Lipid disorders
Lipids and lipoproteins
Lipid and lipoprotein metabolism is complex and the following account is a very brief summary, sufficient to establish the mechanism of action of drugs used to correct lipid abnormalities.
Cholesterol and triglycerides
Cholesterol is a sterol that is a vital structural component of cell membranes and a precursor of many steroids, including bile salts and steroid hormones. About 20–25% of daily production of cholesterol is by the liver; the rest is synthesised in the intestines, adrenal glands and reproductive organs or ingested in the diet from eggs, cheese, meat or fish. The synthesis of cholesterol involves several enzymes, but the rate-limiting step is catalysed by β-hydroxy-β-methylglutaryl-coenzyme A (HMG-CoA) reductase. Intracellular cholesterol is sensed by the cell and produces negative feedback on HMG-CoA reductase to reduce further cholesterol synthesis.
Cholesterol leaves hepatocytes either by transport into the circulation (see below) or by secretion into the bile after incorporation into bile salt micelles (Figs 48.1 and 48.2). Virtually all of the cholesterol secreted in bile is reabsorbed and taken up by the liver, which also retains about 50% of cholesterol that is not incorporated into bile salts.
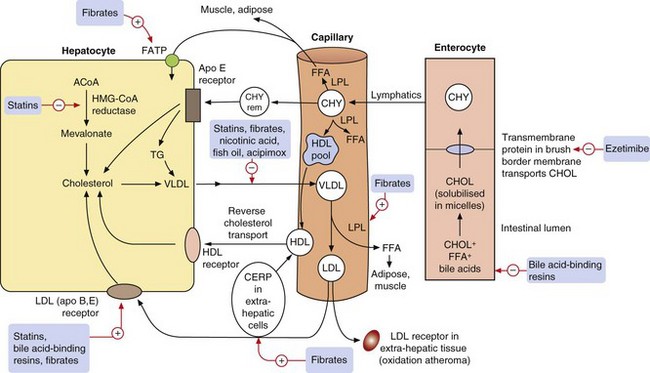
Fig. 48.1 Major pathways in lipoprotein formation and metabolism.
Dietary lipids including cholesterol (CHOL) and free fatty acids (FFA) in the gut are emulsified by bile acids and transported within chylomicrons (CHY) to the liver. They are then circulated as cholesterol and triglycerides (TG) to tissues in very-low-density lipoproteins (VLDL), where endothelial lipoprotein lipase (LPL) liberates FFA in adipose and muscle for storage or metabolism. The resulting low-density lipoproteins (LDL) are returned to hepatocytes via LDL receptors, or taken up by LDL receptors in extrahepatic tissues where they are oxidised and contribute to atherogenesis. The high-density lipoprotein (HDL) pool (nascent HDL) is derived from the chylomicrons following the action of LPL, and the reverse cholesterol pathway returns HDL to the liver via HDL receptors. Lipid-lowering drugs and their principal targets are shown with red arrows. ACoA, acetyl coenzyme A; Apo, apolipoprotein; CERP, cholesterol efflux regulatory protein; CHY rem, chylomicron remnant; FATP, fatty acid transport protein; HMG-CoA reductase, β-hydroxyl-β-methylglutaryl-coenzyme A reductase.
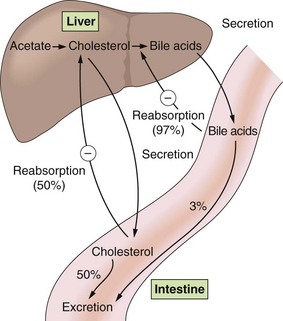
Fig. 48.2 Enterohepatic cycling of cholesterol and bile acids.
Bile acids are secreted via the bile duct into the duodenum, where they aid in dietary lipid absorption, and are then returned to the liver by the portal circulation. A circled minus sign indicates a negative-feedback effect. Percentages are in relation to the amount excreted in bile.
Triglycerides (fatty acids esterified with glycerol) are the major dietary fat, and can also be synthesised from intermediary metabolites formed in the liver from excess carbohydrate in the diet. Triglycerides are stored in adipose tissue, from where they can be mobilised as non-esterified free fatty acids to act as an energy substrate during periods of fasting.
The basis of lipoprotein metabolism
Lipids (triglycerides and esters of cholesterol) have low water solubility. They circulate in plasma encased in a coat of apolipoproteins within a phospholipid monolayer, creating lipoproteins that are water soluble and which make the triglycerides and cholesterol esters transportable. The lipoproteins can be differentiated according to the triglyceride/cholesterol ratio they carry, their apolipoprotein constituent and their density (Table 48.1). They are usually classified according to their density into very-low-density (VLDL), low-density (LDL), intermediate-density (IDL) and high-density (HDL) lipoproteins. There are specific cell-surface receptors for processing different apolipoproteins and these determine where and how particular fractions of circulating cholesterol and triglyceride will be handled (Table 48.1). In healthy individuals, about 70% of plasma cholesterol is carried by LDL and 20% by HDL. The least-dense and largest-diameter particles, known as chylomicrons, are exclusively concerned with the transport of dietary lipid from the intestine to the liver. Their low density and large size reflect their high content of triglycerides (Table 48.1), and they are almost completely removed from blood after a 12 h fast. VLDL carries about 60% of plasma triglyceride in the fasting state. The ratio of cholesterol to triglyceride carried is greatest in the HDL fraction.
