Pituitary and hypothalamic hormones
Anterior pituitary and hypothalamic hormones
Thyrotropin and thyrotropin-releasing hormone are considered in Chapter 41.
Growth hormone
Growth hormone (GH), or somatotropin, is a 191-amino acid peptide that is synthesised in specific cells in the anterior pituitary. Its secretion is controlled by the hypothalamus by a balance between growth hormone-releasing hormone (GHRH) via specific receptors and somatostatin (SST; also called growth hormone release-inhibiting hormone, GHRIH) via specific somatostatin receptors (or SSTRs) (Fig. 43.1). Many other factors modulate this balance such as the stimulant effects of ghrelin, sex hormones, sleep and exercise or the inhibitory effects of plasma GH, glucose and glucocorticoids.
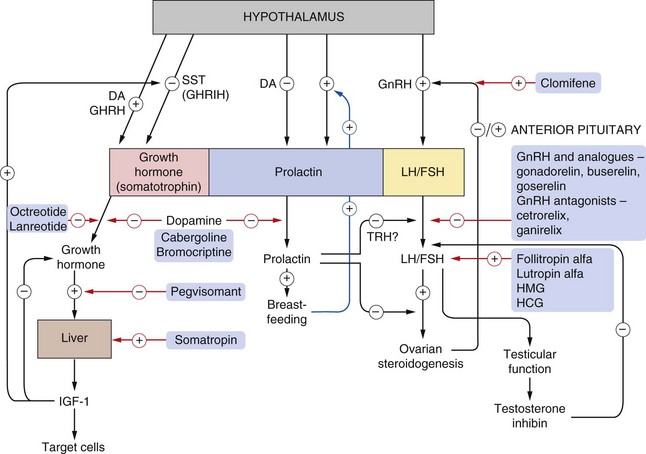
Fig. 43.1 Control mechanisms for the release of growth hormone, gonadotropins and prolactin from the anterior pituitary.
For control of other hormones, see Ch. 41 (thyroid) and Ch. 44 (adrenocorticotropic hormones). Oestrogen effects on gonadotropin-releasing hormone (GnRH) are shown as both positive and negative because oestrogen suppresses luteinizing hormone (LH) secretion in the early follicular phase but enhances secretion around ovulation (Ch. 45). The actions of drugs are shown by red arrows. Gonadorelin is a synthetic GnRH and buserelin and goserelin are GnRH analogues. Pulsatile administration of GnRH or its analogues enhances LH/follicle-stimulating hormone (FSH). On continuous administration, they downregulate the GnRH receptors, inhibiting LH/FSH release, an effect produced more rapidly by GnRH antagonists (cetrorelix, ganirelix). Somatropin is synthetic growth hormone, and octreotide and lanreotide are somatostatin analogues that suppress growth hormone release. For details of dopamine (DA) agonists, see Ch. 24. GHRH, growth hormone-releasing hormone; HMG, human menopausal gonadotropins (menotrophin); HCG, human chorionic gonadotropin; IGF-1 insulin-like growth factor 1; SST, somatostatin (also called GHRIH, growth hormone release-inhibiting hormone); ? = regulating hormone not yet established. Note: dopamine stimulates growth hormone release in healthy individuals, but paradoxically in acromegaly it inhibits release.
GH is released in pulses repeatedly throughout the day and night. Like other peptide hormones, GH binds to cell surface receptors and activates adenylyl cyclase, and has direct metabolic effects on several tissues. In addition it produces other effects via the production of insulin-like growth factor 1 (IGF-1, a somatomedin). IGF-1 is synthesised by the liver in response to GH stimulation, and is highly protein bound in plasma.
The effects of GH are anabolic in relation to protein metabolism, especially in skeletal muscle leading to increased muscle mass, and in epiphyseal cartilage, where the proliferating effects stimulate bone growth. These actions are mediated by IGF-1. IGF-1 also has effects on the liver via the insulin receptor, and promotes hepatic gluconeogenesis. However, GH has an opposing direct effect on carbohydrate metabolism, reducing glucose uptake by skeletal muscle and adipose tissue, creating an insulin-resistant state (Box 43.1).
The effect of GH on fat is catabolic, with a direct action on adipocytes that promotes lipolysis and reduces lipogenesis.
Growth hormone for therapeutic use
Somatropin has several therapeutic uses in children, which include:
 to improve linear growth in children with proven GH deficiency (who usually lack GHRH; pituitary dwarfism),
to improve linear growth in children with proven GH deficiency (who usually lack GHRH; pituitary dwarfism),
To be effective, the hormone must be given before the closure of the epiphyses in long bones. Treatment should be stopped if growth velocity does not increase by at least 50% from baseline.
