Corticosteroids (glucocorticoids and mineralocorticoids)
Structure and synthesis of steroid hormones
Steroid hormones comprise several compounds synthesised mainly in the adrenal cortex and the gonads. They are derived from cholesterol and share a distinctive core structure of four conjoined rings (Fig. 44.1). The pathways of steroid hormone synthesis are shown in Figure 44.2. The steroid hormones responsible for phenotypic gender differences are known as the sex hormones; these compounds are considered in Chapters 45 and 46.
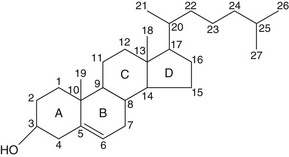
Fig. 44.1 The core structure of steroid hormones is derived from the cholesterol molecule shown.
The four rings are each identified by a letter A–D, and each carbon atom by a number.
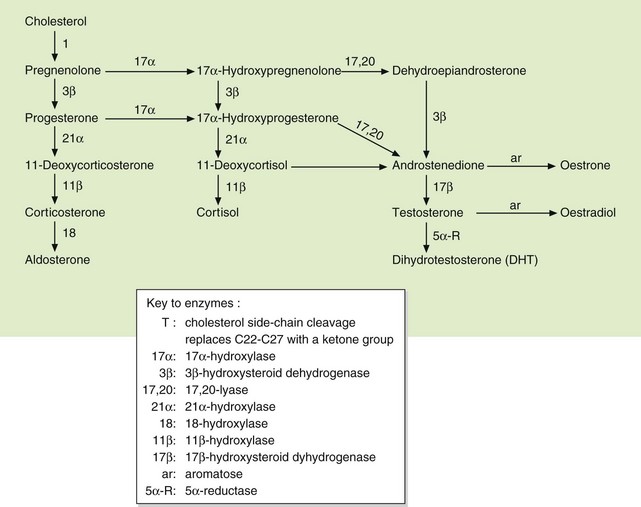
Fig. 44.2 Pathways of biosynthesis of steroid hormones, including progestogens, oestrogens, androgens, mineralocorticoids and glucocorticoids.
This chapter considers steroid hormones derived predominantly from the adrenal cortex that are known as adrenal corticosteroids. They have two distinct classes of action (see below and Table 44.1):
Table 44.1
Relative glucocorticoid and mineralocorticoid activities of some natural and synthetic corticosteroid hormones
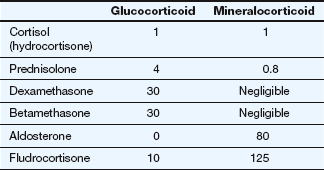
All potencies are relative to the glucocorticoid and mineralocorticoid activities of cortisol, each assigned an arbitrary value of 1. Due to intracellular metabolism by 11β-hydroxysteroid dehydrogenase in aldosterone-sensitive cells, cortisol has about one-thousandth of the mineralocorticoid activity of aldosterone in vivo.
 glucocorticoid activity, which affects carbohydrate and protein metabolism,
glucocorticoid activity, which affects carbohydrate and protein metabolism,
mineralocorticoid activity, which affects water and electrolyte balance.
The natural glucocorticoid is cortisol (also known as hydrocortisone), which has a hydroxyl grouping at position 17 and approximately equal affinity for glucocorticoid and mineralocorticoid receptors (but see below). The natural mineralocorticoid is aldosterone, which has an aldehyde grouping at position 18 and has little glucocorticoid activity. Synthetic corticosteroids that have been modified structurally to enhance either the glucocorticoid or mineralocorticoid activity are widely used therapeutically.
Although hydrocortisone and various synthetic derivatives are used for their glucocorticoid activity, they are frequently referred to as ‘corticosteroids’ or much less accurately as ‘steroids’. In this chapter, the distinction between glucocorticoid and mineralocorticoid is emphasised. In the rest of the book drugs with mainly glucocorticoid activity are usually referred to as corticosteroids.
