Chemotherapy of infections
Antimicrobial drugs are natural or synthetic chemical substances that suppress the growth of, or destroy, micro-organisms including bacteria, fungi, helminths, protozoa and viruses. The term antibiotic is widely used, but strictly should be reserved for those antimicrobial drugs that are derived from micro-organisms. The term antimicrobial or the more restrictive terms antibacterial, antifungal, antihelminthic, antiprotozoal and antiviral are used in this book.
Effective antimicrobial drugs have certain key attributes. In order to minimise unwanted effects in humans, most are designed to act on processes that are unique to the target pathogen. They must also be able to penetrate human tissues to reach the site of infection. Micro-organisms can acquire resistance to various antimicrobial drugs and will then no longer be affected by the drug, so there is a continuing effort to discover and develop antimicrobial drugs that avoid or overcome the evolving mechanisms of resistance.
Bacterial infections
Classification of antibacterial drugs
Antibacterial drugs can be classified in several overlapping ways.
Firstly, they can be bacteriostatic or bactericidal. This categorisation depends largely on the concentration of drug that can be achieved safely in plasma without causing significant toxicity in the person who takes the drug. Bacteriostatic antibacterials inhibit bacterial growth but do not destroy the bacteria at concentrations in plasma that are safe for humans; following inhibition of growth, the natural immune mechanisms of the body are able to eliminate the bacteria. Such drugs will be less effective in immunocompromised individuals or when the bacteria are dormant and not dividing. At plasma concentrations that are safe for humans, bactericidal antibacterials kill bacteria but, even then, immune mechanisms will play a role in the final elimination of the bacteria. Some bactericidal drugs are more effective when bacterial cells are actively dividing, and may therefore be less effective if taken together with a bacteriostatic drug. For antibacterials to be bactericidal they must be present at adequate concentration; too low a concentration may render them bacteriostatic.
Secondly, antibacterials can be grouped according to their mechanisms of action (Fig. 51.1):
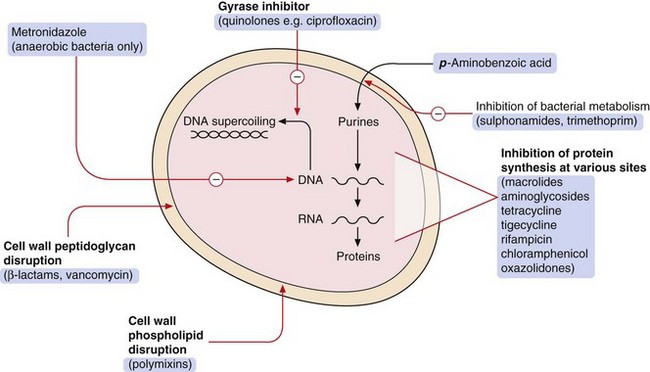
 inhibition of the synthesis of bacterial cell wall peptidoglycans or activation of enzymes that disrupt the cell wall (e.g. β-lactams),
inhibition of the synthesis of bacterial cell wall peptidoglycans or activation of enzymes that disrupt the cell wall (e.g. β-lactams),
increased permeability of the bacterial cell phospholipid membrane, leading to leakage of intracellular contents (e.g. polymyxins),
impaired bacterial ribosome function, producing a reversible inhibition of protein synthesis (e.g. aminoglycosides, macrolides). Such drugs can show selectivity because bacterial 70S ribosomes differ structurally from the 80S ribosomes in humans,
selective block of bacterial metabolic pathways (e.g. trimethoprim),
interference with replication of bacterial DNA or RNA (e.g. quinolones).
Thirdly, antibacterials may be classified according to whether their spectrum of activity against bacteria is limited (narrow-spectrum) or extensive (broad-spectrum).
Finally, they can be classified by chemical structure. In the following text, the antimicrobial drugs are grouped according to their mechanism of action and then by their chemical structure. Cross-referencing to other methods of classification may be necessary. The drug compendium at the end of the chapter is organised to accord with the British National Formulary.
Antimicrobial resistance
When an antibacterial is ineffective against a bacterium, the organism is said to be resistant to the antibacterial drug. Resistance to antibacterial drugs can be intrinsic to the bacterium (innate resistance) or can be acquired by modification of its genetic structure (acquired resistance).
Resistance is a major problem in treating infections with bacteria, and also for many protozoa (e.g. malaria) and viruses (e.g. HIV), but is less significant in fungal infections (unless the person has immunodeficiency).
Antibacterial drug resistance
There are four general processes by which a bacterium can acquire resistance to antibacterial drugs (Fig. 51.2):
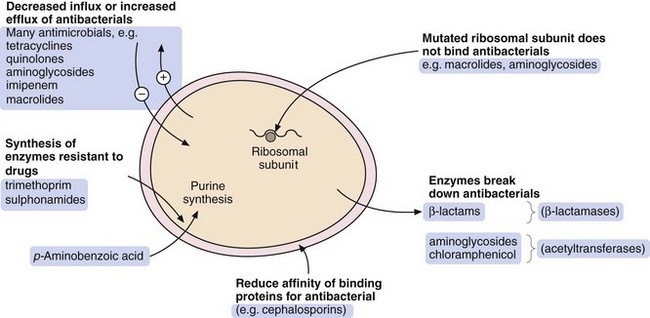
modification of the bacterium such that it produces enzymes that inactivate the drug; examples are β-lactamase enzymes, which inactivate some penicillins, and acetylating enzymes, which can inactivate aminoglycosides,
modification of the bacterium so that penetration of the drug is reduced; an example is the absence of the membrane protein D2 porin in resistant Pseudomonas aeruginosa, which prevents penetration of the β-lactam antibacterial imipenem,
acquisition of efflux pumps that remove the antibacterial drug from the cell faster than it can enter; an example is quinolone efflux pumps in Staphylococcus aureus,
structural change in the target molecule for the antibacterial drug; examples are mutated penicillin-binding proteins in resistant enterococci that have a low affinity for binding of cephalosporins, and mutated dihydrofolate reductase that is not inhibited by trimethoprim (see Fig. 51.4, below).
The major mechanisms by which bacteria acquire resistance to antibacterial drugs are spontaneous mutation, conjugation, transduction and transformation.
Spontaneous mutation
In this process, a single-step genetic mutation in a bacterial population leads to resistant organisms that selectively survive and grow while sensitive bacteria are killed by an antibacterial drug. This is termed vertical evolution.
The other three mechanisms involve acquisition from other resistant organisms of genetic material that confers resistance. This is termed horizontal evolution.
Conjugation
Direct cell-to-cell contact is a way of exchanging genetic material that confers antibacterial resistance. It usually involves transfer of self-replicating circular fragments of DNA (plasmids), which can contain multiple resistance genes. A transposon (a DNA sequence that can change its relative position within the genome) may facilitate transfer of sections of DNA from one organism to another by jumping to plasmid DNA. Transfer of the plasmid occurs via a connecting structure called a pilus. The plasmid can remain outside the genome of the bacterium or can be incorporated into it, when it is more stable but less transmissible. Conjugation is by far the most important source of extrinsic DNA transfer between bacteria.
Transduction
Bacteria are susceptible to infection by viruses known as bacteriophages. During replication of the bacteriophages, the host bacterial cell's DNA (containing resistance genes) may be replicated along with bacteriophage DNA and taken into the virus. The phage carrying the resistance genes can then infect other bacterial cells and spread resistance. This method of acquired resistance is rare.
Antibacterial drugs
The antibacterial drugs in this section are grouped by their mechanism of action and then by their chemical structure.
Drugs affecting the cell wall: β-lactam antibacterials
The drugs in this class all have a β-lactam ring which must be intact for them to be active (Fig. 51.3). The β-lactam antibacterials include penicillins, cephalosporins and cephamycins, monobactams and carbapenems. Some are susceptible to attack by bacterial enzymes (β-lactamases, also known as penicillinases) that split the β-lactam ring, but others have structural modifications that confer resistance to β-lactamase inactivation.
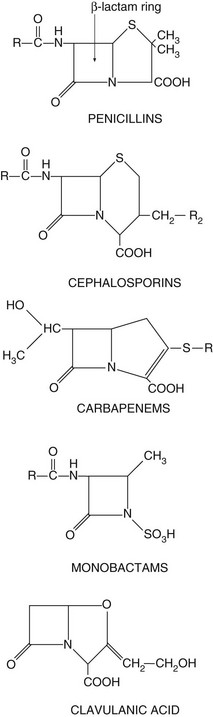
Fig. 51.3 The structural backbones of β-lactam antibacterial drugs.
Also shown is the β-lactamase inhibitor, clavulanic acid.
Mechanism of action of β-lactam antibacterials: Beta-lactam antibacterials bind to several penicillin-binding proteins in bacteria. Some of these proteins are transpeptidases that are required for crosslinking of the peptidoglycan layer of the cell wall which surrounds certain bacteria and is essential for their survival. Inhibition of transpeptidase activity by β-lactam antibacterials prevents the bacterium synthesising an intact cell wall when it divides. The transmembrane osmotic gradient then leads to swelling, rupture and death of the bacterium.
Some bacterial cells also contain enzymes that cause cell lysis when activated. The binding of β-lactam antibacterials to other specific penicillin-binding proteins within bacteria reduces the production of natural inhibitors of lysis-inducing enzymes, promoting lysis of the bacterial cell wall.
Spectrum of activity: Penicillins consist of a thiazolidine ring connected to a β-lactam ring, to which is in turn attached a side-chain (Fig. 51.3). The side-chain determines many of the antibacterial and pharmacological characteristics of particular penicillins (Table 51.1).
Table 51.1
Examples of penicillins and their properties
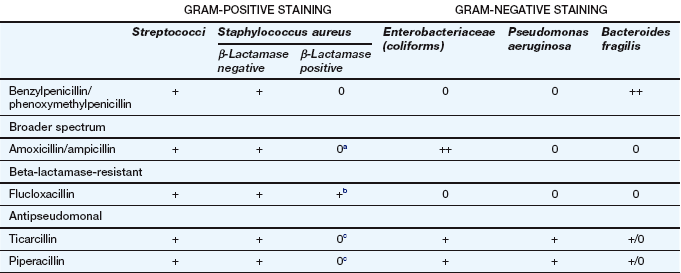
+, Active; +/0, variable activity; 0, inactive or poor activity.
aCan be used combined with a β-lactamase inhibitor, e.g. amoxicillin plus clavulanic acid (co-amoxiclav).
bResistance is increasing.
cTicarcillin only available with clavulanic acid. Piperacillin is combined with the β-lactamase inhibitor tazobactam.
Benzylpenicillin (penicillin G) and phenoxymethylpenicillin (penicillin V) are active against many aerobic Gram-positive bacteria, a more limited range of Gram-negative bacteria, for example cocci (e.g. gonococci and meningococci), and many anaerobic micro-organisms. Gram-negative bacilli are not sensitive to these drugs. Benzylpenicillin and phenoxymethylpenicillin are only effective against organisms that do not produce β-lactamases (see below).
Flucloxacillin has an acyl side chain attached to the β-lactam ring which prevents access of β-lactamase to the ring and makes the drug resistant to inactivation by the enzyme. Flucloxacillin is generally less effective than benzylpenicillin against bacteria that do not produce β-lactamase, and is usually reserved for treating β-lactamase-producing staphylococci.
Ampicillin and amoxicillin are aminopenicillins that have an extended spectrum of activity to include many Gram-negative bacilli. However, they are less effective than benzylpenicillin against Gram-positive cocci. Both drugs are inactivated by β-lactamase.
Other extended-spectrum penicillins include ureidopenicillins (e.g. piperacillin), which are active against P. aeruginosa, and amidinopenicillins (e.g. pivmecillinam), which are active mainly against Gram-negative bacteria. Carboxypenicillins (e.g. ticarcillin) are not widely used, but have activity against Pseudomonas species, Proteus species and Bacteroides fragilis.
Clavulanic acid is a potent inhibitor of several β-lactamases. It is structurally related to the β-lactam antibiotics, although it has little intrinsic antibacterial activity (Fig. 51.3). It is available in combined formulations with penicillins that are destroyed by β-lactamase, such as amoxicillin (as co-amoxiclav) or ticarcillin (Table 51.1); the combination drugs can be used to treat infections caused by some β-lactamase-producing organisms that would otherwise be resistant to the antibacterial. Tazobactam has similar properties to clavulanic acid and is used in combination with piperacillin.
Resistance: Resistance to penicillins is most often due to the production of β-lactamases which hydrolyse the β-lactam ring (Fig. 51.3). There are hundreds of β-lactamases, many of which are closely related to penicillin-binding proteins, but some are structurally different metalloenzymes. The β-lactamases produced by various organisms have widely differing spectra of activity. Some bacteria release extracellular β-lactamases, particularly S. aureus. In Gram-negative bacteria the β-lactamases are located between the inner and outer cell membranes in the periplasmic space. Extended-spectrum β-lactamases (ESBLs) are β-lactamases that also hydrolyse extended-spectrum ‘third-generation’ cephalosporins, such as cefotaxime and ceftriaxone, and monobactams such as aztreonam (see below). ESBLs are most often produced by enterobacteria. The genetic information for β-lactamase production is often encoded in a plasmid and this may be transferred to other bacteria by conjugation. By contrast, the broader-spectrum β-lactamases are often encoded by chromosomal genes.
An alternative type of penicillin resistance occurs in gonococci and in meticillin-resistant S. aureus (MRSA), which develop mutated penicillin-binding proteins that do not bind β-lactam antibacterials. Meticillin has now been discontinued, but the name MRSA is still used.
Pharmacokinetics: Only about one-third of an oral dose of benzylpenicillin (penicillin G) is absorbed; the rest is destroyed by acid in the stomach. Benzylpenicillin is therefore restricted to intramuscular or intravenous administration. The phenoxymethyl derivative (penicillin V) is more acid-stable and better absorbed from the gut; it has a similar spectrum of activity as benzylpenicillin but is generally less active. Penicillins are widely distributed through the body, although transport across the meninges is poor unless they are acutely inflamed (e.g. in meningitis). The half-lives of benzylpenicillin and phenoxymethylpenicillin, in common with most penicillins, are short (about 1 h) because they are rapidly eliminated by the kidney, mainly by active secretion at the proximal tubule. Flucloxacillin and amoxicillin are rapidly and almost completely absorbed from the gut, but ampicillin is incompletely absorbed. These drugs can also be given intramuscularly or intravenously. They are eliminated by the kidney in a similar way to benzylpenicillin. The amidinopenicillin pivmecillinam is a prodrug for oral use which is hydrolysed to mecillinam. The carboxypenicillin ticarcillin is only available in combination with clavulanic acid for intravenous use. The ureidopenicillin piperacillin is given intravenously in combination with the β-lactamase inhibitor tazobactam.
Unwanted effects: Penicillins are normally well tolerated and have a high therapeutic index.
Allergic reactions in 1–10% of exposed individuals. Penicillins and their breakdown products bind to proteins and act as haptens, stimulating the production of antibodies that mediate the allergic response (Chs 38 and 53). Manifestations of allergy to penicillins include fever, vasculitis, serum sickness, exfoliative dermatitis, Stevens–Johnson syndrome and anaphylactic shock. Cross-allergenicity is widespread among various penicillins and to a lesser extent with cephalosporins.
Aminopenicillins (e.g. amoxicillin) frequently produce a non-allergic maculopapular rash in people with glandular fever. This does not recur if another type of penicillin is given.
Reversible neutropenia and eosinophilia with prolonged high doses.
Encephalopathy with excessively high cerebrospinal fluid (CSF) concentrations of penicillin. This occurs in severe renal failure or after inadvertent intrathecal injection (which should never be given).
Diarrhoea or Clostridium difficile-related colitis as a result of disturbance of normal colonic flora, especially with broad-spectrum penicillins.
Cholestatic jaundice, especially with flucloxacillin or clavulanic acid.
Spectrum of activity: Cephalosporins, like penicillins, have a β-lactam ring, to which is fused a dihydrothiazine ring, which makes them more resistant to hydrolysis by β-lactamases (Fig. 51.3). Cephalosporins are often classified by ‘generations’, the members within each generation sharing similar antibacterial activity. Successive generations tend to have increased activity against Gram-negative bacilli, usually at the expense of Gram-positive activity, and an increased ability to cross the blood–brain barrier (Table 51.2).
Table 51.2
Examples of β-lactams other than penicillins and their spectra of activity
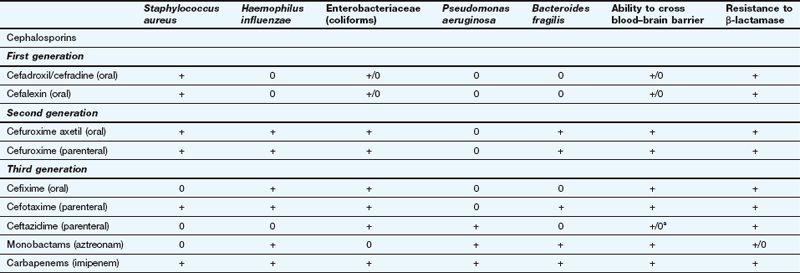
This table is a general guide to selected drugs and susceptibilities of organisms can vary widely.
Staphylococcus aureus is a Gram-positive staining organism. Other illustrative bacteria are Gram-negative staining.
aSome cephalosporins penetrate better into the CNS in the presence of inflamed meninges.
First-generation cephalosporins (e.g. cefadroxil or cefalexin) have activity against staphylococci and most streptococci, but not enterococci.
Second-generation cephalosporins (e.g. cefuroxime) have additional activity against some Gram-negative bacteria such as Haemophilus influenzae and Neisseria gonorrhoeae. They are able to penetrate the blood–brain barrier.
Third-generation cephalosporins have improved β-lactamase stability and are able to penetrate the CSF in useful quantities. They also have greater Gram-negative activity than the other two generations. Cefixime adds Proteus and Klebsiella species to its spectrum, but it has no activity against staphylococci (Table 51.2). Ceftazidime has good activity against Pseudomonas species.
Resistance: The later generations are more resistant to β-lactamase-mediated enzymatic hydrolysis of the β-lactam ring than are the earlier generations (Table 51.2). However, ESBLs can be acquired by Escherichia coli and and other enterobacteria, which confers resistance to third-generation cephalosporins.
Pharmacokinetics: First-generation oral cephalosporins are well absorbed from the gut. Several second- and third-generation drugs, for example cefuroxime and cefotaxime, are acid-labile and must be given by a parenteral route. Cefuroxime has been formulated as a prodrug for oral use (cefuroxime axetil), which is hydrolysed to cefuroxime at first pass through the liver. Most cephalosporins are excreted primarily by the kidney and have short half-lives (less than 3 h), but cefixime is mainly eliminated by biliary excretion. Ceftriaxone has a longer half-life (6–9 h), probably as a result of extensive plasma protein binding.
Nausea, vomiting, abdominal discomfort.
Rashes, including erythema multiforme and toxic epidermal necrolysis.
Cephalosporins can produce allergic reactions similar to those observed with the penicillins. Fewer than 10% of people who are allergic to penicillins show cross-allergy to cephalosporins, but a history of a serious reaction to penicillins precludes the use of cephalosporins.
Diarrhoea or C. difficile-related colitis can be caused by disturbance of normal bowel flora. This is more common with oral cephalosporins, and is more frequent than with many other antimicrobials.