Adult GH deficiency may warrant treatment with somatropin if all the following criteria are fulfilled:
Pharmacokinetics: Somatropin is usually given by subcutaneous injection, although the intramuscular route can be used. Plasma concentrations fluctuate widely following both routes, although the latter gives more stable levels because of slower uptake into the circulation. Somatropin has a very short half-life (0.5 h), but the plasma concentration of IGF-1 is much more constant due to its high protein binding. As a consequence, three doses of somatropin per week give good clinical results, although daily dosing is often used.
Headache, occasionally associated with visual problems, nausea and vomiting, and papilloedema from benign intracranial hypertension.
Fluid retention with peripheral oedema.
Arthralgia, myalgia, carpal tunnel syndrome.
A transient insulin-like action occasionally produces hypoglycaemia.
If excessive doses are used (as may happen during illicit use by athletes to build muscle mass) there is a risk of diabetes mellitus in predisposed individuals.
Acromegaly
Acromegaly results from excessive production of GH, almost always by an adenoma in the anterior pituitary which also secretes prolactin in one-third of cases (Fig. 43.1). The most common clinical features arise from excessive growth of bone and soft tissue. Complex metabolic consequences include insulin resistance with diabetes mellitus and hypertension.
The morbidity and mortality of acromegaly vary according to its severity. Untreated acromegalic individuals have a life expectancy approximately half that of people without acromegaly, due to an excess incidence of cardiovascular and respiratory disease and of carcinoma of the colon. Acromegaly is therefore usually treated actively.
Drugs for acromegaly
Mechanisms of action and uses: The synthetic derivatives of SST are both more potent and longer-acting inhibitors of GH secretion than the native compound. They are selective for the SST receptor subtypes that are highly expressed on GH-secreting adenomas. Like SST, they also inhibit the release of gastro-entero-pancreatic peptide hormones, such as insulin, glucagon, cholecystokinin, gastrin and vasoactive intestinal peptide (VIP), via intestinal SST receptors, which generate intracellular cAMP and modulate Ca2+ influx into the cell. These actions make SST analogues useful also for the treatment of a variety of conditions associated with excess secretion of gut hormones.
Uses of SST analogues include:
management of other endocrine tumours, for example carcinoid tumours (to reduce flushing and diarrhoea), VIPoma (to reduce diarrhoea) and glucagonoma (to improve the characteristic necrolytic rash),
management of medullary thyroid tumours (lanreotide) and prevention of complications following pancreatic surgery (octreotide),
octreotide is sometimes used to stop bleeding from oesophageal varices (Ch. 36).
Pharmacokinetics: Octreotide is given by subcutaneous injection. It has a short half-life (1–2 h) but suppresses GH secretion for up to 8 h so it is used three times daily. A depot preparation is available in which octreotide is adsorbed onto microspheres; given by deep intramuscular injection it has a duration of action of about 4 weeks. The depot is used once control has been achieved by the use of the conventional formulation. Lanreotide also has a short half-life (1–2 h) and is formulated as a sustained-release preparation given by subcutaneous or intramuscular injection every 1–4 weeks.
Gastrointestinal upset is common, especially anorexia, nausea, vomiting, abdominal pain, bloating and diarrhoea. It usually resolves with continued treatment.
Gallstones, due to suppression of cholecystokinin secretion with decreased gallbladder motility. In addition, an increase in bowel transit time alters colonic flora and makes bile salts more lithogenic.
Growth hormone receptor antagonist:
Mechanism of action: Pegvisomant is a pegylated synthetic analogue of GH that acts as a highly selective GH receptor antagonist.
Dopamine receptor agonists: In healthy people, dopaminergic receptor stimulation increases the secretion of GH, but in acromegaly there is a paradoxical decrease. Bromocriptine was originally used to treat acromegaly, but the clinical response was unpredictable and control of plasma IGF-1 was achieved in only about 20% of cases. It has been superseded by better-tolerated drugs such as cabergoline, which adequately suppress IGF-1 concentrations in about 40% of people with acromegaly. Further details of these drugs can be found in Ch. 24.
Treatment of acromegaly
Surgery by the trans-sphenoidal route is the usual treatment of choice, sometimes followed by external radiotherapy if the tumour is large.
Three groups may be suitable for drug treatment:
those in whom an excess of GH persists despite surgery and radiotherapy; after radiotherapy the plasma GH concentration can take 1–2 years to fall,
SST analogues are the first-line treatment, with pegvisomant used when there is intolerance or failure to respond. Cabergoline is sometimes used with a SST analogue when there is resistance to other treatments. The effectiveness of treatment is monitored by the plasma concentration of IGF-1.
