Cerebrovascular disease and dementia
Stroke
Strokes are a major cause of morbidity and mortality, particularly in older people. They present as a transient or permanent neurological disturbance caused by ischaemic infarction or haemorrhagic disruption of neuronal pathways in the brain.
Ischaemic strokes and transient ischaemic attacks
Ischaemic strokes and transient ischaemic attacks (TIAs) account for about 85% of events. Cerebral infarction can result from intracerebral arterial thrombosis or from emboli travelling to the cerebral arteries, typically from an unstable atheromatous plaque in an internal carotid artery (see Ch. 5) or from the heart. The extent and duration of the resulting functional deficit following a stroke are very variable.
Transient (cerebral) ischaemic attacks arise from small cerebral arterial emboli that disperse rapidly. They produce short-lived neurological signs and symptoms but leave no functional deficit 24 h later. A completed stroke results from more severe cerebral ischaemia, which produces cerebral infarction. The neurological disturbance persists for more than 24 h, and frequently there is some permanent loss of function. Following a TIA there is a 5% risk of a completed stroke in the subsequent 24 h, and a 30% risk of a completed stroke in the subsequent 5 years. However, if there is a significant carotid artery stenosis at the time of the TIA the risk of completed stroke is 30% in the first month.
Prevention and treatment
Current treatments produce only a modest limitation of the neurological deficit in acute stroke. Most management is directed to:
 primary prevention of a first event (ischaemic or haemorrhagic stroke),
primary prevention of a first event (ischaemic or haemorrhagic stroke),
prevention of recurrence of stroke or of other cardiovascular events,
About one-third of strokes are recurrent. The recurrence rate for ischaemic stroke is about 3–7% per year for individuals who are in sinus rhythm and about 12% per year for those in atrial fibrillation.
Primary prevention of ischaemic stroke
Blood pressure reduction. Hypertension is the single most powerful predictor of stroke. Pooled trial results indicate that a reduction in diastolic blood pressure by 5–6 mmHg reduces the risk of stroke by about 40% (Ch. 6). For isolated systolic hypertension, a similar reduction in risk has been shown after an average 11 mmHg reduction in systolic blood pressure.
Smoking cessation. The risk of ischaemic stroke is reduced by up to 40% by 2–5 years after smoking cessation. This is probably due to slower progression of arterial atherothrombotic disease.
Reduced platelet aggregation. Low-dose aspirin has not been shown to prevent a first ischaemic stroke when taken by healthy individuals who are in sinus rhythm. For people with atrial fibrillation, aspirin produces a modest reduction in the risk of a first ischaemic stroke by about one-quarter. However, aspirin is much less effective than warfarin (or other oral anticoagulants) for stroke reduction in atrial fibrillation (Ch. 8).
Inhibition of blood clotting. Oral anticoagulation (Chs 8 and 11) in people with atrial fibrillation reduces the risk of a first ischaemic stroke by 70%. Warfarin, at a dosage giving an international normalised ratio (INR) of 2–3, or one of the newer anticoagulants such as apixaban, dabigatran or rivaroxaban, can be used. There is no advantage of warfarin over aspirin for people in sinus rhythm. Warfarin reduces the risk of stroke following myocardial infarction if there is intracardiac clot associated with an akinetic area of the left ventricular wall.
Cholesterol reduction. Reduction of a raised plasma cholesterol with a statin (Ch. 48) produces a 25% reduction in the risk of a first stroke, although much of the evidence for this effect derives from trials in people who already have clinical evidence of vascular disease or who have diabetes mellitus.
Carotid endarterectomy or stenting. This is sometimes recommended for asymptomatic carotid artery disease when the stenosis exceeds 60%, but the annual risk of an ischaemic stroke is low in this situation.
