton in the isomerization to the dimethylallyl pyrophosphate is also stereospecific (Fig. 18.20A).
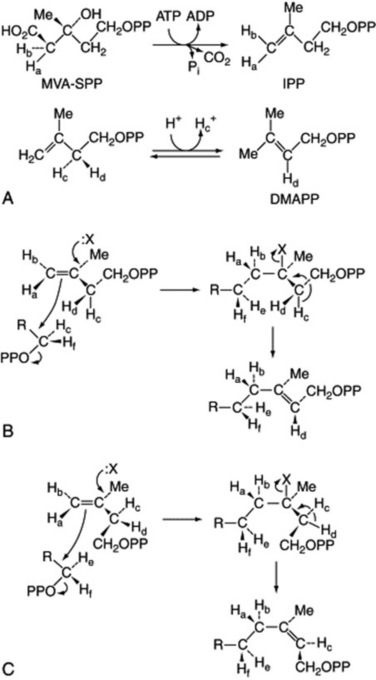
Fig. 18.20 Stereospecific reactions in terpenoid biogenesis. A, Formation from mevalonic acid (MVA) of isopentenyl pyrophosphate (IPP) by trans elimination; isomerization to dimethylallyl pyrophosphate (DMAPP). B, Association of the two 5-C units with trans elimination of hydrogen. C, As B but involving formation of cis double bonds as found in rubber.
In the subsequent additions of the C5 isopentenyl pyrophosphate units to form the terpenoids the elimination of hydrogen is trans. Figure 18.20B shows the stereochemistry of the addition of one isopentenyl pyrophosphate unit. In the biogenesis of rubber, however, the hydrogen elimination produces a cis double bond (Fig. 18.20C).
It is considered that a simple change in orientation of the isopentenyl pyrophosphate on the enzyme surface could produce this change without altering the reaction mechanism. In neither rubber nor gutta are hybrid molecules containing both types of bond detectable. The first direct evidence for the presence of isopentenyl diphosphate isomerase in rubber latex was reported in 1996 (T. Koyama et al., Phytochemistry, 1996, 43, 769).
In recent years the enzymology of the isoprenoid pathway has been extensively studied and for details the reader is referred to a standard text on plant biochemistry. One key regulatory enzyme is hydroxymethylglutaryl-CoA reductase (EC 1.1.1.34, mevalonate kinase); it has been extensively studied in animals and more recently in plants. As with many enzymes the situation is complicated by the existence of more than one species of enzyme and a plant may possess multiple forms each having a separate subcellular location associated with the biosynthesis of different classes of terpenoids. For a review of the functions and properties of the important isomerase enzyme isopentenyl diphosphate isomerase see ‘Further reading’.
It should be noted that some metabolites of mixed biogenetic origin involve the mevalonic acid pathway; prenylation for example is common, with C5, C10 and C15 units associated with flavonoids, coumarins, benzoquinones, cannabinoids, alkaloids, etc.
The validity of the mevalonate pathway in the formation of all the major groups noted in Fig. 18.19 has been shown, and until recently no other biosynthetic route to isoprenoids had been discovered.
The 1-deoxy-D-xylulose (triose/pyruvate) pathway
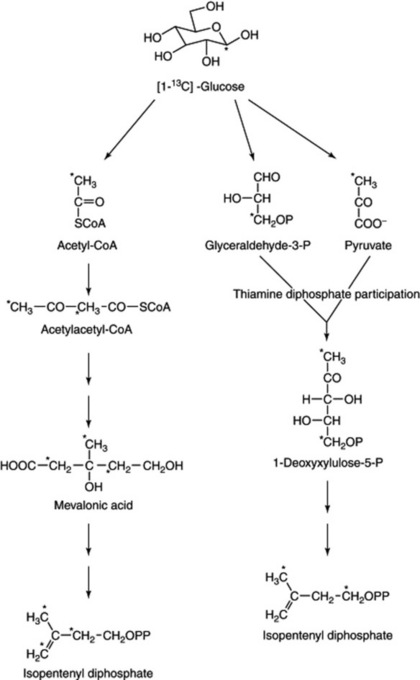
Following its discovery in 1956 mevalonic acid came to be considered the essential precursor for all isoprenoid syntheses. However, in 1993 M. Rohmer et al., (Biochem. J., 1993, 295, 517) showed that a non-mevalonate pathway existed for the formation of hopane-type triterpenoids in bacteria. The novel putative precursor was identified as 1-deoxy-D-xylulose-5-phosphate, formed from glucose via condensation of pyruvate and glyceraldehyde-3-phosphate. Subsequent steps including a skeletal rearrangement afford isopentenyl pyrophosphate—the same methyl-branched isoprenoid building block as formed by the MVA route.