Cortisol (hydrocortisone) is released from the zona fasciculata of the adrenal cortex, and its secretion is controlled by the hypothalamo–pituitary–adrenal axis (Fig. 44.3). An increase in the plasma glucocorticoid concentration feeds back negatively to the hypothalamus and pituitary to reduce the release of corticotropin-releasing hormone (CRH) and adrenocorticotropic hormone (ACTH; corticotropin). Glucocorticoid receptors are found in most tissues, giving cortisol a wide range of actions.
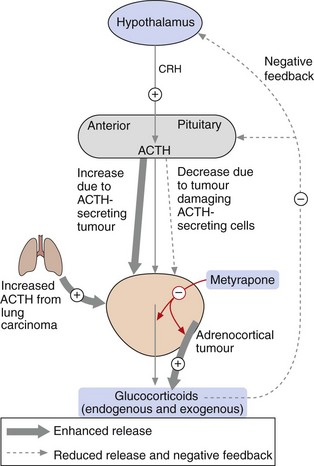
Fig. 44.3 Control of secretion of glucocorticoids.
Corticotropin-releasing hormone (CRH) from the hypothalamus stimulates adrenocorticotropic hormone (ACTH; corticotropin) from the anterior pituitary, which increases the release of glucocorticoids from the adrenal cortex. The level of glucocorticoids in the blood feeds back negatively to control the release of CRH and ACTH. Synthetic glucocorticoids have the same suppressive action on the hypothalamo–pituitary axis. In conditions in which excess glucocorticoids are released, for example in ACTH-secreting tumours or adrenocortical tumours, glucocorticoid synthesis and release can be reduced by metyrapone (red arrows). In people with tumours that result in hormone-induced reduction in glucocorticoids, synthetic glucocorticoids can be administered.
Aldosterone is secreted from the zona glomerulosa of the adrenal cortex. Aldosterone secretion is regulated by several factors, of which angiotensin II (Ch. 6), low plasma Na+ and high plasma K+ are the most important. Angiotensin II acts via specific AT1 receptors (see Chs 1 and 6) to induce aldosterone release. ACTH has a modest stimulatory effect on aldosterone secretion. Mineralocorticoid receptors are found in several tissues including the kidney, colon and heart. Important target cells are in the distal renal tubule and cortical collecting duct, where aldosterone increases the permeability of the luminal tubular membrane to Na+ by increasing the number of epithelial Na+ channels. It also stimulates the Na+/K+-ATPase pump in the basolateral membrane, which leads to active Na+ reabsorption and loss of K+ into tubular urine (Ch. 14). Water is passively reabsorbed with Na+, so that extracellular fluid volume and blood pressure are both increased. Target cells for aldosterone, especially in the renal tubule, contain 11β-hydroxysteroid dehydrogenase which metabolises cortisol to cortisone. Cortisone has very low affinity for the mineralocorticoid receptor and this ensures that most aldosterone-responsive tissues are not stimulated by endogenous glucocorticoid.
Mode of action of steroid hormones
All steroid hormones have similar intracellular receptor mechanisms, but there are distinct receptors for the different structural variants (Ch. 1). The distribution of the various receptors among tissues gives tissue specificity to each type of steroid hormone and defines its activity. In the circulation, steroid hormones are bound to specific globulins, including transcortin and sex hormone-binding globulin. Steroids are highly lipophilic and cross cell membranes by diffusion and bind to a specific cytoplasmic receptor (Ch. 1). In the absence of a steroid molecule, the receptor is retained in the cytoplasm and prevented from migrating to the cell nucleus because it is associated with a heat-shock protein (HSP). Binding of the steroid to the receptor dissociates the complex from the HSP, and the steroid–receptor complex then enters the nucleus and binds to a steroid-response element in the promoter region of the target genes (see Fig. 1.8). The binding usually involves the presence of other proteins, called chaperone proteins, and can lead to either increased or decreased transcription of proteins, depending on the target cell. Some genes are activated by simple interaction of the steroid receptor with the steroid-response element, but the rate of gene transcription is modulated by recruitment of various intranuclear co-regulator proteins and complexes.