Aztreonam is a β-lactam antibacterial related to the penicillins but with a single ring structure (‘monocyclic β-lactam’) (Fig. 51.3). It has little cross-allergenicity with the penicillins and has been successfully given to people with proven penicillin allergy. Its spectrum of activity is limited to Gram-negative bacteria, including P. aeruginosa, Neisseria meningitidis, N. gonorrhoeae and H. influenzae, with no activity against Gram-positive bacteria or anaerobes. Aztreonam is given intramuscularly or intravenously and is resistant to most β-lactamases. However, ESBLs can be acquired by E. coli and other enterobacteria, which confer resistance to aztreonam. Aztreonam is excreted by the kidney and has a half-life of about 2 h. Unwanted effects are similar to those of other β-lactam antibacterials.
Imipenem is a β-lactam drug that has an extremely broad spectrum of bactericidal activity. It has potent activity against Gram-positive cocci, including some β-lactamase-producing pneumococci (Table 51.2), Gram-negative bacilli, including P. aeruginosa, Neisseria suppurans and Bacteroides species, and also many anaerobic bacteria. Imipenem can penetrate the blood–brain barrier and is resistant to β-lactamases. Narrow-spectrum resistance to imipenem in P. aeruginosa occurs from a mutation that results in loss of a specific cell membrane uptake pathway.
Meropenem and ertapenem are structurally related and have broad spectra of activity against Gram-positive and Gram-negative bacteria, but ertapenem is inactive against Pseudomonas species. Imipenem, meropenem and ertapenem are given intravenously; imipenem can also be given by deep intramuscular injection. Imipenem is rapidly hydrolysed by dihydropeptidases in the kidney and so is given in combination with the dihydropeptidase inhibitor cilastatin. Meropenem and ertapenem are not inactivated by the renal dihydropeptidase and can be given alone.
These drugs are mainly excreted by the kidney and have short half-lives (1–5 h). Unwanted effects are similar to those of other β-lactam antibacterials, except for neurotoxicity with seizures, which is more common with imipenem than with other carbapenems.
Other drugs affecting the cell wall
Mechanism of action: Vancomycin and teicoplanin are high-molecular-weight glycopeptide compounds that inhibit bacterial cell wall synthesis by preventing the linking of peptidoglycan constituents (Fig. 51.1). Glycopeptides are bactericidal.
Spectrum of activity: Vancomycin and teicoplanin are active only against Gram-positive bacteria, particularly meticillin-resistant staphylococci. They do not penetrate the cell wall of Gram-negative bacteria. Both are usually reserved for treatment of serious Gram-positive bacterial infection or for bacterial endocarditis that is not responding to other treatments. Vancomycin given orally is also effective against C. difficile, which colonises the colon when the normal gut flora is disturbed by antibacterial drugs, causing diarrhoea and colitis. Metronidazole (see below) is preferred for this indication, but resistance to metronidazole is increasingly common.
Resistance: Acquired resistance to vancomycin is uncommon. In S. aureus it arises as a result of a multi-step genetic acquisition of a thickened peptidoglycan cell wall. This traps the drug and prevents it reaching its target on the cytoplasmic membrane. For other bacteria, plasmid-mediated resistance involves incorporation of D-lactate into the cell wall in place of D-alanine. This modification prevents binding of the glycopeptide.
Pharmacokinetics: Both vancomycin and teicoplanin are poorly absorbed orally and are given by intravenous infusion for systemic infection. Teicoplanin can also be given by intramuscular injection. Oral vancomycin is only used for treating C. difficile-related colitis. Both drugs are excreted by the kidney; vancomycin has a shorter half-life (5–11 h) than teicoplanin (32–176 h). Plasma concentration monitoring of vancomycin is used to minimise the risk of toxicity.
Unwanted effects: Dose adjustment based on monitoring of the trough plasma concentration of vancomycin may reduce the risk of toxic effects.
Nephrotoxicity, which may be enhanced if used in combination with an aminoglycoside.
Thrombophlebitis at the site of infusion.
Rashes, including Stevens–Johnson syndrome and toxic epidermal necrolysis. Rapid intravenous injection of vancomycin produces upper body flushing, the ‘red man’ syndrome.
Blood disorders, including neutropenia, thrombocytopenia.
Mechanism of action: Daptomycin is a lipopeptide antibacterial with a unique mode of action. It binds to the cell wall of Gram-positive bacteria, and creates transmembrane channels that allow leakage of intracellular ions, destroying the membrane potential across the cell.
Spectrum of activity: Daptomycin does not penetrate the membrane of Gram-negative bacteria. It is bactericidal against a similar spectrum of organisms as vancomycin and is used to treat complicated skin and soft tissue infections.
Resistance: Resistance occurs when the bacterial membrane structure changes to prevent binding of the drug.
Mechanism of action: Polymyxins bind to membrane phospholipids in susceptible bacteria and alter the permeability of the membrane to K+ and Na+ ions. The cell's osmotic barrier is lost and the bacteria are killed by lysis (Fig. 51.1).
Spectrum of activity: Polymyxins have bactericidal action against Gram-negative bacteria, including Pseudomonas species, but are inactive against Gram-positive bacteria.
Pharmacokinetics: Colistimethate sodium is very poorly absorbed from the gut and is usually given by inhalation or topically to the skin. Penetration into joint spaces and CSF is poor. It is excreted unchanged by the kidney and has a half-life of 4–8 h. It is occasionally given by mouth for bowel sterilisation.
Drugs affecting bacterial DNA
Quinolones (fluoroquinolones):
Mechanism of action: Quinolones inhibit replication of bacterial DNA. They block the activity of bacterial DNA gyrase and DNA topoisomerase, the enzymes that form DNA supercoils and are essential for DNA replication and repair (Fig. 51.1). The effect is bactericidal.
Spectrum of activity: Ciprofloxacin has a broad spectrum of activity and is active against many micro-organisms resistant to penicillins, cephalosporins and aminoglycosides. Its spectrum includes Gram-positive bacteria, but with only moderate activity against Streptococcus pneumoniae and Enterococcus faecalis. It is active against most Gram-negative bacteria, including H. influenzae, P. aeruginosa, N. gonorrhoeae, and Enterobacter and Campylobacter species. Its spectrum extends to chlamydia and some mycobacteria, but not anaerobes.
Moxifloxacin has a broad spectrum of activity against Gram-positive and Gram-negative bacteria, but is inactive against P. aeruginosa. It has greater activity than ciprofloxacin against pneumococci. Norfloxacin is mainly useful for urinary tract pathogens.
Resistance: Resistance to quinolones is relatively uncommon but can be produced by a mutation that results in a DNA gyrase that is less susceptible to the drug's action, or by increased active drug efflux from the cell (Fig. 51.2).
Pharmacokinetics: Oral absorption of ciprofloxacin is variable but adequate. Intravenous formulations of some quinolones are available, including ciprofloxacin and moxifloxacin. Ciprofloxacin is widely distributed but CSF penetration is poor unless there is meningeal inflammation. Ciprofloxacin and norfloxacin are eliminated mainly by the kidney and have short half-lives (3–4 h). Moxifloxacin is well absorbed from the gut, is metabolised in the liver and has a longer half-life (12 h).
Nausea, vomiting, abdominal pain, diarrhoea.
Central nervous system (CNS) effects: dizziness, headache, tremor, seizures (especially in those with a history of epilepsy).
Pain and inflammation in tendons, occasionally with tendon rupture (especially in the elderly or with concomitant use of corticosteroids).
Moxifloxacin prolongs the Q–T interval on the electrocardiogram (ECG) and predisposes to ventricular arrhythmias. The risk is greater if used in combination with other proarrhythmic drugs (Ch. 8).
Drug interactions: inhibition of hepatic cytochrome P450 by ciprofloxacin and norfloxacin increases the plasma concentrations of theophylline (Ch. 12), warfarin (Ch. 11) and ciclosporin (Ch. 38), which can produce toxicity. The absorption of quinolones from the gut is decreased by oral iron salts.
Mechanism of action: Metronidazole and tinidazole are bactericidal only after they have been converted to an intermediate transient toxic metabolite, which inhibits bacterial DNA synthesis and breaks down existing DNA. Only some anaerobes and some protozoa contain the oxidoreductase enzyme that converts these drugs to their antibacterial derivatives. The intermediate metabolite is not produced in human cells, or in aerobic bacteria. These drugs are equally active against dividing and non-dividing cells.
Spectrum of activity: Metronidazole and tinidazole are mainly active against anaerobic bacteria and protozoa, including B. fragilis, Clostridium species, Gardnerella vaginalis and Giardia lamblia. Metronidazole is an important drug for treating C. difficile-related colitis caused by broad-spectrum antimicrobial use. Metronidazole or tinidazole are important constituents of the triple or quadruple therapy utilised for the elimination of Helicobacter pylori (Ch. 33). They are also amoebicidal, with activity against Entamoeba histolytica.
Resistance: Acquired resistance is becoming more common. For example, in some countries a significant percentage of strains of H. pylori are resistant to metronidazole, as are some strains of C. difficile. Resistance can result from the development of oxidoreductases that do not act on metronidazole, or from the induction of oxidative stress mechanisms that inhibit the action of the drug. Resistance to tinidazole is less common.
Pharmacokinetics: Metronidazole is well absorbed orally and can also be given intravenously or by rectal suppositories. Metronidazole penetrates well into body fluids, including vaginal, pleural and cerebrospinal fluids, and can cross the placenta. Metronidazole and tinidazole are eliminated mainly by metabolism in the liver and have half-lives of 6–9 h and 12–14 h, respectively.
Nausea, vomiting, metallic taste.
Intolerance to alcohol can occur by a mechanism similar to the disulfiram reaction (Ch. 54).
Mechanism of action: Nitrofurantoin is activated inside bacteria by reduction via the flavoprotein nitrofurantoin reductase to unstable metabolites, which disrupt ribosomal RNA, DNA and other intracellular components. It is bactericidal, especially to bacteria present in acid urine.
Spectrum of activity: Nitrofurantoin is active against most Gram-positive cocci and E. coli. Pseudomonas species are naturally resistant, as are many Proteus species. Its use is confined to infections of the lower urinary tract.
Pharmacokinetics: Nitrofurantoin is well absorbed from the gut. Its half-life in plasma is very short (<1 h) and therapeutic plasma concentrations are not achieved. It is excreted largely unchanged in the urine, giving urinary concentrations high enough to treat lower urinary tract infections, but the low tissue concentrations are inadequate for the treatment of acute pyelonephritis.
Drugs affecting bacterial protein synthesis
Mechanism of action: Macrolides interfere with bacterial protein synthesis by binding reversibly to the 50S subunit of the bacterial ribosome. This causes dissociation of the aminoacyl-transfer RNA (tRNA) from its translocation site. The action is primarily bacteriostatic (Fig. 51.1).
Spectrum of activity: Erythromycin has a similar spectrum of activity to broad-spectrum penicillins and is often used for treating individuals who are allergic to penicillin. It is effective against Gram-positive bacteria and gut anaerobes, but has poor activity against H. influenzae. It is also used for infections by Legionella, Mycoplasma, Chlamydia, Mycobacterium and Campylobacter species and for Bordetella pertussis. Although erythromycin is primarily bacteriostatic, it is bactericidal at high concentrations for some Gram-positive species, such as group A streptococci and pneumococci.
Azithromycin has less activity than erythromycin against Gram-positive bacteria, but enhanced activity against H. influenzae. Clarithromycin has slightly greater activity than erythromycin and is also used as part of the multidrug treatment of H. pylori (Ch. 33). Telithromycin is a derivative of erythromycin active against penicillin- and erythromycin-resistant S. pneumoniae.
Resistance: Bacteria become resistant to macrolides by activation of an efflux mechanism. A less common mechanism is a mutation in the gene encoding a methyltransferase that modifies the drug target site on the ribosome.
Pharmacokinetics: Erythromycin is destroyed at acid pH and is therefore given as an enteric-coated tablet or as an acid-stable ester prodrug (erythromycin ethyl succinate). It can also be administered intravenously. Clarithromycin is acid-stable and well absorbed from the gut, but undergoes first-pass metabolism in the liver. Erythromycin and clarithromycin are metabolised in the liver and have short half-lives (1–3 h). Azithromycin is poorly absorbed from the gut. It is widely distributed and released slowly from the tissues, and then excreted unchanged in the bile. It has a long half-life of about 2 days. Telithromycin is well absorbed from the gut, is metabolised in the liver and has a half-life of 10 h.
Epigastric discomfort, nausea, vomiting and diarrhoea are common with the oral preparation of erythromycin. Other macrolides are better tolerated.
Cholestatic jaundice with erythromycin, usually if treatment is continued for more than 2 weeks.
Prolongation of the Q–T interval on the ECG, with a predisposition to ventricular arrhythmias (Ch. 8).
Drug interactions: erythromycin and clarithromycin inhibit P450 drug-metabolising enzymes (CYP1A2, CYP3A4) and can increase the plasma concentration of other drugs metabolised by these enzymes, including carbamazepine (Ch. 23) and ciclosporin (Ch. 38).
Mechanism of action: The aminoglycosides have similar properties, but with some important differences that can be exploited in particular clinical circumstances, as illustrated below. Aminoglycosides inhibit protein synthesis in bacteria by binding irreversibly to the 30S ribosomal subunit (Fig. 51.1). This inhibits transfer of aminoacyl-tRNA to the peptidyl site, causing premature termination of the peptide chain, and also increases the frequency of misreading of mRNA. Aminoglycosides may also damage bacterial cell membranes, causing leak of intracellular contents. They are bactericidal.
Spectrum of activity: Aminoglycosides are active against many Gram-negative bacteria (including Pseudomonas species) and some Gram-positive bacteria. They are inactive against anaerobes, which are unable to take up the aminoglycosides. Aminoglycosides are particularly useful for serious Gram-negative infections, when they have a synergistic action with drugs that disrupt cell wall synthesis (e.g. penicillins). Gentamicin is the most widely used aminoglycoside. Streptomycin is used rarely, but is occasionally used as a component of the drug regimen to treat Mycobacterium tuberculosis (see below).
Resistance: Resistance to aminoglycosides is transferred by plasmids and is an increasing problem. It can occur by several mechanisms, the most frequent being production of enzymes that acetylate, phosphorylate or adenylyl aminoglycosides in the bacterial periplasmic space, with poor uptake of the modified drug (Fig. 51.2). Resistance resulting from reduced penetration of the drug can be overcome by co-administration of antibacterials that disrupt cell wall synthesis, such as penicillins. Netilmicin is less susceptible to these enzymes and is effective against many gentamicin-resistant bacteria. Changes in the ribosomal proteins in bacteria can also reduce drug binding and antibacterial effectiveness, particularly for streptomycin.
Pharmacokinetics: Aminoglycosides are poorly absorbed from the gut and are given parenterally. They are rapidly excreted by the kidney and have short half-lives (1–4 h). They do not cross the blood–brain barrier, but they cross the placenta. Blood concentrations should always be measured to guide dosing. With multiple daily doses the peak plasma concentration (measured 1 h after dosing) and the trough concentration immediately before the next dose are important, both to ensure bactericidal efficacy and to minimise the risk of toxic effects. Once-daily dosage regimens for aminoglycosides are increasingly used and are no more toxic than multiple daily dosages.
Tobramycin is also available in a preservative-free solution for administration by nebuliser for the management of people with bronchiectasis (including cystic fibrosis) whose respiratory tracts are colonised by P. aeruginosa.
Unwanted effects: Most unwanted effects of aminoglycosides are dose-related and many are reversible; they are most closely related to high trough concentrations of the drug.
Ototoxicity can lead to both vestibular and auditory dysfunction. Prolonged treatment or high plasma drug concentrations lead to accumulation of the aminoglycoside in the inner ear, resulting in disturbances of balance or deafness that are often irreversible. Mutations in the human gene encoding mitochondrial 12S ribosomal RNA predispose to ototoxicity. Netilmicin causes less ototoxicity than the other aminoglycosides.
Renal damage occurs through retention of aminoglycosides in the proximal tubular cells of the kidney. It is usually reversible and is manifest initially by a defect in the concentrating ability of the kidney, with mild proteinuria followed by a reduction in the glomerular filtration rate.
Acute neuromuscular blockade can occur, usually if the aminoglycoside is used with anaesthetic drugs (Ch. 17), and aminoglycosides can enhance the effects of other neuromuscular-blocking drugs (Ch. 27). This action is the result of inhibition of pre-junctional acetylcholine release and also reduced postsynaptic sensitivity. It is reversed by intravenous Ca2+ salts.
Mechanism of action: Tetracyclines enter bacteria mainly by an active uptake mechanism that is not found in human cells. They are bacteriostatic and inhibit bacterial protein synthesis by binding reversibly to the 30S subunit of ribosomes, inhibiting the binding of aminoacyl-tRNAs.
Spectrum of activity: Tetracyclines have a broad spectrum of activity against many Gram-positive and Gram-negative bacteria and in infections caused by rickettsiae, amoebae, Chlamydia psittaci, Chlamydia trachomatis, Coxiella burnetii, Vibrio cholerae and Mycoplasma, Legionella and Brucella species. They are useful in acne (Ch. 49). Minocycline is active against N. meningitidis, unlike other tetracyclines.
Resistance: Resistance is carried by plasmids and is usually due to increased transport of the drug out of the bacterium (Fig. 51.2). An alternative mechanism is decreased binding of tetracyclines to bacterial ribosomes. Resistance to the tetracyclines develops slowly, but in the UK is now widespread among most Gram-positive and several Gram-negative bacteria. Micro-organisms that have developed resistance to one tetracycline frequently display resistance to the others.
Pharmacokinetics: Tetracyclines are incompletely absorbed from the gut, particularly if taken with food. Absorption of oxytetracycline is further impaired by milk, antacids (Ch. 33), iron and increased intestinal pH; tetracyclines bind to divalent and trivalent cations, forming inactive chelates (Ch. 56).
The tetracyclines diffuse reasonably well into sputum, urine, and peritoneal and pleural fluid, and cross the placenta, but penetrate the CSF poorly. Tetracyclines are concentrated in the liver and some is excreted via the bile into the small intestine, from where it is partially reabsorbed. Drug concentrations in the bile may be three to five times higher than in the plasma.
Tetracyclines are mainly eliminated unchanged in the urine, with the exception of doxycycline, which is largely eliminated in the bile. All of the tetracyclines have half-lives within the range 8–22 h.
Nausea, vomiting, epigastric discomfort and diarrhoea.
In children, tetracyclines produce permanent yellow–brown discoloration of growing teeth by chelating with Ca2+, and can also cause dental hypoplasia. Tetracyclines should be avoided during the latter half of pregnancy and in children during the first 12 years of life.
Anti-anabolic effects can occur in human cells from inhibition of protein synthesis (not seen with doxycycline or minocycline). If there is pre-existing impairment of renal function it can lead to uraemia.
Idiopathic intracranial hypertension, with headache and visual disturbances.
Mechanism of action: Tigecycline has structural similarities to the tetracyclines and also binds to the 30S ribosomal subunit of bacteria.
Spectrum of activity: Tigecycline has a broad spectrum of activity against Gram-positive and Gram-negative bacteria, including some anaerobes. It is used for complicated skin and soft tissue infections, and for complicated abdominal infections caused by resistant bacteria. Tigecycline is active against MRSA, vancomycin-resistant enterococci, Proteus species and P. aeruginosa.
Resistance: Many strains of Proteus species and P. aeruginosa are resistant, usually as a result of possessing a drug-efflux pump.
Mechanism of action: Chloramphenicol inhibits protein synthesis in bacteria by binding reversibly to the 50S subunit of bacterial ribosomes (Fig. 51.1), where it blocks peptide chain elongation by inhibiting peptidyl transferase activity. The effect is mainly bacteriostatic, but can be bactericidal in some bacteria.