Treatment of acute ischaemic stroke
Fibrinolytic therapy with recombinant tissue plasminogen activator (rt-PA, alteplase; Ch. 11) can reduce the long-term neurological deficit after an ischaemic stroke. If treatment is started within 3 h of the onset of symptoms, thrombolysis reduces the risk of death or dependency at 3 months, with greater benefit the earlier that treatment is given. Overall, 7% more people who are given thrombolysis after ischaemic stroke will have no or minimal disability 3 months later. However, more than half of those who are treated with an intravenous fibrinolytic drug do not have complete or near-complete recovery. There is an increased risk of intracerebral haemorrhage after thrombolysis, particularly in those with a blood pressure above 185/110 mmHg or when treatment is delayed, which can outweigh the benefit from neuronal salvage. About 6% of people who are treated will have a symptomatic intracranial haemorrhage.
Meta-analysis of several studies shows that intravenous alteplase is moderately effective from 3 h until 4.5 h after the event. Intra-arterial alteplase is effective in large ischaemic strokes up to 6 h after the onset of symptoms.
Indications and usual contraindications for thrombolytic therapy in acute stroke are shown in Box 9.1. Anticoagulants or antiplatelet drugs should not be given for 24 h after thrombolysis.
There have been many recent advances in treatment of acute ischaemic stroke. These include the use of mechanical thrombectomy devices to reduce the size of the thrombus, and ultrasonography to improve penetration of the fibrinolytic drug into the thrombus. However, a major advance may be the use of sensitive brain imaging to identify those who have potentially recoverable neurological injury from those with irreversible infarction.
Secondary prevention of recurrent ischaemic stroke
Many treatments are similar to those used for primary prevention of ischaemic stroke (see above).
Blood pressure reduction. Lowering blood pressure after a stroke will reduce the risk of recurrence by 30–40%. There is considerable reluctance to reduce blood pressure in the first few days after a stroke, because of concern that cerebral perfusion pressure may fall too much if the normal cerebral arterial autoregulation has been disturbed by the stroke. However, there is some evidence that early treatment (after the first 24 h) may be advantageous.
Reduced platelet aggregation. Low-dose aspirin with dipyridamole can be used following a TIA for people who are in sinus rhythm. The combination reduces the risk of a subsequent non-fatal stroke by about 35%, and is more effective than aspirin alone. This combination is recommended for up to 2 years after a TIA, when the risk of recurrent stroke is highest, after which aspirin is often used alone. Clopidogrel alone (Ch. 11) is as effective as aspirin and dipyridamole after ischaemic stroke, and is now the preferred treatment. By contrast, the combination of aspirin and clopidogrel is no more effective than aspirin alone for long-term prevention (unlike in acute coronary syndromes; see Ch. 5), and increases the risk of serious bleeds. However, there may be some benefit from the combination for short-term treatment (7–30 days) immediately after a stroke or TIA in those at high risk of recurrence. Warfarin has no role in preventing recurrent stroke in people who are in sinus rhythm.
Inhibition of blood clotting. After a first stroke in people with atrial fibrillation, oral anticoagulation reduces the risk of a further stroke by two-thirds. In contrast, aspirin has no protective effect in this situation (see Ch. 11).
Cholesterol reduction. Cholesterol reduction with a statin is effective in secondary prevention of ischaemic stroke, reducing recurrent stroke by 21% for every 1 mmol⋅L−1 reduction in low-density lipoprotein (LDL) cholesterol. However, the greatest advantage of cholesterol reduction in this situation is in the prevention of ischaemic cardiac events, since coronary artery disease often coexists with atheromatous cerebrovascular disease.
Carotid endarterectomy or stenting. This reduces the risk of recurrent stroke if there have already been transient focal neurological symptoms in the cerebral territory served by a diseased carotid artery. If the stenosis is ≥70% of the vessel diameter (but without near total occlusion), then endarterectomy reduces the risk of recurrent stroke by about two-thirds over the subsequent 2 years (despite a perioperative risk of stroke or death of 3–5%). There is no benefit from surgery if the occlusion is less than 50%, and only marginal benefit if the occlusion is between 50 and 69%, unless the surgery is carried out soon after the event (usually within 2 weeks), when the risk of recurrence is highest.