The corticosteroid receptor only interacts with the steroid-response element for a matter of seconds before dissociating, and appears to have a ‘hit-and-run’ effect on gene transcription. When corticosteroids are given for a therapeutic effect, the response is delayed by many hours due to the time taken for modulation of protein synthesis. However, some actions of glucocorticoids are relatively rapid in onset and do not require gene transcription (non-genomic signalling pathways). They may produce direct activation of various kinases in the cell cytoplasm through a ligand-activated glucocorticoid receptor or through G-protein-coupled receptors, leading to effects such as vasodilation and regulation of cell growth.
Glucocorticoids
Actions of glucocorticoids
Immunosuppressant and anti-inflammatory actions
Glucocorticoids have important immunomodulatory actions that underpin many of their therapeutic uses. A major action of glucocorticoids is to silence pro-inflammatory genes (gene transrepression). These genes are activated by pro-inflammatory transcription factors such as nuclear factor κB (NF-κB) and activator protein 1 (AP-1) produced in response to inflammatory cytokines. NF-κB and AP-1 attach to the promoter region of pro-inflammatory genes and recruit co-activator molecules such as cAMP response element-binding protein (CBP) that have histone acetyltransferase activity. CBP acetylates core histones at the transcription complex which in turn activates pro-inflammatory gene transcription.
When glucocorticoid receptors are activated they bind to CBP at the glucocorticoid response element of inflammatory genes and inhibit histone acetyltransferase activity by recruiting co-repressor molecules. The glucocorticoid receptors also recruit histone deacetylase to suppress the pro-inflammatory genes. As a consequence, glucocorticoids inhibit the synthesis of many inflammatory cytokines, chemokines, adhesion molecules, inflammatory enzymes and receptors for inflammatory mediators.
Glucocorticoids also activate anti-inflammatory genes (gene transactivation). This is achieved by stimulation of core histone acetylation at the promoter regions of anti-inflammatory genes.
Anti-inflammatory effects
As a result of the above actions, glucocorticoids:
reduce T-lymphocyte proliferation and increase T-cell apoptosis, which impairs cell-mediated immunity (Ch. 38). They also inhibit humoral immunity by reducing T-cell activation, B-lymphocyte proliferation and immunoglobulin production, particularly IgG (Ch. 38),
inhibit mononuclear cell and neutrophil leucocyte migration and their adhesion to inflamed capillary endothelium. The ability of these inflammatory cells to phagocytose and destroy micro-organisms and to release oxygen free radicals is also reduced through downregulation of their surface receptors (Ch. 38),
reduce the synthesis of inflammatory prostaglandins by inhibition of phospholipase A2 activity (via induction of annexin-1) and by suppression of cyclo-oxygenase expression (Ch. 29),
impair fibroblast activity with reduced collagen synthesis and inhibition of matrix metalloproteinases, which impairs wound repair,
decrease capillary permeability, which has a protective effect on blood volume and raises blood pressure. The sensitivity of vascular walls to the vasoconstrictor actions of catecholamines and angiotensin II is also enhanced.
Metabolic effects
Gluconeogenesis is increased, particularly in the liver, using amino acids and glycerol from triglycerides. Storage of glycogen in the liver and, to a lesser extent, in muscle is increased, and uptake and utilisation of glucose in muscle and adipose tissue are impaired. These actions promote hyperglycaemia.
Protein is degraded, particularly in muscle, to release amino acids for synthesis of glucose while protein synthesis is inhibited. As a result there is an overall negative nitrogen balance.
Fat is redistributed from the glucocorticoid-sensitive fat stores in the limbs to the glucocorticoid-resistant stores in the face, neck and trunk. This action results from enhancement of the lipolytic response to catecholamines, and releases glycerol for glucose synthesis.