Adrenocorticotropic hormone
Adrenocorticotropic hormone (ACTH; corticotropin) is a single-chain polypeptide with 39 amino acids, of which the 24 that form the N-terminal region are essential for full biological activity. It promotes steroidogenesis in adrenocortical cells by occupying cell surface receptors and stimulating adenylyl cyclase. Release of ACTH occurs in response to the hypothalamic peptide corticotropin-releasing hormone (CRH). CRH secretion is pulsatile and has a diurnal rhythm, with maximal release in the morning around the time of waking (see further detail in Ch. 44). The release of CRH is affected by other factors, including chemical (e.g. antidiuretic hormone, opioid peptides), physical (e.g. heat, cold, injury) and psychological influences. The main inhibitory influence on ACTH release is negative feedback control by circulating glucocorticoids. This occurs at both hypothalamic and pituitary levels. Adrenal androgens, although stimulated by ACTH, play no part in feedback control.
ACTH for therapeutic use
ACTH preparations of animal origin have been replaced by a less allergenic synthetic peptide analogue, tetracosactide, which consists of the active N-terminal amino acids 1–24 of the ACTH molecule.
Pharmacokinetics: There are two formulations of tetracosactide:
a rapid-acting form that increases steroidogenesis for about an hour and is suitable for tests of adrenocortical function. In adrenal insufficiency there is a subnormal or no rise of plasma cortisol 30 min after intramuscular or intravenous injection of tetracosactide,
a depot form that is absorbed slowly into the circulation over several hours and can be used as an alternative to exogenous corticosteroid therapy. However, the unpredictable corticosteroid response means that the therapeutic value of this form is limited.
Once absorbed into the circulation, tetracosactide is metabolised rapidly with a very short half-life (0.2 h).
Unwanted effects: Prolonged use will produce all the features of corticosteroid excess (Ch. 44).
Prolactin
Prolactin is a glycoprotein similar in structure to GH but secreted by distinct cells in the anterior pituitary (Fig. 43.1). The major hypothalamic control mechanism is inhibition by dopamine via D2 receptors on the prolactin-secreting cells of the anterior pituitary (Ch. 45). Thyrotropin-releasing hormone (or TRH) is involved in stimulating prolactin release, and oestrogen increases prolactin production. The main target tissue for prolactin is the breast, which secretes milk in response to prolactin if the mammary glands have been primed by ovarian and other hormones. At delivery, the maternal plasma prolactin concentration is high. Release of further prolactin continues as long as suckling continues. A high plasma concentration of prolactin suppresses follicle-stimulating hormone (FSH) release from the pituitary and leads to a failure of ovarian follicle growth. This may explain the relative subfertility of women who are breastfeeding.
Prolactin has other functions, including producing sexual gratification after intercourse and contributing to maturation of the fetal lung and proliferation of oligodendrocytes that form the neural myelin sheath.
Hyperprolactinaemia
Persistent hyperprolactinaemia is usually caused by a microadenoma of the anterior pituitary or by the action of dopamine receptor antagonist drugs such as phenothiazines (Ch. 21). In younger women hyperprolactinaemia can produce amenorrhoea, infertility and signs and symptoms of oestrogen deficiency (e.g. vaginal dryness and dyspareunia, galactorrhoea and osteoporosis). In men it may cause hypogonadism. Withdrawal of a provoking drug should be considered. For a microadenoma, a dopamine D2 receptor agonist such as cabergoline (Ch. 24) can be used to suppress prolactin secretion. Pituitary surgery may be considered for treatment failure.
Gonadotropin-releasing hormone
Gonadotropin-releasing hormone (GnRH) is a decapeptide that is synthesised in the hypothalamus and is transported by neuronal axons to the pituitary. It is then released in pulses into the capillaries of the pituitary-portal circulation and positively controls the synthesis and release of both luteinizing hormone (LH) and FSH from the anterior pituitary (Fig. 43.1). The cell surface receptors for GnRH are G-protein-linked and are found widely in the body, although their role is poorly understood, as well as on the gonadotropic cells in the anterior pituitary. These receptors are upregulated by repeated stimulation with GnRH, but pulsatile exposure is essential to maintain responsiveness. Low-frequency pulses stimulate FSH release, and high-frequency pulses stimulate LH release. In males, pulse frequency remains constant, whereas in females it varies through the menstrual cycle with a surge just before ovulation (see Ch. 45).
There is rapid tolerance to constant-rate infusions of GnRH because of downregulation of cell surface receptors. Therapeutic administration of GnRH can mimic pulsatile stimulation or produce receptor downregulation, and these have different clinical uses, as described below. There is negative feedback control of GnRH release via neural pathways and sex steroids (Fig. 43.1).
GnRH-related products for therapeutic use
Synthetic GnRH (gonadorelin): Synthetic GnRH is available for assessing hypothalamic-pituitary function and is given as a subcutaneous or intravenous injection. Unwanted effects are unusual, but include nausea, headaches and abdominal pain.
Mechanism of action: Structurally similar to the natural hormone, gonadorelin analogues (Ch. 52) initially stimulate GnRH receptors, but rapidly promote receptor downregulation, which then inhibits further gonadotropin production. The result is reduced production of oestrogen or androgen. This latter action underlies their clinical uses.
