Substance abuse and dependence
Substance abuse is characterised by compulsive drug-seeking and drug-taking behaviour and an inability to control intake (addiction); there may also be symptoms of withdrawal when the drug becomes unavailable (dependence).
Dependence-inducing drugs are mind modifying substances. They may be taken recreationally, initially because of the pleasurable effect they produce, but later to avoid unpleasant withdrawal symptoms. Dependence produces different degrees of need for the drug, from mild desire to an intense craving.
In other cases, the drug is initially prescribed to treat a medical problem.
The biological basis of dependence
The mechanisms of drug dependence are relatively poorly understood but involve complex dysfunctional adaptations of the neurocircuits in the brain that subserve physiological motivation and reward processes. The mesolimbic pathway is central to these processes and, depending upon the particular stimulus and the functional status of the individual, its activation can result in a spectrum of response from slight mood elevation to intense pleasure or euphoria. Stimulation can result from a plethora of factors that are very personal, for example food intake, sexual activity and the controlled and occasional use of lifestyle drugs such as alcohol and nicotine.
Acute activation of the mesolimbic dopamine reward pathways
The mesolimbic pathway consists of several structures:
 the ventral tegmental area, which communicates with the nucleus accumbens via the medial forebrain bundle,
the ventral tegmental area, which communicates with the nucleus accumbens via the medial forebrain bundle,
the amygdala (associated with emotions, especially fear and anxiety),
The reward pathway is activated by impulses arising in the ventral tegmental area of the brain. These impulses are relayed through the medial forebrain bundle, via the nucleus accumbens, to the prefrontal cortex (Fig. 54.1). Activation of the ventral tegmental area results in dopamine release in the nucleus accumbens and stimulation of postsynaptic D2 receptors (acting via inhibitory Gi proteins to inhibit the generation of intracellular cAMP). This is thought to be central to the processes of motivation and reward.
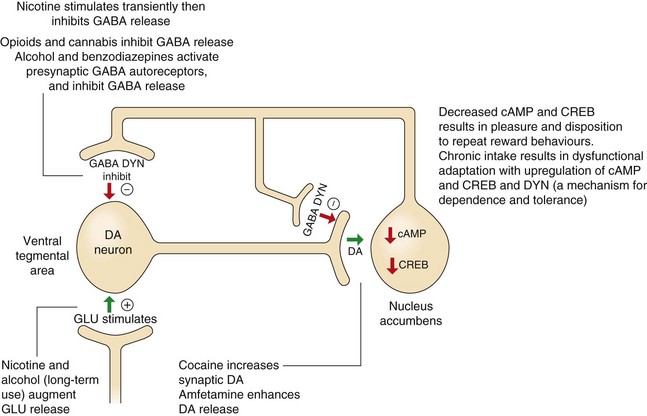
Fig. 54.1 The role of dopamine and cAMP in reward pathways in the mesolimbic system and the relevance of these pathways to substance abuse.
This diagram illustrates only a small part of the complex mechanisms involved in the processes of reward and the ways that substances of abuse influence these pathways, leading to dependence and withdrawal effects. Dopaminergic neurons in the ventral tegmental area release dopamine (DA) at the nucleus accumbens, which decreases cAMP and its activation of the transcription factor, cAMP response element-binding protein (CREB). Many substances of abuse when administered acutely increase DA and consequently inhibit cAMP, providing the pleasurable and rewarding effects of the drug. However, chronic persistent intake eventually increases CREB and dynorphin (DYN), which will dampen reward mechanisms in the nucleus accumbens, providing a possible mechanism for drug dependence and tolerance (see further explanations in the text). GABA, γ-aminobutyric acid; GLU, glutamate.
Occasional and limited administration of most drugs of potential abuse directly or indirectly releases dopamine in the nucleus accumbens (Fig. 54.1). For example, morphine enhances dopaminergic input to the nucleus accumbens by stimulating opioid receptors in the ventral tegmental area. This effect may eventually drive the processes, resulting in drug dependence, because with repeated use, the drug-related dopamine release becomes essential to maintain a ‘normal’ level of pleasure.
Chronic stimulation of the mesolimbic dopamine reward pathways
The complexities of the changes involved are daunting and only a limited description of these events is given here. Chronic exposure of the mesolimbic system to drugs such as opioids, alcohol and cocaine eventually leads to neuro-adaptive changes and sensitisation of the mesolimbic system to further drug administration. Upregulation of neuronal cAMP in the nucleus accumbens results from chronic exposure to many drugs of abuse, with increased intraneuronal cAMP response element-binding protein (CREB). Increased CREB in the nucleus accumbens is probably important for the development of tolerance and dependence. CREB also activates dysphoria-inducing κ-opioid receptors that bind the opioid peptide dynorphin (Ch. 19) on dopamine- and glutamate-releasing neurons in the prefrontal cortex. The long-term actions of dependence-inducing drugs also affect plasticity in the neural circuits of the reward pathway. Upregulation of transcription factors such as CREB and ΔFosB leads to long-term changes in brain-derived neurotrophic factor (BDNF), which regulates the number of dendrites on various neurons in the pathway. Therefore, the changes in the reward and stress systems in the brain that arise with addiction may become ‘imprinted’ even if the causative drug is stopped for long periods. This would explain the vulnerability to relapse after detoxification. There are probably genetic influences on the neurochemical events involved in the reward pathways and stress systems that also increase susceptibility to addiction.
Drug craving is also influenced by neural inputs to the ‘reward’ pathway from the amygdala, which are involved in emotion and conditioned responses. In particular, the amygdala is central to the reinforcing effects of drug binges and also the anxiety and negative effect involved in acute withdrawal. Conditioned responses provide powerful cues to drug-taking in specific social circumstances, and the conditioning is reinforced by aspects of the drug-taking process. Eventually, learning that a drug withdrawal can produce adverse effects that are relieved by the drug may lead to any source of stress or frustration becoming a cue for drug use. Dependence is associated with recruitment of stress systems in the brain on drug withdrawal, probably in an attempt to restore normal neuronal function. There is an elevation of corticotropin-releasing hormone (CRH) and noradrenaline, with suppression of the anti-stress neuropeptide Y.
In contrast, physical dependence on a drug is unrelated to activity in the mesolimbic system and arises from excessive noradrenergic output from the locus ceruleus, a structure in the base of the brain that is involved in arousal and vigilance.
This chapter covers drugs that are encountered in clinical practice primarily because of their abuse, such as ecstasy and cannabis, or because of their potential to cause dependence, such as nicotine and ethanol (Box 54.1).