Effects on bone metabolism
Osteoblast formation is decreased, and apoptosis of mature osteoblasts is increased. The function of mature osteoblasts is inhibited by reducing production of osteocalcin, a key extracellular matrix protein in bone that promotes bone mineralisation. Osteocyte apoptosis is increased which reduces bone strength. By contrast, survival of osteoclasts is prolonged. These actions lead to bone resorption and demineralisation.
Inhibition of the proliferation and differentiation of chondrocytes at the epiphyseal end of the growth plate of long bones reduces bone growth in children.
Central nervous system effects
Plasma cortisol concentrations rise to a peak at the time of awakening and are lowest during sleep. In general, high circulating concentrations of cortisol are associated with alertness, but severe disturbances of mood may occur with abnormally high levels of glucocorticoid. Low concentrations produce a feeling of lethargy.
Mineralocorticoid effects
Natural glucocorticoids also have mineralocorticoid activity, although this has minimal impact at physiological doses (see above). Synthetic glucocorticoid compounds are altered structurally to minimise the amount of mineralocorticoid activity (Table 44.1).
Pharmacokinetics of glucocorticoids
Both hydrocortisone and synthetic glucocorticoids are used in clinical practice. They are readily absorbed from the gut. Hydrocortisone binds to corticosteroid-binding globulin (transcortin) and to albumin in the blood, and is extensively metabolised in the gut wall and liver. Synthetic glucocorticoids are more potent than hydrocortisone and bind to albumin but not to transcortin. They are more slowly metabolised in the liver, giving them a longer duration of action. Of the many synthetic glucocorticoids, dexamethasone is the most potent and has the least mineralocorticoid activity.
Most glucocorticoids are available in formulations for parenteral use. This does not appreciably shorten the time to onset of action, since most effects are delayed by up to 8 h while protein synthesis is modulated intracellularly. Some glucocorticoids are available in formulations for topical use (e.g. beclometasone, budesonide and fluticasone by inhaler for asthma). This reduces their systemic actions although systemic unwanted effects can still occur, particularly with high doses (see also Chs 12 and 34).
The plasma half-lives of glucocorticoids vary, but, because their mechanism of action depend on gene transcription and changes in protein synthesis, their biological (i.e. effective) half-lives are long (varying from 8 h for hydrocortisone to 2 days for dexamethasone).
Clinical uses of systemically administered glucocorticoids
The anti-inflammatory and immunosuppressant effects of glucocorticoids are used for control of various inflammatory diseases (especially those which are immunologically mediated) and treatment of neoplastic conditions, particularly when they involve lymphoid tissue (Box 44.1). Powerful glucocorticoids with little mineralocorticoid activity, such as prednisolone, are usually chosen. Dexamethasone is used to reduce oedema around malignant tumours in the brain and those compressing the spinal cord. It is also used in some anti-emetic regimens during cancer chemotherapy (Ch. 32).
Resistance to the anti-inflammatory and immunosuppressant effects of glucocorticoids occurs in some individuals. The mechanisms are complex, and arise from a variety of excessively activated intracellular inflammatory pathways. There are no reliable strategies to overcome glucocorticoid resistance, and alternative immunosuppressant drugs may need to be used (Ch. 38).
Physiological replacement therapy for corticosteroid deficiency
Hydrocortisone or an equivalent synthetic glucocorticoid is given orally twice or three times daily in doses as close as possible to the amount normally secreted by the adrenal cortex. The dose must be doubled or tripled in stressful situations, for example intercurrent infection. Acute adrenal insufficiency requires immediate treatment with high-dose intravenous hydrocortisone. Conditions that can give rise to glucocorticoid deficiency are shown in Box 44.2.
Box 44.2 Causes of glucocorticoid excess (Cushing's syndrome) and corticosteroid deficiency
Excessive secretion of ACTH by the anterior pituitary (pituitary adenoma) (Cushing's disease)
Excessive secretion of ACTH from an ectopic source (most commonly carcinoma of the bronchus)
A tumour of the adrenal cortex secreting predominantly cortisol
Iatrogenic: administration of glucocorticoid or ACTH in pharmacological doses
Causes of corticosteroid deficiency
Primary adrenal insufficiency (Addison's disease):
Secondary adrenocortical failure (deficient ACTH from the anterior pituitary):
Suppression of the hypothalamo–pituitary–adrenal axis by prolonged glucocorticoid treatment at pharmacological doses
Various enzyme defects in cortisol synthesis (congenital adrenal hyperplasia)
For uses of ACTH and its analogues, see Chapter 43.