Secondary prevention of recurrent haemorrhagic stroke
Blood pressure reduction. Lowering blood pressure after a haemorrhagic stroke will reduce the risk of recurrence by up to 40%. The reduction in risk from treating hypertension is greater than for ischaemic stroke, and even lowering a ‘normal’ blood pressure may be effective. As for ischaemic stroke, treatment is usually delayed to allow return of autoregulation of cerebral blood flow, unless the blood pressure exceeds 185/105 mmHg.
Subarachnoid haemorrhage
Most subarachnoid haemorrhages are caused by rupture of a saccular (or berry) aneurysm on an intracranial artery, usually on or close to the circle of Willis. These aneurysms are acquired during life and the cause is unknown, although there is an association with hypertension and conditions that increase cerebral blood flow such as arteriovenous malformations. About 5% of all strokes are caused by subarachnoid haemorrhage. Sudden onset of severe occipital headache is the most common presenting symptom, but focal neurological signs or progressive confusion and impaired consciousness can occur. Rebleeding is a significant cause of disability and death, and early surgical intervention in survivors of the initial bleed reduces this risk. A more common cause of permanent neurological disability or later death is delayed cerebral ischaemia. This is produced by cerebral vasospasm, which develops in about 25% of cases, usually at least 3 days after the haemorrhage. The mechanism is poorly understood, but involves activation of voltage-dependent L-type Ca2+ channels in intracranial arteries. It presents with confusion, decreased consciousness and new focal neurological deficit.
Drugs for subarachnoid haemorrhage
Nimodipine is a dihydropyridine L-type calcium channel blocker (for mechanism of action see Ch. 5) that is an arterial vasodilator with some selectivity for cerebral arteries. It reduces the risk of vasospasm following subarachnoid haemorrhage, but probably produces most of its benefits by protecting ischaemic neurons from Ca2+ overload. There is a theoretical risk that cerebral vasodilation may actually facilitate further bleeding, but this does not appear to be a problem in practice.
Management of subarachnoid haemorrhage
Initial treatment of subarachnoid haemorrhage aims to reduce ischaemic cerebral damage. Nimodipine is usually given intravenously immediately after the event, followed by oral dosing for a total of 5–10 days. Intravenous fluids are given to avoid hypotension. The optimum blood pressure in the early period after the haemorrhage is not known, but hypotension should be avoided and blood pressure lowered modestly in those who present with significant hypertension. Dexamethasone (Ch. 44) may be used to reduce cerebral oedema.
The definitive management of subarachnoid haemorrhage is surgical, with endovascular coil occlusion of the aneurysm or clipping of the neck of the aneurysm that produced the bleeding. In the last 20 years early surgical intervention, combined with medical therapy, has reduced mortality from 20% to about 5–10%.
Dementia
Dementia usually begins with forgetfulness and is characterised by disorientation in unfamiliar surroundings, variable mood, restlessness and poor sleep. Deterioration in social behaviour with self-neglect often follows, and may be accompanied by personality change with loss of inhibition. Most dementia results from Alzheimer's disease or from cerebrovascular disease (multi-infarct dementia), but there are other causes (Box 9.2). Memory impairment in dementia tends to be associated with bilateral hippocampal damage.

Alzheimer's disease
Alzheimer's disease is the commonest cause of dementia in people over the age of 65 years. About 10% of people over the age of 65 and about 30% of those over the age of 85 have some signs of Alzheimer's disease. The onset of symptoms is gradual, with progressive deterioration, unlike vascular dementia. It is a neurodegenerative disorder that begins pathologically 20–30 years before the clinical onset.
The cause of Alzheimer's disease is unknown, but there are several distinct factors associated with the disease.
Amyloid protein. β-Amyloid is deposited in the medial temporal lobe and cerebral cortex of people with Alzheimer's disease as senile plaques. The initiating factor in Alzheimer's disease may be an imbalance between the production and clearance of β-amyloid in the brain, leading to toxic effects on neuronal synapses. Deposition of β-amyloid may be the driver for hyperphosphorylation of tau protein (found in axons, where it promotes microtubule assembly and vesicle transport), which aggregates into neurofibrillary tangles that are characteristic of Alzheimer's disease.