Table 48.1
Apolipoprotein and lipid composition of major lipoproteins and their sources
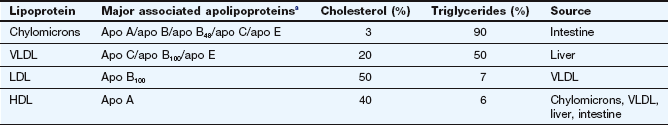
HDL, high-density lipoproteins; LDL, low-density lipoproteins; VLDL, very-low-density lipoproteins.
aThe apolipoproteins supply structural integrity and also have roles in controlling lipoprotein metabolism and as receptor ligands.
Processing of lipids absorbed from the gut
Cholesterol and free fatty acids are solubilised by bile acids in the gut lumen to facilitate absorption into enterocytes (see Fig. 48.1). Soluble cholesterol is transported into the enterocyte from the intestinal lumen by a specific lipid transmembrane transporter called Nieman-Pick C1-like 1 protein (NPC1L1). Triglyceride absorption does not require a specific transporter. Cholesterol and triglycerides are then incorporated into chylomicrons within the enterocytes (see Table 48.1). Chylomicrons pass into the lymphatic system and then into the circulation. Hydrolysis of triglycerides to free fatty acids in the chylomicrons is carried out by a lipoprotein lipase attached to the endothelium of capillaries in muscle and adipose tissue, and requires the chylomicron-associated apolipoprotein C (subtype C2) as a cofactor (Table 48.1). Free fatty acids are utilised by muscle and liver as an energy source or stored as triglycerides in adipose tissue. After removal of triglycerides from the chylomicrons, the remaining surface lipoprotein and lipid fractions leave the particles to enter the HDL pool as ‘nascent HDL’ (Fig. 48.1). The chylomicron remnants are taken into hepatocytes by specific chylomicron remnant (apolipoprotein E, or apo E) receptors.
Plasma transport and liver processing of lipids
Cholesterol is transported to tissues in chylomicrons, VLDL and LDL; it is transported from tissues to the liver by HDL. A high plasma concentration of LDL is associated with atheromatous disease.
Liver cholesterol (as esters) and any triglycerides in the liver that are surplus to synthetic and oxidative requirements are released into the circulation complexed to VLDL. Peripheral lipoprotein lipase acts on VLDL to release free fatty acids, leaving IDL (not shown in Fig. 48.1); triglycerides in IDL are also hydrolysed by hepatic lipase to release free fatty acids, which generates LDL. LDL therefore contains a higher concentration of cholesterol and a lower concentration of triglyceride compared with VLDL (Table 48.1). LDL is removed from the circulation by uptake into liver cells (75%) and peripheral tissues (25%). Some 70% of this uptake is by specific receptors for the apolipoproteins type B and E (Fig. 48.1), while the rest is by non-receptor-mediated pathways. The circulating concentration of LDL rises if there is either excess production of LDL or deficient LDL receptor numbers. When plasma LDL rises, non-receptor-mediated uptake of cholesterol in peripheral tissues such as arterial walls will increase. Within arterial walls, LDL cholesterol undergoes oxidation which leads to formation of lipid-rich deposits and atheromatous plaques (see below).
HDL carries cholesterol mobilised from peripheral tissues (and particularly oxidized cholesterol derivatives), and transports it to the liver (reverse cholesterol transport). The efflux of cholesterol from peripheral cells is facilitated by cholesterol efflux regulatory protein (CERP). This cholesterol is bound to nascent HDL, and then the cholesterol is esterified by the circulating enzyme lecithin-cholesterol acyltransferase (LCAT) to create mature HDL. HDL is believed to protect against atheroma by this reverse cholesterol transport from peripheral tissues to the liver. The enzyme cholesterol ester transfer protein (CETP) can transfer cholesterol from HDL to VLDL in exchange for triglycerides. The extent of this exchange depends on the concentration of circulating triglycerides.
Hypercholesterolaemia and atheroma
Abnormalities of plasma lipoprotein metabolism produce excessive concentrations of circulating cholesterol and/or triglyceride. Their clinical importance lies in their relationship to the production of atheroma (mainly raised plasma LDL cholesterol with a contribution from triglycerides) and pancreatitis (plasma triglycerides >12 mmol⋅L−1). Atheroma is focal thickening of the intima of arteries, produced by a combination of cells, elements of connective tissue, lipids and debris.
Excess LDL accumulates in the arterial wall, where its cholesterol undergoes enzymatic and free radical oxidation to produce a cytotoxic and chemotactic lipid that can activate the endothelium. Activated endothelium (a state which is also initiated by other atherogenic factors such as smoking, diabetes or hypertension) expresses adhesion molecules that attract platelets, monocytes and some T-lymphocytes. These cells migrate into the sub-endothelial space, where the monocytes differentiate into macrophages under the influence of endothelial cytokines. The macrophages take up oxidised LDL cholesterol via scavenger receptors, and the cholesterol accumulates as droplets in the cytosol, creating lipid-rich foam cells. Foam cells initiate fatty streaks that are the precursor of atheroma. T-cells in the developing atheromatous lesion recognise lipid antigens and release various cytokines that attract further inflammatory cells and initiate a T-helper cell type 1 inflammatory response (Ch. 38). These processes also result in formation of a cap of smooth muscle cells and collagen-rich matrix over the lesion. The extent of the inflammatory response determines whether the cap becomes fibrous and stable, or is destabilised by infiltration of inflammatory cells that make the cap prone to rupture or surface erosion. Plaque destabilisation underlies the development of acute coronary syndromes and many cases of ischaemic stroke (see Chs 5 and 9).