Clinical uses of gonadorelin analogues:
The main use is to reduce testosterone secretion to castration levels in men with prostatic cancer. An initial rise in testosterone from receptor stimulation can produce tumour ‘flare’ in the first 1–2 weeks of treatment (Ch. 52). An anti-androgen such as cyproterone acetate (Ch. 46) is usually given to counteract this effect.
Treatment of endometriosis by reducing oestrogen secretion (for up to 6 months only).
Treatment of advanced breast cancer in women, by reducing oestrogen secretion.
To reduce endometrial thickness for 3–4 months prior to intra-uterine surgery.
Preparation of women for assisted conception by methods such as in vitro fertilisation (IVF) (see below).
Suppression of precocious puberty.
‘Hormonal castration’ of males with severe sexual deviation.
Pharmacokinetics: Buserelin can be given by either subcutaneous injection or intranasal spray and has a short half-life (1–1.5 h). Goserelin, which has a half-life of 4 h, must be given by subcutaneous injection and is available as an oily depot preparation. Depot formulations inhibit gonadotropin production for up to 4 weeks after a single injection.
Menopause-like symptoms in women, with hot flushes, sweating, vaginal dryness and loss of libido.
Orchidectomy-like effects in men, with loss of libido, gynaecomastia and vasomotor instability.
Hypersensitivity reactions, including skin rashes, asthma and anaphylaxis.
Osteoporosis with prolonged use.
Local reactions at injection sites, or intranasally with spray.
Mechanism of action and uses: These drugs are competitive GnRH receptor antagonists that produce immediate, reversible suppression of gonadotropin secretion. They are used in assisted reproduction techniques in the management of female infertility (IVF; see below). They have advantages compared with gonadorelin analogues in this role, since there is no initial surge of LH release (which can lead to cancellation of the IVF in about 20% of cycles).
Gonadotropins
LH and FSH are glycoproteins that are released from the anterior pituitary when it is stimulated by pulsatile exposure to GnRH. Negative feedback by inhibin, a hormone produced by the gonads, selectively inhibits FSH secretion. In addition, both gonadotropins are subject to negative feedback from gonadal steroids, including progesterone (Ch. 45).
In males, LH acts on specific receptors on the surface of the Leydig cells in the testes and stimulates adenylyl cyclase, leading to the production of testosterone. FSH acts in a similar way on the Sertoli cells of the seminiferous tubules, stimulating the formation of a specific androgen-binding protein.
In females, receptors for FSH and LH are found in granulosa cells of ovarian follicles. FSH is responsible for follicular development. The rising oestradiol concentration in the late follicular phase has a positive-feedback effect on secretion of LH, and produces a short-lived surge of LH release. This triggers rupture of the follicle, release of the ovum and formation of the corpus luteum (Ch. 45). Both FSH and LH, like human chorionic gonadotropin (HCG), are also produced in large quantities by the placenta during pregnancy.
Gonadotropins for therapeutic use
Human menopausal gonadotropins (HMGs) are FSH and LH (in a 1: 1 ratio, also known as menotrophin) extracted from urine obtained from postmenopausal women.
HCG contains large quantities of LH with little FSH. It is secreted by the placenta and extracted from the urine of pregnant women. An alternative preparation is human choriogonadotropin alfa (recombinant human chorionic gonadotropin).
Follitropin alfa and beta (recombinant human FSH) (Fig. 43.1) and corifollitropin alfa (modified recombinant FSH).
Lutropin alfa (recombinant human LH) (Fig. 43.1).
Gonadotropins are given by intramuscular or subcutaneous injection.
Breast, abdominal and pelvic pain.
In women the most serious problem is ovarian hyperstimulation syndrome, in which the ovaries can become grossly enlarged as a result of multiple follicle stimulation, leading to considerable abdominal pain, ascites and even pleural effusions.
In men the commonest problem is gynaecomastia or oedema with prolonged use.
Clinical uses of gonadotropins
Treatment of infertility in women with hypopituitarism.
Treatment of infertility in women after failure of clomifene treatment (see below).
For superovulation treatment for assisted conception (such as IVF).
In men with hypogonadotropic hypogonadism and oligospermia. This requires long courses of gonadotropin injections, initially to achieve external sexual maturation and then to maintain satisfactory sperm production. Spermatozoa take 70–80 days to mature, and a year or more of treatment may be needed to achieve optimal response. A combination of HMG and HCG is usually given.
Infertility
Infertility is said to be present after 1 year of unprotected intercourse without conception. It has several causes, which need full evaluation of both partners before treatment is given or IVF is considered.
Clomifene
Mechanism of action and use: Clomifene is an agent with both oestrogenic and anti-oestrogenic properties. The latter are related to its ability to block pituitary oestrogen receptors and increase gonadotropin secretion. It is used to stimulate ovulation in anovulatory infertility.