Box 54.1 Common drugs of abuse
Central nervous system depressants
Opioids (see Ch. 19)
Drugs of abuse
Several drugs that have central stimulant properties are abused and produce dependence. Those more commonly encountered are considered here.
Cocaine
Cocaine is an alkaloid obtained from the leaves of the coca plant. It is usually taken as the hydrochloride salt. ‘Crack’ cocaine is the free-base form, named after the crackling sound produced when it is smoked.
Mechanism of action and effects: The psychomotor effects of cocaine are due to inhibition of reuptake transporters for monamines in presynaptic nerve terminals, particularly inhibiting the dopamine reuptake transporter (DAT), and to a lesser extent those for noradrenaline (NET) and serotonin (SERT). This in turn may activate opioid systems in the brain, with upregulation of µ-receptors (Ch. 19). Increased dopaminergic activity in the reward pathway promotes dependence. Changes in various pituitary neuroendocrine functions occur with more prolonged use; in particular, the release of corticotropin and luteinizing hormone (LH) are enhanced. Tolerance to the psychomotor effects of cocaine is limited. One of the metabolites of cocaine, norcocaine, has direct vasoconstrictor activity.
increased confidence and strength,
indifference to concerns and cares,
anxiety, paranoia, restlessness and tactile hallucinations especially with habitual use,
severe psychological, but not physical, dependence, brought about by the reinforcing effect of the rapid onset and brief duration of action; this develops particularly rapidly with ‘crack’ cocaine,
despondency and despair rapidly follow withdrawal (the ‘crash’ or ‘come down’). After chronic use, withdrawal can produce a dysphoric mood with fatigue, vivid dreams, insomnia, exhaustion, increased appetite and either psychomotor retardation or agitation, irritability and aggressive and stereotyped behaviour,
toxic paranoid psychosis, with delusions of great stamina, occurs with chronic use,
in overdose, excessive catecholamine concentrations produce convulsions, hypertension, cardiac rhythm disturbances and hyperthermia (due to excessive muscle activity and reduced heat loss); if severe, death can occur from respiratory depression and circulatory collapse. The cardiovascular toxicity can be treated with combined α- and β-adrenoceptor blockade, and seizures by intravenous diazepam,
cocaine snuff produces necrosis of the nasal septum through its vasoconstrictor action,
exposure in utero leads to impaired brain development and other teratogenic effects.
Pharmacokinetics: Cocaine, as the hydrochloride salt, is used orally, intranasally (cocaine snuff) or by intravenous injection; the intravenous route gives an intense and rapid onset of effect. ‘Crack’ cocaine is prepared by mixing cocaine hydrochloride with sodium bicarbonate or ammonia and water, then heating to volatilise the free base. The product is smoked and produces intense effects similar to intravenous use. Cocaine is metabolised by plasma and liver esterases and its half-life is very short.
Amfetamine and derivatives
Amfetamine, dexamfetamine, methamfetamine and 3,4-methylenedioxymetamfetamine (MDMA, ‘ecstasy’) have little medical value. Dexamfetamine is sometimes used as a treatment for attention deficit hyperactivity disorder (ADHD) (Ch. 22).
Mechanism of action and effects: Amfetamine and related drugs have indirect sympathomimetic effects, releasing monoamines from central nervous system (CNS) neurons (Ch. 4). They are taken up into presynaptic nerve terminals where they block vesicular uptake of dopamine, serotonin and noradrenaline by the vesicular monamine transporters (VMATs), increasing their concentrations in the cytoplasm. This induces release of the monoamines into the synapse by reversing the respective reuptake transporters for dopamine (DAT), serotonin (SERT) and noradrenaline (NET) in the neuronal membrane. CNS stimulation by amfetamine is most marked in the reticular formation although it also occurs in many other areas of the brain including the reward pathway. The D-isomer (dexamfetamine) is twice as potent as the L-isomer of amfetamine in its central stimulant activity. Effects of amfetamine include:
euphoria, similar to that experienced with cocaine; this is particularly intense after intravenous use,
increased self-confidence, reduced fatigue and increased alertness for repetitive tasks,
psychotic behaviour during repeated use over a few days or with acute intoxication, causing hallucinations, delusions of grandiosity, paranoia and aggressive behaviour and repetitive actions,
acute intoxication can cause hyperthermia, cerebral haemorrhage. Other symptoms include panic, tremor, confusion, hallucinations and aggressiveness,
peripheral sympathomimetic effects can lead to hypertension and cardiac arrhythmias,
tolerance develops rapidly to some of the central effects of amfetamine, such as anorexia, presumably through central monoamine depletion; tolerance to the euphoric effects and motor stimulation is slower,
withdrawal leads to prolonged sleep, followed by fatigue, depression and increased appetite. Other symptoms include anxiety, craving, headaches, restlessness and vivid dreams.
MDMA (ecstasy) is more selective than amfetamine for serotonin release, and produces euphoria similar to that of amfetamine but with less stimulant activity. Direct agonism of serotonin (5-hydroxytryptamine) 5-HT1 or 5-HT2 receptors may contribute to its effects, including release of oxytocin associated with the euphoric action. Disturbance of thermoregulatory homeostasis occurs, leading to a syndrome resembling heat stroke with hyperthermia and dehydration, usually after exertion in hot environments. Stimulation of antidiuretic hormone release can cause thirst and water retention with subsequent water intoxication and hyponatraemia. The toxic effects of MDMA include cardiac arrhythmias, seizures, muscle damage and severe metabolic acidosis, which may be fatal. The long-term toxicity may include memory impairment, anxiety or depression.
Pharmacokinetics: Although amfetamine is sometimes used intravenously or via nasal inhalation, absorption from the gut is rapid and complete. Amfetamine readily crosses the blood–brain barrier. About half is excreted unchanged in the urine and the rest is metabolised in the liver. Amfetamine is a basic drug and its half-life varies according to urine flow and pH; at low urine pH, greater ionisation of amfetamine increases its excretion, producing a shorter half-life, whereas at high urine pH the half-life is longer because of greater reabsorption of the non-ionised drug in the renal tubule. Metabolites of amfetamine are believed to contribute to the psychotic effects seen with long-term use.
Ecstasy is usually taken orally. It undergoes hepatic metabolism via CYP2D6, and polymorphism of this enzyme may explain some of the serious intoxication that occurs with the drug, although the half-life does not differ much between poor and extensive metabolisers (about 5 h in both).
Cathinone stimulants
Mechanism of action: Cathinone and cathine are derived from the khat plant, and there are several similar synthetic compounds such as mephedrone, methylone and flephedrone that are used recreationally. They probably stimulate release of monoamines from neuronal vesicles and inhibit their reuptake. Their psychoactive and physical effects resemble those of amfetamines. They are used intranasally or orally, although they can be injected, smoked or taken rectally.