Atherogenic patterns of lipoproteins can result from the following:
 high dietary intake of saturated fat,
high dietary intake of saturated fat,
primary (inherited) disorders of enzymes or receptors involved in lipid metabolism. Most inherited hyperlipidaemias are polygenic, but an important inherited defect is familial hypercholesterolaemia, a single recessive gene disorder that affects 1 in 500 of the population, who have reduced synthesis of LDL receptors,
secondary lipid disorders, when hyperlipidaemia results from diseases that affect lipid metabolism; for example liver disease, nephrotic syndrome, hypothyroidism.
A classification for the various phenotypic patterns of primary hyperlipidaemia adopted by the World Health Organization is shown in Table 48.2.
Table 48.2
The Fredrickson classification of dyslipidaemias
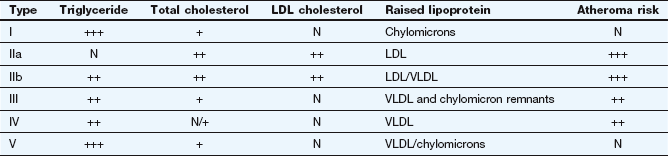
N, normal; +, slightly raised; ++, moderately raised; +++, extremely raised.
Drugs for hyperlipidaemias
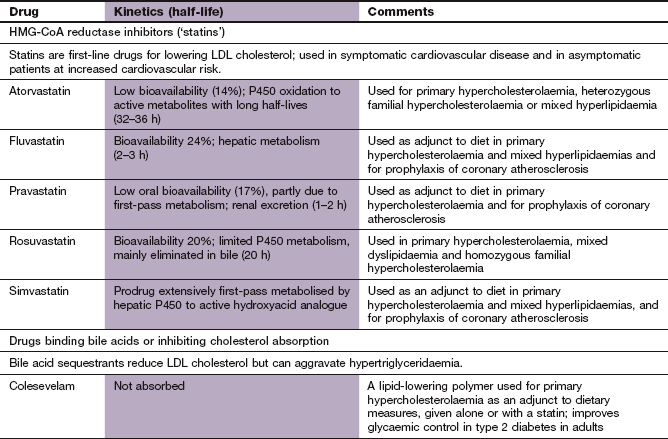
HMG-CoA reductase inhibitors (‘statins’)
Mechanism of action and effects: HMG-CoA reductase inhibitors competitively inhibit the enzyme that catalyses the rate-limiting step in the synthesis of cholesterol (Fig. 48.1). Their most important action is in the liver, where the fall in hepatic cholesterol levels produces a compensatory upregulation in the number of LDL receptors on hepatocytes, with increased clearance of circulating LDL cholesterol. In the liver, the cholesterol is reprocessed to form bile salts. The extent of the reduction in plasma LDL cholesterol depends on the specific drug and the dose of the drug ranges from 25 to 50%. Short-acting statins, such as simvastatin, are most effective when taken at night, which is the time when most cholesterol synthesis occurs. Statins also reduce the circulating concentration of VLDL by stimulating lipoprotein lipase, and therefore reduce circulating triglycerides. A modest increase in HDL cholesterol is usually seen, due to increased synthesis of the constituent apolipoprotein A1. This is due to activation of peroxisome proliferator-activated receptor α (PPAR-α) (see also fibrates, below).
Statins have several other actions, which may be distinct from their ability to reduce plasma lipids (Box 48.1). There is increasing evidence that some of these may contribute significantly to the beneficial actions of statins in reducing clinical events in people with atherothrombotic disease.
Box 48.1 Non-lipid effects of statinsa
Improved function of vascular endothelium damaged by hypercholesterolaemia, either by a direct effect or as a consequence of reduction in plasma LDL cholesterol
Stabilisation of atherosclerotic plaques by altered smooth muscle proliferation and migration
Changes in haemostasis: decreased plasma fibrinogen and enhanced fibrinolysis
Reduction of inflammatory cell infiltration into atherosclerotic plaques
Reduced plasma C-reactive protein (CRP), reflecting anti-inflammatory action
aStatins vary in these non-lipid effects.
Pharmacokinetics: The statins are well absorbed from the gut. Simvastatin is a prodrug (Ch. 2) that is activated in first-pass metabolism in the liver by cytochrome P450 (CYP3A4). Further metabolism inactivates the drug and only 5% of the active compound reaches the circulation. Atorvastatin undergoes first-pass metabolism, in part to active derivatives, and has a very long half-life. Pravastatin is a hydrophilic drug that is eliminated mainly by the kidneys; its half-life is 1–2 h. Rosuvastatin has a low oral bioavailability and is eliminated mainly in the bile, with a half-life of 20 h.