Spectrum of activity: Chloramphenicol is a broad-spectrum antibacterial, active against many Gram-positive cocci (both aerobic and anaerobic) and Gram-negative bacteria. The sensitivities of all these bacteria are variable, but it has a bactericidal effect on E. coli, S. pneumoniae, H. influenzae, N. meningitidis, B. pertussis, V. cholerae, and Salmonella, Shigella and Bacteroides species. It is bacteriostatic for some streptococci and staphylococci.
Because of its toxicity, chloramphenicol is reserved for life-threatening infections, particularly with H. influenzae or Salmonella typhi. It is used topically for conjunctivitis (Ch. 50).
Resistance: Resistance is transferred by plasmids and involves the production of an acetyltransferase that inactivates the drug by acetylation, preventing it from binding to the ribosome. The acetyltransferase is produced by many Gram-negative bacteria but can also be induced in Gram-positive bacteria. Resistant bacteria may also show reduced uptake of the drug.
Pharmacokinetics: Chloramphenicol is well absorbed orally and can also be given intravenously. It is widely distributed, including into CSF and the biliary tree; it crosses the placenta and is present in breast milk. Chloramphenicol is metabolised in the liver and has a half-life of 2–12 h.
The most important unwanted effect is bone marrow toxicity. Reversible anaemia, thrombocytopenia or neutropenia can occur, particularly in those receiving high or prolonged dosing. Aplastic anaemia is rare, but usually fatal.
Peripheral neuritis, optic neuritis, headache.
Premature infants and babies less than 2 weeks old have immature glucuronyl transferase and reduced drug elimination. Chloramphenicol can accumulate in neonates, causing the ‘grey baby syndrome’. Initial symptoms include vomiting and cyanosis, followed by hypothermia, vasomotor collapse and an ashen grey discoloration of the skin. There is a high mortality.
Mechanism of action: Clindamycin inhibits bacterial protein synthesis in a similar manner to the macrolide antibacterials.
Spectrum of activity: Clindamycin is used for staphylococcal bone infection such as osteomyelitis and sometimes for soft tissue infections.
Mechanism of action: Fusidic acid is a steroid compound that inhibits bacterial protein synthesis. It forms a complex with the ribosome and inhibits elongation of the peptide chain.
Spectrum of activity: Fusidic acid is a narrow-spectrum antibacterial, mainly active against Gram-positive bacteria. It is most commonly used for treatment of penicillin-resistant S. aureus, especially in the treatment of osteomyelitis. It is bactericidal.
Resistance: Resistance occurs usually transferred by plasmids, and involves an altered ribosomal binding site for the drug. Resistance develops rapidly when fusidic acid is used alone, so it is usually given in combination with another drug.
Mechanism of action: The oxazolidinones are active against non-replicating bacteria. They have a unique mechanism of action, binding to the ribosomal 50S subunit and preventing initiation of protein synthesis, unlike many other antibacterials which inhibit chain elongation.
Spectrum of activity: Linezolid is active against a range of Gram-positive organisms, including MRSA and also vancomycin-resistant Enterococcus faecium.
Resistance: Resistance is due to mutation, leading to modification of the ribosomal target for the drug. This can develop with prolonged treatment, or with inadequate doses.
Pharmacokinetics: Linezolid is well absorbed orally. It is mainly metabolised in the liver; it has a half-life of 5 h.
Nausea, vomiting, taste disturbances, diarrhoea.
Myelosuppression with anaemia, neutropenia and thrombocytopenia.
Optic neuropathy with prolonged use (over 28 days).
Linezolid is a weak non-selective monoamine oxidase inhibitor, and dietary restriction of tyramine is advisable (see Ch. 22).
Drugs affecting bacterial metabolism
The therapeutic importance of the sulphonamides has diminished because of the spread of resistance and there are now only a few situations (nonetheless important) in which they are first-choice drugs. Sulfamethoxazole is only used in combination with trimethoprim, as co-trimoxazole (see below).
Mechanism of action: Folate is a nutrient essential for cell growth and is used to manufacture purines and thymidine for incorporation into DNA. Unlike humans, who obtain folate from the diet, bacteria cannot utilise pre-formed folate and must synthesise it from p-aminobenzoic acid (PABA). Sulphonamides are structurally similar to PABA and inhibit dihydropteroate synthetase in the synthetic pathway for folic acid (Fig. 51.4).
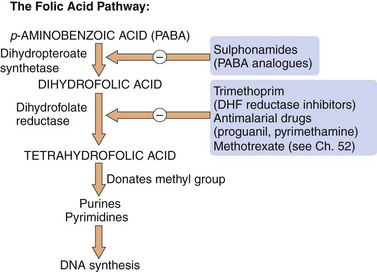
Fig. 51.4 Sites of action of sulphonamides and dihydrofolate (DHF) reductase inhibitors in the folic acid pathway.
Selective inhibitors of bacterial, plasmodial and human DHF reductase isozymes are used respectively as antibacterial (trimethoprim), antimalarial (proguanil, pyrimethamine) and anti-cancer (methotrexate) drugs.
Spectrum of activity: Sulphonamides have a bacteriostatic action against a wide range of Gram-positive and Gram-negative bacteria and are also active against Toxoplasma, Chlamydia and Nocardia species. Because of the frequency of resistance in many of these micro-organisms, sulphonamides are given as sole therapy only for the treatment of nocardiosis or toxoplasmosis.
Resistance: Resistance is common and occurs through the production of a mutated dihydropteroate synthetase with reduced affinity for sulphonamide binding (Figs 51.2 and 51.4). Resistance is transmitted among Gram-negative bacteria by plasmids. Resistance in S. aureus occurs as a result of excessive synthesis of PABA. Some resistant bacteria have reduced uptake of sulphonamides.
Pharmacokinetics: Sulphonamides are well absorbed orally, and a parenteral preparation of sulfadiazine is available. They are widely distributed and cross the blood–brain barrier and placenta.
Sulphonamides are metabolised in the liver, initially by acetylation, which shows genetic polymorphism (Ch. 2). The acetylated products have no antibacterial action but retain a risk of toxicity. The parent drugs and their N-acetyl metabolites are excreted by the kidney. Most sulphonamides have half-lives of about 12 h.
Rashes, including toxic epidermal necrolysis and Stevens–Johnson syndrome.
Haemolysis in individuals with glucose-6-phosphate dehydrogenase deficiency (Chs 47 and 53).
Neutropenia, thrombocytopenia.
Sulphonamides should not be used in the last trimester of pregnancy or in neonates, because the drug competes for bilirubin-binding sites on albumin; this can raise the concentration of unconjugated bilirubin and increases the risk of kernicterus.
Trimethoprim: Trimethoprim can be used alone or, less commonly, combined with the sulphonamide sulfamethoxazole as co-trimoxazole.
Mechanism of action: Trimethoprim inhibits dihydrofolate reductase, which converts dihydrofolate to tetrahydrofolate (Fig. 51.4). The bacterial enzyme is inhibited at much lower concentrations of trimethoprim than its mammalian counterpart. The combination of trimethoprim with sulfamethoxazole (co-trimoxazole) acts synergistically to prevent folate synthesis by bacteria. However, resistance to the sulfamethoxazole component and the incidence of unwanted effects limit the value of this combination.
Spectrum of activity: Trimethoprim has broad-spectrum bacteriostatic activity against Gram-positive and Gram-negative bacteria. In many urinary and respiratory tract infections trimethoprim alone gives results similar to the combination with sulfamethoxazole. Co-trimoxazole is effective against the protozoan Pneumocystis jirovecii, which causes pneumonia in people with AIDS or other immunodeficiencies, and this is now its major indication (see below).
Resistance: Resistance to trimethoprim occurs in a variety of ways, including the production of mutated dihydrofolate reductase that is insensitive to trimethoprim.
Drugs used for tuberculosis
Tuberculosis is usually treated with a multidrug regimen because of the rapid development of resistance. Some drugs used to treat mycobacterial infections also have other clinical uses.
Mechanism of action and spectrum of activity: Rifamycins act by inhibition of bacterial DNA-dependent RNA polymerase and inhibit mRNA transcription. They have a bactericidal action. Rifampicin (rifampin) has a broad spectrum of activity and is used in combination with other drugs for the treatment of mycobacterial infections (M. tuberculosis and M. leprae), brucellosis, Legionella infections, serious staphylococcal infections and endocarditis. In the UK, rifampicin is considered an essential drug for treatment of tuberculosis. Rifampicin is also used for prophylaxis against meningococcal meningitis and H. influenzae type b infection. Rifabutin is used for treatment of tuberculosis and for prophylaxis against Mycobacterium avium complex infection (which most commonly occurs in people who are infected with HIV) and other mycobacterial infections. Rifaximin is a non-absorbable rifamycin used to treat traveller's diarrhoea (Ch. 35).
Resistance: Resistance develops rapidly, which limits the wider use of rifampicin as an antibacterial drug, other than as part of combination treatment for tuberculosis. It is acquired by a one-step genetic mutation of the bacterial DNA-dependent RNA polymerase.
Pharmacokinetics: Oral absorption of rifampicin and rifabutin is good and an intravenous formulation of rifampicin is also available. The bioavailability of rifabutin is low (20%) compared with rifampicin. Rifampicin and rifabutin are metabolised in the liver and have half-lives of 1–6 h and 35–40 h, respectively.
Diarrhoea and antibiotic-associated colitis with rifampicin.
Hepatotoxicity, usually only producing a transient rise in plasma transaminases but occasionally more severe; regular monitoring is recommended.
Orange coloration of tears, sweat, urine.
Leucopenia, thrombocytopenia or anaemia with rifabutin.
Various symptoms with intermittent use of rifampicin, which include influenza-like symptoms, respiratory symptoms, renal failure, shock, disseminated intravascular coagulation and acute haemolytic anaemia.
Drug interactions of rifamycins: induction of drug-metabolising enzymes in the liver (Ch. 2) can reduce plasma concentrations of oestrogen in those taking oral contraceptives (Ch. 45) and of several other drugs including phenytoin (Ch. 23), warfarin (Ch. 11) and sulfonylureas (Ch. 40).
Mechanism of action: Isoniazid is an important and specific drug for the treatment of M. tuberculosis. It is a prodrug activated by catalase–peroxidase activity within the mycobacteria. The activated drug acts on enzymes in the cell to inhibit the synthesis of long-chain mycolic acids, which are unique to the cell wall of mycobacteria. It is bactericidal against dividing organisms, but bacteriostatic on resting organisms. In the UK it is considered an essential drug for treatment of tuberculosis along with rifampicin.
Resistance: Resistance occurs rapidly if isoniazid is used alone and may be due to mutations in the enzymes responsible for the synthesis of mycolic acids, making them less susceptible to the drug. Resistance is currently uncommon in developed countries, but can be troublesome in developing countries.
Pharmacokinetics: Oral absorption of isoniazid is good but reduced by food. It is metabolised by acetylation in the liver, which is subject to genetic polymorphism. Rapid acetylators show extensive first-pass metabolism and plasma isoniazid concentrations are half of those in slow acetylators. The half-life is 0.5–2 h in rapid acetylators and 2–6.5 h in slow acetylators.
Nausea, vomiting, constipation, dry mouth.
Peripheral neuropathy with high doses. This can be prevented by prophylactic use of oral pyridoxine supplements in people at high risk, for example those with diabetes, alcoholism, chronic renal failure, malnutrition or HIV infection. Neuropathy is more common in slow acetylators.
Hepatitis is rare, but regular monitoring with liver function tests is recommended.
Systemic lupus erythematosus-like syndrome. Positive antinuclear antibodies are found in 20% of people during long-term treatment, but fewer develop symptoms.
Mechanism of action: Pyrazinamide is a prodrug that acts through metabolites formed by pyrazinamidase, an enzyme found in M. tuberculosis. The product pyrazinoic acid lowers intracellular pH, inactivates a vital enzyme in fatty acid synthesis and destroys the bacterium. It is bactericidal to dividing cells.
Resistance: Resistance results from a point mutation in the gene which codes for pyrazinamidase. It develops rapidly if pyrazinamide is used as a sole treatment for tuberculosis.
Mechanism of action: Ethambutol probably functions as an arabinose analogue and inhibits arabinosyl transferase, resulting in impaired synthesis of the cell wall of mycobacteria. Ethambutol is primarily bacteriostatic. It is effective against M. tuberculosis and several other mycobacteria, including M. avium complex.
Resistance: Resistance may be due to gene mutations that inhibit the binding of ethambutol to its target enzyme. It develops slowly, but is common during prolonged treatment of tuberculosis if ethambutol is used alone.
Other drugs used in the treatment of tuberculosis: Other drugs can be used as second-line treatments in multidrug-resistant tuberculosis. These include cycloserine, capreomycin, amikacin, ciprofloxacin, moxifloxacin, azithromycin, clarithromycin, streptomycin and p-aminosalicylic acid. Drugs used in countries other than the UK include thiacetazone and protionamide.
Drugs used for leprosy
The drugs recommended for treatment of leprosy, which is caused by Mycobacterium leprae, are rifampicin (see above), dapsone and clofazimine.
Mechanism of action and use: Dapsone is similar to the sulphonamides and acts by inhibition of folate synthesis. It is the most active drug against M. leprae. Dapsone is also used to treat pneumocystis pneumonia and dermatitis herpetiformis.
Pharmacokinetics: Dapsone is well absorbed from the gut. It is metabolised in the liver and undergoes enterohepatic cycling. It has a long half-life (27 h).
Blood disorders: haemolysis and methaemoglobinaemia (Ch. 53), although these are rare at the doses used for treatment of leprosy.
Mechanism of action and use: Clofazimine is a dye that interferes with DNA replication and is used as a second-line drug in the event of dapsone intolerance in people with leprosy. It is given orally.
Principles of antibacterial therapy
Antibacterial therapy is widely misused, which encourages selection of resistant organisms. In particular, use of antibacterials for viral illnesses such as the common cold or sore throats is to be discouraged. The following guidelines outline the principles that should be considered in the choice of a safe and effective antibacterial therapy.
Empirical treatment
Most antibacterial therapy is started without prior identification of the organism and its antibacterial drug sensitivities. Such treatment should be guided by the clinical diagnosis and knowledge of the most common pathogenic bacteria responsible for the infection to be treated. Local information about patterns of antibacterial resistance is an important consideration.
Spectrum of antibacterial activity
A drug with a narrow spectrum of activity should be used in preference to a broad-spectrum drug whenever possible. The unnecessary use of broad-spectrum antibacterials encourages the development of resistant bacteria. This can present problems for the person treated, due to the selection of resistant pathogens or colonisation by resistant bacteria from the environment. For the community, the selection of resistant pathogens can create problems by rendering standard antibacterial therapy less reliable. Broad-spectrum antibacterial cover is sometimes appropriate, for example in a seriously ill person when the infecting bacterium is unknown and a variety of bacteria could be causing the condition being treated.
Combination therapy
Treatment with more than one antibacterial drug should not be used routinely. It may, however, be valuable to provide broad-spectrum cover in serious illness when the organism is unknown, for example the combination of cefotaxime and metronidazole to cover aerobic and anaerobic organisms in suspected Gram-negative septicaemia. When resistance is likely to develop readily to the first-choice drug during prolonged treatment, the use of combination therapy can minimise that risk, for example in the treatment of infective endocarditis or tuberculosis.
Bactericidal versus bacteriostatic drugs
In some situations, bactericidal drugs are preferred to bacteriostatic drugs, for example for the treatment of infective endocarditis (when bacteria divide infrequently) or when the person being treated is immunocompromised (and host defences are ineffective for assisting eradication). In most other situations the choice is not important.
Site of infection
This may determine the choice of drug; for example, some antibacterials only achieve low concentrations in the biliary tree, urine, bone or cerebrospinal fluid.
Mode of administration
Oral therapy is usually preferred to parenteral treatment. Exceptions include the treatment of serious infections for which reliable plasma drug concentrations are essential, when the drug is only available in parenteral formulation, or when gastrointestinal absorption may be unreliable, for example after abdominal surgery.
Duration of therapy
This should be as short as is compatible with adequate treatment of the infection. The decision is often arbitrary, for example 7–10 days in many infections. Some infections can be effectively treated over much shorter periods; for example, courses of 1–3 days are usually adequate for uncomplicated lower urinary tract infections in women. There is evidence that the conventional longer courses of treatment for many other infections may be unnecessary. For a few infections, long periods of treatment may be essential to eliminate semi-dormant organisms or those in ‘privileged sites’ to which antibacterial drug penetration is poor. Examples include infective endocarditis, osteomyelitis and tuberculosis. Some antibacterials produce a ‘post-antibiotic effect’, in which there is delayed regrowth of surviving bacteria following exposure to the drug. This is most marked with aminoglycosides such as gentamicin, but also occurs with other drugs, including β-lactam antibacterials.
Chemoprophylaxis
The use of chemoprophylaxis to prevent infection is important in many situations. Common examples include prevention of meningococcal meningitis, H. influenzae type b infection or pertussis in close contacts of an infected person, and pre-operative prophylaxis before many surgical procedures. More prolonged prophylaxis is used to prevent pneumococcal infection after splenectomy or in people with sickle cell disease.
Treatment of selected bacterial infections
This section is not intended to be comprehensive. It will outline the approach to antibacterial therapy in several common bacterial infections. The choice of antibacterial drug for these infections will depend on factors such as local patterns of bacterial resistance or the risk of C. difficile infection, which make universal recommendations impossible.
Upper respiratory tract infections
Most upper respiratory tract infections are caused by viruses, producing symptoms of the common cold. Symptomatic treatment is all that should be offered, with an antihistamine (e.g. chlorphenamine; Ch. 39) or an anti-muscarinic spray (e.g. ipratropium; Ch. 12) to reduce rhinorrhoea and sneezing. An α-adrenoceptor agonist given orally or nasally (e.g. xylometazoline) can reduce nasal congestion, but prolonged use can provoke a rebound effect (rhinitis medicamentosa) (Ch. 39). A non-steroidal anti-inflammatory drug (NSAID; Ch. 29) can be used to reduce associated headache and malaise. Antibacterial drugs are widely prescribed for upper respiratory tract symptoms but have no benefit.
Sinusitis and otitis media
Sinusitis and otitis media accompany catarrhal conditions in childhood and frequently follow an upper respiratory tract infection. Sinusitis produces headache, facial pain, fever and purulent rhinorrhoea. A nasal decongestant such as an α-adrenoceptor agonist can be helpful, in conjunction with an analgesic. An antibacterial is often not beneficial in acute sinusitis unless there is marked facial swelling and pain, or failure to resolve after 7 days. The most common infecting organisms are H. influenzae (which often produces β-lactamase), S. pneumoniae and Moraxella catarrhalis. Suitable antibacterial drugs include amoxicillin (or amoxicillin plus clavulanic acid if there is no improvement after 48 h), doxycycline and clarithromycin. Chronic sinusitis usually requires correction of an anatomical obstruction in the nose.
Otitis media is very common in childhood. When associated with an effusion in the middle ear, increased pressure causes pain and perforation of the eardrum. The organisms responsible are similar to those causing acute sinusitis. In more than 80% of affected children the condition is self-limiting over 2–3 days without treatment. An antibacterial such as amoxicillin or clarithromycin should be used if symptoms have not resolved after 72 h, or if there are systemic symptoms. Surgery is occasionally necessary for recurrent infections.
Lower respiratory tract infections
Acute bronchitis: This is characterised by new-onset, often productive cough without evidence of pneumonia. It is usually caused by a viral infection and the cough often takes 2–4 weeks to resolve without treatment. Antibacterial treatment is inappropriate and does not alter the course of the illness. Even if there is underlying chronic obstructive airways disease, the evidence for benefit from antibacterial drugs is small, although they may slightly shorten the duration of symptoms. In such cases S. pneumoniae (pneumococcus), H. influenzae or M. catarrhalis are commonly found in the sputum, but these micro-organisms are often isolated in remissions as well. If an antibacterial drug is used then 5 days' treatment with amoxicillin, doxycycline or clarithromycin will be effective against the most likely pathogens. Co-amoxiclav (amoxicillin with clavulanic acid) can be used for resistant H. influenzae.