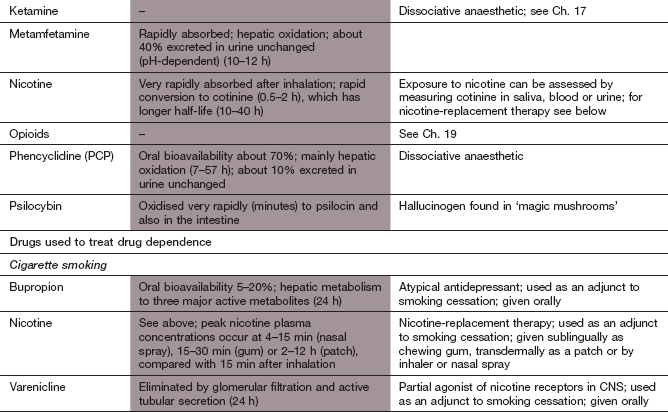
Nicotine and tobacco
Mechanism of action: Over 300 chemical compounds are present in tobacco smoke, but the actions of nicotine are central to the addictive pharmacological effects of smoking. Nicotine has dose-related peripheral actions. At low doses, stimulation of aortic and carotid chemoreceptors enhances sympathetic nervous system activity, and at higher doses there is direct stimulation of the nicotinic N1 receptors in autonomic ganglia (Ch. 4). At even higher doses, nicotine acts as a ganglion-blocking agent. Initial stimulation of autonomic nervous tissue is therefore followed by depression. Effects on the CNS are mediated by presynaptic nicotinic receptors structurally distinct from those in the periphery. Stimulation of CNS nicotinic receptors increases neuronal permeability to Na+ or Ca2+ and enhances the release of glutamate, which promotes dopamine release. With prolonged use nicotine inhibits the release of γ-aminobutyric acid (GABA). Nicotinic receptors are found in the mesocortical and mesolimbic dopaminergic systems, in projections from the ventral forebrain to the cortex that mediate arousal and in hippocampal projections where stimulation enhances learning and short-term memory. Tolerance to the CNS effects of nicotine is rapid due to receptor desensitisation.
Effects of nicotine and tobacco: Tobacco components, including nicotine, have effects on a number of organ systems.
Respiratory effects: The lungs are the first area to be in contact with the chemical components of tobacco smoke and are also exposed to particles and gases. Tars and other irritants, rather than nicotine, are responsible for the chronic damage to the lungs.
An increase in blood carboxyhaemoglobin concentration (from carbon monoxide in tobacco smoke) decreases oxygen-carrying capacity. This may be important in ischaemic heart disease, increasing the chance of provoking angina.
Increased mucus secretion, with reduced activity of bronchial cilia and consequent decreased clearance of lung secretions, leads to chronic bronchitis.
Progressive destruction of the supporting tissue in the bronchioles produces emphysema and chronic obstructive pulmonary disease (COPD). Smoking is now the major cause of this condition.
The risk of lung cancer is increased to about 20 times that of a non-smoker. Inhalation of tobacco smoke is a major contributory factor and explains the greater risk in cigarette smokers. Giving up smoking reduces the risk progressively over about 10 years of abstinence. The constituent of tobacco smoke responsible for altering DNA structure and initiating the cancer process remains controversial, but the relationship between smoking and lung cancer has been confirmed by numerous epidemiological studies. Compared with non-smokers, passive smokers also have a 20–25% increased risk of lung cancer.
Stimulation of the autonomic nervous system and sensory receptors in the heart increases heart rate, blood pressure and cardiac output.
The risk of cardiovascular disease is increased by smoking cigarettes, but not by pipe and cigar smoking, and it occurs at a younger age. The overall risk of death from coronary artery disease is doubled in smokers compared with non-smokers, and the magnitude of the effect is related to the numbers of cigarettes smoked. Peripheral vascular disease and stroke are also increased. Even passive smokers have an excess risk of vascular disease of 25%. The major reason for the excess of events is accelerated formation of atheromatous plaques and enhanced platelet aggregability. The risk of vascular disease falls over the first 3–5 years after stopping smoking to a level close to that of non-smokers.
Psychological effects: The psychological effects of smoking are substantial, as indicated by the difficulties experienced by those attempting to quit.
Decreased appetite, with weight gain on stopping smoking.
Emotional dependence on nicotine and the physical act of smoking is powerful. Physical withdrawal is less marked than psychological withdrawal but includes restlessness, irritability, anxiety, depression, difficulty concentrating, sleep disturbance and increased appetite.
Other effects: Nicotine and smoking have a number of other effects.
Peptic ulceration is twice as common in smokers.
Smoking is a risk factor for osteoporosis.
Smoking in pregnancy, especially during the second half, has several effects. The most important are an increased risk of a low-birth-weight child and increased perinatal mortality. The vasoconstrictor effects of nicotine are responsible. Physical and mental development is slowed in children born to mothers who smoked during pregnancy.
Smoking induces several hepatic cytochrome P450 isoenzymes and increases the clearance of CYP1A2 substrates such as theophylline (Ch. 12) and imipramine (Ch. 22).
Pharmacokinetics of nicotine: Nicotine can be absorbed from the mouth in its non-ionised form, which is found in the less acidic environment of cigar and pipe tobacco smoke. Cigarette smoke, which is acidic, ionises nicotine, which can only be absorbed in significant amounts from the lungs. About 10% of the nicotine from a cigarette is absorbed, but at a faster rate than from cigars or a pipe owing to the larger surface area of the lungs, and results in a higher but less prolonged peak plasma concentration. Nicotine can also be absorbed transdermally. It is metabolised in the liver; the major metabolite, cotinine, has a much longer half-life (about 10–40 h) than nicotine (0.5–2 h) and its concentration in plasma, saliva or urine can be used as a monitor of smoking behaviour.
Dependence on and withdrawal from nicotine: Withdrawal is often difficult to achieve unless motivation is high. Smokers should be supported by counselling about the health benefits of quitting and advice on overcoming problems such as weight gain. Behavioural therapy as an aid to quitting has a success rate of 20% at 1 year. Pharmacotherapy is often used to reduce the intensity of withdrawal symptoms.
Nicotine-replacement therapy: Smokers usually adjust their smoking habit to maintain plasma nicotine concentrations just above a threshold that averts withdrawal symptoms. The plasma concentration falls rapidly within 1–2 h of the last cigarette, and rather more slowly after smoking a cigar or pipe. The resultant craving for nicotine can be reduced by nicotine replacement, delivered via transdermal patches, sublingual tablets, chewing gum, an inhaler (with most absorption occurring in the mouth) or a nasal spray. The delivery method determines the rate at which plasma nicotine concentrations increase, and is most rapid after the nasal spray. The individual can choose the most appropriate vehicle for his or her needs and preferences. Established cardiovascular disease is a caution for, but not a contraindication to, nicotine-replacement therapy. Behavioural therapy enhances the success rate achieved by nicotine-replacement therapy. Use of nicotine-replacement therapy doubles the chance of achieving abstinence.