Gastrointestinal upset, including nausea, vomiting, abdominal pain, flatulence and diarrhoea.
Central nervous system effects, such as dizziness, blurred vision and headache.
Transient disturbance of liver function tests and, rarely, hepatitis.
Myalgia (muscle pain) or myositis (muscle inflammation) and rarely rhabdomyolysis. In some people there is a raised serum creatine kinase concentration suggesting muscle damage, but no muscle symptoms. The mechanism of the myopathy remains uncertain but it is likely that some people have minor metabolic abnormalities in their striated muscle that makes the muscle more susceptible to reduction of its fat substrate. There is an increased risk when a statin is used in combination with a fibrate, nicotinic acid, fusidic acid, ciclosporin and several other drugs (Ch. 38).
Specific cholesterol absorption inhibitors
Mechanism of action: Ezetimibe acts at the brush border of the small intestinal mucosa to specifically inhibit the NPC1L1 transporter and reduces cholesterol absorption by about 50%. It has no effect on the absorption of triglycerides, bile acids or fat-soluble vitamins. Given alone, ezetimibe reduces plasma total cholesterol by about 15% and LDL cholesterol by about 20%. When taken with a low dose of statin the combination is as effective as three doublings of the statin dose in reducing plasma total cholesterol.
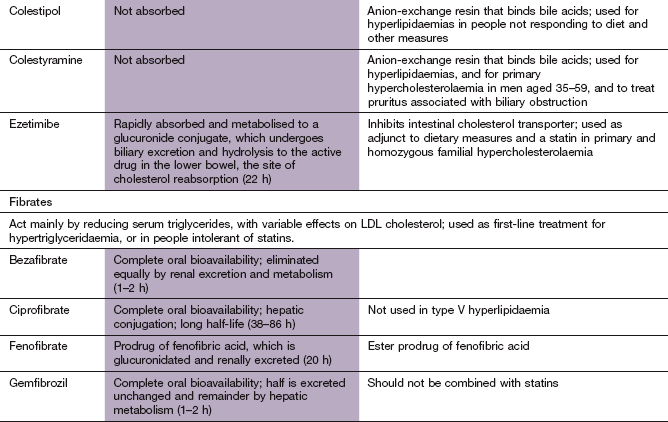
Bile acid-binding (anion-exchange) resins
Mechanism of action: Bile acids are synthesised from cholesterol in the liver, and are secreted into the duodenum to aid absorption of dietary fat. They are then reabsorbed in the terminal ileum and returned to the liver in the portal circulation (Fig. 48.2). Bile acid-binding resins are insoluble, non-absorbable polymers that bind bile salts in the gut and prevent enterohepatic circulation of bile acids.
When reabsorption of bile acids is impaired by binding to the resin, bile acid synthesis is increased from cholesterol in the liver. This reduces intrahepatic cholesterol, and there is compensatory upregulation of hepatic LDL receptors in order to replenish liver cholesterol. LDL cholesterol is cleared more rapidly from plasma, with a fall in circulating levels of 15–20%. Stimulation of VLDL synthesis produces a small rise in plasma triglycerides. There is a small rise in HDL cholesterol, but the mechanism for this is unclear.
Unpalatability. Sachets containing several grams of powder have to be taken, usually mixed with food. The taste and texture limit acceptability and for this reason resins are no longer widely used.
Constipation or, occasionally, diarrhoea.
Interference with the absorption of certain acidic drugs, for example digoxin (Ch. 7), warfarin (Ch. 11) and levothyroxine (Ch. 41). These drugs should be given at least 1 h before or 4 h after taking a resin.
Fibrates
Mechanism of action: The main mechanism of fibrate drugs is activation of gene transcription factors known as PPARs, particularly PPAR-α, which regulate the expression of genes that control lipoprotein metabolism. Fibrates are related to thiazolidinediones and their PPAR-mediated actions are described in Chapter 40. PPAR-α is expressed in several tissues, including the liver, heart and kidney. There are several consequences of PPAR-α activation:
increased free fatty acid uptake by the liver due to induction of the fatty acid transporter protein in the cell membrane. In the liver, fatty acid conversion to acyl-coenzyme A is enhanced as a result of increased acyl-CoA synthetase activity. The esterified fatty acids are less available for hepatic triglyceride synthesis,
increased lipoprotein lipase activity, which enhances the clearance of triglycerides from lipoproteins in the plasma (Fig. 48.1),
formation of LDL with increased affinity for its receptors, thus removing it more readily from the circulation,
increased plasma HDL because of enhanced apolipoprotein A1 and A2 production in the liver.
Pharmacokinetics: Fibrates are well absorbed from the gut and highly protein bound in the plasma. Fenofibrate is an ester prodrug that undergoes complete first-pass metabolism to the active form. Excretion is primarily by the kidney, although some metabolism occurs in the liver. The half-lives of bezafibrate and gemfibrozil are short, and other fibrates have longer half-lives.
Increased lithogenicity of bile theoretically increases the risk of gallstones, but this has not been a problem with the clinical uses of these drugs.
Myalgia and myositis are uncommon, unless there is impaired renal function or the fibrate is used in combination with a statin (especially gemfibrozil with simvastatin).