Pneumonia: Primary community-acquired pneumonia is most commonly caused by S. pneumoniae, and less commonly by H. influenzae and staphylococci. ‘Atypical’ micro-organisms can also cause pneumonia, such as Legionella species, Mycoplasma pneumoniae or Chlamydia pneumoniae. Appropriate antibacterial treatment will be dictated by the most likely infecting agent.
Amoxicillin is the treatment of choice if pneumococcus is suspected. For people who are penicillin-allergic, clarithromycin will cover most likely micro-organisms, including ‘atypical’ ones. Oral amoxicillin combined with clarithromycin is often used for community-acquired pneumonia requiring admission of the person to hospital. Doxycycline is increasingly used as an alternative oral treatment because of a lower risk of C. difficile-related colitis. Severe community-acquired pneumonia (defined by the CURB-65 score; Table 51.3) is usually treated with intravenous therapy comprising benzylpenicillin with intravenous clarithromycin or doxycycline. Co-amoxiclav is often used in place of benzylpenicillin for life-threatening pneumonia, or when Gram-negative organisms are suspected, to cover β-lactamase-producing organisms. A quinolone with activity against pneumococci, such as moxifloxacin, would be an alternative choice. Adjunctive treatment of pneumonia may include supplemental oxygen via a facemask, pain relief for pleurisy and ensuring adequate hydration.
Table 51.3
CURB-65 score for predicting mortality in community-acquired pneumonia
Confusion of new onset (abbreviated mental test score ≤8/10)
Serum urea >7 mmol⋅L−1
Respiratory rate ≥30 breaths⋅min−1
Blood pressure: systolic <90 mmHg or diastolic ≤60 mmHg
Age ≥65 years
Each risk factor above scores one point.
Total score: 0–1 points, mortality 1.5%; 2 points, mortality 9.2%; ≥3 points, mortality 22%.
Secondary pneumonias occur in patients with other concurrent diseases, often during a stay in hospital (nosocomial or hospital-acquired pneumonia). A wide range of pathogens may be involved and parenteral drug treatment is usually necessary. A cephalosporin (e.g. cefuroxime) or co-amoxiclav are often used for early-onset infections, or an antipseudomonal penicillin (e.g. piperacillin with tazobactam) or ciprofloxacin for late-onset infection. An aminoglycoside such as gentamicin can be added for severe infection where P. aeruginosa infection is suspected.
Chronic lung sepsis: This encompasses lung abscess, empyema and bronchiectasis. The pathogens in lung abscesses vary according to the immune status of the individual. Ideally, the antibacterial treatment should be directed by isolation and sensitivity testing of the bacteria. Empyema requires drainage and then specific antibacterial therapy directed at the cultured pathogen. Bronchiectasis is most frequently associated with colonisation by H. influenzae, and less often Pseudomonas species or S. pneumoniae. A quinolone such as moxifloxacin or a macrolide such as azithromycin are suitable empirical treatment choices. Increasing use is being made of inhaled nebulised antibacterial drugs, such as tobramycin, to treat frequent exacerbations. Adjunctive treatment with bronchodilators, mucolytics and physiotherapy may be useful (see also cystic fibrosis, Ch. 13).
Urinary tract infections
Urinary tract infections are more common in women than men, because of their shorter urethra. Infections can occur in structurally normal urinary tracts or in association with a structural genitourinary abnormality that impairs drainage of urine or acts as a focus for infection, such as a stone in the kidney or bladder. An indwelling urinary catheter is often associated with bacterial colonisation of the urine that is almost impossible to eradicate, but often does not cause any symptoms.
The most frequent bacterial cause of urinary tract infection is E. coli. Hospital-acquired infections are often caused by Klebsiella, Enterobacter and Serratia species or by P. aeruginosa, because these organisms can be selected as resistant bacteria following antibacterial usage. Proteus mirabilis is often found if there are stones in the urinary tract. Less commonly, staphylococci, especially Staphylococcus saprophyticus, are responsible.
Uncomplicated urinary tract infection is confined to the bladder (cystitis) and in women can be treated by a short course (3 days) of an aminopenicillin such as amoxicillin. Alternative drugs include a first-generation cephalosporin (e.g. cefalexin), trimethoprim and nitrofurantoin. A quinolone such as ciprofloxacin can be useful for P. aeruginosa infections. Men should be treated for longer (usually 7 days).
Complicated urinary tract infections also involve the kidney (pyelonephritis), or the prostate in males, and require longer courses of treatment. For pyelonephritis, initial intravenous therapy is usually started with broad-spectrum drugs such as aztreonam, ciprofloxacin or cefuroxime, sometimes with an initial dose of gentamicin; treatment is usually continued for 10–14 days. For acute prostatitis, oral treatment with trimethoprim or ciprofloxacin for at least 4 weeks is recommended.
Treatment of infection with an indwelling urinary catheter is only recommended if there are systemic symptoms of infection such as fever or rigors.
Long-term antibacterial prophylaxis against urinary tract infections may be necessary to prevent recurrent infection if there are underlying urinary tract abnormalities. Suitable drugs, usually given at low dosage, include trimethoprim, nitrofurantoin and cefalexin.
Gastrointestinal infection
In the UK, infectious diarrhoea is usually caused by viruses and is self-limiting. However, gastroenteritis (a syndrome that includes nausea, vomiting, diarrhoea and abdominal discomfort) can result from ingestion of bacterial pathogens.
‘Food poisoning’ of bacterial origin can occur from ingestion of a pre-formed bacterial toxin (e.g. from Clostridium botulinum or S. aureus), with onset of symptoms usually within hours, or it can be caused by ingested bacteria infecting the bowel. Severe bacterial infection of the large intestine can cause dysentery, an inflammatory disorder often associated with fever, abdominal pain, and blood and pus in the faeces. The most common cause of bacterial diarrhoea (especially in children in developing countries) is E. coli, which produces powerful enterotoxins. In other circumstances, Salmonella species, Campylobacter species, V. cholerae, Shigella species or various other organisms are responsible.
If diarrhoea is severe, fluid replacement is often necessary. Antibacterial treatment is not usually recommended even if bacterial infection is suspected, unless there are systemic symptoms such as fever, rigors and hypotension. Ciprofloxacin and clarithromycin are effective for Campylobacter enteritis and shigellosis. Salmonella infections can be treated with ciprofloxacin, unless S. typhi is suspected, when cefotaxime is preferred.
Antibacterial drugs can cause diarrhoea due to alteration of bowel flora, which usually resolves rapidly when the drug is withdrawn. However, if it is complicated by colonisation with C. difficile then oral metronidazole or oral vancomycin will be necessary to eliminate the pathogen.
Biliary tract infection
Acute cholecystitis and cholangitis are often caused by E. coli and most often occur if there is biliary obstruction. Supportive treatment with fluid and electrolyte replacement is usually required. Antibacterial therapy with a cephalosporin, ciprofloxacin or gentamicin is usually effective. Combination therapy is recommended if the infection is severe; alternatively, a ureidopenicillin such as piperacillin with tazobactam can be given alone. Treatment is usually given for 7–10 days.
Osteomyelitis
Infection of bone produces necrotic tissue and generates an avascular privileged site for bacteria that antibacterial drugs penetrate to only a limited extent. Organisms involved include S. aureus, which adheres readily to bone matrix, various streptococci, Serratia species, P. aeruginosa and enteric Gram-negative rods.
Early antibacterial treatment is essential and surgical intervention may be necessary to remove necrotic tissue. The choice of drug depends on the suspected organisms. First-line treatment is often with flucloxacillin or clindamycin, combined with fusidic acid or rifampicin for the first 2 weeks if a prosthesis is present or the infection is severe. Amoxicillin or cefuroxime is usually used if H. influenzae is identified, or vancomycin for MRSA. Acute infections are treated with intravenous antimicrobials for 6 weeks, but chronic infections are treated for at least 12 weeks. If long-term therapy is necessary for chronic refractory osteomyelitis an oral quinolone such as ciprofloxacin can be substituted.
Septic arthritis
The most common organism is S. aureus, or less frequently streptococci. The standard treatment is with flucloxacillin or clindamycin for individuals who are penicillin-allergic. Vancomycin is used for MRSA. Treatment should be continued for 4–6 weeks.
Cellulitis
This usually complicates a wound, ulcer or dermatosis. In most cases the infecting organisms are S. aureus or streptococci. Treatment is usually with a β-lactam antibacterial that is active against β-lactamase-producing S. aureus. Flucloxacillin is normally used. Clarithromycin or clindamycin is used for individuals who are penicillin-allergic.
Septicaemia
Septicaemia is a bacterial infection involving the bloodstream and can present with fever or, if more severe, result in circulatory collapse from vasodilation, capillary leak and impaired myocardial contractility. Gram-positive organisms are a more frequent cause than Gram-negative organisms, with about 60% of infections arising from respiratory, intra-abdominal and urinary tract sources. Septicaemia is a medical emergency requiring intensive fluid replacement, plasma volume expansion and electrolyte correction. Noradrenaline (norepinephrine) or other vasopressors may be used to support the blood pressure (Ch. 4). There have been many advances in our understanding of the pathogenesis of sepsis and the associated immune activation, but little improvement in our ability to manage the complications of sepsis. Adrenal insufficiency is common in severe sepsis and treatment with low-dose hydrocortisone may reduce the duration of shock.
If the source of the infection is not clinically apparent then empirical antibacterial therapy is given to cover as wide a range of potential infecting organisms as possible. The prognosis is much worse if first-line drugs are ineffective. Suitable treatment would be with an antipseudomonal penicillin such as piperacillin with tazobactam or a broad-spectrum cephalosporin (e.g. cefotaxime, or ceftazidime if pseudomonal infection is suspected). A carbapenem such as meropenem or imipenem (with cilastatin) can be used if the infection was hospital-acquired. Metronidazole is added if anaerobic infection is suspected, or vancomycin if MRSA is suspected.
Immunocompromised and neutropenic individuals are at particularly high risk from septicaemia. A combination of gentamicin with a broad-spectrum penicillin, cephalosporin, piperacillin with tazobactam or meropenem can be given. Metronidazole is usually added if anaerobic infection is suspected; flucloxacillin or vancomycin is added if Gram-positive infection is suspected. Failure to respond to such triple therapy within 48 h may indicate a fungal infection, for which amphotericin can be added (see below).
Infective endocarditis
The majority of cases of infective endocarditis are caused by bacterial pathogens, most commonly oral streptococci, followed by enterococci, S. aureus and coagulase-negative staphylococci. Endocarditis usually arises on the endothelial surface of a pre-existing heart defect (e.g. valvular heart defect, ventricular septal defect) or on a prosthetic heart valve. It arises when micro-organisms enter the bloodstream and become established on the endocardium, where they may adhere to pre-existing fibrin–platelet vegetations. Bacteria enter the blood during dental procedures, vigorous teeth cleaning or some surgical procedures.
Untreated infection can destroy the infected heart valve and produce severe haemodynamic disturbance. Systemic complications can also arise from embolisation of vegetation from the valve, from bacteraemia, or through immune complexes that form in response to the infection.
When infection is suspected, blood cultures must be taken and empirical antimicrobial treatment started. Prior to identification of the organism, treatment is usually started with intravenous flucloxacillin or benzylpenicillin combined with low-dose gentamicin. If the organism is sensitive to penicillin, then the benzylpenicillin is continued for 4 weeks and the gentamicin stopped after 2 weeks. Vancomycin and rifampicin are substituted for the penicillin in people with penicillin allergy when there is infection on a prosthetic heart valve or if MRSA is suspected.
Antibacterial prophylaxis is no longer recommended prior to procedures such as dental treatment, since there is no good evidence that it prevents endocarditis.
Meningitis
Bacterial meningitis is a medical emergency. The most likely organism depends on the age of the person (Table 51.4). Empirical selection of therapy is usually necessary and treatment should be started at the first suspicion of bacterial meningitis. A single dose of benzylpenicillin can be given if the person is outside hospital, but cefotaxime (with the addition of amoxicillin for those over 50 years old) is the preferred treatment in hospital. Chloramphenicol is an option for those who have an allergy to both penicillin and cephalosporins. Treatment is given for 7 days for meningococcus, and 10 days for H. influenzae or pneumococcus. Rifampicin is given for 2–4 days before hospital discharge if the meningitis was caused by meningococcus or H. influenzae. Close contacts of people with meningococcal or H. influenzae type b meningitis are usually given rifampicin as prophylaxis against infection.
Table 51.4
Organisms causing bacterial meningitis
| Age | Organism |
| <1 month | Group B streptococci |
| 1 month–4 years | Haemophilus influenzae |
| >4 years to young adult | Neisseria meningitidis (meningococcus) |
| Older adults | Streptococcus pneumoniae (pneumococcus) |
Dexamethasone should be considered immediately, and no later than 12 h after starting the antibacterial. This reduces the frequency of neurological complications.
Tuberculosis
M. tuberculosis readily develops resistance to single-drug therapy. Three or four drugs are used for the first 2 months (‘initial phase’) to rapidly reduce the bacterial population prior to information on bacterial sensitivities becoming available, following which treatment is continued with two drugs for a further 4 months (‘continuation phase’) to achieve a cure. In some cases, more prolonged treatment may be necessary, especially for tuberculous meningitis or for resistant mycobacteria.
A standard regimen in the UK includes rifampicin, isoniazid, ethambutol and pyrazinamide for the initial phase (or until bacterial sensitivities are known), followed by rifampicin and isoniazid (preferably in a combination preparation) for a further 4 months. More prolonged treatment is sometimes necessary for cavitating lung disease or slow clearance of bacteria from the sputum. Ethambutol is not used for treatment of young children because of difficulty in monitoring for eye toxicity.
Streptomycin is used in some countries in the initial phase of treatment or if resistance to isoniazid is known. Thiacetazone is often used with isoniazid and initially streptomycin in countries that cannot afford rifampicin. Adherence to the treatment regimen can be a major problem in the treatment of tuberculosis, and combination tablets are often used to maximise this. In developed countries, directly observed treatment (or DOT) has been instituted to improve adherence. This can result in major improvements in eradication rates.
Multidrug-resistant tuberculosis (with resistance to at least rifampicin and isoniazid) is becoming more common, and new treatment strategies are needed to deal with emerging strains of M. tuberculosis. There are also increasing problems with the treatment of tuberculosis in people with HIV infection. This reflects the propensity for interactions among the antiretroviral drugs and antituberculous therapy, and overlapping unwanted effects.
Fungal infections
Fungi (including yeasts) usually infect skin or superficial mucous membranes, but can more rarely involve internal organs. Most fungal infections occur because of an underlying defect in host resistance. Fungi grow more readily in immunosuppressed individuals or following the suppression of normal flora with antibacterials. Good hygiene and the avoidance of sources of infection are important complementary approaches to the use of antifungal drugs.
Compared with antibacterial drugs, fewer drugs have been developed that have activity against fungi, and many of these are toxic to humans. A simplified outline of the ways in which antifungal drugs work is shown in Figure 51.5.
Antifungal drugs
Drugs that impair membrane barrier function
Mechanism of action: Polyenes bind to ergosterol in the cell wall of fungi and form aqueous pores that promote leakage of intracellular ions and disruption of active transport mechanisms in the membrane. They can be fungistatic or fungicidal.
Spectrum of activity: Nystatin is particularly effective for infections with Candida species. Amphotericin is active against all common fungi that cause systemic infection (Candida, Aspergillus, Mucor and Cryptococcus species).
Resistance: Acquired resistance is rare but can occur in immunosuppressed people. Fungi develop a mutation that permits synthesis of the cell membrane without using ergosterol.
Pharmacokinetics: Nystatin is too toxic for systemic use and is not absorbed from the gastrointestinal tract. It is therefore used topically for Candida albicans infections, for example as cream for skin infection, as vaginal pessaries or orally for buccal and bowel infections.
Amphotericin is poorly absorbed from the gut and is usually given intravenously for treatment of serious systemic fungal infections. An oral formulation is used for buccal and intestinal candidal infections. Amphotericin can also be given intrathecally for fungal meningitis. Amphotericin binds to steroid molecules in human tissue, and is released slowly and eliminated via the biliary tract and kidney. It has a very long half-life (about 2 days).
Lipid delivery vehicles for amphotericin have been developed to reduce its nephrotoxicity. These formulations alter drug distribution and help to concentrate the drug at the site of infection. Formulations include liposomal spheres (in which the drug is dissolved in phospholipid membrane vesicles), lipid complexes (in which the lipid exists in ribbons interspersed with amphotericin) and a colloidal dispersion of lipid discs that incorporate the drug. The lipid component is probably cleared from the blood by mononuclear phagocytes.
Unwanted effects: Nystatin is virtually free of both toxic and allergic unwanted effects when used topically. Used orally for intestinal infection, it can cause gastrointestinal upset. Host toxicity with amphotericin is due to binding to cholesterol rather than ergosterol. Intravenous infusion of amphotericin is commonly associated with the following:
fever and rigors during the first week of therapy,
anorexia, nausea, vomiting, diarrhoea,
headache, muscle and joint pain,
anaphylaxis, which makes a test dose advisable,
dose-related nephrotoxicity, which is the major limiting factor in treatment. It presents with reduced glomerular filtration rate and produces hypokalaemia and hypomagnesaemia through tubular leakage of K+ and Mg2+. Lipid formulations substantially reduce the risk of nephrotoxicity and are particularly useful to treat people with pre-existing renal impairment,
Drugs that inhibit cell wall synthesis
Mechanism of action: The imidazoles alter fungal cell membrane fluidity by inhibiting lanosterol 14α-demethylase, a form of cytochrome P450 that participates in the conversion of lanosterol to ergosterol. Reduced ergosterol synthesis alters fungal cell membrane fluidity, reduces the activity of membrane-associated enzymes and increases cell wall permeability. Accumulation of ergosterol precursors in the cell causes growth arrest. Although the equivalent human enzyme is much less sensitive to the effects of the drug, inhibition of cytochrome P450 isoenzymes can occur in human tissues, especially with ketoconazole (Ch. 2).
Spectrum of activity: The imidazoles are active against a wide variety of filamentous fungi including many Candida species. They are less active against Candida krusei. Clotrimazole is used for vaginal candidiasis and for dermatophyte infections, such as ringworm (tinea), the causative fungi of which vary geographically, but which generally are Trichophyton, Microsporon or Epidermophyton species. Ketoconazole can be used for systemic mycoses, resistant mucocutaneous candidiasis, resistant vaginal candidiasis and resistant dermatophyte infections.
Resistance: The development of resistance is rare, except during long-term use in people with AIDS. The mechanism involves a point mutation in the target enzyme, lanosterol 14α-demethylase, or development of an active pump that removes drug from the cell, especially in Candida species.
Pharmacokinetics: Absorption of imidazoles from the gastrointestinal tract is poor, but oral ketoconazole achieves plasma concentrations high enough to treat systemic infection. Clotrimazole is only used in topical formulations for superficial infections, for example skin and vagina. The imidazoles are metabolised in the liver. Ketoconazole has a half-life of about 6–10 h.