It was soon demonstrated that this novel route to IPP was also operative in the formation of monoterpenes (Mentha piperita, Thymus vulgaris), diterpenes (Ginkgo biloba, Taxus chinensis) and carotenoids (Daucus carota). This raised the question of to what extent the two pathways co-existed in the plant and it was hypothesized that the classical acetate/mevalonate pathway was a feature of cytoplasmic reactions whereas the triose/pyruvate sequence was a characteristic of the plastids. This did not exclude either the movement of plastid-synthesized IPP and DMAPP from the organelle to the cytoplasm or the translocation of a suitable C5-acceptor to the plastid. Evidence accumulating indicates a cooperative involvement of both pathways. Indeed recent work on the biosynthesis of the isoprene units of chamomile sesquiterpenes (K.-P. Adam and J. Zapp, Phytochemistry, 1998, 48, 953) has shown that for the three C5 units of both bisaboloxide A and chamazulene, two were mainly formed by the non-mevalonate pathway and the third was of mixed origin.
The deoxyxylulose (DOX) pathway has helped explain the previously reported rather poor incorporations of MVA into certain isoprenoids. Thus V. Stanjek et al. (Phytochemistry, 1999, 50, 1141) have obtained a good incorporation of labelled deoxy-D-xylulose into the prenylated segment of furanocoumarins of Apium graveolens leaves, suggesting this to be the preferred intermediate.
Fig. 18.21 illustrates how [1-13C]-glucose, when fed to plants, can be used to differentiate IPP and subsequent metabolites, formed either by the MVA pathway or the DOX route.
SECONDARY METABOLITES
As indicated earlier in Fig. 18.2, the basic metabolic pathways constitute the origins of secondary plant metabolism and give rise to a vast array of compounds; some of these are responsible for the characteristic odours, pungencies and colours of plants, others give a particular plant its culinary, medicinal or poisonous virtues and by far the greatest number are, on current knowledge, of obscure value to the plant (and to the human race). However, a number of modern authors suggest that secondary metabolites, rather than constituting waste products of metabolism, are biosynthesized to aid the plant’s survival. Notwithstanding, it may be that in some instances the purpose for which these compounds were produced no longer exists but the biosynthetic pathway has survived. For an interesting discussion and references, see E. J. Buenz and S. J. Schepple, J. Ethnopharmacol, 2007, 114, 279. The possible functions in the plant of one large group of secondary metabolites, alkaloids, are discussed in Chapter 26.
Recently, considerable attention has been directed to the possible ecological implications of secondary metabolites not only in relation to plant–plant interaction but also concerning the interrelationship of plants and animals. Various insects sequester specific alkaloids, iridoids, lactones and flavonoids which serve as defensive agents or are converted to male pheromones. The literature concerning chemical ecology is regularly reviewed and the volatile isoprenoids that control insect behaviour and development have been reported on (J. A. Pickett, Nat. Prod. Rep., 1999, 16, 39). The enzymology associated with secondary metabolism is now receiving considerable attention and, with respect to alkaloid formation, a number of enzymes associated with the biosynthesis of the tropane, isoquinoline and indole groups have been prepared. The biosynthetic origins of those metabolites of medicinal interest are considered in more detail in Part 5, which is arranged principally according to biogenetic groups.