Drug interactions include inhibition of the effect of warfarin (Ch. 11).
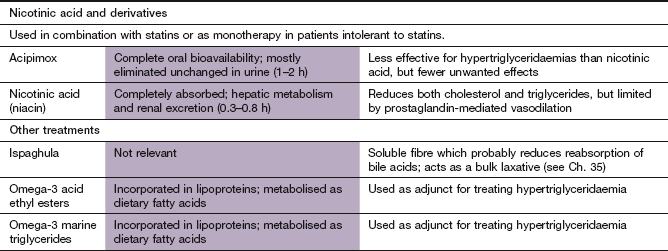
Nicotinic acid and derivatives
Mechanism of action: Nicotinic acid is a B vitamin which has effects on lipids at pharmacological doses. It inhibits hepatic diacylglycerol acyltransferase-2, which is a key enzyme for triglyceride synthesis. As a result, hepatic apo B degradation is enhanced and the hepatic secretion of VLDL and LDL is reduced. The action of nicotinic acid at a specific niacin receptor on adipocytes, skin and immune cells appears to be less important in lipid regulation. However, activation of the receptor may contribute to an anti-atherogenic action and the flushing caused by the drug. Nicotinic acid reduces circulating triglycerides by up to 35% and LDL cholesterol modestly by up to 15%. HDL cholesterol is increased by up to 25% as a result of reduced hepatic uptake of the HDL molecule. A decrease in the activity of hepatic lipase also shifts the distribution of HDL subfractions, with a predominant elevation of HDL2, which has greater protective effect than HDL3.
Pharmacokinetics: Nicotinic acid is well absorbed from the gut. Hepatic metabolism occurs via two pathways. Oxidation is a high-affinity, low-capacity pathway that generates metabolites which are thought to be responsible for the hepatotoxicity that can occur with high doses of nicotinic acid. The other pathway is a low-affinity, high-capacity conjugation pathway. Large doses of nicotinic acid are excreted unchanged in the urine. Acipimox is a synthetic derivative of nicotinic acid that is longer-acting but less effective for lowering LDL cholesterol.
Unwanted effects: Nicotinic acid is often poorly tolerated, but unwanted effects can be reduced by gradual dosage increases. A modified-release formulation that minimises the risk of flushing, and acipimox, are better tolerated.
Cutaneous vasodilation is particularly troublesome and causes flushing and itching. The action of nicotinic acid on specific G-protein-coupled receptors in the skin increases the production of prostaglandin (PG) D2 and PGE2, which cause the flushing. The flushing can be reduced by taking a small dose of aspirin 30 min before nicotinic acid, by taking the drug with food, or by using a modified-release formulation.
Gastrointestinal upset and peptic ulceration.
Glucose intolerance with high doses of nicotinic acid (not with acipimox).
Hepatotoxicity, which is less common with a modified-release formulation of niacin.
Omega-3 fatty acids
Omega-3 fatty acids are long-chain polyunsaturated acids such as α-linolenic acid, which is found in plants, and eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA), which are found in high quantities in oily fish such as mackerel and sardines. They have several potential cardioprotective effects:
because they are poor substrates for the enzymes that synthesise triglycerides, they lead to the production of triglyceride-poor LDL and reduce triglycerides in plasma, although total cholesterol is increased. They also increase conversion of VLDL to LDL, and increase circulating HDL cholesterol,
reduction of plasma fibrinogen, decreasing thrombogenesis,
they substitute for arachidonic acid in platelet phospholipids, which results in increased production of the prostanoid thromboxane A3 in platelets. This has a lower ability to induce platelet aggregation compared with the thromboxane A2 usually formed (Ch. 11),
retardation of growth of atherosclerotic plaques by reduced expression of endothelial adhesion molecules and an anti-inflammatory action,
promotion of nitric oxide-mediated vasodilation,
membrane stabilisation in heart muscle, with reduced susceptibility to ventricular arrhythmias and sudden cardiac death.
Management of hyperlipidaemias
Cardiovascular disease is the major risk associated with raised plasma LDL cholesterol. The relationship with raised LDL cholesterol is strongest for coronary atherosclerosis and peripheral vascular atherosclerosis, and to a lesser extent for cerebrovascular disease and atherothrombotic stroke. HDL cholesterol is protective against atherosclerosis and a low HDL cholesterol (<1.0 mmol⋅L−1) is associated with the highest risk of disease. The ratio of total cholesterol to HDL cholesterol provides a much more sensitive indicator of the relative risk of developing cardiovascular disease than total cholesterol alone.
While a high ratio of total cholesterol to HDL cholesterol predicts the relative risk of cardiovascular disease, the absolute risk (i.e. the overall number of individuals in the population who will develop disease in a particular time period) will be determined by the coexistence of other risk factors (see below).
Raised plasma triglycerides are an independent predictor of the risk of atherosclerosis, but less so than raised plasma cholesterol. Nevertheless, when raised triglycerides coexist with an atherogenic cholesterol profile, the overall risk is enhanced. A markedly raised plasma triglyceride concentration (>12 mmol⋅L−1) confers an increased risk of acute pancreatitis. Isolated hypertriglyceridaemia should be treated intensively for this reason alone.