Unwanted effects: These are unusual with topical formulations, although oral miconazole can cause gastrointestinal upset. Oral ketoconazole can cause the following:
nausea, vomiting, abdominal pain,
hepatitis: asymptomatic elevation of liver enzymes is common; more severe hepatic reactions are unusual but can be fatal. Liver function must be monitored during systemic use of ketoconazole,
high doses of ketoconazole suppress androgen production and in males can cause oligospermia or gynaecomastia,
drug interactions: ketoconazole can inhibit metabolism of drugs that are eliminated by cytochrome P450; examples of drugs affected include ciclosporin, tacrolimus (Ch. 38) and warfarin (Ch. 11).
Mechanism of action and spectrum of activity: The triazoles have a similar mechanism of action and spectrum of activity to the imidazoles (see above). Fluconazole is used for candidiasis and for cryptococcal infection. Itraconazole is used for mucocutaneous candidiasis and for dermatophyte infections, such as pityriasis versicolor (caused by an organism known as Malassezia furfur or Pityosporum orbiculare) and tinea corporis or pedis (ringworms). Voriconazole is an ‘extended-spectrum’ triazole used for invasive aspergillosis, and serious infections caused by Scedosporium species, Fusarium species, or invasive fluconazole-resistant C. krusei and Candida glabrata.
Resistance: As with imidazoles, the development of resistance is rare, except during long-term use in people with AIDS. The mechanism involves development of an active pump that removes drug from the cell, especially in Candida species.
Pharmacokinetics: Oral absorption of triazoles is good. Formulations are also available for intravenous (fluconazole, voriconazole) and topical (itraconazole) use. The triazoles are metabolised in the liver and have long half-lives (6–30 h). Fluconazole penetrates well into cerebrospinal fluid, which is useful for treatment of cryptococcal meningitis.
Nausea, abdominal pain and diarrhoea.
Abnormalities of liver function and, occasionally, hepatitis or cholestasis. These are more common during prolonged treatment with itraconazole. Monitoring of liver function during systemic treatment is essential.
Increased risk of heart failure with itraconazole. The mechanism of its negative inotropic effect is not known.
Mechanism of action and use: Terbinafine is an allylamine that inhibits squalene epoxidase, the enzyme that converts squalene to ergosterol in the cell wall. It impairs fungal cell wall synthesis, and the intracellular accumulation of squalene is probably cytotoxic (Fig. 51.5). It is used topically for treatment of dermatophyte infections of the nails, and systemically for ringworm infections.
Mechanism of action: Caspofungin inhibits fungal cell wall synthesis, targeting the glucans that are found in fungal cell walls but not in human cells (Fig. 51.5). It inhibits the enzyme β(1,3)-D-glucan synthase, and prevents production of the main structural polymer in the fungal cell wall. Caspofungin is used to treat invasive aspergillosis as a second-line drug, and invasive candidiasis.
Resistance: This is uncommon at present, but can occur from a point gene mutation coding for a structural change in the target enzyme, which no longer binds the drug.
Drugs that inhibit macromolecule synthesis
Mechanism of action and use: Flucytosine is converted to 5-fluorouracil (5-FU) selectively in fungal cells by cytosine deaminase. 5-FU is an antimetabolite that competes with uracil for incorporation into fungal RNA, and is metabolised to compounds that inhibit enzymes involved in DNA synthesis (Fig. 51.5).
Flucytosine is only active against yeasts such as Candida, Aspergillus and Cryptococcus species and is used for systemic infections.
Resistance: Resistance occurs readily and arises through a mutation that produces a deficiency of cytosine deaminase or through excessive synthesis of uracil, which competes with the antimetabolite. For this reason, flucytosine is only used in combination with amphotericin or fluconazole.
Drugs that interact with microtubules
Mechanism of action and use: Griseofulvin inhibits dermatophyte mitosis by impairing the polymerisation of microtubule protein (Fig. 51.5). It is active against dermatophytes such as Microsporum, Epidermophyton and Trichophyton species.
Treatment of specific fungal infections
Aspergillus species can cause an invasive fungal infection that most commonly affects the lung. However, in immunocompromised individuals it can invade more widely and infect the heart, brain, sinuses and skin. The treatment of choice is voriconazole, which is more effective and less toxic than amphotericin. If this fails then itraconazole can be used. Caspofungin is reserved for infections that have failed to respond to standard treatments, or for those people who cannot tolerate the other drugs.
Candida
Amphotericin or nystatin is often used for oral candidiasis. If there is oropharyngeal disease that is refractory to topical treatment, then an absorbed drug such as fluconazole or itraconazole is used orally. Vulvovaginal infection is treated with cream and pessaries, and imidazole drugs such as clotrimazole are usually the first choice. Nystatin is an alternative for vulvovaginal candidiasis, but stains clothing yellow. For recurrent vulvovaginal infections, oral fluconazole or itraconazole should be taken once a week for 6 months. Superficial candidal infections of the skin are treated topically with cream, usually containing an imidazole. Terbinafine or nystatin can also be used topically.
Invasive candidiasis mainly occurs as a hospital-acquired infection, particularly in people who have had abdominal surgery, parenteral nutrition, multiple antibacterial drugs or central vascular lines. C. albicans is still the single most common organism, but C. krusei, C. glabrata and Candida parapsilosis are increasingly common. Amphotericin is the treatment of choice, sometimes combined with fluconazole. Caspofungin is an alternative.
Cryptococcus
Infection with Cryptococcus species usually occurs in people who are immunocompromised. It can cause life-threatening meningitis and is treated with intravenous amphotericin with flucytosine. Fluconazole is an alternative and can be given orally for prophylaxis against relapse.
Skin and nail infections
Topical therapy is usually suitable for infections with most dermatophytes. Fungal infection of the scalp (tinea capitis), body (tinea corporis), groin (tinea cruris), hand (tinea manuum), foot (tinea pedis) or nail (tinea unguium) will respond to most azoles. Griseofulvin is usually reserved for treatment of scalp infections. Nail infection usually requires systemic treatment with terbinafine or itraconazole.
Pityriasis versicolor can be treated topically or orally with itraconazole, or with oral fluconazole.
Viral infections
Viruses are small infective particles consisting of either DNA or RNA inside a protein coating (capsule), which in some viruses may be further surrounded by a lipoprotein coating. The proteins can have antigenic properties. Viruses lack any inherent metabolic machinery and must use the host's metabolic processes to replicate. Viruses access host cells after binding to recognition sites that are endogenous receptors for normal cellular constituents; for example, adrenoceptors, cytokine receptors, glycoproteins, etc. (Fig. 51.6). Drugs that interfere with host cell membrane cytokine receptors, such as CCR5 (see below), and surface glycoproteins will inhibit viral entry and interrupt virus replication.
The host will normally eliminate the virus by killing the infected cell. Cytotoxic T-lymphocytes recognise the viral surface proteins that are expressed by infected cells. The host can also produce antibodies that bind to and inactivate virus particles extracellularly. Vaccination is designed to generate this response.
Viruses utilise the host's metabolic processes and this makes it difficult to damage the virus without damaging the host. Importantly, antiviral drugs are only effective while the virus is replicating, so the earlier they are given in the course of the infection the more likely they are to work. An outline of the replication of RNA and DNA viruses is shown in Figures 51.6 and 51.7. Since the replicative mechanisms involved may be distinctive to one type of virus, some antiviral drugs are specific for a particular class of virus.
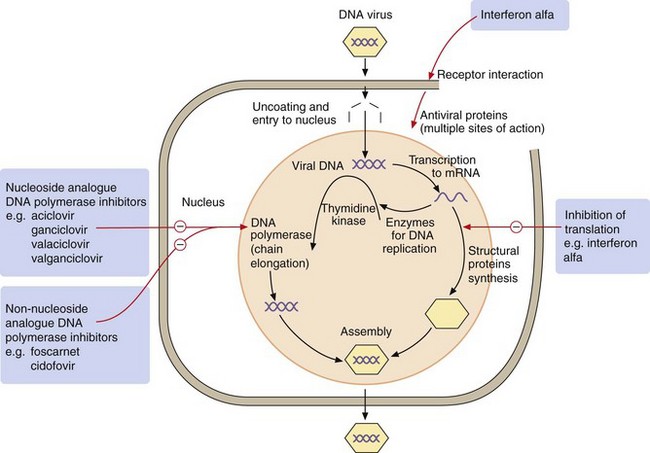
New antiviral drugs are being introduced into clinical practice at an increasing rate. Drugs are available to treat infection by RNA viruses (e.g. HIV, hepatitis C and influenza viruses) and DNA viruses (e.g. herpesviruses, cytomegalovirus and hepatitis B virus). Most of these drugs work by disturbing various steps in the replicative pathways of the virus. Because of the development of resistance and the variability in viral sensitivity to drugs it is sometimes necessary to use concurrently drugs that target different processes in the virus replication.
Resistance to antiviral drugs occurs readily. This relates to the high rate of natural occurrence of mutations in the viral genome and production of quasi-species of the virus. Viral polymerases have a high inherent error rate (especially RNA viruses) and viruses tolerate a large number of nucleoside mutations without losing their infectivity. Normally the large variety of viral quasi-species will be dominated by the variant most selected for survival and therefore use of an antiviral drug will select for growth of resistant variants.
Antiviral drugs
Nucleoside analogue HIV reverse transcriptase inhibitors:
Mechanism of action: Reverse transcriptase inhibitors are active against human immunodeficiency virus (HIV), an RNA virus. These drugs inhibit RNA virus replication by reversible inhibition of the viral enzyme HIV reverse transcriptase, which reverse transcribes viral RNA into viral DNA for insertion into the host DNA sequence. The nucleoside analogue reverse transcriptase inhibitor drugs (Fig. 51.6) are activated by phosphorylation inside the virus to the 5′-triphosphate form. Inhibition of viral replication is achieved by competitive binding of the activated drug to the enzyme–template–primer complex in place of the natural 5′-deoxynucleoside triphosphates, thus terminating further DNA chain elongation.
The nucleoside reverse transcriptase inhibitors are analogues of precursors of the natural purines and pyrimidines involved in DNA transcription initiated by the virus. Zidovudine is an analogue of thymidine, emtricitabine and lamivudine are analogues of cytidine, abacavir is an analogue of deoxyguanosine and tenofovir is an analogue of adenosine.
Resistance: Resistant quasi-species emerge within weeks or months by mutation of the drug-binding site on reverse transcriptase, resulting in an increase in the affinity for the natural substrate compared with the drug. Because of the rapid development of resistance, multiple drug therapy is used for treatment of HIV infection.
Pharmacokinetics: These drugs are almost completely absorbed from the gut. Elimination of abacavir and zidovudine is mainly by hepatic metabolism and the half-lives are short (1–2 h). Emtricitabine, lamivudine and tenofovir (the metabolite of tenofovir disoproxil) are mainly eliminated unchanged by the kidney, with half-lives in the range 1–17 h.
Unwanted effects: These are often so severe that they lead to withdrawal of therapy. They are probably related to inhibition of host mitochondrial enzymes, with impaired generation of intracellular ATP.
Neutropenia and anaemia are the most frequent unwanted effects (usually occurring in individuals with advanced AIDS).
Nausea, vomiting, diarrhoea, abdominal pain.
Myalgia or myositis, especially with high doses.
Severe, potentially life-threatening, hepatomegaly with steatosis and lactic acidosis.
Lipodystrophy syndrome with fat redistribution (loss of subcutaneous fat, buffalo hump, breast enlargement), insulin resistance and dyslipidaemia; this may be due to inhibition of regulatory proteins in adipocytes.
Non-nucleoside HIV reverse transcriptase inhibitors:
Mechanism of action and resistance: The non-nucleoside drugs inhibit HIV reverse transcriptase by binding remotely from the enzyme active site to produce a conformational change that prevents substrate binding. They have greater antiviral activity than nucleoside analogue inhibitors and are better tolerated. Resistance still emerges rapidly by single point mutations, unless they are used in combination with at least two other antiretroviral drugs.
Pharmacokinetics: Oral absorption of nevirapine and rilpivirine is good, while that of efavirenz is variable and incomplete. They are metabolised by hepatic CYP3A4, and also induce the enzyme. They have very long half-lives of about 2 days.
Rash (severe in 10%), especially with nevirapine and efavirenz.
Nausea, vomiting, abdominal pain, diarrhoea.
Headache, drowsiness, fatigue.
Depression with efavirenz and rilpivirine.
Hepatotoxicity with nevirapine, which can cause potentially fatal fulminant hepatitis.
Drug interactions with drugs metabolised by hepatic cytochrome P450.
Mechanism of action: In HIV infection there are some steps in viral replication that differ from the RNA translation processes in host cells. In the virus, RNA is translated into inert polyproteins rather than the functional proteins that are the products of host cells. Proteases found only in the virus cleave these polyproteins to the functionally active proteins required by the virus for its continued existence (Fig. 51.6). Protease inhibitors are specific for the enzymes found in HIV. They block the infectivity of the virus but do not affect virus activity in host cells that are already infected.
Resistance: Resistance occurs by mutation in the amino acid sequence of the HIV proteases that form the targets for the drug. Multiple mutations are required for high-level resistance, but over one-third of the amino acid residues in HIV protein can be changed without altering viral function. High plasma drug concentrations delay the onset of resistance, as does the combination of a protease inhibitor with two reverse transcriptase inhibitors. Sequential use of more than one protease inhibitor encourages high-level resistance.
Pharmacokinetics: Oral absorption varies between the protease inhibitors. All are metabolised by, and inhibit, CYP3A4 in the liver. They have half-lives in the range 2–10 h.
Nausea, vomiting, abdominal pain, diarrhoea.
Lipodystrophy syndrome (see nucleoside analogue HIV reverse transcriptase inhibitors).
Circumoral and peripheral paraesthesiae with ritonavir.
Drug interactions (Ch. 56): inhibition of the P450 enzyme CYP3A4 can enhance the unwanted effects of protease inhibitors; the concurrent use of inducers of CYP3A4 can lower plasma concentrations of the protease inhibitor and encourage viral resistance. Inhibition of CYP3A4 by a low dose of ritonavir can increase the clinical effect of other protease inhibitors (boosted protease inhibition), a useful action that allows less frequent dosing with the other drug. Ritonavir also inhibits the metabolism of drugs such as warfarin (Ch. 11) and carbamazepine (Ch. 23). The use of protease inhibitors with simvastatin (Ch. 48) should be avoided because of an increased risk of myopathy.
HIV binding–fusion–entry inhibitors:
Mechanism of action: To enter a host cell, HIV fuses with the host cell membrane. This fusion is facilitated by a conformational change in a viral glycoprotein, gp41, in the viral cell membrane. Enfuvirtide is a 36-amino-acid peptidomimetic that binds to the gp41 glycoprotein and prevents the conformational change, blocking HIV entry into host cells. Enfuvirtide is used when there is resistance or intolerance to other antiretroviral drugs.
Mechanism of action: Chemokine (C-C motif) receptor 5 (CCR5) is a receptor for several chemokines, including RANTES (regulated upon activation, normal T-cell expressed and secreted) and macrophage inflammatory proteins MIP-1α and MIP-1β. It is expressed on many cells, including T-cells, macrophages, dendritic cells and microglia. CCR5 acts as a viral co-receptor that facilitates entry of HIV into the cell. Some HIV strains use other chemokine receptors such as CCR4, or both CCR4 and CCR5, to access cells. HIV strains which use CCR5 are predominant early in the infection. Maraviroc selectively binds to CCR5 and prevents interaction with CCR5-tropic strains of HIV, inhibiting their entry into the cell. It has no effect on viruses that use CCR4 or are dual-tropic for CCR4 and CCR5. Maraviroc is used in combination with other antiretroviral drugs for individuals who have already received other antiretroviral treatment.
Mechanism of action: Once inside a host cell, HIV integrates its DNA into the host genome. This requires the action of a specific viral integrase. Raltegravir inhibits the integrase and prevents DNA strand transfer from the viral genome. Raltegravir is used when there is resistance to other antiretroviral drugs.
Drugs for herpesvirus and cytomegalovirus infections
Nucleoside analogue inhibitors of viral DNA polymerase:
Mechanism of action: Aciclovir and the other drugs in this class are guanosine analogues that are active against many DNA viruses and inhibit the synthesis of viral DNA (Fig. 51.7). Before they can exert their antiviral activity they all require phosphorylation by viral enzymes that are not present in uninfected host cells. However, the phosphorylating enzymes are not present in all DNA viruses. This dependency on viral enzymes prevents cytotoxic effects in human tissue.
Aciclovir and ganciclovir are activated by phosphorylation to a monophosphate by viral thymidine kinase. The monophosphate is then converted to a triphosphate derivative by other intracellular enzymes. The triphosphate derivatives are potent inhibitors of viral DNA polymerase. This terminates viral DNA synthesis and thus inhibits viral replication (Fig. 51.7).
Spectrum of activity: Aciclovir is most active against herpesviruses (both simplex and zoster). It is only active against cytomegalovirus (CMV) at high doses. Ganciclovir is also active against herpesviruses and has much greater activity than aciclovir against CMV, possibly because it is a better substrate for the CMV protein kinase which activates it.
Resistance: Viral mutants are selected that are unable to phosphorylate the drugs. Thymidine kinase-deficient mutants of herpesvirus usually develop in immunocompromised individuals, for example those with AIDS or after bone marrow transplantation, when resistance rates average 5–10%.
Pharmacokinetics: Aciclovir can be given orally, intravenously or topically to the skin or eye. Absorption from the gut is poor. The drug is widely distributed, but concentrations in the cerebrospinal fluid are low compared with those in plasma. Most is eliminated by the kidney, and the half-life is 3 h. Valaciclovir is an ester of aciclovir with higher oral bioavailability.
Ganciclovir is given intravenously for acute infections since it is poorly absorbed from the gut; it penetrates into CSF moderately well. It is eliminated by the kidney and has a half-life of 4 h. Valganciclovir is an oral prodrug of ganciclovir with better absorption.
Unwanted effects: Most unwanted effects occur with intravenous use, and are much more frequent with ganciclovir than with aciclovir.
Severe local phlebitis at an infusion site.
Nausea, vomiting, abdominal pain, diarrhoea.
Rashes, including photosensitivity.
Headache, dizziness, confusion, convulsions.
Nephrotoxicity is caused by crystallisation of aciclovir in the kidney. It can be limited by a high fluid intake.
Bone marrow suppression is the most frequent serious unwanted effect with ganciclovir, with neutropenia occurring in up to 40% of people, and thrombocytopenia less frequently.
Non-nucleoside analogue inhibitors of viral DNA polymerase:
Mechanism of action: Foscarnet is an inorganic pyrophosphate compound that binds to the pyrophosphate-binding sites of viral DNA polymerase, preventing DNA chain elongation. Affinity for the viral DNA polymerase is a hundred times greater than for the host cell DNA polymerase. Cidofovir is similar in structure to aciclovir but contains a phosphate moiety. Its action is similar to that of foscarnet. Foscarnet and cidofovir do not rely on intracellular activation for their antiviral activities (Fig. 51.7). These drugs are reversible inhibitors of CMV and herpes simplex replication.
Pharmacokinetics: Because both foscarnet and cidofovir are highly polar molecules, they are only given intravenously. Foscarnet and cidofovir are eliminated by the kidney and have half-lives of about 5 h and 3 h, respectively. Cidofovir is given with probenecid (and adequate hydration), which inhibits renal tubular secretion of cidofovir and minimises its nephrotoxicity.
Drugs for treating hepatitis viruses
The nucleoside analogues entecavir, lamivudine, ribavirin and tenofovir disoproxil, and the protease inhibitors bocepravir and telaprevir as well as interferon alfa are used in the treatment of infections with hepatitis B and C viruses. Ribavirin is also used to treat respiratory syncytial virus (RSV). These drugs are discussed in Chapter 36.