Genetic predisposition. This accounts for about 70% of the risk. Mutations of the apolipoprotein E ε4 allele (APOE ε4; essential for amyloid β clearance) confer a higher risk of late-onset Alzheimer's disease, while mutations in genes coding for amyloid precursor protein and presenilin 1 and 2 explain the rare familial disease.
Inflammatory factors. Activated microglia and reactive astrocytes surround the senile plaques, and there is local increase in pro-inflammatory mediators.
Glutamate excitotoxicity. Amyloid deposits may promote neuronal damage by increasing neuronal release of glutamate. This acts at N-methyl-D-aspartate (NMDA) receptors to generate glutamate-induced excitotoxicity of cholinergic neurons. Hyperactivity of glutamatergic neurons is a common finding in Alzheimer's disease.
Oxidative stress. There is evidence for excessive free radical production in Alzheimer's disease. Increased oxidative stress may contribute to the condition by producing vascular damage, reducing β-amyloid clearance and promoting metabolic derangement in neurons.
Loss of cholinergic neurotransmission. There is a marked loss of acetylcholine neurotransmitter synthesis in the cerebral cortex and the hippocampus, particularly affecting the areas involved in cognition and in memory that are impaired in Alzheimer's disease. There is also loss of cholinergic neurons. Activity at both muscarinic and nicotinic receptors is reduced. Depletion of other neurotransmitters is a late and inconsistent finding.
Drugs for Alzheimer's disease
Mechanisms of action and effects: The basis of the cholinergic hypothesis of Alzheimer's disease is that loss of cholinergic neurons in the basal forebrain nuclei results in abnormal function at cholinergic terminals in the hippocampus and neocortex, which are involved in memory and cognition. Anticholinesterases increase cholinergic transmission in the brain by inhibition of acetylcholinesterase in the synaptic cleft (Ch. 4).
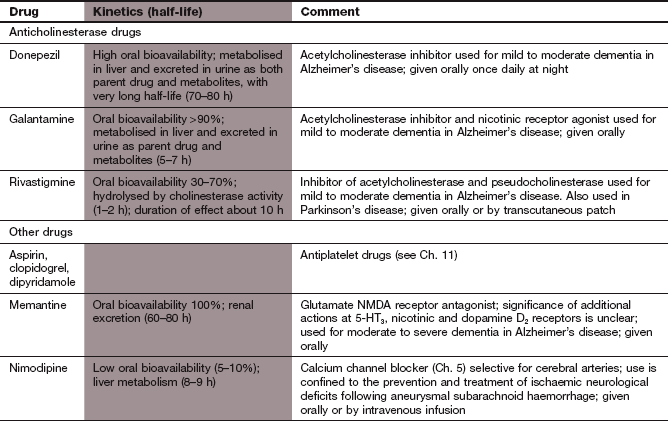
Donepezil is a reversible inhibitor of acetylcholinesterase with a high degree of selectivity for the central nervous system (CNS).
Galantamine is a reversible competitive inhibitor of acetylcholinesterase that also has agonist activity at presynaptic nicotinic receptors by allosterically enhancing the receptor response to acetylcholine.
Rivastigmine is a slowly reversible inhibitor of acetylcholinesterase with selectivity for the CNS, and also inhibits pseudocholinesterase (butyrylcholinesterase) present in tissues and plasma.
Pharmacokinetics: Donepezil, galantamine and rivastigmine are well absorbed from the gut. Donepezil and galantamine are metabolised in the liver, and galantamine has a half-life of 5–7 h while that of donepezil is very long, at 70–80 h. Rivastigmine is rapidly inactivated by cholinesterase-mediated hydrolysis and has a short half-life of 1–2 h.