Bupropion: This is an atypical antidepressant. Most antidepressants are ineffective for smoking cessation, but the use of bupropion gives smoking cessation rates equal to, or slightly greater than, nicotine-replacement therapy. Treatment should be started 1–2 weeks before a ‘quit date’. Used together with nicotine-replacement therapy bupropion produces a modest increase in the chance of stopping. An additional benefit is that smokers who use bupropion as an aid to quitting are less likely to gain weight. Bupropion is a weak inhibitor of neuronal reuptake of noradrenaline and dopamine, and probably works by enhancing mesolimbic dopaminergic activity. It is given as a modified-release formulation and has a long half-life (24 h). Elimination is by hepatic metabolism, which also generates active metabolites. Unwanted effects include anxiety, headache, insomnia and dry mouth. There is an increased risk of epileptic seizures, and bupropion should be avoided if there is a past history of seizures. Nortriptyline is probably as effective as bupropion for smoking cessation.
Varenicline: This is a partial agonist at nicotine receptors, with high selectivity for the CNS receptor subtype involved in addiction. It produces about 30–45% of the response expected from nicotine, and blocks the effect of added nicotine. The modest release of dopamine reduces craving and nicotine withdrawal symptoms. Treatment should be started 1–2 weeks before a ‘quit date’, and combined with behavioural support. The oral bioavailability of varenicline has not been defined; it is excreted unchanged by the kidney and has a half-life of 24 h. Unwanted effects include gastrointestinal disturbances, dry mouth, headache, dizziness, drowsiness and sleep disturbance. Depression with suicidal thoughts has also been reported.
Psychotomimetic agents
Lysergic acid diethylamide (LSD), psilocybin (‘magic mushrooms’), mescaline (from peyote cactus) and the synthetic drug dimethyltryptamine (DMT) are adrenergic hallucinogens that have structural similarities to monoamine neurotransmitters. LSD is the most potent hallucinogen.
Mechanism of action and effects: The actions of hallucinogens on the brain are probably related to postsynaptic 5-HT2 receptor stimulation in the cerebral cortex and locus coeruleus, a region of the midbrain that receives sensory signals. LSD also produces presynaptic 5-HT1A receptor blockade in the dorsal raphe neurons, inhibiting firing of neuronal projections to the forebrain. Tolerance to LSD occurs rapidly, and appears to be related to downregulation of these receptors. The actions of LSD, psilocybin and mescaline are similar, and they share several properties, including cross-tolerance.
Visual hallucinations are frequent, especially with high doses, and auditory acuity is accentuated. There may be an overlap of sensory impressions such that music is ‘seen’ or colours ‘heard’, which can produce severe anxiety. Time appears to pass slowly. Emotions are altered, with either elation or depression, and rapid mood swings can occur. The overall experience can produce a good or a bad ‘trip’, and can vary in the same individual on different occasions.
Serious psychotic reactions can occasionally occur, and long-term psychotic disorders can be precipitated. The other unpleasant persistent effect in some individuals is ‘flashback’, seeing bright flashes, or halos or trails attached to moving objects.
Physical consequences of CNS stimulation include dizziness, weakness, drowsiness and paraesthesiae.
Excessive sympathetic nervous system stimulation with large doses produces nausea, salivation, lacrimation, dizziness, mydriasis, tremor, hyperthermia, tachycardia and hypertension.
Tolerance can occur within 5 days.
Emotional dependence is frequent, but physical dependence is not seen.
Pharmacokinetics: Oral absorption of these drugs is good. Physical effects begin after about 20 min, but psychoactive effects are delayed for 2–4 h and then last up to 12 h. DMT has a rapid onset of hallucinogenic action, within 15–30 min, but the duration is only 1–2 h. Elimination is by hepatic metabolism and the half-lives are short.
Cannabis
Cannabis can be smoked as marijuana, which consists of dried leaves or flowers of the Cannabis sativa (hemp) plant, or as a resin extracted from the leaves of the plant and then dried, known as hashish. Solvent extraction of the resin produces cannabis oil, which can be added to tobacco. The hallucinogenic effects of cannabis are much less marked than those of the aminergic hallucinogens such as LSD.
Mechanism of action and effects: The constituent compounds (cannabinoids) interact with specific CB1 receptors in the brain. These receptors are coupled to Gi proteins that reduce intracellular cAMP production, and inhibit cell membrane Ca2+ and K+ channels. The natural ligands are the arachidonic acid derivatives anandamide, 2-arachidonylglycerol and noladin ether. CB1 receptors are found in greatest density in areas of the brain involved in cognition and pain recognition (cerebral cortex), memory (hippocampus), reward (mesolimbic system) and motor coordination (substantia nigra and cerebellum).
The psychomotor effects result largely from tetrahydrocannabinol (THC) and one of its metabolites, 11-hydroxy-THC, which produce euphoria, heightened intensity of sensations, and relaxation. Occasionally panic reactions, hallucinations and depersonalisation can occur. Psychotic reactions are rare except in predisposed individuals, but the use of cannabis increases the risk of developing schizophrenia. Recent memory is markedly impaired and complex mental tests are executed less well, although the user may perceive that their performance is enhanced. Motor incoordination may affect driving ability.
Effects on the cardiovascular system include tachycardia and increased systolic blood pressure with a postural fall.
The tars inhaled during chronic use of cannabis predispose to heart disease, chronic bronchitis and lung cancer.
THC has an anti-emetic action (Ch. 32), which may be useful during cancer chemotherapy (Ch. 52).
Cannabinoids have analgesic effects that are used by some people with multiple sclerosis to control neuropathic pain.
Tolerance to the psychomotor effects of cannabis occurs with regular use, and there is evidence of dependence.
Pharmacokinetics: Metabolism of THC is extensive, with some active metabolites being produced. The high lipid solubility of THC means that absorption from the lung or gut is high, and it has a large apparent volume of distribution; however, because of its very rapid metabolism its half-life is only 1.5 h. The psychomotor effects last for 2–3 h after inhalation.
Dissociative anaesthetics
Phencyclidine (PCP) and ketamine differ from adrenergic hallucinogens in their mode of action. Both drugs were developed as anaesthetics, but PCP was withdrawn because of severe adverse effects (hallucinations, mania, delirium and disorientation).