Drugs for treating influenza virus
Amantadine is active only against influenza A and inhibits the transmembrane M2 ion channel that permits H+ entry into the viral particle. This is required for uncoating of the virus once it has penetrated the host cell, so viral replication is inhibited. Amantadine is discussed in Chapter 24.
Mechanism of action: Influenza viruses carry two surface glycoproteins, a haemagglutinin and a neuraminidase. The haemagglutinin mediates entry of the virus into the host cell. Neuraminidase cleaves cellular-receptor sialic acid residues to which newly formed viruses are attached as they bud from the infected cell. The released virions can then infect new cells. Neuraminidase inhibitors inhibit the neuraminidases of both influenza A and B, and are effective against isolates resistant to amantadine.
Pharmacokinetics: Zanamivir is administered by inhalation; only 2% of the inhaled drug is absorbed, and then excreted unchanged. Oseltamivir is a more lipophilic molecule that is taken orally, and is converted by hepatic esterases to the active oseltamivir carboxylate, which is excreted by the kidneys and has a half-life of 6–10 h.
Immunomodulators
Interferon alfa: Interferon alfa is most often used in the treatment of chronic hepatitis B infection and is discussed in Chapter 36. Other clinical uses of interferon alfa include the treatment of:
AIDS-related Kaposi's sarcoma,
recurrent or metastatic renal cell carcinoma (Ch. 52).
Mechanism of action and use: Palivizumab is a humanised monoclonal antibody produced by recombinant DNA technology. It has potent neutralising and fusion-inhibiting activity against RSV. It reduces the ability of RSV to replicate and infect cells by binding to an antigenic site on the surface of RSV. RSV is a common cause of mild respiratory illness in infants but can produce more severe illness in premature infants or those with congenital heart disease or bronchopulmonary dysplasia. Palivizumab can be given to at-risk children under the age of 2 years prior to commencement of the RSV season (October to April in the northern hemisphere) and monthly thereafter.
Treatment of specific viral infections
Multiple drug therapy is essential for HIV infection because of the rapid emergence of resistant strains. The most frequently used treatment regimens include two nucleoside analogue reverse transcriptase inhibitors (e.g. tenofovir with emtricitabine, or abacavir with lamivudine) combined with either a protease inhibitor (e.g. atazanavir, darunavir or raltegravir) or a non-nucleoside reverse transcriptase inhibitor (e.g. efavirenz). A low dose of ritonavir is often added to the protease inhibitor (boosted protease therapy) to prolong its action and simplify the dosing regimen. Such combinations are referred to as highly active antiretroviral therapy (HAART). HAART involves complex regimens that require adherence by the individual and careful assessment of the progress of viral suppression. Key principles include the following.
Combination drug treatment should be started before substantial immunodeficiency is present. The goal is to suppress the virus before resistant mutants emerge or irreversible immune damage occurs. The optimal time to start treatment is not known, but should definitely be started when the CD4+ T-lymphocyte count falls below 200 × 106 L−1. If this stage is missed, then treatment is given when symptoms arise as a result of HIV infection.
When resistance occurs, modification in drug therapy should involve the addition or change of at least two drugs. Resistance of the HIV virus persists indefinitely. However, if toxicity limits the tolerability of one drug, a single substitution of a similar drug is a reasonable option.
Optimal treatment should reduce the viral load to below detectable limits and achieve a rise in CD4+ lymphocyte count. This may take 6 months of optimal therapy.
Failure to achieve full suppression of viral load should prompt a change in therapy if adherence is believed to be good. Poor adherence with therapy is likely to encourage the development of drug resistance (see above) and thus treatment failure. Drug therapy is ideally guided by patterns of resistance in the virus.
The optimal duration of treatment is unclear, but withdrawal even when the CD4+ T-cell count rises is probably associated with less favourable long-term outcomes.
Prophylaxis after accidental exposure to HIV may be required. The regimen depends on the level of risk: two drugs are often used for moderate-risk exposures, or three drugs if the risk is high.
Varicella–zoster virus infections
Varicella–zoster virus (VZV) is a herpesvirus responsible for both chickenpox and shingles (herpes zoster). Shingles arises from reactivation of the virus, which lies dormant in a dorsal root ganglion after the primary chickenpox infection. Chickenpox is rarely treated with antiviral therapy, although the use of oral aciclovir reduces lesion formation and results in quicker healing.
Herpes zoster is most commonly found in the elderly and in immunosuppressed people. The rash is often preceded by pain for 1–4 days. Complications of infection occur in 15–20% of people who develop herpes zoster and include meningoencephalitis, motor nerve paralysis, ocular complications and postherpetic neuralgia. Oral antiviral drug therapy reduces pain and accelerates healing. It must be given while the virus is still replicating, and therefore started within 72 h of the onset of the rash. Oral aciclovir or valaciclovir are often used and are particularly indicated for those over 50 years (who are at greater risk of complications), for ophthalmic infections or in immunosuppressed people. The use of aciclovir has little effect on the risk of developing postherpetic neuralgia but does reduce the risk of motor nerve damage. Corticosteroids such as prednisolone as an adjunctive treatment to antiviral therapy reduce pain and produce more rapid healing of lesions, but do not prevent the development of post-herpetic neuralgia. Analgesics are often required in the early phases of zoster, and postherpetic neuralgia may require specific therapy (Ch. 19).
Herpes simplex virus infections
Herpes simplex virus exists in two forms: type 1 produces either oral or genital ulceration, and type 2 produces genital ulceration. Oral herpes simplex infection will respond to early topical application of aciclovir. Primary genital herpes produces multiple painful lesions and responds to oral aciclovir or valaciclovir given for 7–10 days. Recurrent lesions occur from reactivation of latent virus in the dorsal root ganglia, producing symptoms that are usually less severe than with primary episodes. After initial therapy with aciclovir or valaciclovir for up to 3 days, continuous suppressive therapy can be given to prevent further relapses.
Cytomegalovirus infection
CMV infection is common and usually produces mild symptoms. However, it can be devastating in immunosuppressed individuals. Troublesome complications in this group include retinitis (which can threaten sight), gastrointestinal manifestations (including oesophagitis, gastritis, cholecystitis or colitis), pneumonia and CNS involvement.
Intravenous ganciclovir is the treatment of choice for severe manifestations of CMV infection. Foscarnet can be used as an alternative to ganciclovir, or cidofovir if both are contraindicated. For CMV retinitis, oral valganciclovir is used both for treatment and to prevent relapse. Oral valganciclovir or valaciclovir are given for the prevention of CMV infection, especially in renal transplant and bone marrow transplant recipients in whom CMV pneumonia is a major potential complication. Combined therapy with ganciclovir and CMV immunoglobulin may be more effective than ganciclovir alone for treatment of pneumonia in this situation.
Influenza
The use of a neuraminidase inhibitor, such as zanamivir or oseltamivir, reduces the duration of uncomplicated influenza by about 1 day. Treatment must be started within 48 h of the onset of an influenza-like illness. People who are at greater risk of complications of influenza include those with chronic respiratory disease, significant cardiovascular disease, chronic renal disease, diabetes mellitus or those who are immunocompromised. However there is little evidence that these drugs prevent the complications of influenza. Neuraminidase inhibitors are also effective for the prevention of influenza during an epidemic, reducing the likelihood of developing the illness by 70–90%. They should be considered for high-risk individuals who have not been vaccinated and who can be treated within 48 h of contact with someone who has an influenza-like illness. Such prophylaxis is not a substitute for an effective vaccination campaign.
Respiratory syncytial virus
RSV most commonly causes respiratory infections in children under 2 years old. It causes bronchiolitis and increased airway reactivity with wheezing. Treatment is usually symptomatic, but severe infection is sometimes treated with ribavirin. Palivizumab is given to prevent serious lower respiratory tract disease from RSV in children under 2 years old who are at high risk of complications.
Protozoal infections
Five species of the protozoan Plasmodium produce malaria in humans: Plasmodium vivax, Plasmodium ovale (two subspecies), Plasmodium knowlesi, Plasmodium malariae and Plasmodium falciparum. The motile infective form of the parasite, sporozoites, are formed by repeated division of oocysts in the body of the infected female Anopheles mosquito (the vector) and transferred in the mosquito's saliva into the host circulation during a blood meal (only female mosquitoes feed on blood). The parasite is rapidly sequestered in the liver and matures to tissue schizonts which divide asexually to form merozoites (Fig. 51.8). When the pre-erythrocytic (liver) cycle is complete after 5–16 days, 20 000–40 000 merozoites escape into the blood and invade erythrocytes. P. vivax and P. ovale may continue to multiply in the liver, but P. malariae, P. knowlesi and P. falciparum do not.
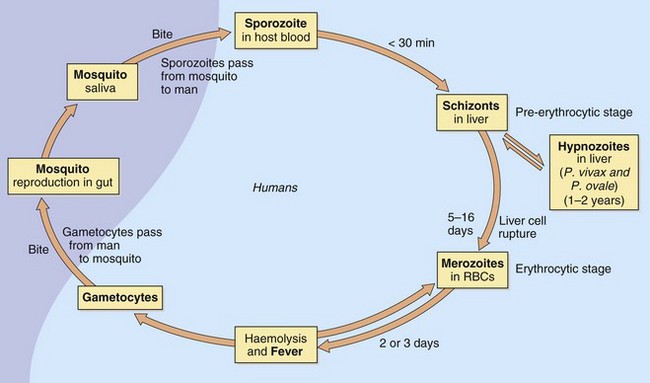
Fig. 51.8 Life-cycle of the malarial parasite.
Most antimalarial drugs kill the Plasmodium parasites that cause repeated cycles of red blood cell (RBC) lysis and fever (erythrocytic stage). Primaquine kills parasites of P. vivax and P. ovale (hypnozoites) dormant in the liver (pre-erythrocytic stage).
Merozoites released from the liver then undergo the erythrocytic cycle, multiplying asexually in erythrocytes and using haemoglobin as the main source of nutrition. Infected erythrocytes rupture (haemolyse) and release merozoites to invade other erythrocytes. Some merozoites in the plasma differentiate into male and female gametocytes. A mosquito biting an infected individual ingests gametocytes, which then go through a sexual development cycle in the mosquito to form sporozoites.
Release of merozoites from erythrocytes every 2 or 3 days causes repeated bouts of tertian or quartan fever in humans. Premonitory clinical symptoms of malaria include chills, and nausea, vomiting and headache are common. A fever follows, and the attack concludes with sweating. P. falciparum produces the most severe symptoms (malignant tertian malaria) because high levels of parasitaemia cause agglutination of red cells, which produces capillary thrombosis, especially in the brain, leading to cerebral malaria.
Because P. vivax and P. ovale continue to multiply in the liver as hypnozoites, they form a reservoir of parasites that are difficult to eradicate and can emerge to give relapses months or years after the initial infection. Drugs that treat only the erythrocytic phase will not produce a radical cure (elimination of all parasites), and relapsing infection can occur. Although P. falciparum, P. malariae and P. knowlesi do not have persistent liver forms, disease can recrudesce if parasites are not completely eliminated from the blood.
Antimalarial drugs
Mechanism of action: Erythrocytes infected by malaria parasites concentrate chloroquine more than 100-fold compared to uninfected erythrocytes, since it binds to a breakdown product of haemoglobin (haemin) induced by the parasite.
Chloroquine is then ingested by the erythrocyte-resident parasite and this raises lysosomal pH, which reduces the ability of the parasite to digest haemoglobin and thereby inhibits its growth.
Chloroquine interacts with haemin (ferriprotoporphyrin IX) formed during digestion of haemoglobin, an action that prevents further degradation by the parasite.
Chloroquine (and its close relative hydroxychloroquine) also possess slow-onset anti-inflammatory activity, which is useful in the treatment of rheumatoid arthritis (Ch. 30).
Pharmacokinetics: Chloroquine is completely absorbed from the gut or can be given intravenously. It has a very high volume of distribution because of selective concentration in melanin-containing tissues, for example the retina of the eye, and in the liver, spleen and kidney. Approximately half is converted in the liver to active metabolites and the rest is excreted unchanged by the kidney. The half-life is very long during chronic dosing; an initial half-life of up to 6 days is followed by a second slow phase of tissue elimination with a half-life of greater than 1 month.
Pharmacokinetics: Mefloquine is well absorbed from the gut and has a high affinity for lung, liver and lymphoid tissue. Extensive metabolism occurs in the liver, but the elimination half-life is extremely long (2–4 weeks).
Mechanism of action: Unlike the structurally related drugs chloroquine and mefloquine, primaquine only affects the exoerythrocytic parasite. It enters the parasite in the liver and may inhibit mitochondrial respiration. It is used after treatment with chloroquine, to eradicate P. vivax or P. ovale from the liver (a radical cure).
Pharmacokinetics: Primaquine is completely absorbed from the gut and rapidly metabolised in the liver, producing active compounds. The half-life is 4–10 h.
Intravascular haemolysis in people with glucose-6-phosphate dehydrogenase (G6PD) deficiency (Ch. 47). G6PD activity in erythrocytes produces NADPH, which keeps glutathione in the reduced state, thereby maintaining cell wall integrity and preventing haemolysis (see Ch. 53). G6PD activity should be checked before initiating treatment.
Gastrointestinal effects are similar to those seen with chloroquine.
Pyrimethamine with sulfadoxine:
Mechanism of action: Selective inhibition of dihydrofolate reductase in the malaria parasite by pyrimethamine reduces folic acid synthesis (Fig. 51.4). Pyrimethamine should only be given in combination with the sulphonamide sulfadoxine because of the widespread emergence of resistance.
Mechanism of action: Proguanil inhibits plasmodial dihydrofolate reductase (Fig. 51.4), mainly through its active metabolite, cycloguanil, which inhibits folate production in both pre-erythrocytic and erythrocytic parasites. It is often used for malaria prophylaxis in combination with chloroquine. It can also be used in the prophylaxis and treatment of P. falciparum in combination with atovaquone (see below).
Mechanism of action: Artemether is a herb extract that is activated by complexing with iron in the haem ingested by the malarial parasite. The resulting compound produces reactive oxygen species that disrupt Ca2+ transporter function in the parasite. It is used with lumefantrine, which may work by inhibiting the production of β-haematin in the erythrocyte. The combination reduces the emergence of resistance.
Treatment of malaria
Chemotherapy of malaria falls into three categories:
rapid-acting blood schizonticides (to kill schizonts in acute malaria): chloroquine, mefloquine, quinine, artemether with lumefantrine,
slow-acting blood schizonticides (to suppress blood infections): pyrimethamine with sulfadoxine, proguanil,
tissue schizonticide (to eliminate liver parasites): primaquine.
The recommended drug to use within each category depends on the type of parasite and the pattern of resistance where the infection was acquired. If the infecting organism is unknown, it is assumed to be P. falciparum, which carries the greatest risk. The latest drug recommendations should be obtained from tropical disease advisory centres.
Examples are given for current recommended treatments for acute attacks of high- and low-risk malaria.
For P. falciparum, chloroquine and mefloquine resistance is common. Oral quinine (or intravenous quinine for serious infections) for 5–7 days is followed by pyrimethamine with sulfadoxine for 7 days or by doxycycline or clindamycin (see antibacterials, above) if the plasmodia are resistant to sulfadoxine. Alternatively, proguanil with atovaquone or artemether with lumefantrine are used for 3 days.
For benign malaria, chloroquine is taken for 3 days. For P. vivax and P. ovale primaquine is then taken for 14 days to destroy hepatic parasites.
Prophylaxis against malaria
The recommendations for prophylaxis depend on patterns of resistance in the area to be visited. Chloroquine or proguanil is often recommended for areas where resistance is low. For many areas a combination of both drugs is desirable. Mefloquine, doxycycline, or proguanil with atovaquone are recommended in some areas where there is a high risk of chloroquine-resistant malaria. Prophylaxis must also take into account the unwanted effects of the drugs and other factors such as pregnancy and renal or hepatic impairment. Prophylaxis must be started 1 week before travel (2–3 weeks for mefloquine; 1–2 days for proguanil with atovaquone, or for doxycycline) and continued for 4 weeks after leaving a malarial area (7 days for proguanil with atovaquone), to protect against infection acquired immediately prior to departure.
Other protozoal infections
Details of the natural history of other protozoal infections are not given in this book. Important drugs available in the UK for these conditions are discussed below. An outline of therapeutic uses is given in Table 51.5.
Table 51.5
Selected protozoan infections and antiprotozoal drugs
| Protozoa | Disease | Drug examples |
| Plasmodium species | Malaria | Chloroquine, mefloquine, primaquine, quinine, proguanil, pyrimethamine with sulfadoxine, atovaquone, artemether with lumefantrine |
| Entamoeba histolytica | Amoebic dysentery | Metronidazole, tinidazole, diloxanide |
| Trichomonas vaginalis | Vaginitis | Metronidazole, tinidazole |
| Giardia lamblia | Gastrointestinal dysfunction | Metronidazole, tinidazole, mepacrine |
| Leishmania species | Cutaneous or visceral (kala-azar) leishmaniasis | Stibogluconate, pentamidine |
| Trypanosoma species | Trypanosomiasis, Chagas' disease, sleeping sickness | Suramina, nifurtimaxa, benznidazolea, eflornithine, pentamidine, melarsoprola |
| Toxoplasma gondii | Encephalomyelitis, toxoplasmosis | Pyrimethamine plus sulfadiazine, trimetrexatea |
| Pneumocystis jirovecii | Pneumocystis pneumonia | Co-trimoxazole, pentamidine, atovaquone, trimetrexatea |
Atovaquone
Mechanism of action and uses: Atovaquone interferes with DNA synthesis by inhibiting pyrimidine synthesis. It is selective for protozoa that cannot utilise pre-formed pyrimidines. It affects mitochondrial electron transport and ATP synthesis.
Atovaquone is used as a second-line drug for treatment of P. jirovecii infections and for treating Toxoplasma gondii infection. It is also active against Plasmodium species, E. histolytica and Trichomonas vaginalis.
Pentamidine
Mechanism of action and uses: Pentamidine undergoes active uptake into the cell, where it probably inhibits DNA synthesis and ribosomal synthesis of protein and phospholipid.
Pentamidine is used in pneumocystis pneumonia, leishmaniasis and trypanosomiasis. It is cytotoxic to P. jirovecii in the non-replicating state, but because of its toxicity it is usually reserved for people who are intolerant of co-trimoxazole.
Sodium stibogluconate
Mechanism of action and use: Sodium stibogluconate is an organic antimony derivative that may act by binding to thiol groups in the parasite and inhibiting the formation of high-energy phosphates. Sodium stibogluconate is used to treat visceral leishmaniasis.
Diloxanide furoate
Mechanism of action and use: The mechanism of action is unknown. Diloxanide is used to treat chronic amoebiasis in asymptomatic individuals who are excreting cysts of E. histolytica in the stool. Acute infection is treated with metronidazole or tinidazole (Table 51.5).
Pharmacokinetics: It is given orally and hydrolysed in the gut to diloxanide and furoic acid; 90% of the diloxanide is then absorbed and rapidly conjugated in the liver. There is little information about its fate in the body. The unabsorbed fraction of diloxanide may contribute to the drug's effectiveness in amoebic dysentery.
Helminthic infections
Details of the natural history of helminth infections are not given here, but an outline of the more commonly encountered conditions and drug treatments is given in Table 51.6. Drugs specifically for helminth infections are discussed below.