Drug treatment of female infertility
If there is hyperprolactinaemia then a dopamine agonist such as cabergoline should suppress prolactin levels and permit ovulation in 70–80% of women. The management of polycystic ovary syndrome is considered below.
When deficiency of gonadal stimulation by gonadotropin is the limiting factor, FSH (follitropin) can be given with LH (HMG), or in combination as HCG to encourage the development of a single mature ovarian follicle (see Ch. 45). Ovarian hyperstimulation can be a problem.
If the hypothalamic–pituitary axis is normal, the anti-oestrogen clomifene blocks oestrogen receptors in the pituitary, which decreases the negative feedback on FSH (Fig. 43.1), giving increased FSH concentrations that stimulate follicle growth. There is a small risk of ovarian hyperstimulation, and multiple fetuses develop in about 11% of those who become pregnant.
Preparation for assisted conception (in vitro fertilisation)
Ovulation is targeted on a particular date, and initial inhalation of a gonadorelin analogue or use of a GnRH antagonist will ‘switch off’ natural cyclical menstrual activity. Ovarian stimulation treatment is then begun to achieve maturation of oocytes at the time chosen for egg recovery prior to IVF. This involves giving large doses of HCG or HMG to stimulate the maturation of several follicles (superovulation treatment). These ova are then ‘harvested’ by aspiration of the follicles.
Polycystic ovary syndrome
This is a common cause of infertility, affecting 5–10% of women of reproductive age. It is characterised by abnormal ovarian function with hyperandrogenism. Other complaints include menstrual disturbance, hirsutism and acne. Polycystic ovary syndrome is often associated with obesity and insulin resistance in adipose and muscle tissue, conferring an increased risk of diabetes mellitus and cardiovascular disease in later life. Increased insulin secretion is also a factor in stimulating ovarian androgen production. Assessment and treatment of cardiovascular risk is an important component of management.
Weight loss (especially if the body mass index is greater than 29 kg⋅m−2) may restore ovulatory cycles if infertility is the main concern. Metformin (Ch. 40) can be used if this is inadequate, and the resulting improvement in insulin sensitivity reduces androgen concentrations. This leads to weight loss, reduction of the consequences of hyperandrogenisation, and improved fertility. Clomifene can be added if fertility is not restored. Because of the absence of good safety data metformin is usually stopped during pregnancy.
Alternative approaches to treatment include a combined oral hormonal contraceptive for management of amenorrhoea or oligomenorrhoea (Ch. 45) or anti-androgen therapy with cyproterone acetate (often used in combination with an oestrogen) for hirsutism (Ch. 46). Topical therapies are available for managing hirsutism associated with polycystic ovary syndrome. These include minoxidil cream (Ch. 6) to reverse male-pattern hair loss, and eflornithine cream (an ornithine decarboxylase inhibitor) to slow facial hair growth by inhibiting cell division in hair follicles.
Posterior pituitary hormones
Vasopressin (antidiuretic hormone)
Vasopressin is a nonapeptide, sometimes referred to as arginine-vasopressin (AVP) because human vasopressin has an arginine residue in position 8. It is also known as antidiuretic hormone (ADH). Vasopressin is released from neurosecretory cells of the hypothalamus and transported down the axons of the nerve cells that form the pituitary stalk. It is stored in the nerve endings in the posterior pituitary and released in response to stimulation of the hypothalamus via osmoreceptors, sodium receptors and volume receptors in response to reduced plasma volume or increased plasma osmolality. Vasopressin has two main target tissues.
Stimulation of vascular smooth muscle via V1 receptors leads to Ca2+ influx and vasoconstriction. Vasoconstriction sufficient to raise blood pressure only occurs at high plasma vasopressin concentrations.
At the collecting ducts of the kidney nephron, stimulation of V2 receptors increases intracellular cAMP production which leads to expression of aquaporin-2 channels that allow water reabsorption down an osmotic gradient to produce more concentrated urine. Expression of urea transporters in the collecting duct is also enhanced with consequent urea reabsorption from the renal filtrate.
Vasopressin is metabolised in many tissues, including the liver and kidney, and has a very short half-life of about 10 min. It is given therapeutically by subcutaneous or intramuscular injection or by intravenous infusion.
Vasopressin analogues
Vasopressin has a short duration of action. Desmopressin (DDAVP, des-amino-D-arginine-vasopressin) has an increased antidiuretic potency and reduced pressor activity compared with vasopressin. It is absorbed through the nasal mucosa and is most conveniently administered by a metered-dose nasal spray or sublingually. It can also be given by subcutaneous, intramuscular or intravenous injection. An additional action of parenteral desmopressin is to increase clotting factor VIII concentration in blood (Ch. 11).
Terlipressin is also a vasopressin analogue that is used to treat bleeding oesophageal varices. It is discussed in Chapter 36.