NMDA receptor antagonists
Mechanism of action and effects: Memantine is a derivative of the antiviral drug amantadine (Chs 24 and 51), and is a non-competitive antagonist at glutamate NMDA receptors. This may prevent glutamate-induced excitotoxicity (by limiting long-lasting influx of Ca2+ into neurons), but without interfering with the actions of glutamate that are involved in memory and learning. Memantine is also a 5-HT3 receptor antagonist and a non-competitive nicotinic receptor antagonist, but the significance of these actions for the treatment of dementia is unknown. It can be taken together with an anticholinesterase.
Treatment of Alzheimer's disease
In the UK, drug treatment for Alzheimer's disease is started only for people who have a Mini-Mental State Examination (MMSE) score of 10–20 points (moderate dementia). The diagnosis of Alzheimer's disease should be first confirmed in a specialist memory clinic.
Cholinesterase inhibitors produce modest improvement in symptoms, and a delay in the decline of cognitive function and memory, in up to 40% of those who are treated. Efficacy should be assessed after 2–4 months of treatment at a suitable dose. Treatment should be continued only if the ‘global, functional and behavioural condition remains at a level where the drug is considered to be having a worthwhile effect’. Treatment should be stopped in non-responders. The decline in mental function is delayed by about 3–6 months but not arrested. Rapid progression resumes when the drugs are stopped, but there may be limited benefit from restarting treatment more than a month after withdrawal. Anticholinesterases produce some improvement in other functional measures and behaviour that also affect the quality of life.
Memantine produces moderate improvement in cognition and reduction in functional decline, and is usually well tolerated. However, it may be ineffective in the early stages of Alzheimer's disease. Combination of a cholinesterase inhibitor with memantine can give additive benefits.
Current treatments for Alzheimer's disease do not alter the progression of the underlying disease. New treatment strategies are being developed that are directed either at enhancing the various neurotrophic proteins which protect neuronal systems, or at altering the production or clearance of amyloid protein or tau protein found in neurofibrillary tangles. Such strategies may offer a more fundamental approach to retarding the progress of Alzheimer's disease.
Neuropsychiatric complications such as depression and severe aggression may require treatment, but the value of antidepressant therapy is uncertain and antipsychotic drugs can produce significant unwanted effects with only modest benefit.
Vascular dementia
Cerebrovascular disease is a particularly common cause of dementia over the age of 85 years, and overall is the second most frequent cause of dementia. The deterioration in mental function is produced by multiple cerebral infarcts (multi-infarct dementia), particularly if they affect the white matter. The risk of dementia is increased ninefold in people with stroke. In some of these, dementia may be produced by specific, strategically located infarcts, especially in the angular gyrus of the inferior parietal lobule. In contrast to Alzheimer's disease, the initial presentation is usually more acute and cognitive decline has a stepwise course arising from recurrent cerebrovascular events.
Treatment
Prophylaxis against cerebral emboli with aspirin or warfarin depending on the heart rhythm (see prevention of stroke, above). However, the Cochrane database finds that there is no evidence, as yet, that aspirin is of benefit in vascular dementia.
Control of hypertension (Ch. 6). Trials have shown that calcium channel blockers are effective for reducing the risk of vascular dementia, but it is likely to be an effect related to blood pressure reduction rather than a more specific effect of this class of drug.
Anticholinesterases or memantine may produce some improvement in vascular dementia.
Immunosuppressant drugs (Ch. 38) can be used in the rare cases caused by cerebral vasculitis.
True/false questions
1. Aspirin reduces the risk of a first stroke in healthy individuals.
2. Aspirin cannot prevent a first stroke in people with persistent atrial fibrillation.
3. Approximately 85% of all strokes have a haemorrhagic aetiology.
4. If thrombolysis with recombinant tissue plasminogen activator (rt-PA; alteplase) is given in acute stroke, antiplatelet and anticoagulant therapies should not be given concurrently.
5. Anticoagulation with warfarin and antiplatelet therapy with aspirin are equal first-choice drugs for secondary prevention of recurrent ischaemic strokes in the presence of sinus rhythm.