Secondary cause of hyperlipidaemia, such as diabetes, hypothyroidism and nephrotic syndrome, should be excluded or treated before embarking on other aspects of management.
Primary prevention of cardiovascular disease
Atherothrombotic disease has a multifactorial aetiology, and any strategy for primary prevention must consider all relevant treatable factors. Drug treatment of hyperlipidaemia with drugs for primary prevention should only be considered if there is a sufficiently high absolute risk of disease, and should not be based on the cholesterol level alone. Important factors to consider in risk management include the following.
Smoking: smoking doubles the risk of coronary artery disease. Stopping smoking reduces the risk close to that of a non-smoker in 3–5 years (see Ch. 5).
Physical activity: a physically active lifestyle reduces the risk of myocardial infarction by up to 50% compared with a sedentary lifestyle.
Maintaining ideal body weight: obesity (see Ch. 37) increases the risk of myocardial infarction by up to 50%.
Mild-to-moderate alcohol consumption: a modest alcohol intake (see Ch. 54) can reduce the risk of myocardial infarction by about one-third. A high alcohol intake increases blood pressure, and thus increases cardiovascular risk.
Treating hypertension (see Ch. 6): although this is more effective for the prevention of stroke, it also reduces the risk of myocardial infarction, especially in older people.
Control of diabetes: there is conflicting evidence on whether close control of plasma glucose reduces vascular events. However, since the risk of ischaemic heart disease in diabetes is at least twice that of people without diabetes, intensive management of coexistent risk factors should be undertaken.
Modifying the diet: dietary management should be advised for all people with hypercholesterolaemia, with a reduction in saturated fat intake (saturated fat decreases hepatic LDL receptors) and an increase in monounsaturated fats (which increases hepatic LDL cholesterol receptors). Eating a diet containing fresh fruit and vegetables reduces oxidation of LDL cholesterol and therefore makes it less atherogenic.
Treating raised LDL cholesterol: this is a powerful predictor of future cardiovascular disease, especially in young people. The greatest risk is present when there is familial hypercholesterolaemia (FH), a dominantly inherited genetic defect that predisposes to premature coronary heart disease even in the absence of other risk factors. Heterozygous FH is associated with reduced LDL receptors on liver cells, and the total serum cholesterol is usually greater than 7.5 mmol⋅L−1 in adult life. Lipid-lowering therapy in FH, usually with a statin, should normally begin before the age of 10 years with the goal of reducing plasma LDL cholesterol by 50%.
For other forms of multigenic and acquired hypercholesterolaemia, the risk of cardiovascular disease should first be estimated using risk tables that assess the contribution of the total cholesterol/HDL ratio, systolic blood pressure, smoking, family history and other risk factors such as rheumatoid arthritis and socioeconomic deprivation. The question exercising the minds of health economists is not whether treatment is effective, but when it becomes cost-effective. As part of a multiple risk factor intervention strategy, drug therapy for raised LDL cholesterol has a role for those at higher absolute risk, usually because several other risk factors coexist or there is a history of premature coronary artery disease in a first-degree relative. In the UK, lipid-lowering drugs are recommended if the predicted 10-year cardiovascular disease risk (a combination of coronary heart disease and stroke risk) is greater than 20%. There is no target cholesterol recommendation for primary prevention, except for those with FH for whom a 50% reduction in LDL cholesterol is recommended. For other people, using a dose of statin that has been shown to reduce cardiovascular events is recommended, regardless of the achieved reduction in plasma LDL cholesterol.
The ability of cholesterol-lowering drugs (and particularly statins) to prevent ischaemic heart disease has been demonstrated in many trials. Reducing plasma total cholesterol by 25–30% (with a reduction in LDL cholesterol of 30–35%) with a statin reduces the subsequent risk of myocardial infarction or vascular death by about 30%.
Secondary prevention of cardiovascular disease
Once cardiovascular disease is clinically apparent the subsequent risk of death from vascular events is increased. People with clinical evidence of vascular disease are at much greater absolute risk of a further event than are those without clinical coronary artery disease but who have similar, or even higher, plasma cholesterol concentrations. A recent myocardial infarction or an episode of unstable angina confers the highest risk. At slightly lower absolute risk are those with stable angina pectoris, peripheral vascular disease or ischaemic stroke. Reduction of even ‘normal’ plasma cholesterol concentrations (to as low as 4.0 mmol⋅L−1) in people with established vascular disease reduces the subsequent risk of both fatal and non-fatal cardiovascular events.
Current evidence supports the use of statins as first-line therapy. Trials with fibrates have shown less marked benefit unless the major lipid abnormality is low HDL cholesterol (see also Ch. 5). The target cholesterol concentration is total cholesterol below 4.0 mmol⋅L−1 (LDL cholesterol <2 mmol⋅L−1). When this is not achieved with a statin alone, then combinations of drugs, such as a statin with ezetimibe or a fibrate, may be used, although there is little evidence of improved clinical outcomes. The use of omega-3 fatty acids after myocardial infarction has a cardioprotective effect, which may not entirely relate to their effects on plasma lipids.
Lowering plasma cholesterol for secondary prevention of coronary artery disease should be only one aspect of a comprehensive strategy for improving prognosis (see Ch. 5).