Clinical uses of topically administered glucocorticoids
Topical use of glucocorticoids can deliver high concentrations to a target site and reduce systemic unwanted effects. However, significant absorption into the blood occurs at higher doses. Examples of the clinical uses of topical glucocorticoids are given in Table 44.2.
Table 44.2
Examples of topical corticosteroid administration
| Disease | Mode of administration | Chapter in this book |
| Asthma | Aerosol | 12 |
| Vasomotor rhinitis | Aerosol | 39 |
| Eczema | Ointment or cream | 49 |
| Superficial ocular inflammation | Aqueous solution | 50 |
| Ulcerative colitis | Aqueous solution or foam enema | 34 |
| Proctitis | Suppository | 34 |
| Arthritis | Aqueous solution by intra-articular injection | 30 |
Unwanted effects of glucocorticoids
Pharmacological doses of systemic glucocorticoids given over long periods will produce the typical features of adrenocortical overactivity (Cushing's syndrome). Unwanted glucocorticoid actions are shown in Box 44.3. These effects do not occur when physiological replacement doses of a glucocorticoid are used.
Box 44.3 Unwanted effects of glucocorticoids
Central obesity with ‘buffalo hump’, moon face and abdominal striae
Loss of supporting tissue in skin with skin atrophy, bruising and poor wound healing; local atrophy can be marked at the site of topical corticosteroid application
Osteoporosis due to catabolism of protein matrix in the bone and defective mineralisation
Proximal (i.e. shoulder and hip girdle) muscle wasting and weakness
Hyperglycaemia, which may lead to overt diabetes mellitus (Ch. 40)
Peptic ulceration due to inhibition of gastrointestinal prostaglandin (Ch. 33)
Mood changes, including euphoria and occasionally psychosis
Posterior capsular cataracts in the eye, and exacerbation of glaucoma
Increased susceptibility to infection with bacteria, viruses or fungi; activation of latent infections such as tuberculosis can also occur
Growth retardation in children, with reduced linear bone growth and premature epiphyseal closure
After long-term treatment, sudden withdrawal can produce an acute adrenal crisis due to suppression of the hypothalamic–pituitary–adrenal axis and adrenal atrophy. Recovery of adrenal responsiveness can take several months. Basal cortisol secretion is restored before maximal responses, leaving patients at risk during stress and intercurrent infection.
Mineralocorticoid effects (which vary between the different drugs)
Cushing's syndrome
Cushing's syndrome is characterised by excessive glucocorticoid effects (Box 44.3). There are four possible causes (Box 44.2).
Surgery is the definitive treatment for excessive pituitary secretion of ACTH (usually from an adenoma) and for unilateral adrenal tumours, with subsequent radiotherapy for some pituitary tumours.
Drug treatment to reduce glucocorticoid secretion is desirable for several weeks before surgery, in order to reverse the excessive tissue catabolism and correct the metabolic disturbances. This is usually achieved with metyrapone, which reduces glucocorticoid biosynthesis by competitive inhibition of 11β-hydroxylase (Fig. 44.2). Metyrapone also inhibits cytochrome P450 in the liver, which can produce important drug interactions. Gastrointestinal upset is the main unwanted effect. The antifungal agent ketoconazole (Ch. 51) also reduces cortisol synthesis by inhibition of 11β-hydroxylase, but its onset of action is slower than that of metyrapone. In addition, it inhibits sex steroid production and therefore often causes gynaecomastia and decreased libido in males and hirsutism in females. Ketoconazole can be used alone or in combination with metyrapone. All these drugs have relatively short-lived benefit, with rapid loss of control (escape) when increased ACTH secretion is the cause of the syndrome.