Clinical uses of vasopressin and its analogues
Vasopressin can be given acutely for treatment of cranial diabetes insipidus (see below), using the longer-acting derivative desmopressin for maintenance treatment.
Desmopressin can be given sublingually for primary nocturnal enuresis.
Terlipressin will control bleeding from oesophageal varices in portal hypertension (Ch. 36). Vasopressin is sometimes used for this indication.
Desmopressin by injection is used to boost factor VIII concentration and reduce bleeding in mild to moderate haemophilia.
Desmopressin can be given to test for urine-concentrating ability in suspected diabetes insipidus (see below).
Vasopressin is given for its pressor activity in the treatment of shock associated with hypotension, when it also increases vascular sensitivity to noradrenaline.
Diabetes insipidus
Diabetes insipidus is usually caused by a failure of secretion of vasopressin in the hypothalamus (‘cranial’ diabetes insipidus). Tumours, inflammatory conditions, granulomatous conditions such as sarcoidosis, and trauma to the hypothalamus are the main causes. A distinct condition known as nephrogenic diabetes insipidus occurs when the kidney is unresponsive to vasopressin. It can result from a hereditary deficiency of renal vasopressin receptors, or from drug therapy, particularly with lithium (Ch. 22) or the tetracycline demeclocycline. Diabetes insipidus presents clinically with thirst, polyuria and a tendency to high plasma osmolality together with an inappropriately low urine osmolality.
Vasopressin produces concentrated urine in people with cranial diabetes insipidus; desmopressin is used for long-term treatment. Treatment of nephrogenic diabetes insipidus is more difficult, because the kidney does not respond to vasopressin. Paradoxically, thiazide diuretics (Ch. 14) can reduce the polyuria. This may be due to initial contraction in extracellular volume, with subsequent increase in proximal tubular salt and water retention. Carbamazepine (Ch. 23) is also effective, by sensitising the renal tubule to the effect of vasopressin.
Syndrome of inappropriate antidiuresis
This is a condition caused by inappropriately high secretion of vasopressin, resulting in excess water retention and a dilutional hyponatraemia. There are many causes, including malignant tumours that secrete vasopressin, pulmonary disorders (including infection) and various disorders of the central nervous system. Drugs such as antidepressants (Ch. 22), carbamazepine (Ch. 23) and various cytotoxic agents can also produce the syndrome.
Vasopressin V2 receptor antagonist:
Mechanism of action and uses: Tolvaptan is a competitive antagonist at vasopressin V2 receptors. Its major action is in the renal collecting ducts to reduce water reabsorption and produce aquaresis without sodium loss, thus increasing free water clearance, and correcting dilutional hyponatraemia. In the UK, tolvaptan is currently licensed for the treatment of hyponatraemia secondary to inappropriate ADH secretion. However, it is also effective for correction of hyponatraemia in cirrhosis and in heart failure when it arises from diuretic use. Tolvaptan is not suitable for urgent treatment of severe hyponatraemia, when there is a risk of significant neurological complications. When initiating treatment, it is important to monitor the rise in serum sodium to avoid rapid correction and precipitation of osmotic demyelination syndrome.
Treatment of syndrome of inappropriate antidiuresis: Severe hyponatraemia can cause confusion, seizures and coma. Slow correction of the serum Na+ concentration with intravenous saline 0.9% is the mainstay of treatment. Rapid correction can disturb the osmotic balance across neurons and cause irreversible damage known as central pontine myelinolysis, which produces dysarthria, spastic quadriparesis and pseudobulbar palsy.
Less severe hyponatraemia may respond to fluid restriction. Demeclocycline, a tetracycline antimicrobial agent that reduces the sensitivity of the collecting ducts to vasopressin, can also be used. The vasopressin V2 receptor antagonist tolvaptan is sometimes used, but not for urgent treatment due to the risk of neurological complications.
Hypopituitarism
Pituitary insufficiency can arise from traumatic brain injury and subarachnoid haemorrhage, when a single hormonal axis is often affected. Pituitary irradiation or surgery, by contrast, often affects multiple axes. Less common causes include both pituitary and non-pituitary intracranial tumours, and ischaemic damage.
Replacement of a deficiency of glucocorticoid (Ch. 44) and thyroid (Ch. 41) hormonal function is essential and urgent. Female sex hormone (Ch. 45) or testosterone (Ch. 46) replacement can be important at a later stage to restore libido and bone mass. In some individuals deficiency of GH or vasopressin may also require replacement.
True/false questions
1. The release of growth hormone (GH, somatotropin) is reduced by somatostatin.
2. Somatostatin is only produced by the hypothalamus.
3. Octreotide is a useful drug for the treatment of acromegaly.
4. Pegmisovant is a pegylated GH analogue that stimulates GH receptors.
5. Prolactin suppresses ovarian steroidogenesis.
6. Cabergoline reduces prolactin secretion.
7. Continuous administration of gonadorelin analogues stimulates sex steroid synthesis.
8. The gonadorelin analogue buserelin stimulates gonadotropin-releasing hormone (GnRH) receptors and is used for the treatment of endometriosis, but has no clinical use in males.