Table 51.6
| Helminth | Common name | Drug examples |
| Enterobius vermicularis | Threadworm | Mebendazole, piperazine |
| Ascaris lumbricoides | Roundworm | Mebendazole, piperazine, levamisole |
| Toxocara canis | Dog roundworm | Tiabendazole, diethylcarbamazine |
| Taenia species | Tapeworm | Niclosamide, praziquantel |
| Ancylostoma species, Necator species | Hookworm | Mebendazole, ivermectin, albendazole |
| Microfilariae (e.g. Loa loa, Wuchereria bancrofti, Brugia malayi) | Diethylcarbamazine, ivermectin | |
| Strongyloides stercoralis | Tiabendazole, albendazole, ivermectin | |
| Echinococcus granulosa | Hydatid disease | Albendazole |
Antihelminthic drugs
Mechanism of action and use: Ivermectin is used to treat filariasis (especially onchocerciasis, for which it is the drug of choice), hookworm and Strongyloides stercoralis infection. Treatment with a single dose reduces microfilarial levels for several months and can be repeated every 6–12 months if necessary. It is available in the UK on a ‘named-patient’ basis.
Ivermectin produces an influx of Cl− ions via an action on glutamate-gated membrane ion channels, generating muscle hyperpolarisation and paralysis of the filariae.
Diethylcarbamazine
Mechanism of action and use: Diethylcarbamazine is a first-line treatment for filariasis. Its mechanism of action is not well understood. It may inhibit arachidonic acid metabolism in the filariae. It also triggers exposure of antigens on the surface coat, leading to antibody-mediated phagocytosis. Treatment is usually required for 2–3 weeks to eliminate the microfilariae. Diethylcarbamazine is not marketed in the UK.
Pharmacokinetics: Oral absorption is good, and approximately half the drug is metabolised in the liver; the rest is excreted unchanged by the kidney. The half-life is not well established.
Benzimidazoles
Mechanism of action and uses: The benzimidazoles bind to tubulin, preventing its polymerisation into the cytoskeletal microtubules. The effect is selective for parasitic tubulin and the drugs are active against the adults, larvae and eggs. Tiabendazole and albendazole are available in the UK on a ‘named-patient’ basis.
Uses include (see also Table 51.6):
Piperazine
Mechanism of action and uses: Piperazine is given orally to treat threadworm and roundworm infections. It competitively inhibits the effect of acetylcholine on the smooth muscle of the worm, producing a reversible flaccid paralysis.
Niclosamide
Mechanism of action and use: Niclosamide is given orally to treat tapeworm infection. It inhibits generation of ATP by preventing phosphorylation of ADP in mitochondria. It is ineffective against larval worms. Purgatives are usually given after niclosamide to remove viable ova from the gut. Niclosamide is available in the UK on a ‘named-patient’ basis.
Praziquantel
Mechanism of action and uses: Praziquantel is given orally to treat tapeworm infection and schistosomiasis. It is not well understood how praziquantel acts; it is known to increase the permeability of the cell membrane of sensitive helminths to Ca2+, causing muscular contraction and paralysis. It may also reduce cellular transport of adenosine, which cannot be synthesised by the parasites. Praziquantel is available in the UK on a ‘named-patient’ basis.
True/false questions
1. Resistance to antibacterials may be due to mutations in bacterial ribosomes.
2. Benzylpenicillin has a short half-life as it is rapidly excreted by the kidneys.
3. Broad-spectrum penicillins do not disturb normal colonic flora.
4. Penicillins are bactericidal by binding to bacterial ribosomal sites.
5. The antipseudomonal penicillin ticarcillin is resistant to β-lactamase.
6. Cefotaxime is a third-generation cephalosporin.
7. Individuals who are allergic to penicillins cannot be given cephalosporins.
8. Imipenem is rapidly metabolised in the kidney.
9. Imipenem has only bacteriostatic activity.
10. Ciprofloxacin is ineffective for Pseudomonas aeruginosa infections in cystic fibrosis.
11. Ciprofloxacin interacts with theophylline used in asthma management.
12. Erythromycin commonly causes gastrointestinal disturbances.
13. Gentamicin has a low incidence of unwanted effects.
14. Gentamicin is not active when given orally.
15. Metronidazole can be used for eradicating Helicobacter pylori.
16. Antibacterials are effective for the majority of people with sore throat.
17. Tetracyclines should be avoided during pregnancy and in young children.
18. Vancomycin is active against β-lactamase-producing Gram-positive bacteria.
19. Rifampicin is an important drug for the treatment of tuberculosis.
20. Isoniazid is active against a wide range of bacteria.
21. Co-trimoxazole is the drug of choice for hospital-acquired acute urinary tract infection.
22. Trimethoprim administration can result in folate deficiency.
23. Imidazoles and triazoles have the same mechanism of antifungal action.
One-best-answer (OBA) questions
1. Choose the most accurate statement about HIV:
A Binding of HIV to the chemokine receptor CCR5 in the host cell membrane inhibits entry of the virus.
B Low doses of ritonavir reduce the activity of other protease inhibitors.
C Once resistance of HIV has developed, it persists indefinitely.
D Zidovudine acts in HIV by preventing viral entry to the host cells.
E The non-nucleoside reverse transcriptase inhibitors treat HIV by preventing insertion of the viral DNA into the host genome.
2. Identify the least accurate statement regarding the treatment of malaria:
A Primaquine kills parasites in the liver (hypnozoites) in Plasmodium falciparum infections.
B Chloroquine concentrates in erythrocytes and prevents the erythrocytic stage of malarial parasite reproduction.
C In many areas prophylaxis against malaria requires administration of two drugs.
D Primaquine can induce haemolysis in people with glucose-6-phosphate dehydrogenase (G6PD) deficiency.
E Proguanil acts to inhibit malarial parasite folate production.
Case-based questions
Mr JW, age 40 years, lives at home and was previously healthy, but saw his GP in August, 5 days after returning from a conference abroad, where he had stayed in a large hotel and indulged his passion for frequent whirlpool baths. He had characteristic symptoms of pneumonia, including pleuritic chest pain and sudden development of fever and cough, producing yellow sputum. He was disorientated. Physical examinations and chest radiograph supported the diagnosis.
Case 2
Mr RH, age 80 years, had influenza and was admitted to hospital when he developed symptoms similar to Mr JW (see above) and became seriously ill. A chest radiograph showed multiple abscesses.
Case 3
A 31-year-old man suffering from AIDS was admitted with shortness of breath, cough and generalised chest discomfort. Chest radiograph revealed diffuse bilateral opacities and a blood gas analysis demonstrated an arterial partial oxygen pressure (PaO2) of 8.0 kPa (normal range 11.0–14.0 kPa). Sputum culture was uninformative. A bronchoalveolar lavage was performed and transbronchial biopsies taken.
A What was the likely clinical diagnosis?
B What microscopic investigation could have been useful and what might it have shown?
C How could this man have been managed and what factors needed to be considered?
D What drug treatment unrelated to the infection could have further exposed him to the increased risk of opportunistic infection?
Case 4
Twenty-four hours after attending a convention, a 36-year-old man became ill with a temperature, abdominal pain, vomiting and diarrhoea. Faeces were collected and inoculated onto culture plates with several different types of culture medium. Pale-coloured colonies that were non-lactose fermenting were identified.
1. True. Mutations in bacterial ribosomes can reduce binding of many antibacterials that inhibit protein synthesis.
2. True. Benzylpenicillin is actively secreted into the proximal tubule by the organic acid transporter (Ch 2). This can be inhibited by probenecid or by other drugs that use the same secretory mechanism, such as aspirin.
3. False. Broad-spectrum penicillins in particular may alter the balance of gut flora, allowing overgrowth of pathogenic bacteria such as Clostridium difficile.
4. False. Penicillins disrupt cell membrane peptidoglycans.
5. False. Ticarcillin is broken down by β-lactamase.
6. True. Cefotaxime is a third-generation cephalosporin that penetrates the CNS and is resistant to β-lactamases.
7. False. Cephalosporins can be given, but with caution, as 8–16% of penicillin-allergic people will also have allergy to cephalosporins.
8. True. Imipenem (but not other carbapenems) is rapidly metabolised by renal dihydropeptidase and is always given in combination with cilastatin, a dihyhdropeptidase inhibitor.
9. False. Like all β-lactam antibacterials given in correct doses, imipenem is bactericidal.
10. False. The quinolones have good activity against Pseudomonas species.
11. True. Ciprofloxacin inhibits hepatic metabolism of theophylline and increases its toxicity.
12. True. Erythromycin often causes nausea and diarrhoea. Azithromycin is better tolerated.
13. False. Gentamicin is nephrotoxic and ototoxic, and its plasma levels should be monitored.
14. True. Gentamicin is poorly absorbed from the gastrointestinal tract and is given parenterally.
15. True. Metronidazole can be used with a proton pump inhibitor and other antibacterials to eliminate H. pylori.
16. False. Most sore throats are caused by viruses; those with bacterial causes usually resolve without antibacterials.
17. True. Tetracyclines can chelate with Ca2+ and form permanent yellow–brown deposits on developing teeth.
18. True. Vancomycin is reserved for meticillin-resistant Staphylococcus aureus (MRSA) and metronidazole-resistant C. difficile, which causes colitis.
19. True. Rifampicin is a broad-spectrum antibacterial also used in some serious diseases caused by Gram-negative bacteria such as Legionella and mycobacteria, and for MRSA.
20. False. Isoniazid is a highly selective inhibitor of the production of mycolic acids, which are unique to the cell wall of Mycobacterium species.
21. False. Trimethroprim alone is usually preferred to co-trimoxazole in urinary tract infections due to the lower risk of severe side effects compared to the combination drug.
22. True. Trimethoprim inhibits the role of folate in the synthesis of nucleotides; folate deficiency can result in megaloblastic anaemia, preventable during long-term treatment by giving folinic acid.
23. True. Imidazoles and triazoles suppress ergosterol synthesis by inhibiting lanosterol 14α-demethylase and they also share a similar spectrum of antifungal activity.
24. False. Griseofulvin inhibits dermatophyte mitosis by impairing microtubule formation.
25. True. Liposomes and other lipid delivery vehicles reduce the nephrotoxicity of amphotericin.
OBA answers
1. Answer C is the most accurate.
A Incorrect. The virus binds to a number of cell surface molecules including CCR5 receptors and glycoproteins to facilitate entry.
B Incorrect. Ritonavir inhibits breakdown of other protease inhibitors by CYP3A4 enzymes and is used to enhance their activity.
C Correct. Resistance of HIV persists indefinitely and drug treatment should be modified.
D Incorrect. Zidovudine is an RNA reverse transcriptase inhibitor and prevents the formation of viral DNA from viral RNA.
2. Answer A is the least accurate.
A Incorrect. Only infections by Plasmodium vivax and Plasmodium ovale result in schizonts that remain in the liver as hypnozoites, and they require primaquine for their eradication.
B Correct. Chloroquine concentrates 100-fold in red cells and interferes with the ability of the parasite to digest haemoglobin.
C Correct. Resistance is a problem in many areas, where prophylaxis with chloroquine plus proguanil or atovaquone plus one of mefloquine, doxycycline or proguanil is recommended.
D Correct. Toxic metabolites of primaquine can induce haemolysis in people with G6PD deficiency.
E Correct. Proguanil inhibits dihydrofolate (DHF) reductase.
Case-based answers
A The most common cause of community-acquired infection is Streptococcus pneumoniae, but other ‘atypical’ organisms could be involved. In Mr JW, who was previously well, a recent stay in a hotel abroad might indicate the involvement of Legionella species, which multiply in warm water, for example in the tanks of air-conditioning systems. The incubation time is 5–10 days. Co-amoxiclav (amoxicillin and clavulanic acid) plus erythromycin or another macrolide should be given orally before the diagnosis is confirmed. If his condition is severe, rifampicin should also be given. The treatment should be reviewed immediately the microbiology sensitivities are known.
B Amoxicillin is bactericidal and acts by interfering with bacterial cell wall peptidoglycan synthesis. It also allows greater activity of enzymes that lyse bacterial cells. Clavulanic acid inhibits β-lactamase, thus extending the spectrum of activity of amoxicillin. Erythromycin inhibits bacterial protein synthesis by acting on the bacterial ribosome. Rifampicin, perhaps better known for its role in treating tuberculosis, inhibits DNA-dependent RNA polymerase in many Gram-positive and Gram-negative bacteria.
Case 2
A Staphylococcus aureus is a likely cause of acute pneumonia following an attack of influenza. Although the treatment must be guided by the sensitivity tests, most S. aureus strains are sensitive to flucloxacillin and this is the most appropriate antibiotic to start with. In the circumstances, it may be combined with fusidic acid or gentamicin. S. aureus commonly produces abscesses in the lungs. Pulmonary infection with S. aureus may also occur in people with cystic fibrosis. A Gram stain of the sputum would demonstrate Gram-positive cocci in clusters, typical of staphylococci. The production of coagulase and DNase would identify the organism as S. aureus. Many different species of coagulase-negative staphylococci exist and are found as part of the normal skin flora. The coagulase-negative staphylococci are typical causes of prosthetic valve endocarditis, joint prostheses and infected venous catheters. Of concern is the large number of meticillin-resistant staphylococci (MRSA), which pose a threat to people who are frail or immunocompromised.
B A range of other second-choice antibacterials effective against β-lactamase-producing S. aureus might be useful. For example, fusidic acid, gentamicin, a cephalosporin, a quinolone or erythromycin may be effective. There are increasing concerns about MRSA. Vancomycin, a bactericidal glycopeptide that inhibits cell wall synthesis, is effective against some MRSA, or linezolid may be used if the glycopeptide is unsuitable.
Case 3
A Clinically, the man is likely to have Pneumocystis jirovecii pneumonia (PCP), which is the predominant respiratory illness in people with AIDS. The causative organism, which is a protozoan, is believed to be acquired at a young age and reactivates with waning immunity. The organism is endemic in the community and multiplies within the lungs and causes symptoms. There is often a seasonal prevalence of PCP. Symptoms can be scant and, if present, consist of breathlessness and cough. Induced sputum or bronchoalveolar lavage specimens should be sent to laboratory for detection of PCP and routine culture.
B PCP can be detected in sputum or lavage by staining with methenamine silver stain for typical casts. It can also be detected by use of the polymerase chain reaction, and on lung biopsy.
C The treatment of choice is high-dose co-trimoxazole. Many people with AIDS have hypersensitivity reactions to sulphonamides and are taking multiple drug combinations. Alternative treatments for PCP are aerosolised or parenteral pentamidine, dapsone and trimethoprim, primaquine, etc.
D Long-term treatment with corticosteroids or other immunosuppressants.
E After an attack of PCP, a prophylactic regimen should be taken, for example nebulised pentamidine or oral trimethoprim 3 days per week.
Case 4
A Salmonella, Shigella, Proteus and Pseudomonas species are non-lactose fermenting and produce pale-coloured colonies on this medium. All were contenders.
B Antibacterials have no role to play in the management of the majority of cases of Salmonella gastroenteritis. Exceptions are when the gastroenteritis occurs in an individual who is immunocompromised or if there is evidence of systemic invasion. Ciprofloxacin would be the antibacterial of choice; it can be given orally and is cheaper than intravenous preparations. Dehydration and electrolyte imbalance should be corrected by fluid replacement. Control of the diarrhoea by antidiarrhoeal drugs is contraindicated because of the risk of inducing paralytic ileus and causing septicaemia.
C Food poisoning, as this case would seem to be from the history, is a notifiable condition and should be reported to the consultant in Communicable Disease Control. Because the man has attended a convention, it is possible that this is part of an outbreak and all persons attending the convention should be contacted to find out if they have been symptomatic and to collect faecal specimens for culture. Specimens of food, if still available, should also be collected for culture.