Mechanism of action and effects: Both drugs block the excitatory effects of glutamate at N-methyl-D-aspartate (NMDA) receptors. These receptors are abundant in the cortex, basal ganglia and sensory pathways of the CNS. PCP also releases dopamine from nerve terminals in a manner similar to amfetamine. The term dissociative anaesthetic refers to the feelings of detachment (dissociation) from the environment and self that are produced by the drugs. These are not true hallucinations.
Acute effects include euphoria, decreased inhibition, a feeling of immense power, analgesia, altered perception of time and space, and depersonalisation. Ketamine creates a ‘mellow, colourful wonderworld’.
Catatonic rigidity can occur, followed by ataxia and slurring of speech.
Adverse experiences include confusion, restlessness, disorientation and impaired judgement. Irritability, paranoia, depression and anxiety are also common. Psychotic reactions are precipitated in susceptible people.
Ketamine can produce near-death experiences.
Persistent abuse of PCP leads to memory loss, speech and thought difficulties, and depression that persist for months after the last use.
Pharmacokinetics: PCP is rapidly absorbed from the gut, nose or lungs after smoking. Effects are seen within minutes of ingestion and usually last 4–6 h. It is a weak base that is excreted in the urine. It is also excreted into the stomach, and reabsorbed by the small intestine. The half-life is variable and can be up to 2 days. Ketamine is abused intravenously. It is metabolised in the liver and has a short half-life (see Ch. 17).
CNS depressants
Alcohol (ethyl alcohol, ethanol)
Mechanism of action and effects: Alcohol has multiple actions on the CNS. Non-specific actions such as increased fluidity of neuronal cell membranes (cf. general anaesthetics) may be important by reducing Ca2+ flux across the cell membrane, but several other actions have been described (Box 54.2). Overall, alcohol facilitates central inhibitory neurotransmission, particularly enhancing the effects of GABA, and therefore it is a general CNS depressant. With acute alcohol intake there is an initial depression of inhibitory neurons, particularly in the mesolimbic system, which produces a sense of relaxation, but this is followed by progressive depression of all CNS functions. Mental processes that are modified by education, training and previous experience are affected first, while relatively ‘mechanical’ tasks are less impaired. Despite subjective impressions, there is no increase in mental or physical capabilities, unless anxiety has previously reduced performance. All effects are closely related to blood alcohol concentration (Table 54.1).
Table 54.1
The effects of alcohol at various plasma concentrations
| Plasma concentration (mg⋅100 mL−1) | Effects |
| 30 | Mild euphoria owing to suppression of social inhibitory pathways in the cortex; the individual is more talkative and emotionally labile with loss of self-control; the risk of accidental injury is increased |
| 80 | Delayed reactions and reduced comprehension; memory impairment; the risk of serious injury in a road accident is more than doubled (80 mg⋅100 mL−1 is the legal limit for driving in the UK) |
| 100–200 | Speech becomes slurred and motor coordination is impaired |
| >300 | Often produces loss of consciousness |
| >400 | Frequently fatal as a result of respiratory and vasomotor centre depression |
In people who regularly use large amounts of alcohol, tolerance is seen to many of its psychological effects. Alcohol increases dopamine release in the nucleus accumbens indirectly by activating presynaptic GABA receptors that actually inhibit GABA release. This is explained by the different alpha subunits on the presynaptic receptors that are sensitive to alcohol, whereas the postsynaptic receptors are not. Long-term use of alcohol produces long-lasting adaptive changes in the NMDA receptor that enhance their function. Opioid and serotonin receptor stimulation is also involved in the reinforcing effects of alcohol on the brain.
Alcohol intake is usually measured in units (Box 54.3).
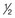 pint of normal-strength beer, lager, cider
pint of normal-strength beer, lager, cider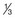 pint of strong beer, lager, cider
pint of strong beer, lager, cider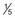 pint of extra-strong beer, lager, cider
pint of extra-strong beer, lager, cider bottle of ‘alcopop’
bottle of ‘alcopop’Other effects of alcohol: Alcohol has a range of effects.
A modest alcohol intake may have protective effects on the circulation by inhibiting platelet aggregation and increasing high-density lipoprotein cholesterol. The form in which the alcohol is taken is probably not important. The extent of this beneficial effect is probably greatest at 1 unit per day and is lost when intake exceeds 3–4 units per day.
Higher intake of alcohol has pressor effects that raise blood pressure, possibly through increased vascular sensitivity to catecholamines. This increases the risk of coronary artery disease and stroke.
Cardiac arrhythmias can be provoked by high alcohol intake, particularly atrial fibrillation. This can occur after an alcoholic binge (‘holiday heart’ syndrome) or following more chronic abuse (Ch. 8).
Alcoholic cardiomyopathy is a dilated cardiomyopathy that is only partially reversible with abstinence, and can lead to heart failure. An average intake of 10 units of alcohol daily for 8–10 years can produce this condition.
Hypoglycaemia occurs as a consequence of the metabolism of alcohol in the liver. The metabolic process generates excess protons, which enhance the conversion of glucose, via pyruvate, to lactate and predisposes to lactic acidosis. Alcoholics often have a low-carbohydrate diet, which compounds the hypoglycaemia. Hypoglycaemia tends to occur several hours after heavy alcohol intake and can contribute to seizures on alcohol withdrawal.
The lactic acidosis created by alcohol metabolism in the liver impairs the renal excretion of uric acid, which predisposes to gout.
Lactic acidosis also facilitates the synthesis of saturated fatty acids, which accumulate in the liver, leading to a fatty liver, possibly with altered liver function. Plasma triglycerides are also increased.
Alcoholic hepatitis is usually a consequence of short-term heavy alcohol abuse. It can be fatal.
Cirrhosis occurs with prolonged alcohol abuse, but individual susceptibility varies widely. On average, consumption of more than 8 units per day for at least 10 years is required for cirrhosis to occur in men. About two-thirds of this amount creates the same risk for women. Established cirrhosis reduces the first-pass metabolism and clearance of drugs eliminated by the liver (Ch. 56).
Chronic intake of alcohol induces hepatic drug-metabolising enzymes, especially CYP2E1, which decreases the effectiveness of some therapeutic drugs, for example warfarin, phenytoin and carbamazepine.
Sexual desire is often increased by alcohol, but the ability to sustain penile erection is reduced, possibly because of the vasodilator actions of alcohol.
Direct damage to the Leydig cells of the testis reduces the circulating testosterone, leading to reduced libido, infertility and a loss of the male distribution of body hair. Altered steroid metabolism in the liver leads to an increase in circulating oestrone in males, which causes gynaecomastia.