Mitotane is a chemotherapeutic drug (Ch. 52) that causes more profound suppression of glucocorticoid synthesis with no escape. It can be used for long-term control of symptoms for people who are unwilling or too unfit to undergo surgery. Glucocorticoid-replacement therapy is usually necessary during treatment with mitotane.
Ectopic ACTH secretion is not usually amenable to surgical cure, but palliative drug treatment can be helpful (Fig. 44.3).
Mineralocorticoids
Fludrocortisone
Aldosterone is almost completely inactivated on its first passage through the liver and is therefore unsuitable for oral use. Fludrocortisone (9α-fluorohydrocortisone) is a synthetic alternative but only about 10% escapes first-pass metabolism. Its half-life is short (0.5 h) due to rapid hepatic metabolism.
Clinical uses
Fludrocortisone is given as replacement therapy for defective aldosterone production. This is usually the result of primary adrenal pathology with destruction of all three zones of the adrenal cortex (Addison's disease).
Expansion of blood volume by fludrocortisone can be used to raise blood pressure in postural hypotension resulting from autonomic neuropathy. However, it often produces supine hypertension without fully eliminating the postural fall in blood pressure.
Unwanted effects
Excessive Na+ retention and K+ loss can occur with pharmacological doses of fludrocortisone. Hypertension can result, but the expansion of blood volume stimulates cardiac stretch receptors, leading to secretion of natriuretic peptides. This results in an ‘escape’ natriuresis which establishes a new equilibrium between Na+ intake and excretion at a higher blood volume. Consequently, oedema does not usually occur.
Primary hyperaldosteronism (Conn's syndrome)
Autonomous oversecretion of aldosterone causes hypertension and a hypokalaemic alkalosis. Some cases are caused by an adenoma in the zona glomerulosa of the adrenal cortex and are treated surgically. The majority are caused by hyperplasia of both zonae glomerulosa. This usually causes less marked clinical consequences, and a potassium-sparing diuretic (usually spironolactone) is the treatment of choice to preserve the plasma K+ concentration and reduce blood pressure (Ch. 14).
1. Glucocorticoids take many hours to produce their clinical effects.
2. Inhaled glucocorticoids do not cause systemic unwanted effects.
3. Oral fludrocortisone is a useful anti-inflammatory corticosteroid in severe asthma.
4. Fluticasone is not used orally.
5. Metyrapone inhibits glucocorticoid synthesis.
6. Hypoglycaemia is common during glucocorticoid administration.
7. If prolonged administration of prednisolone results in unwanted effects, it should be withdrawn immediately.
8. Mineralocorticoid secretion is decreased in Addison's disease.
9. Aldosterone secretion is inhibited by angiotensin II.
10. Glucocorticoids do not affect inflammatory responses provoked by infection.
A 35-year-old woman showed signs of cortisol excess, including centripetal obesity, muscle weakness, easy bruising and amenorrhoea.
A What were the possible causes?
B She was not taking corticosteroids, eliminating an iatrogenic cause. The cortisol level in a 24-h urine sample was elevated, and the plasma adrenocorticotropic hormone (ACTH) level was also high.
What did these results indicate?
C A single high dose of dexamethasone was administered and resulted in only marginal suppression of plasma cortisol.
What did this result indicate?
D A computed tomography scan and other tests showed an inoperable carcinoma of the bronchus.
Mr BFG, a 69-year-old man, had late-onset asthma, which was poorly controlled by β2-adrenoceptor agonists. His GP prescribed a low-dose inhaled corticosteroid that helped him initially.
A Which glucocorticoids could have been given by aerosol inhaler?
B What were the possible unwanted effects of the low-dose inhaled glucocorticoid?
C Mr BFG then had a particularly severe attack (acute severe asthma or status asthmaticus) which led to his admission to hospital as an emergency.
Among the drugs he was given was an intravenous corticosteroid. Which drug was likely to have been given and what was the objective of its use?