6. Nimodipine reduces risk of vasospasm following subarachnoid haemorrhage.
7. Donepezil is an acetylcholinesterase inhibitor with selectivity for the CNS.
8. Cerebral ischaemia depolarises neurons and causes the release of large amounts of glutamate.
9. The NMDA receptor antagonist memantine reduces neurotoxicity caused by the excitatory transmitter glutamate.
10. Cerebral emboli arising from the heart are invariably caused by atrial fibrillation.
One-best answer (OBA) question
Choose the one correct statement from the following.
A Alzheimer's disease is associated with a relative lack of cholinergic and glutamatergic neurotransmission.
B It is recommended that rivastigmine is prescribed in Alzheimer's disease irrespective of the Mini-Mental State Examination (MMSE) score.
C Memantine is a muscarinic receptor agonist.
D Anticholinesterases should not be co-prescribed with memantine.
E Unlike donepezil, rivastigmine inhibits both acetylcholinesterase and butyrylcholinesterase.
Case-based questions
A 70-year-old man had a blood pressure of 190/110 mmHg despite intensive antihypertensive drug treatment. He was admitted to hospital 6 h after the acute onset of unilateral weakness and sensory loss. At the time of admission to hospital, most of the neurological signs had resolved. He had no headache or vomiting and remained conscious. He was in sinus rhythm. Following clinical examination and a CT brain scan, this episode was diagnosed as a TIA.
1. False. Aspirin does not reduce the risk of a first stroke in healthy individuals in sinus rhythm.
2. False. Aspirin reduces the risk of a first embolic stroke in atrial fibrillation by about 25%, but is less effective than warfarin.
3. False. About 85% of strokes have an ischaemic aetiology; up to 15% are caused by intracerebral haemorrhage.
4. True. The immediate risk of intracranial haemorrhage with alteplase is high and could be compounded by simultaneous administration of antiplatelet or anticoagulant agents. These should be considered later, when the effect of the thrombolytic has waned.
5. False. In people in sinus rhythm, aspirin alone (or together with dipyridamole) reduces the risk of stroke; warfarin is no more effective and there is a greater risk of major bleeding.
6. True. By blocking L-type calcium channels, nimodipine reduces vasospasm and also protects ischaemic neurons from Ca2+ overload.
7. True. Anticholinesterase drugs enhance cholinergic activity in the hippocampus and neocortex, improving memory and cognition.
8. True. The excitatory transmitter glutamate can cause a substantial rise in intracellular Ca2+, causing Ca2+overload and cell death by generation of free radicals.
9. True. Memantine blocks NMDA (glutamate) receptors and reduces glutamate-induced excitotoxicity of cholinergic neurons.
10. False. Cerebral emboli arising from the heart can also be caused by infected or damaged prosthetic valves or following damage to the myocardium
OBA answer
A Incorrect. Alzheimer's disease is associated with reduced cholinergic transmission and increased glutamatergic transmission.
B Incorrect. In the UK, it is advised that the anticholinesterases should not be prescribed if the MMSE is below 10.
C Incorrect. Memantine is a glutamate NMDA receptor antagonist.
D Incorrect. Anticholinesterases and memantine can be useful co-prescribed.
E Correct. But it is unclear whether additional inhibition of pseudocholinesterase (butyrylcholinesterase) by rivastigmine provides additional clinical benefit.
Case-based answers
A Thrombolysis is inappropriate in this situation. His blood pressure is high and it is a considerable time since the onset of symptoms. Thrombolysis has been approved for use within 3 h of the onset of symptoms. The rapid resolution of signs indicates a TIA, for which thrombolysis is not given. (Although thrombolysis has been shown in some trials to be useful in the treatment of stroke, safe and effective use is determined by a rigid set of criteria as there is a significant risk of intracranial haemorrhage.)
B His blood pressure must be brought under control. Reduction in blood pressure has a major effect on the prevention of a recurrent stroke. He should also be given low-dose aspirin and dipyridamole.