9. Cetrorelix and ganirelix are GnRH receptor antagonists used in female infertility.
10. Follitropin alfa is a recombinant follicle-stimulating hormone (FSH) analogue used in preparation for in vitro fertilisation.
11. The production of vasopressin is impaired in nephrogenic diabetes insipidus.
12. Vasopressin increases expression of aquaporin channels.
13. Desmopressin is administered by intranasal spray in cranial diabetes insipidus.
14. Tolvaptan is used in the urgent treatment of severe hyponatraemia.
15. In nephrogenic diabetes insipidus, thiazide diuretics increase the polyuria.
1. True. Somatostatin is also known as growth hormone release-inhibiting hormone (GHRIH).
2. False. Somatostatin is also produced from intestinal and pancreatic cells.
3. True. Octreotide is a long-acting analogue of somatostatin and therefore inhibits GH release.
4. False. Pegvisomant is a pegylated GH analogue but it selectively blocks GH receptors; it is used when somatostatin analogues such as octreotide are poorly tolerated or ineffective.
5. True. Prolactin inhibits FSH release and suppresses ovarian follicle growth and steroidogenesis.
6. True. Cabergoline is a dopamine receptor agonist and inhibits prolactin release from the anterior pituitary; it can be used to improve fertility in women with hyperprolactinaemia.
7. False. Although brief administration of gonadorelin analogues stimulates sex steroid synthesis, on continued administration gonadotropin receptors are rapidly downregulated and sex steroid synthesis declines.
8. False. Gonadorelin analogues downregulate GnRH receptors and are used to inhibit testosterone synthesis in prostate cancer, as well as reducing synthesis of ovarian hormones in endometriosis.
9. True. GnRH receptor antagonists produce immediate suppression of gonadotropin secretion without the initial surge in LH caused by gonadorelin analogues.
10. True. Follitropin alfa is a synthetic FSH analogue and promotes follicle growth.
11. False. In nephrogenic diabetes insipidus the kidney is unresponsive to vasopressin; vasopressin secretion is impaired in cranial diabetes insipidus.
12. True. Vasopressin (antidiuretic hormone) acting at V2 receptors enhances water reabsorption by increasing aquaporin channels in the renal collecting duct.
13. True. Desmopressin is a modified vasopressin with a longer duration of action and selectivity for V2 receptors in the kidney.
14. False. The vasopressin V2 antagonist tolvaptan is licensed for maintenance treatment of hyponatraemia caused by excessive ADH secretion, but if used for acute treatment of severe hyponatraemia it can cause neurological complications.
15. False. Paradoxically, in diabetes insipidus, the response to thiazide diuretics is a beneficial reduction in polyuria.
OBA answer
Answer A is the inaccurate statement.
A Incorrect. GH is released in a pulsatile manner and is higher during deep sleep, particularly in children.
B Correct. GH stimulates IGF-1 release from the liver, which then acts on receptors in many tissues and in concert with other hormones.
C Correct. In normal individuals dopamine receptor agonists such as cabergoline stimulate GH release, but in acromegaly they paradoxically inhibit GH release.
D Correct. IGF-1 inhibits GH release and also stimulates somatostatin release from the hypothalamus, which further inhibits GH release.
Case-based answers
A Epiphyseal closure occurs much later than 10 years of age, so treatment of this girl with short stature can increase her growth.
B Low levels of GHRH and GH suggest pituitary dwarfism and therefore synthetic GH (somatropin) would be appropriate. Although somatropin has a half-life of only 25 min, it generates IGF-1 which is highly protein-bound, so three subcutaneous injections of somatropin a week are sufficient to maintain IGF-1 levels.
C Transient insulin-like effects of somatropin can produce hypoglycaemia and there may be pain at the site of injection. Headache and oedema can also occur.