D Mr BFG's status asthmaticus resolved and he was then prescribed a course of oral corticosteroid while in hospital and subsequently sent home with high-dose inhaled corticosteroid.
Which corticosteroid could have been used for oral therapy?
E Mr BFG's asthma was poorly controlled by a higher dose of inhaled corticosteroid and it was decided to recommence oral therapy.
Why might oral therapy be better than inhaled therapy in severe chronic asthma?
F How would you have determined the dose to be used and monitor its appropriateness?
G After many months of this therapy, Mr BFG started to complain of apparently unrelated problems, including: recurrent minor infections, minor epigastric discomfort, especially on an empty stomach, weight gain and increased appetite, a tendency to bruise easily and severe back pain after a minor fall. Examination revealed a cushingoid appearance, and investigation showed a raised plasma glucose level and a decreased plasma level of cortisol and ACTH. Discuss the reasons for Mr BFG's symptoms.
1. True. Glucocorticoids act principally by modulating gene transcription so most of their clinical effects are not seen for several hours.
2. False. In maximum recommended doses, inhaled glucocorticoids can cause systemic effects.
3. False. Fludrocortisone is a synthetic mineralocorticoid with weak anti-inflammatory effects; glucocorticoids such as prednisolone are used orally in severe asthma.
4. True. Inhaled fluticasone is used for topical effects on the airways in the treatment of asthma.
5. True. Metyrapone inhibits synthesis of cortisol, and to a lesser extent that of aldosterone, by inhibiting steroid 11β-hydroxylase.
6. False. Corticosteroids cause hyperglycaemia by reducing tissue uptake of glucose and increased gluconeogenesis (‘steroid-induced diabetes’).
7. False. Prolonged administration of a systemic glucocorticoid may suppress the hypothalamo–pituitary–adrenal axis. The drug should be withdrawn slowly to allow the adrenals to recover their normal cortisol secretion and avoid corticosteroid deficiency.
8. True. Addison's disease can be caused by autoimmune disease or drugs such as ketoconazole.
9. False. Angiotensin II is produced by the action of renin, which is secreted when plasma sodium concentrations are low; angiotensin II then stimulates aldosterone release from the adrenal cortex and this increases Na+ reabsorption in the renal tubule.
>10. False. Glucocorticoids suppress all inflammatory responses including those produced by infection.
>11. False. Dexamethasone can inhibit vomiting and will add to the anti-emetic actions of agents such as ondansetron used in cancer chemotherapy.
>12. True. Glucocorticoids delay wound healing because of their catabolic effect on proteins.
A Possible causes include a tumour of the adrenal cortex secreting cortisol; excess secretion of adrenocorticotropic hormone (ACTH) by a pituitary tumour, or by a non-pituitary tumour (commonly small-cell carcinoma of the lung, medullary or thyroid carcinoma); or therapeutic administration of glucocorticoids or ACTH (iatrogenic).
B The cause could not be a primary cortisol-secreting adrenocortical tumour or glucocorticoid administration, as the plasma ACTH level would then be low due to negative feedback of glucocorticoid on the anterior pituitary and hypothalamus. The possibilities are an ACTH-secreting pituitary or non-pituitary tumour.
C Dexamethasone suppresses ACTH of pituitary origin but not from ectopic ACTH-producing tumours or adrenocortical tumours. The result might therefore exclude a tumour of pituitary origin.
D Metyrapone, an inhibitor of adrenal corticosteroid synthesis, could be given. She would probably also show signs of excess mineralocorticoid activity, which should be treated concomitantly with spironolactone.
A Inhaled glucocorticoids include beclometasone, budesonide and fluticasone (see Ch. 12).
B Systemic unwanted effects are unlikely at low doses of inhaled glucocorticoids. Local problems such as oral candidiasis could be managed with a spacer device and good oral hygiene.
C Intravenous hydrocortisone is likely to have been used to reduce airway inflammation in this acute exacerbation of asthma, although its onset of action would be delayed by several hours.