C Antiplatelet therapy should be continued. The antiplatelet drug dipyridamole has additional benefit when given together with aspirin. Cholesterol reduction with a statin is effective in secondary prevention of ischaemic stroke. It would be worth treating this man with a statin even if his cholesterol is not raised. An important reason for cholesterol reduction is prevention of ischaemic cardiac events, since coronary artery disease often coexists with atheromatous cerebrovascular disease.
Claiborne, JS. Transient ischemic attack. N Engl J Med. 2002;347:1687–1692.
Davis, SM, Donnan, GA. Secondary prevention after ischaemic stroke or transient ischaemic attack. N Engl J Med. 2012;366:1914–1922.
Donnan, GA, Fisher, M, MacLeod, M, et al. Stroke. Lancet. 2008;371:1612–1623.
Feher, A, Pusch, G, Koltai, K, et al. Statin therapy in the primary and secondary prevention of ischaemic cerebrovascular disease. Int J Cardiol. 2011;148:131–136.
Hacke, W, Donnan, G, Fieschi, C, et al. Association of outcome with early stroke treatment: pooled analysis of ATLANTIS, ECASS, and NINDs rt-PA stroke trials. Lancet. 2004;363:768–774.
Lansberg, MG, Bluhmki, E, Thijs, VN. Efficacy and safety of tissue plasminogen activator 3 to 4.5 hours after acute ischemic stroke: a meta-analysis. Stroke. 2009;40:2438–2441.
Rashid, P, Leonardi-Bee, J, Bath, P. Blood pressure reduction and secondary prevention of stroke and other vascular events: a systematic review. Stroke. 2003;34:2741–2748.
Rothwell, PM, Algra, A, Amarenco, P. Medical treatment in acute and long-term secondary prevention after transient ischaemic attack and ischaemic stroke. Lancet. 2011;377:1681–1692.
Salman, RA, Labovitz, DL, Stapf, C. Spontaneous intracerebral haemorrhage. BMJ. 2009;339:b2586.
Van der Worp, HB, van Gijn, J. Acute ischemic stroke. N Engl J Med. 2007;357:572–579.
Wardlaw, JM, Murray, V, Berge, E, et al. Recombinant tissue plasminogen activator for ischaemic stroke: an updated systematic review and meta-analysis. Lancet. 2012;379:2364–2372.
Wechsler, LR. Intravenous thrombolytic therapy for acute ischaemic stroke. N Engl J Med. 2011;364:2138–2146.
Al-Shahi, R, White, PM, Davenport, RJ, et al. Subarachnoid haemorrhage. BMJ. 2006;333:235–240.
van Gijn, J, Kerr, RS, Rinkel, GJ. Subarachnoid haemorrhage. Lancet. 2007;369:306–318.
Ballard, C, Gauthier, S, Corbett, A, et al. Alzheimer's disease. Lancet. 2011;377:1019–1031.
Farlowe, MR. Use of antidementia agents in vascular dementia: beyond Alzheimer disease. Mayo Clin Proc. 2006;81:1350–1358.
Farlowe, MR, Cummings, JL. Effective pharmacologic management of Alzheimer's disease. Am J Med. 2007;120:388–397.
Mayeux, R. Early Alzheimer's disease. N Engl J Med. 2010;362:2194–2201.
O'Brien, Erkinjuntti, T, Roman, G, et al. Vascular cognitive impairment. Lancet Neurol. 2003;2:89–98.
Raina, P, Santaguida, P, Ismalia, A, et al. Effectiveness of cholinesterase inhibitors and mementine for treating dementia: evidence review for a clinical practice guideline. Ann Intern Med. 2008;148:379–397.
Ritchie, K, Lovestone, S. The dementias. Lancet. 2002;360:1759–1766.
Rodda, J, Carter, J. Cholinesterase inhibitors and memantine for symptomatic treatment of dementia. BMJ. 2012;344:e2986.
Salomone, S, Caraci, F, Leggio, GM, et al. New pharmacological strategies for the treatment of Alzheimer's disease: focus on disease modifying drugs. Br J Clin Pharmacol. 2012;73:504–517.