Mechanisms of prevention of coronary events by lipid-lowering drugs
There is a close relationship between the degree of LDL cholesterol reduction and the reduced risk of coronary events, especially when it is achieved with a statin. Overall there is a 2–3% reduction in risk for every 1% reduction in plasma total cholesterol concentration. Reducing plasma cholesterol probably stabilises existing atheromatous plaques by preventing lipid accumulation in their core and therefore reduces the risk of plaque rupture. Statins prevent the growth of existing coronary artery plaques and reduce the formation of new plaques. High doses of a statin may even produce some regression of existing plaque. An anti-inflammatory effect of statins may be important, measured by a reduction in plasma high-sensitivity C-reactive protein. Other actions of statins, such as reduction in thrombogenicity of blood and inhibition of smooth muscle proliferation in atheromatous plaques, may contribute to the clinical benefit, but their roles remain speculative.
There is uncertainty whether lipid-lowering drugs other than statins have the same ability to reduce cardiovascular events. Although it is widely accepted that lowering LDL cholesterol should provide some protection against atheroma, however it is achieved, there is little evidence to confirm this.
Management of hypertriglyceridaemia
When triglycerides are markedly raised, control of diabetes, weight loss and reduction of alcohol intake should be considered when appropriate. When drug therapy is necessary, modest hypertriglyceridaemia in association with hypercholesterolaemia will usually respond to a statin. Extremely high plasma triglyceride concentrations usually respond well to a fibrate or to nicotinic acid. Combination therapy with a statin and a fibrate may be necessary in some high-risk individuals to achieve an acceptable lipid profile.
True/false questions
1. The risk of coronary disease is strongly related to the plasma triglyceride concentration.
2. Genetic factors contribute to the development of hypercholesterolaemia.
3. Anion-exchange resins enhance the absorption of bile acids from the gut.
4. Statins inhibit β-hydroxy-β-methylglutaryl-coenzyme A (HMG-CoA) reductase activity.
5. Decreased hepatic cholesterol synthesis results in increased numbers of high-density lipoprotein (HDL) receptors.
6. Pravastatin lowers plasma low-density lipoprotein (LDL) cholesterol by about 10%.
7. Co-administration of statins and fibrates has no greater effect than giving each drug separately.
8. Ezetimibe blocks cholesterol absorption in the intestinal mucosa.
9. Gemfibrozil inhibits lipoprotein lipase by activating peroxisome proliferator-activated receptor α (PPAR-α).
10. Nicotinic acid reduces triglyceride synthesis in the liver.
11. Skin flushing is a common unwanted effect of nicotinic acid.
One-best-answer (OBA) questions
1. Identify the most accurate statement below concerning lipids and cardiovascular disease.
A A high total plasma cholesterol/HDL cholesterol ratio reduces cardiovascular risk.
B Very-low-density lipoprotein (VLDL) has a greater percentage of cholesterol than HDL.
C The outer coat of lipoproteins is a phospholipid bilayer.
D Reducing dietary saturated fat reduces coronary disease risk.
E Even moderate alcohol consumption increases the risk of myocardial infarction.
2. Identify the inaccurate statement below concerning the actions of statins.
1. False. Cardiovascular risk is mostly associated with high low-density lipoprotein (LDL) cholesterol, which leads to lipid peroxidation and formation of foam cells in atheromatous plaque.
2. True. The best understood genetic risk is familial hypercholesterolaemia, in which a recessive gene disorder predisposes to premature coronary disease.
3. False. The anion-exchange resins sequester bile acids in the gut, decreasing the absorption of dietary cholesterol. They also increase incorporation of hepatic cholesterol into bile acids, leading to further loss of cholesterol in bile secretions into the gut.
4. True. HMG-CoA reductase is the rate-limiting enzyme for cholesterol synthesis in the liver.
5. False. Reducing cholesterol synthesis in the liver results in increased LDL receptors and hence increased LDL clearance from the plasma.
6. False. Statins reduce plama LDL cholesterol by 25–50%.
7. False. Statins and fibrates act mainly by different mechanisms, and their co-administration can help achieve target plasma cholesterol concentrations, although the additional benefit on clinical outcomes is unclear.
8. True. Ezetimibe reduces cholesterol absorption by blocking the intestinal NPC1L1 cholesterol transporter.
9. False. Fibrates activate PPAR-α but this increases lipoprotein lipase activity and clears triglycerides from the plasma.
10. True. Nicotinic acid inhibits triglyceride synthesis by diacylglycerol acyltransferase-2 in the liver, hence reducing production of VLDL and LDL.
11. True. Nicotinic acid may cause skin flushing by stimulating prostaglandin synthesis and it can be reduced by a non-steroidal anti-inflammatory drug (NSAID).
12. False. Arachidonic acid is an omega-6 fatty acid and the precursor of prothrombotic thromboxane A2. Omega-3 fatty acids include eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) found in fish oils, which may have cardioprotective effects.
OBA answers
A Incorrect. A low ratio of total cholesterol to HDL cholesterol is associated with lower cardiovascular risk.
B Incorrect. VLDL carries a greater load of triglycerides, so the proportion of cholesterol is lower in VLDL (20%) than in HDL (40%).