D A short course (5 days) of oral prednisolone could have been used.
E A high concentration of glucocorticoid is needed at inflammatory cells in the airways, and poor inhaler technique or airway obstruction by inflammation and mucus might prevent inhaled drugs reaching their target. Systemic prednisolone reaches airway inflammatory leucocytes more effectively than after inhalation in these circumstances, and may also reduce bone marrow proliferation and airway recruitment of eosinophils.
F The lowest possible doses of glucocorticoid should be used in chronic asthma management, using peak expiratory flow measurements and a symptom diary at home to monitor asthma control and to step the dosages up or down as appropriate.
G Glucocorticoids have a wide range of metabolic effects in addition to their anti-inflammatory and immunomodulatory actions. The cushingoid symptoms described can all be attributed to actions on carbohydrate, protein and lipid metabolism and suppression of the hypothalamo–pituitary–adrenal axis.
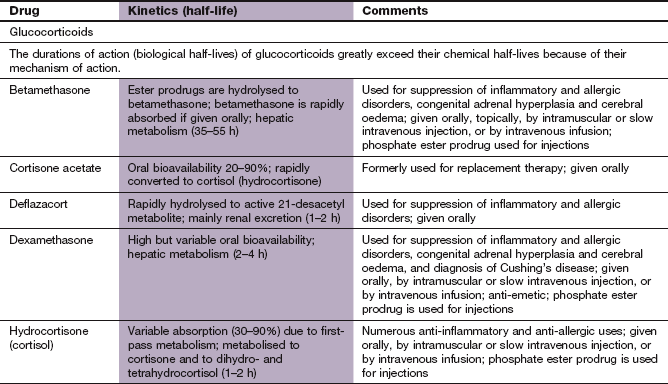
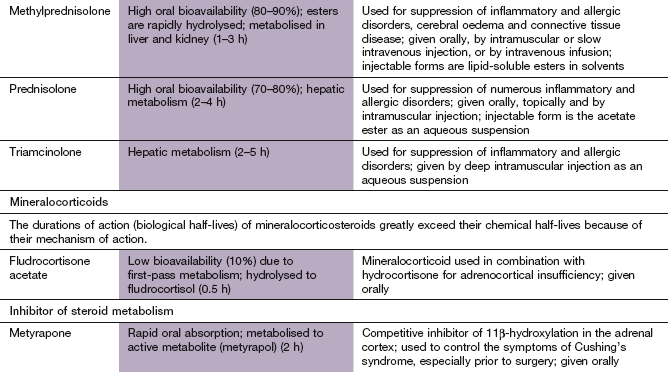
Additional corticosteroids used for specific purposes are covered in other chapters (see Table 44.2).
Barnes, PJ, Adcock, IM. Glucocorticoid resistance in inflammatory disease. Lancet. 2009;373:1905–1917.
Feldman, RD, Gros, R. Unravelling the mechanisms underlying the rapid vascular effects of steroids: sorting out the receptors and the pathways. Br J Pharmacol. 2011;163:1163–1169.
Lipworth, BS. Systemic adverse effects of inhaled corticosteroid therapy. Arch Intern Med. 1999;159:941–955.
Lovas, K, Husebye, ES. Replacement therapy in Addison's disease. Expert Opin Pharmacother. 2003;4:2145–2149.
Newell-Price, J, Bertagna, X, Grossman, AB, et al. Cushing's syndrome. Lancet. 2006;367:1605–1617.
Nieman, LK, Ilias, I. Evaluation and treatment of Cushing's syndrome. Am J Med. 2005;118:1340–1346.
Rhen, T, Cidlowski, JA. Antiinflammatory effects of glucocorticoids–new mechanisms for old drugs. N Engl J Med. 2005;353:1711–1723.
Weinstein, RS. Glucocorticoid-induced bone disease. N Engl J Med. 2011;365:62–70.
Young, WF, Jr. Minireview: primary aldosteronism – changing concepts in diagnosis and treatment. Endocrinology. 2003;144:2208–2213.