Although a number of the biogenetic groups are characterized by particular skeletal structures, the actual chemical properties of particular compounds are determined by the acquisition of functional groups. Thus, terpenes may occur as alcohols (menthol), ethers (cineole), ketones (carvone), etc., and as such have similar chemical properties to non-terpenoid compounds possessing the same group; aldehydes, as an example of a functional group, may be of aliphatic origin (citronellal), aromatic (cinnamic aldehyde), steroidal (some cardioactive glycosides); and resulting from the introduction of a heterocyclic system one biogenetic group may possess some of the chemical properties of another (e.g. steroidal alkaloids).
A particular group of compounds may also involve different biogenetic entities; thus, the complex indole alkaloids contain moieties derived from both the shikimate and isoprenoid pathways. In contrast, the same structure, as it occurs in different compounds, may arise from different pathways, as has been previously indicated with the formation of aromatic systems.
Stress compounds
These are compounds which accumulate in the plant to a higher than normal level as a result of some form of injury, or disturbance to the metabolism; they may be products of either primary or secondary metabolism. Common reactions involved in their formation are the polymerization, oxidation or hydrolysis of naturally occurring substances; many however, are entirely secondary in their formation. A number of environmental and biological factors promote the synthesis of stress compounds and these include mechanical wounding of the plant, exposure to frost, ultraviolet irradiation, dehydration, treatment with chemicals, and microbial infection (see phytoalexins below). The production of such compounds has also been observed in cell cultures subjected to antibiotic treatment and in cells immobilized or brought into contact with calcium alginate. Examples of the latter include the formation of acridone alkaloid epoxides by Ruta graveolens, and the increased production of echinatin and the novel formation of a prenylated compound by Glycyrrhiza echinata cultures.
Stress compounds are of pharmaceutical interest in that they may be involved in various crude drugs formed pathologically (e.g. some gums and oleoresins) and potential drugs (gossypol); they are implicated in the toxicity of some diseased foodstuffs and they play a role in the defensive mechanism of the plant. In the latter area, the phytoalexins have received considerable attention in recent years and can be regarded as antifungal compounds synthesized by a plant in greatly increased amounts after infection. The antifungal isoflavonoid pterocarpans produced by many species of the Leguminosae are well known, see ‘Spiny restharrow’. Other phytoalexins produced in the same family are hydroxyflavanones, stilbenoids, benzofurans, chromones and furanoacetylenes. Sesquiterpene phytoalexins have been isolated from infected Ulmus glabra and Gossypium. In the vine (Vitis vinifera) the fungus Botrytis cinerea acts as an elicitor for the production of the stilbenes resveratrol (q.v.) and pterostilbene.
Chemically, stress compounds are, in general, of extreme variability and include phenols, resins, carbohydrates, hydroxycinnamic acid derivatives, coumarins, bicyclic sesquiterpenes, triterpenes and steroidal compounds. For the promotion of stress compounds in cell cultures see Chapter 13 and Table 13.1.
Facchini PJ, Huner-Allnach KL, Tari LW. Plant aromatic L-amino acid decarboxylases: evolution, biochemistry, regulation and metabolic engineering applications. Phytochemistry. 2000;54(2):121-138.
Grayson DH. Monoterpenoids (a review covering mid-1997 to mid-1999). Natural Product Reports. 2000;17(4):385.
Knaggs AR. 2000 The biosynthesis of shikimate metabolites. Natural Product Reports. 1999;16(4):525-560. 17(3): 269–292
Kruger NT, Hill SA, Ratcliffe RG, editors. Regulation of primary metabolic pathways in plants. Kluwer, Dordrecht, Netherlands, 1999.
Ramos-Valdivia AC, Van der Heijden R, Verpoorte R. Isopentenyl diphosphate isomerase: a core enzyme in isoprenoid biosynthesis. Natural Product Reports. 1997;14(6):591-604.
Rohmer M. The discovery of a mevalonate-independent pathway for isoprenoid biosynthesis in bacteria, algae and higher plants. Natural Product Reports. 1999;16(5):565-574.
Seigler DS. Plant secondary metabolism. Kluwer, Dordrecht, Netherlands, 1998.
Singh BK, editor. Plant amino acids—biochemistry and biotechnology. New York: Marcel Dekker Inc, 1999.
Wink M, editor. Biochemistry of plant secondary metabolism. Sheffield Academic Press, 1999.