A combination of alcohol toxicity with deficiencies of vitamin B6 and thiamine in the diet of alcoholics predisposes to peripheral neuropathy and dementia. Specific mid-brain damage can result and produces the syndromes of Wernicke's encephalopathy and Korsakoff's psychosis.
Alcohol has anticonvulsant properties and withdrawal predisposes to seizures, even in individuals without a history of epilepsy.
Alcohol can disturb sleep patterns, with decreased rapid eye movement (REM) sleep and increased stage 4 sleep during intoxication. Withdrawal increases REM sleep, with associated nightmares (Ch. 20).
Dose-related memory impairment can be caused by suppressed hippocampal function.
Subdural haematoma is more common after head injury in heavy drinkers, perhaps as a consequence of cerebral atrophy.
Depression or anxiety states are more common in heavy drinkers.
Carcinogenesis and teratogenesis:
Cancer of the mouth, oesophagus and liver are more common with heavy alcohol use. Colon and breast cancer may also be increased.
The fetal alcohol syndrome is believed to be caused by the effects of alcohol on neuronal adhesion molecules that regulate neuronal migration. Heavy maternal drinking during pregnancy leads to impaired learning and memory in the child. Genetic factors may be involved in the susceptibility of the fetus to these problems.
Pharmacokinetics: Although ethanol is absorbed from the stomach, the majority is absorbed from the small intestine, due to its larger surface area. High concentrations of alcohol (above 20%) and large volumes inhibit gastric emptying and delay absorption, as do foods high in fat or carbohydrate. Therefore, peak blood alcohol concentrations depend on the dose and strength of the alcohol and on whether or not it was taken with food. Following absorption, alcohol undergoes substantial first-pass metabolism in the liver. The extent of first-pass metabolism of alcohol is related to the speed of absorption; thus, with slower absorption, such as when alcohol is taken with food, less alcohol will reach the systemic circulation. Distribution of alcohol is fairly uniform and the ready passage across the blood–brain barrier and high cerebral blood flow ensure rapid access to the CNS. The effects on the brain are more marked when the concentration is rising, indicating a degree of acute tolerance. Metabolism occurs mainly in the liver (Fig. 54.2), more than 90% being oxidised, mainly by alcohol dehydrogenase, while the rest is removed unchanged in expired air (in direct proportion to the blood concentration, which is the basis of the alcohol breath test) or in the urine. Alcohol metabolism shows saturation kinetics due to the limited supply of nicotine adenine nucleotide (NAD+), which is the cofactor for the oxidative process. The maximum rate of alcohol metabolism averages 8 g·h−1. The initial metabolic reaction mediated by alcohol dehydrogenase produces acetaldehyde, which is subsequently metabolised by aldehyde dehydrogenase to acetic acid (Fig. 54.2). Genetic variability in alcohol and aldehyde dehydrogenases occurs among ethnic groups, leading to different capacities for alcohol or aldehyde metabolism. Accumulation of acetaldehyde in the circulation is responsible for many of the unpleasant effects of a hangover. Small amounts of alcohol are metabolised via the microsomal ethanol oxidising system (CYP2E1), the activity of which is increased by enzyme inducers such as alcohol itself (which does not affect the activity of alcohol dehydrogenase) (Ch. 36).
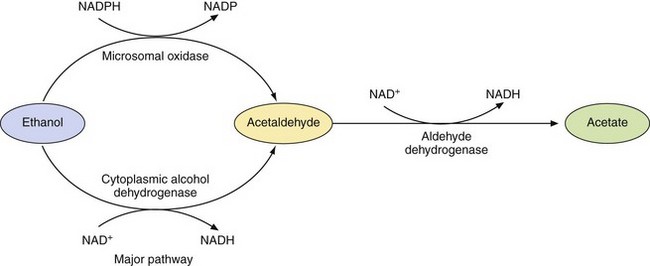
Fig. 54.2 The metabolism of alcohol.
Alcohol dehydrogenase is responsible for 80–90% of the metabolism of ethanol. The microsomal oxidase is a minor pathway dependent on CYP2E1, the activity of which is increased by enzyme inducers such as alcohol itself.
Some drugs, such as metronidazole (Ch. 51), inhibit aldehyde dehydrogenase, leading to acetaldehyde accumulation if alcohol is taken with them. Typical ‘hangover’ effects of flushing, sweating, headache and nausea then occur after even small amounts of alcohol.
Alcohol abuse and dependence: There are no reliable estimates of the number of people in the UK with alcohol-related problems, although it has been suggested that 1–2% of the population are affected. The distribution curve for alcohol consumption is continuous but skewed at higher alcohol intakes. The risk of alcohol-related problems rises with the average alcohol intake. Hazardous drinking is defined as a level or pattern of alcohol intake that will probably eventually cause harm. It applies to anyone drinking more than the recommended limits (21 units per week for men; 14 units per week for women). Harmful drinking is at a level that is already causing damage to physical or mental health. Dependent drinking is identified by features common to all drug dependence.
Up to 30% of hospital admissions are for alcohol-related problems, although the contribution of heavy drinking is often unrecognised. Screening for alcohol abuse can be carried out by obtaining a complete history of alcohol intake and using either the Alcohol Use Disorders Identification Test (AUDIT) or the Fast Alcohol Screening Test (FAST) (Box 54.4). Abnormal measurements of both the mean corpuscular volume (MCV) of red cells (which is raised with increasing alcohol intake because of an effect of alcohol on the cell membrane) and the liver enzyme γ-glutamyl transpeptidase (γGT) will identify about 75% of people with an alcohol problem.
Box 54.4 The Fast Alcohol Screening Test (FAST) for alcohol problemsa

aOne drink =  pint of beer or 1 glass of wine or 1 single spirits. On question 1, if the answer is Never (score=0), then the patient is not misusing alcohol. If the answer is Weekly (3) or Daily/almost daily (4) then the patient is a hazardous, harmful or dependent drinker. If the answer is Less than monthly (1) or Monthly (2), then questions 2, 3 and 4 should be considered. If the aggregate score is 3 or more for questions 1–4, out of the possible total of 16, the patient is misusing alcohol. Based on Hodgson et al. (2002) The FAST Alcohol Screening Test. Alcohol and Alcoholism 37, 61–66.
pint of beer or 1 glass of wine or 1 single spirits. On question 1, if the answer is Never (score=0), then the patient is not misusing alcohol. If the answer is Weekly (3) or Daily/almost daily (4) then the patient is a hazardous, harmful or dependent drinker. If the answer is Less than monthly (1) or Monthly (2), then questions 2, 3 and 4 should be considered. If the aggregate score is 3 or more for questions 1–4, out of the possible total of 16, the patient is misusing alcohol. Based on Hodgson et al. (2002) The FAST Alcohol Screening Test. Alcohol and Alcoholism 37, 61–66.