Compendium: pituitary and hypothalamic hormones
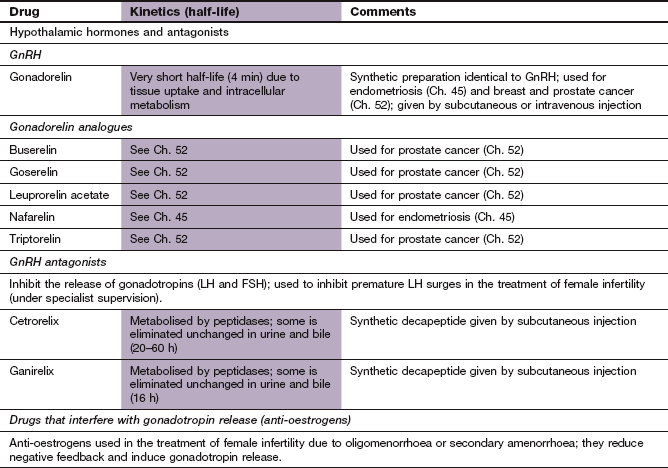
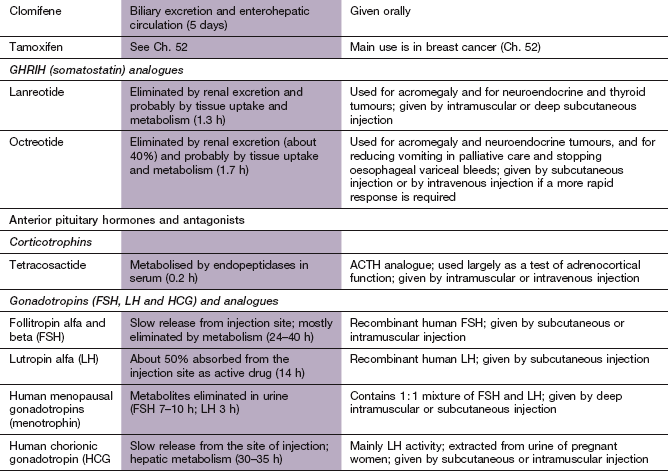
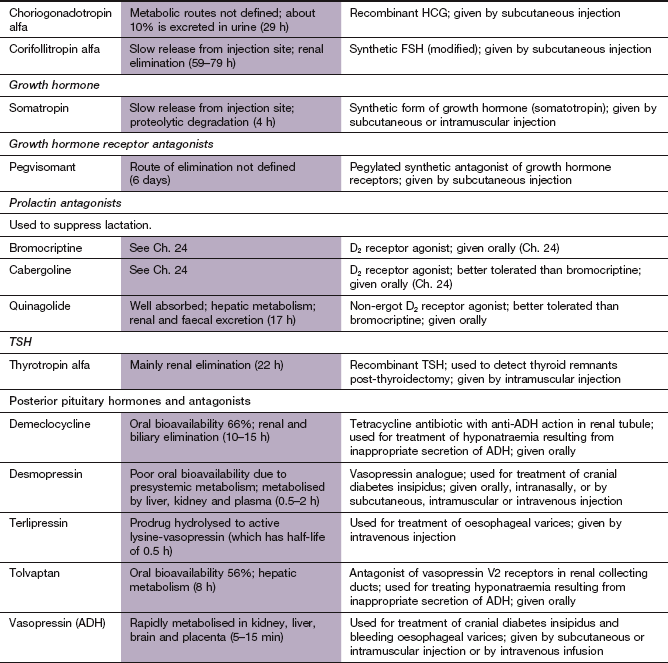
ACTH, adrenocorticotropic hormone; ADH, antidiuretic hormone; FSH, follicle-stimulating hormone; GHRIH, growth hormone-release inhibiting hormone; GnRH, gonadotropin-releasing hormone; HCG, human chorionic gonadotropin; LH, luteinizing hormone; TSH, thyroid-stimulating hormone.
Balen, AH, Rutherford, AJ. Management of infertility. BMJ. 2007;335:608–611.
Balen, AH, Rutherford, AJ. Managing anovulatory infertility and polycystic ovary syndrome. BMJ. 2007;335:663–666.
Danzig, J. Acromegaly. BMJ. 2007;335:824–825.
Dattani, M, Preece, M. Growth hormone deficiency and related disorders: insights into causation, diagnosis, and treatment. Lancet. 2004;363:1977–1987.
Ehrmann, DA. Polycystic ovary syndrome. N Engl J Med. 2005;352:1223–1236.
Ellison, DH, Beri, T. The syndrome of inappropriate antidiuresis. N Engl J Med. 2007;356:2064–2072.
Khanna, A. Acquired nephrogenic diabetes insipidus. Semin Nephrol. 2006;26:244–248.
Melmed, S. Acromegaly. N Engl J Med. 2006;355:2558–2573.
Norman, RJ, Dewailly, D, Legro, RS, et al. Polycystic ovary syndrome. Lancet. 2007;370:685–697.
Olive, DL. Gonadotropin–releasing hormone agonists for endometriosis. N Engl J Med. 2008;359:1136–1142.
Sands, JM, Bichet, DG. Nephrogenic diabetes insipidus. Ann Intern Med. 2006;144:186–194.
Schneider, HJ, Almaretti, G, Kreitschmann-Andermahr, I, et al. Hypopituitarism. Lancet. 2007;369:1461–1470.
Setji, TJ, Brown, AJ. Polycystic ovary syndrome: diagnosis and treatment. Am J Med. 2007;120:128–132.
Van Voorhis, BJ. In vitro fertilization. N Engl J Med. 2007;356:379–388.