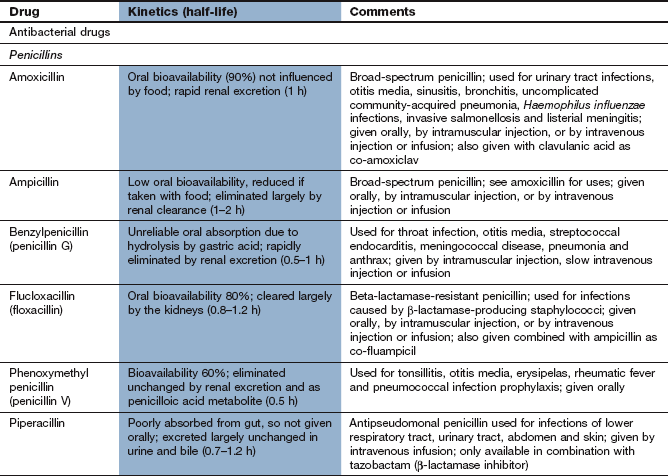
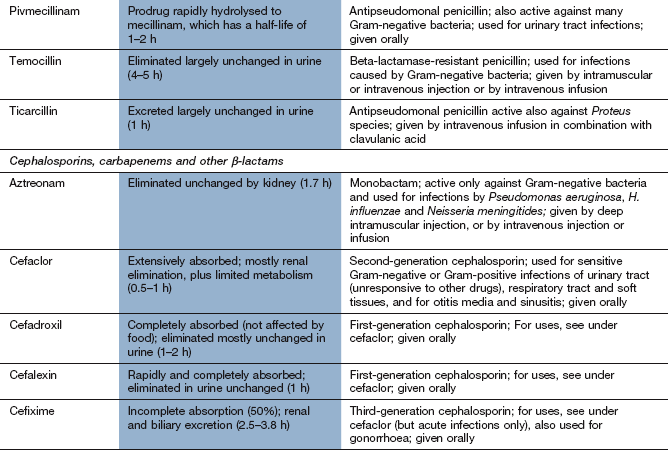
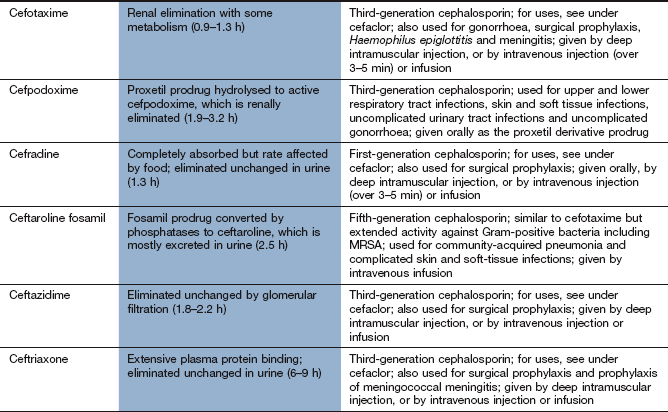
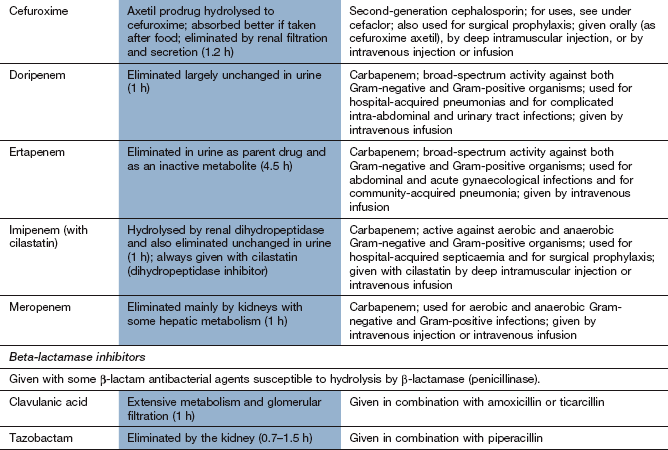
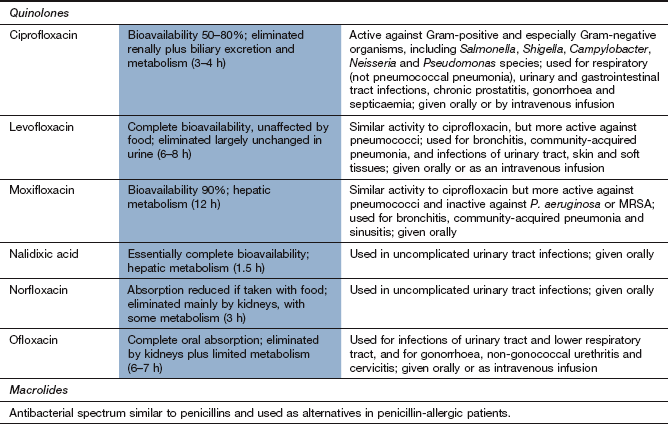
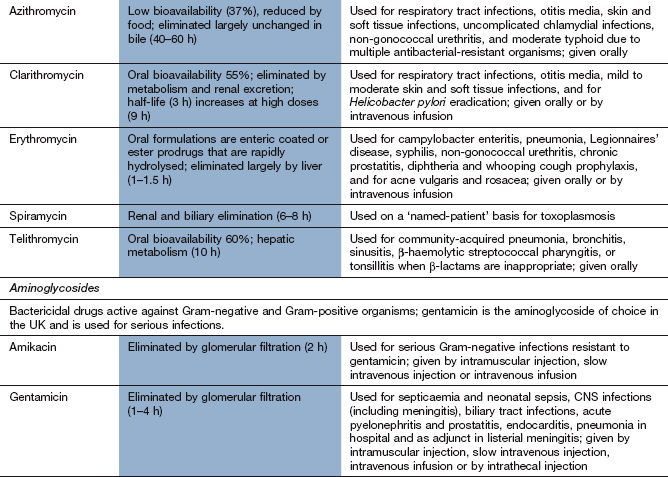
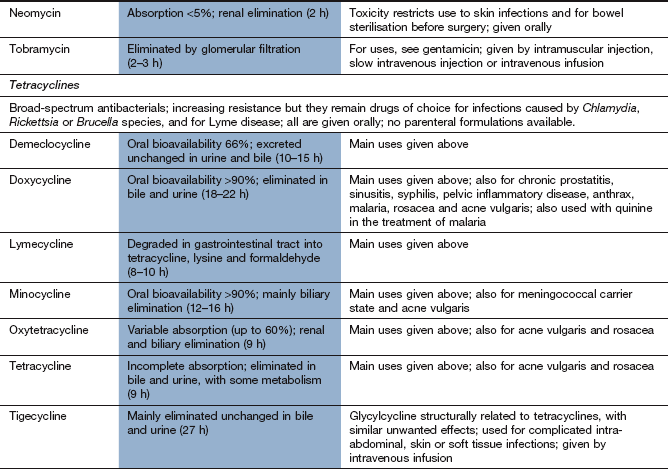
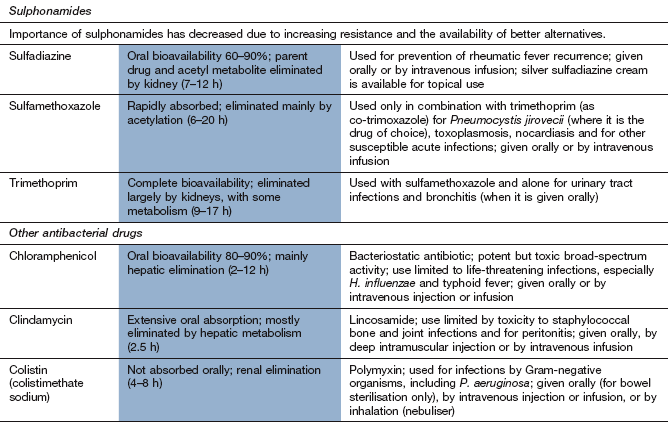
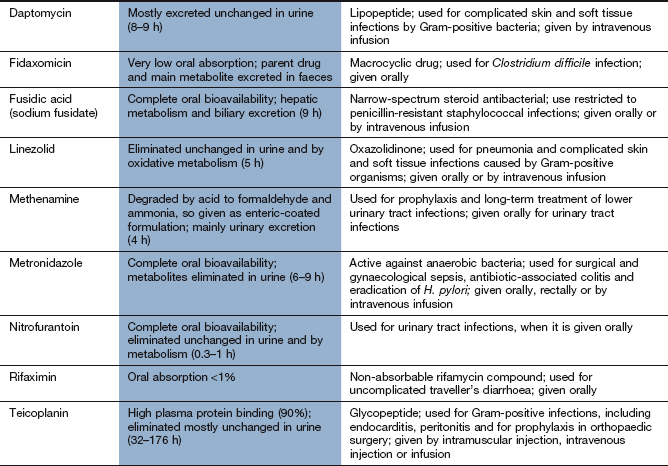
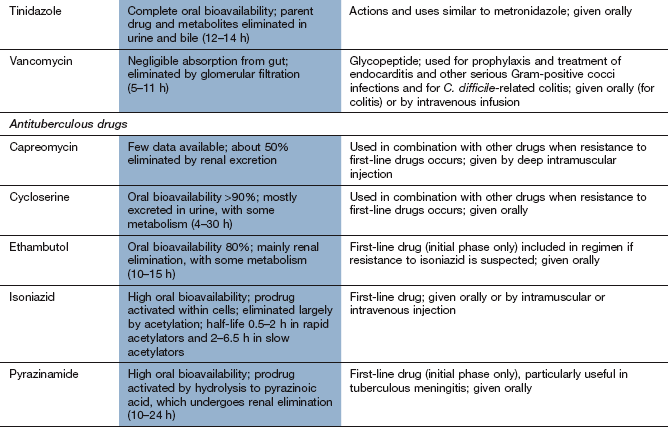
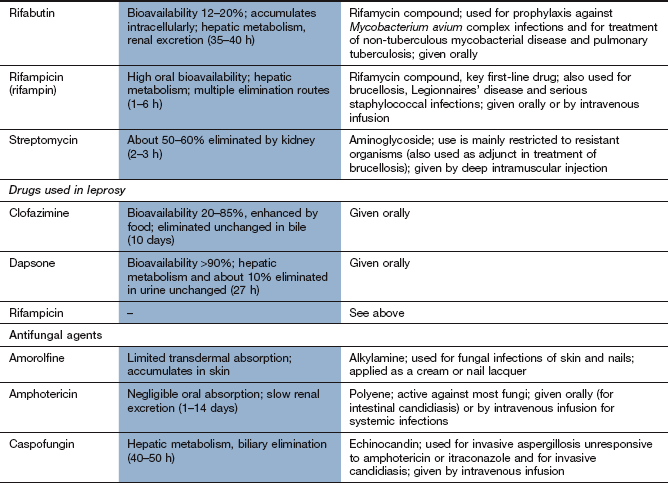

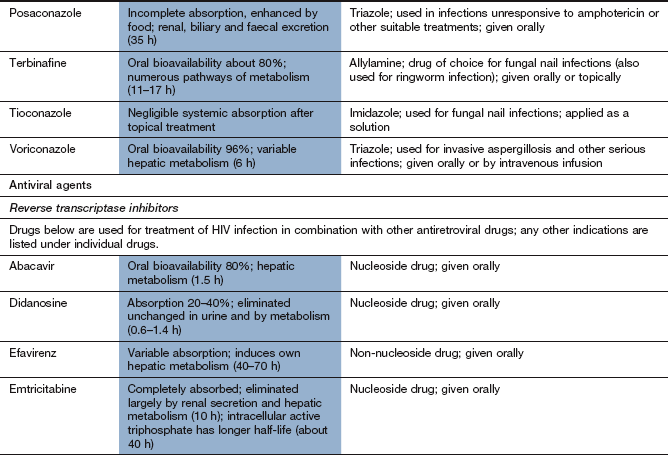
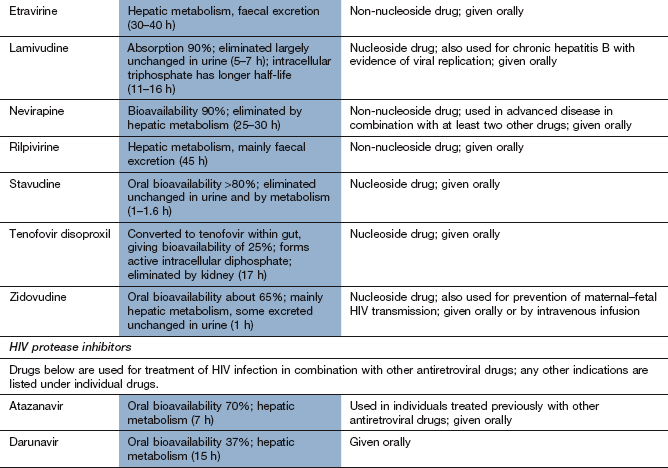
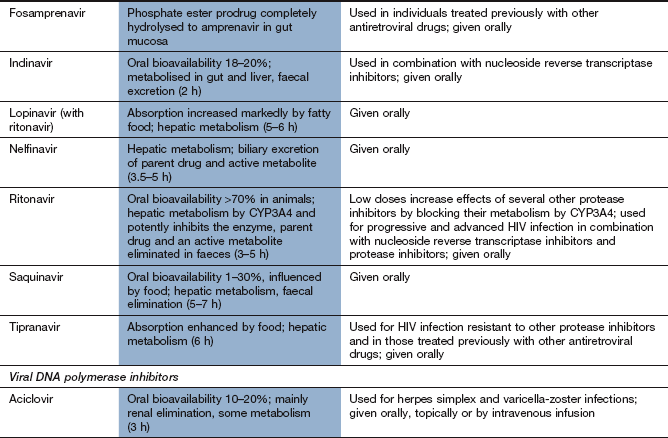
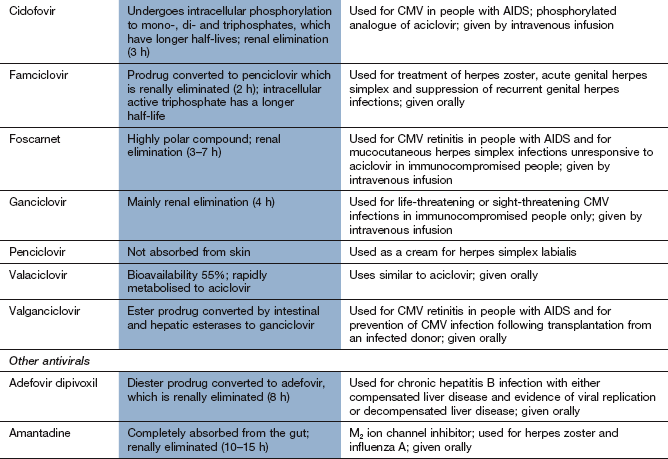
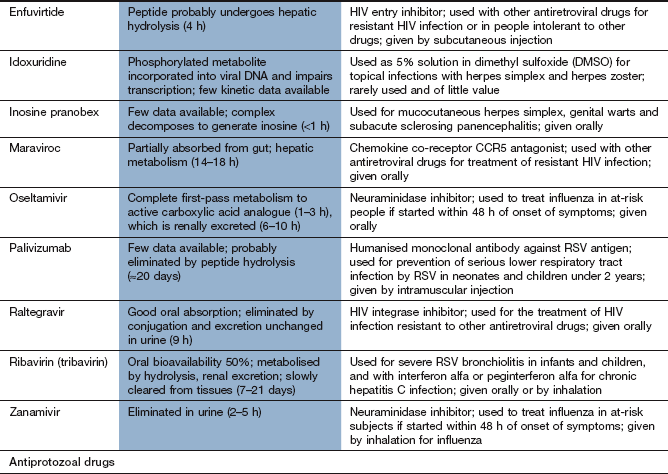
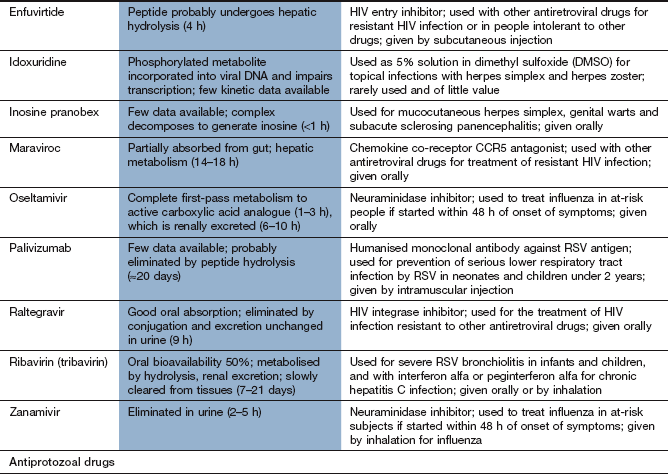
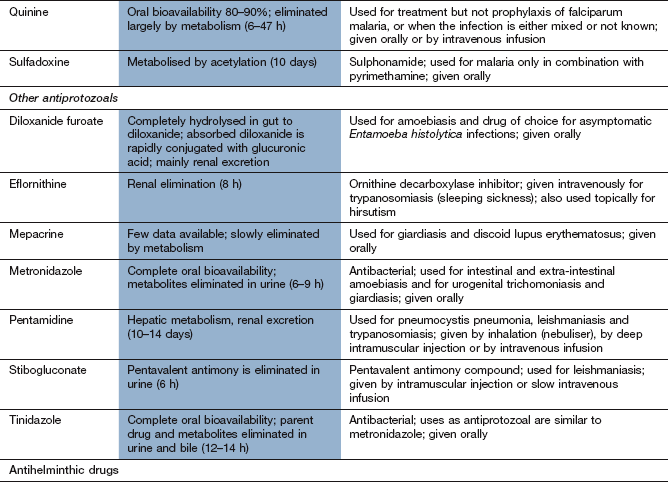
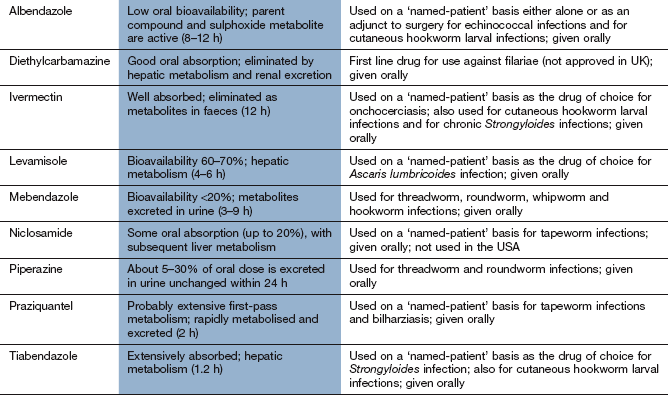
Bartlet, JG. Antibiotic-associated diarrhoea. N Engl J Med. 2002;346:334–339.
Calhoun, JH, Manring, MM. Adult osteomyelitis. Infect Dis Clin North Am. 2005;19:765–786.
Durrington, HJ, Summers, C. Recent changes in the management of community acquired pneumonia. BMJ. 2008;336:1429–1433.
Fihn, SD. Acute uncomplicated urinary tract infection in women. N Engl J Med. 2003;349:259–266.
Fitch, MT, Abrahamian, FM, Moran, GJ, et al. Emergency department management of meningitis and encephalitis. Infect Dis Clin North Am. 2008;22:33–52.
Grant, A, Gothard, P, Thwaites, G. Managing drug resistant tuberculosis. BMJ. 2008;337:564–569.
Gruchella, RS, Pirmohamed, M. Antibiotic allergy. N Engl J Med. 2006;354:601–609.
Hawkwy, PM, Livermore, DM. Carbapenem antibiotics for serious infections. BMJ. 2012;344:e3236.
Hirschmann, JV. Antibiotics for common respiratory tract infections in adults. Arch Intern Med. 2002;162:256–264.
Jacoby, GA, Munoz-Price, LS. The new β-lactamases. N Engl J Med. 2005;352:380–391.
Lawn, SD, Zumia, AI. Tuberculosis. Lancet. 2011;378:57–72.
Mathews, CJ, Weston, VC, Jones, A, et al. Bacterial septic arthritis in adults. Lancet. 2010;375:846–855.
Mackenzie, I, Lever, A. Management of sepsis. BMJ. 2007;335:929–932.
Mansharamani, NG, Koziel, H. Chronic lung sepsis: lung abscess, bronchiectasis and empyema. Curr Opin Pulm Med. 2003;9:181–185.
Moreillon, P, Que, Y-A. Infective endocarditis. Lancet. 2004;363:139–149.
Niederman, MS. Recent advances in community-acquired pneumonia: inpatient and outpatient. Chest. 2007;131:1205–1215.
Phoenix, G, Das, S, Joshi, M. Diagnosis and management of cellulitis. BMJ. 2012;345:e4955.
Picazo, JJ. Management of the febrile neutropenic patient: a consensus conference. Clin Infect Dis. 2004;15(suppl 1):S1–S6.
Rovers, MM, Schilder, AGM, Zielhuis, GA, et al. Otitis media. Lancet. 2004;363:465–473.
ten Hacken, NHT, Wijkstra, PJ, Kerstjens, HAM. Treatment of bronchiectasis in adults. BMJ. 2007;335:1089–1093.
Tenover, FC. Mechanisms of antimicrobial resistance in bacteria. Am J Med. 2006;119(suppl 1):S3–S10.
Thuny, F, Grisoli, D, Collart, F, et al. Infective endocarditis. Lancet. 2012;379:965–975.
Turnidge, J. Responsible prescribing for upper respiratory tract infections. Drugs. 2001;61:2065–2077.
Westphal, J-F, Brogard, J-M. Biliary tract infections. Drugs. 1999;57:81–91.
Moriarty, B, Hay, R, Morris-Jones, R. The diagnosis and management of tinea. BMJ. 2012;345:e4380.
Patterson, TF. Advances and challenges in management of invasive mycoses. Lancet. 2005;366:1013–1025.
Pfaller, MA. Antifungal drug resistance mechanisms, epidemiology, and consequences for treatment. Am J Med. 2012;125(suppl):S3–S13.
Sobel, JD. Management of patients with recurrent vulvovaginal candidiasis. Drugs. 2003;63:1059–1066.
Clavel, F, Hance, AJ. HIV drug resistance. N Engl J Med. 2004;350:1023–1035.
Esté, JA, Telenti, A. HIV entry inhibitors. Lancet. 2007;370:81–88.
Glezen, WP. Prevention and treatment of seasonal influenza. N Engl J Med. 2008;359:2579–2585.
Gupta, R, Warren, T, Wald, A. Genital herpes. Lancet. 2007;370:2127–2137.
Moscana, A. Neuraminidase inhibitors for influenza. N Engl J Med. 2005;353:1363–1373.
Volberding, PA, Deeks, SG. Antiretroviral therapy and management of HIV infection. Lancet. 2010;376:49–62.
Wareham, DW, Breuer, J. Herpes zoster. BMJ. 2007;334:1211–1215.
Davies, AP, Chalmers, RM. Cryptosporidiosis. BMJ. 2009;339:b4168.
Freedman, DO. Malaria prevention in short-term travellers. N Engl J Med. 2008;359:603–612.
Kremsner, PG, Krishna, S. Antimalarial combinations. Lancet. 2004;364:285–294.
Lalloo, DG, Hill, DR. Preventing malaria in travellers. BMJ. 2008;336:1362–1366.
Montoya, JG, Liessenfeld, O. Toxoplasmosis. Lancet. 2004;363:1965–1977.
Stanley, SL, Jr. Amoebiasis. Lancet. 2003;361:1025–1034.
Whitty, CJM, Lalloo, D, Ustianowski, A. Malaria: an update on treatment of adults in non-endemic countries. BMJ. 2006;333:241–245.