Psychological dependence on alcohol is common, but physical dependence also occurs. Withdrawal symptoms occur 6–24 h after the last drink in dependent persons. If mild, these are related to autonomic hyperactivity and include anxiety, agitation, tremor, sweating, anorexia, nausea and retching. Convulsions can occur through neuronal excitation. Insomnia, tachycardia and hypertension are common with more severe withdrawal reactions. The most severe form of withdrawal is delirium tremens, with confusion, paranoia and visual and tactile hallucinations. Delirium tremens can cause death from respiratory and cardiovascular collapse.
If an individual is drinking excessively, controlled drinking may be an option. However, if there is alcohol dependence or alcohol-related problems, then abstinence is usually preferable.
Controlled detoxification is usually undertaken with a sedative agent, such as a benzodiazepine (Ch. 20), to attenuate withdrawal symptoms. Chlordiazepoxide or diazepam is usually used, decreasing the dose over 7–10 days. Clonidine (a presynaptic α2-adrenoceptor agonist at the vasomotor centre in the brain; Ch. 6) can be useful, by reducing the excessive sympathetic stimulation that accompanies withdrawal. Beta-adrenoceptor antagonists (Ch. 5) may be helpful for the same reason. Multivitamin preparations containing an adequate amount of thiamine should be given for 1 month to prevent Wernicke's encephalopathy. Relapse is common after withdrawal from alcohol.
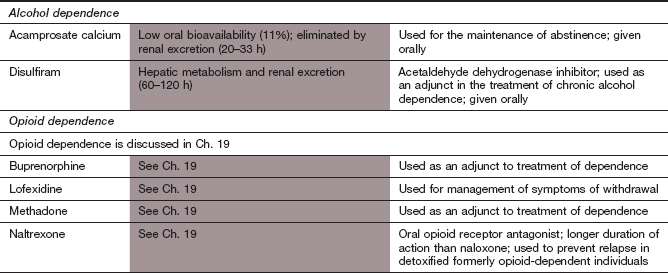
Two drugs are licensed in the UK to assist in the management of chronic alcoholism. Disulfiram, an inhibitor of acetaldehyde dehydrogenase, causes unpleasant hangover symptoms after small amounts of alcohol. Given alone, or with psychosocial rehabilitation, it can help to maintain abstinence. Acamprosate may activate GABAA receptors and block glutamate NMDA receptors, although several other contributory effects have been suggested. It has few unwanted effects, is non-addictive and can be used to reduce the craving for alcohol. Naltrexone, a long-acting opioid receptor antagonist, can reduce the craving associated with alcohol withdrawal. It is not licensed for this indication in the UK. Other potential agents to reduce the urge to drink include ondansetron (Ch. 32), topiramate and gabapentin (Ch. 23).
Gamma-hydroxybutyric acid
Mechanism of action and effects: Gamma-hydroxybutyric acid (GHB) was originally introduced as a general anaesthetic, but is now used illegally as an intoxicant, a ‘date rape’ drug or by athletes to improve performance. Its precursors γ-butyl-lactone (GBL) and 1,4-butanediol are also abused. GHB acts as an agonist at a specific inhibitory GHB receptor in the cortex and hippocampus of the brain, and also as an agonist at GABAB receptors, which mediate its sedative effects. GHB receptor activation stimulates dopamine release. GHB receptor stimulation also increases growth hormone release, which is the basis of its abuse by athletes and bodybuilders. In a similar manner to alcohol, GHB produces euphoria, increased libido and increased sociability. At high doses it produces nausea, dizziness, drowsiness, agitation, visual disturbances, amnesia and coma. Both psychological and physical dependence occur. Withdrawal can be treated with baclofen (Ch. 24).
Inhaled solvents
Various organic solvents are abused as recreational drugs. Examples include butane, toluene and diethyl ether. Inhalation via a plastic bag held over the mouth or from an open container produces rapid intoxication resembling that produced by alcohol. These compounds probably act in a similar way to volatile general anaesthetics (Ch. 17). Death can occur from asphyxiation during inhalation, while long-term use produces brain damage by increasing neuronal apoptosis.
True/false questions
1. Cocaine causes mydriasis by inhibiting the reuptake of noradrenaline into nerve terminals.
2. ‘Crack’ cocaine is the free-base form of cocaine.
3. Cocaine use has little damaging effect on the cardiovascular system.
4. Tolerance to the euphoric and anorexic effects of cocaine develops rapidly.
5. MDMA (ecstasy) blocks the release of serotonin from nerve endings.
6. In some individuals MDMA causes hyperthermia and dehydration.
8. Amfetamines are used to treat attention deficit hyperactivity disorder (ADHD).
9. Cannabis impairs driving ability.
10. The euphoria caused by cannabis lasts for 24 h.
11. Tetrahydrocannabinol (THC), the main active ingredient of cannabis, causes nausea and vomiting.
12. Cannabis acts on specific receptors in the brain.
13. Nicotine causes tachycardia and reduced gut motility.
14. Tolerance to the effects of nicotine develops slowly.
15. Varenicline is a partial agonist of nicotine receptors in the central nervous system (CNS).
16. Cotinine has a long half-life and can be measured in serum to determine smoking habits.
17. Nicotine patches given alone are the optimum method for someone giving up smoking.
18. A physical withdrawal symptom does not occur when giving up smoking.
19. Alcohol is initially metabolised in the liver to acetaldehyde.
20. Chronic intake of alcohol induces hepatic drug-metabolising enzymes.
21. Even moderate alcohol intake increases the incidence of cardiovascular disease.
22. Some individuals have a genetically determined low ability to metabolise alcohol.
23. Disulfiram produces acute sensitivity to alcohol by blocking its conversion to acetaldehyde.
24. Acamprosate, which is used to encourage abstinence, acts on alcohol metabolism in a similar way to disulfiram.
25. The symptoms of alcohol withdrawal (detoxification) cannot be controlled by pharmacological means.
26. Alcohol can cause a macrocytosis.
27. Alcohol enhances antidiuretic hormone secretion.
28. Plasma levels of γ-glutamyl transpeptidase (γGT) are depressed with heavy alcohol intake.
29. Cathinone derivatives in the khat plant produce sedation.
30. Gamma-hydroxybutyric acid (GHB) may be abused by athletes.
1. True. Cocaine blocks monoamine reuptake; in the eye, the resulting increase in noradrenaline causes mydriasis by contraction of radial pupillary muscles.