C Incorrect. The lipoprotein coat is a phospholipid monolayer, in which apolipoproteins are embedded, and provides a lipophilic interior for the transport of triglycerides.
D Correct. High dietary intake of saturated fat is associated with coronary disease.
E Incorrect. Moderate alcohol consumption reduces myocardial infarction risk by 30–40%.
2. Answer E is the inaccurate statement.
A Correct. The effect on smooth muscle proliferation may improve plaque stability.
B Correct. Statins can cause muscle pain and, rarely, rhabdomyolysis.
C Correct. The reduced plasma CRP reflects an anti-inflammatory action of statins.
D Correct. This action on lipoprotein lipase reduces plasma triglycerides.
E Incorrect. Statins increase plasma HDL concentration by 5–15%.
Case-based answers
A General advice would include a low-fat diet, exercise and stopping smoking. Concomitant risk factors including obesity, diabetes and hypertension should be investigated.
B A statin would be recommended as first-choice drug in preventing cardiovascular events. Statins are of proven benefit based on data from many studies and they are well tolerated.
C The response would depend upon the chosen statin, its dosage and additional lifestyle changes, but the recommended targets are a total cholesterol concentration less than 4 mmol⋅L−1 and LDL cholesterol less than 2 mmol⋅L−1. Several weeks of treatment may be required.
D Gastrointestinal upsets are common. Use of statins is not recommended in people with hypothyroidism or liver disease. Thyroid and liver function tests should be performed. He should be advised to report unexplained muscle pain.
Afilalo, J, Majdan, AA, Eisenberg, MJ. Intensive statin therapy in acute coronary syndromes and stable coronary heart disease: a comparative meta-analysis of randomised controlled trials. Heart. 2007;93:914–921.
Almuti, K, Rimawi, R, Spevack, D, et al. Effects of statins beyond lipid lowering: potential for clinical benefits. Int J Cardiol. 2006;109:7–15.
Armitage, J. Safety of statins in clinical practice. Lancet. 2007;370:1781–1790.
Baber, U, Toto, RD, de Lemos, J. Statins and cardiovascular risk reduction in patients with chronic kidney disease and end-stage renal failure. Am Heart J. 2007;153:471–477.
Cholesterol Treatment Trialists’ Collaborators. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90 056 participants in 14 randomised trials of statins. Lancet. 2005;366:1267–1278.
Cholesterol Treatment Trialists’ Collaborators. Efficacy of cholesterol-lowering therapy in 18 686 people with diabetes in 14 randomised trials of statins: a meta-analysis. Lancet. 2008;371:117–125.
Durrington, P. Dyslipidaemia. Lancet. 2003;362:717–731.
Gami, AS, Montori, VM, Erwin, PJ, et al. Systematic review of lipid lowering for primary prevention of coronary heart disease in diabetes. BMJ. 2003;326:528–529.
Gutierrez, J, Ramirez, G, Rundek, T, et al. Statin therapy in the prevention of recurrent cardiovascular events: a sex-based meta-analysis. Arch Intern Med. 2012;172:909–919.
Hachem, SB, Mooradian, AD. Familial dyslipidaemias. An overview of genetics, pathophysiology and management. Drugs. 2006;66:1949–1969.
Joy, TR, Hegele, RA. Narrative review: statin-related myopathy. Ann Intern Med. 2009;150:858–868.
Kwak, SM, Nyung, S-K, Lee, YJ, et al. Efficacy of omega-3 fatty acid supplements (eicosapentaenoic acid and docasahexaenoic acids) in the secondary prevention of cardiovascular disease: a meta-analysis of randomized, double-blind, placebo-controlled trials. Arch Intern Med. 2012;172:686–694.
Lee, C-H, Olson, P, Evans, RM. Minireview: lipid metabolism, metabolic diseases, and peroxisome proliferator-activated receptors. Endocrinolog y. 2003;144:2201–2207.
Minder, CM, Blaha, MJ, Horne, A, et al. Evidence-based use of statins for primary prevention of cardiovascular disease. Am J Med. 2012;125:440–446.
Nicholls, SJ, Tuczu, EM, Sipahi, I, et al. Statins, high-density lipoprotein cholesterol, and regression of coronary atherosclerosis. JAMA. 2007;297:499–508.
Nissen, SE, Tuzcu, EM, Schoenhagen, P, et al. Statin therapy, LDL cholesterol, C-reactive protein, and coronary artery disease. N Engl J Med. 2005;352:29–38.
Rallidis, LS, Lekakis, J, Kremastinos, DT. Current questions regarding the use of statins in patients with coronary heart disease. Int J Cardiol. 2007;122:188–194.
Saravanan, P, Davidson, NC, Schmidt, EB, et al. Cardiovascular effects of marine omega-3 fatty acids. Lancet. 2010;376:540–550.
Sathasivam, S, Lecky, B. Statin induced myopathy. BMJ. 2008;337:a2286.
Schillinger, M, Exner, M, Mlekusch, W, et al. Statin therapy improves cardiovascular outcome of patients with peripheral vascular disease. Eur Heart J. 2004;25:742–748.
Walsh, JME, Pignone, M. Drug treatment of hyperlipidaemia in women. JAMA. 2004;291:2243–2253.