2. True. Unlike the salt form of cocaine, the free base can be illicitly smoked.
3. False. Acute effects include cardiac arrhythmias and chronic use can lead to heart failure.
4. True. Tolerance develops to euphoria and appetite suppression in only a few days.
5. False. MDMA (ecstasy) increases release of serotonin and other monamines by preventing their uptake into synaptic vesicles and promoting their release from the cytoplasm into the synapse. It may also be an agonist at serotonin receptors.
6. True. Malignant hyperthermia resembling heat stroke and dehydration is observed in some individuals after ingesting MDMA.
7. True. Like other amfetamines, MDMA has a short-term effect to suppress appetite.
8. True. Amfetamines can be used to treat ADHD (but not an approved use in UK).
9. True. Cannabis impairs driving ability and the performance of complex mental tasks, and may give rise to psychotic reactions in predisposed individuals.
10. False. The euphoric effects last only 2–3 h.
11. False. The related cannabinoid, nabilone, is used to inhibit nausea and vomiting in patients taking cytotoxic drugs.
12. True. Cannabis acts on cannabinoid receptors CB1 and CB2 in the brain and periphery. The natural ligands for these receptors include anandamide.
13. True. These effects are caused by stimulation of nicotinic N1 receptors in autonomic ganglia.
14. False. Tolerance to nicotine develops rapidly.
15. True. Varenicline reduces tobacco cravings by partial agonism at CNS nicotinic receptors, and it partially blocks the additional effect of nicotine if tobacco is smoked.
16. True. Cotinine is a stable and inactive nicotine metabolite; it can be measured in saliva, serum or urine.
17. False. Nicotine-replacement therapy should be supplemented with counselling.
18. False. Irritability, sleep disturbances and reduced psychomotor test performance occur on giving up smoking.
19. True. Alcohol is mainly metabolised to acetaldehyde (by alcohol dehydrogenase) and then to acetic acid (by aldehyde dehydrogenase).
20. True. The induction of CYP2E1 by alcohol can decrease the effectiveness of some drugs such as warfarin and phenytoin.
21. False. Moderate alcohol intake (below 3–4 units per day for men) has cardiovascular protective effects.
22. True. Some individuals have a genetically determined variant of alcohol dehydrogenase that has a reduced capacity to metabolise alcohol. Its incidence is low in white people but higher in people from some Far Eastern countries.
23. False. Disulfiram blocks conversion of acetaldehyde to acetic acid by aldehyde dehydrogenase; the accumulation of acetaldehyde causes sickness, headache and hangover symptoms following even a small amount of alcohol intake.
24. False. Acamprosate acts to reduce the craving for alcohol and not by affecting its metabolism.
25. False. Benzodiazepines can attenuate withdrawal symptoms but there is a risk of dependence to these agents.
26. True. Alcohol intake is a common cause of macrocytosis (increased red cell volume) in the absence of anaemia.
27. False. The diuresis resulting from alcohol intake is partly caused by inhibition of release of antidiuretic hormone.
28. False. Plasma γ-glutamyl transpeptidase (γGT) is elevated in heavy alcohol intake.
29. False. Cathinone and its derivatives have amfetamine-like properties and cause wakefulness and insomnia.
30. True. GHB has sedative activity but also increases growth hormone release, which is the basis of its abuse by athletes and bodybuilders.
Anton, RF. Naltrexone for the management of alcohol dependence. N Engl J Med. 2008;359:715–721.
Aveyard, P, West, R. Managing smoking cessation. BMJ. 2007;335:37–41.
Barlecchi, CE, MacKenzie, TD, Schrier, RW. The human cost of tobacco. N Engl J Med. 1994;330:907–912.
Benowitz, NL. Nicotine addiction. N Engl J Med. 2010;362:2295–2303.
Berke, JD, Hyman, SE. Addiction, dopamine, and the molecular mechanisms of memory. Neuron. 2000;25:515–532.
Cami, J, Farre, M. Drug addiction. N Engl J Med. 2003;349:975–986.
Farrell, M, Wodak, A, Gowing, L. Maintenance drugs to treat opioid dependence. BMJ. 2012;344:e2823.
Gerdeman, GL, Partridge, JG, Lupica, CR, et al. It could be habit forming: drugs of abuse and striatal synaptic plasticity. Trends Neurosci. 2003;26:184–192.
Hall, W, Solowij, N. Adverse effects of cannabis. Lancet. 1998;352:1611–1616.
Hatsukami, DK, Stead, LF, Gupta, PC. Tobacco addiction. Lancet. 2008;371:2027–2038.
Hays, JT, Ebbert, JO. Varenicline for tobacco dependence. N Engl J Med. 2008;359:2018–2024.
Hodgson, RJ, Alwyn, T, John, B, Thom, B, Smith, A. The FAST Alcohol Screening Test. Alcohol and Alcoholism. 2002;37:61–66.
Johnson, BA. Medication treatment of different types of alcoholism. Am J Psychiatry. 2010;167:630–639.
Koob, GF. The neurobiology of addiction: a neuroadaptational view relevant for diagnosis. Addiction. 2006;101(suppl 1):23–30.
Lancaster, T, Hajek, P, Stead, LF, et al. Prevention of relapse after quitting smoking. Arch Intern Med. 2006;166:828–835.
Mendelson, JH, Mello, NK. Management of cocaine abuse and dependence. N Engl J Med. 1996;334:965–972.
Moore, THM, Zammit, S, Lingford-Hughes, A, et al. Cannabis use and risk of psychotic or affective mental health outcomes: a systematic review. Lancet. 2007;370:319–328.
Nides, M. Update on pharmacologic options for smoking cessation treatment. Am J Med. 2008;121(suppl 4A):S20–S31.
Parker, AJ, Marshall, EJ, Ball, DM. Diagnosis and management of alcohol use disorder. BMJ. 2008;336:496–501.
Rigotti, A. Treatment of tobacco use and dependence. N Engl J Med. 2002;346:506–512.
Saitz, R. Unhealthy alcohol use. N Engl J Med. 2005;352:596–607.
Schippenberg, TS, Zapata, A, Chefer, VI. Dynorphin and the pathophysiology of drug addiction. Pharmacol Ther. 2007;116:306–321.
Schuckit, MA. Alcohol-use disorders. Lancet. 2009;373:492–501.
Snead, OC, Gibson, KM. γ-Hydroxybutyric acid. N Engl J Med. 2005;352:2721–2732.
Sullivan, LE, Fiellin, DA. Narrative review: Buprenorphine for opioid-dependent patients in office practice. Ann Intern Med. 2008;148:662–670.
Winstock, AR, Mitcheson, L. New recreational drugs and the primary care approach to patients who use them. BMJ. 2012;344:e288.